

Pseudomonas aeruginosa is an opportunistic bacterial pathogen capable of causing chronic lung infections. Phenotypes important for the long-term persistence and adaption to this unique lung ecosystem are largely regulated by the AlgT sigma factor. Chronic infection isolates often contain mutations in the anti-sigma factor mucA, resulting in uncontrolled AlgT and continuous production of alginate in addition to the expression of ∼300 additional genes. Here, we report that in the absence of wild-type MucA, AlgT overproduction is lethal and that suppressors of toxic AlgT production have mutations in the MucA protease, MucP. Since AlgT contributes to the establishment of chronic infections, understanding how AlgT is regulated will provide vital information on how P. aeruginosa is capable of causing long-term infections.
KEYWORDS: Pseudomonas, gene regulation, lethality, proteolysis, sigma factor
ABSTRACT
Pseudomonas aeruginosa isolates from chronic lung infections often overproduce alginate, giving rise to the mucoid phenotype. Isolation of mucoid strains from chronic lung infections correlates with a poor patient outcome. The most common mutation that causes the mucoid phenotype is called mucA22 and results in a truncated form of the anti-sigma factor MucA that is continuously subjected to proteolysis. When a functional MucA is absent, the cognate sigma factor, AlgT, is no longer sequestered and continuously transcribes the alginate biosynthesis operon, leading to alginate overproduction. In this work, we report that in the absence of wild-type MucA, providing exogenous AlgT is toxic. This is intriguing, since mucoid strains endogenously possess high levels of AlgT. Furthermore, we show that suppressors of toxic AlgT production have mutations in mucP, a protease involved in MucA degradation, and provide the first atomistic model of MucP. Based on our findings, we speculate that mutations in mucP stabilize the truncated form of MucA22, rendering it functional and therefore able to reduce toxicity by properly sequestering AlgT.
IMPORTANCE Pseudomonas aeruginosa is an opportunistic bacterial pathogen capable of causing chronic lung infections. Phenotypes important for the long-term persistence and adaption to this unique lung ecosystem are largely regulated by the AlgT sigma factor. Chronic infection isolates often contain mutations in the anti-sigma factor mucA, resulting in uncontrolled AlgT and continuous production of alginate in addition to the expression of ∼300 additional genes. Here, we report that in the absence of wild-type MucA, AlgT overproduction is lethal and that suppressors of toxic AlgT production have mutations in the MucA protease, MucP. Since AlgT contributes to the establishment of chronic infections, understanding how AlgT is regulated will provide vital information on how P. aeruginosa is capable of causing long-term infections.
INTRODUCTION
Alginate production by the opportunistic pathogen Pseudomonas aeruginosa results in a mucoid phenotype and correlates with the establishment of a chronic infection (1–6). Production of alginate is highly regulated and is usually a response to external and internal membrane stressors (7–9). In nonmucoid strains, the anti-sigma factor MucA is inserted into the inner membrane where the cytosolic N terminus interacts with the AlgT sigma factor (also called σ22 or AlgU) (10). AlgT is the master regulator of alginate biosynthesis genes as well as approximately 300 other genes controlling phenotypes, including pigment production, motility, and very long O antigen production (11–16). When AlgT is sequestered by MucA, the alginate biosynthesis operon is not transcribed and these strains produce little to no alginate.
AlgT is made available and alginate biosynthesis is triggered by a regulated intermembrane proteolysis (RIP) cascade that is activated by proteases (17). Briefly, activated AlgW cleaves the periplasmic C terminus of MucA, making the truncated inner membrane portion of MucA available for cleavage by MucP (18, 19). This releases the cytosolic N terminus AlgT-bound fragment of MucA where it is finally completely degraded by the ClpPX proteasome. Once free of MucA, AlgT initiates transcription of its regulon (8, 20–22). The gene encoding AlgT itself is the first in an operon followed by mucA, mucB, mucC, and mucD. This operon is highly similar to the rpoE, rseA, rseB, and rseC operon in Escherichia coli (17, 23). AlgT and RpoE (also called σE) are 66% similar and functionally interchangeable, with RpoE capable of complementing algT null mutants of P. aeruginosa (24). The upstream region of algT contains five promoters, with two of these promoters themselves being AlgT dependent (25, 26); therefore, algT is positively autoregulated.
While nonmucoid strains are capable of producing and regulating alginate in certain environments, P. aeruginosa isolates from cystic fibrosis lung infections often overproduce alginate, termed the mucoid phenotype (1, 2, 27–29). The most common mutations that result in the mucoid phenotype are found in mucA (1, 30). The mucA22 mutation, which is a deletion of a G in a string of 5 Gs, is the most common mutation observed in clinical isolates and results in a form of MucA with a truncated C-terminal domain (31). As a result, MucA22 is continuously subjected to RIP, AlgT is always free, and the alginate biosynthesis operon is always expressed (32, 33). Since AlgT promotes transcription of itself, mucoid strains have high levels of AlgT.
Despite the many discoveries surrounding the regulation of AlgT in nonmucoid strains, it is still unclear if mucoid strains are capable of regulating AlgT in the absence of wild-type MucA. Therefore, we sought to primarily study AlgT in the context of mucoid strain PDO300, a derivative of the nonmucoid laboratory strain PAO1 that contains the mucA22 allele (31). However, we report here that the expression of exogenous algT is lethal in PDO300. Furthermore, we show that suppressors of toxic AlgT production have mutations in mucP, encoding a protease involved in MucA degradation. Our findings support a model where mutations in mucP stabilize the MucA22-AlgT complex, ultimately rendering MucA22 functional. Since it has been reported that a complete deletion of mucA is lethal (34), we propose that deletion of mucA is likely lethal due to deregulation and overproduction of AlgT.
RESULTS AND DISCUSSION
Overexpression of algT is lethal in mucA22 strains.
To study the expression of AlgT in mucoid P. aeruginosa, we began by making an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible copy of algT (Ptac-algT) and inserted this construct in single copy at the attTn7 site in PDO300. However, we found it difficult to grow PDO300 in the presence of inducer. Serial dilutions of PDO300 on IPTG revealed that increasing ectopic expression of algT reduced growth of PDO300 compared to growth without inducer (Fig. 1). Furthermore, high levels of algT expression were entirely lethal (Fig. 1). To determine if alginate production impacts this lethality, we monitored growth of PDO300 ΔalgD, which has a clean deletion of the first gene in the alginate biosynthesis operon and does not produce alginate. At low concentrations of IPTG and therefore low levels of algT, growth of PDO300 ΔalgD containing Ptac-algT was only partially inhibited. However, when plated on high levels of inducer in which algT expression was higher, no growth was observed (Fig. 1). These results indicate that alginate modestly impacts the toxicity caused by the overexpression of algT. We also monitored growth of PDO300 and PDO300 ΔalgD containing Ptac-algT over time in liquid broth culture; without inducer, both strains grew to high density (see Fig. S1 in the supplemental material). Conversely, when grown in the presence of inducer, both strains had decreased growth rates compared to those under uninduced conditions (Fig. S1), corroborating what we observed by serial plate dilutions (Fig. 1). In all scenarios, the algD mutant grew slightly better than the wild-type strain. Ultimately, both PDO300 and PDO300 ΔalgD reached the same final density, likely due to the selection of mutants that can suppress toxic AlgT production. Altogether, these data suggest that in mucA22 strains, such as PDO300, tightly controlled expression of algT is essential for growth.
FIG 1.

Overproduction of algT is lethal in mucA22 strains and is partially rescued when wild-type mucA is provided in trans. The algT coding sequence was cloned downstream of an IPTG-inducible tac promoter and inserted, in single copy, at the attTn7 site of each strain (Tn7::Ptac-algT). Overnight cultures were grown without inducer, normalized to the same optical density, and then serially diluted onto LA containing no inducer, 0.1 mM IPTG, or 1 mM IPTG and/or 1% arabinose. IPTG induces expression of algT, while arabinose induces expression of mucA. Strains were serially diluted onto increasing concentrations of IPTG to induce algT expression. Overexpression of algT inhibited growth. Each strain on the right also contains an arabinose-inducible copy of mucA on multicopy plasmid pHERD20T (pHERD20T-mucA). Corresponding dilution factors are shown on top. The strains shown are PAC539, PAC543, PAC559, and PAC561.
Complementation with wild-type MucA partially rescues algT lethality.
We hypothesized that algT lethality is due to the fact that PDO300 contains a mucA mutation. To investigate this, we monitored growth of PAO1 and PAO1 ΔalgD (35, 36). PAO1 is the nonmucoid parent strain of PDO300 and contains wild-type mucA. When grown on lysogeny agar (LA), PAO1 is nonmucoid, since algT expression is low. When the IPTG-inducible copy of algT was inserted into both strains and these strains were grown on increasing concentrations of IPTG to induce algT expression, PAO1 became mucoid, as expected (37), while PAO1 ΔalgD remained nonmucoid (see Fig. S2). Both PAO1 and PAO1 ΔalgD grew equally well when algT expression was induced, indicating that in the presence of wild-type MucA, algT overexpression and accumulation are not lethal (Fig. S2).
If wild-type MucA is required to circumvent AlgT lethality, we hypothesized that expression of wild-type mucA, in trans, would rescue PDO300 from the toxic effects of algT overexpression. To test this, we expressed mucA from an arabinose-inducible plasmid and transferred this plasmid to the PDO300 and PDO300 ΔalgD strains containing the IPTG-inducible promoter driving algT. On low levels of IPTG (to slightly express algT) and on high levels of arabinose (to overexpress mucA), growth of PDO300 and PDO300 ΔalgD was no longer inhibited by the ectopic expression of algT (Fig. 1). Overexpression of mucA also rendered PDO300 nonmucoid, indicating that AlgT is being sequestered by MucA. Still, on higher levels of IPTG (1 mM), providing wild-type mucA in trans failed to rescue growth of either PDO300 or PDO300 ΔalgD (Fig. 1). This is likely because not enough MucA was produced, even at this high level of induction, to sufficiently sequester AlgT to rescue these strains. In PAO1, the expression of mucA is positively autoregulated by algT; therefore, expression is equally very high when algT is overexpressed. This is unlike that in PDO300, where the chromosomal copy of mucA is mutated to mucA22, which is very unstable. Therefore, the autoregulation of mucA is lost, which is very important for maintaining high levels of MucA when algT expression is induced.
Identification of mutations that inactivate AlgT.
Over the course of our experiments, we observed some PDO300 and PDO300 ΔalgD colonies that were able to grow when algT was overexpressed. We isolated 8 suppressors (4 from PDO300 and 4 from PDO300 ΔalgD) for further analysis. These strains grew on high levels of inducer and therefore were able to avoid the lethality caused by overexpression of algT. The most likely way to suppress algT toxicity would be to acquire mutations in one of the two copies of algT. To determine if this was the case, we sequenced both the native and inducible copies of algT and found that 2 of the 8 suppressors had mutations in the cloned copy of algT. One had acquired a 9-bp duplication of nucleotides 136 to 144 (GAA GCC CAG), while the other had a C-to-T transition at nucleotide 400 that would result in a stop codon.
It has been observed that mucoid strains frequently revert to nonmucoid by acquiring mutations in algT (19, 25, 38). To determine what other mutations we could identify in algT that result in nonmucoid reversion, we grew mucoid PDO300 under low aeration, a stressful condition that selects for reversion (19). We isolated 18 PDO300 nonmucoid revertants and then sequenced algT to determine what proportion had acquired mutations in this gene. We found that 10 of the 18 (56%) nonmucoid revertants had mutations in algT. These data are similar to previous studies that estimated 41% to 83% of nonmucoid revertants contain algT mutations (14, 38, 39). Of interest to us, we found that the same 9-bp nucleotide insertion as described above had occurred in 6 of our nonmucoid revertants. This insertion represented a duplication of nucleotides 127 to 135 (corresponding to amino acids DAQ) or nucleotides 136 to 144 (corresponding to amino acids EAQ), the insertion representing amino acids 43 to 45 or 46 to 48 (see Fig. S3). Two studies have described a 9-bp insertion in this region, but whether this was a similar duplication was not indicated, as the sequence of the insertion was not defined (27, 39).
How the duplication we observed affects AlgT is unclear, since the insertion of 3 amino acids results in an in-frame mutation. To begin to assess how these mutations might affect function, we constructed a model of AlgT in a DNA-bound form (Fig. 2A and B and S4) using a recent cryo-electron microscopy (cryo-EM) structure of the E. coli transcription initiation complex with RpoE (40). This model shows that domain I of AlgT comprising ∼1 to 100 residues interacts with the −10 element of the promoter and plays a crucial role in promoter melting. AlgT domain II binds to −35 element of the promoter. We also note that AlgT binds to MucA in a more compact structure (PBD 6IN7), wherein domain I and II come closer together (Fig. S4, only AlgT conformation shown). The flexibility of the 25-residue linker between the two domains might play a critical role in its promoter DNA binding. To determine how the 3-amino-acid insertion alters AlgT domain I binding to DNA, we mapped the location of the DAQ and EAQ duplications to a highly conserved loop region in the AlgT model (Fig. 2C). To structurally interpret the effect of this duplication, we modeled the DAQ duplication in the loop region (Fig. 2D). The duplication distorts the −10 promoter binding region, and we also speculate that adding an additional negatively charged residue (Asp or Glu) further alters the binding strength between the loop and the crucial promoter melting region, thus changing AlgT function and introducing a possible steric clash. Therefore, in this configuration, AlgT would be unable to efficiently promote transcription, consistent with the nonmucoid phenotype and loss of alginate production observed in these mutants. In E. coli, suppressors of high RpoE levels contain mutations in rpoE, but these are mostly near the −35 promoter-binding region and are proposed to weaken binding to promoters but still support some transcription initiation (41), which is not what we observed for this P. aeruginosa mutant.
FIG 2.

Model of AlgT bound to promoter DNA and the effect of the 3-amino-acid insertion. (A) Linear schematic of AlgT structure highlighting domain I and domain II, connected by a linker (gray). The loop insertion in the AlgT domain I occurs between D45 and E48 (triangle). (B) Homology model of AlgT bound to promoter DNA modeled from RpoE sigma factor bound to DNA (from PDB 6JBQ). (C) Residues in the DNA-binding loop in domain I (D43 to Q48, shown as sticks) interact with DNA. (D) The DAQ insertion (green) increases the size of the loop, thus disrupting the optimal distance between the loop and the DNA, introducing steric clash.
Suppressor mutations in mucP rescue AlgT-mediated toxicity.
We performed whole-genome sequencing to determine what mutations in the remaining 6 suppressor mutants were acquired that allowed for survival when algT was overexpressed. We identified a number of alginate gene mutations that were unique to PDO300 ΔalgD compared to PDO300. These were located upstream of algD in the promoter and in alg8, which is located just downstream of algD. Although there was only a modest growth difference between PDO300 and PDO300 ΔalgD when algT was overexpressed (Fig. 1), these PDO300 ΔalgD-specific suppressor mutations in or near the alginate operon further suggest a potential role for alginate production in promoting algT toxicity. We also identified a mutation in PA4059, a hypothetical protein, in all of the suppressors and a synonymous mutation in purA in one of the suppressors. It is not clear how mutations in these genes would suppress algT lethality, and we did not pursue them further.
Of most interest were the single nucleotide polymorphisms (SNPs) found in the MucA protease encoded by mucP in 3 of the 6 suppressors (Table S1). MucP is a zinc metalloprotease that participates in the proteolytic cascade that degrades MucA (18, 19, 42). Delgado et al. analyzed the MucP sequence and found four possible transmembrane domains, one beta-loop domain, a metalloprotease zinc-binding motif, two PDZ binding domains, and a RIP motif (19). Many previous studies have predicted the secondary structure of the membrane-bound region of MucP but failed to provide a three-dimensional atomistic model. The membrane-bound region of MucP does not share overall sequence similarity to any protein for which a structure has been determined, and so our initial trials for generating homology modeling or threading using the Swiss-Model, I-TASSER, and LOMETS (43–45) servers were unsuccessful. Next, we attempted ab initio structure determination using QUARK (46, 47), which again failed to produce a model with a conserved zinc-binding domain. We next searched for distant homologs of MucP using InterPro database (IPR008915; www.ebi.ac.uk/interpro), and the resulting sequences were further sorted based on the zinc-binding HXXXH and DGGH motifs using the PROSITE database (https://prosite.expasy.org). Among these sequences, we found one homologous protease, from Methanocaldococcus jannaschii, for which the membrane bound structure was determined (PDB 3B4R). We used this model to generate a de novo MucP membrane-bound structural model using helical constraints and active site geometry constraints from the HXXXH and DGGH motifs (see Fig. S5A). As structural motifs are more conserved than sequences for M50 family proteases, we expected MucP to fold in a locally similar manner with the HXXXH and DGGH motifs, which coordinate a catalytically critical zinc ion, coming within a 5-Å distance. As structure determination of membrane proteins is difficult and the membrane region of MucP does not share any significant sequence similarity with known homologs for which structure is known, we used a combination of evolutionary and structural constraints and the partial template from M. jannaschii peptidase to generate the first atomistic model of Pseudomonas MucP.
The periplasmic region is composed of two PDZ domains that are connected by linkers to the membrane-bound N-terminal and C-terminal domains of MucP (Fig. S5B). The membrane-bound structure illustrates that any deletion, even in the C-terminal domain, would render the protein inactive, as the complete catalytic site needs the 21HXXXH25 motif from the N terminus and the RIP motif 403DGGH406 from the C terminus. Mutations destabilizing the helices of MucP would also destabilize the fold and would inactivate the protein.
One mucP suppressor mutant, found in PDO300, had an insertion of a “C” after nucleotide 358 (PDO300 mucP358C_ins), leading to early truncation of the protein after 122 amino acids (Fig. 3). This truncates the protein before the first PDZ domain. The second mucP suppressor mutant was in PDO300 ΔalgD, which had a deletion of nucleotides 910 to 916 (PDO300 ΔalgD mucPΔ910–916), GCG GGG G, that also led to early truncation of the protein after 316 amino acids (Fig. 3). Our model shows that this would truncate the protein just before the C-terminal membrane-bound domain. Delgado et al. found the same 7-nucleotide deletion in MucP when looking for mutations that cause nonmucoid reversion (19). This mutation also would make the peptidase function of MucP inactive due to loss of the 403DGGH406 RIP motif. The third mucP suppressor, also found in PDO300 ΔalgD, had an in-frame deletion of nucleotides 1022 to 1036, CGC TCG ACT CCA TAA (PDO300 ΔalgD mucPΔ1022–1036), resulting in a protein that is only 445 instead of 450 amino acids long (Fig. 3). This would again disrupt the catalytic site of MucP, making it inactive for its peptidase function. Overall, we predict that each of these mutations interferes with the ability of MucP to degrade MucA22.
FIG 3.

Suppressor mutations in mucP rescue AlgT-mediated toxicity. Depiction of the primary structure of wild-type MucP and suppressors (top). PDO300 mucP358C_ins has an insertion of a “C” after nucleotide 355, leading to truncation of the protein after 122 amino acids. PDO300 ΔalgD mucPΔ910–916 suppressor has a deletion of nucleotides 906 to 912 (GGG GGC G), leading to truncation of the protein after 316 amino acids. PDO300 ΔalgD mucPΔ1022–1036 has an in-frame deletion of nucleotides 1019 to 1033 (TAA CGC TCG ACT CCA), resulting in a 445-amino-acid protein. The location of each mutation is shown using a homology model of MucP (bottom). MucP, depicted in the inner membrane (IM), contains 4 transmembrane helices and a periplasmic region composed of two PDZ domains that are connected by linkers to the N-terminal (NTD) and C-terminal (CTD) membrane-bound domains of MucP. The strains shown are PAC577, PAC579, and PAC582.
Complementation of mucP suppressor mutants restores sensitivity to algT overexpression.
Multiple attempts at constructing clean deletions of mucP in PAO1 and PDO300 were unsuccessful; therefore, we could not test the hypothesis that deletion of mucP from PDO300 or PDO300 ΔalgD allows for growth when algT is overexpressed. In E. coli, the mucP homolog rseP is essential, because the absence of RseP leads to loss of RpoE activity and the accumulation of unfolded outer membrane proteins (48, 49). In P. aeruginosa, however, algT does not appear to be essential under standard conditions, as algT deletion strains have been reported (23, 50). If mucP is essential in P. aeruginosa and algT is not, this implies that MucP may have targets other than MucA-AlgT that are critical for survival.
Since we could not delete mucP, we chose to overexpress mucP in the suppressor mutants PDO300 mucP358C_ins and PDO300 ΔalgD mucPΔ910–916. We hypothesized that providing wild-type MucP in trans would render the suppressors sensitive to the overproduction of AlgT. To test this, we cloned the mucP gene on a multicopy arabinose-inducible plasmid (pHERD20T-mucP) and inserted this into both suppressor mutants. First, we confirmed that overexpression of algT in the presence of this plasmid was not lethal to the PDO300 and PDO300 ΔalgD suppressors (Fig. 4). Likewise, overexpression of mucP by itself also did not inhibit growth of either strain. When algT and mucP were both induced, however, growth of both suppressor mutants was inhibited (Fig. 4). In summary, the suppressors were unable to grow due to complementation with wild-type mucP, confirming that mutations in mucP are a mechanism to circumvent toxicity due to AlgT overexpression.
FIG 4.

Complementation of mucP suppressor mutants restores sensitivity to algT overexpression. Strains were grown as described for Fig. 1. Each strain contains an IPTG-inducible algT at the attTn7 site (Tn7::Ptac-algT) and an inducible copy of mucP on multicopy plasmid pHERD20T (pHERD20T-mucP). IPTG induces expression of algT, while arabinose induces expression of mucP. Corresponding dilution factors are shown on top. Growth was inhibited only when both algT and mucP were overexpressed. The strains shown are PAC667 and PAC678.
Inactivation of MucP reduces expression of algT.
We postulated that mutations in mucP ultimately reduce toxic AlgT levels, likely due to stabilization the MucA22-AlgT complex. If this is the case, we expect that expression of genes controlled by AlgT would be reduced in mucP mutants. Since algT is autoregulated, we used an algT reporter as a readout of AlgT activity and production. To do this, the algT promoter region was transcriptionally fused to a promoterless optRBS-lacZ and then inserted in single copy at the attCTX site in each of the strains. In mucoid PDO300, algT promoter activity was ∼2.5 times higher than in nonmucoid PAO1, which contains wild-type MucA (Fig. 5). When algT promoter activity was measured in the mucP suppressor mutant PDO300 mucP358C_ins, algT promoter activity was significantly decreased and more comparable to PAO1 levels (Fig. 5). We complemented mucP back in trans under an inducible promoter and monitored algT promoter activity under uninduced and induced conditions. When mucP was uninduced, algT promoter activity was restored to wild-type PDO300 levels, indicating that leaky expression of mucP from the arabinose promoter is sufficient for complementation (Fig. 5). When mucP was further induced, there was a 2.3-fold increase in activity of the algT promoter, well beyond wild-type levels. This demonstrates a direct relationship between mucP expression and algT promoter activity in PDO300 strains. Overall, our results suggest that inactivation of MucP renders MucA22 functional, and MucA22 is then able to sequester AlgT and reduce AlgT activity. As a result, mucP mutants lower the toxicity caused by the overexpression of algT.
FIG 5.
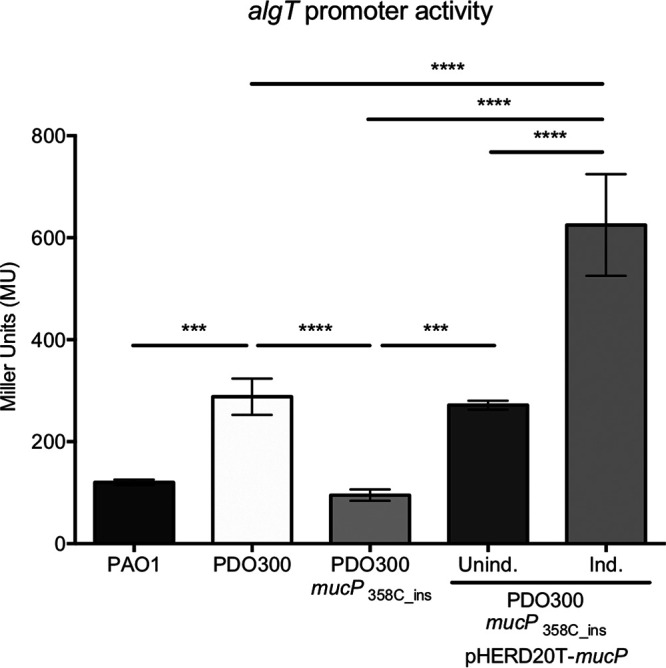
Inactivation of MucP reduces expression of algT. The algT promoter region was transcriptionally fused to a promoterless lacZ containing an optimized RBS to construct a P5algT-optRBS-lacZ reporter. The reporter was inserted at the attCTX site of the P. aeruginosa chromosome of each of the strains. β-Galactosidase activity was determined during exponential-phase growth in LB. Significance was determined using a one-way analysis of variance (ANOVA) with multiple comparisons. Error bars represent standard deviations (SDs) from the means from at least four biological replicates. ***, P < 0.001; ****, P < 0.0001. The strains shown are PAC541, PAC539, and PAC577.
Conclusions.
In mucoid strains containing a truncated form of the anti-sigma factor MucA, called MucA22, the cleavage site for the MucP protease is available and MucA22 is continuously degraded. As a result, MucA22 is no longer able to sequester the AlgT sigma factor and AlgT is available to direct transcription of its regulon, including the alginate biosynthesis operon. Here, we show that overexpression of AlgT in the presence of MucA22, but not wild-type MucA, is lethal. Suppressors of toxic AlgT contain mutations in the gene encoding the MucA protease, MucP. Based on these results, we hypothesize that when MucP is inactivated, MucA22 is not degraded and the truncated form of MucA22 is functional and able to sequester AlgT, reducing expression of the AlgT regulon and the subsequent downstream toxic effects.
It is well documented in E. coli that fine-tuning of RpoE is necessary to maintain cell envelope integrity (41, 51, 52). Deletion of the anti-sigma factor gene rseA results in high RpoE activity, leading to cell lysis in stationary phase (41, 53). Similarly, it is also not possible to make a complete deletion of mucA in P. aeruginosa (34). We propose that this is due to the toxic accumulation of AlgT. If the mechanism is similar to that in E. coli, this is hypothesized to be due to destabilization of the outer membrane. The mucA22 mutation appears to be selected over a mucA null mutation, likely because this form can be functional. Mutations in mucP are likely only one of the possible mechanisms for circumventing toxic AlgT accumulation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacteria were maintained on lysogeny agar (LA) containing 1.5% agar and grown in lysogeny broth (LB). When appropriate, medium was supplemented with 15 μg/ml gentamicin, 10 μg/ml tetracycline, or 100 μg/ml carbenicillin for E. coli and 60 μg/ml gentamicin, 100 μg/ml tetracycline, or 100 μg/ml carbenicillin for P. aeruginosa. Vogel-Bonner minimal medium (VBMM) plates supplemented with gentamicin and no-salt LB (10 g/liter tryptone and 5 g/liter yeast extract) containing 15% sucrose were used for allelic exchange (54). Conjugations and sucrose counterselections were performed at 30°C. A list of strains, plasmids, and primers used is available in Table S2 in the supplemental material.
Construction of PDO300 ΔalgD.
SM10 containing pEXG2-mucA22 (14) was conjugated with PAO1 ΔalgD according to the puddle-mating protocol described by Hmelo et al. (54). After sucrose counterselection, single colonies were patched onto LA and LA containing gentamicin. Next, mucA was amplified by single-colony PCR from gentamicin-sensitive colonies using oAC089/oAC090 and sent to Genewiz for sequencing to identify colonies that contained the mucA22 mutation.
Construction of pHERD20T-mucP and miniCTX-P5algT-optRBS-lacZ and generation of mutants.
The mucP gene was amplified using oAC276/oAC277. This fragment was PCR purified (Qiagen) and ligated into HindIII-digested gel-purified pHERD20T (37) using isothermal assembly (Gibson assembly master mix; New England BioLabs). The reaction product was transformed into chemically competent DH5α and selected for on carbenicillin. Plasmids were miniprepped (Qiagen) from carbenicillin-resistant colonies, and the mucP insertion was screened for by using oAC039/oAC040. To generate miniCTX-P5algT-optRBS-lacZ, 480 bp of the upstream algT region starting 20 bp upstream of the start codon, to incorporate the five algT promoters but not the native ribosome binding site (RBS), was amplified using oAC158/oAC159 and inserted into BamHI-digested miniCTX1-optRBS-lacZ (14) using isothermal assembly (Gibson assembly master mix; New England BioLabs). The reaction products were transformed into chemically competent DH5α and selected for on tetracycline. Plasmids from single colonies were isolated (Qiagen Miniprep kit), and the algT promoter insertion was screened for by using oAC32/oAC33. All plasmids in this study were electroporated into P. aeruginosa strains, as previously described (55). miniTn7-Ptac-algT (14) was selected for on gentamicin, miniCTX-P5algT-optRBS-lacZ was selected for on tetracycline, and pHERD20T-mucA (23) and pHERD20T-mucP were selected for on carbenicillin.
Isolation of nonmucoid revertants.
A 1-ml culture of LB was inoculated with a single colony and incubated for 2 to 5 days, shaking vertically, at 37°C. Each day, the culture was serially diluted on LB plates and incubated overnight at 37°C. Nonmucoid revertants were picked when the plate still contained both nonmucoid and mucoid colonies; therefore, nonmucoid colonies could be compared to the mucoid ones. Using single-colony PCR, algT was amplified using oAC107/oAC108, column purified (Qiagen miniprep kit), and sent to Genewiz for Sanger sequencing.
Isolation of suppressors, whole-genome sequencing, and analysis.
Overnight cultures of each suppressor strain grown without inducer were normalized to an optical density at 600 (OD600) of 0.5 and serially diluted on plates with increasing concentrations of IPTG. Colonies that grew on high concentrations were considered suppressors of toxic algT expression. Genomic DNA was isolated using a DNeasy blood and tissue kit (Qiagen) and sent to the Microbial Genome Sequencing (MiGS) Center (Pittsburgh, PA). Genomes were quality trimmed using TrimGalore! v0.6.2, and reads having quality of >20 were assembled using SPAdes v3.13.1 (56, 57). Variant calling was performed using Snippy v4.4.0 (https://github.com/tseemann/snippy) using PAO1 as the reference genome (assembly accession ASM676v1).
β-Galactosidase assays.
Overnight cultures of each strain were back diluted 1:100 into fresh LB and allowed to grow, rolling at 37°C, until exponential phase, when samples were collected. Reporter assays were performed as previously described (58) with modifications (14).
Computational methods for modeling of AlgT.
Homology modeling of Pseudomonas AlgT was performed using Swiss Model (43) to generate a DNA-bound conformation of the protein. The DNA-bound structure of RpoE (PDB 6JBQ) from E. coli was taken as the template to model a DNA-bound conformation of AlgT. The model was further examined in Coot (59) to confirm favorable side chain conformations near the DNA-binding interface and to minimize clashes before a round of energy minimization in UCSF Chimera (60). The loop insertions 43DAQ45 and 46EAQ48 in AlgT were modeled in UCSF Chimera. We determined the sequence conservation of AlgT with the protocol described by Kuiper et al. (61). AlgT homologous sequences were retrieved by BLAST in UniProt. A set of ∼200 unique amino acid sequences was then selected by applying a 90% sequence identity cutoff in CD-HIT (62).
Computational methods for modeling of MucP.
Calculations of site-specific conservation were made using Geneious Prime (63) for all MucP homolog sequences. Pseudomonas MucP has two regions: (i) membrane bound and (ii) periplasmic, where the membrane-bound M50 peptidase domain was predicted previously to be formed by both N-terminal (∼1 to 100 residues) and C-terminal (∼340 to 450 residues) sequences. NCBI BLAST searches identified two periplasmic PDZ domains, which were then modeled using Swiss Model with template PDB 2ZPL. The model was further examined in Coot (59) to confirm favorable side chain conformations before five rounds of energy minimization were performed in UCSF Chimera (60).
Supplementary Material
ACKNOWLEDGMENTS
We thank Graeme Conn for critical reading of the manuscript.
A.R.C. was supported by a predoctoral fellowship from the Cystic Fibrosis Foundation (CFF)-funded CF@LANTA RDP Center (MCCART15R0) and an NRSA predoctoral fellowship from the National Institutes of Health (NIH) under award number F31 AI136310. Additional funding was provided under award numbers NIH-R21AI122192 (J.B.G.), CFF-GOLDBE16G0 (J.B.G.), and CFF-DEY18F0 (D.D.).
The content of the paper does not represent the official views of the NIH or CFF, who had no role in study design, data collection, or interpretation of the data.
A.R.C. designed the experiments and wrote the paper; A.R.C., V.R., Z.W., and D.D. performed research and analyzed data; V.R., Z.W., D.D., and J.B.G. provided feedback on the experiments and the manuscript.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Boucher JC, Yu H, Mudd MH, Deretic V. 1997. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect Immun 65:3838–3846. doi: 10.1128/IAI.65.9.3838-3846.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doggett RG, Harrison GM, Carter RE Jr. 1971. Mucoid Pseudomonas aeruginosa in patients with chronic illness. Lancet 297:236–237. doi: 10.1016/S0140-6736(71)90973-1. [DOI] [PubMed] [Google Scholar]
- 3.Doggett RG, Harrison GM, Wallis ES. 1964. Comparison of some properties of Pseudomonas aeruginosa isolated from infections in persons with and without cystic fibrosis. J Bacteriol 87:427–431. doi: 10.1128/JB.87.2.427-431.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun 28:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Govan JRW, Deretic V. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60:539–574. doi: 10.1128/MMBR.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilligan PH. 1991. Microbiology of airway disease in patients with cystic fibrosis. Clin Microbiol Rev 4:35–51. doi: 10.1128/cmr.4.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood LF, Ohman DE. 2009. Use of cell wall stress to characterize sigma22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol Microbiol 72:183–201. doi: 10.1111/j.1365-2958.2009.06635.x. [DOI] [PubMed] [Google Scholar]
- 8.Wood LF, Leech AJ, Ohman DE. 2006. Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: roles of sigma (AlgT) and the AlgW and Prc proteases. Mol Microbiol 62:412–426. doi: 10.1111/j.1365-2958.2006.05390.x. [DOI] [PubMed] [Google Scholar]
- 9.Damron FH, Davis MR Jr, Withers TR, Ernst RK, Goldberg JB, Yu G, Yu HD. 2011. Vanadate and triclosan synergistically induce alginate production by Pseudomonas aeruginosa strain PAO1. Mol Microbiol 81:554–570. doi: 10.1111/j.1365-2958.2011.07715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Lou X, Xu Y, Teng X, Liu R, Zhang Q, Wu W, Wang Y, Bartlam M. 2019. Structural basis for the recognition of MucA by MucB and AlgU in Pseudomonas aeruginosa. FEBS J 286:4982–4994. doi: 10.1111/febs.14995. [DOI] [PubMed] [Google Scholar]
- 11.Tart AH, Blanks MJ, Wozniak DJ. 2006. The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J Bacteriol 188:6483–6489. doi: 10.1128/JB.00636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tart AH, Wolfgang MC, Wozniak DJ. 2005. The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ. J Bacteriol 187:7955–7962. doi: 10.1128/JB.187.23.7955-7962.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CJ, Newsom D, Kelly B, Irie Y, Jennings LK, Xu B, Limoli DH, Harrison JJ, Parsek MR, White P, Wozniak DJ. 2014. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog 10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cross AR, Goldberg JB. 2019. Remodeling of O antigen in mucoid Pseudomonas aeruginosa via transcriptional repression of wzz2. mBio 10:e02914-18. doi: 10.1128/mBio.02914-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lizewski SE, Schurr JR, Jackson DW, Frisk A, Carterson AJ, Schurr MJ. 2004. Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J Bacteriol 186:5672–5684. doi: 10.1128/JB.186.17.5672-5684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firoved AM, Deretic V. 2003. Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol 185:1071–1081. doi: 10.1128/JB.185.3.1071-1081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damron FH, Goldberg JB. 2012. Proteolytic regulation of alginate overproduction in Pseudomonas aeruginosa. Mol Microbiol 84:595–607. doi: 10.1111/j.1365-2958.2012.08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damron FH, Yu HD. 2011. Pseudomonas aeruginosa MucD regulates the alginate pathway through activation of MucA degradation via MucP proteolytic activity. J Bacteriol 193:286–291. doi: 10.1128/JB.01132-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgado C, Florez L, Lollett I, Lopez C, Kangeyan S, Kumari H, Stylianou M, Smiddy RJ, Schneper L, Sautter RT, Smith D, Szatmari G, Mathee K. 2018. Pseudomonas aeruginosa regulated intramembrane proteolysis: protease MucP can overcome mutations in the AlgO periplasmic protease to restore alginate production in nonmucoid revertants. J Bacteriol 200:e00215-18. doi: 10.1128/JB.00215-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC, Deretic V. 1996. Control of AlgU, a member of the sigmaE-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J Bacteriol 178:4997–5004. doi: 10.1128/jb.178.16.4997-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathee K, McPherson CJ, Ohman DE. 1997. Posttranslational control of the algT (algU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J Bacteriol 179:3711–3720. doi: 10.1128/jb.179.11.3711-3720.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood LF, Ohman DE. 2012. Identification of genes in the σ22 regulon of Pseudomonas aeruginosa required for cell envelope homeostasis in either the planktonic or the sessile mode of growth. mBio 3:e00094-12. doi: 10.1128/mBio.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damron FH, Qiu D, Yu HD. 2009. The Pseudomonas aeruginosa sensor kinase KinB negatively controls alginate production through AlgW-dependent MucA proteolysis. J Bacteriol 191:2285–2295. doi: 10.1128/JB.01490-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H, Mudd M, Boucher JC, Schurr MJ, Deretic V. 1995. Functional equivalence of Escherichia coli sigmaE and Pseudomonas aeruginosa AlgU: E. coli rpoE restores mucoidy and reduces sensitivity to reactive oxygen intermediates in algU mutants of P aeruginosa. J Bacteriol 177:3259–3268. doi: 10.1128/JB.177.11.3259-3268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeVries CA, Ohman DE. 1994. Mucoid-to-nonmucoid conversion in alginate-producing Pseudomonas aeruginosa often results from spontaneous mutations in algT, encoding a putative alternate sigma factor, and shows evidence for autoregulation. J Bacteriol 176:6677–6687. doi: 10.1128/jb.176.21.6677-6687.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schurr MJ, Yu H, Boucher JC, Hibler NS, Deretic V. 1995. Multiple promoters and induction by heat shock of the gene encoding the alternative sigma factor AlgU (sigmaE) which controls mucoidy in cystic fibrosis isolates of Pseudomonas aeruginosa. J Bacteriol 177:5670–5679. doi: 10.1128/jb.177.19.5670-5679.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Candido Caçador N, Paulino da Costa Capizzani C, Gomes Monteiro Marin Torres LA, Galetti R, Ciofu O, da Costa Darini AL, Høiby N. 2018. Adaptation of Pseudomonas aeruginosa to the chronic phenotype by mutations in the algTmucABCD operon in isolates from Brazilian cystic fibrosis patients. PLoS One 13:e0208013. doi: 10.1371/journal.pone.0208013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deretic V, Schurr MJ, Yu H. 1995. Pseudomonas aeruginosa, mucoidy and the chronic infection phenotype in cystic fibrosis. Trends Microbiol 3:351–356. doi: 10.1016/S0966-842X(00)88974-X. [DOI] [PubMed] [Google Scholar]
- 29.Evans LR, Linker A. 1973. Production and characterization of the slime polysaccharide of Pseudomonas aeruginosa. J Bacteriol 116:915–924. doi: 10.1128/JB.116.2.915-924.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin DW, Schurr MJ, Mudd MH, Govan JRW, Holloway BW, Deretic V. 1993. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci U S A 90:8377–8381. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JIA, Jensen P, Johnsen AH, Givskov M, Ohman DE, Søren M, Høiby N, Kharazmi A. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145:1349–1357. doi: 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- 32.Deretic V, Gill JF, Chakrabarty AM. 1987. Gene algD coding for GDPmannose dehydrogenase is transcriptionally activated in mucoid Pseudomonas aeruginosa. J Bacteriol 169:351–358. doi: 10.1128/jb.169.1.351-358.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wozniak DJ, Ohman DE. 1994. Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J Bacteriol 176:6007–6014. doi: 10.1128/jb.176.19.6007-6014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panmanee W, Su S, Schurr MJ, Lau GW, Zhu X, Ren Z, McDaniel CT, Lu LJ, Ohman DE, Muruve DA, Panos RJ, Yu HD, Thompson TB, Tseng BS, Hassett DJ. 2019. The anti-sigma factor MucA of Pseudomonas aeruginosa: dramatic differences of a mucA22 vs. a ΔmucA mutant in anaerobic acidified nitrite sensitivity of planktonic and biofilm bacteria in vitro and during chronic murine lung infection. PLoS One 14:e0216401. doi: 10.1371/journal.pone.0216401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hancock REW, Carey AM. 1979. Outer membrane of Pseudomonas aeruginosa: heat- and 2-mercaptoethanol-modifiable proteins. J Bacteriol 140:902–910. doi: 10.1128/JB.140.3.902-910.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, Singh PK, Chopp DL, Packman AI, Parsek MR. 2013. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ Microbiol 15:2865–2878. doi: 10.1111/1462-2920.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sautter R, Ramos D, Schneper L, Ciofu O, Wassermann T, Koh CL, Heydorn A, Hentzer M, Hoiby N, Kharazmi A, Molin S, Devries CA, Ohman DE, Mathee K. 2012. A complex multilevel attack on Pseudomonas aeruginosa algT/U expression and algT/U activity results in the loss of alginate production. Gene 498:242–253. doi: 10.1016/j.gene.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciofu O, Lee B, Johannesson M, Hermansen NO, Meyer P, Hoiby N. 2008. Investigation of the algT operon sequence in mucoid and non-mucoid Pseudomonas aeruginosa isolates from 115 Scandinavian patients with cystic fibrosis and in 88 in vitro non-mucoid revertants. Microbiology 154:103–113. doi: 10.1099/mic.0.2007/010421-0. [DOI] [PubMed] [Google Scholar]
- 40.Fang C, Li L, Shen L, Shi J, Wang S, Feng Y, Zhang Y. 2019. Structures and mechanism of transcription initiation by bacterial ECF factors. Nucleic Acids Res 47:7094–7104. doi: 10.1093/nar/gkz470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicoloff H, Gopalkrishnan S, Ades SE. 2017. Appropriate regulation of the sigma E-dependent envelope stress response is necessary to maintain cell envelope integrity and stationary-phase survival in Escherichia coli. J Bacteriol 199:e00089-17. doi: 10.1128/JB.00089-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu D, Eisinger VM, Rowen DW, Yu HD. 2007. Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 104:8107–8112. doi: 10.1073/pnas.0702660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S, Zhang Y. 2007. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu D, Zhang Y. 2012. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu D, Zhang Y. 2013. Toward optimal fragment generations for ab initio protein structure assembly. Proteins 81:229–239. doi: 10.1002/prot.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Las Penas A, Connolly L, Gross CA. 1997. SigmaE is an essential sigma factor in Escherichia coli. J Bacteriol 179:6862–6864. doi: 10.1128/jb.179.21.6862-6864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konovalova A, Grabowicz M, Balibar CJ, Malinverni JC, Painter RE, Riley D, Mann PA, Wang H, Garlisi CG, Sherborne B, Rigel NW, Ricci DP, Black TA, Roemer T, Silhavy TJ, Walker SS. 2018. Inhibitor of intramembrane protease RseP blocks the σE response causing lethal accumulation of unfolded outer membrane proteins. Proc Natl Acad Sci U S A 115:E6614–E6621. doi: 10.1073/pnas.1806107115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marko VA, Kilmury SLN, MacNeil LT, Burrows LL. 2018. Pseudomonas aeruginosa type IV minor pilins and PilY1 regulate virulence by modulating FimS-AlgR activity. PLoS Pathog 14:e1007074. doi: 10.1371/journal.ppat.1007074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hart EM, O’Connell A, Tang K, Wzorek JS, Grabowicz M, Kahne D, Silhavy TJ. 2019. Fine-tuning of σE activation suppresses multiple assembly-defective mutations in Escherichia coli. J Bacteriol 201:e00745-18. doi: 10.1128/JB.00745-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayden JD, Ades SE. 2008. The extracytoplasmic stress factor, σE, is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3:e1573. doi: 10.1371/journal.pone.0001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nitta T, Nagamitsu H, Murata M, Izu H, Yamada M. 2000. Function of the σE regulon in dead-cell lysis in stationary-phase Escherichia coli. J Bacteriol 182:5231–5237. doi: 10.1128/jb.182.18.5231-5237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Tolker-Nielsen T, Parsek MR, Schweizer HP, Harrison JJ. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10:1820–1841. doi: 10.1038/nprot.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 57.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JH. 1973. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 59.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuiper EG, Dey D, LaMore PA, Owings JP, Prezioso SM, Goldberg JB, Conn GL. 2019. Substrate recognition by the Pseudomonas aeruginosa EF-Tu-modifying methyltransferase EftM. J Biol Chem 294:20109–20121. doi: 10.1074/jbc.RA119.011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li W, Godzik A. 2006. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 63.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.