

ABSTRACT
Most sporadic colorectal cancer reflects acquired mutations in the adenomatous polyposis coli (APC) tumor suppressor gene, while germline heterozygosity for mutant APC produces the autosomal dominant disorder Familial Adenomatous Polyposis (FAP) with a predisposition to colorectal cancer. In these syndromes, loss of heterozygosity (LOH) silences the remaining normal allele of APC, through an unknown mechanism, as the initiating step in transformation. Guanylyl cyclase C receptor (GUCY2C) and its hormones, uroguanylin and guanylin, have emerged as a key signaling axis opposing mutations driving intestinal tumorigenesis. Indeed, uroguanylin and guanylin are among the most commonly repressed genes in colorectal cancer. Here, we explored the role of APC heterozygosity in mechanisms repressing hormone expression which could contribute to LOH. In genetic mouse models of APC loss, uroguanylin and guanylin expression were quantified following monoallelic or biallelic deletion of the Apc gene. Induced biallelic loss of APC repressed uroguanylin and guanylin expression. However, monoallelic APC loss in Apcmin/+ mice did not alter hormone expression. Similarly, in FAP patients, normal colonic mucosa (monoallelic APC loss) expressed guanylin while adenomas and an invasive carcinoma (biallelic APC loss) were devoid of hormone expression. Thus, uroguanylin and guanylin expression by normal intestinal epithelial cells persists in the context of APC heterozygosity and is lost only after tumor initiation by APC LOH. These observations reveal a role for loss of the hormones silencing the GUCY2C axis in tumor progression following biallelic APC loss, but not in mechanisms creating the genetic vulnerability in epithelial cells underlying APC LOH initiating tumorigenesis.
KEYWORDS: APC, colorectal cancer, FAP, guanylin, guanylyl cyclase C, GUCA2A, GUCA2B, GUCY2C, LOH, loss of heterozygosity
Introduction
Nearly 8% of incident cancers in the United States originate in the colorectum.1 Most sporadic cases begin with mutations that silence one allele of the canonical tumor suppressor gene adenomatous polyposis coli (APC) producing acquired heterozygosity in intestinal epithelial cells.2,3 Alternatively, germline mutations in APC cause a hereditary autosomal dominant cancer predisposition syndrome called familial adenomatous polyposis (FAP.)4 In both sporadic and germline syndromes, subsequent acquired mutations inactivate the second wildtype allele of APC producing loss of heterozygosity (LOH).5–7 Mutations in APC occur most frequently in exon 15, a genetic hot spot in both acquired and hereditary cases. LOH resulting in biallelic loss of APC is generally considered the initiating event in colorectal transformation, creating the premalignant field that supports the development of adenomas and, ultimately, invasive carcinoma.3,5 The role of LOH in this process can best be appreciated by considering FAP patients, in whom all epithelial cells are heterozygous for APC. In these patients, APC LOH can produce hundreds of adenomatous polyps4,8,9 and, without intervention, they have a ~ 100% lifetime risk of colorectal cancer.10,11
Loss of APC, and its associated functions, through LOH results in cytosolic accumulation of β-catenin which translocates to the nucleus where it regulates transcriptional programs driving epithelial dysfunction, an essential step for tumor initiation.12–15 While the role of APC heterozygosity in the pathophysiology of colorectal cancer is established, there is an incomplete understanding of the molecular steps contributing to the genetic vulnerability in epithelial cells leading to inactivation of the second APC allele and LOH. Defining mechanisms underlying epithelial cell vulnerability to LOH, and the resultant evolution of the premalignant field, could provide insights to mitigate tumorigenesis and improve cancer prevention.
Guanylyl cyclase C (GUCY2C) is an intestine-specific receptor which plays a role in maintaining epithelial homeostasis.16–21 Uroguanylin and guanylin, peptide paracrine hormones produced in small intestine and colon, respectively,22–24 activate GUCY2C to produce the second messenger cyclic guanosine monophosphate (cGMP).25,26 In turn, cGMP signaling regulates homeostatic processes in intestine that are canonically disrupted in cancer,27 including maintenance of DNA integrity and genomic stability.19,28 In that context, silencing the GUCY2C axis amplifies intestinal epithelial cell vulnerability for APC LOH in mouse models of FAP.19 Indeed, loss of GUCY2C signaling drives intestinal transformation, specifically at the step of tumor initiation, induced by genetic mutations or carcinogens.17,19,28–30 Importantly, uroguanylin and guanylin are the most commonly lost gene products in colorectal cancer.29,31,32 Moreover, loss of these paracrine hormones occurs universally at the earliest stages of tumorigenesis in mice and humans coincident with APC LOH.29,31–34 In contrast, GUCY2C expression appears to be preserved in most primary and metastatic colorectal tumors.33–37
In the context of these observations, we explored the hypothesis that paracrine hormone loss, which in turn silences GUCY2C, is an obligatory step contributing to the genetic vulnerability underlying LOH which establishes the premalignant field. Specifically, these studies determined whether APC heterozygosity alters GUCY2C paracrine hormone expression. Here, we reveal that uroguanylin and guanylin expression is maintained after monoallelic loss of APC but eliminated following APC LOH. Thus, APC heterozygosity does not suppress guanylin and uroguanylin expression to create epithelial genetic vulnerability underlying LOH that initiates tumorigenesis. Rather, loss of hormone expression silencing GUCY2C is a direct effect of APC LOH, contributing to tumor progression.
Results
Development of uroguanylin and guanylin knockout mice using CRISPR/Cas9 gene editing
To establish controls for loss of hormone expression, knockout mice were developed using single guide (sg)RNAs targeting exons 2 and 3 of the Guca2b and Guca2a genes, respectively (Figure 1a,b). Following microinjection, mouse embryos were screened by PCR, using primers that spanned the target sites. Knockouts were confirmed by sequencing analyses (Supplementary Figure 1). Mice chosen for the Guca2b knockout line harbor a ~ 1.5 Kb deletion in the Guca2b gene, while those chosen for the Guca2a knockout line harbor a ~ 1.7 Kb deletion in the Guca2a gene. Genotypes were confirmed by PCR, which indicated a 3 Kb PCR product for the wildtype allele and a ~ 1.5 Kb and ~1.3 Kb PCR product for the Guca2b and Guca2a knockout deletion mutations, respectively (Figure 1c,d). Analysis of Guca2b and Guca2a transcript levels by qRT-PCR confirmed elimination of Guca2b and Guca2a expression in small intestine and colon, compared to control mice (Figure 2a,b).
Figure 1.
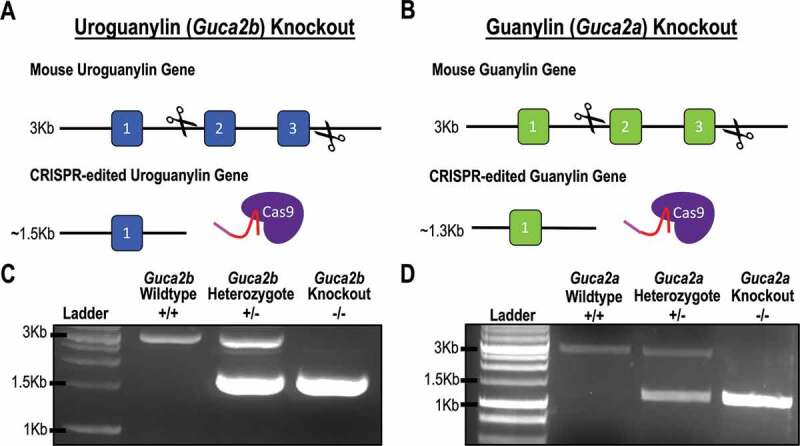
(a and b) CRISPR/Cas9 gene editing approach used to generate the uroguanylin (Guca2b) and guanylin (Guca2a) knockout mice. The targeting strategy was designed to delete exons 2 and 3 of Guca2b and Guca2a, respectively. (c) Representative PCR genotyping for Guca2b wildtype (3Kb), homozygous knockout (1.5Kb), and heterozygous knockout mice (3Kb and 1.5Kb). (d) Representative PCR genotyping for Guca2a wildtype (3Kb), homozygous knockout (1.3Kb), and heterozygous knockout mice (3Kb and 1.3Kb).
Figure 2.
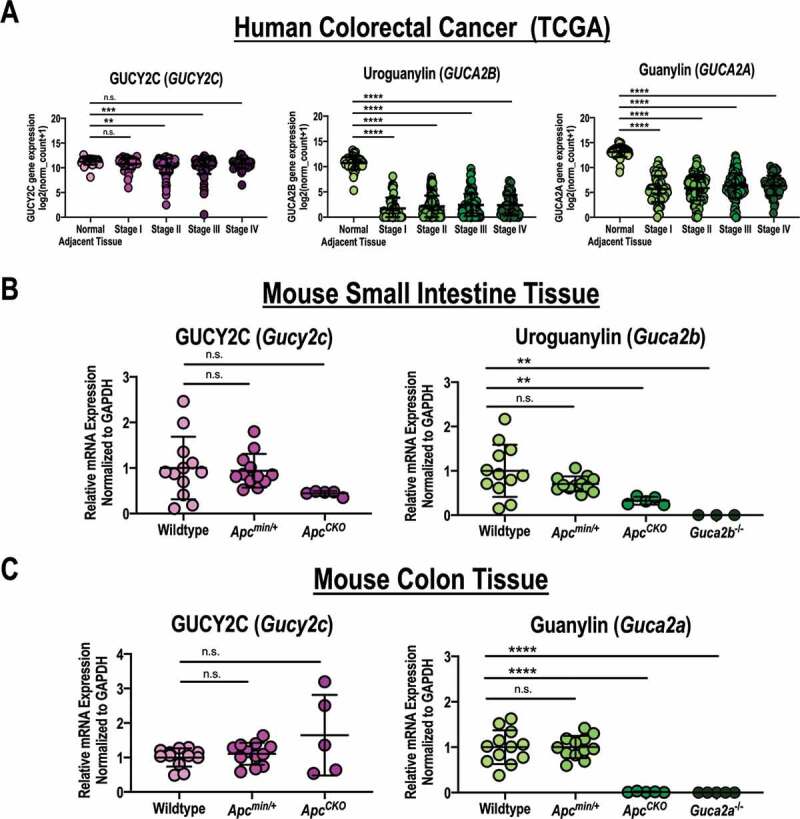
GUCY2C ligand mRNA expression is retained after monoallelic loss of APC. (a) GUCY2C, uroguanylin, and guanylin mRNA expression quantified by RNAseq in colorectal carcinomas, classified by stage of disease [stage I (n = 57), stage II (n = 136), stage III (n = 113), stage IV (n = 52)], and normal colorectum (n = 51) from the TCGA database.38 (b) GUCY2C and uroguanylin mRNA quantified by qRT-PCR from jejunum in wildtype (n = 12), Apcmin/+ (n = 11), Guca2b−/- (n = 3) and ApcCKO (n = 5) mice. (c) GUCY2C and guanylin mRNA quantified by qRT-PCR from proximal colon in wildtype (n = 12), Apcmin/+ (n = 11), Guca2a−/- (n = 3) and ApcCKO (n = 5) mice. n.s., not significant, * p < .05, ** p < .01, *** p < .001, **** p < .0001.
GUCY2C hormone expression is maintained after monoallelic APC loss, but eliminated by APC LOH
Loss of GUCY2C hormone expression is an early event along the continuum of colorectal cancer that aligns with mutations in the APC-β-catenin signaling pathway.2,31,34 Previously, we demonstrated that biallelic loss of APC increases Wnt signaling which, in turn, represses GUCY2C hormone expression, supporting a role for this process in tumor progression.34 Here, we explored the effect of monoallelic APC loss (APC heterozygosity) on hormone expression as a potential mechanism contributing to genetic vulnerability, by silencing the GUCY2C axis, that could promote APC LOH and tumor initiation.
Analysis of data from The Cancer Genome Atlas (TCGA)38 confirmed that expression of uroguanylin and guanylin mRNA is reduced ~250-fold in human colorectal tumors at all disease stages (Figure 2a).33,34,38 In contrast, expression of GUCY2C mRNA was retained in most of those tumors (Figure 2a).33,34,38 Small differences (~1.75-fold) in GUCY2C mRNA at some stages likely reflect a contribution of tumors arising from the serrated adenoma pathway, in which receptor expression is substantially reduced.33,38 In close agreement with human samples, uroguanylin and guanylin mRNA and protein were reduced in small intestine and colon, respectively, in mice with biallelic Apc loss (ApcCKO) (Figures 2b,c, 3a,b), and in adenomas (Apc LOH) in Apcmin/+ mice, which are heterozygous for Apc (Figure 3c). Conversely, GUCY2C mRNA (Figure 2b,c) and protein (Figure 3a,b) were preserved in ApcCKO mice, and in adenomas in Apcmin/+ mice (Figure 3c), recapitulating observations with patient tumor samples (Figure 2a). Importantly, uroguanylin, guanylin, and GUCY2C mRNA (Figure 2b,c) and protein (Figure 3a-d) were quantitatively similar in normal mucosa in small intestine and colon of Apcmin/+ mice (Apc heterozygosity), compared to wildtype mice.
Figure 3.
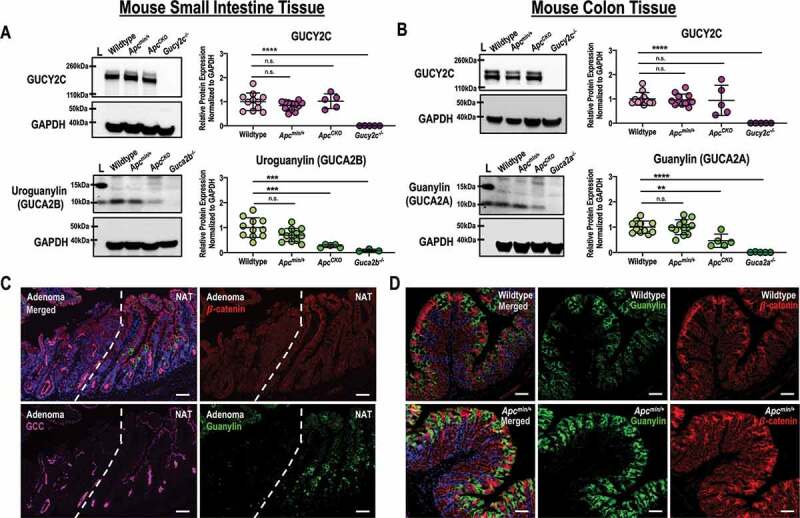
GUCY2C ligand protein expression is retained after monoallelic loss of APC. (a) GUCY2C and uroguanylin protein quantified by immunoblot (representative shown) from jejunum in wildtype (n = 12), Apcmin/+ (n = 11), Guca2b−/- (n = 3) and ApcCKO (n = 5) mice. (b) GUCY2C and guanylin protein quantified by immunoblot (representative shown) from proximal colon in wildtype (n = 12), Apcmin/+ (n = 11), Guca2a−/- (n = 3) and ApcCKO (n = 5) mice. (c) Single field immunofluorescence of Apcmin/+ small intestine containing an adenoma and normal adjacent tissue (NAT). (d) Immunofluorescence of wildtype and Apcmin/+ proximal colon for guanylin. β-catenin (red), guanylin (green), GUCY2C (magenta), DAPI (blue). n.s., not significant, * p < .05, ** p < .01, *** p < .001, **** p < .0001. Scale bar = 100 µm.
Suppression of GUCY2C hormones requires APC LOH in FAP patients
Like Apcmin/+ mice, FAP patients have a germline mutation in one allele of APC which creates a predisposition for LOH, transformation, and the formation of intestinal polyps.10 Here, we obtained fresh specimens from colon resections of FAP patients to explore the expression of components of the GUCY2C signaling axis in the context of APC mutations. Guanylin protein (Figure 4a) and mRNA expression (Figure 4b) were maintained in normal epithelium (APC heterozygosity), but were absent in adjacent premalignant polyps and invasive adenocarcinoma reflecting APC LOH. Importantly, GUCY2C protein and mRNA expression were maintained at each stage of transformation, in the context of monoallelic or biallelic loss of APC. Moreover, these data further support our previous findings in FAP patients.34 Taken together, these observations support a mechanism in which paracrine hormone loss silencing the GUCY2C tumor suppressor in intestinal transformation reflects biallelic inactivation of APC in mice and humans (Figure 4c).
Figure 4.
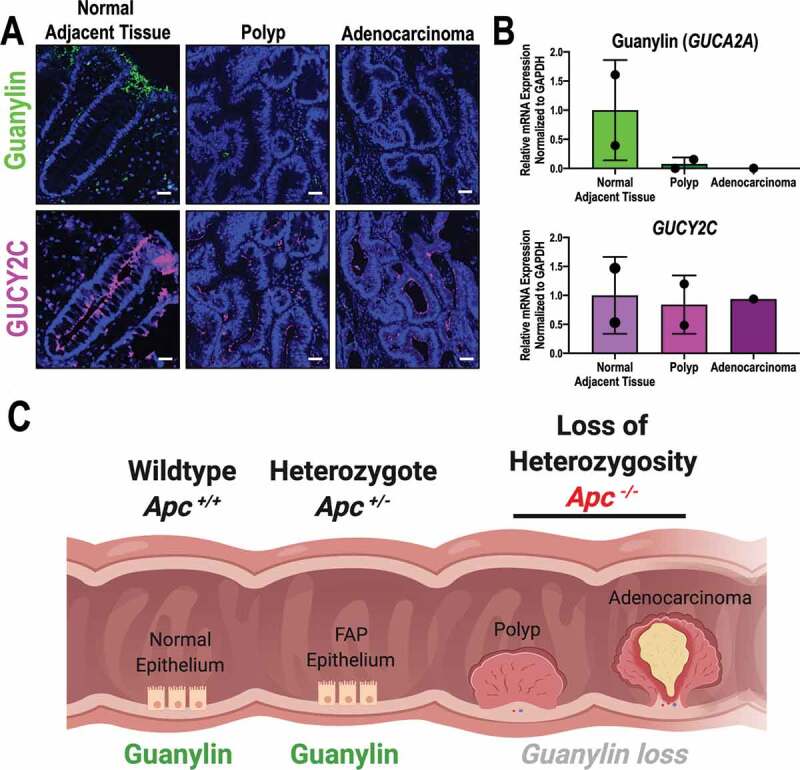
Suppression of guanylin requires APC LOH in an FAP patient. (a) Representative immunofluorescence of guanylin and GUCY2C in an FAP patient (Biobank ID: #027). Samples were obtained from the colon of this patient that captured the progression of transformation from normal adjacent tissue (NAT; monoallelic APC loss), through premalignant polyp and invasive adenocarcinoma (biallelic APC loss). Representative images shown, guanylin (green), GUCY2C (magenta), DAPI (blue). (b) Quantification of guanylin and GUCY2C mRNA by qRT-PCR in NAT (n = 2 patients), polyp (n = 2 patients) and adenocarcinoma (n = 1 patient). (c) Graphical representation for APC-dependent mechanism of guanylin suppression created with Biorender.com. Scale bar = 100 µm.
Discussion
APC mutations occur in ~80% of colorectal tumors.3,12,13 Although an essential role for biallelic loss of APC gene function in the development of colorectal cancer is established, mechanisms linking inactivation of the first APC allele with the genetic vulnerability that promotes loss of the second allele in the same cell remain to be defined. Here, we tested whether APC heterozygosity produced loss of uroguanylin and guanylin, which could silence GUCY2C and downstream signaling mechanisms maintaining genomic integrity, as a potential pathophysiological step underlying formation of the premalignant field.
The intestine is a complex continuously regenerating organ renewing the epithelial layer every 3–5 days.39–41 Continuous epithelial replacement requires tight coordination of regenerative processes including proliferation, migration, differentiation, and apoptosis.39,41–43 Beyond coordinating normal regeneration, these homeostatic processes are canonically dysregulated in mechanisms underlying tumorigenesis.27 GUCY2C, an intestinal epithelial cell-specific membrane receptor, regulates a number of homeostatic process that organize the intestinal epithelium, including proliferation, differentiation, metabolism, DNA damage repair, and epithelial-mesenchymal crosstalk.17–21 GUCY2C has emerged as a key intestinal tumor suppressor, and silencing this receptor disrupts those canonical homeostatic mechanisms, promoting tumorigenesis in mouse models of colorectal cancer.16–21,44,45 In that context, loss of guanylin and uroguanylin, which silences GUCY2C signaling, occurs at the earliest stage of intestinal transformation by a mechanism that is conserved across species.31,32,34
Previously, we demonstrated that GUCY2C hormone expression is eliminated by a dysregulated nuclear β-catenin-dependent transcriptional mechanism reflecting APC LOH in mice and humans.34 Indeed, we have identified a pathophysiological model in which guanylin hormone expression is eliminated as a direct downstream consequence of mutant APC-β-catenin-TCF nuclear transcriptional reprogramming.34 These studies demonstrated that oncogenic APC-β-catenin signaling silences guanylin mRNA and loss of guanylin expression was dependent on a canonical TCF-dependent mechanism.34 These observations underscore the established pathophysiological association of hormone loss with colorectal tumorigenesis across species.29,31–34 They suggest a role for hormone repression silencing GUCY2C in mechanisms contributing to tumor progression, for example through hyperproliferation, desmoplasia, chromosomal instability, and DNA hyper-mutation,17–21,33 reflecting dependence of this repression on LOH and APC biallelic loss.34
Beyond progression, eliminating GUCY2C expression in the Apcmin/+ mouse model of genetic transformation increased the incidence and burden of tumors in the colorectum. Similarly, administering the colorectal pro-carcinogen azoxymethane to mice devoid of intestinal GUCY2C increased the incidence and burden of tumors in the colorectum.19 Surprisingly, increased tumor burden in both genetic and carcinogen models of transformation reflected a predominant effect on tumor number (initiation), rather than tumor size (progression).17,19 Conversely, oral administration of GUCY2C ligands to Apcmin/+ mice reduced tumor burden through a primary effect on tumor number, rather than on tumor size.29,30 Similarly, chronic colonization of mice with bacteria constitutively secreting GUCY2C ligand opposed azoxymethane-induced colorectal tumorigenesis primarily by reducing tumor number, with a minor effect on tumor size.28
These observations suggest that, beyond progression after APC LOH, hormone repression silencing the GUCY2C signaling axis might contribute to creating the epithelial genetic vulnerability underlying tumor initiation through APC LOH. In that context, the established link between dysregulated nuclear APC-β-catenin signaling and transcriptional repression of hormone expression34 suggested a model in which APC heterozygosity suppressed hormone expression silencing GUCY2C leading to genetic vulnerability. Indeed, recent preliminary studies suggested that hormone mRNA expression was suppressed in normal epithelium (monoallelic APC loss), but not in tumors (biallelic APC loss), in Apcmin/+ mice.30 Here, we confirmed that uroguanylin and guanylin mRNA and protein expression was quantitatively reduced in small intestine and colon, respectively, in mice with tissue-specific inducible biallelic APC loss (ApcCKO).34 However, in striking contrast to previous reports,30 uroguanylin and guanylin mRNA and protein expression was quantitatively maintained in normal epithelia in small intestine and colon, respectively, in mice with monoallelic APC loss (Apcmin/+). Hormone expression in normal epithelia was prominently juxtaposed with the elimination of GUCY2C hormone expression in immediately adjacent intestinal tumors. Moreover, observations in genetic mouse models were confirmed in patient samples. Thus, in FAP patients, normal colon epithelia (APC heterozygosity) exhibited robust guanylin expression while adenomas and an invasive carcinoma (biallelic APC loss) were devoid of guanylin expression, confirming previous reports.33 It is noteworthy that the sample size of FAP patients remains a limitation of this study, and it will be important to confirm these observations in future studies with larger cohorts.
Taken together, these observations establish a previously unappreciated detailed mechanistic sequence along the pathophysiological continuum of intestinal transformation linking APC loss-of-function mutational dynamics with paracrine hormone loss silencing GUCY2C. They reveal that elimination of paracrine hormone expression requires biallelic APC inactivation, positioning GUCY2C loss of signaling after tumor initiation, contributing to progression of intestinal transformation. These results align closely with the ability of gain-of-function mutations in β-catenin, which phenocopy the dysregulated nuclear transcriptional programming and tumorigenic potential of biallelic APC loss, to eliminate guanylin expression in colorectum.34 In contrast, these studies reveal that monoallelic APC loss, and its associated heterozygosity, does not suppress uroguanylin and guanylin mRNA and protein expression in small intestine and colon, respectively. They suggest that APC heterozygosity suppressing hormone expression silencing GUCY2C does not contribute to molecular mechanisms establishing the genetic vulnerability of individual intestinal cells underlying APC LOH and tumor initiation, in contrast to earlier reports.30 However, the present observations notwithstanding, mechanisms beyond APC inactivation may entrain the GUCY2C tumor suppressor signaling axis in initiation of intestinal transformation. Thus, obesity and inflammation, processes with well-established links to colorectal cancer risk, suppress paracrine hormone expression silencing GUCY2C and contributing to intestinal tumorigenesis, through mechanisms that are independent of APC.46,47 In close agreement, hormone replacement opposes intestinal tumorigenesis induced by inflammation or obesity.47,48 Moreover, the substantial impact of GUCY2C signaling on tumor initiation, compared to progression, in both genetic and carcinogen mouse models of intestinal transformation17-19,29,30 underscore the potential of leveraging GUCY2 C signaling as a novel chemoprevention target for colorectal cancer.
Materials and methods
Animal models
All animal protocols were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee (Protocol 00914, approved July 11th, 2019). Apcmin/+ mice contain a transversion point mutation converting codon 850 from leucine to a premature stop codon, truncating one copy of APC (Jackson Laboratories, Stock #002020). Apcmin/+ mice were used as a model for monoallelic Apc loss. ApcCKO mice contain a conditional knockout allele of Apc with loxP sites flanking exon 14, producing a truncated APC protein in the context of Cre-mediated recombination (NCI Mouse Repository, #01XAA). ApcCKO mice were crossed with vil-Cre-ERT2 mice to induce biallelic Apc inactivation (vil-Cre-ERT2-ApcCKO/CKO) in intestinal epithelial cells.49 Conditional mouse models were induced with an intraperitoneal administration of a 100 mg/kg dose of tamoxifen and harvested 5 days later. Genotyping for wildtype, Apcmin/+ and ApcCKO mice was performed by Transnetyx using real-time PCR. Guca2b and Guca2a knockout mice were generated using the CRISPR/Cas9 system at the University of Pennsylvania Transgenic and Chimeric Mouse Facility. For Guca2b knockouts, Cas9 mRNA and sgRNAs were microinjected into fertilized embryos of C57Bl/6 J mice. Mutations in Guca2b were confirmed by sequencing analyses. Guca2b knockout mice were genotyped using PCR with specific primers (forward, 5ʹ- GTG TGA GCT TGG AAG CGA G − 3ʹand reverse, 5ʹ- CTT GTG TCC TGT AAG TAC TCT GC −3ʹ). Guca2a knockouts were developed by microinjection of Cas9 mRNA and sgRNAs into fertilized embryos of SJl/J mice. Mutations in Guca2a were confirmed by sequencing analyses and Guca2a knockout mice were genotyped using PCR with specific primers (forward, 5ʹ- TGG CTG TCC TGG TAG AAG GGG TCA C − 3ʹ and reverse, 5ʹ- ACC CTG GTA CCT CTG TGT TCC CAC C − 3ʹ). In order to establish the mouse line, a Guca2a knockout founder was backcrossed to C57Bl/6 J for 5 generations using Jackson Laboratories SNP Genome Scanning Analysis for speed congenics. Gucy2c knockout mice were described previously.50
Human samples
This study was approved by the local Institutional Review Board (control #18D.495, approved August 23rd, 2018). Patients gave written informed consent for their participation in the study. Tissue specimens from two FAP patients were provided in a de-identified fashion by the Department of Pathology at Thomas Jefferson University Hospital. All tumors underwent routine profiling in the Department of Pathology. Patients carried a diagnosis of FAP based on clinical and histopathological criteria. Expression of GUCY2C, uroguanylin, and guanylin mRNA in human colorectal adenocarcinomas classified by disease stage was obtained from The Cancer Genome Atlas (TCGA).38
Immunofluorescence
Tissues were fixed in 4% paraformaldehyde, processed and embedded in paraffin. Antigens were retrieved using the Dako Target Retrieval Solution pH 9.0 (Agilent, S2367) for 15 min and then stained. Antibody to β-catenin was from BD Biosciences (Purified Mouse anti-β-catenin clone 14, 1:800) and anti-human GUCA2A antibody (#HPA018215, 1:500) was from Sigma. Guanylin antisera (#2538, 1:1000) used for mouse tissue staining was gifted from Dr. M. Goy.51 Ms20 monoclonal antibody to GUCY2C (1:1000) was previous validated and used for both mouse and human samples.47 Background autofluorescence was reduced with sudan black (0.5% solution in 70% ethanol). Secondary antibodies were from Jackson ImmunoResearch. GUCY2C and GUCA2A were detected using tyramide signal amplification. Fluorescence images were captured with an EVOS FL auto cell imaging system (Thermo Fischer Scientific).
Western blots
Protein was extracted from intestinal mucosa dissected from mouse small intestine and colon (20–30 µg protein per lane). Tissue lysates were extracted in T-Per (Thermo Fisher Scientific), supplemented with protease and phosphatase inhibitors (Roche, St. Louis, MO). Protein was quantified by immunoblot analysis employing antibodies to: GAPDH (cat. #2118, 1:3000), guanylin antisera (#2538, 1:1000), uroguanylin antisera (#6910, 1:1000) and GUCY2C (1:500). Immunoblot images were captured on the BioRad ChemiDoc MP imaging station and bands were quantified by densitometry normalized to that of GAPDH using ImageJ. Average relative intensity reflects at least two independent experiments each with at least three biological replicates. Uroguanylin antisera (#6910, 1:1000) used for immunoblot detections was also gifted from Dr. M. Goy.51
Messenger RNA analysis
Tissue samples were flash frozen in liquid nitrogen and stored at −80°C until use. RNA was extracted and purified using the RNeasy kit (Qiagen, Germantown, MD). Following isolation, RNA concentration and purity were measured using the Nanodrop 1000 (Thermo Fisher Scientific) and two-step quantitative (q)RT-PCR used to interrogate gene expression. Complementary DNA was produced using the Taqman RT-PCR kit (Life Technologies, Carlsbad, CA) according to the manufacturer’s specifications and then quantified by PCR (Applied Biosystems, Foster City, CA) using Taqman primer probes (Life Technologies).
Statistical analysis
Statistical significance was determined by one-way ANOVA for multiple comparisons, for analyses of immunoblot and relative mRNA expression determinations in both humans (TCGA) and mice. For animal studies, minimum cohort sizes were computed using a power of 80% and a significance level of 0.05 (2-tailed test) employing a priori predictions of effect size and variance established by preliminary studies or literature review. All statistical tests were calculated using GraphPad Prism (La Jolla, CA). Analyses represent mean ± SD with n = 3–12 mice per cohort, and * p < .05, ** p < .01, *** p < .001, ****, p < .0001.
Supplementary Material
Acknowledgments
We would like to acknowledge the University of Pennsylvania Transgenic and Chimeric Mouse Facility for the CRISPR/Cas9 design and technical development of the Guca2b and Guca2a knockout mouse lines. RNA expression results for the GUCY2C signaling axis in the various stages of colorectal cancer published here are in whole based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Illustration was created with Biorender.com. S.A.W. is the Samuel MV Hamilton Professor at Thomas Jefferson University.
Funding Statement
Supported by grants to S.A.W. from NIH [1R01 CA204881, 1R01 CA206026, P30 CA56036], Department of Defense Congressionally Directed Medical Research Program W81XWH-17-PRCRP-TTSA, The Courtney Ann Diacont Memorial Foundation, and Targeted Diagnostic & Therapeutics, Inc., and to A.E.S. (Department of Defense Congressionally Directed Medical Research Program W81XWH-17-1-0299, PhRMA Foundation, the W.W. Smith Charitable Trust, and Margaret Q. Landenberg Foundation). A.M.P. and J.A.R. were supported by Ruth Kirschstein Individual Research Fellowship Awards [F31 CA225123 and F30 CA232469, respectively]. J.A.R and J.B. were supported by pre-doctoral fellowships from the PhRMA Foundation. A.Z. was supported by NIH institutional award T32 GM008562 for Postdoctoral Training in Clinical Pharmacology.
Author contributions
A.M.P. conceived and designed the experiments. A.M.P. performed the biochemical experiments and analyzed the data. A.M.P., J.R.B., A.A.E. and J.A.R. performed mouse colony maintenance and genotyping. A.M.P. wrote the paper with supervision, review and editing by S.A.W. All authors have read and agreed to the published version of the manuscript.
Disclosure of Potential Conflicts of Interest
S.A.W. is a member of the Board and Chair of the Scientific Advisory Board of, and A.E.S. is a consultant for, Targeted Diagnostics & Therapeutics, Inc. which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, SaHSRACSA G, Miller KD, SaHSRACSA G, Jemal A, SaHSRACSA G.. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW.. Apc mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 3.Fodde R, Smits R, Clevers H.. Apc, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 4.Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, Mori T, Utsunomiya J, Baba S, Petersen G, et al. Germ-line mutations of the apc gene in 53 familial adenomatous polyposis patients. Proc Nat Acad Sci. 1992;89:4452. doi: 10.1073/pnas.89.10.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 6.Yamada Y, Hata K, Hirose Y, Hara A, Sugie S, Kuno T, Yoshimi N, Tanaka T, Mori H. Microadenomatous lesions involving loss of apc heterozygosity in the colon of adult apc(min/+) mice. Cancer Res. 2002;62:6367–6370. [PubMed] [Google Scholar]
- 7.Sameer AS. Colorectal cancer: molecular mutations and polymorphisms. Front Oncol. 2013;3:114. doi: 10.3389/fonc.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 9.Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y, Koike M, Shitara N, Iwama T, Kuroki T. Frequent mutation of beta-catenin and apc genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res. 1999;59:4506. [PubMed] [Google Scholar]
- 10.Nieuwenhuis MH, Vasen HFA. Correlations between mutation site in apc and phenotype of familial adenomatous polyposis (fap): A review of the literature. Crit Rev Oncol Hematol. 2007;61:153–161. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Corley DA, Jensen CD, Marks AR, Zhao WK, Lee JK, Doubeni CA, Zauber AG, de Boer J, Fireman BH, Schottinger JE, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014;370:1298–1306. doi: 10.1056/NEJMoa1309086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-tcf signaling in colon cancer by mutations in beta-catenin or apc. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 13.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the apc/β-catenin/tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130. [PubMed] [Google Scholar]
- 14.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-tcf complex in apc-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 15.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Arshad N, Visweswariah SS. The multiple and enigmatic roles of guanylyl cyclase c in intestinal homeostasis. FEBS Lett. 2012;586:2835–2840. doi: 10.1016/j.febslet.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 17.Lin JE, Li P, Snook AE, Schulz S, Dasgupta A, Hyslop TM, Gibbons AV, Marszlowicz G, Pitari GM, Waldman SA, et al. The hormone receptor gucy2c suppresses intestinal tumor formation by inhibiting akt signaling. Gastroenterology. 2010;138:241–254. doi: 10.1053/j.gastro.2009.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P, Lin JE, Chervoneva I, Schulz S, Waldman SA, Pitari GM. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase c restricts the proliferating compartment in intestine. Am J Pathol. 2007;171:1847–1858. doi: 10.2353/ajpath.2007.070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li P, Schulz S, Bombonati A, Palazzo JP, Hyslop TM, Xu Y, Baran AA, Siracusa LD, Pitari GM, Waldman SA, et al. Guanylyl cyclase c suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133(2):599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 20.Lin JE, Snook AE, Li P, Stoecker BA, Kim GW, Magee MS, Garcia AVM, Valentino MA, Hyslop T, Schulz S, et al. Gucy2c opposes systemic genotoxic tumorigenesis by regulating akt-dependent intestinal barrier integrity. PLoS One. 2012;7(2):e31686. doi: 10.1371/journal.pone.0031686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbons AV, Lin JE, Kim GW, Marszalowicz GP, Li P, Stoecker BA, Blomain ES, Rattan S, Snook AE, Schulz S, et al. Intestinal GUCY2C prevents TGF-β secretion coordinating desmoplasia and hyperproliferation in colorectal cancer. Cancer Res. 2013;73(22):6654–6666. doi: 10.1158/0008-5472.CAN-13-0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brenna O, Furnes MW, Munkvold B, Kidd M, Sandvik AK, Gustafsson BI. Cellular localization of guanylin and uroguanylin mrnas in human and rat duodenal and colonic mucosa. Cell Tissue Res. 2016;365(2):331–341. doi: 10.1007/s00441-016-2393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikpa PT, Sleddens HF, Steinbrecher KA, Peppelenbosch MP, de Jonge HR, Smits R, Bijvelds MJC. Guanylin and uroguanylin are produced by mouse intestinal epithelial cells of columnar and secretory lineage. Histochem Cell Biol. 2016;146(4):445–455. doi: 10.1007/s00418-016-1453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitaker TL, Witte DP, Scott MC, Cohen MB. Uroguanylin and guanylin: distinct but overlapping patterns of messenger rna expression in mouse intestine. Gastroenterology. 1997;113:1000–1006. doi: 10.1016/S0016-5085(97)70197-5. [DOI] [PubMed] [Google Scholar]
- 25.Vaandrager AB. Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase c. Mol Cell Biochem. 2002;230:73–83. doi: 10.1023/A:1014231722696. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol Rev. 2016;96:751–804. doi: 10.1152/physrev.00022.2015. [DOI] [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 28.Li P, Lin JE, Snook AE, Waldman SA. St-producing e. Coli oppose carcinogen-induced colorectal tumorigenesis in mice. Toxins (Basel). 2017;9:279. doi: 10.3390/toxins9090279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shailubhai K, Yu HH, Karunanandaa K, Wang JY, Eber SL, Wang Y, Joo NS, Kim HD, Miedema BW, Abbas SZ, et al. Uroguanylin treatment suppresses polyp formation in the apc(min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic gmp. Cancer Res. 2000;60:5151–5157. [PubMed] [Google Scholar]
- 30.Sharman SK, Islam BN, Hou Y, Singh N, Berger FG, Sridhar S, Yoo W, Browning DD. Cyclic-GMP–Elevating agents suppress polyposis in Apc min mice by targeting the preneoplastic epithelium. Cancer Prev Res (Phila). 2018;11:81–92. doi: 10.1158/1940-6207.CAPR-17-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinbrecher KA, Tuohy TM, Heppner Goss K, Scott MC, Witte DP, Groden J, Cohen MB. Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem Biophys Res Commun. 2000;273(1):225–230. doi: 10.1006/bbrc.2000.2917. [DOI] [PubMed] [Google Scholar]
- 32.Wilson C, Lin JE, Li P, Snook AE, Gong J, Sato T, Liu C, Girondo MA, Rui H, Hyslop T, et al. The paracrine hormone for the gucy2c tumor suppressor, guanylin, is universally lost in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(11):2328–2337. doi: 10.1158/1055-9965.EPI-14-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bashir B, Merlino DJ, Rappaport JA, Gnass E, Palazzo JP, Feng Y, Fearon ER, Snook AE, Waldman SA. Silencing the guca2a-gucy2c tumor suppressor axis in cin, serrated, and msi colorectal neoplasia. Hum Pathol. 2019;87:103–114. doi: 10.1016/j.humpath.2018.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blomain ES, Rappaport JA, Pattison AM, Bashir B, Caparosa E, Stem J, Snook AE, Waldman SA. Apc-β-catenin-tcf signaling silences the intestinal guanylin-gucy2c tumor suppressor axis. Cancer Biol Ther. 2020;21(5):441–451. doi: 10.1080/15384047.2020.1721262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD, Waldman SA. Guanylyl cyclase c is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci U S A. 1996;93(25):14827–14832. doi: 10.1073/pnas.93.25.14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrithers SL, Parkinson SJ, Goldstein S, Park P, Robertson DC, Waldman SA. Escherichia coli heat-stable toxin receptors in human colonic tumors. Gastroenterology. 1994;107:1653–1661. doi: 10.1016/0016-5085(94)90804-4. [DOI] [PubMed] [Google Scholar]
- 37.Danaee H, Kalebic T, Wyant T, Fassan M, Mescoli C, Gao F, Trepicchio WL, Rugge M. Consistent expression of guanylyl cyclase-c in primary and metastatic gastrointestinal cancers. PLoS One. 2017;12:e0189953. doi: 10.1371/journal.pone.0189953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darwich AS, Aslam U, Ashcroft DM, Rostami-Hodjegan A. Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab Dispos. 2014;42:2016. doi: 10.1124/dmd.114.058404. [DOI] [PubMed] [Google Scholar]
- 40.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 41.Creamer B, Shorter RG, Bamforth J. The turnover and shedding of epithelial cells. I. The turnover in the gastro-intestinal tract. Gut. 1961;2:110–118. doi: 10.1136/gut.2.2.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P, Waldman SA. Corruption of homeostatic mechanisms in the guanylyl cyclase c signaling pathway underlying colorectal tumorigenesis. Cancer Biol Ther. 2010;10:211–218. doi: 10.4161/cbt.10.3.12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer. 2014;14:468–480. doi: 10.1038/nrc3744. [DOI] [PubMed] [Google Scholar]
- 44.Steinbrecher KA, Harmel-Laws E, Garin-Laflam MP, Mann EA, Bezerra LD, Hogan SP, Cohen MB. Murine guanylate cyclase c regulates colonic injury and inflammation. J Immunol. 2011;186:7205–7214. doi: 10.4049/jimmunol.1002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brierley SM. Guanylate cyclase-c receptor activation: unexpected biology. Curr Opin Pharmacol. 2012;12:632–640. doi: 10.1016/j.coph.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Lan D, Niu J, Miao J, Dong X, Wang H, Yang G, Wang K, Miao Y. Expression of guanylate cyclase-c, guanylin, and uroguanylin is downregulated proportionally to the ulcerative colitis disease activity index. Sci Rep. 2016;6:25034. doi: 10.1038/srep25034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin JE, Colon-Gonzalez F, Blomain E, Kim GW, Aing A, Stoecker B, Rock J, Snook AE, Zhan T, Hyslop TM, et al. Obesity-induced colorectal cancer is driven by caloric silencing of the guanylin-gucy2c paracrine signaling axis. Cancer Res. 2016;76:339–346. doi: 10.1158/0008-5472.CAN-15-1467-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang WL, Masih S, Thadi A, Patwa V, Joshi A, Cooper HS, Palejwala VA, Clapper ML, Shailubhai K. Plecanatide-mediated activation of guanylate cyclase-C suppresses inflammation-induced colorectal carcinogenesis in Apc +/Min-FCCC mice. World J Gastrointest Pharmacol Ther. 2017;8:47–59. doi: 10.4292/wjgpt.v8.i1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S, et al. Tissue-specific and inducible cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 50.Schulz S, Lopez MJ, Kuhn M, Garbers DL. Disruption of the guanylyl cyclase-c gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J Clin Invest. 1997;100:1590–1595. doi: 10.1172/JCI119683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian X, Prabhakar S, Nandi A, Visweswariah SS, Goy MF. Expression of gc-c, a receptor-guanylate cyclase, and its endogenous ligands uroguanylin and guanylin along the rostrocaudal axis of the intestine*. Endocrinology. 2000;141:3210–3224. doi: 10.1210/endo.141.9.7644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.