

Abstract
Background and Purpose
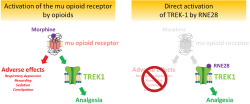
Opioids are effective painkillers. However, their risk–benefit ratio is dampened by numerous adverse effects and opioid misuse has led to a public health crisis. Safer alternatives are required, but isolating the antinociceptive effect of opioids from their adverse effects is a pharmacological challenge because activation of the μ opioid receptor triggers both the antinociceptive and adverse effects of opioids.
Experimental Approach
The TREK1 potassium channel is activated downstream of μ receptor and involved in the antinociceptive activity of morphine but not in its adverse effects. Bypassing the μ opioid receptor to directly activate TREK1 could therefore be a safer analgesic strategy.
Key Results
We developed a selective TREK1 activator, RNE28, with antinociceptive activity in naive rodents and in models of inflammatory and neuropathic pain. This activity was lost in TREK1 knockout mice or wild‐type mice treated with the TREK1 blocker spadin, showing that TREK1 is required for the antinociceptive activity of RNE28. RNE28 did not induce respiratory depression, constipation, rewarding effects, or sedation at the analgesic doses tested.
Conclusion and Implications
This proof‐of‐concept study shows that TREK1 activators could constitute a novel class of painkillers, inspired by the mechanism of action of opioids but devoid of their adverse effects.
Keywords: analgesics, drug‐related side effects and adverse reactions, K2P, opioids, pain, TREK1
Abbreviations
- %MPE
percentage of the maximum possible effect
- KO
knockout
- NCBI
National Center for Biotechnology Information
- WT
wild‐type
What is already known
The μ opioid receptor drives both the antinociceptive and adverse effects of opioids.
TREK1 is activated downstream of μ receptor and contributes to the antinociceptive effect of morphine.
What this study adds
Direct pharmacological activation of TREK1 induces antinociception without opioid adverse effects.
What is the clinical significance
TREK1 activators could constitute a new class of analgesic drugs with improved risk–benefit ratio.
1. INTRODUCTION
Although opioids are reference analgesics, particularly for nociceptive pain, their risk–benefit ratio is not optimal because of frequent and potentially serious adverse effects. The risk of dependence, for example, has led to the opioid crisis in some western countries, with a rise in the misuse of prescription opioids and deaths by overdose. Dependence on opioids has colossal repercussions both for public health in general and for the economy (Volkow & Collins, 2017). Unfortunately, the pharmacopoeia of analgesics is scarce and no other class of painkiller available on the market can be substituted for opioids, which remain the therapy of reference, particularly for nociceptive pain.
From a pharmacological point of view, the main obstacle to improving this situation lies in the fact that the analgesic and adverse effects (constipation, nausea, vomiting, sedation, respiratory depression,and dependence) of opioids are due to the activation of the same μ opioid receptor whose ubiquitous location is the origin of the wide diversity of the effects of opioids (Marrone et al., 2017; Matthes et al., 1996). Several pharmacological strategies have been developed with the aim of preserving the analgesic activity of opioids while reducing their adverse effects. The design of biased agonists is undoubtedly the most advanced initiative with products in clinical trials that seem to provide a satisfactory analgesic effect and to induce fewer adverse effects than opioids. However, respiratory and digestive functions still seem to be affected (DeWire et al., 2013; Manglik et al., 2016) and these molecules might retain abuse potential through G‐protein signalling (Negus & Freeman, 2018). The only biased opioid tested in humans is oliceridine (TRV130), which completed phase 3 clinical trials. In October 2018, however, the US Food and Drug Administration decided not to grant it approval owning to doubts whether the benefits associated with the drug outweighed the risks (Mores, Cummins, Cassell, & van Rijn, 2019).
Against this background, we initiated a different strategy involving direct activation of an analgesic effector protein downstream of the μ receptor. The strategy is based on the finding that TREK1 potassium channels, activated downstream of the μ receptor, play an important part in the antinociceptive effect of morphine and fentanyl without being involved in opioid adverse effects (Devilliers et al., 2013). TREK1 is a background two‐pore potassium (K2P2.1) channel broadly expressed in humans in the peripheral nervous system and CNS (with high expression in the cerebellum and putamen) and, to a lesser extent, in other regions such as the heart, lungs, smooth muscle (myometrium), pancreas and prostate (Fink et al., 1996; Medhurst et al., 2001; Schwingshackl, Teng, Ghosh, & Waters, 2013; Wu, Singer, & Buxton, 2012). Its role in pain perception as well as in anaesthesia, neuroprotection and depression is well documented (Alloui et al., 2006; Heurteaux et al., 2004, 2006).
The involvement of TREK1 in the antinociceptive effect of opioids together with the role of this channel in polymodal nociceptors prompted us to develop TREK1 activators that could have an antinociceptive activity devoid of opioid‐related adverse effects (Rodrigues et al., 2014; Vivier et al., 2017). Here, we show that one of these molecules, (2E)‐2‐cyano‐3‐(furan‐3‐yl)prop‐2‐enoic acid (RNE28), is a specific TREK1‐activator with antinociceptive properties in naive mice and rodent pain models. The antinociceptive activity of RNE28 is strongly reduced by the pharmacological blockade of TREK1 and in TREK1 knockout (KO) animals. Most interestingly, RNE28 treatment does not induce constipation, respiratory depression, sedation or rewarding effects at the analgesic doses tested, which validates the concept of TREK1 activation as an alternative to opioids.
2. METHODS
2.1. Animals and models
Procedures were evaluated by a regional ethics committee (CEMEA Auvergne) before approval by the French Ministry of Research and Education (project TREKANALGESIA) under the European 2010/63/UE directive. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). We provide the Extended Methods Form recommended for uniformity and transparency in the reporting of animal studies (Rice et al., 2008) (Data S1).
Experiments were conducted on 20–25 g (5–8 weeks old) male C57Bl/6J mice (RRID:MGI:5650797) and male Sprague Dawley rats (RRID:MGI:5651135) weighting 150–175 g (about 4 weeks old, neuropathic pain model) or 175–200 g (about 5 weeks old, carrageenan pain model), purchased from Janvier Labs and kept under standard conditions (21–22°C, 12/12‐h light/dark cycle, 55% humidity). RNE28 is inspired by the mechanism of action of opioids and because sex differences have been observed in opioid analgesia (Craft, 2003), we did not include female animals in the study. Upon arrival, animals were given a week to acclimatize before any experimentation. Treatment groups were randomized according to the method of equal blocks and experimenters were blind to the treatment and genotype. TREK1−/− mice were generated by crossing mice carrying an allele of the Kcnk2 gene in which exon 3 was floxed (Heurteaux et al., 2006) (RRID:MGI:3050295) with ZP3‐Cre mice (de Vries et al., 2000) (RRID:MGI:3835429), both in C57Bl6/J background. TREK1+/− F2 offspring not carrying the ZP3‐Cre transgene were crossed to generate TREK1−/− and TREK1+/+ littermates, weaned in separate cages after genotyping using 2‐mm tail samples taken at 10 days of age. Identification was made by digital tattoos.
The post‐operative pain model was induced in the mice by a sterile 4‐mm incision of the skin, fascia and muscle in the midline of the left hind paw, from the base of the heel, before suture of the skin at two sites. The procedure was performed under 3% isoflurane anaesthesia and pain thresholds were measured 24 and 48 h post‐surgery. The chronic constriction injury (CCI) model was induced in rats as previously described (Chaumette et al., 2018) and consists in tying four loose ligatures around the common sciatic nerve under pentobarbital anaesthesia so that the epineurial blood flow is decreased but not interrupted.
2.2. Materials
RNE28 (see Supplementary Methods section in the supporting information for synthesis), spadin (Tocris Bioscience), duloxetine (Eli Lily), morphine chlorhydrate (Coopération Pharmaceutique Française), gabapentin (LeanCare), riluzole (Tocris Bioscience) and λ‐carrageenan (Sigma‐Aldrich) were dissolved in 0.9% NaCl on the day of use, or the day before with overnight stirring for λ‐carrageenan and delivered by oral gavage (p.o.), subcutaneous injection (s.c.) or intraperitoneal injection (i.p.) in a volume of 10 ml·kg−1 (mice) or 2 ml·kg−1 (rats). Intracerebroventricular (i.c.v.), intrathecal (i.t.) and intraplantar (i.pl.) injections in volumes of 2, 5 and 20 μl, respectively, were given to mice under brief 2% isoflurane anaesthesia for the first two routes. The intraplantar injection volume in rats was 200 μl. Positive controls were used at the following doses: 5 mg·kg−1 morphine in rats for analgesia (Tsutsui, Wood, & Craft, 2011), 3 mg·kg−1 morphine in mice for analgesia and constipation (Devilliers et al., 2013), 50 mg·kg−1 morphine in mice for respiratory depression (Devilliers et al., 2013), 15 mg·kg−1 morphine in mice for place preference conditioning (Francès, Smirnova, Leriche, & Sokoloff, 2004), 10 mg·kg−1 duloxetine for screening tests of antidepressant drugs (Zomkowski, Engel, Cunha, Gabilan, & Rodrigues, 2012) and 100 mg·kg−1 gabapentin in rats for neuropathic pain relief (Urban et al., 2005).
2.3. Evaluation of pain thresholds
Thermal pain thresholds were assessed in mice by immersion of the left hind paw up to the ankle in a 46°C water bath until shaking or withdrawal of the paw was observed (Lolignier et al., 2011). Animals were manually restrained in a dedicated piece of fabric with only one paw and the tail out. To minimize stress, they were habituated to the test twice a day for 2 weeks prior to testing, with the water bath temperature set at 28°C. The first two latencies measured at no more than a second apart were averaged and assigned as the pain threshold. A cut‐off time of 30 s was applied. Percentages of the maximum possible effect (%MPE) were calculated as follows: 100 * (threshold − baseline)/(cut‐off − baseline).
Mechanical pain thresholds were assessed by applying von Frey filaments (Bioseb) calibrated from 0.02 to 1.40 g perpendicularly to the plantar surface of the left hind paw using the up‐down method (Chaplan, Bach, Pogrel, Chung, & Yaksh, 1994). Results were expressed as the calculated force required to trigger a paw withdrawal 50% of the time. Animals were habituated to the von Frey chambers for 1 h twice the day before the experiment and 1 h immediately prior to the experiment. In rats, mechanical pain thresholds were assessed by the paw pressure test on a Randall–Selitto apparatus (Takesue, Schaefer, & Jukniewicz, 1969) (Ugo Basil, probe tip diameter of 1 mm). The test consists in applying an increasing force to the left hind paw until withdrawal or vocalization. Pain thresholds were the average of two consecutive measurements. The cut‐off was set at 450 g.
2.4. Predictive tests for antidepressant drugs
We assessed the antidepressant‐like and prodepressant‐like effects of drugs with two tests used for the screening of antidepressant drugs: the forced swimming test and the tail suspension test (TST) (Porsolt, Bertin, & Jalfre, 1977; Steru, Chermat, Thierry, & Simon, 1985). The forced swimming test consisted in measuring the immobility time of mice forced to swim for 6 min in a 13‐cm‐diameter glass beaker filled with water at 22 ± 2°C. The tail suspension test consisted in measuring the immobility time of mice taped by the tail to a string, the head pointing down 15 to 20 cm above the bench. Drugs were administered 30 min prior to each test.
2.5. Conditioned place preference
Drug rewarding effects were assessed with an unbiased conditioned place preference protocol (Hajasova, Canestrelli, Acher, Noble, & Marie, 2018; Tzschentke, 2007). The conditioned place preference apparatus (Imetronic) is made up of four identical boxes each of which has two lateral chambers (15 × 15 × 20 cm) connected by a central alley (5 × 15 × 20 cm) and two sliding doors to separate the alley from the chambers. In each chamber, two Plexiglas prisms with triangular bases (5 × 7 × 19 cm) were arranged to form different patterns and to cover the same surface of the chamber. They were used, along with two different types of embossed Plexiglas floors (gridded or striped patterns), as conditioning stimuli. The protocol was performed in three phases. (1) Preconditioning phase during which drug‐naive animals had free access to both chambers for 20 min and the time spent in each chamber was recorded. (2) Conditioning phase lasting 4 days during which the conditioning chambers were closed. In the morning of the first conditioning day, mice received the vehicle and were placed individually in one of the conditioning environments for 20 min. In the afternoon, they were given the drug in the opposite compartment. This sequence was alternated over the next 3 days. (3) Test phase which took place 1 day after the final conditioning session and was carried out similarly to the preconditioning phase. Conditioned place preference scores (in seconds) were calculated as the difference between the time spent in the drug‐paired compartment during the test phase and the time spent in the same compartment during the preconditioning phase.
2.6. Sedation
Sedation was assessed in animals by locomotor activity recordings using an IR actimeter and by motor coordination using the rotarod test (Bourin, Hascoet, Mansouri, Colombel, & Bradwejn, 1992). The IR actimeter (Apelex) is a set of 26 × 21 × 10‐cm dark boxes crossed by two IR beams originating perpendicularly from the centre of two adjacent walls at a height of 1 cm. Thirty minutes after drug administration, mice were placed in the boxes with no prior habituation and the number of beam interruption was counted over 10 min. For the rotarod test (Bioseb), animals were first habituated to stay on the motionless rod for 10 min followed by another 10 min at a rotational speed of 4 rpm. Animals received the drug immediately after the training and the test was performed 30 min later. The test consists in measuring the latency to fall of animals as the rotation increases from 4 to 40 rpm linearly over 5 min. The two closest values of three trials were averaged.
2.7. Whole‐body plethysmography
Respiratory rate was assessed in conscious mice by whole‐body plethysmography (Devilliers et al., 2013). Animals were placed in the chamber (Buxco, unrestrained whole‐body plethysmograph) 15 min before drug administration and their respiratory rate was subsequently measured continuously for 240 min.
2.8. Gastrointestinal transit
Gastrointestinal transit was firstly monitored by oral gavage with 10% methylene blue in a volume of 20 ml·kg−1, 30 min after drug administration (Devilliers et al., 2013). Fifteen minutes after methylene blue gavage, animals were killed and the small intestine was removed to measure the ratio of the length travelled by the dye over the total length of the small intestine, from the stomach to the caecum. To evaluate colonic transit, we used the bead expulsion test (Raffa, Mathiasen, & Jacoby, 1987). After 20‐min habituation to the cage, a 3‐mm bead was inserted into the distal colon with a catheter 2 cm away from the anus. The time to expulsion of the bead was measured before injection of the drug. The test was performed again 30 min after drug administration and the result is given baseline‐subtracted.
2.9. Constructs
Human TREK1 (Moha ou Maati et al., 2011) (KCNK2, National Center for Biotechnology Information [NCBI] RefSeq NM_014217), TREK2 (Lesage, Maingret, & Lazdunski, 2000) (KCNK10, GenBank: BC069462.1), and TRAAK (Lesage, Terrenoire, Romey, & Lazdunski, 2000) (KCNK4, NCBI RefSeq NM_033310.3) and chimeras were cloned into pIRES2‐eGFP vector (Clontech). All the chimeras were obtained by overlapping PCR and inserted into the same vector. All the constructs were verified by DNA sequencing.
2.10. Cell culture and transfection
HEK‐293 cells (RRID:CVCL_0045) were grown in DMEM (Gibco, Life Technologies) supplemented with 10% fetal calf serum (Hyclone, Thermo Fisher Scientific GMBH) and 1% penicillin–streptomycin (Gibco, Life Technologies) in a humidified incubator at 37°C, 5% CO2. For electrophysiology, cells were plated on 35‐mm dishes at a density of 30,000 cells per dish, transfected 2 h after plating with 0.8‐μg DNA per dish using JET PEI according to the manufacturer's instructions (Polyplus transfection). Experiments were performed over the following 1–2 days.
2.11. Electrophysiology
For whole‐cell patch‐clamp recordings, dishes were continuously perfused with control bath solution containing (in mM) 140 NaCl, 10 tetraethylammonium‐Cl, 5 KCl, 3 MgCl2, 1 CaCl2 and 10 HEPES and adjusted to pH 7.4 with NaOH. The pipette solution contained (in mM) 155 KCl, 3 MgCl2, 5 EGTA and 10 HEPES and was adjusted to pH 7.2 with KOH. Currents were recorded with a RK 400 patch‐clamp amplifier (Bio‐Logic Science Instruments), low‐pass filtered at 3 kHz and digitized at 10 kHz with a Digidata‐1322 (Axon Instrument). Clampex 8.2 software was used for current recordings and voltage stimulations. Patch pipettes were double step‐pulled and had resistances of 2–4 MΩ. Whole‐cell patch‐clamp configuration was obtained at a holding potential of −80 mV. Voltage ramp protocols consisted of a step to −100 mV (20 ms in duration) followed by a ramp from −100 to +60 mV over 400 ms, applied every 5 s. Currents were recorded during control bath solution perfusion and during perfusion of bath solution supplemented with RNE28.
2.12. Data and statistical analysis
Data were analysed with GraphPad Prism 6.07 (RRID:SCR_002798). Outliers were removed by the ROUT method with Q = 0.1%. Statistical significance was tested by non‐parametric tests (Mann–Whitney for comparing two groups and Kruskal–Wallis for comparing three groups or more) and by parametric tests when all groups passed the D'Agostino and Pearson omnibus normality test (Student's t‐test for comparing two groups and one‐way ANOVA for comparing three groups or more). Two‐way ANOVA was used for comparing two groups or more with two varying factors. Post hoc tests (Sidak's, Dunnett's, or Dunn's tests) were ran in accordance with the experimental design and are indicated in the figures legends. Median effective doses (ED50) and concentrations (EC50) were calculated following least square fit of the data with sigmoidal four‐parameter dose–response curve and ED50 and Emax were compared between groups with the extra sum‐of‐squares F test. All the data are presented as mean ± SEM, except for non‐linear regression results (Emax, ED50 and EC50), presented as mean ± SD. Significance threshold was * P < 0.05. Details of the statistical analyses (n numbers, outliers, normal distribution, tests and post hoc tests) and their results (F and P values for each factor and their interaction) are given in Table S1. Statistical analysis was undertaken only for studies in which each group size was at least n = 5 and n numbers account for independent values only, not technical replicates. Group sizes were set at the minimum to give reproducible and statistically significant differences between positive and negative control groups. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. RNE28 is a TREK1 activator with antinociceptive activity in naive mice and rodent pain models
Screening of a series of previously synthetized compounds (Vivier et al., 2017) enabled us to identify RNE28 as an activator of the human TREK1 channel. In patch‐clamp experiments, TREK1 current at +60 mV was increased with an Emax of 4.46‐fold and an EC50 of 37.37 μM (Figure 1a). At 100‐μM RNE28, TREK1 activation reached its maximum with a current increase of 4.54‐fold. In contrast, TREK2 and TRAAK currents were only increased by 1.78‐fold and 1.38‐fold, respectively. RNE28 binding to 81 different receptors, channels and transporters was also assessed with no positive hit (Figure S1). We consequently aimed to assess whether RNE28, which has an interesting selectivity for TREK1 over the evolutionary‐related TREK2 and TRAAK channels, could have an antinociceptive activity. Oral administration of RNE28 in naive mice induced a dose‐dependent antinociceptive effect peaking at 30 and 15 min and lasting for 30 and 45 min at 30 and 60 mg·kg−1, respectively (Figure 1b). A low dose of 15 mg·kg−1 had no effect on the pain thresholds of naive mice. However, in animals undergoing carrageenan‐induced paw inflammation, a reduction in pain thresholds was observed from 15 mg·kg−1 (Figure 1c) and the percentage of the maximum possible effect (%MPE) achieved at 30 and 60 mg·kg−1 was higher than in naive animals. At these two doses, the peak of antinociception was observed 30 min after gavage and the antinociceptive effect lasted for 45 to 60 min. Plotting and fitting of the %MPE values obtained for the three RNE28 doses in both naive and inflamed mice at 30 min post‐administration resulted in two separate dose–effect curves (Figure 1d). While ED50 were not different between naive and carrageenan‐injected mice (26.47 ± 3.72 and 28.40 ± 5.65 mg·kg−1, respectively), Emax was higher in inflamed animals (77.50 ± 7.10%) than in naive animals (46.55 ± 8.28%). Compared to morphine, RNE28 was 10 to 5 times less potent in naive animals and in the carrageenan model, respectively (ED50 of morphine: 2.96 ± 0.13 and 6.09 ± 0.14 mg·kg−1, respectively, Figure S2), with a very similar maximum effect (77.60 ± 6.13% and 84.16 ± 3.90%, respectively). In a different post‐operative pain model induced in mice by subplantar incision, we found that the 15 mg·kg−1 dose of RNE28 was also able to reduce hyperalgesia in response to both thermal (heat) and mechanical stimulation modalities (Figure 2a,b). The 30 mg·kg−1 RNE28 dose had an effect of 57% of that of 3 mg·kg−1 morphine for heat‐triggered hypersensitivity, considering AUC for the whole 90‐min time course and 77% for mechanical hypersensitivity. We then assessed the effect of RNE28 on mechanical pain thresholds in rats, in which we had first estimated the plasma half‐life of RNE28 to be 0.2–0.3 h (Figure S3). We used two pain models of different aetiologies: the inflammatory pain model induced by intraplantar carrageenan injection and a neuropathic pain model induced by loose ligation of the sciatic nerve (CCI model). In a rat model of inflammatory pain, hyperalgesia was reduced by RNE28 at 100 and 200 mg·kg−1, with the latter inducing an antihyperalgesic effect similar to that of 5 mg·kg−1 morphine (AUC of 7,636 ± 955 and 7,960 ± 733, respectively, Figure 2c). In neuropathic animals, both 100 and 200 mg·kg−1 doses of RNE28 had an antihyperalgesic activity, with 200 mg·kg−1 RNE28 being as effective in relieving hypersensitivity as 100 mg·kg−1 of gabapentin (AUC of 4,573 ± 934 and 4,519 ± 1,440, respectively), the reference molecule in the treatment of neuropathic pain, a condition particularly resistant to conventional painkillers including opioids. It is noteworthy that both gabapentin and RNE28 only partially reversed hypersensitivity in neuropathic rats, whereas an increase in pain threshold up to baseline values was observed in inflamed rats given morphine or RNE28.
FIGURE 1.
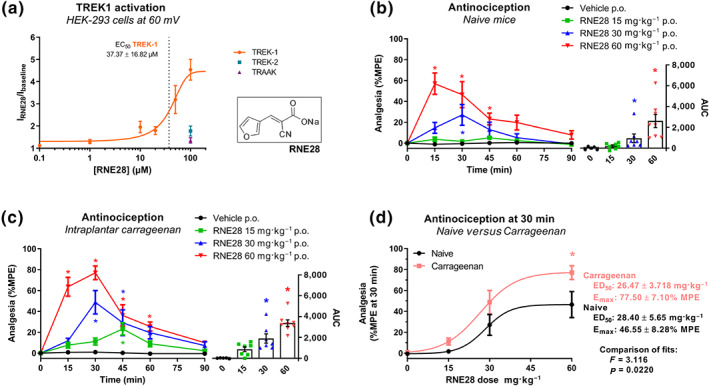
Activation of TREK1 by RNE28 and its antinociceptive activity in mice. (a) Currents recorded at +60 mV in HEK‐293 cells transfected with the human TREK1 channel in response to 0.1 (n = 6), 1 (n = 6), 10 (n = 5), 20 (n = 6), 50 (n = 7), 100 (n = 7) and 200 (n = 5) μM of RNE28, expressed as the ratio of currents recorded after and before RNE28 exposure. TREK1 dose–effect curve was generated by the least square method. Responses to 100‐μM RNE28 of cells transfected with the human TREK2 (n = 10) and TRAAK (n = 5) channels are indicated. The insert shows RNE28 skeletal formula. (b) Left, time course of the antinociceptive effect induced by 0 (n = 5), 15 (n = 8), 30 (n = 7) and 60 (n = 8) mg·kg−1 RNE28 administered orally in naive mice. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (c) Left, time course of the antinociceptive effect induced by 0 (n = 5), 15 (n = 7), 30 (n = 7) and 60 (n = 8) mg·kg−1 RNE28 administered orally in mice 3.5 h following paw inflammation induction by intraplantar carrageenan injection (20 μl, 3%). Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (d) Dose–effect curves of the antinociceptive activity of RNE28 30 min following oral gavage in naive (a) and carrageenan‐injected (b) mice. Datasets were fitted with the least square method and fits were compared with the extra sum‐of‐square F test. ED50, Emax, and P and F values are shown on the right. * P < 0.05 versus vehicle (b, c) or naive (d). Two‐way ANOVA followed by Dunnett's post hoc test (b, c left), Kruskal–Wallis followed by Dunn's post hoc test (b, c right) and two‐way ANOVA followed by Sidak's post hoc test (d). n numbers represent cells (a) or mice (b–d)
FIGURE 2.
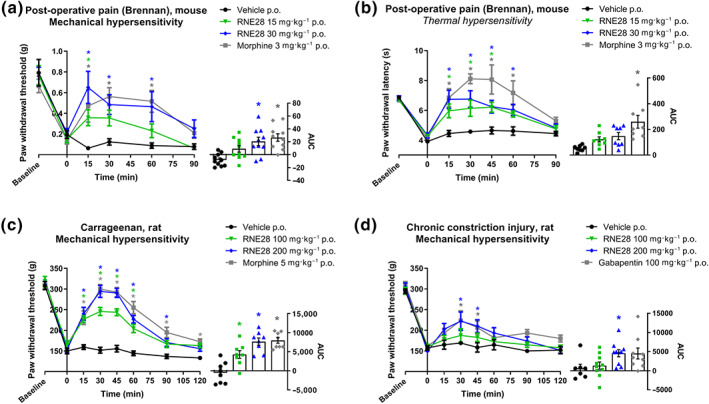
RNE28 effect on thermal and mechanical pain thresholds in inflammatory and neuropathic rodent pain models. (a) Left, time course of the antinociceptive effect induced by 3 mg·kg−1 morphine (n = 8) or 0 (n = 8), 15 (n = 8) and 30 (n = 8) mg·kg−1 RNE28 administered orally in mice 24 h following post‐operative pain induction by paw incision and suture. Pain thresholds were evaluated by the von Frey test. Right, AUCs calculated from 0 to 90 min post‐administration. (b) Left, time course of the antinociceptive effect induced by 3 mg·kg−1 morphine (n = 10) or 0 (n = 10), 15 (n = 10) or 30 (n = 10) mg·kg−1 RNE28 administered orally in mice 48 h following post‐operative pain induction by paw incision and suture. Pain thresholds were assessed by paw immersion in 46°C water. Right, AUCs calculated from 0 to 90 min post‐administration. (c) Left, time course of the antinociceptive effect induced by 5 mg·kg−1 morphine (n = 8) or 0 (n = 8), 100 (n = 8) or 200 (n = 8) mg·kg−1 RNE28 administered orally in rats 4 h following paw inflammation induction by intraplantar carrageenan injection (200 μl, 2%). Pain thresholds were assessed by the paw pressure test. Right, AUCs calculated from 0 to 120 min post‐administration. (d) Left, time course of the antinociceptive effect induced by 100 mg·kg−1 gabapentin (n = 7) or 0 (n = 9), 100 (n = 10) or 200 (n = 10) mg·kg−1 RNE28 administered orally in neuropathic rats 14 days following chronic constriction of the sciatic nerve. Pain thresholds were assessed by the paw pressure test. Right, AUCs calculated from 0 to 120 min post‐administration. * P < 0.05 versus vehicle. Two‐way ANOVA followed by Dunnett's post hoc test (a–d left) and Kruskal–Wallis followed by Dunnett's post hoc test (a–d right). n numbers represent mice (a, b) and rats (c, d)
3.2. The antinociceptive activity of RNE28 is mediated by TREK1
To assess the contribution of TREK1 channels to the antinociceptive effect of RNE28, we treated naive wild‐type (WT) and TREK1 KO mice with 30 mg·kg−1 RNE28 (ED50 RNE28 = 28.4 mg·kg−1, Figure 1d) and followed for 60 min the evolution of pain thresholds in response to paw immersion in 46°C water. As previously reported (Alloui et al., 2006), we observed that TREK1−/− mice have lower basal heat pain thresholds than WT animals (6.4 ± 0.3 s vs. 8.7 ± 0.3 s, respectively, P < 0.05, unpaired two‐tailed Student's t‐test). Thirty minutes after RNE28 administration, the %MPE score was 66.5 ± 18.8% in WT animals but was down to 25.1 ± 5.2% in TREK1−/− mice (Figure 3a). Over the 60‐min time course, the AUC of the antinociceptive effect of RNE28 was reduced by 45.4% in KO mice in comparison to WT animals. To further confirm the contribution of TREK1 to the antinociceptive effect of RNE28, we used pharmacological inactivation of TREK1 with spadin, a known selective blocker of the channel (Mazella et al., 2010) able to block the effect of RNE28 on TREK1 current in vitro (Figure S4). We administered a dose of 1 mg·kg−1 spadin because we found it was sufficient to reduce the antinociceptive activity of 3 mg·kg−1 morphine by 91.8% (Figure 3b). Similarly to what we observed in KO animals, systemic administration of spadin at 1 mg·kg−1 induced a reduction in the antinociceptive effect of 30 mg·kg−1 RNE28 (Figure 3c). At 30 min post‐injection, RNE28 had an antinociceptive effect of 6.07 ± 1.4% MPE when co‐administered with spadin compared to 15.8 ± 1.1% in animals given RNE28 alone. AUC of the antinociceptive effect over 90 min was also decreased by 58% with spadin co‐administration.
FIGURE 3.
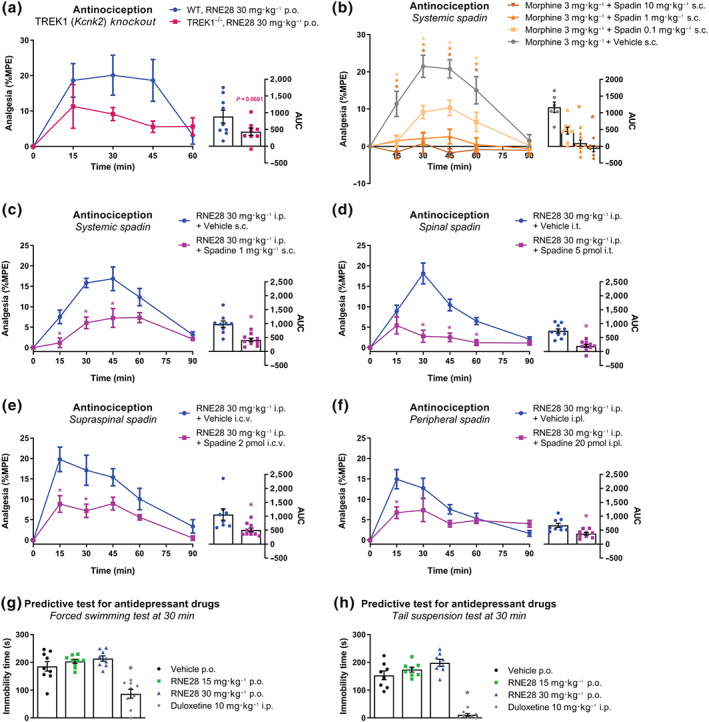
Contribution of TREK1 to RNE28‐induced antinociception in mice. (a) Left, time course of the antinociceptive effect induced by 30 mg·kg−1 RNE28 administered orally in WT (n = 9) and TREK1−/− (n = 8) mice. Spadin was injected 10 min before RNE28. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 60 min post‐administration. (b) Left, time course of the antinociceptive effect induced in mice by co‐treatment with 3 mg·kg−1 subcutaneous morphine and 0.1 (n = 9), 1 (n = 9) and 10 (n = 6) mg·kg−1 subcutaneous spadin or vehicle (n = 6). Spadin was injected 10 min before morphine. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (c) Left, time course of the antinociceptive effect induced in mice by co‐treatment with 30 mg·kg−1 intraperitoneal RNE28 and 1 mg·kg−1 subcutaneous spadin (n = 10) or vehicle (n = 9). Spadin was injected 10 min before RNE28. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (d) Left, time course of the antinociceptive effect induced in mice by co‐treatment with 30 mg·kg−1 intraperitoneal RNE28 and 5‐pmol intrathecal spadin (n = 10) or vehicle (n = 10). Spadin was injected 10 min before RNE28. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (e) Left, time course of the antinociceptive effect induced in mice by co‐treatment with 30 mg·kg−1 intraperitoneal RNE28 and 2‐pmol intracerebroventricular spadin (n = 10) or vehicle (n = 9). Spadin was injected 10 min before RNE28. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (f) Left, time course of the antinociceptive effect induced in mice by co‐treatment with 30 mg·kg−1 intraperitoneal RNE28 and 20‐pmol intraplantar spadin (n = 10) or vehicle (n = 9). Spadin was injected 10 min before RNE28. Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, AUCs calculated from 0 to 90 min post‐administration. (g) Immobility time of mice submitted to the forced swimming test 30 min after administration of 10 mg·kg−1 duloxetine (n = 9) or 0 (n = 9), 15 (n = 9) or 30 (n = 9) mg·kg−1 RNE28. (h) Immobility time of naive mice submitted to the tail suspension test 30 min following administration of 10 mg·kg−1 duloxetine (n = 8) or 0 (n = 8), 15 (n = 8), or 30 (n = 8) mg·kg−1 RNE28. * P < 0.05 versus WT (a) or vehicle (b–h). Two‐way ANOVA followed by Sidak's post hoc test (a–f left), unpaired two‐tailed Student's t‐test (a right, c, d right), unpaired two‐tailed Mann–Whitney test (e, f right), one‐way ANOVA followed by Dunnett's post hoc test (g) and Kruskal–Wallis followed by Dunn's post hoc test (b left, h). n numbers represent mice
To gain further insight into the site of action of RNE28, we administered spadin locally to block TREK1 at the periphery (intraplantar injection), in the spinal cord (intrathecal injection) or at the supraspinal level (intracerebroventricular injection), together with intraperitoneal injections of RNE28 at 30 mg·kg−1. Intrathecal injection of 10 ng (5 pmol) spadin resulted in a strong decrease in RNE28 antinociceptive activity, with a 72% AUC reduction in the spadin‐treated group and an antinociceptive effect at 30 min of 2.8 ± 1.5% versus 18.2 ± 2.6% MPE in animals given RNE28 alone (Figure 3d). Injection of 4 ng (2 pmol) spadin in the lateral ventricular space of the brain resulted in a reduction of the antinociceptive effect of RNE28 by 52% (AUC), with a maximum possible effect at 30 min dropping from 17.1 ± 3.7% to 7.2 ± 1.7% MPE (Figure 3e). Finally, peripheral blockade of TREK1 channels by intraplantar delivery of 40 ng (20 pmol) spadin was also effective in decreasing the antinociceptive activity of RNE28, albeit to a lesser extent. AUC was reduced by 23% in animals injected with spadin, whose pain thresholds at 30 min were 7.3 ± 2.8% MPE versus 12.8 ± 2.6 in the control group (Figure 3f).
Spadin has been described for its antidepressant effect which is characterized in mice by a decreased immobility time during predictive tests for antidepressant drugs such as the forced swimming test and the tail suspension test (Mazella et al., 2010). We therefore decided to look for an effect of RNE28 in the same tests. Naive mice were given RNE28 at 15 or 30 mg·kg−1 and were submitted to the forced swimming test or to the tail suspension test at 30 min post‐administration, which corresponds to the peak of RNE28 antinociceptive activity. In both tests, the average immobility time of mice did not differ between the three groups given RNE28 and the control group while the antidepressant duloxetine given at 10 mg·kg−1 had a strong effect (Figure 3g,h).
3.3. TREK1 activation by RNE28 requires the N‐terminal domain of the channel
RNE28 at 100 μM was observed to strongly activate the human TREK1 channel without activating the evolutionary‐related human TRAAK channel (Figure 1a) and so we generated seven different hTREK1/hTRAAK chimeric clones to determine which of the TREK1 domains was/were involved in the activation by RNE28 (Figure 4a). Clones were expressed in HEK‐293 cells and changes in whole‐cell current in response to 100‐μM RNE28 were explored by patch‐clamp. Voltage ramps from −100 to +60 mV were applied starting from a holding potential of −80 mV. We confirmed the activation of TREK1 (Figure 4b) but not that of TRAAK (Figure 4c) by 100‐μM RNE28. We then compared the average current produced by different chimeras at +60 mV (Figure 4d). Replacing the first (chimera 2), second (chimera 3), or both TRAAK extracellular loops (chimera 1) by those of TREK1 did not affect the sensitivity of the channel to RNE28 and nor did the replacement of the C‐terminal tail of the channel (chimera 5), even with the addition of the third and fourth transmembrane domains (chimera 7). However, we found that chimera 4, in which the N‐terminal domain of TRAAK was replaced by that of TREK1, was activated by RNE28 (Figure 4e). The increase in current at +60 mV produced by RNE28 in cells transfected with chimera 4 was 73%. In chimera 6, carrying a TREK1 sequence including the N‐terminus and the first two transmembrane domains, the effect of 100‐μM RNE28 was more pronounced, with an increase in current of 130% (Figure 4f).
FIGURE 4.
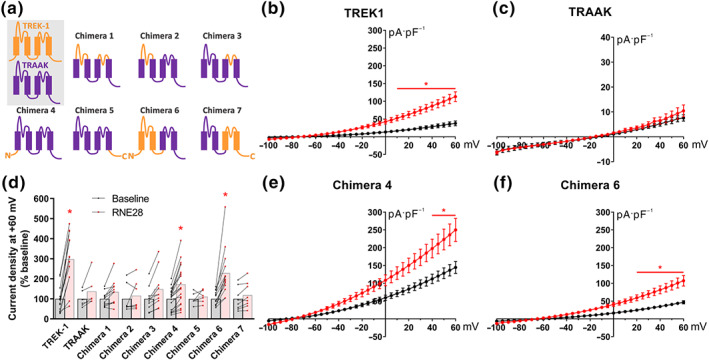
Responses of TREK1/TRAAK chimeras to RNE28. (a) Schematic description of human TREK1 (orange), TRAAK (purple) and TREK1/TRAAK chimeras constructions. (b, c) Current–voltage curves obtained from HEK‐293 cells transfected with TREK1 (b) or TRAAK (c) clones in response to a voltage ramp protocol from −100 to +60 mV before (black) and after (red) application of 100‐μM RNE28. (d) Relative current increase at +60 mV in presence of 100‐μM RNE28 (red), expressed as percentage of the baseline recorded with the control perfusion (black). (e, f) Current–voltage curves obtained from HEK‐293 cells transfected with chimera 4 (e) or chimera 6 (f) clones in response to a voltage ramp protocol from −100 to +60 mV before (black) and after (red) application of 100‐μM RNE28. * P < 0.05 versus baseline. Two‐way ANOVA followed by Sidak's post hoc test (b–f left). n = 13 TREK1, n = 6 TRAAK, n = 11 chimera 1, n = 7 chimera 2, n = 10 chimera 3, n = 20 chimera 4, n = 6 chimera 5, n = 14 chimera 6 and n = 8 chimera 7. n numbers represent cells
3.4. RNE28 does not induce opioid‐related adverse effects
The rationale behind the development of TREK1 activators with an analgesic perspective resides in part in the fact that morphine loses its antinociceptive activity in TREK1‐deficient mice while opioid‐related adverse effects are unaffected by the absence of functional TREK1 channels (Devilliers et al., 2013). TREK1 is therefore only involved in the antinociceptive effect of morphine and its activation could result in antinociception without the induction of opioid‐related adverse effects. Thus, we investigated for constipation, sedation, respiratory depression and rewarding effects in mice treated with the same 15, 30 and 60 mg·kg−1 doses given orally.
We first assessed the gastrointestinal transit of RNE28‐treated mice by two different tests, with the aim of quantifying transit in the colon as well as in the stomach and small intestine. First, we monitored the expulsion time of a 3‐mm bead inserted into the distal colon of mice, 2 cm from the anus (Figure 5a). While the latency to bead expulsion was increased 30 min after gavage with 3 mg·kg−1 morphine, RNE28 up to 60 mg·kg−1 had no effect on colonic transit. We then administered methylene blue orally to mice 30 min after 3 mg·kg−1 morphine or RNE28 at 15, 30 and 60 mg·kg−1. Fifteen minutes after gavage with methylene blue, the dye had covered 61 ± 3% of the stomach–caecum distance in vehicle‐treated animals (Figure 5b). In animals given morphine, the dye progression was only 36 ± 4% while in RNE28‐treated mice, no difference in methylene blue transit was observed in comparison to the control group.
FIGURE 5.
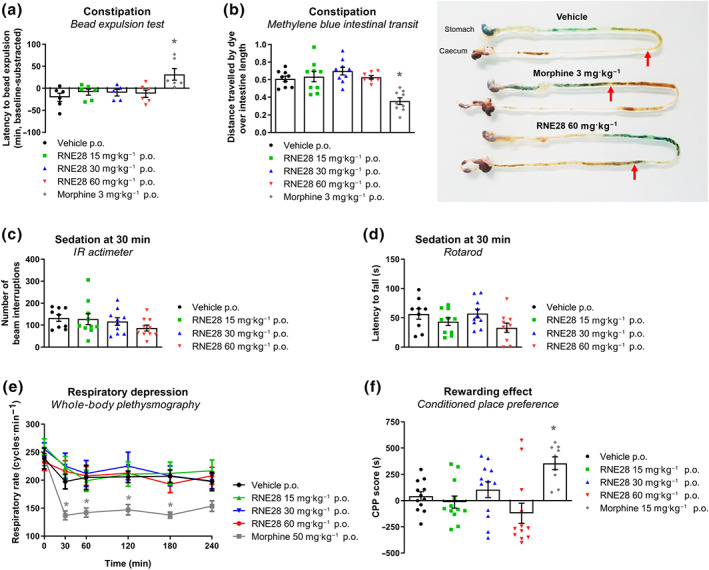
Opioid‐related adverse effects monitoring in RNE28‐treated mice. (a) Latency to expulsion of a 3‐mm‐diameter bead inserted intrarectally in mice 30 min after oral gavage with 3 mg·kg−1 morphine (n = 6 mice) or 0 (n = 6), 15 (n = 6), 30 (n = 5) or 60 (n = 6) mg·kg−1 RNE28. Results are shown in minutes as differences between post‐gavage and baseline latencies (baseline‐subtracted). (b) Left, intestinal progression of methylene blue administered by oral gavage in mice 30 min after oral gavage with 3 mg·kg−1 morphine (n = 9 mice) or 0 (n = 9), 15 (n = 9), 30 (n = 9) or 60 (n = 8) mg·kg−1 RNE28. Mice were killed 15 min after methylene blue gavage. Results are expressed as percentage of the stomach to caecum distance covered by methylene blue. Right, representative intestines of mice treated with the vehicle, 3 mg·kg−1 morphine or 60 mg·kg−1 RNE28. Red arrows indicate the distance travelled by the dye. (c) Locomotor activity of mice, monitored by IR actimetry 30 min after oral gavage with 0 (n = 9), 15 (n = 10), 30 (n = 10), or 60 (n = 10) mg·kg−1 RNE28. (d) Motor coordination of mice, assessed by the rotarod test 30 min after oral gavage with 0 (n = 9), 15 (n = 10), 30 (n = 10) or 60 (n = 10) mg·kg−1 RNE28. (e) Respiratory rate of conscious mice monitored by whole‐body plethysmography for 240 min after oral gavage of 50 mg·kg−1 morphine or 0, 15, 30, or 60 mg·kg−1 RNE28 (n = 6 mice per group). (f) Place preference of mice for the drug compartment after 4 days of conditioning consisting in daily oral gavage with the vehicle in the control compartment and 15 mg·kg−1 morphine (n = 9 mice) or 0 (n = 12), 15 (n = 12), 30 (n = 12) or 60 (n = 12) mg·kg−1 RNE28 in the drug compartment. * P < 0.05 versus vehicle. Two‐way ANOVA followed by Dunnett's post hoc test (a), one‐way ANOVA followed by Dunnett's post hoc test (b, d, g) and Kruskal–Wallis followed by Dunn's post hoc test (c, f). n numbers represent mice
RNE28‐induced sedation was assessed by locomotor activity recordings (IR actimeter, Figure 5c) and motor coordination (rotarod test, Figure 5d) in mice following RNE28 administration. Thirty minutes after oral gavage with RNE28 up to 60 mg·kg−1, no impairment of motor function was observed in either test.
The respiratory rate of mice was also followed over 240 min after oral gavage of RNE28 by whole‐body plethysmography (Figure 5e). While 50 mg·kg−1 morphine induced a strong respiratory depression, characterized by a reduction of the respiratory rate in mice lasting for 180 min, the respiratory rate of mice given 15, 30 or 60 mg·kg−1 RNE28 was undistinguishable from that of mice treated with the vehicle.
We also treated animals twice daily with 30 mg·kg−1 RNE28 for 9 days to look for the development of a tolerance to the analgesic activity of RNE28. We observed a non‐significant reduction in the analgesic effect of RNE28, suggesting that tolerance to the product could develop over time (Figure S5).
Finally, using the conditioned place preference test, we assessed whether RNE28 could induce rewarding effects (Figure 5f). After 4 days of conditioning, during which mice received a daily administration of the drug in one compartment and of vehicle in the other one, animals were allowed to explore both compartments freely. Mice treated daily with 10 mg·kg−1 morphine developed conditioned place preference, characterized by an increase in the conditioned place preference score (time spent in the drug‐paired compartment minus the time spent in the same compartment before the conditioning phase) compared to that of the vehicle‐treated group. In RNE28‐treated animals, no preference for or aversion to the drug‐paired compartment was observed, whatever the dose. In parallel, we tried to induce naloxone‐precipitated withdrawal in mice given RNE28 twice a day for 9 days and observed no jumping behaviour, which is characteristic of opioid withdrawal (Figure S6).
4. DISCUSSION
This study provides a proof‐of‐concept of the antinociceptive activity and safety of selective TREK1 pharmacological activation in mice using the newly developed compound RNE28. RNE28 activates the human TREK1 channel with an EC50 of 37.4 μM, which is higher than that of other known TREK1 activators such as BL‐1249 (Pope et al., 2018) (EC50 = 5.5 μM), ML335 (Lolicato et al., 2017) (EC50 = 5.2 μM), ML402 (Lolicato et al., 2017) (EC50 = 5.9 μM), ML67‐33 (Bagriantsev et al., 2013) (EC50 = 9.7 μM) and GI‐530159 (Loucif et al., 2018) (EC50 = 0.9 μM). However, these compounds are all described as dual TREK1/TREK2 activators, in addition to being TRAAK activator for ML67‐33. RNE28 is the first compound shown to activate TREK1 specifically among TREK1, TREK2 and TRAAK channels. No data regarding the antinociceptive effect of BL‐1249, ML335, ML402, ML67‐33 and GI‐530159 have yet been generated, but we show that activation of TREK1 alone by RNE28 is able to produce an antinociceptive effect in naive animals. RNE28 was even more effective in relieving hypersensitivity in an animal model of inflammatory pain, as shown by the increased maximum possible effect and the lower minimum effective dose in mice after injection of carrageenan into the paw. In this model, RNE28 increased mechanical pain thresholds and totally reversed thermal hyperalgesia. In these two conditions of nociceptive pain, RNE28 was 5 to 10 times less potent than morphine, depending on the model. In a traumatic model of neuropathic pain, a condition that, because it is generally resistant to opioids, is best treated by antidepressants or antiepileptic drugs, RNE28 also decreased hypersensitivity, but in a less marked way since neuropathic rats only partially recovered pre‐injury pain thresholds. The antinociceptive profile of RNE28 is therefore similar to that of morphine, whose antinociceptive activity was shown to rely on TREK1 channels (Devilliers et al., 2013). The contribution of these channels to the antinociceptive effect of RNE28 is confirmed by the marked decrease in its antinociceptive effect in TREK1 KO mice or in WT animals co‐treated with the TREK1 blocker spadin. Spadin was able to reduce the antinociceptive effect of RNE28 at the peripheral, spinal and supraspinal levels, showing that TREK1 is a signal modulator at different levels of the pain pathway on which RNE28 can have an effect. This observation is in accordance with TREK1 broad expression, observed in peptidergic and non‐peptidergic nociceptive peripheral sensory neurons (Alloui et al., 2006), in the spinal cord (with a strong lamina II staining) and in several brain regions such as cerebral cortexes, hippocampus, thalamus, basal ganglia and periaqueductal grey (Hervieu et al., 2001). Given the dual central/peripheral effect of RNE28, future efforts for pharmacological improvement of this molecule should not only focus on increased affinity for TREK1 but should also promote blood–brain barrier crossing, especially as we have observed no supraspinal adverse effect with RNE28 treatment. The interest of pharmacological activation of TREK1 as an analgesic strategy has been finally further reinforced by a recent study (Poupon et al., 2018) in which we showed that the non‐specific and transient TREK1 activator riluzole was effective in relieving hypersensitivity in a peripheral neuropathic pain model induced by the antineoplastic agent oxaliplatin. In this study, spadin treatment was able to suppress the antinociceptive effect of an acute or chronic riluzole treatment and riluzole was further shown to be ineffective in reducing pain in TREK1 KO mice. We have conducted similar experiments in the carrageenan‐induced plantar inflammatory pain model, which showed that riluzole also reduces hypersensitivity in inflamed animals (Figure S7). The antinociceptive effect of riluzole was absent in TREK1 KO animals but still present in TREK2 KOs, supporting again the potential of TREK1 pharmacological activation as an analgesic strategy.
Our data show that the N‐terminus domain of TREK1 is necessary and sufficient for RNE28 response. This is another difference with known TREK activators. GI‐530159 was shown to be active on a TREK1 clone lacking the N‐terminus (Loucif et al., 2018). ML67‐33 action requires the C‐type gate near the selectivity filter of the channel (Bagriantsev et al., 2013) and a cryptic binding site for ML335 and ML402 has been identified behind the selectivity filter at the interface between P1 and M4 segments, which allow these compounds to activate the C‐type gate (Lolicato et al., 2017; Şterbuleac, 2019). BL‐1249 also stimulates the C‐type gate but binds to a different site. While the M2/M3 intracellular region of the channel is involved in the selectivity of BL‐1249 for the two TREK channels and the cytoplasmic C‐terminal region is important for the response of TREK1 to the molecule (Pope et al., 2018), key residues for BL‐1249 response were identified in M2 and M4 helices (Schewe et al., 2019). The C‐terminus of TREK1 is responsible for the polymodal activation of the channel by proton, lipids, membrane stretch, temperature, or phosphorylation signals by PKA and PKC, through its crosstalk with the C‐type gate (Bagriantsev et al., 2013; Chemin et al., 2005; Honoré, Maingret, Lazdunski, & Patel, 2002; Patel et al., 1998). Involvement in activation by RNE28 of the N‐terminal of the channel, a region of poor homology with TREK2, could account for the selectivity of RNE28 for TREK1, whether the gating mechanism is direct or indirect. Of note, a TREK2 activator with good selectivity for TREK2 versus TREK1, pranlukast, was also found to bind a different site to that of BL‐1249 and ML335/ML402 (Wright et al., 2019).
We previously showed that TREK1 channels are activated by morphine, downstream of the μ receptor activation and are involved in its antinociceptive effects but not in its adverse effect (Devilliers et al., 2013) and so we speculated that TREK1 activators might be devoid of opioid‐like adverse effects. Hence, we looked for opioid‐induced adverse effects in RNE28‐treated mice. RNE28 produced robust antinociception from 30 mg·kg−1 in naive mice and 15 mg·kg−1 in inflamed mice, so we studied the occurrence of these adverse effects at doses ranging from 15 to 60 mg·kg−1. At none of these doses did we observe an effect of the product on respiratory rate or gastrointestinal transit. RNE28 was not sedative and did not have rewarding effects, whatever the dose tested. This is particularly important given the current rise in prescription opioids misuse. RNE28 also had no effect on predictive tests for antidepressant drugs in naive animals. This observation is in agreement with a previous finding that riluzole has no prodepressant effects in mice and is even able to reverse the depressive phenotype of WT but not TREK1 KO mice with chronic neuropathic pain induced by oxaliplatin (Poupon et al., 2018). This is an interesting result because TREK1 is blocked by fluoxetine, a serotonin‐specific recapture inhibitor antidepressant which loses its activity in TREK1 KO mice (Heurteaux et al., 2006; Kennard et al., 2005), while the TREK1‐selective blocker spadin, along with shorter analogues, has an antidepressant effect in WT mice (Djillani et al., 2017; Mazella et al., 2010). The mechanism by which TREK1 activation produces antinociception without depression, while TREK1 inhibition has an antidepressant effect without inducing pain hypersensitivity (Moha ou Maati et al., 2012), remains to be elucidated. Note that this study was performed on male mice and rats and these findings cannot be assumed to apply to females.
Dependence on prescription opioids, together with their unsatisfactory safety profile, has prompted the development of different new compounds with an improved risk–benefit ratio. Successful strategies, such as biased μ agonists (DeWire et al., 2013), activators of specific μ splice variants (Majumdar et al., 2011), a pH‐dependent μ agonist (Spahn et al., 2017) and a dual nociceptin/μ agonist (Ding et al., 2018), have been developed. The originality of our concept resides in its independence from the μ opioid receptor, since RNE28 acts on one of its downstream effectors specifically involved in antinociception. As suggested by the previously observed loss of antinociceptive effect of morphine in TREK1 KOs, while adverse effects are not affected, we indeed show that targeting TREK1 allows the antinociceptive and adverse effects of morphine to be completely dissociated, including constipation and abuse potential which are the most challenging for alternative strategies. In view of these results, TREK1 selective pharmacological activation is potentially a powerful and safe strategy to relieve pain.
AUTHOR CONTRIBUTIONS
Conditioned place preference test was done by N.M. (designed the experiment and analysed the results), F.N. (designed the experiment) and L.C. (performed the experiment). S.R. provided the respiratory rate measurements performed at CERB under his supervision as part of a contract of services. Patch‐clamp studies were performed and analysed by I.B.S., F.L., M.M. and S.L. Other experiments involving live animals were performed by L.P., C.J., J.S., M.D. and E.C. and analysed by J.B. and S.L. S.D. supervised the production of RNE28. J.B., I.B.S., N.M., F.N., F.L., S.D., A.E. and S.L. were involved in the manuscript writing and proofreading. Project was supervised by J.B., A.E. and S.L.
CONFLICT OF INTEREST
S.R. is a board member of the INNOPAIN Company, which holds a license on the RNE28 patent (WO/2013/098416). The respiratory rate measurements were performed at CERB under his supervision. He did not contribute to the study design, data analysis or writing of the manuscript. The remaining authors have nothing to disclose.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Animal Experimentation and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Data S1 Extended Method Form for the reporting of analgesic drugs testing in animal models.
Figure S1 RNE28 binding assay. Percentage of inhibition of specific binding of radiolabeled ligands for various receptors, channels and transporters, by 10 μM RNE28 (scintillation counting). Assay performed by Eurofins‐Cerep (Celle l'Evescault, France)
Figure S2 Analgesic activity of morphine in naive and inflamed animals. Dose‐effect curves of the antinociceptive activity of morphine 30 min following oral gavage in naive (a) and carrageenan‐injected (b) mice. Datasets were fitted with the least‐square method and fits were compared with the extra sum‐of‐square F test. ED50, Emax, and p and F values are shown on the right. n = 10 and represent mice
Figure S3 RNE28 pharmacokinetics. Rats were injected with 10, 20 or 40 mg/kg RNE28 via the intravenous route. Blood samples were taken before and 10, 30, 60 and 90 min after injection and RNE28 plasmatic concentration was determined by HPLC. Assay performed by CERB (Baugy, France). n = 5 and represent rats
Figure S4 Blockade of the effect of RNE28 on TREK1 currents by spadin. Current densities recorded in HEK‐293 cells transfected with the human TREK1 channel in response to 100 μM RNE28 (n = 29), 1 μM spadin (n = 10), both drugs (n = 14) or their respective vehicles (n = 24). *p <0.05 vs Vehicle + Vehicle. One‐way ANOVA followed by Dunnett's post‐hoc test. n numbers represent cells
Figure S5 Tolerance to the antinociceptive effect of RNE28. Mice were given RNE28 at 30 mg/kg or morphine at 5 mg/kg per os, twice a day for 9 days. Pain thresholds were assessed at days 0, 3, 5 and 8 by paw immersion in 46°C water and plotted as %MPE. *p < 0.05 vs Day 0 of the same treatment. Two‐way ANOVA followed by Dunnett's post‐hoc test. n = 12 for both groups and represent mice
Figure S6 Naloxone‐precipitated withdrawal in RNE28‐treated mice. Mice were given RNE28 at 30 mg/kg (n = 5) or morphine at 5 mg/kg (n = 6) per os, twice a day for 9 days. 2 h after the last administration, animals received an intraperitoneal injection of 2 mg/kg naloxone. Jumping behaviour, which is characteristic of a precipitated withdrawal syndrome, was monitored for 30 min. *p < 0.05 vs morphine. Mann‐Whitney test. n represent mice
Figure S7 Analgesic activity of riluzole in TREK‐1−/− and TREK‐2−/− mice. a Left, time‐course of the analgesic effect induced by 0 or 7.5 mg/kg riluzole administered intraplantarly in WT and TREK‐1−/− mice 2 h following paw inflammation induction by intraplantar carrageenan injection (20 μl, 2%). Pain thresholds were evaluated by paw immersion in 46°C water and plotted as %MPE. Right, areas under curves calculated from 0 to 60 min post‐administration. b Left, time‐course of the analgesic effect induced by 0 or 7.5 mg/kg riluzole administered intraplantarly in WT and TREK‐2−/− mice 2 h following paw inflammation induction by intraplantar carrageenan injection (20 μl, 2%). Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, areas under curves calculated from 0 to 60 min post‐administration. *p < 0.05, **p < 0.01, ****p < 0.0001 vs vehicle of the same genotype. Two‐way ANOVA followed by Sidak's post‐hoc test (a‐b left) and Kruskal‐Wallis followed by Dunn's post‐hoc test (a‐b right). n = 6 (a, in all groups) and n = 7 (b, in all groups) and represent mice
Table S1 Data analysis summary
ACKNOWLEDGEMENTS
This project was funded by the Fondation Fyssen, the Auvergne Regional Council (Conseil Régional d'Auvergne),and the European Fund for Regional Economic Development (FEDER) through the smart specialization strategy for research and innovation (S3, project AV0014790) and the French government IDEX‐ISITE initiative 16‐IDEX‐0001 (CAP 20‐25). We would like to thank Claire Chazaud (GReD, Université Clermont Auvergne, Clermont‐Ferrand, France) for providing the ZP3‐Cre mice.
Busserolles J, Ben Soussia I, Pouchol L, et al. TREK1 channel activation as a new analgesic strategy devoid of opioid adverse effects. Br J Pharmacol. 2020;177:4782–4795. 10.1111/bph.15243
REFERENCES
- Alexander, S. P. H. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , … CGTP Collaborators . (2019). The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176, S142–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloui, A. , Zimmermann, K. , Mamet, J. , Duprat, F. , Noël, J. , Chemin, J. , … Lazdunski, M. (2006). TREK‐1, a K+ channel involved in polymodal pain perception. The EMBO Journal, 25, 2368–2376. 10.1038/sj.emboj.7601116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev, S. N. , Ang, K.‐H. , Gallardo‐Godoy, A. , Clark, K. A. , Arkin, M. R. , Renslo, A. R. , & Minor, D. L. Jr. (2013). A high‐throughput functional screen identifies small molecule regulators of temperature‐ and mechano‐sensitive K2P channels. ACS Chemical Biology, 8, 1841–1851. 10.1021/cb400289x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourin, M. , Hascoet, M. , Mansouri, B. , Colombel, M. C. , & Bradwejn, J. (1992). Comparison of behavioral effects after single and repeated administrations of four benzodiazepines in three mice behavioral models. Journal of Psychiatry & Neuroscience: JPN, 17, 72–77. [PMC free article] [PubMed] [Google Scholar]
- Chaplan, S. R. , Bach, F. W. , Pogrel, J. W. , Chung, J. M. , & Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods, 53, 55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chaumette, T. , Chapuy, E. , Berrocoso, E. , Llorca‐Torralba, M. , Bravo, L. , Mico, J. A. , … Sors, A. (2018). Effects of S 38093, an antagonist/inverse agonist of histamine H3 receptors, in models of neuropathic pain in rats. European Journal of Pain, 22, 127–141. 10.1002/ejp.1097 [DOI] [PubMed] [Google Scholar]
- Chemin, J. , Patel, A. J. , Duprat, F. , Lauritzen, I. , Lazdunski, M. , & Honoré, E. (2005). A phospholipid sensor controls mechanogating of the K+ channel TREK‐1. The EMBO Journal, 24, 44–53. 10.1038/sj.emboj.7600494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft, R. M. (2003). Sex differences in opioid analgesia: ‘From mouse to man’. The Clinical Journal of Pain, 19, 175–186. 10.1097/00002508-200305000-00005 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, W. N. , Binns, L. T. , Fancher, K. S. , Dean, J. , Moore, R. , Kemler, R. , & Knowles, B. B. (2000). Expression of Cre recombinase in mouse oocytes: A means to study maternal effect genes. Genesis, 26, 110–112. 10.1002/(SICI)1526-968X(200002)26:2<110::AID-GENE2>3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- Devilliers, M. , Busserolles, J. , Lolignier, S. , Deval, E. , Pereira, V. , Alloui, A. , … Eschalier, A. (2013). Activation of TREK‐1 by morphine results in analgesia without adverse side effects. Nature Communications, 4 10.1038/ncomms3941 [DOI] [PubMed] [Google Scholar]
- DeWire, S. M. , Yamashita, D. S. , Rominger, D. H. , Liu, G. , Cowan, C. L. , Graczyk, T. M. , … Violin, J. D. (2013). A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. The Journal of Pharmacology and Experimental Therapeutics, 344, 708–717. 10.1124/jpet.112.201616 [DOI] [PubMed] [Google Scholar]
- Ding, H. , Kiguchi, N. , Yasuda, D. , Daga, P. R. , Polgar, W. E. , Lu, J. J. , … Ko, M.‐C. (2018). A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Science Translational Medicine, 10, eaar3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djillani, A. , Pietri, M. , Moreno, S. , Heurteaux, C. , Mazella, J. , & Borsotto, M. (2017). Shortened spadin analogs display better TREK‐1 inhibition, in vivo stability and antidepressant activity. Frontiers in Pharmacology, 8, 643 10.3389/fphar.2017.00643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink, M. , Duprat, F. , Lesage, F. , Reyes, R. , Romey, G. , Heurteaux, C. , & Lazdunski, M. (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. The EMBO Journal, 15, 6854–6862. 10.1002/j.1460-2075.1996.tb01077.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francès, H. , Smirnova, M. , Leriche, L. , & Sokoloff, P. (2004). Dopamine D3 receptor ligands modulate the acquisition of morphine‐conditioned place preference. Psychopharmacology, 175, 127–133. 10.1007/s00213-004-1807-9 [DOI] [PubMed] [Google Scholar]
- Hajasova, Z. , Canestrelli, C. , Acher, F. , Noble, F. , & Marie, N. (2018). Role of mGlu7 receptor in morphine rewarding effects is uncovered by a novel orthosteric agonist. Neuropharmacology, 131, 424–430. 10.1016/j.neuropharm.2018.01.002 [DOI] [PubMed] [Google Scholar]
- Hervieu, G. J. , Cluderay, J. E. , Gray, C. W. , Green, P. J. , Ranson, J. L. , Randall, A. D. , & Meadows, H. J. (2001). Distribution and expression of TREK‐1, a two‐pore‐domain potassium channel, in the adult rat CNS. Neuroscience, 103, 899–919. 10.1016/S0306-4522(01)00030-6 [DOI] [PubMed] [Google Scholar]
- Heurteaux, C. , Guy, N. , Laigle, C. , Blondeau, N. , Duprat, F. , Mazzuca, M. , … Lazdunski, M. (2004). TREK‐1, a K+ channel involved in neuroprotection and general anesthesia. The EMBO Journal, 23, 2684–2695. 10.1038/sj.emboj.7600234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux, C. , Lucas, G. , Guy, N. , El Yacoubi, M. , Thümmler, S. , Peng, X.‐D. , … Lazdunski, M. (2006). Deletion of the background potassium channel TREK‐1 results in a depression‐resistant phenotype. Nature Neuroscience, 9, 1134–1141. 10.1038/nn1749 [DOI] [PubMed] [Google Scholar]
- Honoré, E. , Maingret, F. , Lazdunski, M. , & Patel, A. J. (2002). An intracellular proton sensor commands lipid‐ and mechano‐gating of the K+ channel TREK‐1. The EMBO Journal, 21, 2968–2976. 10.1093/emboj/cdf288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard, L. E. , Chumbley, J. R. , Ranatunga, K. M. , Armstrong, S. J. , Veale, E. L. , & Mathie, A. (2005). Inhibition of the human two‐pore domain potassium channel, TREK‐1, by fluoxetine and its metabolite norfluoxetine. British Journal of Pharmacology, 144, 821–829. 10.1038/sj.bjp.0706068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage, F. , Maingret, F. , & Lazdunski, M. (2000). Cloning and expression of human TRAAK, a polyunsaturated fatty acids‐activated and mechano‐sensitive K+ channel. FEBS Letters, 471, 137–140. 10.1016/S0014-5793(00)01388-0 [DOI] [PubMed] [Google Scholar]
- Lesage, F. , Terrenoire, C. , Romey, G. , & Lazdunski, M. (2000). Human TREK2, a 2P domain mechano‐sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein‐coupled receptors. The Journal of Biological Chemistry, 275, 28398–28405. 10.1074/jbc.M002822200 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology. https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato, M. , Arrigoni, C. , Mori, T. , Sekioka, Y. , Bryant, C. , Clark, K. A. , & Minor, D. L. Jr. (2017). K2P2.1(TREK‐1):activator complexes reveal a cryptic selectivity filter binding site. Nature, 547, 364–368. 10.1038/nature22988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolignier, S. , Amsalem, M. , Maingret, F. , Padilla, F. , Gabriac, M. , Chapuy, E. , … Busserolles, J. (2011). Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE, 6(8), e23083 10.1371/journal.pone.0023083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loucif, A. J. C. , Saintot, P. , Liu, J. , Antonio, B. M. , Zellmer, S. G. , Yoger, K. , … Mathie, A. (2018). GI‐530159, a novel, selective, mechanosensitive two‐pore‐domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. British Journal of Pharmacology, 175, 2272–2283. 10.1111/bph.14098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar, S. , Grinnell, S. , Le Rouzic, V. , Burgman, M. , Polikar, L. , Ansonoff, M. , … Pasternak, G. W. (2011). Truncated G protein‐coupled mu opioid receptor MOR‐1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proceedings of the National Academy of Sciences, 108, 19778–19783. 10.1073/pnas.1115231108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik, A. , Lin, H. , Aryal, D. K. , McCorvy, J. D. , Dengler, D. , Corder, G. , … Shoichet, B. K. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature, 537, 185–190. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone, G. F. , Le Rouzic, V. , Varadi, A. , Xu, J. , Rajadhyaksha, A. M. , Majumdar, S. , … Pasternak, G. W. (2017). Genetic dissociation of morphine analgesia from hyperalgesia in mice. Psychopharmacology, 234, 1891–1900. 10.1007/s00213-017-4600-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes, H. W. , Maldonado, R. , Simonin, F. , Valverde, O. , Slowe, S. , Kitchen, I. , … Kieffer, B. L. (1996). Loss of morphine‐induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu‐opioid‐receptor gene. Nature, 383, 819–823. 10.1038/383819a0 [DOI] [PubMed] [Google Scholar]
- Mazella, J. , Pétrault, O. , Lucas, G. , Deval, E. , Béraud‐Dufour, S. , Gandin, C. , … Borsotto, M. (2010). Spadin, a sortilin‐derived peptide, targeting rodent TREK‐1 channels: A new concept in the antidepressant drug design. PLoS Biology, 8, e1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhurst, A. D. , Rennie, G. , Chapman, C. G. , Meadows, H. , Duckworth, M. D. , Kelsell, R. E. , … Pangalos, M. N. (2001). Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Molecular Brain Research, 86, 101–114. 10.1016/S0169-328X(00)00263-1 [DOI] [PubMed] [Google Scholar]
- Moha ou Maati, H. , Peyronnet, R. , Devader, C. , Veyssiere, J. , Labbal, F. , Gandin, C. , … Borsotto, M. (2011). A human TREK‐1/HEK cell line: A highly efficient screening tool for drug development in neurological diseases. PLoS ONE, 6, e25602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moha ou Maati, H. , Veyssiere, J. , Labbal, F. , Coppola, T. , Gandin, C. , Widmann, C. , … Borsotto, M. (2012). Spadin as a new antidepressant: Absence of TREK‐1‐related side effects. Neuropharmacology, 62, 278–288. [DOI] [PubMed] [Google Scholar]
- Mores, K. L. , Cummins, B. R. , Cassell, R. J. , & van Rijn, R. M. (2019). A review of the therapeutic potential of recently developed G protein‐biased kappa agonists. Frontiers in Pharmacology, 10 10.3389/fphar.2019.00407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus, S. S. , & Freeman, K. B. (2018). Abuse potential of biased mu opioid receptor agonists. Trends in Pharmacological Sciences, 39, 916–919. 10.1016/j.tips.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. J. , Honoré, E. , Maingret, F. , Lesage, F. , Fink, M. , Duprat, F. , & Lazdunski, M. (1998). A mammalian two pore domain mechano‐gated S‐like K+ channel. The EMBO Journal, 17, 4283–4290. 10.1093/emboj/17.15.4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope, L. , Arrigoni, C. , Lou, H. , Bryant, C. , Gallardo‐Godoy, A. , Renslo, A. R. , & Minor, D. L. Jr. (2018). Protein and chemical determinants of BL‐1249 action and selectivity for K2P channels. ACS Chemical Neuroscience, 9, 3153–3165. 10.1021/acschemneuro.8b00337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt, R. D. , Bertin, A. , & Jalfre, M. (1977). Behavioral despair in mice: A primary screening test for antidepressants. Archives Internationales de Pharmacodynamie et de Thérapie, 229, 327–336. [PubMed] [Google Scholar]
- Poupon, L. , Lamoine, S. , Pereira, V. , Barriere, D. A. , Lolignier, S. , Giraudet, F. , … Busserolles, J. (2018). Targeting the TREK‐1 potassium channel via riluzole to eliminate the neuropathic and depressive‐like effects of oxaliplatin. Neuropharmacology, 140, 43–61. 10.1016/j.neuropharm.2018.07.026 [DOI] [PubMed] [Google Scholar]
- Raffa, R. B. , Mathiasen, J. R. , & Jacoby, H. I. (1987). Colonic bead expulsion time in normal and μ‐opioid receptor deficient (CXBK) mice following central (ICV) administration of μ‐ and δ‐opioid agonists. Life Sciences, 41, 2229–2234. 10.1016/0024-3205(87)90520-0 [DOI] [PubMed] [Google Scholar]
- Rice, A. S. C. , Cimino‐Brown, D. , Eisenach, J. C. , Kontinen, V. K. , Lacroix‐Fralish, M. L. , Machin, I. , … Stöhr, T. (2008). Animal models and the prediction of efficacy in clinical trials of analgesic drugs: A critical appraisal and call for uniform reporting standards. Pain, 139, 243–247. 10.1016/j.pain.2008.08.017 [DOI] [PubMed] [Google Scholar]
- Rodrigues, N. , Bennis, K. , Vivier, D. , Pereira, V. , Chatelain, F. C. , Chapuy, E. , … Ducki, S. (2014). Synthesis and structure–activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK‐1 channel. European Journal of Medicinal Chemistry, 75, 391–402. 10.1016/j.ejmech.2014.01.049 [DOI] [PubMed] [Google Scholar]
- Schewe, M. , Sun, H. , Mert, Ü. , Mackenzie, A. , Pike, A. C. W. , Schulz, F. , … Baukrowitz, T. (2019). A pharmacological master key mechanism that unlocks the selectivity filter gate in K+ channels. Science, 363, 875–880. 10.1126/science.aav0569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwingshackl, A. , Teng, B. , Ghosh, M. , & Waters, C. M. (2013). Regulation of monocyte chemotactic protein‐1 secretion by the two‐pore‐domain potassium (K2P) channel TREK‐1 in human alveolar epithelial cells. American Journal of Translational Research, 5, 530–542. [PMC free article] [PubMed] [Google Scholar]
- Spahn, V. , Del Vecchio, G. , Labuz, D. , Rodriguez‐Gaztelumendi, A. , Massaly, N. , Temp, J. , … Stein, C. (2017). A nontoxic pain killer designed by modeling of pathological receptor conformations. Science, 355, 966–969. 10.1126/science.aai8636 [DOI] [PubMed] [Google Scholar]
- Şterbuleac, D. (2019). Molecular determinants of chemical modulation of two‐pore domain potassium channels. Chemical Biology & Drug Design, 94, 1596–1614. 10.1111/cbdd.13571 [DOI] [PubMed] [Google Scholar]
- Steru, L. , Chermat, R. , Thierry, B. , & Simon, P. (1985). The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology, 85, 367–370. 10.1007/BF00428203 [DOI] [PubMed] [Google Scholar]
- Takesue, E. I. , Schaefer, W. , & Jukniewicz, E. (1969). Modification of the Randall‐Selitto analgesic apparatus. The Journal of Pharmacy and Pharmacology, 21, 788–789. 10.1111/j.2042-7158.1969.tb08175.x [DOI] [PubMed] [Google Scholar]
- Tsutsui, K. T. , Wood, R. I. , & Craft, R. M. (2011). Anabolic–androgenic steroid effects on nociception and morphine antinociception in male rats. Pharmacology, Biochemistry, and Behavior, 99, 500–508. 10.1016/j.pbb.2011.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke, T. M. (2007). Review on CPP: Measuring reward with the conditioned place preference (CPP) paradigm: Update of the last decade. Addiction Biology, 12, 227–462. 10.1111/j.1369-1600.2007.00070.x [DOI] [PubMed] [Google Scholar]
- Urban, M. O. , Ren, K. , Park, K. T. , Campbell, B. , Anker, N. , Stearns, B. , … Bristow, L. (2005). Comparison of the antinociceptive profiles of gabapentin and 3‐methylgabapentin in rat models of acute and persistent pain: Implications for mechanism of action. The Journal of Pharmacology and Experimental Therapeutics, 313, 1209–1216. 10.1124/jpet.104.081778 [DOI] [PubMed] [Google Scholar]
- Vivier, D. , Soussia, I. B. , Rodrigues, N. , Lolignier, S. , Devilliers, M. , Chatelain, F. C. , … Ducki, S. (2017). Development of the first two‐pore domain potassium channel TWIK‐related K+ channel 1‐selective agonist possessing in vivo antinociceptive activity. Journal of Medicinal Chemistry, 60, 1076–1088. 10.1021/acs.jmedchem.6b01285 [DOI] [PubMed] [Google Scholar]
- Volkow, N. D. , & Collins, F. S. (2017). The role of science in addressing the opioid crisis. The New England Journal of Medicine, 377, 391–394. 10.1056/NEJMsr1706626 [DOI] [PubMed] [Google Scholar]
- Wright, P. D. , McCoull, D. , Walsh, Y. , Large, J. M. , Hadrys, B. W. , Gaurilcikaite, E. , … Mathie, A. (2019). Pranlukast is a novel small molecule activator of the two‐pore domain potassium channel TREK2. Biochemical and Biophysical Research Communications, 520, 35–40. 10.1016/j.bbrc.2019.09.093 [DOI] [PubMed] [Google Scholar]
- Wu, Y.‐Y. , Singer, C. A. , & Buxton, I. L. O. (2012). Variants of stretch‐activated two‐pore potassium channel TREK‐1 associated with preterm labor in humans1 . Biology of Reproduction, 87, 96 10.1095/biolreprod.112.099499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomkowski, A. D. E. , Engel, D. , Cunha, M. P. , Gabilan, N. H. , & Rodrigues, A. L. S. (2012). The role of the NMDA receptors and l‐arginine–nitric oxide–cyclic guanosine monophosphate pathway in the antidepressant‐like effect of duloxetine in the forced swimming test. Pharmacology, Biochemistry, and Behavior, 103, 408–417. 10.1016/j.pbb.2012.09.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Extended Method Form for the reporting of analgesic drugs testing in animal models.
Figure S1 RNE28 binding assay. Percentage of inhibition of specific binding of radiolabeled ligands for various receptors, channels and transporters, by 10 μM RNE28 (scintillation counting). Assay performed by Eurofins‐Cerep (Celle l'Evescault, France)
Figure S2 Analgesic activity of morphine in naive and inflamed animals. Dose‐effect curves of the antinociceptive activity of morphine 30 min following oral gavage in naive (a) and carrageenan‐injected (b) mice. Datasets were fitted with the least‐square method and fits were compared with the extra sum‐of‐square F test. ED50, Emax, and p and F values are shown on the right. n = 10 and represent mice
Figure S3 RNE28 pharmacokinetics. Rats were injected with 10, 20 or 40 mg/kg RNE28 via the intravenous route. Blood samples were taken before and 10, 30, 60 and 90 min after injection and RNE28 plasmatic concentration was determined by HPLC. Assay performed by CERB (Baugy, France). n = 5 and represent rats
Figure S4 Blockade of the effect of RNE28 on TREK1 currents by spadin. Current densities recorded in HEK‐293 cells transfected with the human TREK1 channel in response to 100 μM RNE28 (n = 29), 1 μM spadin (n = 10), both drugs (n = 14) or their respective vehicles (n = 24). *p <0.05 vs Vehicle + Vehicle. One‐way ANOVA followed by Dunnett's post‐hoc test. n numbers represent cells
Figure S5 Tolerance to the antinociceptive effect of RNE28. Mice were given RNE28 at 30 mg/kg or morphine at 5 mg/kg per os, twice a day for 9 days. Pain thresholds were assessed at days 0, 3, 5 and 8 by paw immersion in 46°C water and plotted as %MPE. *p < 0.05 vs Day 0 of the same treatment. Two‐way ANOVA followed by Dunnett's post‐hoc test. n = 12 for both groups and represent mice
Figure S6 Naloxone‐precipitated withdrawal in RNE28‐treated mice. Mice were given RNE28 at 30 mg/kg (n = 5) or morphine at 5 mg/kg (n = 6) per os, twice a day for 9 days. 2 h after the last administration, animals received an intraperitoneal injection of 2 mg/kg naloxone. Jumping behaviour, which is characteristic of a precipitated withdrawal syndrome, was monitored for 30 min. *p < 0.05 vs morphine. Mann‐Whitney test. n represent mice
Figure S7 Analgesic activity of riluzole in TREK‐1−/− and TREK‐2−/− mice. a Left, time‐course of the analgesic effect induced by 0 or 7.5 mg/kg riluzole administered intraplantarly in WT and TREK‐1−/− mice 2 h following paw inflammation induction by intraplantar carrageenan injection (20 μl, 2%). Pain thresholds were evaluated by paw immersion in 46°C water and plotted as %MPE. Right, areas under curves calculated from 0 to 60 min post‐administration. b Left, time‐course of the analgesic effect induced by 0 or 7.5 mg/kg riluzole administered intraplantarly in WT and TREK‐2−/− mice 2 h following paw inflammation induction by intraplantar carrageenan injection (20 μl, 2%). Pain thresholds were assessed by paw immersion in 46°C water and plotted as %MPE. Right, areas under curves calculated from 0 to 60 min post‐administration. *p < 0.05, **p < 0.01, ****p < 0.0001 vs vehicle of the same genotype. Two‐way ANOVA followed by Sidak's post‐hoc test (a‐b left) and Kruskal‐Wallis followed by Dunn's post‐hoc test (a‐b right). n = 6 (a, in all groups) and n = 7 (b, in all groups) and represent mice
Table S1 Data analysis summary