

In response to starvation, cells tune protein synthesis to promote autophagy, thus ensuring nutrient recycling and survival.
Abstract
Cells respond to starvation by shutting down protein synthesis and by activating catabolic processes, including autophagy, to recycle nutrients. This two-pronged response is mediated by the integrated stress response (ISR) through phosphorylation of eIF2α, which represses protein translation, and by inhibition of mTORC1 signaling, which promotes autophagy also through a stress-responsive transcriptional program. Implementation of such a program, however, requires protein synthesis, thus conflicting with general repression of translation. How is this mismatch resolved? We found that the main regulator of the starvation-induced transcriptional program, TFEB, counteracts protein synthesis inhibition by directly activating expression of GADD34, a component of the protein phosphatase 1 complex that dephosphorylates eIF2α. We discovered that GADD34 plays an essential role in autophagy by tuning translation during starvation, thus enabling lysosomal biogenesis and a sustained autophagic flux. Hence, the TFEB-GADD34 axis integrates the mTORC1 and ISR pathways in response to starvation.
INTRODUCTION
Amino acid starvation leads to repression of cap-dependent translation through the integrated stress response (ISR) pathway, thus decreasing global protein synthesis in the cell (fig. S1, A and B). Starvation also inhibits the mammalian target of rapamycin complex 1 (mTORC1), thus initiating autophagy and triggering a transcriptional program required for lysosomal biogenesis and a sustained autophagic flux (1). Translation of the starvation-induced transcriptional program, however, does require protein synthesis, thus conflicting with the repression of general translation. We set to investigate how this conflict is resolved.
Activation of the ISR results in phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α) by the GCN2 kinase. This event impairs cap-dependent translation initiation but favors selective translation of a subset of stress-responsive mRNAs containing inhibitory upstream open reading frames (uORFs), such as the transcription factor ATF4, which drives expression of essential autophagy genes such as LC3 and Atg5 (2). However, of the 513 genes annotated to lysosomal and autophagic processes according to the literature (3), only 65 (12.7%) contain multiple uORFs, and 21 (4.1%) an intra ribosomal entry sequence (4, 5), suggesting that a different mechanism must be at play.
Inactivation of the mTORC1 complex leads to the nuclear translocation of the TFEB/TFE3/MiTF-TFE (transcription factor EB/transcription factor binding To IGHM enhancer 3/microphthalmia associated transcription factor/transcription factor ebox) family of transcription factors and activation of a specific transcriptional program (1, 6) that itself may incorporate a strategy to overcome protein synthesis inhibition. In this study, we demonstrated that this is the case: TFEB/TFE3 fine-tunes protein synthesis during starvation by transcriptionally regulating GADD34 to enable lysosomal biogenesis and autophagic flux.
RESULTS
GADD34 is an early and direct TFEB target
We analyzed the global transcriptional response to increasing concentrations of TFEB upon its overexpression by means of a tetracycline-responsive promoter (Fig. 1, A to C, fig. S2, table S1, and Methods) and concomitantly performed genome-wide TFEB chromatin immunoprecipitation followed by sequencing (ChIP-seq) (Fig. 1D and table S2).
Fig. 1. Identification of TFEB direct targets following TFEB overexpression.
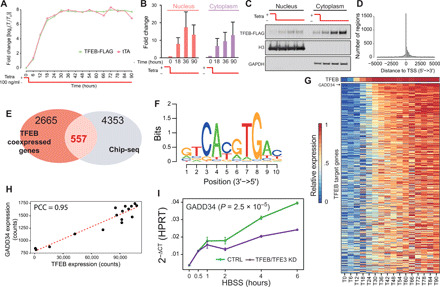
(A) Characterization of inducible expression system in human embryonic kidney (HEK) 293 cells with genomic integration of the expression cassette in fig. S2. Exogenous TFEB-FLAG and tTA transactivator levels measured at the indicated time points by quantitative real-time polymerase chain reaction (qRT-PCR). (B) Quantification by densitometry of (C) immunoblotting assay (WB) of TFEB-FLAG in nuclear and cytoplasmatic fractions at the indicated time points expressed as fold change relative to time 0. (D) Distribution of TFEB binding sites detected by ChIP-seq relative to the transcription start site (TSS). (E) Selection of 557 bona fide TFEB direct targets and (F) de novo motif finding in their proximal promoter revealing the CLEAR binding site. (G) Expression levels of the 557 TFEB direct targets at increasing TFEB-FLAG levels at the indicated time points. Genes are ordered according to their correlation with TFEB-FLAG expression. (H) Scatter plot of GADD34 versus TFEB-FLAG expression levels. (I) qRT-PCR of the indicated gene during amino acid deprivation in untreated cells (control, CTRL) or following small interfering RNA–mediated knockdown (KD) of TFEB and TFE3 (TFEB/TFE3 KD); P value refers to two-way analysis of variance (ANOVA) after post hoc correction.
TFEB direct targets should be expressed in response to its induction in a dose-dependent manner and should have a TFEB binding site in their promoter. A total of 557 genes satisfied both conditions (Fig. 1, E to G), as they were both positively coexpressed with TFEB [Pearson correlation coefficient, false discovery rate (FDR) < 10%] and contained TFEB ChIP-seq binding sites within ±2.5 Kb from their transcription start site (table S1). Bioinformatics analysis of their binding sites revealed the previously reported CLEAR motif (Fig. 1F). Gene Ontology Enrichment Analysis (table S3) (7) of the 557 direct targets highlighted “canonical” TFEB-regulated pathways, including endolysosomal genes (e.g., AP1G1, ATP6V0D1, ATPV1F, ATP6V1H, CD68, CLN3, CTSK, GNS, LAMP1, M6PR, NAGLU, NEU1, PSAP, RRAGC, and RRAGD) and autophagic genes (ATG14, GABARAPL2, LANCL2, PLIN2, RAB1A, SQSTM1, and TP53INP2). Genes classified as “endoplasmic reticulum (ER) stress related” were also significantly enriched, including the protein phosphatase 1 (PP1) phosphatase regulator PPP1R15A (aka GADD34), ranked as the third most correlated as shown in Fig. 1H, in addition to DDIT3 (aka CHOP, ranked 52nd) and PPP1R15B (aka CREP, ranked 384th). GADD34 undergoes selective translation upon eIF2α phosphorylation, and it is required for the activity of PP1 phosphatase that dephosphorylates eIF2α (8). Its role in the unfolded protein response (UPR) is well established: Upon eIF2α phosphorylation triggered by the accumulation of misfolded proteins, GADD34 is translated and acts to terminate the response either by restoring protein synthesis, if the ER stress is resolved, or by sensitizing cells to apoptosis, if not (9). To exclude any role of ER stress in our experimental condition, that is, TFEB overexpression in nutrient-rich medium, we verified the absence of canonical markers of the UPR, i.e., no increase in BiP levels, no alternative splicing of XBP, and no transcription of ER-associated degradation (ERAD) genes (fig. S3). We next showed that the endogenous TFEB regulates GADD34 expression early during starvation. GADD34 transcript and protein levels increased quickly in response to amino acid deprivation in a TFEB-dependent manner, as concomitant knockdown (KD) of TFEB and TFE3 strongly attenuated GADD34 expression (Fig. 1I and fig. S4, C to F), while TFEB overexpression increased it (fig. S4, A and B). This is in contrast with the delayed activation of GADD34 observed after prolonged ER stress (16 hours) (9–11).
GADD34 is required to maintain autophagic flux
We thus hypothesized that GADD34 may be necessary to initiate response to starvation by enabling translation of starvation-induced genes including lysosomal and autophagic components, under conditions (i.e., starvation) that would otherwise be not permissive for de novo protein synthesis. We assessed the impact of genetically ablating GADD34 on the autophagic flux. We thus first evaluated the number of LC3-positive structures in wild-type (WT) and GADD34 knockout (KO) cells (Fig. 2A) (12). GADD34 KO cells showed significantly more LC3-positive structures than WT cells both in growth medium and following amino acid deprivation (Fig. 2B). This effect may be explained by either an increase in autophagosome biogenesis or a decrease in autophagic flux: To distinguish between these possibilities, we treated cells with bafilomycin A1, an inhibitor of lysosome function that induces the accumulation of undigested autophagosomes—the higher the accumulation of autophagosomes induced by bafilomycin A1 treatment, the higher the rate of autophagic flux. While bafilomycin A1 induced a marked increase in LC3-positive structures in WT cells (Fig. 2, A and C), its effect was either dampened (basal and 6 hours in amino acid starvation) or absent (20 hours in amino acid starvation) in GADD34 KO cells (Fig. 2, A and C), indicating an impairment of autophagic flux in these cells.
Fig. 2. GADD34 activity is required for sustained autophagic flux in starved cells.
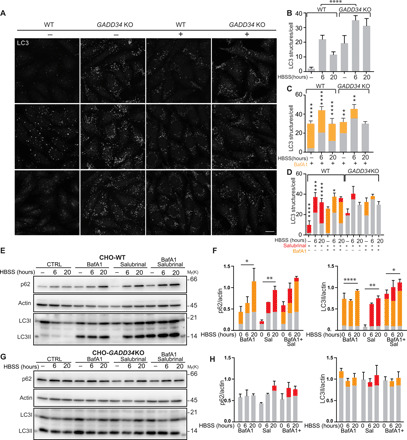
(A) Cells lacking GADD34 have an impaired autophagy flux. WT cells or knocked out for GADD34 (GADD34 KO) in growth medium or under amino acid starvation [Hanks’ balanced salt solution (HBSS)] with or without bafilomycin A1 treatment (BafA1, 100 nM) and immunostained for LC3. Scale bar, 10 μm. (B) LC3 puncta for untreated WT and GADD34 KO cells; means ± SD. n = 150 cells per condition from three experiments; GADD34 KO cells have more LC3 puncta than WT cells (P = 1.96 × 10−12, two-way ANOVA after post hoc correction). (C and D) LC3 puncta for WT and GADD34 KO cells with BafA1 in (C), or with 50 μM salubrinal in (D), or BafA1 + salubrinal in fig. S5. P values refer to t tests between salubrinal-treated and untreated cells, or between salubrinal + Baf1–treated cells and Baf1-treated cells. (E) Western blot (WB) of Chinese hamster ovary (CHO)–WT cells following amino acid deprivation alone (CTRL), or with 100 nM BafA1, or with 50 μM salubrinal, or with BafA1 + salubrinal. (F) Densitometry of p62 and LC3-II. Means ± SD. n = 2 independent experiments; P values refer to two-way ANOVA after post hoc correction. (G) WB of GADD34 KO cells treated as in (E). (H) Densitometry of WBs. Means ± SD. n = 2 independent experiments. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. The BafA1-dependent and salubrinal-dependent increase are highlighted in orange and red, respectively.
To evaluate whether GADD34’s role in autophagic flux was indeed mediated by its ability to promote dephosphorylation of p-eIF2α, we used salubrinal, an inhibitor of eIF2α phosphatase enzymes including GADD34 (13). We first assessed the specificity of salubrinal in this context by counting LC3-positive structures in WT and GADD34 KO cells treated either with salubrinal alone or in combination with bafilomycin A1 during starvation (Fig. 2D and fig. S5). Salubrinal increased the number of LC3-positive structures in WT cells as expected, with no substantial additional effect following cotreatment with bafilomycin A1 (Fig. 2D and fig. S5). Salubrinal alone, or in combination with bafilomycin, was ineffective in GADD34 KO cells, thus indicating that salubrinal’s action on autophagy is mediated by GADD34 inhibition (Fig. 2D and fig. S5).
To further assess the autophagic flux, we also measured p62 and LC3II protein levels in WT (Fig. 2, E and F) and GADD34 KO cells (Fig. 2, G and H) during starvation in the presence of either bafilomycin A1 or salubrinal, or a combination of both drugs. In agreement with the results of the immunofluorescence studies, GADD34 KO cells exhibited higher levels of p62 and LC3II (Fig. 2, E to H). Salubrinal or bafilomycin A1 significantly increased p62 and LC3II levels in WT cells, while their combination did not induce a significant effect when compared with salubrinal alone (Fig. 2, E and F). In contrast, neither salubrinal nor bafilomycin induced significant effects in GADD34 KO cells (Fig. 2, G and H). Together, these data further support a key role of GADD34 in sustaining the autophagic flux in starved cells.
We additionally confirmed the role of GADD34 in sustaining autophagy flux also in HeLa cells by monitoring p62 and LC3II protein levels during amino acid starvation following salubrinal and/or bafilomycin A1 treatment (fig. S6, A and B) and GADD34 depletion by small interfering RNA (siRNA) (fig. S6, C and D).
Last, we measured p-eIF2α levels during amino acid starvation. As shown in fig. S7 (A and B), the levels of p-eIF2α increased upon amino acid deprivation, as previously reported (10), and this increase was amplified by salubrinal. TFEB overexpression significantly decreased p-eIF2α (in a salubrinal-sensitive fashion) in agreement with its role in increasing GADD34 expression and, thus, PP1 phosphatase activity (fig. S7, A and B), while TFEB/TFE3 or GADD34 depletion had the opposite effect (fig. S7, C and D). These results show that increased levels of GADD34 during starvation are required to prevent excessive eIF2α phosphorylation (p-eIF2α).
Overall, our data demonstrate that GADD34 inhibition causes excessive phosphorylation of eIF2α leading to a reduced autophagic flux. Phosphorylation of eIF2α is required to promote and sustain autophagy during starvation, as cells with a permanently unphosphorylated eIF2α mutant (S51A) exhibit a reduced autophagic response to starvation (fig. S8, A to D). Thus, both excessive phosphorylation and a lack thereof are detrimental to the autophagic flux, and both impair the cell ability to survive prolonged starvation (fig. S8E).
GADD34 is required for lysosome biogenesis in response to starvation
We next assessed the role of GADD34 on TFEB-mediated lysosome biogenesis, a process necessary to sustain the autophagic flux during starvation. To this end, we developed an assay in a cell model of Fabry disease (14), a lysosomal storage disorder due to the loss of function of galactosidase alpha (encoded by the GLA gene): the GLA-KO cells. As depicted in Fig. 3A, “newly formed” lysosomes can be distinguished from “old” ones in GLA-KO cells as they are smaller and devoid of storage material [Gb3, stained with Shiga Toxin (15)]. Following amino acid deprivation, the overall number of lysosomes in GLA-KO cells increased significantly (Fig. 3, B and C), and, specifically, of “new” lysosomes as indicated by the increase in small storage-free lysosomes (Fig. 3D, red bars). Salubrinal treatment completely abolished the effect of starvation on lysosomal biogenesis as evidenced by the lack of change in total lysosomal number (Fig. 3, B and C) and specifically of small new lysosomes (Fig. 3D, red bars). To further confirm these results, we analyzed lysosomal biogenesis also in WT and GADD34 KO cells by counting the total number of lysosomes under growth and starvation conditions (Fig. 3, E and F). Only in WT cells, but not in GADD34 KO cells, did starvation induce an increase in lysosome number. Together, these results support the role of GADD34 as an enabler of lysosomal biogenesis by dephosphorylating p-eIF2α.
Fig. 3. GADD34 activity is required for lysosomal biogenesis following starvation.
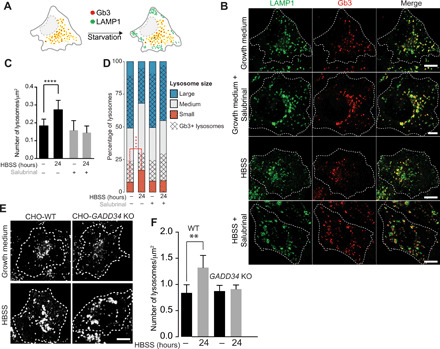
(A) Lysosomal biogenesis assay in a model of Fabry disease, i.e., HeLa cells knocked out for α-galactosidase (GLA) gene. Storage accumulation [globotriaosylceramide (Gb3)] discriminates small newly formed storage-free lysosomes from older and larger storage-filled lysosomes. (B) Lysosomes and Gb3 labeled with anti-LAMP1 antibody (green) and CY3-conjugated Shiga Toxin Subunit B (CY3-ShTxB) in GLA-KO cells in growth medium or starved (HBSS) for 24 hours in the absence or presence of salubrinal (50 μM). (C) Total number of lysosomes in GLA-KO cells across conditions in (B); means ± SD. n = 60 cells per condition from three experiments; P values from t tests. (D) Distribution of lysosome size and storage (Gb3) content across the indicated conditions as a percentage of their total number; n = 60 cells per condition from three experiments. Lysosomes were binned into three classes according to their area [small, S = area < 0.1 μm2; medium, M = area in (0.1 μm2, 1 μm2); large, L = area > 1 μm2] (P = 7.06 × 10−16 from Fisher’s exact test). (E) Lysosomes were labeled with anti-LAMP1 antibody in CHO-WT and GADD34 KO cells in growth medium or starved (HBSS) for 24 hours. (F) Total number of lysosomes in WT and GADD34 KO cells across conditions in (E); means ± SD. n = 50 cells per condition from three experiments; P value from t tests. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
GADD34 activity is required to translate the starvation-induced transcriptional program
We next asked whether GADD34 is necessary to enable translation of transcripts during starvation. To this end, we performed high-resolution quantitative proteomics in control conditions (growth medium) and following amino acid deprivation [Hanks’ balanced salt solution (HBSS)] in cells with either an active or blocked (HBSS + salubrinal) GADD34 activity. By applying bio-orthogonal amino acid tagging with acid azidohomoalanine (AHA), we focused on proteins that are actively synthesized during starvation (16). As shown in fig. S1B, the pattern of labeling with AHA confirmed that de novo protein synthesis is lower in starved than in control cells. This finding is consistent with the puromycin labeling pattern following amino acid deprivation (fig. S1A).
By quantitative proteomics, we identified 3667 proteins (table S4) that were present in at least one of the three conditions (growth medium, HBSS, and HBSS + salubrinal). As shown in Fig. 4A, amino acid deprivation alone (HBSS versus growth) significantly [one-way analysis of variance (ANOVA) post hoc test, P < 0.1] changed 968 proteins (up-regulating 477 and down-regulating 491). Down-regulated proteins were enriched for highly expressed proteins related to protein anabolism such as ribosomal components, in addition to mitochondrial proteins, and included autophagy substrates (e.g., SQSTM1/p62, GABARAP2L) (Fig. 4A and table S5). These proteins belong to classes known to be preferentially degraded early in response to amino acid deprivation (17).
Fig. 4. GADD34 activity is required for the starvation-induced transcriptional program to be implemented at the protein level.
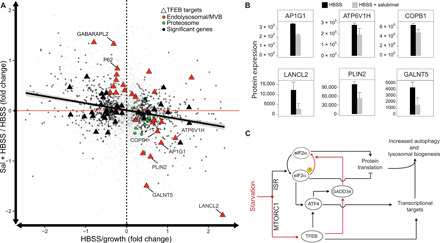
(A) Quantification of neosynthesized proteins by LC-MS following bio-orthogonal amino acid tagging with AHA for 6 hours in the following conditions: full medium (growth) and 6-hour amino acid starvation (HBSS) with or without salubrinal (Sal). Each protein is shown as a dot whose coordinates are the relative (fold) change following starvation alone (HBSS/growth – x axis) or starvation in the presence of 100 μM salubrinal (HBSS + Sal/HBSS – y axis). For each condition, the average fold change was computed from five independent samples. The trend line is shown as a solid black line. The dashed vertical line separates proteins increasing following starvation (right) from those decreasing (left). The red horizontal line separates proteins for which Sal treatment in starvation resulted in increased protein levels with respect to starvation alone (top) from those which resulted in decreased protein levels (bottom). Of 3667 proteins detected in all the samples (gray dots), HBSS treatment alone significantly changed 968 proteins (black dots). (B) Quantification of changes in protein levels measured as described in (A) for selected TFEB targets following starvation in the absence (black) or presence (gray) of salubrinal (P < 0.1). (C) Model of TFEB-mediated regulation of protein translation during starvation.
Up-regulated proteins belonged to classes related to protein catabolism, including lysosomal and autophagic proteins (ATP6V1H, CAT, CTSB, CTSC, CTSL, CTSZ, LANCL2, GNS, and PLIN2), endosome/multivesicular body proteins (AP1G1, CHMP1B, CHMP2B, EEA1, RAB7A, and VPS35), Golgi proteins (COPB1 and GALNT5), and the proteasome (PSMA1-5, PSMB2-6, PSMC2-5, PSMD2, PMSD11, and PMSD14) (Fig. 4B and table S6). These observations are in line with the previously reported increase in proteasome subunits and activity, and lysosomal biogenesis in response to nutrient deprivation (18, 19). Blocking GADD34 activity during starvation (HBSS + salubrinal versus HBSS) resulted in a significant (one-way ANOVA post hoc test, P < 0.1) decrease in the synthesis of the very proteins belonging to the endolysosomal system, including TFEB direct targets, as reported in Fig. 4 (A and B). On the contrary, proteins increasing the most included those degraded by autophagy (e.g., SQSTM1/p62 and GABARAPL2 in Fig. 4A), consistently with the reduced autophagic flux caused by salubrinal treatment. Together, these results confirm that GADD34 is necessary for the translation of the starvation-induced transcriptional program.
DISCUSSION
Regulation of translation under starvation is a complex process that requires cross-talk between the mTOR and ISR pathways. The ISR induces eIF2α phosphorylation, thus inhibiting CAP-dependent translation and favoring translation of eIF2α-sensitive mRNAs, while acute mTOR inhibition reduces translation of selected mRNAs via LARP1- and 4E-BP–dependent mechanisms (20) and activates TFEB (1, 6). Our work demonstrates a novel and essential interplay between these two pathways during starvation involving TFEB-mediated regulation of GADD34, a key effector of the ISR. Our results support the model depicted in Fig. 4C. Upon nutrient deprivation, the kinase GCN2 phosphorylates eIF2α, thus reducing global protein synthesis and inducing specific translation of the transcription factor ATF4, a known activator of GADD34 transcription following ER stress (9, 10). Concomitantly, inhibition of mTORC1 induces TFEB nuclear accumulation, which in turn activates a transcriptional program to promote lysosomal biogenesis and increase autophagic flux. TFEB also directly binds the ATF4 promoter, thus enhancing its expression following prolonged ER stress and starvation (11). Here, we show that to enable an effective translation of the starvation-induced transcriptional program, TFEB directly drives early expression of GADD34, which is selectively translated in the presence of phosphorylated eIF2α (21). GADD34 activity prevents excessive eIF2α phosphorylation, thus allowing translation of the starvation-induced transcriptional program to occur. This role of GADD34 in starvation has some parallelisms to its function in infected cells in response to double-stranded RNAs, where it is needed to allow cytokine production in the face of a general mRNA translation block to prevent viral replication (22). A link between GADD34 and autophagy has been previously reported, but it was related to mTOR inactivation [by starvation (23), by ER stress (24, 25), or by the expression of mutant huntingtin proteins (26)], and never associated with either the starvation-induced TFEB transcriptional response or a role in lysosome biogenesis, the two key findings of our work.
Previous studies have shown that eIF2α phosphorylation is necessary for protracted autophagy during starvation, but the mechanisms remain unclear (27). Here, we show that the two extremes, no phosphorylation or excessive phosphorylation of eIF2α, are both deleterious (fig. S8E). In the first case, general protein synthesis is not reduced, thus preventing catabolism of amino acids for energy production. In the latter case, excessive phosphorylation causes an extreme reduction in protein synthesis, preventing translation of starvation-responsive genes. Our work raises the question of how a cell determines the optimal level of eIF2α phosphorylation in response to starvation. Understanding this regulatory mechanism may yield novel ways to modulate autophagic flux, which do not directly depend on the mTOR pathway, and may potentially benefit those disorders where autophagy induction is thought to be beneficial, including neurodegenerative diseases and aging.
MATERIALS AND METHODS
Cell cultures
Human embryonic kidney (HEK) 293, HEK-293FT (for lentivirus production), and HeLa (WT and GLA-KO) cells were grown at 37°C in an atmosphere of 5% CO2 and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific–Gibco). The media for HEK-293 and HeLa cells’ growth were supplemented with 10% fetal bovine serum (FBS) (catalog number 0270-098, Thermo Fisher Scientific–Gibco), 2 mM l-glutamine, and penicillin/streptomycin (100 U/ml) (catalog number 15140122, Thermo Fisher Scientific–Gibco), while the media for the HEK-293FT cells’ growth were supplemented with 10% heat-inactivated FBS (catalog number A3840001, Thermo Fisher Scientific–Gibco), 2 mM l-glutamine, 1× MEM nonessential amino acids, 1 mM sodium pyruvate, and penicillin/streptomycin (100 U/ml) (catalog number 15140122, Thermo Fisher Scientific–Gibco). Chinese hamster ovary (CHO) cells (WT, GADD34 KO, and p-eIF2α S51A were grown in Ham’s F-12 supplemented with 10% FBS (catalog number 0270-098, Thermo Fisher Scientific–Gibco), 2 mM l-glutamine, and penicillin/streptomycin (100 U/ml) (catalog number 15140122, Thermo Fisher Scientific–Gibco). When confluent, the cells were trypsinized with 0.25% trypsin-EDTA (catalog number T4049, Sigma-Aldrich) for 1 min and then plated at a density of 104 cells/cm. HBSS with calcium and magnesium (Gibco catalog number 24020117) was used as starvation medium.
Cloning strategy
The cassettes comprising the genetic circuits were implemented by using the ViraPower Promoterless Lentiviral Gateway expression system (catalog number 442050, Thermo Fisher Scientific). Entry vectors for Gateway cloning of the PFL were designed as described in (28) and synthesized by Geneart. The blunted-end polymerase chain reaction (PCR) product of TFEB3xFlag sequence was amplified from pCMV_TFEB3xFlag by using the High-Fidelity Taq Phusion (catalog number F553, Fynnzimes) and then cloned into pCR-Blunt II TOPO vector by using the Zero Blunt TOPO PCR Cloning Kit (catalog number 450245, Thermo Fisher Scientific–Invitrogen). We digested both pCR-Blunt II TOPO-TFEB and pMAtTA-IRES-d2EYFP, already available in the laboratory (29), with Nhe I and Eco RV restriction enzymes (NEB) to ligate TFEB in place of the d2EYFP, thus obtaining pMAtTA-IRES-TFEB. To stabilize the transcripts, we kept the WPRE sequence also present in the original vector. The pMAtTA-IRES-TFEB-WPRE cassette was amplified by High-Fidelity Taq Phusion PCR and cloned into the pENTR directional TOPO vector (catalog number 450218, Thermo Fisher Scientific–Invitrogen) with the specific recombination sites to generate pENTR-tTA-IRES-TFEB vector. To generate the lentiviral vector containing the gene expression cassette PFL-TFEB, the recombination reaction was performed between the pENTR-tTA-IRES-TFEB-WPRE, the pENTR59-TOPO-CMV-TET already available in the laboratory, and the pLenti/R4R2/V5-DEST (catalog number V498-10, Thermo Fisher Scientific) according to the manufacturer’s instructions. According to the manufacturer’s instructions, the lentivirus was then produced in 293FTcells as previously described (29).
Transfection
When confluent (80%), the HEK-293 cells were transfected in six-well plates with Lipofectamine LTX (catalog number 15338100, Thermo Fisher Scientific–Life Technologies): 1000 ng of DNA was mixed with Opti-MEM I reduced serum medium (catalog number 3198506, Thermo Fisher Scientific) to a final volume of 100 μl followed by addition of 1 μl of PLUS reagent; 5 min later, 1.8 μl of Lipofectamine LTX and of transfection complex was added dropwise to the seeded cells; 6 hours later, the transfection medium was replaced with fresh medium. HeLa cells were transfected in a six-well plate with TFEB–green fluorescent protein plasmid (gift from A.B.’s laboratory) with Transit-LT1 (Mirusbio LLC) according to the manufacturer’s instructions and incubated for 18 to 24 hours before fixation.
siRNA treatment
siRNA duplexes against TFEB and TFE3 were purchased as smartpool from Dharmacon. siRNA duplexes against ATF4 and GADD34 were purchased from Sigma-Aldrich. Three siRNA duplexes were used to knock down ATF4 and GADD34. The sequences of the siRNA used are listed below:
siRNA-hGADD34 #1 GUGGAUAGUGAGGAUAAGGAA[dT][dT]
siRNA-hGADD34 #2 GACCAACUGGUUUGCCUAUAA[dT][dT]
siRNA-hGADD34 #3 GGACACUGCAAGGUUCUGA[dT][dT]
siRNA-hATF4 #1 GUGAGAAACUGGAUAAGAA[dT][dT]
siRNA-hATF4 #2 GCCUAGGUCUCUUAGAUGA[dT][dT]
siRNA-hATF4 #3 CCCUGUUGGGUAUAGAUGA[dT][dT]
HeLa cells were transfected with siRNAs for 96 hours using Oligofectamine (Thermo Fisher Scientific) according to the manufacturer’s instructions. The siRNA duplexes were used at 25 pmol for TFEB, TFE3, ATF4, and GADD34. Mock-treated or nontargeting siRNA-treated HeLa cells are referred to as controls (CTRL).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was carried out with the LightCycler 480 SYBR Green I mix (Roche) using the LightCycler 480 II detection system (Roche) with the following conditions: 95°C, 5 min; (95°C, 10 s; 60°C, 10 s; 72°C, 15 s) × 20 cycles. For expression studies, the qRT-PCR results were normalized against an internal control (HPRT). The primers used in this study are the following:
GADD34:
Forward 5′-TGAGACTCCCCTAAAGGCCA-3′
Reverse 5′-CCAGACAGCCAGGAAATGGA-3′
ATF4:
Forward 5′-ATGGGTTCTCCAGCGACAAG-3′
Reverse 5′-GAAGGCATCCTCCTTGCTGT-3′
HPRT:
Forward 5′-TGCTGACCTGCTGGATTACA-3′
Reverse 5′-CCTGACCAAGGAAAGCAAAG-3′
TFEB:
Forward 5′-AGGAGTTGGGAATGCTGATC-3′
Reverse 5′-TGTAATCCACAGAGGCCTTG-3′
TFE3:
Forward 5′-GAACTGGGCACTCTCATCCC-3′
Reverse 5′-CCGGCTCTCCAGGTCTTTG-3′
XBP1: Amplicon length, condizioni PCR, C + C-, %gel.
hXBP1Fw: AAACAGAGTAGCAGCTCAGACTGC
mXBP1Rev: TCCTTCTGGGTAGACCTCTGGGA
Lentivirus production and monoclonal cell population
Lentiviral particles carrying PFL-TFEB construct were produced in HEK-293FT cells with ViraPower Lentiviral Expression System (catalog number K497500, Thermo Fisher Scientific) according to the manufacturer’s instructions. Subconfluent HEK-293 cells were transduced with viral particles, and stable clones were selected with Blasticidin S (4 μg/ml; catalog number R21001, Thermo Fisher Scientific). A monoclonal cell population stably expressing the PFL-TFEB gene expression cassette (HEK_PFL-TFEB) was isolated with serial dilutions and used to perform farther time series experiment.
Compounds
Tetracycline (catalog number T7660) and doxycycline (catalog number D3447) were purchased from Sigma-Aldrich and used at the concentrations of 100 ng/ml and 1 μg/ml, respectively, in the culture media. Salubrinal (catalog number SML0951) and bafilomycin A1 (catalog number B1793) were purchased from Sigma-Aldrich.
Antibodies
ANTI-FLAG (M2) purchased from Sigma-Aldrich (catalog number F1804) was used at 1:1000 dilution. Anti–β-actin was purchased from Sigma-Aldrich (catalog number A2066) and was used at 1:5000 dilution. Anti–GAPDH (glyceraldehyde-3-phosphate dehydrogenase)–mouse purchased from Santa Cruz Biotechnology (catalog number sc-32233) was used at 1:5000 dilution. Anti-histone H3 antibody is a rabbit polyclonal antibody purchased from Merck Millipore (catalog number 06-755) and used at dilution 1:1000. Anti–epidermal growth factor recptor (EGFR) (catalog number SC-03, Santa Cruz Biotechnology, Santa Cruz, CA) is a polyclonal antibody epitope mapping to the C-terminal of human EGFR and was used at 1:1000 dilution. Anti-BIP (catalog number 31-77), anti-p-eIF2α (catalog number 3597), and anti-eif2α (catalog number 5324) were purchased from Cell Signaling Technology and used at 1:1000 dilution. The bead-conjugated antibody ANTI-FLAG M2 Affinity Gel (catalog number A2220) was purchased from Sigma-Aldrich. Anti-LC3 (catalog number NB100-2220) antibody was purchased from Novus Biologicals and used at 1:1000 [western blot (WB)] or 1:200 (Immunofluorescence). Anti SQSTM1/p62 was purchased from Abnova (catalog number H00008878) and used at 1:1000 dilution. Anti-GADD34 used in WB (1:1000) was purchased from Proteintech (catalog number 10449-1-AP), and anti-GADD34 used in immunofluorescence (1:100) was purchased from Invitrogen (catalog number PA1-139). Anti-Sar1 antibody used in WB (1:2000) was purchased from Millipore (catalog number 07-692). Anti-Sec31A antibody used in immunofluorescence (1:200) was purchased from BD (catalog number 612350). Anti-LAMP1 antibody used in immunofluorescence (1:2000) was purchased from Developmental Studies Hybridoma Bank (catalog number H4A3).
Immunoblot experiments
Whole-cell lysates were extracted in lysis buffer [150 mM NaCl, 25 mM tris-HCl (pH7.4), 0.5% Triton X-100, and 0.5 mM EDTA] supplemented with proteases and phosphatases inhibitors (Protease Inhibitor Cocktail P8340 and Phosphatase Inhibitor Cocktail 2 P5726 from Sigma-Aldrich). After clarification by centrifugation at 10,000g at 4°C for 10 min, protein quantification was performed by a modified Bradford assay (Bio-Rad, Munich, Germany), and equal micrograms of protein extract were loaded onto gradient gels (catalog number 4561086, Bio-Rad). Nitrocellulose or polyvinylidene difluoride membranes were probed with the indicated antibodies. Secondary antibodies (catalog numbers NA931V and NA934V) coupled to horseradish peroxidase were from GE Healthcare, United Kingdom. Immune complexes were detected with the enhanced chemiluminescence kit (ECL Western Blotting Detection Reagents, catalog number GERPN2209 from Sigma-Aldrich). Signal intensity was analyzed with UVP ChemiDoc Imaging System (LCC Jena USA). Quantification of WB data was performed using Fiji software (30) by drawing a rectangle around the band to quantify and measure the signal intensity. Background subtraction was performed by subtracting the intensity of a rectangle of the same size used for the quantification of the specific band, located in a signal-free area of the membrane. In cases of membranes with variable background, background subtraction was performed for each lane independently.
Puromycin incorporation assay
HeLa cells (∼80% confluent) in normal growth conditions (full medium) and amino acids starved (HBSS, Gibco catalog number 14025092) for 1 and 3 hours were incubated with puromycin (1 μg/ml) for the last 30 min of the treatment to label nascent polypeptides. Puromycin-containing proteins were resolved by denaturing SDS–polyacrylamide gel electrophoresis (PAGE) and detected by Western blotting using anti-puromycin antibody (catalog number MABE343, Millipore).
AHA-protein labeling and click reaction for quantitative proteomics
HeLa cells seeded in petri dishes (150 mm) were washed three times with HBSS and incubated for 30 min in DMEM without methionine and cysteine or in HBSS to deplete the internal methionine storages to improve the incorporation of the AHA. Cells (growth) were then incubated in DMEM without methionine and cysteine with the addition of cysteine (63 mg/ml), dialyzed amino acid–free FBS (10%), and AHA (50 μM). Starved cells were incubated in HBSS and AHA (50 μM) in the presence or in the absence of salubrinal (50 μM) for 6 hours. Cell lysis, click reaction, and AHA-labeled protein enrichment and preparation for quantitative mass spectrometry were performed, on 6 mg of protein lysate/sample according to the Click-iT Protein Enrichment Kit, for click chemistry capture of azide-modified proteins following the manufacturer’s instruction (catalog number C10416, Thermo Fisher Scientific).
Quantitative mass spectrometry
For quantitative proteomics, the AHA-labeled enriched proteins were processed for mass spectrometry coupled to liquid chromatography (LC-MS/MS) analysis. The samples were incubated for 30 min at 56°C with 10 μl of dithiothreitol (100 mM) in digestion buffer (50 mM NH4HCO3 in MilliQ water, pH 8) for the reduction of disulfide bonds of cysteine residues. An incubation for 20 min in the dark with 30 μl of IAA (indole-3-acetic acid; 100 mM) in digestion buffer was performed to carbamidomethylate the cysteine residues. Protein content was then precipitated overnight at −20°C by adding 1 ml of cold acetone. After a centrifugation step (20,000g at 4°C for 30 min), the supernatant was discarded and the protein pellet was dried under nitrogen stream. The pellet was then dissolved in 200 μl of digestion buffer, and 1 μg of trypsin was added for an overnight digestion at 37°C. The resulting tryptic peptides were dried under vacuum and then dissolved in 150 μl of 3% ACN + 0.1% FA for LC-MS/MS analysis.
LC-MS/MS analysis and protein quantification
Five microliters of the obtained peptide solution was injected on a nanoACQUITY chromatographic system (Waters, Milford, MA, USA) and then analyzed with a TripleTOF 5600+ mass spectrometer equipped with a NanoSpray III ion source (SCIEX, Ontario, Canada). The peptides were first loaded on a trapping column (180 μm by 20 mm Acquity C18 column), desalted for 4 min at 4 μl/min at 1% ACN + 0.1% FA, and then moved to a Picofrit column (C18 column, 75 μm by 25 cm, from New Objective Inc., Woburn, MA, USA). A 2-hour linear gradient from 3 to 45% ACN in H2O, both added with 0.1% FA, was used to elute peptides at 300 nl/min. The column was then washed with 90% ACN for 10 min and the reequilibrated to 3% ACN for 18 min. Analysis was then performed in positive ion mode with the following parameters: ion spray voltage, 2500 V; spray gas 1, 10; curtain gas, 30; declustering potential, 80 V; and source temperature, 90°C. Spectra were acquired in data-independent acquisition mode following the SWATH protocol for label-free proteomics (31). The mass/charge ratio (m/z) range of precursor ions went from 400 to 1250, with a variable window width from 7 to 50 Da. The instrument first acquired a full range scan of 250 ms, and then 100 consecutive SWATH experiments, each lasting 25 ms, were performed within the 100 to 1500 m/z range. The obtained SWATH spectra were imported in PeakView software and then searched against the PanHuman ion library (32). For the protein quantification, the following settings were used: use only nonshared and nonmodified peptides, minimum peptide confidence 90%, 50 ppm maximum mass tolerance, 30 min maximum RT tolerance, and six MRM transitions per peptide. A total of 3667 proteins were quantified, and the corresponding raw data files were imported in MarkerView software to perform a normalization using the most likely ratio method (33). Differentially expressed proteins across conditions were identified using one-way ANOVA, and post hoc test was performed using the function aov and TukeyHSD in R statistical environment.
Confocal fluorescence microscopy, image processing, lysosomal size measurement, and colocalization analysis
HeLa (WT and GLA-KO) cells were grown to subconfluence on glass coverslips, and immunofluorescence microscopy and quantitative image analysis were performed as reported in (34). Briefly, confocal images of at least 100 to 150 cells per condition were acquired at the same laser power and photomultiplier gain. Images were then processed using ImageJ software. Single channels from each image were converted into 8-bit grayscale images and thresholded to subtract background. The ImageJ “Analyze Particles” plugin was then used to identify and count the total number of the structures (with an area above 0.01 μm2) in channel 1 (i.e., LAMP1). The structures in channel 2 (i.e., GB3, stained using CY5-conjugated Shiga Toxin B subunit) were used to build a mask that was then overlapped with the LAMP1 structures to subtract, and thus count, the structures containing both markers. The remaining structures, positive only for LAMP1, were counted, and by difference, the number of structures containing both LAMP1 and GB3 was calculated. Furthermore, given the small threshold used for the size of LAMP1 structures, we divided LAMP1 structures in three classes (small = area < 0.1 μm2, medium = 0.1 < area, 1 μm2, and large = area > 1 μm2). Within each class, we calculated the number of LAMP1 structures that were also positive for GB3. Note that this quantitative analysis procedure does not use merged images and is not affected by the fluorescence intensity. For conventional confocal microscopy, a confocal laser microscope (Zeiss LSM800 confocal microscope systems; Carl Zeiss, Gottingen, Germany) with a 63× 1.4 numerical aperture oil objective was used.
Time series experiment, qRT-PCR, and RNA extraction
Inducible HEK_PFL-TFEB monoclonal cells (2.5 × 105) were plated in each well of a six-well cluster plate. After o/n incubation in medium supplemented with tetracycline (100 ng/ml), the antibiotic was removed from the culture medium at time 0 hour to induce TFEB expression, and samples were collected every 6 hours up to 90 hours for RNA extraction. Total RNA extraction was performed using the Qiagen RNeasy Kit (catalog number 74106, Qiagen) according to the manufacturer’s instructions. When necessary, retrotranscription of 1 μg of the total RNA extracted was performed using the QuantiTect Reverse Transcription Kit (catalog number 205313, Qiagen) according to the manufacturer’s instructions. qRT-PCRs were set up in duplicates using the LightCycler 480 SYBR green master mix (catalog number 04707516001, Roche), and the amplification was performed using a LightCycler 480 RT-PCR instrument (Roche).
RNA-seq, alignment, and data normalization
Total RNA was isolated using Qiagen RNeasy Kit (catalog number 74106, Qiagen). RNA was quantified using a NanoDrop ND-8000 spectrophotometer (NanoDrop Technologies), and the integrity was evaluated using an RNA ScreenTape chip on an Agilent 2200 TapeStation. The RNA of the 16 samples had RNA integrity number above 9.5. Libraries were prepared according to manufacturer’s instructions (TruSeq RNA Sample Preparation kit) with an initial amount of 1 μg of total RNA. Quality control of library templates was performed using a High Sensitivity D1000 ScreenTape (catalog number 5067-5584, Agilent Technologies) on an Agilent 2200 TapeStation. Qubit quantification platform was used to normalize samples for the library preparation using Qubit double-stranded DNA HS Assay Kit (catalog number Q32854, Thermo Fisher Scientific–Life Technologies) on Qubit 2.0 Fluorometer, Life Technologies. Libraries were sequenced via a paired-end chemistry on an Illumina HiSeq 1000 platform. Sequencing reads from each one of the 16 samples were aligned to GENCODE Human transcripts on HG19 genome using bowotie2 aligner (35). The expression of each gene then was estimated in the form of expected counts using RSEM (36) and normalized for sequencing depth using the method developed in DESeq2 package (37) in the R statistical environment with default parameters.
DNA extraction and ChIP-seq
ChIP-seq experiment was carried out in duplicate on samples collected at 18, 36, and 90 hours after TFEB induction. In brief, 5 × 107 to 10 × 107 HEK_PFL TFEB cells were fixed with 1% formaldehyde. Formaldehyde was quenched with 2.5 M glycine, and nuclear fractions were isolated and lysed. After chromatin shearing by sonication, lysed nuclei were purified (QIAquick PCR purification kit, 28106), and the quality of sonicated samples was checked on 1.5% agarose gel. Lysed nuclei were incubated overnight at 4°C with bead-conjugated antibody (ANTI-FLAG M2 Affinity Gel, catalog number A2220, Sigma-Aldrich). DNA was then eluted with 2% SDS Elution buffer and de–cross-linked, together with INPUT, overnight at 65°C. Immunoprecipitated and INPUT samples were purified on QIAquick column (Extraction PCR purification kit, catalog number 28706). For validation by ChIP-qPCR, 1 μl of purified DNA was amplified on an Applied Biosystems 7500 Fast Real-time PCR system (with Applied Biosystems SYBR Green). One to five nanograms of each DNA sample was used for ChIP-seq library preparation. The sequencing was performed at the IFOM-IEO Genomics Unit following the standard HT-ChIP protocol with 50–base pair (bp) single end generated on the Illumina HiSeq 2000 as previously described (38).
Sequence alignment and identification of TFEB binding regions
Short reads from each sample were aligned to the human genome (GRCh37/hg19) using Bowtie2 with --very-sensitive parameter option. PCR duplicates, multimapping reads, and reads having a mapping quality (MAPQ) score smaller than 5 were discarded using SAM tools (39). Peak calling was performed by using MACS2 (40), comparing each sample in which TFEB was overexpressed with the corresponding input sample. MACS2 was used with default parameters and bin width = 200. Regions identified by MACS2 as bound by TFEB (P < 5 × 10−3) in at least two conditions (i.e., time points) and overlapping for more than 10 bp were merged together. Binding regions were then annotated using the ChIPseeker package (41) in R statistical environment.
Identification of TFEB target genes
TFEB-FLAG expression measured across the 16 time points was correlated with all the other 17,488 genes measured from RNA sequencing (RNA-seq) experiment using the function cor.test of R statistical environment. P values were then corrected for FDR using Benjamin and Hackenberg correction of p.adjust function of R statistical environment. After correction for FDR, only genes with an FDR less than 10% were considered correlated with TFEB expression. Last, direct targets of TFEB were defined as genes positively correlated with TFEB expression and having a binding site of TFEB in the ±2500 bp of their promoter region identified as described above.
Identification of TFEB consensus binding sequence
TFEB consensus binding sequence was identified using the Weeder tool 2.0 (42). Weeder tool was run using human motifs (-O HS parameter) in single-strand mode (−ss parameter) and discarding from the output those motifs that were too similar to other motifs already found (−sim 0.1 parameter). Ten cycles of expectation maximization (EM) instead of only one as in the default run mode were performed to obtain more robust results (−em 10 parameter). The tool was run using as input the sequences of the identified binding regions associated with the 557 direct targets of TFEB identified in this study. The estimated position weight matrix of the TFEB consensus binding sequence was lastly plotted using the seqLogo function of the seqLogo package in R statistical environment.
Identification of lysosome and autophagic genes with multiple uORFs or intra ribosomal entry sequence
The list of 513 bona fide genes involved in lysosomal and autophagic processes from Di Fruscio et al. (3) was intersected with the list of genes congaing multiple uORFs in their promoter regions collected from Lee et al. (4) or the list of 583 genes having IRES sequences from Weingarten-Gabbay et al. (5).
Protein extraction, nucleo-cytoplasm fractionation, and Western blot
HEK_PFL-TFEB cells were cultured in the absence or presence of tetracycline (100 ng/ml) (Sigma-Aldrich, catalog number T7660), and proteins were collected at 18, 36, and 90 hours following TFEB-FLAG induction. Cell culture dishes were incubated on ice for 15 min. Cells were then washed twice and collected in ice-cold 1× phosphate-buffered saline (PBS) containing the cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (Roche, catalog number 04693159001) and 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich catalog number P7626). Cells were centrifuged for 5 min at 4°C at 2000 RPM, and pellets were resuspended in lysis buffer [50 mM tris-HCl (pH 8), 0.5% Triton, 137.5 mM NaCl, 5 mM EDTA, 10% glycerol, 1 mM PMSF, and cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail] for 15 min on ice. Cell lysates were then centrifuged 5 min at 4°C at 2000 RPM, and supernatants were collected as cytosolic protein fractions. Cell pellets were washed twice in lysis buffer, centrifuged for 5 min at 4°C at 2000 RPM, resuspended in lysis buffer containing 0.5% SDS, and sonicated for 10 min (10× 30″on/30″off cycles) in a Bioruptor Plus sonication device (Diagenode, catalog number B01020001). Sonicated samples were centrifuged for 5 min at 4°C at 13,000 RPM, and store supernatants were collected as nuclear protein fractions. Protein fractions were quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, catalog number 23225) according to the manufacturer’s instructions.
For Western blot analysis, 20 μg of protein fraction samples was boiled and loaded onto a precast NuPAGE 4–12% bis-tris protein gel (Thermo Fisher Scientific, catalog number NP0321BOX) and run in NuPAGE MES SDS Running Buffer (Thermo Fisher Scientific, catalog number NP0002). After electrophoresis, proteins were transferred to a nitrocellulose membrane using the iBlot 2 Gel Transfer Device (Thermo Fisher Scientific, catalog number IB21001). Membranes were blocked in PBS containing 1% Triton (PBS-T) and 5% Blotting-Grade Blocker (Bio-Rad, catalog number N170-6404) for 1 hour at room temperature (RT) with constant agitation and incubated with indicated primary antibody overnight at 4°C with agitation. The membrane was then washed three times with PBS-T, each time for 5 min, followed by incubation with horseradish peroxidase–conjugated secondary antibody for 1 hour at RT. Following three washes in PBS-T for 5 min, the Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, catalog number RPN2232) was used to initiate the chemiluminescence of horseradish peroxidase. The chemiluminescent signal was captured using an ImageQuant LAS3000 system (GE Healthcare). Protein expression quantification was lastly performed with the ImageJ Software (https://imagej.nih.gov/ij/download.html).
Supplementary Material
Acknowledgments
We thank D. Ron for providing GADD34-KO CHO cells and eIF2αS51A CHO cells; G. Natoli for ChIP-seq protocols; the TIGEM Bioinformatics Core for help with bioinformatics analysis; the TIGEM NGS facility for RNA-seq; C. Settembre and G. Diez Roux for the helpful discussions; and L. Patanella and M. Santoro for technical support. Funding: This work was supported by Fondazione Telethon grant TMDDSB116TT to D.d.B., and TGM11CB1 to M.A.D.M. M.A.D.M. acknowledges the support of the Italian Association for Cancer Research (AIRC, grant IG2013_14761) and European Research Council Advanced Investigator grant number 670881 (SYSMET). G.G. was supported by a Junior PI STAR grant from the University of Naples “Federico II.” A.B. was supported by the European Research Council H2020 AdG “LYSOSOMICS 694282.” A.B. is a cofounder of CASMA Therapeutics Inc. Author contributions: G.G. performed bioinformatics and statistical data analysis; L.S. performed all the cell biology experiments; M.N.M. performed TFEB ChIP-seq, with the help of S.P., and RNA-seq; R.D.C. helped with bioinformatics analysis and literature review; L.F. and A.Z. validated the TFEB o.e. assay; G.D.T. helped with protein analyses; C.B. and A.A. performed quantitative proteomics; A.B. revised the manuscript and suggested experiments, M.A.M.D. and D.d.B. supervised the work and wrote the manuscript; and D.d.B. conceived the original idea. Competing interests: A.B. is a cofounder of CASMA Therapeutics Inc. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. The mass spectrometry proteomics data related to this project have been deposited to the ProteomeXchange Consortium via the PRIDE (43) partner repository with the dataset identifier PXD016149, while RNA-seq and Chip-seq data are available on the GEO database with the dataset identifier GSE98248. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabb0205/DC1
REFERENCES AND NOTES
- 1.Napolitano G., Ballabio A., TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B., Kroemer G., Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Fruscio G., Schulz A., De Cegli R., Savarese M., Mutarelli M., Parenti G., Banfi S., Braulke T., Nigro V., Ballabio A., Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 11, 928–938 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee S., Liu B., Lee S., Huang S.-X., Shen B., Qian S.-B., Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 109, E2424–E2432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weingarten-Gabbay S., Elias-Kirma S., Nir R., Gritsenko A. A., Stern-Ginossar N., Yakhini Z., Weinberger A., Segal E., Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 351, aad4939 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Medina D. L., Paola S. D., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., Settembre C., Wang W., Gao Q., Xu H., Sandri M., Rizzuto R., De Matteis M. A., Ballabio A., Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Lee Y.-Y., Cevallos R. C., Jan E., An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2α phosphorylation. J. Biol. Chem. 284, 6661–6673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A. M., The integrated stress response. EMBO Rep. 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novoa I., Zeng H., Harding H. P., Ron D., Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 153, 1011–1021 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martina J. A., Diab H. I., Brady O. A., Puertollano R., TFEB and TFE 3 are novel components of the integrated stress response. EMBO J. 35, 479–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crespillo-Casado A., Chambers J. E., Fischer P. M., Marciniak S. J., Ron D., PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. eLife 6, e26109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyce M., Bryant K. F., Jousse C., Long K., Harding H. P., Scheuner D., Kaufman R. J., Ma D., Coen D. M., Ron D., Yuan J., A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science 307, 935–939 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Bernstein H. S., Bishop D. F., Astrin K. H., Kornreich R., Eng C. M., Sakuraba H., Desnick R. J., Fabry disease: Six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J. Clin. Invest. 83, 1390–1399 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ling H., Boodhoo A., Hazes B., Cummings M. D., Armstrong G. D., Brunton J. L., Read R. J., Structure of the shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37, 1777–1788 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M., Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Mejlvang J., Olsvik H., Svenning S., Bruun J.-A., Abudu Y. P., Larsen K. B., Brech A., Hansen T. E., Brenne H., Hansen T., Stenmark H., Johansen T., Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 217, 3640–3655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rousseau A., Bertolotti A., An evolutionarily conserved pathway controls proteasome homeostasis. Nature 536, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pechmann S., Willmund F., Frydman J., The ribosome as a hub for protein quality control. Mol. Cell 49, 411–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gandin V., Masvidal L., Cargnello M., Gyenis L., Laughlan S. M., Cai Y., Tenkerian C., Morita M., Balanathan P., Jean-Jean O., Stambolic V., Trost M., Furic L., Larose L., Koromilas A. E., Asano K., Litchfield D., Larsson O., Topisirovic I., mTORC1 and CK2 coordinate ternary and eIF4F complex assembly. Nat. Commun. 7, 11127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young S. K., Wek R. C., Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 291, 16927–16935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dalet A., Argüello R. J., Combes A., Spinelli L., Jaeger S., Fallet M., Manh T.-P. V., Mendes A., Perego J., Reverendo M., Camosseto V., Dalod M., Weil T., Santos M. A., Gatti E., Pierre P., Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 36, 761–782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uddin M. N., Ito S., Nishio N., Suganya T., Isobe K.-i., Gadd34 induces autophagy through the suppression of the mTOR pathway during starvation. Biochem. Biophys. Res. Commun. 407, 692–698 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Holczer M., Bánhegyi G., Kapuy O., GADD34 keeps the mTOR pathway inactivated in endoplasmic reticulum stress related autophagy. PLOS ONE 11, e0168359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holczer M., Besze B., Zámbó V., Csala M., Bánhegyi G., Kapuy O., Epigallocatechin-3-Gallate (EGCG) promotes autophagy-dependent survival via influencing the balance of mTOR-AMPK pathways upon endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2018, 6721530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyrskyluoto A., Reijonen S., Kivinen J., Lindholm D., Korhonen L., GADD34 mediates cytoprotective autophagy in mutant huntingtin expressing cells via the mTOR pathway. Exp. Cell Res. 318, 33–42 (2012). [DOI] [PubMed] [Google Scholar]
- 27.B’chir W., Maurin A.-C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., Bruhat A., The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siciliano V., Menolascina F., Marucci L., Fracassi C., Garzilli I., Moretti M. N., di Bernardo D., Construction and modelling of an inducible positive feedback loop stably integrated in a mammalian cell-line. PLOS Comput. Biol. 7, e1002074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siciliano V., Garzilli I., Fracassi C., Criscuolo S., Ventre S., di Bernardo D., MiRNAs confer phenotypic robustness to gene networks by suppressing biological noise. Nat. Commun. 4, 2364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Q., Yang L., Luo J., Guo L., Wang Z., Yang X., Jin W., Fang Y., Ye J., Shan B., Zhang Y., SWATH enables precise label-free quantification on proteome scale. Proteomics 15, 1215–1223 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Rosenberger G., Koh C. C., Guo T., Röst H. L., Kouvonen P., Collins B. C., Heusel M., Liu Y., Caron E., Vichalkovski A., Faini M., Schubert O. T., Faridi P., Ebhardt H. A., Matondo M., Lam H., Bader S. L., Campbell D. S., Deutsch E. W., Moritz R. L., Tate S., Aebersold R., A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data. 1, 140031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambert J.-P., Ivosev G., Couzens A. L., Larsen B., Taipale M., Lin Z.-Y., Zhong Q., Lindquist S., Vidal M., Aebersold R., Pawson T., Bonner R., Tate S., Gingras A.-C., Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 10, 1239–1245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Leo M. G., Staiano L., Vicinanza M., Luciani A., Carissimo A., Mutarelli M., Campli A. D., Polishchuk E., Tullio G. D., Morra V., Levtchenko E., Oltrabella F., Starborg T., Santoro M., Bernardo D. D., Devuyst O., Lowe M., Medina D. L., Ballabio A., De Matteis M. A., Autophagosome–lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balestrieri C., Alfarano G., Milan M., Tosi V., Prosperini E., Nicoli P., Palamidessi A., Scita G., Diaferia G. R., Natoli G., Co-optation of tandem DNA repeats for the maintenance of mesenchymal identity. Cell 173, 1150–1164.e14 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G., Wang L.-G., He Q.-Y., ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Zambelli F., Pesole G., Pavesi G., Using Weeder, Pscan, and PscanChIP for the discovery of enriched transcription factor binding site motifs in nucleotide sequences. Curr. Protoc. Bioinformatics 47, 2.11.1–2.11.31 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabb0205/DC1