

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease in which motor neurons throughout the brain and spinal cord progressively degenerate resulting in muscle atrophy, paralysis and death. Recent studies using animal models of ALS implicate multiple cell-types (e.g., astrocytes and microglia) in ALS pathogenesis in the spinal motor systems. To ascertain cellular vulnerability and cell-type specific mechanisms of ALS in the brainstem that orchestrates oral-motor functions, we conducted parallel single cell RNA sequencing (scRNA-seq) analysis using the high-throughput Drop-seq method. We isolated 1894 and 3199 cells from the brainstem of wildtype and mutant SOD1 symptomatic mice respectively, at postnatal day 100. We recovered major known cell types and neuronal subpopulations, such as interneurons and motor neurons, and trigeminal ganglion (TG) peripheral sensory neurons, as well as, previously uncharacterized interneuron subtypes. We found that the majority of the cell types displayed transcriptomic alterations in ALS mice. Differentially expressed genes (DEGs) of individual cell populations revealed cell-type specific alterations in numerous pathways, including previously known ALS pathways such as inflammation (in microglia), stress response (ependymal and an uncharacterized cell population), neurogenesis (astrocytes, oligodendrocytes, neurons), synapse organization and transmission (microglia, oligodendrocyte precursor cells, and neuronal subtypes), and mitochondrial function (uncharacterized cell populations). Other cell-type specific processes altered in SOD1 mutant brainstem include those from motor neurons (axon regeneration, voltage-gated sodium and potassium channels underlying excitability, potassium ion transport), trigeminal sensory neurons (detection of temperature stimulus involved in sensory perception), and cellular response to toxic substances (uncharacterized cell populations). DEGs consistently altered across cell types (e.g., Malat1), as well as cell-type specific DEGs, were identified. Importantly, DEGs from various cell types overlapped with known ALS genes from the literature and with top hits from an existing human ALS genome-wide association study (GWAS), implicating the potential cell types in which the ALS genes function with ALS pathogenesis. Our molecular investigation at single cell resolution provides comprehensive insights into the cell types, genes and pathways altered in the brainstem in a widely used ALS mouse model.
Keywords: ALS, Motor neurons, Glial cells, Brainstem, Drop-seq, Single cell RNA-seq, scRNA-seq, Transcriptome, SOD1 (G93A) mutant
1. Introduction
Amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease, is a devastating motor neuron disease that currently has no cure, and the mechanisms for initiation and progression of the disease are still poorly understood (Chia et al., 2018). Most previous studies have focused on motor neurons, but it is now clear that the disease is not strictly cell autonomous, and other cell types, including interneurons and non-neuronal cells such as astrocytes and microglia (Baufeld et al., 2018; Maniatis et al., 2019; Taylor et al., 2016), contribute to the disease. To provide further insight into the ALS pathology and to identify new cellular/molecular targets that could be used for more effective therapeutic treatment strategies than presently exist, one must determine, for all cell types, the unique sets of expressed genes (the transcriptome) responsible for cell function and phenotype during the progression of ALS. Furthermore, we must characterize, quantitatively, the differentially expressed genes (DEGs) using various animal models representative of different mutational causes, and the pathways they regulate, to fully reveal disease mechanisms (Gama-Carvalho et al., 2017; Poulin et al., 2016). Therefore, an objective, comprehensive understanding of all the vulnerable cell types, cell-cell interactions, as well as, the causal genes and cellular pathways altered during ALS progression is necessary to guide the development of more effective therapeutic strategies.
To explore the molecular mechanisms underlying ALS, previous transcriptomic studies characterized changes in gene expression profiles and cellular pathways affected in susceptible CNS regions such as spinal cord and brainstem that could contribute to the complicated pathology of the disease in a variety of cell types (Heath et al., 2013; Krokidis and Vlamos, 2018; Taylor et al., 2016). The majority of those studies sequenced RNA samples from whole tissue that inevitably contained mixed cell types (Ferraiuolo et al., 2007), or targeted specific cells within a CNS nucleus, such as motor neurons, using laser capture methods (Bandyopadhyay et al., 2013). However, most anatomically defined nuclei within the spinal cord and brainstem are heterogeneous, including motor nuclei (Kanning et al., 2010), with varied genetic profiles and susceptibility to disease (Hedlund et al., 2010). Although important information has emerged, those studies that sequenced RNA from sampled aggregates of cells, as opposed to each individual cell, produced a type of “average” of the gene expression profiles, thus masking the cellular heterogeneity and origin, and possibly confounding the interpretation of the results.
In this study, we report for the first time the results obtained from single cell RNA sequencing (scRNA-seq) technology, Drop-seq (Macosko et al., 2015), applied to pontine brainstem from an established ALS transgenic mouse model. Most of the previous studies on ALS focused on spinal cord (Krokidis and Vlamos, 2018). However, in the present study we sample tissue from pontine brainstem since this region contains neuronal circuitry responsible for oral-motor functions (Chandler and Tal, 1986; Kogo et al., 1996; Westberg and Kolta, 2011), which are affected during ALS (Riera-Punet et al., 2018). Furthermore, we (Seki et al., 2019; Venugopal et al., 2015) and others (Lever et al., 2009) showed, previously, significant changes in either physiological or anatomical properties of trigeminal motor and proprioceptive sensory neurons using the well-established SOD1 mouse model. The advantage of scRNA-seq is that it uses parallel sequencing of thousands of cells in an unbiased manner from a defined region of brainstem to uncover both known and novel cell types, as well as, the differentially expressed genes in ALS SOD1(G93A) transgenic mice. Furthermore, as opposed to sampling one cell type from a defined nucleus, we can profile numerous cell types from a region of CNS simultaneously. To exploit this methodology, we focus on a comparison between wildtype and ALS mice at postnatal day 100 (p100), a time when animals are symptomatic for the disease (Gurney et al., 1994) and would be expected to display changes in gene expression profiles in not only motor neurons, but other cell types as well.
Our cell type specific investigation of mutant SOD1 mice forms the foundation for developing a comprehensive map of the cell types, genes, and pathways altered in this classic ALS model in a defined region of brainstem involved in oral-motor control, and will serve as a basis for comparison with changes in varied spinal cord cell types (Bandyopadhyay et al., 2013; Maniatis et al., 2019) and in other ALS models in future studies.
2. Material and methods
2.1. SOD1 mice and tissue dissection
Transgenic mice expressing high levels of human SOD1(G93A) (mutant) and their age-matched non-carrier wild-type (WT) controls were used for the experiments (JAX Strain: 002726 B6SJL-Tg (SOD1*G93A)1Gur/J) (Gurney et al., 1994). All animal protocols were approved by the Institutional Animal Care and Use Committee at UCLA. Experiments were performed in two symptomatic and two non-transgenic WT female animals at P100 in parallel. The genotype of mice was determined using a standard PCR technique from tail samples (Laragen, Inc., CA). Mice were anesthetized using isoflurane vapor inhalation, and decapitated. The head was immediately immersed in carboxygenated (95% O2, 5% CO2), ice-cold modified ACSF (artificial cerebrospinal fluid) composed of (in mM): 124 NaCl, 4.5 KC1, 1.2 NaH2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, 1 MgCl2, with pH 7.28 ± 0.2. The pontine brainstem was rapidly extracted and adhered to the cutting chamber of a vibratome platform at the rostral end (DSK Microslicer; Ted Pella, Redding, CA); the brainstem was vertically supported by an agar block. The cutting chamber was filled with ice-cold carboxygenated ACSF as above. Beginning at the caudal level where the exit of the facial nerve was markedly visible, a pontine brain block was cut (~1200 μm thick) and was collected. This section contained trigeminal sensory, interneuronal, and motor nuclei essential for rhythmical jaw movements (Kogo et al., 1996). Additionally, the peripheral sensory nuclei in the trigeminal ganglia (TG) were also extracted and collected along with the brainstem sample. The TG were bilaterally removed with the aid of a dissection microscope and transferred into ice-cold (4 °C) carboxygenated ACSF with composition as above. With the aid of a dissection microscope, the ganglia were carefully dissected and separated from the nerve roots and collected and placed along with the brainstem section in RNAase free 1 × phosphate buffered saline (PBS) in 1 ml vials. Subsequent tissue dissociation protocols were then immediately performed.
2.2. Tissue dissociation for Drop-seq
Single cells within the pontine brainstem containing masticatory related neurons (Kogo et al., 1996; Westberg and Kolta, 2011) were dissociated using a modified version of a previously published protocol (Brewer and Torricelli, 2007). In brief, each tissue was finely dissected and incubated in Hibernate A without calcium (BrainBits, Springfield, IL, USA) supplemented with 12 mg papain (Worthington, Lakewood, NJ, USA). This solution is then transferred to a shaker-incubator (set to 30 °C and 60 rpm) for 30 min. After 30 min, using a Pasteur pipette, the solution is triturated three times to release cells from the tissue remnants, and transferred to a new collection tube. The media containing the cells is then gently added to the OptiPrep density gradient (Sigma Aldrich, St. Louis, MO, USA) and centrifuged at 800 g for 15 min at 22 °C. The layers consisting of cells were collected, and this solution was passed through a 40-μm strainer (Fisher Scientific, Hampton, NH, USA). The cells were counted and suspended at a concentration of 100 cells/μl in 0.01% BSA-PBS.
2.3. Drop-seq single cell barcoding and library preparation
Barcoded single cells, or STAMPS (single-cell transcriptomes attached to microparticles), and cDNA libraries were prepared as described previously (Macosko et al., 2015) and in version 3.1 of the Drop-Seq Laboratory Protocol available online (http://mccarrolllab.com/download/905/). Briefly, single cell suspensions at 100 cells/μl, EvaGreen droplet generation oil (BIO-RAD, Hercules, CA, USA), and ChemGenes barcoded microparticles (ChemGenes, Wilmington, MA, USA) which contain unique molecular identifiers (UMIs) and cell barcodes were co-flowed through a FlowJEM aquapel-treated Drop-seq microfluidic device (FlowJEM, Toronto, Canada) at recommended flow speeds (oil: 15,000 μl/h, cells: 4000 μl/h, and beads 4000 μl/h) to generate STAMPS. The following modifications were made to the online protocol to obtain sufficient cDNA as quantified by a high sensitivity BioAnalyzer (Agilent, Santa Clara, CA, USA) to continue the protocol: (1) 6000 beads were apportioned into each PCR tube, (2) the number of PCR cycles was 4 + 11 cycles. (3) multiple PCR tubes were pooled. The libraries were then checked on a BioAnalyzer high sensitivity chip (Agilent, Santa Clara, CA, USA) for library quality, average size, and concentration estimation. The samples were then tagmented using the Nextera DNA Library Preparation kit (Illumina, San Diego, CA, USA) and multiplex indices were added. After another round of PCR, the samples were checked on a BioAnalyzer high sensitivity chip for library quality check before sequencing.
2.4. Illumina high-throughput sequencing of Drop-seq libraries
The Drop-seq library molar concentration was quantified by Qubit Fluorometric Quantitation (ThermoFisher, Canoga Park, CA, USA) and library fragment length was estimated using a Bioanalyzer. Sequencing was performed on an Illumina HiSeq 4000 (Illumina, San Diego, CA, USA) instrument using the Drop-seq custom read 1B primer (GCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGTAC) (IDT, Coralville, IA, USA). 100 bp paired-end reads were generated with an 8 bp index read for multiplexing. Read 1 consisted of the 12 bp cell barcode, followed by the 8 bp UMI. Read 2 was comprised of the single cell transcripts.
2.5. Drop-seq data pre-processing and quality control
A total of 131,035,487 sequencing reads were generated from Drop-seq. Fastq files were converted to BAM format and cell and molecular barcodes were tagged. Reads corresponding to low quality barcodes with quality score less than 10 was removed using DropSeq Tools version 1.12 (http://mccarrolllab.com/download/922/). Next, any occurrence of the SMART adapter sequence or polyA tails found in the reads was trimmed, yielding 113,432,674 cleaned reads. These cleaned reads were converted back to fastq format and were aligned to the mouse reference genome mm10 using STAR-2.5.0c (Dobin et al., 2013). A percentage of the Chemgenes barcoded beads which contain the UMIs and cell barcodes were anticipated to have synthesis errors, which were assessed using the function DetectBeadSynthesisErrors in DropSeq Tools. The bead synthesis error rate was estimated to be 5–10%, within the acceptable range. Finally, a digital gene expression matrix for each sample was generated using DropEst (Petukhov et al., 2018), where each row is the read count of a gene and each column is a unique single cell.
The transcript counts of each cell were normalized by the total number of UMIs for that cell. These values were then multiplied by 10,000 and log10 transformed. Gene counts were obtained by summing up the counts of transcripts for that gene. Transcript and gene counts were evaluated for each potential cell to filter out cells that i) might represent background noise due to ambient RNAs by using a threshold of at least 200 genes and 500 transcripts, 2) undergo apoptosis or cell death or contain ambient mRNAs using the following thresholds: mitochondrial gene percentage more than 0.2, ribosomal percentage higher than 0.075, and predicted gene percentage less than 0.03. Digital gene expression matrices from the four samples (2 wildtype and 2 mutant samples) were combined to create three different pooled digital gene expression matrices for: 1) all wildtype samples containing 1894 cells, 2) all mutant samples containing 3199 cells, and 3) combined wildtype and mutant samples containing 5093 cells.
2.6. Identification of major cell clusters and neuronal sub-clusters
The Seurat R package (version 3; https://github.com/satijalab/seurat) was used to project all sequenced cells onto two dimensions using t-SNE (Van Der Maaten and Hinton, 2008), and assign clusters by Louvain clustering. To further refine the neuronal cell clusters that express classic neuronal markers, we used the self-projection method in the reference component analysis (RCA) package (Li et al., 2017), which can distinguish cell types not captured by traditional t-SNE-based approaches. Briefly, single-cell gene expression count profiles were first normalized using pseudo-counted quantile (pQ) normalization (Sengupta et al., 2016) and then loglO transformed. Then, genes that were highly expressed (i.e., each gene’s expression was greater than log10 50 and also greater than its own median expression value across all cells) in at least one of every 15 cells were selected as features. The counts of the feature genes of each single cell were projected onto the entire set of cells by calculating the Pearson correlation coefficient between the respective expression vector. The vector of correlation coefficients for each cell was z-score transformed to define its projection vector. Single cells were clustered using average-linkage hierarchical clustering of their projection vectors. The cell type clusters within the resulting dendrogram were identified by the cutreeDynamic function from the WGCNA (Langfelder and Horvath, 2008) package, with deepSplit = 2 and minimum group size (the number of cells in each group) = 10.
2.7. Identification of marker genes of individual cell clusters
We defined cell-cluster specific marker genes from our Drop-seq dataset using a Wilcoxon rank sum test (Wilcoxon 1945). Single cells were split into two groups for each test: the cell cluster of interest and all remaining single cells. To be considered in the analysis, the gene had to be expressed in at least 10% of the single cells from the cluster of interest and there had to be at least 1.3-fold higher expression in the cell cluster of interest than in other cells. Multiple testing was corrected using the Benjamini–Hochberg method and genes with a false discovery rate (FDR) < 0.05 were defined as cell marker genes. The use of both FDR and 1.3-fold change cutoffs help select significant markers with reasonable specificity to each cell cluster.
2.8. Resolving cell identities of the cell clusters
We used two methods to resolve the identities of the cell clusters. First, known cell-type specific markers from previous studies were curated and checked for expression patterns in the cell clusters identified in our study. Known markers for major brain cell types and neuronal subtypes were retrieved from previous studies ((Habib et al., 2016; Nguyen et al., 2017; Rosenberg et al., 2018; Zeisel et al., 2015); http://mousebrain.org) and the Allen Brain Atlas (Hawrylycz et al., 2012). A cluster showing distinct high expression levels of a known marker gene specific for a particular cell type was considered to carry the identity of that cell type. These markers were sufficient to define all major cell types as well as neuronal subtypes.
Second, we evaluated the overlap between known marker genes of previously characterized cell types with the marker genes identified in our cell clusters as described in the previous section. Overlap was assessed using a Fisher’s exact test and significance was set to p < .05. A cluster was considered to carry the identity of a cell type if the cluster marker genes showed significant overlap with known markers of that cell type. The two methods showed consistency in cell identity determination.
2.9. Detecting vulnerable cell types in ALS based on global transcriptomic shifts
We adopted the Euclidean distance method (Arneson et al., 2018) to compare the mutant cells with wildtype ones for each cell cluster regarding the vulnerability to the SOD1 mutation based on the global transcriptomic shifts between groups. For each cell type, we generate two representative cells, one for the SOD1 mutant group and the other for the wildtype condition by calculating the average gene expression of each gene for each group within that cell type. We then calculate the Euclidean distance in gene expression between these representative cells as a metric to quantify the effect of SOD1 mutant on each cell type. To determine if the observed Euclidean distance between mutant and wildtype cells within each cell type is significantly larger than that of random cells, we estimated a null distribution by calculating the Euclidean distance between randomly sampled cells of the given cell type. This permutation approach is repeated for a total of 1000 times to generate the null distribution, which is compared to the Euclidean distance generated from the true mutant and wildtype groups to determine an empirical p value. To correct for multiple testing across all the cell types tested, we applied a Bonferroni correction to retrieve adjusted p values.
To visualize the differences between wildtype and mutant cells for individual cell types, the fold change (FC) in the Euclidean distance of mutant cells compared with WT cells in each cell type was normalized by dividing the empirical Euclidean distance of that cell type by the median Euclidean distance of the null distribution of that cell type. The log10(FC) vs. −log10(p value) of each cell type were then plotted to visualize and rank the vulnerable cell types in ALS.
2.10. Identification of differentially expressed genes and pathways between wildtype and mutant cells within each cell type
To identify DEGs within each identified cell type, WT and mutant cells were compared for differential gene expression using a Wilcoxon rank sum test. To be considered in the analysis, a gene had to be expressed in at least 10% of the single cells from at least one of the two groups for that cell type and there had to be at least 1.1-fold change in gene expression between the groups. A 1.1-fold change threshold rather than a higher threshold was used here to help avoid missing genes with subtle yet biologically important changes between WT and mutant (Ben-Shaul et al., 2005; Pedotti et al., 2008). Multiple testing correction was done using the Benjamini–Hochberg method to estimate FDR.
The DEGs were then subject to pathway annotations. Because limited numbers of cells were captured for certain cell types, using a stringent FDR cutoff yielded few DEGs which limits the statistical power for downstream pathway analysis. Previous studies (Bayerlova et al., 2015) showed that pathway enrichment analysis is less sensitive to random noise because the random chance to have multiple genes from the same pathway to be DEGs is low, making a less stringent DEG p-value cutoff feasible for pathway analysis. For this reason, we used DEGs with a p value < .01 for major cell types, and DEGs with a p value <.05 for neuron subtypes (fewer cells in subtypes and hence lower statistical power) in pathway enrichment analyses to identify suggestive pathways. Pathways from KEGG, Reactome, BIOCARTA, and GO Biological Processes were assessed for overlap with the DEGs using Fisher’s exact test, followed by multiple testing correction with the Benjamini–Hochberg method to obtain FDRs. FDR < 5% was used as the cutoff for significance of the pathways.
2.11. Comparison of cell type-specific DEGs with ALS GWAS top hits
Recent ALS GWAS studies have revealed numerous genetic loci associated with ALS (Nicolas et al., 2018; van Rheenen et al., 2016). To assess whether the DEGs identified in our mouse study implicated similar genes as revealed in human GWAS studies, we retrieved the genetic association summary statistics of a recent large scale ALS GWAS (van Rheenen et al., 2016) involving meta-analysis of three cohorts: whole-genome sequenced patients with ALS (n = 1246) and controls (n = 615), GWAS of 12,577 cases and 23,475 controls in a second cohort, and GWAS of 2579 cases and 2767 controls in a third independent cohort. The summary statistics from the linear mixed model analysis from this meta-analysis study was used in our analysis.
To compare our cell type-specific DEGs with the top genetic hits from this ALS GWAS (van Rheenen et al., 2016), we used a previously established method, Mergeomics (Shu et al., 2016), to assess enrichment of top ALS GWAS hits among our DEGs from the SOD1 model. We first extracted the top 10% single nucleotide polymorphisms (SNPs) from the ALS GWAS and then used a marker dependency filter (MDF) in Mergeomics (Shu et al., 2016) to obtain independent SNPs by filtering out redundant SNPs with linkage disequilibrium r2 > 0.5. To map these filtered top GWAS SNPs to genes, we used the union of the following criteria: 1) the distance between GWAS SNPs and the mapped genes are within 100 kb, 2) functional connections between SNPs and genes based on the ENCODE (ENCODE, 2012) regulatory elements database (https://www.encodeproject.org/), and 3) expression quantitative trait loci (eQTL) from brain tissues examined in Gene by Tissue Expression (GTEx) (Consortium, 2015) (https://gtexportal.org/home/). The genes mapped to human GWAS top SNPs were compared with the DEGs from individual cell types of our brainstem single cell study to assess for overlap using the Marker set enrichment analysis (MSEA) in Mergeomics (Shu et al., 2016). In MSEA, significance of enrichment was evaluated using a chi-square-like statistics and multiple testing was corrected using the Benjamini–Hochberg method.
2.12. Comparison of cell type-specific DEGs and pathways with known ALS genes and pathways from the literature
We curated 29 known ALS genes reviewed by Chia et al. (Chia et al., 2018). Known ALS pathways were collected based on literature review of those previously implicated in ALS (Chia et al., 2018; Krokidis and Vlamos, 2018). These known ALS genes or pathways were intersected with the DEGs and pathways derived from the current single cell study.
3. Results
3.1. Quality assessment of Drop-seq data
Using Drop-seq (Macosko et al., 2015), we sequenced 1894 and 3199 brainstem cells (5093 in total) from wildtype and mutant SOD1 pi00 symptomatic mice, respectively. All samples were processed in parallel to exclude batch effects. As shown in Supplementary Fig. 1a–d, the Drop-seq library statistics and quality control features were typical of Drop-seq and comparable among the four samples. The gene-gene correlations between biological samples for individual cell types indicated overall agreement of cell populations in their main transcrip tomic features across samples (Supplementary Fig. 1e).
3.2. Drop-seq recovered major cell types in brainstem
We detected 11 cell clusters each containing cells sharing similar gene expression patterns using t-SNE based on the digital gene expression matrix across the four biological samples (detailed in Methods). Each of the cell clusters was represented by cells from all four samples (Fig. 1a). To resolve the cell-type identities for each of the cell clusters, we first obtained cluster-specific marker genes, which represent uniquely and highly expressed genes in each cluster compared to cells in all the other clusters (Supplementary Table 1). We then compared these cell cluster markers to known reference cell type markers (Supplementary Table 2) of brain and spinal cord cell types derived from previous single cell studies and in situ hybridization images in Allen Brain Atlas (details in Methods). We found distinct patterns of overlap between our cell cluster markers and known markers of brain cell types (Fig. 1b), allowing us to label the cell clusters with their corresponding cell identities. Notably, two clusters expressed classic neuronal markers, and, therefore, were labeled as “Neurons1” and “Neurons2”, respectively. Examination of specific cell type markers confirmed their distinct cluster-specific expression patterns, such as Aldoc, Aqp4, and Agt for astrocytes (Fig. 1c). These analyses support the contention that our data-driven approach reliably recovered and distinguished major known cell types in brainstem including neurons, oligodendrocytes, microglia, endothelial cells, astrocytes, ependymal cells, vascular and leptomeningeal cells (VLMCs), and Schwann cells in TG. We labeled a cell population which expressed marker genes that did not overlap with those of known cell types as “Unknown”. The heatmap on the top 50 cell-type markers for each cell cluster confirmed their cluster-specific expression patterns (Fig. 1d).
Fig. 1.

Major brainstem cell clusters and cell identities, a) t-SNE plot of the major brainstem cell types recovered from our scRNA-Seq data from 2 wildtype and 2 mutant mice, with each cell colored with the genotype: red and orange dots represent mutant cells from two mutant mice; blue and green dots represent wildtype cells from two wildtype mice, b) Significant overlap of marker genes of each cluster with known gene signatures of major brain cell types, c) The violin plots of known cell marker genes in our cell clusters. The width of violin plots shows the probability of the number of cells taking the given expression value, d) Heatmap of top 50 cell-type specific makers (columns) on all 5093 cells (rows ordered by cell clusters with each row representing a cell). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Refinement of the neuronal cell population into 8 sub-clusters
To further characterize neuronal diversity within the brainstem cells, we pooled the cells from the Neurons1 and Neurons2 clusters in Fig. 1a, which express classic neuronal markers, resulting in 254 mutant cells and 141 wildtype cells. We used a more sensitive clustering method, RCA (Li et al., 2017), to detect 8 neuronal sub-clusters (Fig. 2a). Annotation with curated neuronal marker genes (Methods), as well as with in situ hybridization images in Allen Brain Atlas, helped resolve motor neurons, TG sensory neurons, GABAergic interneurons, cerebellar neurons (Purkinje neurons and Gabra6 expressing cerebellar neurons).
Fig. 2.
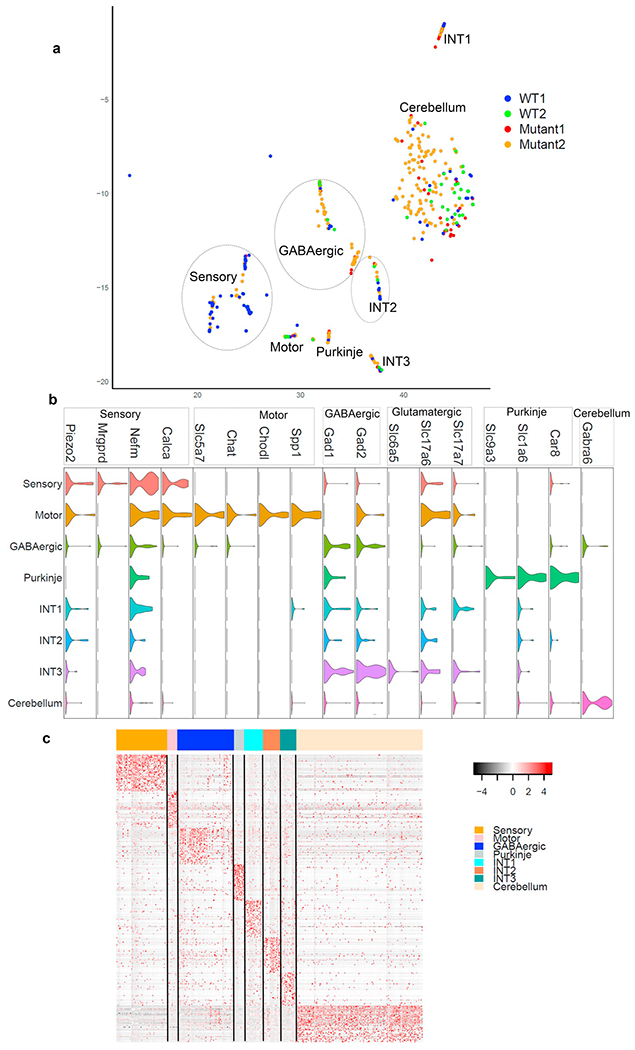
Neuronal sub-clusters and cell identities, a) t-SNE plot of the neuronal sub-clusters recovered from our scRNA-Seq data from 2 wild type and 2 mutant mice, with each cell labeled with the genotype: red and orange dots represent mutant cells from two mutant mice; blue and green dots represent wildtype cells from two wildtype mice, b) The violin plots of known neuronal subtype marker genes in our neuron sub-clusters, c) Heatmap of the top 50 cell-type specific makers (columns) on neuron cells (rows ordered by neuron sub-clusters and each row representing a cell). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Distinct expression patterns of the various cell type markers in our 8 sub-clusters were observed (Fig. 2b). For example, motor neuron populations were identified by the classic motor neuron markers Chat, Chodl, Spp1 and Slc5a7 (Fig. 2b). GABAergic interneurons were identified by the known markers Gad1, Gad2 and Slc6a5. TG sensory neurons were identified based on the expression patterns of known sensory neuron markers Piezo2, Mrgprd, Calca, and Nefm (Nguyen et al., 2017) (Fig. 2b). Cerebellar purkinje neurons were identified by known markers Slcla6, Slc9a3, and Car8 ((Rosenberg et al., 2018); http://mousebrain.org) (Fig. 2b).
Interestingly, three clusters could not be annotated based on known neuronal subtype markers. These clusters might represent potential novel neuronal subtypes or states. We labeled these clusters as “Interneuron 1 (INT1)”, “Interneuron 2 (INT2)” and “Interneuron 3 (INT3)”. The marker genes for each of the neuronal sub-clusters are also listed in Supplementary Table 1 to facilitate future functional examination of these neuronal clusters. The heatmap based upon the top 50 cell type markers for each of the 8 neuron sub-clusters confirmed their cluster-specific expression patterns (Fig. 2c).
3.4. Identification of sensitive cell types in ALS
We examined the shifts in global transcriptome patterns within each cell type from both the general clusters (Fig. 1a) and the neuronal sub-clusters (Fig. 2a) by testing if the Euclidean distance of gene expression profiles of cells between the ALS and wildtype groups is larger than expected by random chance (details in Methods). This analysis revealed that the majority of the cell types including astrocytes, oligodendrocytes, oligodendrocyte precursor cells (OPCs), Schwann cells, microglia, ependymal, endothelial, and neurons, demonstrated significant global transcriptomic shifts between mutant and wildtype cells (Fig. 3a). Among the neuronal subpopulations, two uncharacterized neuronal populations, INT1 and INT2, showed significant global transcriptomic shifts. This analysis indicates that transcriptional programs were altered in the majority of brainstem cell types of SOD1 mutant mice.
Fig. 3.

Cell types that show overall transcriptome shifts between wildtype and mutant cells based on a Euclidian distance-based analysis, a) Scatter plot of normalized logFC vs −log10 (p values) from the Euclidian distance analysis, b) Scatter plot of normalized logFC vs. −log10 (p values) of neuronal subtypes. LogFC represents fold change of Euclidian distance between wildtype and mutant cell over the distance between random pools of cells; −log10(pval) of major brain cell types represents the statistical significance of the Euclidian distance of empirical data compared to randomized data, c) Heatmap of top 50 DEGs in individual major cell clusters, with cells in wildtype (blue sidebar) and mutant groups (red sidebar) arranged in rows and DEGs arranged in columns, d) Heatmap of the top 50 DEGs (columns) in individual neuronal sub-clusters, with cells in wildtype (blue sidebar) and mutant groups (red sidebar) arranged in rows and DEGs arranged in columns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. Identification of genes related to ALS in individual cell types
To determine specific genes that may be involved in ALS pathogenesis in each cell type and to further refine the sensitive cell types, we identified differentially expressed genes (DEGs) between mutant and WT cells within each cell cluster (summary of DEGs in Table 1; all DEGs listed in Supplementary Table 3). At p < .01, the cell types that had hundreds of DEGs between mutant and WT cells are ranked based on the number of DEGs over the number of expressed genes of that cell type, in the following order: astrocytes (4.72% of expressed genes as DEGs) > oligodendrocytes (3.73%) > Microglia (1.63%) > Neurons2 (1.58%) > OPC (1.52%) > endothelial (1.49%) > unknown (1.35%) > Schwann Cells (0.97%) > Neurons1 (0.85%) > Ependymal (0.39%) (Table 1). When neuronal subpopulations were considered, based on the percentages of DEGs among expressed genes at p < .01, cellular sensitivity was ranked in the following order: GABAergic (1.77%) > INT3 (1.17%) > INT1 (1.03%) > cerebellar neurons (0.85%) > TG sensory (0.39%) > INT 2 (0.13%) (Table 2). At a stringent cutoff FDR < 5%, much fewer DEGs were identified due to multiple testing penalties and statistical power issue particularly for neuronal subpopulations. The heatmap based upon the top 50 cell-type specific DEGs confirmed their discrimination between mutant and WT cells (major cell types in Fig. 3c, neuron subtypes in Fig. 3d).
Table 1.
DEGs of major cell types from brainstem cell populations and enriched pathways compared with previous known ALS-related genes (Chia et al., 2018) and pathways (Krokidis and Vlamos, 2018; Poulin et al., 2016; Darmanis et al., 2015). Top DEGs (p < .01) and pathways, as well as those overlapping with previously reported AhS genes and pathways are shown.
| Cell type | No. of DEGs at p < .01 (DEGs at FDR < 5% in parenthesis) | Top 5 DEGs | Known ALS genes | Top enriched pathways from DEGs at p < .01; previously reported ALS-related pathways are in bold |
|---|---|---|---|---|
| Astrocytes | 864 (277) |
Up: Sod1, Mt1, Mt2, Fam107a
Down: Malat1 |
SOD1, PFN1, FUS, ATXN2 | neurogenesis, central nervous system development, head development, regulation of cell projection organization, neuron differentiation |
| Endothelial | 239 (17) |
Up: Sod1, Grid2, Rpl26, Creb3l3
Down: Gm4070 |
SOD1, VCP | interspecies interaction between organisms, regulation of multicellular organismal development, cell junction assembly, platelet degranulation, fc epsilon receptor signaling pathway |
| Ependymal | 35 (0) |
Up: Sod1, Ier3ip1, Cct3
Down: mt-Rnr2, Plac9a |
SOD1, PFN1 | negative regulation of smoothened signaling pathway, response to fluid shear stress, toxin transport, neurogenesis, osteoblast differentiation |
| Microglia | 227 (8) |
Up: Sod1, Camk2b
Down: Nav2, mt-Rnr2, 1700112E06Rik |
SOD1, VCP, ATXN2 | regulation of RAC protein signal transduction, positive regulation of molecular function, peptidyl lysine modification, intracellular signal transduction, immune system |
| Neurons1 | 102 (2) |
Up: Sod1
Down: Ctnna2, Astn2, Faf1, Ptprg |
SOD1 | synaptic signaling, regulation of dendritic spine morphogenesis, regulation of synaptic vesicle transport, cell-cell signaling, regulation of dendrite morphogenesis |
| Neurons2 | 223 (9) |
Up: Sod1, Grid2
Down: Sncg, Nefm, Lxn |
SOD1, NEFH, PFN1 | regulation of nervous system development, establishment of localization in cell, regulation of cell development, neurogenesis, positive regulation of cellular component organization |
| Schwann cells | 113 (5) |
Up: Sod1, Fth1
Down: Malat1, Reps2, Fopn1 |
SOD1 | neurogenesis, MET pathway, positive regulation of pri-miRNA transcription from RNA polymerase II promoter, regulation of neuron projection development, response to organophosphorus |
| MO | 685 (114) |
Up: Sod1, Sgk1, Grid2
Down: mt-Rnr2, Malat1 |
SOD1, VCP, FUS, MATR3, ATXN2, UBQLN2 | head development, neurogenesis, central nervous system development, ensheathment of neurons, protein localization to membrane |
| OPC | 229 (9) |
Up: Sod1
Down: Malat1, mt-Rnr2, Pcdh9, Lrrc4c |
SOD1, HNRNPA2B1, ATXN2, VCP, OPTN | neurogenesis, neuron cell cell adhesion, substantia nigra development, synapse organization, central nervous system development |
| VLMC | 209 (1) |
Up: Sod1
Down: Dcaf7, Slk, Abca8a, Col15a1 |
SOD1, MATR3 | protein localization to chromatin, collagen formation |
| unknown | 161 (7) |
Up: Sod1
Down: Fabp7, mt-Rnr2, Vim, Malat1 |
SOD1 | negative regulation of response to stimulus, negative regulation of multicellular organismal process, regulation of DNA templated transcription in response to stress, response to purine containing compound, negative regulation of protein metabolic process |
Note: MO – mature oligodendrocytes; OPC – oligodendrocyte precursor cells.
Table 2.
DEGs of neuron subtypes and enriched pathways compared with previous known ALS-related genes (Chia et al., 2018) and pathways (Krokidis et al., 2018; Poulin et al., 2016; Darmanis et al., 2015). Top DEGs (p < .01) and pathways, as well as those overlapping with previously reported ALS genes and pathways are shown.
| Cell type | No. of DEGs at p < .01 (GEGs at FDR < 5% in parenthesis) | Top 5 DEGs | Known ALS genes | Top enriched pathways from DEGs at p < .01 (motor pathways enriched from DEGs at p < .05); previously reported ALS-related pathways are in bold |
|---|---|---|---|---|
| Sensory neurons | 36 (1) |
Up: Sod1, Grid2, Mrpl21
Down: Lxn, Prune2 |
SOD1 | intermediate filament based process, negative regulation of myeloid leukocyte differentiation, negative regulation of bone remodeling, detection of temperature stimulus involved in sensory perception |
| Motor | 0 (0) |
Up: Scn2a1, Slc17a6, Nenf
Down: Kcnc3, Nefl |
axon regeneration, neuron projection regeneration, voltage gated potassium channels | |
| GABAergic | 185 (0) | Down: Ret, Nefm, Nefh, Abcg1, Mxd4 | SOD1, FUS, NEFH | response to temperature stimulus, response to heat |
| INT1 | 86 (0) | Down: 1700034P13Rik, Nr3c1, Ptpra, Tmtc2, Golga1 | SOD1, NEK1, SPG11 | |
| INT2 | 10(0) | Down: Dec, Ssbp2, Pnn, Kcnc2, Zeb1 | cellular response to toxic substance, role of dec in regulating apoptosis, role of second messengers in netrin1 signaling, dscam interactions | |
| INT3 | 103 (0) | Down: Rpl39, Nol7, Dctn3, Smim7, Alas1 | SOD1, TUBA4A, CHMP2B | |
| Cerebellum neurons | 91 (1) |
Up: Sod1, mt-Cytb
Down: Bai3, Astn2, Ptprt |
SOD1 | multicellular organismal signaling, circadian rhythm, rhythmic process |
3.6. Pathways altered in ALS in individual cell populations
We next annotated the DEGs with curated biological pathways from KEGG, Reactome, BIOCARTA, and Gene Ontology databases to identify key pathways that could explain fundamental aspects of the ALS pathology and the cell types involved (Tables 1 and 2; full pathway list in Supplementary Table 4).
Among the known pathways previously implicated in ALS (Chia et al., 2018; Krokidis and Vlamos, 2018), our cell-type specific DEGs recapitulated immune system (microglia), neurogenesis (astrocytes, oligodendrocytes, Neurons2), and synaptic organization/transmission (oligodendrocytes, Neurons1) related pathways (Table 1).
More importantly, our analysis revealed potential novel ALS pathways from the major cell types, such as “regulation of RAC protein signal transduction and peptidyl lysine modification” in microglia, “toxin transport and cell differentiation” in ependymal cells, “response to organophosphorus” in Schwann cells within TG, and “collagen formation” in VLMC.
For the neuronal subtypes (Table 2), GABAergic DEGs are related to “response to temperature stimulus” pathway (Sod1, St8sla1, Ano3, Trpv2, Stac, Pirt, Eif2b5, Nfkbia). DEGs from motor neurons were enriched for voltage-gated potassium channels/potassium-calcium transporter (Kcnc3, Kcna1, Slc24a2) and axon regeneration (Nefl, Chl1) pathways. TG sensory neurons showed alterations in “intermediate filament based process” pathway (Sod1, Ndel1, Dst), “negative regulation of myeloid leukocyte differentiation” (Calca, Cartpt, Runx1), and “detection of temperature stimulus involved in sensory perception” (Calca, Lxn) pathways. Specific pathways were also identified for the unknown neuronal subpopulations: “Endocardium development and positive regulation of ERBB signaling” pathway in INT1 subtype; “cellular response to toxic substance” in INT2; “positive regulation of dendritic spine development” in INT3. These additional cell-type specific pathways may also participate in ALS pathogenesis or protection, and warrant further validation.
3.7. Prioritization of global and cell-type specific DEGs
As numerous DEGs were identified from individual cells, prioritization of the DEGs is necessary to pinpoint potential targets, as demonstrated in our previous single cell study (Arneson et al., 2018). To this end, we evaluated the cell-type specificity of DEGs to prioritize genes that either show consistent alterations across cell types or demonstrate cell-type specificity. The genes that are consistently perturbed in brainstem, regardless of cell types, could serve as general targets in a global manner, whereas the cell type-specific DEGs could facilitate precise targeting of pathogenic pathways in specific cell types in ALS.
Notably, and as expected, the most consistent DEG across cell types was SOD1, confirming it is the major genetic perturbation in mutant mice (Table 3 for major cell types and Table 4 for neuronal subtypes). In addition to SOD1, numerous genes (e.g., Malat1, mt-Cytb, mt-Rnr2) also showed significant differential expression in many cell types with mostly consistent directional changes across cell types. There were also DEGs that were highly cell-type specific, indicating unique alterations in individual cell types as exemplified in Tables 3 and 4 (full list in Supplementary Table 5 and 6). For example, Tmem255a is a TG sensory-specific DEG and Scn2a1 is a motor-specific DEG.
Table 3.
DEGs consistent across multiple cell types or specific to each cell type. The log fold changes in each cell type are given (all p-values shown reach <0.01; details Supplementary Table 5).
| Astrocytes | Endothelial | Ependymal | Microglia | Neurons1 | Neurons2 | Schwann cells | OM | OPC | VLMC | unknown | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DEGs consistent across cell types | |||||||||||
| Sod1 | 2.51 | 2.43 | 2.41 | 1.62 | 2.78 | 2.27 | 3.11 | 3.34 | 2.14 | 2.41 | 2.04 |
| Malat1 | −0.76 | −0.25 | – | −0.27 | −0.24 | – | −0.75 | −0.33 | −0.41 | −0.28 | −0.48 |
| mt-Cytb | 0.42 | 0.28 | − | 0.54 | 0.78 | 0.5 | 0.51 | 0.27 | – | 0.84 | – |
| mt-Rnr2 | −0.44 | −0.15 | −0.97 | −0.44 | – | – | – | −0.52 | −0.46 | – | −0.53 |
| Gm26924 | −0.53 | −0.31 | – | −0.55 | −0.74 | – | – | −0.58 | −0.44 | – | −0.62 |
| Ubb | 0.67 | 0.31 | – | – | 0.95 | – | – | 0.39 | 0.32 | – | 0.86 |
| Grid2 | – | 1.1 | 1.58 | – | – | 1.82 | 1.62 | 1.08 | – | – | – |
| Lrrc4c | −1.2 | – | – | – | −0.86 | – | −0.98 | −0.35 | −0.59 | – | −0.96 |
| Fth1 | 0.37 | 0.35 | – | – | – | – | 0.76 | 0.32 | 0.66 | – | 0.71 |
| Rpl26 | 0.38 | 1.24 | – | – | 1.39 | – | – | 0.62 | 1.16 | – | 0.74 |
| Rora | −0.71 | – | – | – | −0.63 | 1.39 | – | – | −0.72 | – | – |
| Tmtc2 | −0.37 | −0.36 | – | – | −0.69 | – | – | −0.42 | – | – | −0.87 |
| Plxdc2 | −0.26 | – | – | −0.39 | −0.69 | – | −1.02 | −0.42 | – | – | – |
| Dst | −0.3 | −0.31 | – | – | – | −0.84 | – | −0.3 | – | – | −0.63 |
| Diap2 | −0.3 | −0.49 | – | −0.71 | −0.59 | – | – | −0.28 | – | – | – |
| Klf7 | −0.25 | – | – | −0.79 | −0.59 | −0.8 | −0.57 | – | – | – | – |
| Mt1 | 0.94 | – | – | – | – | – | 0.45 | 0.47 | – | 1.55 | 0.71 |
| Dbi | 0.9 | – | – | – | 1.44 | – | 1.08 | 0.24 | 0.47 | – | – |
| mt-Rnr1 | 0.32 | 0.36 | – | – | – | 1.04 | 0.6 | – | – | – | – |
| mt-Nd4 | 0.24 | 0.32 | – | – | – | 0.46 | – | 0.23 | – | 0.64 | – |
| H3f3b | 0.37 | 0.19 | – | 0.45 | – | – | – | 0.29 | – | – | 1.07 |
| Cell type specific DEGs | |||||||||||
| Cyp26b1 | −0.90 | – | – | – | – | – | – | – | – | – | – |
| Gbp4 | – | −0.54 | – | – | – | – | – | – | – | – | – |
| Ier3ip1 | – | – | 1.81 | – | – | – | – | – | – | – | – |
| Cd74 | – | – | – | −1.43 | – | – | – | – | – | – | – |
| Psmd7 | – | – | – | – | 1.12 | – | – | – | – | – | – |
| Rgs4 | – | – | – | – | – | −0.91 | – | – | – | – | – |
| Fopnl | – | – | – | – | – | – | −0.72 | – | – | – | – |
| Cldn11 | – | – | – | – | – | – | – | 0.25 | – | – | – |
| Mmp16 | – | – | – | – | – | – | – | – | −0.48 | – | – |
| Dcaf7 | – | – | – | – | – | – | – | – | – | −1.21 | – |
| Mmd2 | – | – | – | – | – | – | – | – | – | – | −1.55 |
Table 4.
DEGs consistent across multiple neuron subtypes or specific to neuronal subtypes. The log fold changes in each cell type are given (all p-values <.01; details Supplementary Table 6).
| Sensory | Motor | GABAergic | INT1 | INT2 | INT3 | Cerebellum | |
|---|---|---|---|---|---|---|---|
| DEGs consistent across cell types | |||||||
| Sod1 | 3.08 | – | 2.34 | 3.42 | – | 2.38 | 2.39 |
| Malat1 | – | – | – | −0.74 | – | – | −0.26 |
| mt-Rnr2 | – | – | – | −0.7 | −0.78 | – | – |
| Grid2 | 1.33 | – | 2.39 | – | – | – | – |
| Rora | – | – | 1.66 | – | – | – | −0.68 |
| Cell type specific DEGs | |||||||
| Tmem255a | 1.22 | – | – | – | – | – | – |
| Scn2a1 | – | 1.99 | – | – | – | – | – |
| Nefh | – | – | −1.91 | – | – | – | – |
| 1700034P13Rik | – | – | – | −1.87 | – | – | – |
| Kcnc2 | – | – | – | – | −2.30 | – | – |
| Rpl39 | – | – | – | – | – | −2.54 | – |
| Gm5914 | – | – | – | – | – | – | −1.13 |
3.8. Overlap of cell-type specific DEGs from the SOD1 mutants with known ALS genes and GWAS top hits in humans
To assess the relevance of the DEGs identified in our mouse study to human ALS, we compared our DEGs with curated ALS genes from the literature (Comley et al., 2015; Darmanis et al., 2015; Poulin et al., 2016), as well as candidate genes mapped to ALS GWAS top hits in human studies (van Rheenen et al., 2016).
As shown in Tables 1 and 2, out of the 29 known ALS genes (Chia et al., 2018), 14 were among our cell-type specific DEGs. These include Sod1, Hnrnpa2b1, Pfn1, Fus, Vcp, Matr3, Nek1, Nefh, Ubqln2, Atxn2, Chmp2b, Tuba4a, Spg11, and Optn. Interestingly, several of the known ALS genes (Nek1, Spg11, Tuba4a and Chmp2b) were DEGs in uncharacterized neuronal cell populations. These neuronal populations might be where these genes function and play a role in ALS pathogenesis (Table 2).
Comparison of our DEGs with the recent human ALS GWAS top hits (shown in Table 5; all results shown in Supplement Table 7) also revealed numerous overlaps, which mostly showed down-regulation in our study. Additionally, there was a statistically significant over-representation of DEGs from endothelial cells, Neurons2 and neuronal subpopulations (TG sensory and INT1) among the top human ALS GWAS signals.
Table 5.
Overlap between cell-type-specific DEGs (pval <0.05) with genes mapped to GW AS hits (pval <10E-5). (details in Supplementary Table 7).
| Cell type DEGs | Enrichment P value | FDR | DEGs (p < .05) overlapping with top GWAS hits (GWAS pval < 10E-5) (up-regulation in SOD1 mutant shown in bold) |
|---|---|---|---|
| INT1 DEG (down) | 1.04E-04 | 0.0012 | WHAMM, AXIN1, TUSC1, CHRNB1, SEC13 |
| Endothelial DEG (down) | 1.20E-04 | 0.0013 | MAP3K3, BAZ2A, ZBTB4 |
| Neurons2 DEG (up) | 5.20E-4 | 0.0044 | |
| Sensory DEG (up) | 0.0023 | 0.0148 | |
| Microglia DEG (up) | 0.0380 | 0.1760 | SYNGR1 |
| MO DEG (down) | 0.0509 | 0.2161 | FNBP1, PTGES3, ACTR2 |
The above comparisons between the DEGs and human ALS genes support the contention that some of the DEGs uncovered in the present study using the SOD1 mouse model have biological relevance to human ALS pathogenesis.
4. Discussion
To better understand the molecular alterations in ALS in individual cell populations in the brainstem, we conducted a single cell investigation using SOD1(G93A) mice, a well-established ALS mouse model (Gurney et al., 1994). This is the first study to investigate single cell RNA expression within pontine brainstem using this high-throughput high-resolution approach. We were able to not only capture most major cell types and neuronal subtypes present in brainstem, but also detect previously uncharacterized cell types, thus exploiting the power of the scRNA-seq method. Based on the overall transcriptomic shifts, as well as DEGs in individual cell clusters, we found that most of the major cell populations (e.g., astrocytes, oligodendrocytes, microglia, Schwann cells, ependymal cells, neurons) and several neuronal subpopulations (e.g., uncharacterized neuronal subtypes INT1 and INT2) show alterations in their gene expression patterns, suggestive of sensitivity in ALS. We further identified numerous genes and pathways altered in the mutant mice in individual cell types, recovering not only previously known ALS genes and pathways, such as immune functions, neurogenesis and synaptic transmission, but new genes and biological processes in specific brainstem cell types such as toxin transport in ependymal cells and response to organophosphorus in Schwann cells. Many DEGs were consistently altered across cell types (e.g., Malat1, mt-Cytb, mt-Rnr2, Grid2), while others were cell type specific (e.g., motor neuron-specific DEG Scn2a1). Our DEGs also overlapped with human GWAS of ALS genes and thus, supports the relevance of our findings from mice to human disease. Overall, our cell-type specific investigation of the mutant SOD1 mice forms a comprehensive map of the cell types, genes, and pathways altered in this ALS model in a defined region of brainstem involved in control of jaw movements, and will serve as a basis for comparison with spinal cord cell types.
Previous ALS studies focused, predominately, on spinal cord and brainstem motor neurons and the genes and pathways altered at different stages of the disease (Krokidis and Vlamos, 2018; Van Damme et al., 2017). More recently, supporting cell types have been studied as well (Serio and Patani, 2018). However, to fully understand the origin of, and underlying mechanism(s) for progression of ALS, as well as the molecular basis for changes in neurons and supportive cells, a more comprehensive catalog of cell types affected within the CNS is necessary. scRNA-seq methodology provides this opportunity (Chen et al., 2017; Sathyamurthy et al., 2018; Saunders et al., 2018; Zeisel et al., 2015). In the present study we examined transcriptional changes at single cell resolution within a defined region of brainstem containing cells involved in jaw movements (Tanaka et al., 1999; Westberg and Kolta, 2011). Previously, we showed significant early alterations in membrane excitability at presymptomatic ages (P8–12) in both masticatory motor neurons and proprioceptive sensory neurons of jaw closer spindle origin (Trigeminal Mesencephalic V (Mes V neurons)) from ALS mice (Seki et al., 2019; Venugopal et al., 2015). Furthermore, we observed that in symptomatic SOD1 mice, at P90 there is severe vacuolization, and reduced motor neuron count in the trigeminal motor nucleus (unpublished observation). Clinically, 20–30% of the ALS cases present with progressive bulbar palsy (Riera-Punet et al., 2018) affecting facial, masticatory and swallowing functions. However, most human ALS cases, regardless of the form of ALS, will involve, eventually, reduction in jaw muscle force production and movement capabilities (Riera-Punet et al., 2018). A benefit of studying this region of brainstem is that the extensively studied sensory, motor and interneuronal nuclei involved in jaw movements (Kogo et al., 1996; Westberg and Kolta, 2011) are well defined, and the use of scRNA-seq provides the opportunity to unravel and sub-classify neurons and supporting cells according to gene expression profiles within these heterogeneous nuclei.
Motor neuron degeneration in ALS most likely has a cell autonomous and non-autonomous contribution from glial cells (astrocytes, microglia, oligodendrocytes, Schwann cells) to the disease process (Rizzo et al., 2014; Serio and Patani, 2018). Our study confirmed the involvement of these cell types in ALS as demonstrated by the global shifts in their transcriptome and the large numbers of DEGs and pathways identified. For example, in mutant SOD1 models, oligodendrocytes are implicated in initiation of disease (Kang et al., 2013), and we found that pathways related to neuron ensheathment and neurogenesis were altered in ALS oligodendrocytes, which could explain how this cell type may contribute to ALS development. We also found that immune pathway genes were perturbed in microglia, which agrees with previous spinal cord data on how inflammatory events elicited by microglia may drive disease progression (Boillée et al., 2006; Frakes et al., 2014; Yamanaka et al., 2008). More importantly, we found that the majority of the brainstem cell types and neuronal subtypes, including previously uncharacterized cell populations, exhibit global transcriptomic shifts and/or significant DEGs. These results significantly broaden the scope of the cell types that may participate in ALS pathogenesis, and open the possibility for investigating the specific roles of each cell type in ALS pathology and for designing targeted treatments more precisely against individual vulnerable cell types.
Previous studies suggested neuronal hyperexcitability and excitotoxicity in upper and lower motor neurons, and subsequent internal calcium dysregulation, as potential causative mechanisms for neuronal death depending on motor neuronal type (slow vs. fast motor units) (reviewed in (Fogarty, 2018)), although this is contentious (Leroy and Zytnicki, 2015). A novel finding in the motor neuron cluster from ALS mice in the present study was the upregulation of the sodium channel gene Scn2a, which codes for the sodium channel protein NaV 1.2 (de Lera and Kraus, 2015), and downregulation of the potassium channel genes, Kcna1 and Kcnc3 (Fig. 4b), important genes which code for the delayed rectifier and transient A type channels, respectively, and contribute to resting potential and spike repolarization, as well as control of discharge frequency. Such changes in both sodium and potassium ion channel function could account for hyperexcitability observed in motor neurons in ALS. The present data provide a molecular basis for previous electrophysiology data regarding hyperexcitability observed in both animal models and patient-derived cellular models of ALS (Fogarty, 2018; Nakata et al., 2006; Wainger et al., 2014), as well as our previous electrophysiological data from postnatal stage of SOD1 mouse (Venugopal et al., 2015) demonstrating hyperexcitability in trigeminal motor neurons, albeit obtained in presymptomatic ALS mice. Additionally, we found Slc24a2, a DEG related to Ca2+ transport and control of internal Ca2+ concentration, was also downregulated in the motor cluster (Fig. 4b), and a number of DEGs relating to synaptic function and calcium ion homeostasis in the GABA interneuron subcluster were altered as well (Fig. 4a). Changes in ion channel function related to control of membrane excitability, synaptic transmission, and calcium homeostasis are likely contributors to ALS pathogenesis (Seki et al., 2019; Starr and Sattler, 2018; Venugopal et al., 2015; Wainger et al., 2014).
Fig. 4.
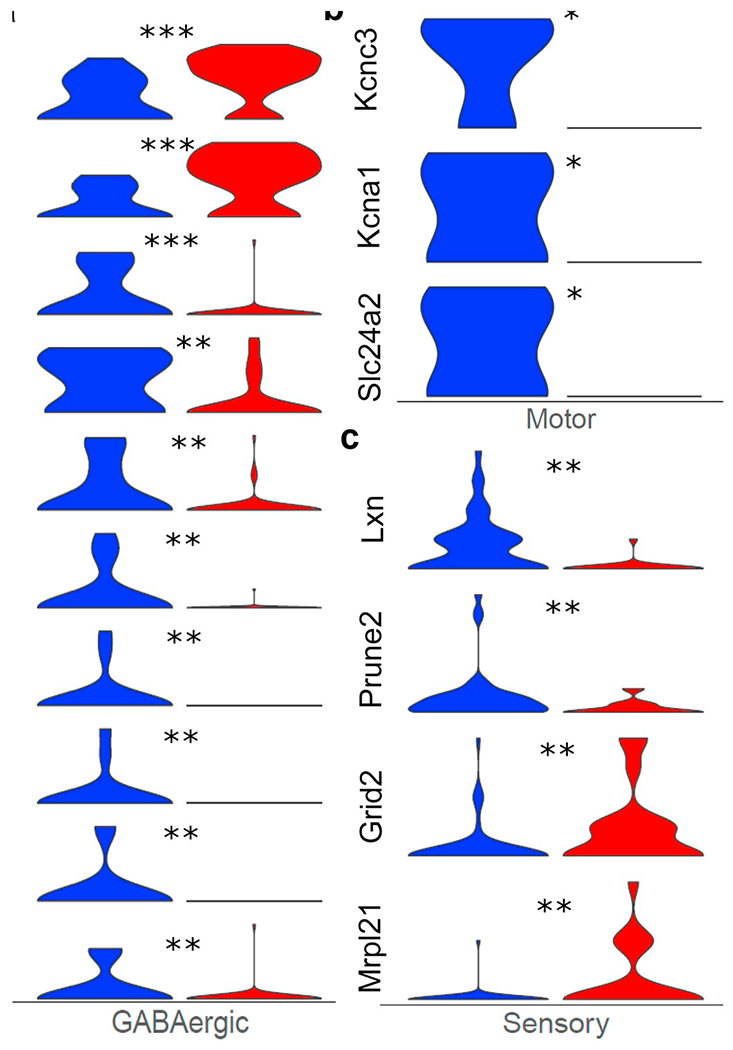
Expression change of select DEGs (wildtype cells in blue and mutant ones in red) in GABAergic, motor and sensory neurons. The stars above the violin plots represent the significance in gene expression changes: *0.01 < p value < .05; **0.001 < p-value < .01; *** p-value <.001. a) Expression changes of DEGs related to synaptic signaling in GABAergic neurons. b) Expression changes of DEGs related to potassium channel function in Motor neurons. c) Expression changes of top DEGs in the peripheral sensory neurons. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Among the DEGs revealed in individual cell types, many were previously known ALS genes. For example, 14 out of the 29 known ALS genes (Comley et al., 2015; Darmanis et al., 2015; Poulin et al., 2016) were among our DEGs in brainstem cell populations. Interestingly, known ALS genes Nek1, Spg11, Tuba4a and Chmp2b were altered in previously uncharacterized neuronal populations in our mouse model. These findings help point to the specific cell populations in which these ALS genes might function and set the stage for future characterization of these neuronal populations, as well as the functions of the ALS genes in these cell types. Additionally, the DEGs from various cell types, including astrocytes, endothelial cell, mature oligodendrocytes, and neuronal subpopulations (TG neurons and the uncharacterized neuronal sub-population INT1), also showed overlap with human ALS GWAS hits. For example, See13, a GWAS gene related to vesicle-mediated transport and signaling by Rho GTPases, is downregulated in INT1 neurons. Similarly, GWAS candidate gene Sez6l, which may contribute to specialized endoplasmic reticulum functions in neurons, is downregulated in the INT3 neuronal cluster. The altered expression of numerous other ALS GWAS genes in the SOD1 mice suggests that ALS genes are tightly connected, as perturbation of the SOD1 gene in this mouse model alters the expression of other known ALS genes. It is possible that despite the heterogeneity in the causal mutations in human patients, similar sets of downstream genes and pathways in tightly connected networks are altered, aligning with the omnigenic model of human complex diseases (Boyle et al., 2017).
In addition to confirming pathways implicated in previous motor neuron studies (Brockington et al., 2013; Krokidis and Vlamos, 2018), our scRNA-seq analysis supports involvement of these pathways in other cell types as well. For instance, DEGs from endothelial cells and microglia were associated with immune pathways; DEGs from ependymal and an unknown cell type were associated with response to stress; the neurogenesis pathway was found to be altered in numerous major cell types (astrocytes, oligodendrocytes, OPC, Neurons2, ependymal, Schwann cells); and synaptic transmission related pathways were enriched among DEGs not only from GABAergic interneurons but also from cerebellar neurons. In the “synaptic signaling” pathway, GABAergic specific DEGs Nrxn3 and Grid2 are upregulated whereas Sncg, Krfn5, Hspa8, Syt7, Grik1, Kcnmb2, Cask, Lrrk2, Prkcg are down-regulated; in the pathway “regulation of synapse structure”, cerebellum specific DEGs Bai3, Ctnna2, Lphn3, Ntrk2, Cntn4, and Fgf14 are down-regulated. Interestingly, in an unknown general cell cluster and an unknown neuron subtype (INT1), specific DEGs (e.g., Sod1 and Ubb, both upregulated) were enriched for the “regulation of mitochondrial membrane potential” pathway. These results suggest that specific cell types in the brainstem, which demonstrate perturbations in known ALS pathways, are not limited to motor neurons and the cell specific DEGs provide potential novel targets for investigating the pathogenic pathways in ALS.
Our analyses showed potential novel cell-type specific ALS pathways, as well. For instance, pathways involved in “toxin transport” and “cell differentiation” in ependymal cells, “response to organophosphorus” in Schwann cells, and “collagen formation” in VLMC were observed. Among these, “toxin transport and organophosphorus responses” have been previously linked to environmental insults and triggers of ALS (Merwin et al., 2017; Yu and Pamphlett, 2017). Therefore, our findings implicate the connection of the genetic insult (SOD1 in this case) with environmentally responsive pathways. The exact roles of these pathways and the associated cell types in ALS warrant further investigation.
Additionally, our study facilitated prioritization of potential target genes that are either independent of cell types or cell-type specific. Top cell-type independent DEGs include the mitochondrial genes mt-Rnr1 (upregulated), mt-Rnr2 (downregulated), and mt-Cytb (upregulated). mt-Rnr1 and mt-Rnr2 encode small mitochondrial peptides MOTS-c and humanin, respectively. MOTS-c is essential for regulation of gene expression and mitochondrial homeostasis (Kim et al., 2018), and humanin (Zapala et al., 2010) is implicated as a therapeutic target for numerous neurodegenerative disorders. Although humanin has been implicated in ALS before (Kovanda et al., 2018), our study is the first to reveal its consistent alteration across most cell populations in brainstem, thereby offering strong evidence that mitochondrial peptides may serve as major biomarkers or targets that are not cell-type specific. Other consistent DEGs across cell types that may also serve as cell-independent global markers or targets for ALS include Malat1, a long noncoding RNA important for cell cycle regulation (Tripathi et al., 2013) and neurorepair (Zhang et al., 2017); Ubb, a proteasome component; Grid2, a non-conventional glutamate receptor; Rora, a key regulator of diverse functions ranging from embryonic development, cellular differentiation, to immunity, circadian rhythm, and metabolism; and Gm26924 with unknown function. Many of these genes have not been implicated in ALS previously. Cell-type specific DEGs include motor-specific Scn2a (upregulated), which could mediate motor neuron hyperexcitability, and TG neuron specific Tmem255a (upregulated) and cerebellar neuron specific Gm5914 (downregulated), whose functions remain poorly understood. These cell specific DEGs provide potential novel targets for investigating the pathogenic pathways in ALS in specific cell types.
5. Conclusions
In this first high-throughput single cell transcriptome study of the brainstem using a SOD1 mutant ALS model, we characterized thousands of cells representing the majority of the brainstem cell types. We identified cell types that show sensitivity to the SOD1 mutation, as well as, cell-type specific genes and pathways altered in the mutant, thus recapitulating both known ALS genes and pathways and revealing novel ones that warrant further investigation. Our findings provide comprehensive molecular insights into ALS pathogenesis in a cell-type specific manner for a defined region of brainstem. The results add to a growing compendium of molecular characterization of CNS cell types and stimulate future studies using additional ALS human and animal models for determination of similarities/dissimilarities in gene expression profiles in both resistant and vulnerable cell types at single cell resolution. Such studies should lead to new insights into the pathogenesis of the disease and provide new targets for development of therapeutic intervention.
Supplementary Material
Acknowledgements
This work was supported by a UCLA academic senate grant (SHC), SCEHSC Pilot Award (XY) and NINDS grant NS095157-01(SV). Graciel Diamante is supported by American Diabetes Association Postdoctoral Fellowship grant # 1-19-007-R. We thank Soju Seki for helping with tissue extraction, and Poonam Sachan for maintaining the mouse colony.
Footnotes
Declaration of Competing Interest
None
Appendix A.: Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.104877.
References
- Arneson D, Zhuang Y, Byun HR, Ahn IS, Ying Z, Zhang G, Gomez-Pinilla F, Yang X, 2018. Single cell molecular alterations reveal pathogenesis and targets of concussive brain injury. Nat. Commun 9, 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U, Cotney J, Nagy M, Oh S, Leng J, Mahajan M, Mane S, Fenton WA, Noonan JP, Horwich AL, 2013. RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS One 8, e53575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baufeld C, OĽoughlin E, Calcagno N, Madore C, Butovsky O, 2018. Differential contribution of microglia and monocytes in neurodegenerative disease. J. Neural Trans 125 (5), 809–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayerlová M, Jung K, Kramer F, Klemm F, Bleckmann A, Beißbarth T, 2015. Comparative study on gene set and pathway topology-based enrichment methods. BMC Bioinform. 16, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shaul Y, Bergman H, Soreq H, 2005. Identifying subtle interrelated changes in functional gene categories using continuous measures of gene expression. Bioinformatics 21, 1129–1133. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW, 2006. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312 (5778), 1389–1392. [DOI] [PubMed] [Google Scholar]
- Boyle EA, Li YI, Pritchard JK, 2017. An expanded view of complex traits: from polygenic to Omnigenic. Cell 169 (7), 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, 2007. Isolation and culture of adult neurons and neurospheres. Nat. Protoc 2 (6), 1490–1498. [DOI] [PubMed] [Google Scholar]
- Brockington A, Ning K, Heath PR, Wood E, Kirby J, Fusi N, Lawrence N, Wharton SB, Ince PG, Shaw PJ, 2013. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler SH, Tal M, 1986. The effects of brain stem transections on the neuronal networks responsible for rhythmical jaw muscle activity in the guinea pig. J. Neurosci 6, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Wu X, Jiang L, Zhang Y, 2017. Single-cell RNA-Seq reveals hypothalamic cell diversity. Cell Rep. 18, 3227–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia R, Chio A, Traynor BJ, 2018. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comley L, Allodi I, Nichterwitz S, Nizzardo M, Simone C, Corti S, Hedlund E, 2015. Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 291, 216–229. [DOI] [PubMed] [Google Scholar]
- Consortium GT. Human genomics, 2015. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR, 2015. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci 112 (23), 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lera Ruiz M., Kraus RL, 2015. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem 58 (18), 7093–7118. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium, 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489 (7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ, 2007. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci 27, 9201–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MJ, 2018. Driven to decay: excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res. Bull 140, 318–333. [DOI] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK, 2014. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81 (5), 1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. , 1994. Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 264 (5166), 1772–1775. 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M, M LG-V FRP, Besse F, Weis, Voigt A, et al. , 2017. Linking amyotrophic lateral sclerosis and spinal muscular atrophy through RNA-transcriptome homeostasis: a genomics perspective. J. Neurochem 141 (1), 12–30. [DOI] [PubMed] [Google Scholar]
- Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, Hession C, Zhang F, Regev A, 2016. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353 (6302), 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz M, Lein E, Guillozet-Bongaarts A, et al. , 2012. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath PR, Kirby J, Shaw PJ, 2013. Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front. Cell. Neurosci 7, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund E, Karlsson M, Osborn T, Ludwig W, Isacson O, 2010. Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain J. Neurol 133, 2313–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE, 2013. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci 16 (5), 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE, 2010. Motor neuron diversity in development and disease. Annu. Rev. Neurosci 33, 409–440. [DOI] [PubMed] [Google Scholar]
- Kim KH, Son JM, Benayoun BA, Lee C, 2018. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 28 (3) 516–524.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogo M, Funk GD, Chandler SH, 1996. Rhythmical oral-motor activity recorded in an in vitro brainstem preparation. Somatosensory Motor Res. 13, 39–48. [DOI] [PubMed] [Google Scholar]
- Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Ristic Kovacic S, Curk T, Rogelj B, 2018. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep 8 (1), 5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokidis MG, Vlamos P, 2018. Transcriptomics in amyotrophic lateral sclerosis. Front. Biosci 10, 103–121. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S, 2008. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Zytnicki D, 2015. Is hyperexcitability really guilty in amyotrophic lateral sclerosis? Neural Regen. Res 10, 1413–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever TE, Gorsek A, Cox KT, O’Brien KF, Capra NF, Hough MS, Murashov AK, 2009. An animal model of oral dysphagia in amyotrophic lateral sclerosis. Dysphagia 24, 180–195. [DOI] [PubMed] [Google Scholar]
- Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, Wong M, Choi PJ, Wee LJK, Hillmer AM, Tan IB, Robson P, Prabhakar S, 2017. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet 49 (5), 708–718. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA, 2015. Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell 161 (5), 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis S, Äijö T, Vickovic S, Braine C, Kang K, Mollbrink A, Fagegaltier D, Andrusivová Ž, Saarenpää S, Saiz-Castro G, Cuevas M, Watters A, Lundeberg J, Bonneau R, Phatnani H, 2019. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364 (6435), 89–93. [DOI] [PubMed] [Google Scholar]
- Merwin SJ, Obis T, Nunez Y, Re DB, 2017. Organophosphate neurotoxicity to the voluntary motor system on the trail of environment-caused amyotrophic lateral sclerosis: the known, the misknown, and the unknown. Arch. Toxicol 91 (8), 2939–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata M, Kuwabara S, Kanai K, Misawa S, Tamura N, Sawai S, Hattori T, Bostock H, 2006. Distal excitability changes in motor axons in amyotrophic lateral sclerosis. Clin Neurophysiol. 117 (7), 1444–1448. 10.1016/j.clinph.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Nguyen MQ, Wu Y, Bonilla LS, von Buchholtz LJ, Ryba NJP, 2017. Diversity amongst trigeminal neurons revealed by high throughput single cell sequencing. PLoS One 12 (9), e0185543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A, et al. , 2018. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97 (6) 1268–1283.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedotti P, THoen PA, Vreugdenhil E, Schenk GJ, Vossen RH, Ariyurek Y, de Hollander M, Kuiper R, van Ommen GJ, den Dunnen JT, Boer JM, de Menezes RX, 2008. Can subtle changes in gene expression be consistently detected with different microarray platforms? BMC Genom. 9, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhov V, Guo J, Baryawno N, Severe N, Scadden DT, Samsonova MG, Kharchenko PV, 2018. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Tasic B, Hjerling-Leffler J, Trimarchi JM, Awatramani R, 2016. Disentangling neural cell diversity using single-cell transcriptomics. Nat. Neurosci 19 (9), 1131–1141. [DOI] [PubMed] [Google Scholar]
- Riera-Punet N, Martinez-Gomis J, Paipa A, Povedano M, Peraire M, 2018. Alterations in the masticatory system in patients with amyotrophic lateral sclerosis. J. Oral Facial Pain Headache 32, 84–90. [DOI] [PubMed] [Google Scholar]
- Rizzo F, Riboldi G, Salani S, Nizzardo M, Simone C, Corti S, Hedlund E, 2014. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell. Mol. Life Sci 71 (6), 999–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. , 2018. Single cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360 (6385), 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ, 2018. Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep. 22, 2216–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA, 2018. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174 (4), 1015–1030 ( e16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki S, Yamamoto T, Quinn K, Spigelman I, Pantazis A, Olcese R, Wiedau-Pazos M, Chandler SH, Venugopal S, 2019. Circuit-specific early impairment of proprioceptive sensory neurons in the SOD1G93A mouse model for ALS. J. Neurosci 39 (44), 8798–8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D, Rayan NA, Lim M, Lim B, Prabhakar S, 2016. Fast, scalable and accurate differential expression analysis for single cells. Preprint at bioRxiv. 10.1101/049734. [DOI] [Google Scholar]
- Serio A, Patani R, 2018. Concise review: the cellular conspiracy of amyotrophic lateral sclerosis. Stem Cells 36, 293–303. [DOI] [PubMed] [Google Scholar]
- Shu L, Zhao Y, Kurt Z, Byars SG, Tukiainen T, Kettunen J, Orozco LD, Pellegrini M, Lusis AJ, Ripatti S, Zhang B, Inouye M, Makinen VP, Yang X, 2016. Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics 17 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr A, Sattler R, 2018. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res 1693, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Kogo M, Chandler SH, Matsuya T, 1999. Localization of oral-motor rhythmogenic circuits in the isolated rat brainstem preparation. Brain Res. 821, 190–199. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr., Cleveland DW, 2016. Decoding ALS: from genes to mechanism. Nature 539 (7628), 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, et al. , 2013. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 9 (3), e1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W, Van Den Bosch L, 2017. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech 10, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Maaten LJP, Hinton GE, 2008. Visualizing high-dimensional data using t-sne. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. , 2016. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet 48 (9), 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Hsiao CF, Sonoda T, Wiedau-Pazos M, Chandler SH, 2015. Homeostatic dysregulation in membrane properties of masticatory motoneurons compared with oculomotor neurons in a mouse model for amyotrophic lateral sclerosis. J. Neurosci 35, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH Jr., Cudkowicz ME, Bean BP, Eggan K, Woolf CJ, 2014. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberg KG, Kolta A, 2011. The trigeminal circuits responsible for chewing. Int. Rev. Neurobiol 97, 77–98. [DOI] [PubMed] [Google Scholar]
- Frank Wilcoxon, 1945. Individual comparisons by ranking methods. Biom. Bull 1 (6), 80–83. [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW, 2008. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci 11 (3), 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Pamphlett R, 2017. Environmental insults: critical triggers for amyotrophic lateral sclerosis. Transl. Neurodegener 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapala B, Kaczynski L, Kiec-Wilk B, Staszel T, Knapp A, Thoresen GH, Wybranska I, Dembinska-Kiec A, 2010. Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties. Pharmacol. Rep 62, 767–777. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S, 2015. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang X, Hamblin MH, Yin KJ, 2017. The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. 14 (12), 1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.