

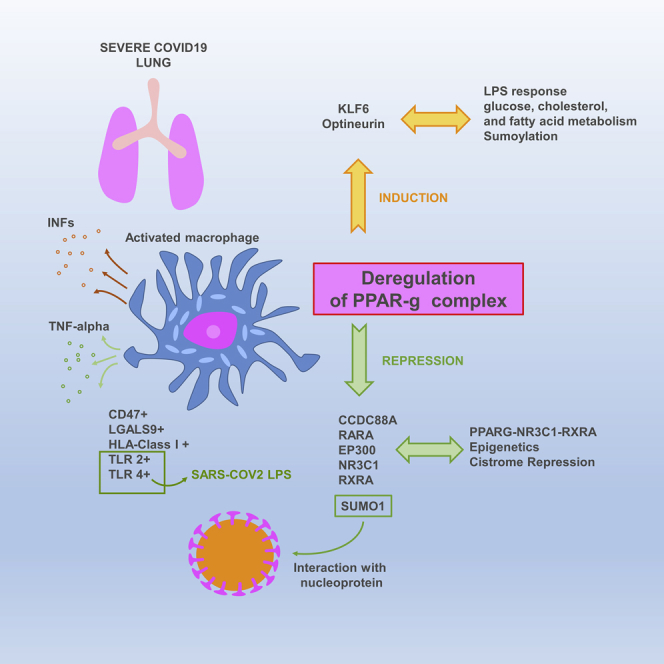
Summary
The molecular mechanisms of cytokine storm in patients with severe COVID-19 infections are poorly understood. To uncover these events, we performed transcriptome analyses of lung biopsies from patients with COVID-19, revealing a gene enrichment pattern similar to that of PPARγ-knockout macrophages. Single-cell gene expression analysis of bronchoalveolar lavage fluids revealed a characteristic trajectory of PPARγ-related disturbance in the CD14+/CD16+ cells. We identified a correlation with the disease severity and the reduced expression of several members of the PPARγ complex such as EP300, RXRA, RARA, SUMO1, NR3C1, and CCDC88A. ChIP-seq analyses confirmed repression of the PPARγ-RXRA-NR3C1 cistrome in COVID-19 lung samples. Further analysis of protein-protein networks highlighted an interaction between the PPARγ-associated protein SUMO1 and a nucleoprotein of the SARS virus. Overall, these results demonstrate for the first time the involvement of the PPARγ complex in severe COVID-19 lung disease and suggest strongly its role in the major monocyte/macrophage-mediated inflammatory storm.
Subject Areas: Immunology, Viral Microbiology, Transcriptomics
Graphical Abstract
Highlights
-
•
PPARγ expression is repressed in inflammatory lungs of patients with severe COVID-19
-
•
PPARγ trajectory is disrupted in bronchoalveolar CD14+/CD16+ cells of patients with COVID-19
-
•
We report here the epigenetics repression of PPARγ-NR3R1-RXRA cistrome in this setting
-
•
SUMO1, as repressed PPARγ partner, interacts with nucleoprotein of the human SARS virus
Immunology; Viral Microbiology; Transcriptomics
Introduction
The pandemic caused by the novel SARS-CoV-2 coronavirus has rapidly become the chief public health challenge for many countries around the world. Respiratory complications have been well documented in patients with this disease (COVID-19). This coronavirus harbors a viral S-protein, which, during infection, binds with the human protein receptor ACE2 (Zhou et al., 2020b). ACE2 is abundant in lung, heart, kidney, and adipose tissue and, thus, the binding of the S-protein to ACE2 enables membrane fusion and the introduction of SARS-CoV-2 RNA into these cells (Turner et al., 2004). The mean incubation period of the disease is about 3–9 days (Li et al., 2020) and about 18% of cases remain asymptomatic (Nishiura et al., 2020). About 41.8% of patients develop acute respiratory distress syndrome (ARDS), for which diabetes mellitus can be a contributing factor. Other comorbidities associated with ARDS include hypertension, cardiovascular disease, and chronic kidney disease (Wu et al., 2020a). The severity of the disease is also age associated, with the risk of the mortality increasing in patients over 60 years old (Wu and McGoogan, 2020). One of the potential consequences of SARS-CoV-2 infection is an uncontrolled immune response in the lungs, which can require treatment in an intensive care unit. This immune response, associated with a cytokine storm, is heterogenetic and variable among individuals and is still not well understood (McGonagle et al., 2020). It also remains unclear why the heterogenetic immune response to COVID-19 worsens in certain patients; as yet, we lack adequate knowledge to predict which cases will evolve from mild to severe infection.
In this work, we investigated the immune infiltration in biopsied lung tissues from patients with COVID-19 using different bioinformatics approaches, including whole transcriptome experiments, single-cell transcriptome characterization, and investigations of the epigenetic landscape. Mechanistically, we observed major deregulation of the innate immune response in these lung samples. In particular, we identified notable dysregulation of the PPARγ-dependent genetic program in macrophages, which is associated with an increase in the inflammation-promoting program in monocyte-macrophage cells. PPARγ repression was found in different independent cohorts and was strongly correlated in multiple patients with disease severity at the single-cell level in the CD14-CD16 myeloid population. PPARγ is a member of peroxisome proliferator-activated receptor family with PPARα and PPARβ.They have common structure comprising an amino terminal activation function-1 domain, a DNA binding domain, a hinge domain, and a conserved activation function-2 domain which allow ligand binding (Nolte et al., 1998). PPARγ was first described as an important regulator of adipocyte differentiation and could have some unsaturated fatty acids, eicosapentaenoic acids, and oxidized lipids as natural ligands (Bell-Parikh et al., 2003). In immune system and lung, PPARγ is expressed in various cell types such as monocytes and macrophages (Standiford et al., 2005), dendritic cells (Gosset et al., 2001), and epithelial airway cells (Hetzel et al., 2003). During lung inflammation, PPARγ could represses transcription factors that regulated expression of pro-inflammatory molecules like NFKB1, STAT1, and AP1 (Honda et al., 2004; Straus and Glass, 2007). In our work, PPARγ repression in lung was associated with the induction of several inhibitory immune checkpoints, the lipopolysaccharide-sensitive TLR2 receptor, and HLA class I molecules, and these relationships were all associated with disease severity. In an epigenetic analysis of patients with severe COVID-19, we detected the repression of components of PPARγ-associated cistrome, including NR3C1 and RXRA, which had RGS12 as target in its second promoter. Finally, we also observed the repression of SUMO1, a partner of PPARγ, which is able to interact with nucleoprotein of human SARS-CoV virus and could potentially affect sumoylation.
Results
Extensive Innate Response and Interferon Gamma Activation in COVID-19 Lung Biopsies
Transcriptome data of human lung biopsy samples from the dataset GSE147507 (Blanco-Melo et al., 2020) were subjected to gene set enrichment analysis using the “hallmarks” gene set from the MSigDB database. As compared with healthy donor samples, the majority of samples from COVID-19 lungs were significantly enriched in genes associated with innate immunity, such as the interferon alpha response (with a normalized enrichment score [NES] of +7.72, p value<0.001; Figure S1A), TNFA signaling via NF-κB (NES of +4.88, p value<0.001; Figure 1), complement cascade (NES of +3.51, p value<0.001; Figure S1A), and reactive oxygen species pathway (NES of +2.78, p value<0.001; Figure S1A). Likewise, these tissues were also enriched in genes linked with IL-6/Jak signaling, which plays a role in both innate and adaptive immunity (NES = +2.49, p value<0.001; Figure S1B). Furthermore, we observed dramatic enrichment in the interferon gamma response, which is involved in the adaptive immune response (NES = +8.11, p value<0.001; Figure S1C). Taken together, changes in the expression patterns of these immune-related genes enabled us to clearly differentiate, via unsupervised classification (Euclidean distances), between lung samples from patients with COVID-19 and those from healthy donors (Figure S1D).
Figure 1.

Immune Network and Immune Checkpoint Regulation in COVID-19 Lung Samples
(A) Functional network of immune genes that were upregulated in lung samples from patients with COVID-19 compared with those from healthy donors; functions are represented as octagons, and their size represents the number of direct edge connections; (B) UMAP dimensionality reduction performed on merged single-cell transcriptome data from bronchoalveolar lavage fluid of healthy donors (green), patients with mild COVID-19 (blue), and patients with severe COVID-19 (red); major markers of cell subpopulations are indicated in black; (C) Pie chart representing the proportions of different cell types in the merged single-cell transcriptome analysis; (D) Dot plot of single-cell expression patterns (bronchoalveolar lavage fluid) of the main markers of each cluster, by patient of origin (HD, mild COVID-19, and severe COVID-19) (percent: percent of cells expressing each marker, expression level: color intensity), (E) Dimensionally reduced (UMAP) single-cell transcriptome (bronchoalveolar lavage fluid) expression patterns for the monocyte markers FCGR3A (CD16) and CD14 (F), by patient of origin (HD, mild COVID-19, and severe COVID-19).
These results suggested that in COVID-19 lung samples there was a dramatic upregulation of the interferon gamma response, which was accompanied by a large innate immune response. Indeed, a network analysis of this immune-activated program revealed centralized connections around the interferon gamma response (Figure 1A), with some targets shared with the interferon alpha response. A large part of the network was organized around aspects of the innate response such as interferon alpha, TNFA, the complement cascade, and reactive oxygen species (ROS). The majority of the immune network (Figure 1A) was composed of connections between enzymes, followed by signaling molecules, receptors, and transcription factors. We detected expression changes for a few ligands including chemokines and interleukins (Figure 1A), which were mainly induced by the IFNα and IFNγ response. To validate these results, we obtained single-cell transcriptome data from the bronchoalveolar lavage fluid of healthy donors (n = 6, green) and patients with mild COVID-19 (n = 3, blue) or severe COVID-19 (n = 3, red) from the dataset GSE145926. These data were merged together for the purpose of UMAP dimensionality reduction (Figure 1B) (Liao et al., 2020); after canonical correlation and filtration, the merged transcriptome analysis comprised 90,696 cells, with a representative proportion of each subgroup (Figure 1C). In this analysis, some of the main subpopulations identified were T lymphocytes that expressed CD3E and/or CD8A (Figure S2, at the top of the UMAP in Figure 1B), with a reduction in the CD8A cluster size in severe COVID-19 samples (Figure S2); natural killer cells that expressed NKG7, for which the cluster size increased in severe disease (Figure 1D, Figure S2, at the top right of the UMAP in Figure 1B); some epithelial cells that expressed KRT8 (Figure S3, on the right of the UMAP in Figure 1B); and some B lymphocytes expressing MS4A1 alias CD20 (Figure S3, on the top of the UMAP Figure 1B). This analysis also confirmed a major increase in the monocyte-macrophage markers CD14 and CD16 (alias FCGR3A; Italiani and Bora schi, 2014) with increased disease severity (Figures 1D–1F). These results suggest a major role for the innate immune response in the lungs of patients with COVID-19, which intensifies with disease severity.
COVID-19 Lung Transcriptome Reveals Defects in Cell Proliferation, Stem Cell Signaling, and Metabolism, with Major Myeloid Infiltration
A differentially expressed gene (DEG) analysis was performed on lung biopsies from patients with COVID-19 and healthy donors; this revealed widespread repression of many gene pathways in COVID-19 lungs (Figures S4A and S4B), which could affect major functionalities of the cells in this organ. These results were consistent with those of a gene set enrichment analysis (Figure S5). Specifically, the gene set enrichment analysis (performed using the “hallmarks” gene set of the MsigDB database) highlighted repression of the mitosis spindle and p53 pathway (cell cycle gatekeeper) in samples of COVID-19 lungs compared with those of healthy donors (NES = −3.45 and −2.77, respectively, with p value<0.001, Figure S5A). We also detected the repression of signaling pathways that are implicated in stem cell functionality, such as the NOTCH pathway (NES = −1.70, p value <0.001, Figure S5B) and the beta-catenin signaling pathway (NES = −2.17, p value<0.001, Figure S5B). Furthermore, heme metabolism was also repressed in these samples (NES = −1.70, p value<0.001, Figure S5C). These results suggest that in COVID-19 lung pathology there are major defects in important functionalities linked with tissue homeostasis, such as the regulation of cell proliferation, stem cell signaling, and metabolism. Of the 108 genes that were found to be upregulated in COVID-19 lung samples (Table S1), the majority were implicated in immune functionalities. In particular, the DEG analysis highlighted IFI6 as the most upregulated molecule in COVID-19 samples, and it was expressed in individual cells at progressively higher levels in more severe cases (Figure S4C). A landscape immune score analysis, performed on lung biopsies with the xcell algorithm and downstream limma analysis (Table S2), revealed a significant increase in immune score in COVID-19 lung samples, representing increases in neutrophil, monocyte, M1 macrophage, and adaptive dendritic cell infiltrations (Figure S4D). At the single-cell level, CD68 macrophage M1 marker was confirmed to be increased in bronchoalveolar lavage fluid of mild COVID-19 as compared with healthy donor samples but it was not case in severe COVID-19. In contrast, expression of CD163 macrophage M2 marker seems to be constant between different groups of samples (Figure S6). In order to characterize the immune infiltration of COVID-19 lungs, transcriptome data from lung biopsies were cross-integrated with transcriptome data from human hematopoietic and non-hematopoietic tissues (GSE76340) (Pont et al., 2016), which were taken to be representative of the micro-environment of the lung samples. The resulting cross-matrix comprised 170 whole transcriptome analyses and was submitted to the cross-batch normalization algorithm Combat to correct batch error (Figure S7 and Table S3). After mathematical correction, the normalized matrix was reduced to the 108 genes that were found to be upregulated in COVID-19 lung samples (Table S1). An unsupervised principal component analysis revealed major differentiation between healthy donor lungs and COVID-19 lungs along the first principal axis (p value = 3.215239 × 10−29, Figure S4E). Mononuclear cells, monocytes, and macrophages were found in positions similar to the COVID-19 lung samples, suggesting major infiltrations in this tissue (Figure S4E) and confirming the results of the “xcell” immune score analysis (Figure S4C). In this principal component analysis, lung samples from healthy donors clustered together with normal primary bronchial epithelial cells, as well as endothelial cells and fibroblasts, suggesting that these cell populations are not implicated in the immune infiltration of COVID-19 lungs. Hematopoietic stem cells, B cells, T cells, and dendritic cells were found in intermediary positions between healthy donor and COVID-19 lung samples, which suggested that these cells played only a minor role in the immune infiltration (Figure S4E). We also confirmed that the microarray batches coming from different datasets used in this analysis did not influence the results, as they were found to be well distributed across the first principal axis (Figure S8).
Induction of Inhibitory Immune Checkpoints and CD48 in COVID-19 Lung Samples
Immune checkpoints have been widely discussed as new therapeutic targets for the regulation of the immune system in cancer therapy (Pardoll, 2012). In the case of COVID-19, a patient's prognosis has been found to depend, at least in part, on the immune response (Xu et al., 2020). For this reason, we next investigated the expression of immune checkpoints in COVID-19 lung biopsies compared with healthy donor samples and constructed an expression heatmap with unsupervised classification that enabled us to differentiate COVID19 samples (COVA and COVB, COVID-19 lung biopsy replicate A and B) from those of healthy donors (HD1 and HD2, lung biopsies from healthy donors 1 and 2) (Figure S9A). The majority of inhibitory immune checkpoints, including CEACAM1, IDO1, LGALS9, CYBB, PDL1 (alias CD274), and CD47, were found to be overexpressed in COVID-19 lung samples (Figure S9A). Some of these, such as CD47, LGALS9, HAVCR2, IDO1, and CD274, were confirmed to be induced during severe disease progression (Figure S9B) at a single-cell level in the CD14-CD16 subpopulation (Figures S9C, 9D and S10). These results suggest a potential modification of communication among immune cells in COVID-19 lungs. Instead, our investigation of stimulatory immune checkpoints did not differentiate among COVID-19 and healthy donor lung samples (Figure S9E). In bronchoalveolar lavage fluid at the single-cell level, CD48 and CD40 were found to be expressed in COVID-19 lung tissue; CD48 was associated with increased disease severity (Figure S9F) but CD40 was not (Figure S11). In cases of severe COVID-19, CD48 was found to be expressed in the CD14-CD16 subpopulation (Figure S9G). These results mainly suggest that immune cell communication, which is regulated by immune checkpoints, may be modified during infection by COVID-19, particularly by the upregulation of inhibitory checkpoints.
Activation of HLA Class I Molecules and Pathogen-Associated Molecular Patterns of Lipopolysaccharide-Sensitive TLRs in COVID-19 Lung Samples
When we examined the transcriptomes of the COVID-19 lung biopsies, we observed that, compared with data from healthy donors, there was a notable increase in antigen-presenting molecules from the HLA class I subcategory, including HLA-A, HLA-B, and HLA-C. We also detected an increase, although lower in magnitude, in antigen-presenting molecules from the class II subcategory: HLA-DPA1, HLA-DQA1, and HLA-DRB5 (Figure 2A). The strong expression of HLA class I molecules was confirmed with the single-cell transcriptome data obtained from bronchoalveolar lavage fluid, especially in the case of severe COVID-19 (Figure 2B). Specifically, HLA-B was highly expressed in all lymphoid and myeloid cell subpopulations in the lungs of the patient with severe COVID-19 (Figure 2C). When we examined the expression of Toll-like receptors (TLRs), we found no evidence for the induction of the TLR7 sensor in COVID-19 lung samples but did observe upregulation of TLR4, which is sensitive to lipopolysaccharides (Figure 2D). In bronchoalveolar lavage fluid at the single-cell level, expression of the lipopolysaccharide (LPS) sensors TLR2 and TLR4 was found to be induced in COVID-19 lungs (Figure 2E) but expression of TLR7 was unchanged. TLR2 expression appeared to increase with disease severity, especially in the CD14-CD16 cell populations (Figure 2F), whereas TLR4 induction was less dramatic (Figure 2G). These results provide evidence for CD14-CD16 myeloid infiltration in COVID-19 lung samples, with a substantial induction of HLA class I-presenting molecules and the TLR2 LPS sensor. To further characterize the transcriptome of COVID-19 lung biopsies, specifically with respect to the lympho-myeloid lineages, we performed transcriptome immune cell deconvolution on this subset of the bulk transcriptome data (Figure 3A). We observed that COVID-19 samples were segregated from those of healthy donors by a notable differentiation in lymphoid and myeloid components along the first principal axis (p value = 2.003047 × 10−17, Figure 3A). COVID-19 samples were distinguished by positive correlations with myeloid markers (blue bar plot, Figure 3B) and a single negative correlation with one lymphoid IFITM1 (pink bar plot, Figure 3B). These results confirmed the strong influence of the myeloid signature, particularly when compared with that of the lymphoid signature, in COVID-19 lung samples. We observed a similar pattern at the single-cell level in the bronchoalveolar lavage liquid of patients with severe COVID-19 (Figure 3C), especially with the markers CCL7, FCERG1, and S100A11. In patients with severe COVID-19, S100A11 was found to be mainly induced in the CD14-CD16 subpopulations (Figure 3D). FCERG1 (Fc Fragment of IgE Receptor Ig) is known to be expressed in monocytes and macrophages (Bournazos et al., 2016), and here its induction was also found to be more restricted to the CD14-CD16 cell subpopulations (Figure 3E). These results suggested that, along with HLA and TLRs, some myeloid markers are also induced in the CD14-CD16 cell subpopulation during severe COVID-19.
Figure 2.

Overexpression of HLA Molecules and TLRs in COVID-19 Lung Samples
(A) Transcriptome expression heatmap of HLA molecules in COVID-19 lung biopsies and samples from healthy donors; (B) Single-cell expression (bronchoalveolar lavage fluid) dot plot of HLA molecules from HDs and patients with mild or severe COVID-19 (percent: percent of cells expressing each marker, expression level: color intensity); (C) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression data for HLA-B from HDs or patients with mild or severe COVID-19; (D) Transcriptome expression heatmap of TLRs in biopsies from COVID-19 and healthy lungs; (E) Single-cell expression (bronchoalveolar lavage fluid) dot plot of TLRs from HDs and patients with mild or severe COVID-19 (percent: percent of cells expressing each marker, expression level: color intensity); (F) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression data from HDs and patients with mild or severe COVID-19 for TLR2 and (G) TLR4.
Figure 3.

Main Monocyte-Macrophage Infiltration in COVID-19 Lung Samples
(A) Transcriptome deconvolution of lymphoid-myeloid populations from biopsies of COVID-19 and healthy lungs (p value: differentiation of groups by Pearson correlation along the first principal axis). Abbreviations: HD_lung, lung biopsy of healthy donor; Lung_COVID-19, lung biopsy of patient with COVID-19; MO, monocyte; M1, M1 macrophage; M2, M2 macrophage; IDC, immature dendritic cell; MDC, mature dendritic cells; LYB, B lymphocyte; LYT, T lymphocyte; BEC, primary bronchial epithelial cells.
(B–E) (B) Bar plot of correlations between genes (Pearson coefficients) and the lymphoid (purple) and myeloid (blue) deconvolution of COVID-19 lung biopsies as compared with those of healthy donors; (C) Single-cell expression (bronchoalveolar lavage fluid) dot plot of markers that were positively correlated to COVID-19, by patient of origin: HD, mild COVID-19, and severe COVID-19 (percent: percent of cells expressing each marker, expression level: color intensity); (D) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression data from HDs or patients with mild or severe COVID-19 for S100A11 and (E) FCER1G.
Monocyte-Macrophage Dysregulation Characterized by PPARγ Repression in COVID-19 Lung Samples
A gene set enrichment analysis performed using the MSigDB immune gene subset revealed that, compared with samples from healthy donors, COVID-19 samples were enriched in genes that had previously been found to be upregulated in macrophages following knockout of PPARγ (NES = +3.20, p value<0.001, Figure 4A). Consistent with this, we determined that PPARγ expression was in fact lower in COVID-19 lung samples compared with those from healthy donors (Figure 4B). By unsupervised classification, an expression heatmap confirmed that the genes involved in this PPARγ-dependent program in macrophages were also overexpressed in COVID-19 lung samples (Figure 4C). We specifically identified interferon-related genes such as IFI6, IFITM2, and IFI44; TNF-related molecules such as TNFAIP3; interleukins such as IL1B; chemokines such as CCL20; molecules associated with reactive oxygen species such as SOD2 (superoxide dismutase 2); and immune checkpoints like CD48 (Figure 4C). Single-cell transcriptome data from bronchoalveolar lavage fluid confirmed the drastic repression of PPARγ in patients with severe COVID-19 (Figure 4D) that was exacerbated in the CD14-CD16 cell subpopulations (Figure 4E). To investigate this further, we examined patterns of PPARγ-dependent gene expression in single-cell transcriptomes of the CD14+/CD16+ cell subset of bronchoalveolar lavage fluid from healthy donors (n = 6) and patients with severe COVID-19 (n = 3, as compared with six healthy donor lungs) (Figure 5A); this subset comprised 559 cells from healthy donors and 1,134 cells from patients with severe COVID-19. In this cell compartment, PPARγ-negative cells were representative of patients with severe COVID-19 and PPARγ-positive cells were representative of samples from healthy donors (Figure 5B). Likewise, unsupervised principal component analysis differentiated, along the first principal axis, between cells from patients with severe COVID-19 and those from healthy donors (Figure 5C), and identified a specific gene expression signature that characterized these myeloid cells (Figure 5D). For example, CD16+/CD14+/PPARγ-negative cells from patients with severe COVID-19 especially repressed some membrane molecules like the tetraspanin CD9, the transferrin receptor TFRC, the cell junction molecule JAML, and the free fatty acid receptor FFAR4 (Figure 5E). In this cell compartment, inflammatory transcription factors such as NFKBIA and MAFB, as well as superoxide dismutase (SOD2), were overexpressed in severe COVID-19 samples (Figure 5F). Upon closer examination of the CD14+/CD16+ cell compartment (Figure 6A), it was also possible to subdivide the cell population based on PPARγ expression: high (over 8 copies of PPARγ per cell), medium (between 3 and 8 copies of PPARγ per cell), and low (fewer than 3 copies of PPARγ per cell) (Figure 6B). We constructed a pseudotime expression trajectory based on PPARγ expression, and this replicated the distribution of cells from severe COVID-19 samples and from healthy donor samples (Figure 6C), as well as the distribution of cells in the PPARγ-expression categories (Figure 6D). An expression heatmap of the pseudotime PPARγ trajectory enabled the identification of six clusters of molecules; clusters 1 and 5, in particular, closely followed the same pattern of pseudotime expression as PPARγ (Figure 6E). Within these clusters, we were intrigued to find RETN alias resistin, which is known to be associated with diseases such as noninsulin-dependent diabetes mellitus and acquired generalized lipodystrophy (Figure 6F), FABP4, a fatty-acid binding molecule (Figure 6F), and MRC1, which is implicated in the recognition of complex carbohydrate structures on glycoproteins (Figure 6F). In the same population of cells, we also confirmed a PPARγ-dependent induction trajectory of the inhibitory immune checkpoint IDO1 (Figure 6G). These results suggest that deregulation of this PPARγ-dependent program plays a central role in the macrophage response during severe COVID-19 lung infection. In particular, the CD14+/CD16+ cell compartment of bronchoalveolar lavage fluids demonstrated a characteristic immune disturbance in samples from patients with severe COVID-19.
Figure 4.

Deregulation of PPARγ Program in COVID-19 Lung Samples
(A) Gene set enrichment analysis revealed that COVID-19 lung samples are enriched in genes that are upregulated in macrophages following PPARγ knockout (NES, normalized enrichment score; FDR, false discovery rate); (B) Boxplot of PPARγ expression in COVID-19 lung biopsies compared with samples from healthy donors, data from two independent datasets (p value from two-sided test with Welch correction); (C) Expression heatmap of PPARγ-knockout-dependent macrophage expression program in the transcriptome of COVID-19 lung biopsies compared with that of healthy donors (Euclidean distances); (D) Violinplot of PPARγ count expression in single-cell transcriptome of bronchoalveolar lavage fluid; (E) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression of PPARγ in HDs and patients with mild or severe COVID-19.
Figure 5.

Single-Cell Expression Signature of CD14+/CD16+ Cell Compartment, in Which PPARγ was Repressed in the Lungs of Patients with Severe COVID-19
(A) Dot plot of PPARγ expression in CD14+/CD16+ (FCGR3A) cells in bronchoalveolar lavage fluid from patients with severe COVID-19 as compared with samples from healthy donors; (B) UMAP dimensionality reduction for CD14, FCGR3A, PPARγ and groups (sc: severe COVID-19, control: healthy donors); (C) Principal component analysis performed on CD14+/CD16+ PPARγ+/- cell compartment (sc: severe COVID-19, control: healthy donors); (D) Expression heatmap of the predictive signature for the first principal axis of the analysis in (C); (E) UMAP representation of the expression of membrane molecules that are repressed in the CD14+/CD16+ cell compartment of bronchoalveolar lavage fluid from severe COVID-19 cases; (F) UMAP representation of the expression of molecules that are upregulated in the CD14+/CD16+ cell compartment of bronchoalveolar lavage fluid from severe COVID-19 cases.
Figure 6.

PPARγ Expression Trajectory in the CD14+/CD16+ Cell Compartment in Lungs
(A) TSNE dimensional reduction of CD14+/CD16+ cells from bronchoalveolar lavage fluid from patients with severe COVID-19 (sc, in blue) as compared with healthy donors (control, in red); (B) TSNE dimensional reduction of CD14+/CD16+ cells with a projection of three expression categories for PPARγ: cells with high expression of PPARγ in red, cells with medium expression of PPARγ in blue, and cells with low expression of PPARγ in green; (C) Pseudotime transformation of single-cell transcriptome of the CD14+/CD16+ cell compartment (dot size represents PPARγ expression); (D) Pseudotime transformation with projection of PPARγ cell expression categories; (E) Pseudotime expression heatmap of PPARγ trajectory in the CD14+/CD16+ cell compartment of bronchoalveolar lavage fluids; (F) TSNE on markers following PPARγ trajectory in the CD14+/CD16+ cell compartment; (G) Pseudotime expression of IDO1 in the CD14+/CD16+ cell compartment of bronchoalveolar lavage fluids.
Deregulation of PPARγ Interactome in COVID-19 Bronchoalveolar Lavage Fluid
To further investigate the dysregulation of the PPARγ-dependent macrophage program in the lungs of patients with COVID-19, we constructed an interactome of the PPARγ network and examined the changes in expression among groups. To do this, PPARγ interactions were collected from the innateDB database of immune network interactions (Breuer et al., 2013) (Figure 7A). Of the gene partners associated with PPARγ, we found evidence in the single-cell transcriptomes (bronchoalveolar lavage fluid) for changes in expression associated with COVID-19 infection (Figure 7B), specifically in SUMO1, NR3C1, KLF6, RXRA, CEBPD, and PLAGL1. At the single-cell level, a strong correlation was found between PPARγ and SUMO1 (r = 0.47, Figure 7C), PPARγ and NR3C1 (r = 0.44, Figure 7C), and PPARγ and RXRA (r = 0.35, Figure 7C). A weaker correlation was detected between PPARγ and KLF6 (r = 0.13, Figure 7D) in patients with COVID-19, and indeed, we did observe a strong activation of KLF6 associated with the severe cases of the disease (Figure 7E) in all lympho-myeloid cell compartments. We constructed another PPARγ interactome using the IntAct protein database (Figure 7F) and, again, found evidence of relationships between PPARγ and some of its partners at the single-cell level in the bronchoalveolar lavage fluid of patients with COVID-19 (Figure 7G). For example, PPARγ expression was strongly correlated with that of EP300 (r = 0.29, Figure 7H), CCDC88A (r = 0.60, Figure 7H), and RXRA (r = 0.45, Figure 7H) in COVID-19 cases. The expression of OPTN did not demonstrate a strong correlation to PPARγ (r = 0.15, Figure 7I); however, its expression did progressively increase in the CD14-CD16 myeloid compartment with increased disease severity (Figure 7J). These results suggest that, along with PPARγ, some of its associated proteins are also affected by COVID-19, particularly in the CD14-CD16 cell population in severe cases of the disease. The corepression of PPARγ-associated DNA-binding partners such as NR3C1, RARA, RXRA, and EP300 could be evidence for coregulation of the entire PPARγ cistrome in the CD14-CD16 cell population during severe COVID-19.
Figure 7.

Deregulation of the PPARγ Interactome in COVID-19 Lung Samples
(A) PPARγ network interactome retrieved from InnateDB database; (B) Single-cell expression (bronchoalveolar lavage fluid) dot plot of PPARγ interactome markers from innateDB in HDs and patients with mild or severe COVID-19 (percent: percent of cells expressing each marker, expression level: color intensity); (C and D) (C) Single-cell expression biplot of PPARγ correlation with SUMO1, NR3C1, RXRA, and (D) KLF6 (healthy donor in green, mild COVID-19 in blue, severe COVID-19 in red); (E) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression of KLF6 from HDs or patients with mild or severe COVID-19; (F) PPARγ network interactome retrieved from IntAct database; (G) Single-cell expression (bronchoalveolar lavage fluid) dot plot of PPARγ interactome markers from IntAct in HDs and patients with mild or severe COVID-19 (percent: percent of cells expressing each marker, expression level: color intensity); (H) Single-cell expression biplot of PPARγ correlation with EP300, CCDC88A, RARA, and (I) OPTN (healthy donor in green, mild COVID-19 in blue, severe COVID-19 in red); (J) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression of OPTN from HDs or patients with mild or severe COVID-19.
Co-repression of PPARγ-NR3C1-RXRA Cistrome during COVID-19 Disease in the Lungs
We retrieved data from a PPARγ ChIP-sequencing (ChIP-seq) experiment performed on the THP-1 monocyte cell line (Gene Expression Omnibus [GEO] sample GSM624141) (Pott et al., 2012) from the Cistrome project database and processed the dataset using version HG38 of the human genome. Promoter mapping was performed with the BETA cistrome application +/− 100 kb around transcription starting sites (Wang et al., 2013). This analysis confirmed the presence of a well-conserved signal in the mammalian promoter database (Figure 8A). When we integrated the PPARγ ChIP-seq data with the PPARγ-repressed signature found in the lungs of patients with COVID-19 (Table S2), we were able to identify a genomic program with a landscape of both distal and proximal promoters (Figure 8B and Table S4). To confirm the relationships between the PPARγ-NR3C1-RXRA cistrome and the PPARγ repression signature of the COVID-19 transcriptomes, CHIP-seq data obtained from the THP-1 monocyte cell line were retrieved for both NR3C1 (GEO sample GSM2661793) (Rollins et al., 2017) and RXRA (GEO sample GSM624142) (Pott et al., 2012) from the Cistrome project database. We also collected THP-1 CHIP-seq data for POLR2A (GEO sample GSM1905827) (Yu et al., 2015) to highlight transcriptional activity in this program. Using the “deeptools” pipeline (Ramírez et al., 2014), all of these CHIP-seq experiments were integrated with the PPARγ-repressed signature from COVID-19 lungs in order to identify common promoter signals +/− 5 kb around transcription starting sites. As shown in Figure 8C, the proximal PPARγ-repressed epigenetic program in monocytes shared its normal transcriptionally active promoters with RXRA and NR3C1. To characterize the active promoters in monocytes, we retrieved THP-1 CHIP-seq experiments for histone H3 lysine 27 acetylation (GEO sample GSM2544237) (Phanstiel et al., 2017) from the Cistrome project database. From the PPARγ-repressed program (Table S4 and Figure 8B), RGS12 was confirmed to present a PPARγ signal in its second promoter; its activity was demonstrated with the H3K27Ac histone mark and binding of RNA polymerase II (Figure 8D), suggesting that this target belongs to the PPARγ-repressed program during COVID-19 in the lungs. At the single-cell level, RGS12 was confirmed to be repressed in the CD14-CD16 cell compartment in COVID-19 lungs, especially in the severe form of the disease (Figure 8E).
Figure 8.

PPARγ-RXRA-NR3C1 Cistrome Repression in COVID-19 Lungs and SUMO1-SARS Interaction
(A) Phastconservation plot of PPARγ CHIP-seq data from the THP-1 cell line (HG38 human genome) mapped on a database of mammalian promoters; (B) Signal-promoter plot of PPARγ CHIP-seq data from THP-1 integrated with the repression signature found in COVID-19 lung transcriptomes (size of the circle is proportional to the negative logRatio found in the COVID-19 lung transcriptome, mapping +/− 100 kb around transcription starting sites [TSS]); (C) Promoter heatmap for PPARγ-RXRA-NR3C1 cistrome and POLR2A from CHIP-seq data from THP-1 adapted to the repression signature found in the COVID-19 lung transcriptome (mapping +/− 5 kb around TSS); (D) PPARγ CHIP-seq signal found in the promoter of RGS12, mapped with POLR2A and Histone H3 K27 acetylation in THP-1 cells; (E) UMAP representation of single-cell transcriptome (bronchoalveolar lavage fluid) expression of RGS12 in HDs or patients with mild or severe COVID-19; (F) Human-SARS protein-protein interactome network, with integration of the PPARγ complex affected in COVID-19 lung infection (edges represent connections found in VirusHostNet database).
SUMO1, from the COVID-19 PPARγ-Repressed Complex, Interacts with Nucleoprotein in Human SARS Virus
In order to investigate the interactions of the PPARγ complex with the human SARS virus, we explored the VirusHostNet 2.0 database (Guirimand et al., 2015) using the deregulated partners of PPARγ that we identified (Table S5). By integrating these interactions with the PPARγ network, we were able to create a virus-host network (Figure 8F). Using this, we determined that, within the PPARγ-repressed complex, only SUMO1 is capable of interacting with a SARS virus, through the nucleoprotein NCAP_CVHSA of the human coronavirus. This result suggests that, during lung infection, repression of the PPARγ complex may be mediated via the interaction of SUMO1 with the virus and that sumoylation function may also be affected.
Discussion
The COVID-19 pandemic has already affected hundreds of thousands of people and has become the greatest health challenge worldwide. The range of clinical presentations varies from asymptomatic and mild clinical symptoms to acute respiratory-distress syndrome (ARDS) and death. Respiratory complications have been well described in this disease, which is caused by infection with the virus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). Diabetes mellitus has been identified as a factor that contributes to the development of ARDS, whereas other associated comorbidities include hypertension, cardiovascular disease, and chronic kidney disease (Wu et al., 2020a). In addition, a link has been demonstrated between obesity and COVID-19 cases that require invasive mechanical ventilation (Simonnet et al., 2020).
In this work, by comparing the transcriptomes of COVID-19 lung biopsies with those of healthy donors, we were able to characterize the immune infiltration in this tissue using different bioinformatics approaches. The major finding of our work is the discovery of a link between the severity of COVID-19 and repression of the PPARγ complex. In general, we observed that the adaptive immune response to the disease appeared to be driven by the induction of interferon gamma, whereas the innate response was largely the result of the induction of interferon alpha, TNF alpha, the complement cascade, and reactive oxygen species. Immune scoring of COVID-19 lung biopsies revealed major myeloid infiltration, specifically by monocytes, M1 macrophages, and neutrophils. Instead, the lymphoid scores for this sample were not significant, which suggests that the role of the adaptive immune response is less important than that of the innate response. This finding was confirmed by immune deconvolution analysis of the transcriptome data from COVID-19 lung samples. The SARS-CoV-2 coronavirus is a single-, positive-stranded RNA virus enveloped in a lipid bilayer (Wu et al., 2020b) (Lu et al., 2020). The lipid bilayer fuses with the host cell membrane, releasing RNA into the cytoplasm and resulting in the translation of various viral proteins. The replicated RNA genome and synthesized viral proteins reassemble into new viruses, which burst out of the cell (Qinfen et al., 2004). When we analyzed the induction of TLRs in COVID-19 lung samples, only the lipopolysaccharide sensor TLR2 was found to be upregulated with respect to controls, which is consistent with detection of the coronavirus envelope. The TLR7 sensor did not seem to be affected by COVID-19 infection in our samples, which suggests a lack of presentation of genetic material from the virus in the lungs; this is consistent with our other data demonstrating inefficiency in the adaptive immune response to this disease.
Compared with the virus that caused the SARS outbreak in 2003, the virus that causes COVID-19 uses the same mechanism to enter host cells, but at a slower speed. However, SARS-CoV-2 accumulates to higher concentrations in the body compared with SARS-CoV. This explains why COVID-19 has a longer incubation period and is more contagious, whereas SARS instead presents with more symptoms and increased disease severity (Guo et al., 2020). Immune scoring of COVID-19 lung biopsies revealed the infiltration of monocytes/macrophages in this tissue, and gene set enrichment analysis confirmed a major role for this type of cells in the immune program described in COVID-19 lungs. Several types of cytokines and chemokines belong to this dysregulated macrophage program. As in macrophage activation syndrome (MAS), macrophages in the lungs of patients with COVID-19 could play a central role in the non-adapted lung immune response through their contributions to cytokine storm. In COVID-19 lung samples, we detected an increase in interleukin 1 (IL1A and IL1B) and its receptor IL1R2. IL-1 is a pro-inflammatory cytokine produced primarily by monocytes and macrophages. It is present in the inactive form, pro-IL-1β, but, upon monocyte/macrophage activation it is cleaved by caspase-1 and becomes biologically active. Via signaling through its receptor, IL-1β causes lymphocyte and endothelial activation as well as the production of other inflammatory cytokines including IL-6 (Pascual et al., 2005b). IL-6 signaling, through the JAK cascade, was found to be enriched in COVID-19 lung samples compared with healthy donor tissues and expression of IL-6ST was likewise upregulated. IL-6 is a pleotropic cytokine produced in the early stages of inflammation and is central in driving the acute-phase response. One study of patients with MAS demonstrated that IL-6 produced activated macrophages in tissue obtained from liver biopsies (Billiau et al., 2005). For this reason, treatment with tocilizumab monoclonal antibody, which blocks IL-6 receptors, has been proposed to neutralize the cytokine storm that can occur during severe cases of COVID-19 (Fu et al., 2020). Within the IL-6 pathway, we found that the SCOC3 (suppressor of cytokine signaling 3) protein was upregulated in COVID-19 lung samples and, at the same time, we detected an induction of inhibitory immune checkpoints such as PDL1. IL-6 was reported to signal, via SCOS3, for the induction of its receptor PD1 during a cytokine storm, which was then found to neutralize the cell-mediated antiviral response (Velazquez-Salinas et al., 2019). Among the inhibitory immune checkpoints that were upregulated in COVID-19 lungs, there is an FDA-approved targeted therapy (consisting of an antagonistic monoclonal antibody) for only one, CD274. However, a potential therapeutic strategy of targeting PDL1/PD1 in order to manipulate the adaptive immune response could be dangerous in COVID-19, because a variety of fatal adverse events related to the respiratory system have been recorded, including but not limited to pneumonia and respiratory failure (Wang et al., 2020).
In the lungs of patients with COVID-19, we detected a strong induction of the TNF-alpha response using gene set enrichment analysis. TNF-alpha is a pleomorphic cytokine that has been implicated in the pathogenesis of several inflammatory diseases. It is produced largely by monocytes and macrophages that are activated by TLR ligands such as endotoxins as well as cytokines such as IL-18 and stimulates local endothelial cells as well as lymphocytes (Shenoi and Wallace, 2010). IL-18 was also found to be upregulated in COVID-19 lung biopsies compared with controls; this is a unique cytokine in the IL-1 family because it is constitutively present in keratinocytes, epithelial cells, and blood monocytes (Puren et al., 1999). In addition to promoting secretion of TNF-α and chemokines by macrophages, IL-18 also induces production of the pro-inflammatory cytokine IFNγ by NK cells and T cells (Dinarello, 2007). Indeed, using gene set enrichment analysis, we observed substantial activation of the IFN-gamma response in COVID-19 lung samples, the primary function of which is to strongly activate monocytes and macrophages (Schroder et al., 2004). Activated macrophages are divided into several general classes based upon their respective stimuli and their resulting polarization, with M1 macrophages driven by IFN-gamma into a classical pro-inflammatory phenotype that is characterized by increased microbicidal ability, heightened responses to TLR ligands, and upregulated antigen processing and presentation. Consistent with this, we found that the immune scores of COVID-19 lung biopsies highlighted significant infiltration of macrophages of the M1 type. These cells are potent producers of pro-inflammatory cytokines, including IL-6, IL-12, and IL-23, as well as the chemokines IP-10, MIG, and ITAC, which recruit polarized Th1 cells in addition to NK cells (Mosser, 2003) (Mantovani et al., 2004). There is also evidence that IFNγ may be a critical driver of hemophagocytosis by these activated macrophages (Zoller et al., 2011). This could be connected to the reduction in heme metabolism-related activity we observed in the transcriptome of COVID-19 lung samples.
This dysregulation in secreted effectors in the lungs of patients with COVID-19 places monocytes/macrophages at the center of the cytokine storm that has been observed in severe cases of this disease. This was further corroborated by our finding that a gene set enrichment analysis performed on COVID-19 lung samples highlighted upregulation in a transcriptional program that was characteristic of macrophage cells in which PPARγ has been knocked out. Specifically, this program involved the activation of interferon-related genes, including several cytokines and chemokines. We determined that the expression of PPARγ was indeed downregulated in COVID-19 lung samples, which confirmed the origin of this macrophage dysregulation. Previous research has shown that PPARγ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection (Huang et al., 2019b). In a murine influenza infection model with genetic-induced obesity (db/db), it was shown that the transcription factor PPARγ was downregulated in the lung macrophages of db/db mice after influenza infection. The treatment of 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2), a PPAR-γ agonist, largely rescued the survival of db/db mice after influenza infection (Huang et al., 2019a). As was demonstrated in H1N1 infection, there could be a link between PPARγ and macrophages that drives inflammation in patients with COVID-19 (Josset et al., 2012). It is possible that restoring higher levels of PPARγ expression in macrophages could attenuate the severe immune response that has been linked with these innate cells. Several PPARγ agonists exist on the market (in phases II/III) for this purpose, and they have already been proposed as an alternative therapy to target the cytokine storm (Ciavarella et al., 2020). In an animal model, it has been shown that loss of PPARγ can delay monocyte differentiation into macrophages and increase a pro-inflammatory phenotype with long-term LPS stimulation, characterized by an elevated production of the pro-inflammatory cytokines TNF-α, IL1-β, IL-6, and IL-12 and reduced production of the anti-inflammatory cytokine IL-10 (Heming et al., 2018). PPARγ has also been implicated in the homeostasis of lipid metabolism in macrophages (Li et al., 2000), and PPAR receptors have been characterized as a crossroad between lipid metabolism and inflammation (Chinetti et al., 2000). In mice, systemic PPARγ deletion provokes severe type-2 diabetes (Gilardi et al., 2019) and PPARγ has been associated with type-2 diabetes in humans via a genome-wide association study (GWAS) of Finns (Scott et al., 2007, p. 2). Within the hospitalized COVID-19 population, individuals with diabetes are over-represented (Zhou et al., 2020a) and this disorder has been defined as a major comorbidity of COVID-19 (Zhu et al., 2020). It is possible that the link between COVID-19 and type-2 diabetes could be PPARγ. Furthermore, genetic polymorphism in PPARγ has also been linked to body mass index (BMI) (Fornage et al., 2005) via GWAS analysis by the Framingham Heart Study 100K project (Fox et al., 2007). PPARγ could also represent a link with the obesity comorbidity found with COVID-19 that could reach 44% (Richardson et al., 2020).
The epigenetically repressed cistrome program identified in COVID-19 lungs extended to proteins of the classical retinoic acid pathway, such as RXRA, and also to the glucocorticoid receptor NR3C1; this result could suggest changes in the sensitivity of macrophages to glucocorticoids via their anti-inflammatory action on this cell type (Mylka et al., 2018). Inside the PPARγ interactome, optineurin was found to be induced during severe COVID-19; this molecule has been characterized as an interleukin-1 receptor-associated kinase 1-binding protein and has been implicated in inflammatory signaling (Tanishima et al., 2017). Finally, within the PPARγ-associated repressed protein network, we identified SUMO1 as capable of interacting with a nucleoprotein of the human SARS virus. PPARγ sumoylation is important for the LPS response and transregulation of proinflammatory cytokines, via NCOR occupancy of kappaB-binding sites (Jennewein et al., 2008). Sumoylation could act as a brake on PPARγ-associated repression of the transcriptional activation of inflammatory response genes in macrophages (Pascual et al., 2005a).
In this work, the immune response to COVID-19 infection in the lungs was investigated; using different approaches, we identified dysfunction in M1 monocytes/macrophages in the innate response process. This dysfunction was characterized by repression of PPARγ, which may play a key role in the cytokine storm of inflammatory monocytes/macrophages in the SARS-CoV-2-infected lung.
Limitations of the Study
Integrative analysis through different bioinformatics methods on lung transcriptomes of patients with COVID-19 enabled the characterization of the lung innate response as major in this pathological tissue. A fundamental deregulation of PPARγ disruption was found in the innate immune response through monocyte-macrophage cells. Repression of PPARγ was confirmed at the single-cell level in the severity of the disease. Network analysis on the PPARγ interacting complex revealed also the disruption of some of its partners. PPARγ is an essential molecule implicated in macrophage activation and glucose, fatty acid, and cholesterol metabolism: these mechanisms could link to the severity of the disease highly correlated to diabetes and obesity. Interestingly, PPARγ is a druggable target. This bioinformatics study on disturbed immune markers and immune cell deconvolution was validated at the single-cell level in an independent cohort of samples. Epigenetic integration in the analysis allowed one to confirm the genome landscape implication of PPARγ-NR3C1-RXRA cistrome repression in severe lung COVID-19 diseases. SUMO1 that represses the PPARγ partner in COVID-19 diseased lungs was also found to interact with the SARS virus. The discovery of this original mechanism could help to better understand the huge innate inflammatory processes in COVID-19 diseased lungs as well as the associated risk factors found in this disease.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank Griscelli (franck.griscelli@gustaveroussy.fr).
Materials Availability
This study did not generate specific reagents.
Data and Code Availability
We obtained transcriptomes from COVID-19 lung biopsies from dataset GSE147507 in the Gene Expression Omnibus database (https://www.biorxiv.org/content/10.1101/2020.03.24.004655v1) and now publish in Cell on 28th May 2020 (https://www.sciencedirect.com/science/article/pii/S009286742030489X?via%3Dihub) (Blanco-Melo et al., 2020). “Volcanoplot” software was developed to analyze the significance of immune scores; the corresponding R functions are available at the web address: https://github.com/cdesterke/volacanoimmune. Chip2heat.sh software was developed in SHELL BASH (Bourne-again SHELL) and implemented with the deeptools pipeline, specifically computeMatrix and plotHeatmap, with the promoter plot saved in different formats: PNG, PDF, SVG. This software can be downloaded at https://github.com/cdesterke/chip2heat. The R code for the single-cell analyses for marker validation is detailed in the Transparent Methods section of the Supplemental Information and also provided at the following address: https://github.com/cdesterke/covid19sc. Additional resources with interactive Web interface of the single cell analysis: to facilitate data exploration of disturb CD14+/CD16+ double population in bronchoalveolar lavage fluid of patients with severe COVID-19, we developed an interactive web interface available at https://cdesterke.shinyapps.io/COVID19LUNG/. This website was built with flexdasboard and shiny application inclusion and with graphical interactivity display by R plotly. This data dashboard allows exploring expression of biomarkers found on PPARγ trajectory in lung monocyte-macrophage disrupted cell population in severe COVID-19. The user needs to select gene ID on the left sidebar, and the application will display expression of this selected marker with interactivity on TSNE graph, number of positive cells for this marker will be displayed in the value box at the top right of the dashboard, also expression by group will be displayed on violinplot, finally statistical summary (mean and standard deviation, SD) will be displayed by group of samples.
Mendeley Dataset associated to this manuscript: 10.17632/3xnypzvcf7.1.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the association “Vaincre le cancer NRB” (https://www.vaincrelecancer-nrb.org/), which made it possible to finance the computer equipment used to carry out these analyses. This work was performed with grants from the ANR “Programme d’Investissements d’Avenir” of the INGESTEM National Infrastructure (ANR-11-INBS-0009-INGESTEM) and INSERM, University Paris Sud.
Author Contributions
C.D., A.B.-G., F.G.: Conceptualization. C.D., F.G., A.G.T.: Methodology. C.D., F.G., A.B.-G., A.G.T.: Validation. C.D., F.G., A.B.-G., A.G.T.: Data analysis. C.D., F.G.: Writing – original draft preparation. F.G., A.B.-G.: Supervision. C.D., A.B.-G., A.G.T., F.G.: Funding acquisition.
Declaration of Interests
The authors do not have any conflict of interest to declare.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101611.
Supplemental Information
https://doi.org/10.17632/3xnypzvcf7.1#file-39267130-458c-41d8-8bf7-26ae18b717d1
https://doi.org/10.17632/3xnypzvcf7.1#file-03b71cec-f317-439c-9176-170edd6ac7e0
https://doi.org/10.17632/3xnypzvcf7.1#file-71c195a0-6834-4a0a-8719-826bae172b31
https://doi.org/10.17632/3xnypzvcf7.1#file-14fb92f7-2a84-478c-a1fc-99d41668f1e4
References
- Bell-Parikh L.C., Ide T., Lawson J.A., McNamara P., Reilly M., FitzGerald G.A. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma. J. Clin. Invest. 2003;112:945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau A.D., Roskams T., Van Damme-Lombaerts R., Matthys P., Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105:1648–1651. doi: 10.1182/blood-2004-08-2997. [DOI] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S., Wang T.T., Ravetch J.V. The role and function of Fcγ receptors on myeloid cells. Microbiol. Spectr. 2016;4 doi: 10.1128/microbiolspec.MCHD-0045-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer K., Foroushani A.K., Laird M.R., Chen C., Sribnaia A., Lo R., Winsor G.L., Hancock R.E.W., Brinkman F.S.L., Lynn D.J. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–D1233. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinetti G., Fruchart J.C., Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- Ciavarella C., Motta I., Valente S., Pasquinelli G. Pharmacological (or synthetic) and nutritional agonists of PPAR-γ as candidates for cytokine storm modulation in COVID-19 disease. Molecules. 2020;25 doi: 10.3390/molecules25092076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin. Nephrol. 2007;27:98–114. doi: 10.1016/j.semnephrol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Fornage M., Jacobs D.R., Steffes M.W., Gross M.D., Bray M.S., Schreiner P.J. Inverse effects of the PPAR(gamma)2 Pro12Ala polymorphism on measures of adiposity over 15 years in African Americans and whites. The CARDIA study. Metab. Clin. Exp. 2005;54:910–917. doi: 10.1016/j.metabol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Fox C.S., Heard-Costa N., Cupples L.A., Dupuis J., Vasan R.S., Atwood L.D. Genome-wide association to body mass index and waist circumference: the Framingham Heart Study 100K project. BMC Med. Genet. 2007;8(Suppl 1):S18. doi: 10.1186/1471-2350-8-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B., Xu X., Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J. Transl. Med. 2020;18:164. doi: 10.1186/s12967-020-02339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardi F., Winkler C., Quignodon L., Diserens J.-G., Toffoli B., Schiffrin M., Sardella C., Preitner F., Desvergne B. Systemic PPARγ deletion in mice provokes lipoatrophy, organomegaly, severe type 2 diabetes and metabolic inflexibility. Metab. Clin. Exp. 2019;95:8–20. doi: 10.1016/j.metabol.2019.03.003. [DOI] [PubMed] [Google Scholar]
- Gosset P., Charbonnier A.S., Delerive P., Fontaine J., Staels B., Pestel J., Tonnel A.B., Trottein F. Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur. J. Immunol. 2001;31:2857–2865. doi: 10.1002/1521-4141(2001010)31:10<2857::aid-immu2857>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Guirimand T., Delmotte S., Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015;43:D583–D587. doi: 10.1093/nar/gku1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.-R., Cao Q.-D., Hong Z.-S., Tan Y.-Y., Chen S.-D., Jin H.-J., Tan K.-S., Wang D.-Y., Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil. Med. Res. 2020;7:11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heming M., Gran S., Jauch S.-L., Fischer-Riepe L., Russo A., Klotz L., Hermann S., Schäfers M., Roth J., Barczyk-Kahlert K. Peroxisome proliferator-activated receptor-γ modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front. Immunol. 2018;9:893. doi: 10.3389/fimmu.2018.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzel M., Walcher D., Grüb M., Bach H., Hombach V., Marx N. Inhibition of MMP-9 expression by PPARgamma activators in human bronchial epithelial cells. Thorax. 2003;58:778–783. doi: 10.1136/thorax.58.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K., Marquillies P., Capron M., Dombrowicz D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J. Allergy Clin. Immunol. 2004;113:882–888. doi: 10.1016/j.jaci.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Huang S., Jiang L., Cheon I.S., Sun J. Targeting peroxisome proliferator-activated receptor-gamma decreases host mortality after influenza infection in obese mice. Viral Immunol. 2019;32:161–169. doi: 10.1089/vim.2019.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Zhu B., Cheon I.S., Goplen N.P., Jiang L., Zhang R., Peebles R.S., Mack M., Kaplan M.H., Limper A.H., Sun J. PPAR-γ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J. Virol. 2019;93 doi: 10.1128/JVI.00030-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein C., Kuhn A.-M., Schmidt M.V., Meilladec-Jullig V., von Knethen A., Gonzalez F.J., Brüne B. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 2008;181:5646–5652. doi: 10.4049/jimmunol.181.8.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josset L., Belser J.A., Pantin-Jackwood M.J., Chang J.H., Chang S.T., Belisle S.E., Tumpey T.M., Katze M.G. Implication of inflammatory macrophages, nuclear receptors, and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. J. Virol. 2012;86:7192–7206. doi: 10.1128/JVI.00563-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A.C., Brown K.K., Silvestre M.J., Willson T.M., Palinski W., Glass C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J. Clin. Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020 doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- McGonagle D., Sharif K., O’Regan A., Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020;102537 doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M. The many faces of macrophage activation. J. Leukoc. Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Mylka V., Deckers J., Ratman D., De Cauwer L., Thommis J., De Rycke R., Impens F., Libert C., Tavernier J., Vanden Berghe W. The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator compound A (CpdA) in macrophages. Autophagy. 2018;14:2049–2064. doi: 10.1080/15548627.2018.1495681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiura H., Kobayashi T., Suzuki A., Jung S.-M., Hayashi K., Kinoshita R., Yang Y., Yuan B., Akhmetzhanov A.R., Linton N.M., Miyama T. Estimation of the asymptomatic ratio of novel coronavirus infections (COVID-19) Int. J. Infect. Dis. 2020 doi: 10.1016/j.ijid.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte R.T., Wisely G.B., Westin S., Cobb J.E., Lambert M.H., Kurokawa R., Rosenfeld M.G., Willson T.M., Glass C.K., Milburn M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G., Fong A.L., Ogawa S., Gamliel A., Li A.C., Perissi V., Rose D.W., Willson T.M., Rosenfeld M.G., Glass C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual V., Allantaz F., Arce E., Punaro M., Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005;201:1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel D.H., Van Bortle K., Spacek D., Hess G.T., Shamim M.S., Machol I., Love M.I., Aiden E.L., Bassik M.C., Snyder M.P. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol. Cell. 2017;67:1037–1048.e6. doi: 10.1016/j.molcel.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont M.J., Honders M.W., Kremer A.N., van Kooten C., Out C., Hiemstra P.S., de Boer H.C., Jager M.J., Schmelzer E., Vries R.G. Microarray gene expression analysis to evaluate cell type specific expression of targets relevant for immunotherapy of hematological malignancies. PLoS One. 2016;11:e0155165. doi: 10.1371/journal.pone.0155165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott S., Kamrani N.K., Bourque G., Pettersson S., Liu E.T. PPARG binding landscapes in macrophages suggest a genome-wide contribution of PU.1 to divergent PPARG binding in human and mouse. PLoS One. 2012;7:e48102. doi: 10.1371/journal.pone.0048102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puren A.J., Fantuzzi G., Dinarello C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. U S A. 1999;96:2256–2261. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qinfen Z., Jinming C., Xiaojun H., Huanying Z., Jicheng H., Ling F., Kunpeng L., Jingqiang Z. The life cycle of SARS coronavirus in Vero E6 cells. J. Med. Virol. 2004;73:332–337. doi: 10.1002/jmv.20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson S., Hirsch J.S., Narasimhan M., Crawford J.M., McGinn T., Davidson K.W., the Northwell COVID-19 Research Consortium. Barnaby D.P., Becker L.B., Chelico J.D., Cohen S.L. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York city Area. JAMA. 2020 doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins D.A., Kharlyngdoh J.B., Coppo M., Tharmalingam B., Mimouna S., Guo Z., Sacta M.A., Pufall M.A., Fisher R.P., Hu X. Glucocorticoid-induced phosphorylation by CDK9 modulates the coactivator functions of transcriptional cofactor GRIP1 in macrophages. Nat. Commun. 2017;8:1739. doi: 10.1038/s41467-017-01569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoi S., Wallace C.A. Tumor necrosis factor inhibitors in the management of juvenile idiopathic arthritis: an evidence-based review. Paediatr. Drugs. 2010;12:367–377. doi: 10.2165/11532610-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Simonnet A., Chetboun M., Poissy J., Raverdy V., Noulette J., Duhamel A., Labreuche J., Mathieu D., Pattou F., Jourdain M., Lille Intensive Care COVID-19 and Obesity study group High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. 2020 doi: 10.1002/oby.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standiford T.J., Keshamouni V.G., Reddy R.C. Peroxisome proliferator-activated receptor-{gamma} as a regulator of lung inflammation and repair. Proc. Am. Thorac. Soc. 2005;2:226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- Straus D.S., Glass C.K. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Tanishima M., Takashima S., Honda A., Yasuda D., Tanikawa T., Ishii S., MaruYama T. Identification of optineurin as an interleukin-1 receptor-associated kinase 1-binding protein and its role in regulation of MyD88-dependent signaling. J. Biol. Chem. 2017;292:17250–17257. doi: 10.1074/jbc.M117.813899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A.J., Hiscox J.A., Hooper N.M. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004;25:291–294. doi: 10.1016/j.tips.2004.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Salinas L., Verdugo-Rodriguez A., Rodriguez L.L., Borca M.V. The role of interleukin 6 during viral infections. Front. Microbiol. 2019;10:1057. doi: 10.3389/fmicb.2019.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sun H., Ma J., Zang C., Wang C., Wang J., Tang Q., Meyer C.A., Zhang Y., Liu X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013;8:2502–2515. doi: 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wu S., Chen Y., Shao E., Zhuang T., Lu L., Chen X. Fatal adverse events associated with programmed cell death ligand 1 inhibitors: a systematic review and meta-analysis. Front. Pharmacol. 2020;11:5. doi: 10.3389/fphar.2020.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020 doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA. 2020 doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Xu X., Han M., Li T., Sun W., Wang D., Fu B., Zhou Y., Zheng X., Yang Y., Li X. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. U S A. 2020;117:10970–10975. doi: 10.1073/pnas.2005615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Yang W., Ni T., Tang Z., Nakadai T., Zhu J., Roeder R.G. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science. 2015;350:1383–1386. doi: 10.1126/science.aad2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., She Z.-G., Cheng X., Qin J.-J., Zhang X.-J., Cai J., Lei F., Wang H., Xie J., Wang W. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020 doi: 10.1016/j.cmet.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller E.E., Lykens J.E., Terrell C.E., Aliberti J., Filipovich A.H., Henson P.M., Jordan M.B. Hemophagocytosis causes a consumptive anemia of inflammation. J. Exp. Med. 2011;208:1203–1214. doi: 10.1084/jem.20102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
https://doi.org/10.17632/3xnypzvcf7.1#file-39267130-458c-41d8-8bf7-26ae18b717d1
https://doi.org/10.17632/3xnypzvcf7.1#file-03b71cec-f317-439c-9176-170edd6ac7e0
https://doi.org/10.17632/3xnypzvcf7.1#file-71c195a0-6834-4a0a-8719-826bae172b31
https://doi.org/10.17632/3xnypzvcf7.1#file-14fb92f7-2a84-478c-a1fc-99d41668f1e4
Data Availability Statement
We obtained transcriptomes from COVID-19 lung biopsies from dataset GSE147507 in the Gene Expression Omnibus database (https://www.biorxiv.org/content/10.1101/2020.03.24.004655v1) and now publish in Cell on 28th May 2020 (https://www.sciencedirect.com/science/article/pii/S009286742030489X?via%3Dihub) (Blanco-Melo et al., 2020). “Volcanoplot” software was developed to analyze the significance of immune scores; the corresponding R functions are available at the web address: https://github.com/cdesterke/volacanoimmune. Chip2heat.sh software was developed in SHELL BASH (Bourne-again SHELL) and implemented with the deeptools pipeline, specifically computeMatrix and plotHeatmap, with the promoter plot saved in different formats: PNG, PDF, SVG. This software can be downloaded at https://github.com/cdesterke/chip2heat. The R code for the single-cell analyses for marker validation is detailed in the Transparent Methods section of the Supplemental Information and also provided at the following address: https://github.com/cdesterke/covid19sc. Additional resources with interactive Web interface of the single cell analysis: to facilitate data exploration of disturb CD14+/CD16+ double population in bronchoalveolar lavage fluid of patients with severe COVID-19, we developed an interactive web interface available at https://cdesterke.shinyapps.io/COVID19LUNG/. This website was built with flexdasboard and shiny application inclusion and with graphical interactivity display by R plotly. This data dashboard allows exploring expression of biomarkers found on PPARγ trajectory in lung monocyte-macrophage disrupted cell population in severe COVID-19. The user needs to select gene ID on the left sidebar, and the application will display expression of this selected marker with interactivity on TSNE graph, number of positive cells for this marker will be displayed in the value box at the top right of the dashboard, also expression by group will be displayed on violinplot, finally statistical summary (mean and standard deviation, SD) will be displayed by group of samples.
Mendeley Dataset associated to this manuscript: 10.17632/3xnypzvcf7.1.