

Abstract
Massive formalin-fixed, paraffin-embedded (FFPE) tissue archives exist worldwide, representing an invaluable resource for clinical proteomics research. However, current protocols for FFPE proteomics lack standardization, efficiency, reproducibility, and scalability. Here we present high-yield protein extraction and recovery by direct solubilization (HYPERsol), an optimized workflow using ultrasonication and S-Trap sample processing that enables proteome coverage and quantification from FFPE samples comparable to that achieved from flash-frozen tissue (average R = 0.936). When applied to archival samples, HYPERsol resulted in high-quality data from FFPE specimens in storage for up to 17 years, and may enable the discovery of new immunohistochemical markers.
Keywords: FFPE, tissue, immunohistochemistry, sample preparation, S-Trap, clinical proteomics
Graphical Abstract
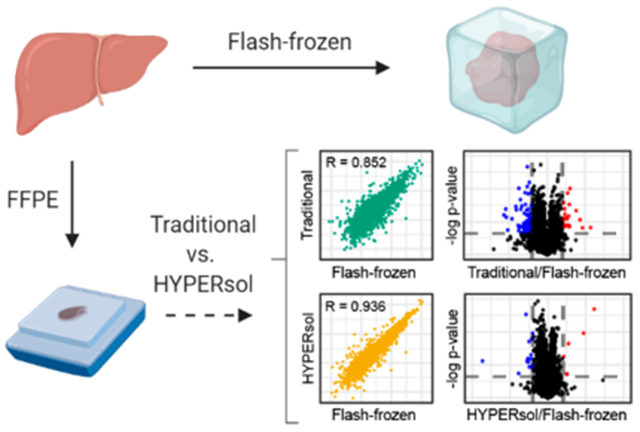
INTRODUCTION
Formalin fixation and paraffin embedding (FFPE) is a tissue preparation method common in experimental research and medicine. It is standard in all pathology departments where pathological diagnosis is based on tissue section staining and immunohistochemistry on FFPE slides. The method is over one hundred years old and yields biologically inactive samples that are stable at room temperature for decades and longer.1–3 The ubiquity of this practice in pathology combined with the unique stability of FFPE samples has resulted in massive numbers of specimens housed in countless historical tissue archives around the world. These collections represent an invaluable resource for retrospective research and translational studies, especially when specimens are paired with medical records describing the diagnosis and course of disease. However, despite this huge potential, proteomic analysis of FFPE samples has yet to be widely adopted.4
Multiple disparate protocols for proteomic analysis of FFPE material exist. Traditionally, paraffin is removed from FFPE tissues by stepwise incubation of samples with xylene, ethanol, multiple aqueous ethanol solutions of gradually decreasing ethanol concentration, and finally an aqueous buffer.5–10 Downstream of this time-consuming process, a variety of alternative methods have been developed to solubilize the protein material, which can broadly be categorized as detergent-free and detergent-based. Detergent-free approaches to protein solubilization include the use of an alternative organic solvent such as the SubX Clearing Agent.11 Alternatively, samples can be resuspended in buffer containing RapiGest, a liquid chromatography–mass spectrometry (LC-MS)-compatible surfactant.12
The detergent-free approaches can appear attractive because detergents are notorious for contaminating both liquid chromatographs and mass spectrometers. However, the omission of detergent raises concerns about the extent of solubilization that is achieved, and whether incomplete sample solubilization might bias proteomic results. Accordingly, a number of detergent-based methods have also been developed, generally using sodium dodecyl sulfate (SDS). Though the inclusion of SDS improves sample dissolution, it necessitates downstream SDS removal. Techniques to remove SDS from FFPE-derived tissue homogenates include filter-aided sample preparation (FASP),13 SDS-PAGE and in-gel digestion,14 and the single-pot, solid-phase enhanced sample preparation (SP3) method, in which peptides are immobilized on a mixture of hydrophobic and hydrophilic paramagnetic beads and extensively washed.15
An alternative technique for the removal of SDS from protein lysates is the use of suspension trapping (S-Trap) technology.16 S-Traps are commercially available and are also readily fabricated in-house, suggesting that they might represent a cost-effective alternative to the SP3-CTP method. S-Traps were also recently shown to outperform FASP in terms of sample processing time, reproducibility, and depth-of-coverage.17 Moreover, the ability to efficiently remove even high concentrations of SDS presents the possibility of directly solubilizing FFPE tissues without laborious, time-consuming deparaffinization steps that may result in sample loss.
We reasoned that S-Traps might be part of an optimized, broadly applicable FFPE workflow for clinical proteomics. To test this, we embarked on an evaluation of a variety of workflows for FFPE proteomics, comparing each to matched flash-frozen tissue as a “gold-standard”. We divide the general workflow into three essential steps: extraction, tissue homogenization, and protein recovery. We evaluate two alternative strategies for tissue extraction: xylene–ethanol deparaffinization (designated by an “X”), and direct solubilization in aqueous buffer with 5% SDS (designated by a “D”). We compare two techniques for homogenization: probe sonication (designated by a “P”) and ultrasonication (designated by a “U”). Finally, we compare two techniques for protein recovery for downstream analysis: methanol–chloroform precipitation (designated by an “M”) and S-Trap sample processing (designated by an “S”).
We find that the combination of direct solubilization, ultrasonication, and S-Trap processing, a method we term HYPERsol (for high-yield protein extraction and recovery by direct solubilization), enables depth-of-coverage, reproducibility, and quantification closely approaching results obtained from flash-frozen tissue. We also demonstrate that this method efficiently solubilizes total protein from 18 distinct human tissue types of clinical interest. Finally, we demonstrate the utility of HYPERsol on a set of 32 archival samples of malignant peripheral nerve sheath tumor (MPNST), melanoma, and synovial sarcoma that had been in storage for up to 17 years. These three tumor types are considered histomorphologic mimics and may be mistaken for one another in routine clinical practice, though these cancer types have different prognoses and therapeutic options. Proteomic analysis both identified known tumor markers and suggested novel markers that may further aid in differentiating these tumors. Moreover, we found that the number of proteins that were able to be quantified in each sample was not significantly associated with the duration of storage, suggesting that this method will enable retrospective analysis of existing specimens in archives around the world.
EXPERIMENTAL PROCEDURES
IRB Statement
Human samples were collected under protocols approved by the Institutional Review Board (IRB) of the University of Pennsylvania or documented as exempt from IRB review. All samples were subjected to histopathologic review for confirmation of diagnosis and selection of the region for tissue isolation.
FFPE Tissue Processing
Automated tissue processing was carried out in a Leica Peloris II processor (Leica Biosystems) with the following incubation settings: 60 min 10% neutral buffered formalin, 60 min formalin 10% neutral buffered formalin, 80% EtOH 20 min, 95% EtOH 60 min, 100% EtOH 30 min, 100% EtOH 50 min, 100% EtOH 60 min, xylene 30 min, xylene 50 min, xylene 60 min, paraffin 60 min, paraffin 60 min, paraffin 60 min. Following processing, samples were embedded in paraffin and stored in blocks at room temperature prior to processing.
Hematoxylin and Eosin Staining
The original diagnostic histologic sections were used for confirmation of diagnosis and for the photomicrographs shown in Figure 3. These were standard 5 μm tissue sections stained with hematoxylin/eosin according to standard histopathology protocols. Digital images were taken on a Leica DMC 4500 camera and captured and processed using the Leica Digital Application Suite v4.12.
Figure 3.
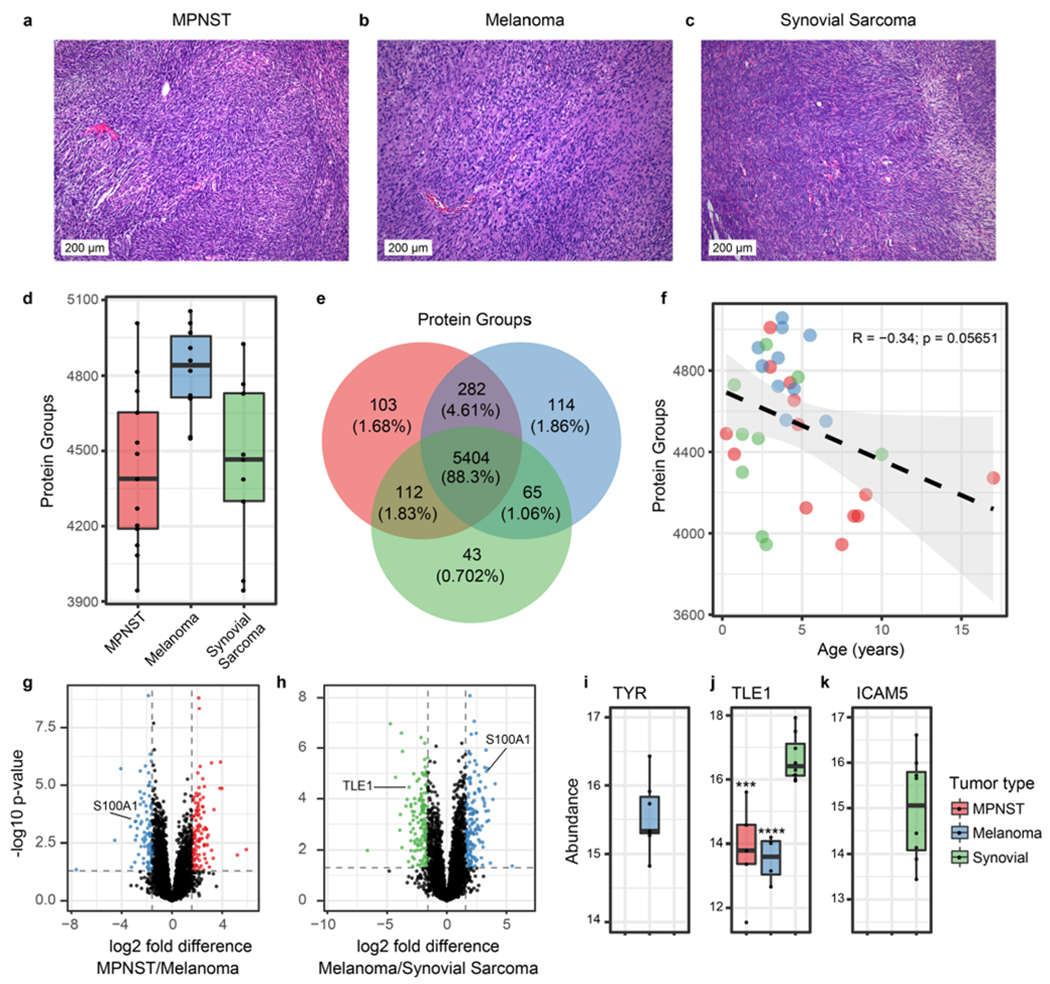
HYPERsol enables proteomic characterization of archival tissue samples. (a–c) Representative hematoxylin and eosin (H&E) stains of malignant peripheral nerve sheath tumor (MPNST), melanoma, and synovial sarcoma. (d) Tukey boxplot depicting the number of proteins identified in each tumor type (n = 13 MPNST, 10 melanoma, 9 synovial sarcoma). (e) Venn diagram illustrating the overlap in detected proteins among the three tumor types. (f) Scatter plot depicting the correlation between the duration of storage (“Age”) of specimens and the number of protein groups that were identified. Dotted line indicates the best-fit line with a 95% confidence interval. R and p-values are based on Pearson’s product moment correlation. (g) Volcano plot illustrating differential protein expression in MPNST and melanoma. 5372 proteins are depicted. (h) Volcano plot illustrating differential protein expression in melanoma and synovial sarcoma. 5090 proteins are depicted. (i) Tukey boxplot depicting the abundance of tyrosinase (TYR) across the three tumor types (detected in 0 MPNSTs, 9 melanomas, and 0 synovial sarcomas). (j) Tukey boxplot depicting the abundance of TLE1 across the three tumor types (detected in 5 MPNSTs, 4 melanomas, and 8 synovial sarcomas). Asterisks indicate statistical significance when compared to synovial sarcoma with p < 0.001 = *** and p < 0.0001 = ****. (k) Tukey boxplot depicting the abundance of ICAM5 across the three tumor types (detected in 0 MPNSTs, 0 melanomas, and 9 synovial sarcomas).
Preparation of Tissue Cores
FFPE cores were obtained from tissue blocks using a 1 mm Kai Biopsy Punch (Miltex-Integra 33–31AA). For liver blocks, which were generated for the purpose of this study, the thickness was approximately 0.3 cm. For archival tumor samples, the thickness of the blocks varied considerably from approximately 0.1 to 0.3 cm. In all cases, cores were trimmed of excess paraffin using a sterile razor blade and, if necessary, pooled, until 5 mg of total FFPE material was available from each sample.
Extraction (Deparaffinization or Direct Solubilization)
For the xylene–ethanol deparaffinization (designated by “X”), 5 mg of tissue cores were diced into small pieces, transferred to 1.5 mL Eppendorf tubes, and resuspended in 10× volume (v/w = μL per mg of dry tissue weight) of xylene and incubated at 37 °C for 10 min with gentle agitation. Following centrifugation xylene removal, the process was repeated with xylene and then twice with 100% ethanol, 95% ethanol, 85%, 70%, 50%, 20%, and then water. Samples were then resuspended in solubilization buffer containing 5% SDS and 100 mM Tris pH 8.5, and homogenized with a micropestle. Samples were then passed through an 18-gauge needle 10×, then a 21-gauge needle 10×. Following sonication (as described below), the process was repeated. The homogenized lysate was then spun down at 16 000g in a benchtop centrifuge for 15 min and the total protein concentration of the soluble fraction was measured using a BCA assay (Pierce, catalog no. 23225). For direct solubilization (designated by “D”), 5 mg of tissue cores were diced into small pieces, transferred to 1.5 mL Eppendorf tubes, and resuspended in 20× volume/weight of solubilization buffer containing 5% SDS and 100 mM Tris pH 8.5, and incubated at 50 °C for 10 min. The pellet was homogenized with a micropestle, and passed through an 18-gauge needle 10×, followed by a 21-gauge needle 10×. Following sonication as described below, the samples were placed on a heat block at 80 °C for 1 h. The samples were removed and the sonication was repeated. The samples were returned to the heating block for 1 h. The homogenized lysate was spun down at 16 000g in a benchtop centrifuge for 15 min and the total protein concentration in the soluble fraction was measured using a BCA assay (Pierce, catalog no. 23225).
Homogenization (Probe Sonication or Covaris AFA Ultrasonication)
For probe sonication, deparaffinized samples were subjected to benchtop sonication with a Fisher Scientific Sonic Dismembrator Model 100 with 3 × 30 s pulses, 20% power, and with a 50% duty ratio at room temperature. Two rounds of sonication were performed as described above. For AFA sonication, deparaffinized samples were transferred to Covaris microTUBE-130 AFA Fiber Screw-Caps (520216) and sonicated in a Covaris S220 AFA in the Screw-Cap microTUBE-130 holder (500339). The general parameters were as follows: water level set point 15, chiller set point 18 °C, peak incident power 175 W, duty factor 10%, cycles per burst 200, instrument temperature 20 °C. Two rounds of sonication were performed: In the first round the treatment time was 300 s. In the second round the treatment time was 360 s.
Tissue Yield
To calculate tissue yield and percent solubilized for each tissue type depicted in Figure S1, 1 mm tissue cores were weighed, then directly placed in solubilization buffer, ground with a micropestle, and allowed to equilibrate for 15 min at room temperature. The sample was spun at 16 000g in a benchtop centrifuge and the initial supernatant was saved for subsequent BCA assay. The hydrated FFPE pellets were weighed again to account for the weight of the buffer. The samples were then directly solubilized as described in the above Extraction section, and subject to AFA sonication as described in the above Homogenization section, and spun down again at 16 000g in a benchtop centrifuge. The supernatant was saved for BCA assay, and the final pellet weighed again. The percent solubilized was calculated as . The yield was calculated as [(protein conc. of supernatant 1 × volume of supernatant 1) + (protein conc. of supernatant 2 × volume of supernatant 2)]/mass of initial dry pellet.
For all samples, the amount of protein in the initial resuspension/equilibration solution contributed negligibly to the overall yield.
Recovery of Protein and Preparation for LC-MS
For methanol/chloroform precipitation, sonicated samples were processed as previously described.18 The sample volume containing 100 μg of total protein was adjusted to 300 μL with water. 300 μL of ice-cold methanol was added, and the sample was vortexed briefly. Next, 75 μL of ice-cold chloroform was added and the sample was vortexed again, then immediately centrifuged for 1 min at 9000g on a benchtop centrifuge. Following centrifugation, the upper phase was removed and discarded. 300 μL of ice cold methanol was added and the sample was vortexed again, then spun at 16 000g in a benchtop centrifuge. The supernatant was removed without disturbing the pellet, and the tube was left with the cap off for 5 min to allow excess methanol to evaporate. The pellet was then resuspended in 20 μL of 6 M urea + 2 M thiourea. Following resuspension, 100 μL of 50 mM ammonium bicarbonate pH 8.0 with 1.2× HALT protease inhibitor cocktail (Pierce catalog no. 87786) was added, and DTT was added to a final concentration of 10 mM. Samples were incubated for 30 min at room temperature. Iodoacetamide was added to a final concentration of 20 mM and samples were incubated in the dark for 30 min. An additional 10 mM DTT was added to quench the derivatization reaction, and the samples were digested with trypsin at a 1:50 ratio overnight at room temperature. Alternatively, for S-Trap recovery, sonicated samples were reduced and alkylated as described above. Trypsin digestion and cleanup was then performed according to the S-Trap manufacturer’s instructions. Briefly, 50 μg protein was loaded on S-Trap micro spin columns (ProtiFi, LLC, catalog no. C02-micro) and washed extensively with 90% methanol containing 100 mM TEAB, pH 7.1. Trypsin was added directly to the microcolumn at a 1:20 ratio in 50 mM TEAB, pH 8, and samples were incubated in a water bath at 47 °C for 1 h. Peptides were eluted by serial addition of 50 Mm TEAB, 0.2% formic acid, and 0.2% formic acid in 50% acetonitrile.
High-pH Reversed-Phase Fractionation for Spectral Library Generation
Small peptide aliquots from each liver sample (paired FFPE and flash-frozen collections) processed by the different sample preparation workflows described above were pooled and acidified with 0.1% trifluoroacetic acid (TFA). The pooled peptide mix was loaded on a Harvard apparatus Micro SpinColumn (Cat# 74–4601), washed with 0.1% TFA, and eluted with 12 serial additions of 100 mM ammonium formate, pH 10, containing increasing concentrations of acetonitrile (10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 35, 60% ACN). Fractions were pooled (1 + 6, 2 + 7, etc.), dried in a centrifugal evaporator, and desalted prior to analysis. The same was done to the 32 tumor samples to allow for creation of a tumor-specific spectral library.
Desalting
All samples were resuspended in 0.1% TFA, loaded on homemade C18 stage-tips (3 M Empore Discs) and desalted as previously described, with minor modifications.19 Briefly, columns were conditioned with 100 μL acetonitrile and equilibrated with 100 μL 0.1% TFA. Samples were loaded and the stage-tip was washed with 100 μL 0.1% TFA before peptides were eluted with 100 μL 0.1% formic acid in 60% acetonitrile.
Liquid Chromatography and Mass Spectrometry
For the initial series of experiments (data in Figure 1 and Figure S2), samples were analyzed on a Thermo Scientific Easy nLC 1000 coupled to a Thermo Fusion Orbitrap Tribrid via a Nanospray Flex Ion Source (Thermo Scientific). The LC was equipped with a 75 μm × 20 cm column packed in-house using Reprosil-Pur C18-AQ (2.4 μm; Dr. Maisch GmbH, Germany). Buffer A was 0.1% formic acid and Buffer B was 0.1% formic acid in 80% acetonitrile. The flow rate was 400 nL/min. For spectral library generation, the gradient was as follows: start at 2% B, 5% B over 2 min, 8% B over 2 min, 10% B over 2 min, 12% B over 3 min, 15% B over 5 min, 18% B over 12.50 min, 32% B over 33 min, 34% B over 5 min, 37% B over 3 min, 40% B over 3 min, 43% B over 2 min, 46% B over 2 min, 49% B over 2 min, 55% B over 3 min, 98% B over 10 s, hold for 10 min. Spectral library samples were analyzed in data-dependent acquisition (DDA) mode with the following settings: MS1 350–1200 m/z, 120 K resolution, AGC target 1 × 106, max inject time 60 ms; MS2 15K resolution, AGC 5 × 104, max inject time 120 ms, Top Speed = 3 s, isolation window = 2 m/z, stepped HCD collision energy 29 ± 5%, include z = 2–5. For samples, the gradient was as follows: start at 2% B, 3% B over 2 min, 24% B over 52 min, 40% B over 20 min, 48% B over 4 min, 55% B over 2 min, 98% B over 10 min, hold for 9 min 50 s. DIA analysis on the Fusion was performed with the following settings: MS1 350–1200 m/z, 120 K resolution, AGC target 1 × 106, max inject time 60 ms, MS2 30K resolution, AGC 1 × 106, max inject time 54 ms, 36 windows of 23.7 m/z, stepped HCD collision energy 30 ± 5%.
Figure 1.
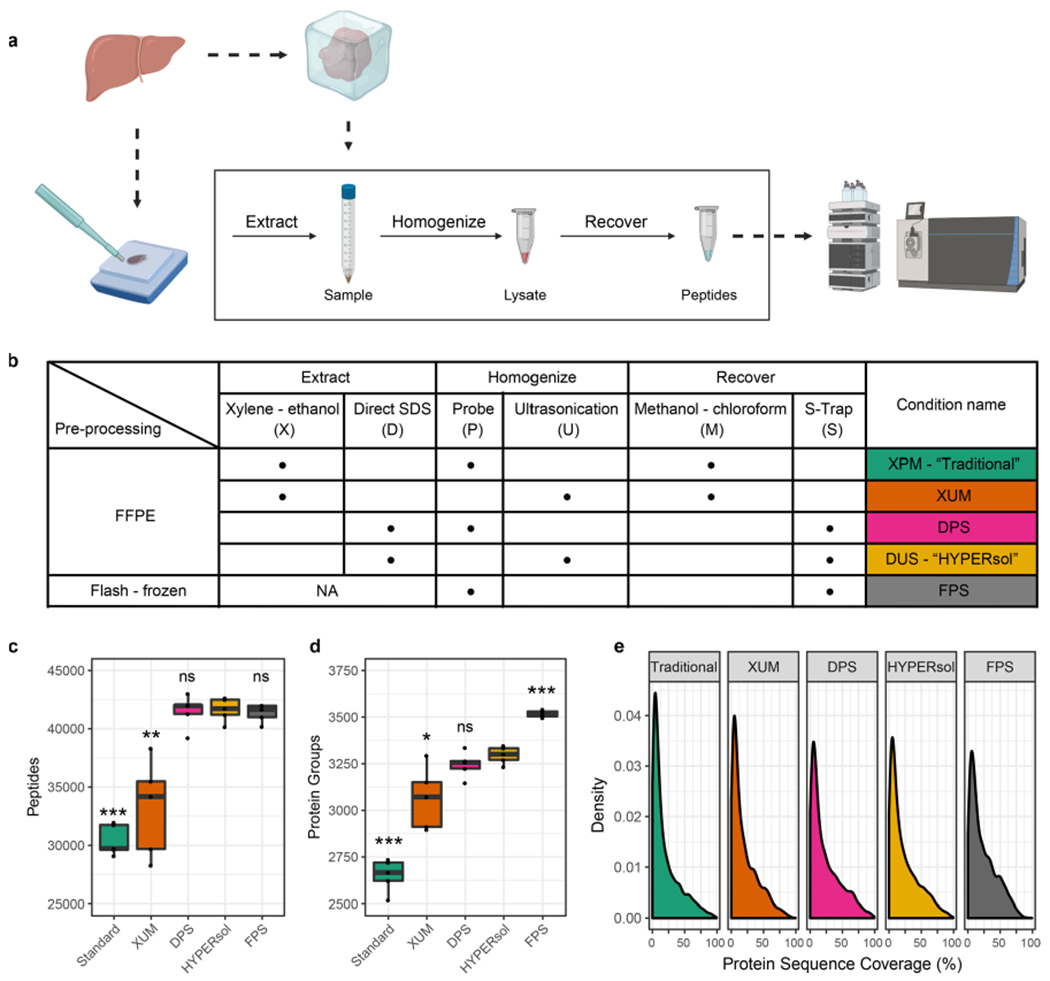
HYPERsol improves the depth-of-coverage achievable from FFPE samples compared to the Traditional workflow. (a) Schematic illustrating study design. Freshly dissected liver samples were either flash-frozen or formalin-fixed and paraffin-embedded prior to proteomic analysis. (b) Table of experimental conditions. (c) Tukey boxplot displaying the number of peptide identifications across conditions. (d) Tukey boxplot displaying the number of protein group identifications across conditions. (e) Density plot illustrating protein sequence coverage across conditions. The conditions were as follows. Traditional: xylene–ethanol, probe, methanol–chloroform. XUM: xylene–ethanol, ultrasonication, methanol–chloroform. DPS: direct, probe, S-Trap. HYPERsol: direct, ultrasonication, S-Trap. FPS: flash-frozen, probe, S-Trap. For box plots, n = 5 and asterisks indicate statistical significance when compared to FPS with Welch’s two-tailed t test and p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***.
For the second series of experiments (data in Figure 2), samples were analyzed on a Thermo Dionex Ultimate 3000 with an equivalent column and buffer setup coupled to a Thermo QE HF-X. The gradient was as follows: start at 2% B, 3% B over 2 min, 24% B over 52 min, 40% B over 20 min, 48% B over 4 min, 55% B over 2 min, 95% B over 6 s, hold for 10 min. This was followed by 10 min of 2% B to re-equilibrate the column for the next run. DIA analysis was performed with the following settings: MS1 350–1200 m/z, 120 K resolution, AGC 3 × 106, max inject time 50 ms, MS2 30K resolution, AGC 3 × 106, max inject time auto, MSX 1, 36 windows with a width of 23.7 m/z and stepped NCE (25.5, 27, 30).
Figure 2.
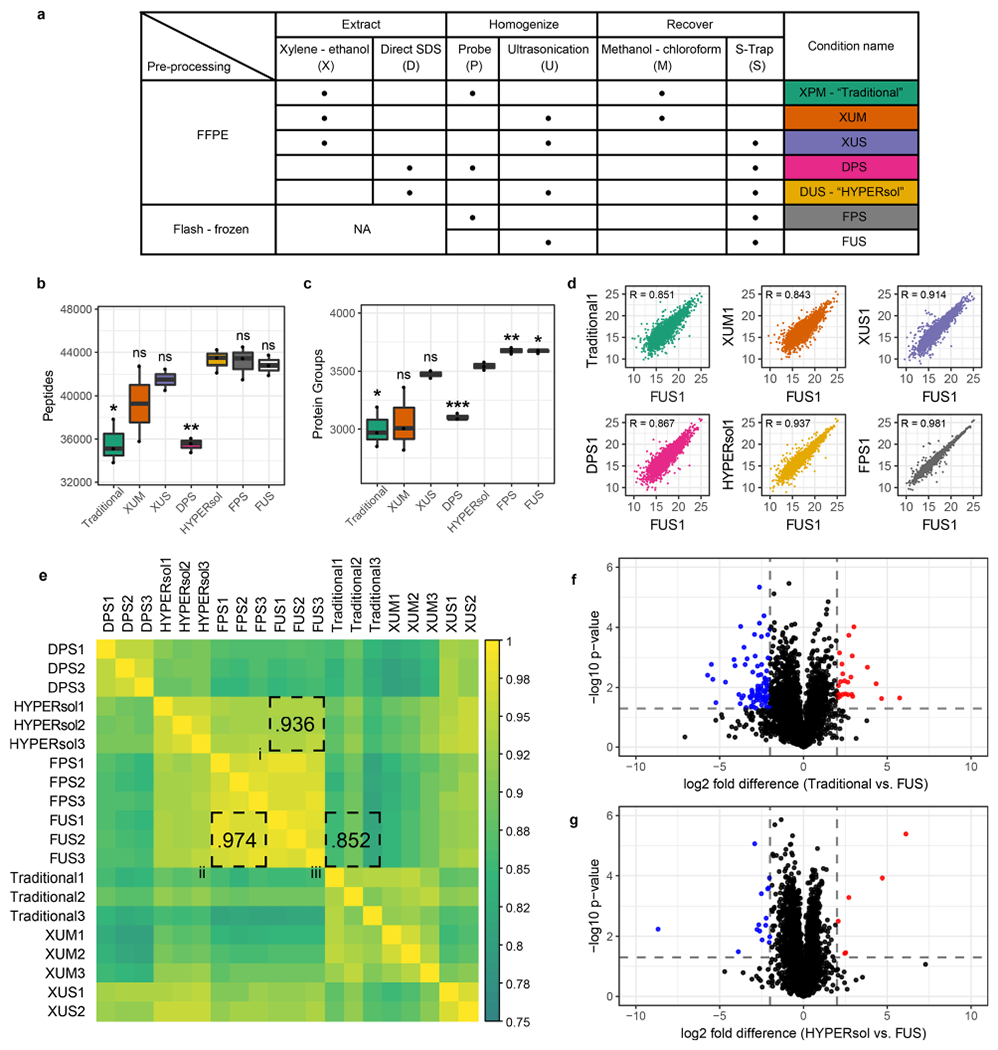
HYPERsol yields proteomic data that closely resemble matched flash-frozen tissue. (a) Table of experimental conditions. The conditions were as follows. Traditional: xylene–ethanol, probe, methanol–chloroform. XUM: xylene–ethanol, ultrasonication, methanol–chloroform. XUS: xylene–ethanol, ultrasonication, S-Trap. DPS: direct, probe, S-Trap. HYPERsol: direct, ultrasonication, S-Trap. FPS: flash-frozen, probe, S-Trap. FUS: flash-frozen, ultrasonication, S-Trap. (b) Tukey boxplot displaying the number of peptide identifications across conditions. (c) Tukey boxplot displaying the number of protein group identifications across conditions. For both (b) and (c), n = 3 for all conditions except XUS, for which n = 2, and asterisks indicate statistical significance when compared to HYPERsol with Studenťs two-tailed t test and p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***. (d) Representative scatter plots depicting the correlation between proteomic data from each experimental condition against data from FUS. R values are Pearson correlation coefficients. (e) Correlation matrix depicting the Pearson correlation coefficients across all pairwise run comparisons. In this panel, shorthand is used for both Traditional (XPM) and HYPERsol (DUS) for the sake of space. (f) Volcano plot comparing the relative abundance of proteins detected in both the Traditional and FUS conditions. (g) Volcano plot comparing the relative abundance of proteins detected in both the HYPERsol and FUS conditions. Dotted lines indicate absolute log2 fold-difference = 2 and p-value = 0.05.
For analysis of tumor samples, (data in Figure 3) samples were analyzed with an Easy nLC with an equivalent column and buffer setup coupled to a Thermo QE HF-X. The gradient was as follows: 1% B to 4% B over 3 min, 6% over 3 min, 8% over 4 min, 10% over 5 min, 12% over 18 min, 17% over 9 min, 26% over 41 min, 28% over 9 min, 30% over 6 min, 32% over 5 min, 34% over 4 min, 36% over 4 min, 38% over 3 min, 41% over 3 min, 52% over 3 min, 90% over 5 min, hold 90% for 10 min. DDA analysis of the fractions of the pooled sample was performed with the following settings: MS1 350–1200 m/z, 120 K resolution, AGC 1 × 105, max inject time 120 ms, MS2 30K resolution, AGC 1 × 105, max inject time 120 ms, Top N = 20, isolation window 1.3 m/z, stepped NCE (25.5, 27, 30), exclude z = unassigned and z = 1. DIA analysis of the individual samples was performed with the following settings: MS1 350–1200 m/z, 120 K resolution, AGC 3 × 106, max inject time 50 ms, MS2 30K resolution, AGC 3 × 106, max inject time auto, MSX 1, 45 windows with a width of 20 m/z, stepped NCE (25.5, 27, 30).
Mass Spectrometry Data Analysis
The liver-specific spectral library was generated from the data-dependent analysis (DDA) runs of the fractions of the pooled liver sample in Spectronaut Pulsar X with default settings (digest type = specific, missed cleavage = 2, min peptide length = 7, max peptide length = 52, toggle N-terminal M = true), and using the 2017–10–25 version of the Homo sapiens [SwissProt TaxID = 9606] proteome. The individual data-independent analysis (DIA) liver runs were also analyzed with directDIA and the resulting search archives were used to improve the Pulsar library search. For the methyl and methylol adduct search, these were included as variable modifications. For tumor samples, spectral libraries were likewise generated in Spectronaut Pulsar X as described above. All DIA runs were analyzed in Spectronaut Pulsar X using BGS Default Factory Settings. Peptide and protein intensities were log2 transformed and processed by a two-step normalization. First, within each run, the run-level median intensity was subtracted from each measured intensity such that the values were normally distributed around 0. Then the global median intensity from the entire sample set was added back such that all intensity values were positive. No imputation was performed. Figures were generated in R using the packages ggplot2, ggthemes, corrplot, and VennDiagram. Figure 1a and the Table of Contents figure were created in BioRender.
Data Availability
The raw mass spectrometry data have been deposited to CHORUS (https://chorusproject.org/pages/index.html) under project #1635.
Immunohistochemistry
Immunohistochemistry of FFPE tissue was performed using an antibody against human Tyrosinase (Thermo Fisher, MS-800-P, clone T311; diluted 1:75) or TLE1 (Cell Marque 401M-18, clone IF5; prediluted). Staining was done on a Leica Bond-III instrument using the Bond Polymer Refine Detection System (Leica Microsystems DS9800). Heat-induced epitope retrieval was done for 20 min with ER2 solution (Leica Microsystems AR9640). Incubation (1:75) was 15 min, followed by 8 min postprimary step, 8 min incubation with polymer HRP, then block endogenous peroxidase for 5 min, followed by 10 min DAB. The experiment was done at room temperature and slides were washed 3× between each step with bond wash buffer or water.
RESULTS
We developed HYPERsol by optimizing the techniques of deparaffinization, protein solubilization, and sample preparation applied to bottom-up proteomics analysis of FFPE samples. We compared the traditional xylene–ethanol deparaffinization procedure (designated by “X”) to a procedure in which FFPE cores were directly solubilized in buffer containing 5% SDS (designated by “D”). Protein extraction was performed with either probe sonication (designated by “P”) or ultrasonication (designated by “U”). Protein was recovered and processed for liquid chromatography–mass spectrometry with either Wessel–Flügge methanol–chloroform precipitation18 and in-solution digestion (designated by “M”) or with S-Traps (designated by “S”) [Figure 1a,b].
We first examined the extent of solubilization of FFPE liver samples achieved by each method. Compared to the traditional workflow employing xylene–ethanol deparaffinization and probe sonication (designated by “XP”),20 direct resuspension in 5% SDS buffer followed by ultrasonication (designated by “DU”) solubilized >2-fold more protein with a corresponding >2-fold decrease in residual insoluble material [Figure S1a–c]. To further benchmark the improved method, we tested its ability to solubilize cores from 18 different FFPE human tissue types. The method solubilized between 64% to 96% of the samples by mass, and the yield of soluble protein ranged from 40–116 μg per mg of dry FFPE, suggesting that this protocol may be compatible with workflows that require significant amounts of starting material such as post-translational modification enrichments (e.g., acetyl- or phosphoproteomics) [Figure S1d,e]. It will be important to determine whether this method is compatible with laser microdissection, as results from bulk tissue are not easily extrapolated to samples consisting of very low cell numbers.9
To directly compare the performance of each sample preparation workflow for proteomic analysis against a standard tissue source, we utilized tissue from five human livers. Each sample was split at the time of autopsy: a portion was immediately flash-frozen, and another portion fixed with formalin, processed and embedded according to standard histopathology protocols.
On average, compared to the combination of xylene–ethanol deparaffinization, probe sonication and methanol–chloroform precipitation previously used in our laboratory (designated by “XPM”; “Traditional”), the combination of direct 5% SDS solubilization, AFA ultrasonication, and S-Trap sample processing (designated by “DUS”; “HYPERsol”) resulted in the identification of 37% more peptides (from 30 432 ± 1324 to 41 643 ± 1012) and 24% more protein groups (from 2653 ± 87 to 3297 ± 46), a depth closely approaching that obtained in a flash-frozen samples processed with SDS, probe sonication and S-Traps (designated by “FPS”; 3517 ± 18) [Figure 1c,d; Table S1, S2]. Among the Traditional, HYPERsol, and FPS data sets the protein group overlap was 84.1%, and an additional 11% of proteins were identified in HYPERsol and FPS, but not Traditional [Figure S2a]. The average protein sequence coverage ranged from 20.0% to 23.9%, with both DPS and HYPERsol enabling statistically significant increases compared to the Traditional workflow [Figure 1e, Figure S2c].
Counterintuitively, both HYPERsol and direct solubilization, probe sonication, S-Trap (designated by “DPS”) workflows yielded slightly more peptide identifications (IDs) than the FPS workflow [Figure 1c]. In addition, 4567 peptides were identified in both Traditional and HYPERsol, but not FPS [Figure S2b]. The distribution of grand average of hydropathy (GRAVY) scores of these unique peptides was similar to that of the peptides that were unique to the flash-frozen samples [Figure S2d–f; Table S3]. Further inspection revealed these unique peptides to be primarily the result of missed tryptic cleavages of highly abundant proteins, an effect presumably resulting from formalin cross-linking blocking trypsin access. When lysine monomethylation and methylolation, two known chemical artifacts of formalin fixation, were included as variable modifications during both spectral library generation and searching, they were observed to be enriched in all FFPE samples, affecting approximately 5% and 0.15% of all detected peptides, respectively [Figure S2g,h, Table S3], which is in line with previous reports.21 Like missed cleavages, these modifications were primarily detected on highly abundant proteins, and omitting them from database searching did not greatly alter the library composition [Figure S2i,j]. The distribution of gene ontology (GO) component terms of detected proteins was similar across all sample preparation methods, but subtle statistically significant differences were observed when comparing FFPE conditions to FPS. Extracellular, cytoplasmic, and membrane proteins were modestly overrepresented in FFPE samples relative to FPS. These differences were minimized by the HYPERsol method [Figure S2k; Table S4]. All together, these results suggest that, despite the minor presence of formalin-induced artifacts, HYPERsol sample processing enables deeper and more reproducible FFPE proteome analysis than the Traditional xylene–ethanol deparaffinization workflow.
Next, in order to evaluate the extent to which protein extracted from FFPE samples matches protein extracted from frozen tissue, we directly compared each method against 2 to 3 replicate samples derived from the same patient. We introduced two additional conditions: “XUS” (xylene–ethanol, ultrasonication, S-Trap) to determine if the combination of ultrasonication and S-Trap processing could improve the data quality from xylene-deparaffinized samples, and “FUS” (flash-frozen, ultrasonication, S-Trap) to determine if ultrasonication would enable deeper proteome coverage than probe sonication on flash-frozen tissue [Figure 2a].
Again, among FFPE conditions, the HYPERsol protocol resulted in the greatest number of peptide and protein group IDs [Figure 2b,c; Table S5, S6]. The numbers of peptides and protein groups identified in the XUS samples were markedly better than those from the Traditional workflow, indicating that the combination of ultrasonication and S-Trap cleanup can partially compensate for the biases introduced through xylene–ethanol deparaffinization. Unexpectedly, whereas in the previous experiment, the DPS condition was nearly equivalent to HYPERsol, in this experiment, the DPS samples more closely resembled XPM. We suspect that this variability is attributable to the inherent variability in manual probe sonication, in which an operator manually dips the probe tip into the sample and estimates 1 s pulses. Substitution of the probe sonication with ultrasonication did not improve the number of peptide or protein group IDs from the flash-frozen tissue, suggesting that the increased sonication energy afforded by ultrasonication is only obligatory when solubilizing protein from physically tougher FFPE. Nevertheless, given the improved performance of ultrasonication relative to probe sonication in the other conditions, we considered the FUS data set as the ground truth data set for subsequent comparisons.
In order to compare the similarity of the proteomic data sets generated via each sample preparation workflow, we examined the Pearson correlation coefficients among the protein quantification tables derived from each workflow [Figure 2d,e; Table S6, S7] against FUS. Overall, the HYPERsol data sets were exceptionally similar to the FUS data sets, with R ranging from 0.928 to 0.951 across any pairwise comparison, and an average R of 0.936 [Figure 2e, box i]. The average correlation between the FUS and FPS data sets was 0.974, indicating that the variability introduced by FFPE and HYPERsol extraction is not substantially greater than that introduced by alternative sample preparation strategies for replicates of the same flash-frozen tissue [Figure 2e, box ii]. Analyses of protein extracted with the Traditional method were less similar (range = 0.811–0.890, average = 0.852), suggesting that in addition to reducing the overall number of IDs, the incomplete solubilization and recovery afforded by the Traditional workflow also distorts the relative abundance of the detected proteins [Figure 2e, box iii]. To further explore this possibility we generated volcano plots comparing the relative abundance of protein groups that were identified across both Traditional and FUS or both HYPERsol and FUS extractions [Figure 2f,g; Table S8]. Whereas use of the Traditional protocol resulted in 120 proteins with absolute log2 estimated expression difference >2 and p-value <0.05 out of the 2956 quantified (101 underestimated, 19 overestimated; representing 4.0% of total proteins), the use of HYPERsol reduced this number to only 24 out of 3517, (6 overestimated, 18 underestimated; representing (0.68% of total proteins) reducing experimental noise and thereby effecting a more faithful representation of the composition of the original tissue.
While many potential applications exist, proteomic analysis of FFPE tissue is especially well-suited to identify new immunohistochemical (IHC) markers to facilitate the diagnosis of tumors for which histomorphology is insufficiently specific. Malignant peripheral nerve sheath tumor (MPNST) is one such tumor which is notoriously difficult to diagnose.22 There are no reliable positive IHC markers for MPNST. The best-established IHC targets are H3 K27 di- or trimethylation, but these marks are only globally altered in approximately half of cases.23,24 It is particularly difficult to distinguish MPNST from histologic mimics including melanoma and synovial sarcoma [Figure 3a–c].25
We therefore applied HYPERsol to 32 archival FFPE tumor samples (13 MPNSTs, 10 melanomas, and nine synovial sarcomas) to identify new candidate IHC markers. Approximately 4000–5000 protein groups were detected per sample [Figure 3d]. Among the 6123 unique protein groups identified overall, nearly 90% were detected in at least one case of each tumor type, a figure which numerically illustrates the inherent difficulty of identifying distinguishing markers [Figure 3e; Table S9]. The time that the archival samples had spent in storage ranged from approximately 6 months to over 17 years, and the correlation between the duration of storage and the number of protein identifications was weak (R = −0.34, p = 0.05651) [Figure 3f; Table S10].
Our analysis revealed proteins that were either specific or highly enriched in each tumor type, and these were largely consistent with known protein expression signatures [Figure 3g–k; Table S9]. For example, the protein S100A was overexpressed in melanoma relative to both MPNST and synovial sarcoma [Figure 3g,h]. This is consistent with the longstanding use of S100 as an IHC marker for melanoma, and the loss of S100 expression seen in MPNST as compared to benign nerve sheath tumors [Figure 3g,h].26,27 Additionally, TLE1, a widely used marker for synovial sarcoma, was indeed highly expressed in 8/9 synovial sarcomas.28 However, in line with reports that TLE1 is not entirely specific for synovial sarcoma, TLE1 expression was also observed in 4/10 melanomas and 5/13 MPNSTs29 [Figure 3h,j]. This experiment also revealed several other proteins that were entirely tumor-type specific (103 in MPNST, 114 in melanoma, and 43 in synovial sarcoma; [Figure 3e; Table S9]). Among these was the known melanoma marker tyrosinase, which was detected in 9/10 melanomas and no other tumors [Figure 3i].30 Intriguingly, the protein ICAM5 was detected in 9/9 synovial sarcomas and no other tumors, suggesting that it might represent a useful IHC marker for synovial sarcoma [Figure 3k]. Future work will establish whether this or any of the other tumor-specific or tumor-enriched proteins that we detected are useful IHC markers for these diagnostically challenging tumors.
In order to the validate of protein markers measured by HYPERsol, we performed IHC for both tyrosinase and TLE1 on a tissue microarray containing a larger set of tumors. The array consisted of 16 MPNSTs, 16 melanomas, and 15 synovial sarcomas [Figure 4], including 10 examples of each tumor type that were not included in the proteomic analysis. As in the proteomics data, tyrosinase was both sensitive and highly specific for melanoma, detected in 14/16 cases, including all of those for which it detected by MS. One case of synovial sarcoma was positive for tyrosinase. TLE1 was 100% sensitive for synovial sarcoma, but it was detected in 37.5% of MPNSTs and 50% of melanomas, consistent with the proteomics results, and again illustrating the imperfect specificity of this commonly used marker.
Figure 4.
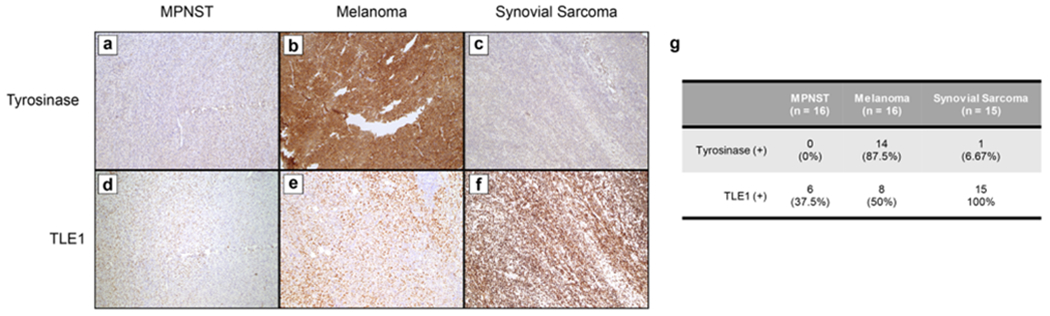
Tyrosinase and TLE1 are immunohistochemical markers for melanoma and synovial sarcoma, respectively. (a–c) Representative images from tyrosinase immunohistochemistry (IHC) on MPNST (a), melanoma (b), and synovial sarcoma (c). (d–f) Representative images from TLE1 IHC on MPNST (d), melanoma (e), and synovial sarcoma (f). (g) Table summarizing IHC results from a tissue microarray.
DISCUSSION
In conclusion, HYPERsol enables highly reproducible protein identification and quantification from FFPE tissue, yielding results that are highly similar to flash-frozen tissue. Direct resuspension of samples in 5% SDS obviates the need for deparaffinization, thereby increasing sample throughput and improving agreement with results obtained from paired flash-frozen tissue. Substitution of AFA ultrasonication for probe sonication ensures that samples are thoroughly and reproducibly solubilized, while also eliminating the potential for sample cross-contamination through contact with the sonication probe. Finally, the use of S-Traps for protein recovery and downstream sample preparation is far superior to incubation with methanol–chloroform, because it does not require precipitation and resolubilization of protein. Together, the combination of direct SDS solubilization, AFA ultrasonication, and S-Trap sample processing, which we call HYPERsol, markedly increases protein yield from FFPE tissues, such that it is possible to obtain tens to hundreds of micrograms of soluble protein per milligram of material. This increased protein yield translates to an increased number of proteins identified, with higher sequence coverage. Moreover, data generated using the HYPERsol technique closely resemble matched flash-frozen tissue, both in terms of the depth-of-coverage achievable and the relative quantification of detected proteins. The fact that proteomic characterization of archival tumor samples revealed several known tumor markers suggests that it may also enable the discovery of novel markers. Finally, the lack of a strong correlation between the duration of sample storage and number of proteins identified (up to at least 17 years) supports the notion that HYPERsol will be applicable to FFPE samples regardless of their age.
This work underscores the reality that flash-freezing is not a strict requirement for reproducible and accurate proteomic characterization of human tissue samples. We anticipate that the reduced variability of HYPERsol sample processing will enhance the capacity of researchers to extract meaningful biological information from both existing FFPE samples and those yet to be generated. With the availability of 96-well plates for AFA and S-Trap sample processing, HYPERsol will be suitable for the automated, high-throughput analysis of clinical specimens. We thus anticipate that the HYPERsol workflow will enable novel discoveries from rich clinically annotated and histologically characterized FFPE biorepositories worldwide, thereby helping to usher in a new era of clinical proteomics.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Li Ping Wang and Amy Ziober in the developmental immunohistochemistry lab at the University of Pennsylvania for their technical help and support in assay implementation. This research was supported by US National Institutes of Health (NIH) grants (T32GM008275 and TL1TR001880 to DMM; and R01-GM110174, R01-AI118891, and P01-CA196539 to BAG). BAG is also supported by a Robert Arceci Scholar Award from the Leukemia and Lymphoma Society. JBW was supported by Institutional Startup Funds from University of Pennsylvania Department of Pathology, and by the University of Pennsylvania Sarcoma Research Program.
ABBREVIATIONS
- AFA
adaptive-focused acoustics
- BCA
bicinchoninic acid
- DDA
data-dependent acquisition
- DIA
data-independent acquisition
- DPS
direct, probe, S-Trap
- DUS
direct, ultrasonication, S-Trap
- FASP
filter-aided sample preparation
- FFPE
formalin-fixed paraffin-embedded
- FPS
fresh, probe, S-Trap
- FUS
fresh, ultrasonication, S-Trap
- H&E
hematoxylin and eosin
- HRP
horseradish peroxidase
- HYPERsol
high-yield protein extraction and recovery by direct solubilization
- IHC
immunohistochemistry
- MPNST
malignant peripheral nerve sheath tumor
- SDS
sodium dodecyl sulfate
- TFA
trifluoroacetic acid
- TLE1
transducin-like enhancer protein 1
- TYR
tyrosinase
- XPM
xylene–ethanol, probe, methanol–chloroform
- XUM
xylene–ethanol, ultrasonication, methanol–chloroform
- XUS
xylene–ethanol, ultrasonication, S-Trap
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00686.
Figure S1: Direct solubilization combined with ultrasonication efficiently solubilizes FFPE samples of various human tissue types; Figure S2: Peptides from FFPE samples contain missed cleavages and modifications (PDF)
Table S1: Experiment 1 Peptide Report (TXT)
Table S2: Experiment 1 Protein Report (TXT)
Table S3: Experiment 1 Unique Peptides and Protein Modifications (XLSX)
Table S4: Experiment 1 Gene Ontology Terms (XLSX)
Table S5: Experiment 2 Peptide Report (XLS)
Table S6: Experiment 2 Protein Report (XLS)
Table S7: Experiment 2 Protein Correlation Matrix (TXT)
Table S8: Experiment 2 Volcano Plots (XLSX)
Table S9: Archival FFPE Tumor Specimen Protein Report (XLSX)
Table S10: Archival FFPE Tumor Specimen Age and Number of Protein IDs (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.9b00686
The authors declare the following competing financial interest(s): JBW, DM, II, KD, DS, and BAG have no conflicts to disclose. JPW and DJP founded ProtiFi, LLC, which has commercialized S-Trap sample processing.
The raw mass spectrometry data have been deposited to CHORUS (https://chorusproject.org/pages/index.html) under project #1635.
Contributor Information
Dylan M. Marchione, University of Pennsylvania, Philadelphia, Pennsylvania.
Ilyana Ilieva, University of Pennsylvania, Philadelphia, Pennsylvania.
Kyle Devins, University of Pennsylvania, Philadelphia, Pennsylvania.
Danielle Sharpe, University of Pennsylvania, Philadelphia, Pennsylvania.
Darryl J. Pappin, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, and ProtiFi, LLC, Huntington, New York
Benjamin A. Garcia, University of Pennsylvania, Philadelphia, Pennsylvania.
John P. Wilson, ProtiFi, LLC, Huntington, New York
John B. Wojcik, University of Pennsylvania, Philadelphia, Pennsylvania.
REFERENCES
- (1).Fox CH; Johnson FB; Whiting J; Roller PP Formaldehyde Fixation. J. Histochem. Cytochem 1985, 33 (8), 845–853. [DOI] [PubMed] [Google Scholar]
- (2).Sanderson C; Emmanuel J; Emmanual J; Campbell P A Historical Review of Paraffin and Its Development as an Embedding Medium. J. Histotechnol 1988, 11 (1), 61–63. [Google Scholar]
- (3).Kokkat TJ; Patel MS; McGarvey D; LiVolsi VA; Baloch ZW Archived Formalin-Fixed Paraffin-Embedded (FFPE) Blocks: A Valuable Underexploited Resource for Extraction of DNA, RNA, and Protein. Biopreserv. Biobanking 2013, 11 (2), 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mason JT Proteomic Analysis of FFPE Tissue: Barriers to Clinical Impact. Expert Rev. Proteomics 2016, 13 (9), 801–803. [DOI] [PubMed] [Google Scholar]
- (5).Nirmalan NJ; Hughes C; Peng J; McKenna T; Langridge J; Cairns DA; Harnden P; Selby PJ; Banks RE Initial Development and Validation of a Novel Extraction Method for Quantitative Mining of the Formalin-Fixed, Paraffin-Embedded Tissue Proteome for Biomarker Investigations. J. Proteome Res. 2011, 10 (2), 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alkhas A; Hood BL; Oliver K; Teng P-N; Oliver J; Mitchell D; Hamilton CA; Maxwell GL; Conrads TP Standardization of a Sample Preparation and Analytical Workflow for Proteomics of Archival Endometrial Cancer Tissue. J. Proteome Res. 2011, 10 (11), 5264–5271. [DOI] [PubMed] [Google Scholar]
- (7).Hembrough T; Thyparambil S; Liao W-L; Darfler MM; Abdo J; Bengali KM; Hewitt SM; Bender RA; Krizman DB; Burrows J Application of Selected Reaction Monitoring for Multiplex Quantification of Clinically Validated Biomarkers in Formalin-Fixed, Paraffin-Embedded Tumor Tissue. J. Mol. Diagn 2013, 15 (4), 454–465. [DOI] [PubMed] [Google Scholar]
- (8).Bronsert P; Weißer J; Biniossek ML; Kuehs M; Mayer B; Drendel V; Timme S; Shahinian H; Küsters S; Wellner UF Impact of Routinely Employed Procedures for Tissue Processing on the Proteomic Analysis of Formalin-Fixed Paraffin-Embedded Tissue. Proteomics: Clin. Appl 2014, 8 (9–10), 796–804. [DOI] [PubMed] [Google Scholar]
- (9).Longuespée R; Alberts D; Pottier C; Smargiasso N; Mazzucchelli G; Baiwir D; Kriegsmann M; Herfs M; Kriegsmann J; Delvenne P; et al. A Laser Microdissection-Based Workflow for FFPE Tissue Microproteomics: Important Considerations for Small Sample Processing. Methods 2016, 104, 154–162. [DOI] [PubMed] [Google Scholar]
- (10).Wojcik JB; Marchione DM; Sidoli S; Djedid A; Lisby A; Majewski J; Garcia BA Epigenomic Reordering Induced by Polycomb Loss Drives Oncogenesis but Leads to Therapeutic Vulnerabilities in Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2019, 79, 3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hood BL; Darfler MM; Guiel TG; Furusato B; Lucas DA; Ringeisen BR; Sesterhenn IA; Conrads TP; Veenstra TD; Krizman DB Proteomic Analysis of Formalin-Fixed Prostate Cancer Tissue. Mol. Cell. Proteomics 2005, 4 (11), 1741–1753. [DOI] [PubMed] [Google Scholar]
- (12).Föll MC; Fahrner M; Oria VO; Kühs M; Biniossek ML ; Werner M; Bronsert P; Schilling O Reproducible Proteomics Sample Preparation for Single FFPE Tissue Slices Using Acid-Labile Surfactant and Direct Trypsinization. Clin. Proteomics 2018, 15, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wiśniewski JR; Ostasiewicz P; Mann M High Recovery FASP Applied to the Proteomic Analysis of Microdissected Formalin Fixed Paraffin Embedded Cancer Tissues Retrieves Known Colon Cancer Markers. J. Proteome Res. 2011, 10 (7), 3040–3049. [DOI] [PubMed] [Google Scholar]
- (14).Fowler CB; O’Leary TJ; Mason JT Improving the Proteomic Analysis of Archival Tissue by Using Pressure-Assisted Protein Extraction: A Mechanistic Approach. J. Proteomics Bioinf. 2014, 7 (6), 151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive Proteome Analysis Using Paramagnetic Bead Technology. Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zougman A; Selby PJ; Banks RE Suspension Trapping (STrap) Sample Preparation Method for Bottom-up Proteomics Analysis. Proteomics 2014, 14 (9), 1006–1000. [DOI] [PubMed] [Google Scholar]
- (17).Ludwig KR; Schroll MM; Hummon AB Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wessel D; Flügge UI A Method for the Quantitative Recovery of Protein in Dilute Solution in the Presence of Detergents and Lipids. Anal. Biochem 1984, 138 (1), 141–143. [DOI] [PubMed] [Google Scholar]
- (19).Rappsilber J; Mann M; Ishihama Y Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc 2007, 2 (8), 1896–1906. [DOI] [PubMed] [Google Scholar]
- (20).Gustafsson OJR; Arentz G; Hoffmann P Proteomic Developments in the Analysis of Formalin-Fixed Tissue. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854 (6), 559–580. [DOI] [PubMed] [Google Scholar]
- (21).Zhang Y; Muller M; Xu B; Yoshida Y; Horlacher O; Nikitin F; Garessus S; Magdeldin S; Kinoshita N; Fujinaka H; et al. Unrestricted Modification Search Reveals Lysine Methylation as Major Modification Induced by Tissue Formalin Fixation and Paraffin Embedding. Proteomics 2015, 15 (15), 2568–2579. [DOI] [PubMed] [Google Scholar]
- (22).Le Guellec S; Decouvelaere A-V; Filleron T; Valo I; Charon-Barra C; Robin Y-M; Terrier P; Chevreau C; Coindre J-M Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am. J. Surg. Pathol 2016, 40 (7), 896–908. [DOI] [PubMed] [Google Scholar]
- (23).Pekmezci M; Cuevas-Ocampo AK; Perry A; Horvai AE Significance of H3K27me3 Loss in the Diagnosis of Malignant Peripheral Nerve Sheath Tumors. Mod. Pathol 2017, 30 (12), 1710–1719. [DOI] [PubMed] [Google Scholar]
- (24).Marchione DM; Lisby A; Viaene AN; Santi M; Nasrallah M; Wang L-P; Williams EA; Larque AB; Chebib I; Garcia BA Histone H3K27 Dimethyl Loss Is Highly Specific for Malignant Peripheral Nerve Sheath Tumor and Distinguishes True PRC2 Loss from Isolated H3K27 Trimethyl Loss. Mod. Pathol 2019, 32, 1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Le Guellec S; Macagno N; Velasco V; Lamant L; Lae M; Filleron T; Malissen N; Cassagnau E; Terrier P; Chevreau C; et al. Loss of H3K27 Trimethylation Is Not Suitable for Distinguishing Malignant Peripheral Nerve Sheath Tumor from Melanoma: A Study of 387 Cases Including Mimicking Lesions. Mod. Pathol 2017, 30 (12), 1677–1687. [DOI] [PubMed] [Google Scholar]
- (26).Nakajima T; Watanabe S; Sato Y; Kameya T; Shimosato Y; Ishihara K Immunohistochemical Demonstration of S100 Protein in Malignant Melanoma and Pigmented Nevus, and Its Diagnostic Application. Cancer 1982, 50 (5), 912–918. [DOI] [PubMed] [Google Scholar]
- (27).Miettinen MM; Antonescu CR; Fletcher CDM; Kim A; Lazar AJ; Quezado MM; Reilly KM; Stemmer-Rachamimov A; Stewart DR; Viskochil D; et al. Histopathologic Evaluation of Atypical Neurofibromatous Tumors and Their Transformation into Malignant Peripheral Nerve Sheath Tumor in Patients with Neurofibromatosis 1-a Consensus Overview. Hum. Pathol 2017, 67, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Terry J; Saito T; Subramanian S; Ruttan C; Antonescu CR; Goldblum JR; Downs-Kelly E; Corless CL; Rubin BP; van de Rijn M; et al. TLE1 as a Diagnostic Immunohistochemical Marker for Synovial Sarcoma Emerging from Gene Expression Profiling Studies. Am. J. Surg. Pathol 2007, 31 (2), 240–246. [DOI] [PubMed] [Google Scholar]
- (29).Kosemehmetoglu K; Vrana JA; Folpe AL TLE1 Expression Is Not Specific for Synovial Sarcoma: A Whole Section Study of 163 Soft Tissue and Bone Neoplasms. Mod. Pathol 2009, 22 (7), 872–878. [DOI] [PubMed] [Google Scholar]
- (30).Clarkson KS; Sturdgess IC; Molyneux AJ The Usefulness of Tyrosinase in the Immunohistochemical Assessment of Melanocytic Lesions: A Comparison of the Novel T311 Antibody (anti-Tyrosinase) with S-100, HMB45, and A103 (anti-Melan-A). J. Clin. Pathol 2001, 54 (3), 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mass spectrometry data have been deposited to CHORUS (https://chorusproject.org/pages/index.html) under project #1635.