

Abstract
Prion disease is a rapidly progressive neurodegenerative disorder caused by misfolding and aggregation of the prion protein (PrP), and there are currently no therapeutic options. PrP ligands could theoretically antagonize prion formation by protecting the native protein from misfolding or by targeting it for degradation, but no validated small-molecule binders have been discovered to date. We deployed a variety of screening methods in an effort to discover binders of PrP, including 19F-observed and saturation transfer difference (STD) NMR spectroscopy, differential scanning fluorimetry (DSF), DNA-encoded library selection, and in silico screening. A single benzimidazole compound was confirmed in concentration-response, but affinity was very weak (Kd > 1 mm), and it could not be advanced further. The exceptionally low hit rate observed here suggests that PrP is a difficult target for small-molecule binders. Whereas orthogonal binder discovery methods could yield high-affinity compounds, non-small-molecule modalities may offer independent paths forward against prion disease.
Keywords: PrP, prion, fragment screening, 19F NMR, STD NMR, TROSY NMR, DNA-encoded library, DSF, in silico screening, binders, prion disease, drug screening, high-throughput screening (HTS), drug discovery, small molecule, nuclear magnetic resonance (NMR), neurodegenerative disease, differential scanning fluorimetry
Prion disease is a rapidly progressive neurodegenerative disorder caused by misfolding and aggregation of the prion protein, or PrP (1). No effective therapeutics currently exist for prion disease, but PrP is a genetically and pharmacologically validated drug target (2). PrP-lowering antisense oligonucleotides (ASOs) are in preclinical development (3–5), and PrP-binding antibodies have been tested preclinically (6) as well as clinically in a compassionate use context (7). Here, we sought to augment the therapeutic pipeline by discovering small molecules that bind PrP.
In principle, small molecules could prevent or treat prion disease by protecting PrP from misfolding or by lowering its abundance. By sterically blocking interactions with misfolded PrP, or simply through the free energy of binding, a chaperone might stabilize PrP against misfolding, following precedents in transthyretin amyloidosis (8, 9) and cystic fibrosis (10, 11). Proofs of principle for this approach include the efficacy of monoclonal antibodies to PrP to clear prion infection in cell culture (12, 13) and in peripheral tissues of animals (14), as well as the stability of PrP “stapled” with nonnative disulfide bonds (15). Alternatively, small-molecule binding events can sometimes directly lead to protein degradation (16, 17), and, if not, a binder could serve as a starting point for engineering a bifunctional molecule to specifically target PrP for degradation (18, 19). Although at present most bifunctional degrader strategies are best suited to intracellular targets because of reliance on cytoplasmic E3 ubiquitin ligases, recent studies suggest alternate routes to targeted degradation of cell surface proteins (20), such as PrP.
Decades of effort have not yet yielded a small-molecule PrP binder suitable for advancement as a drug candidate (21). The development of phenotypic screening for antagonists of misfolded PrP accumulation in cultured cells (22) enabled the identification of several compounds effective in vivo (23–26), but advancement of these compounds has been hindered by lack of activity against human prion strains and unclear mechanisms of action (26–29). Meanwhile, several compounds shown to interact with PrP through biophysical assays have demonstrated antiprion activity in a range of experimental systems (30–34). However, these compounds likewise appear to lack clinical promise, as none are simultaneously specific (35), potent, and drug-like. Certain metallated porphyrins (36, 37) interact with PrP with affinity values comparable to their effective concentrations in cell culture (36, 37), and some exhibit in vivo activity in certain paradigms (38). Similarly, a range of anionic polymers (33, 39–42) also bind PrP and show in vivo antiprion activity in certain contexts (41, 43, 44). However, these binding events may not be monomeric (45) or specific to PrP (35, 46). Still other compounds with demonstrated antiprion activity exhibit interaction with PrP only at concentrations orders of magnitude above their effective concentration in cell culture (47, 48).
Because PrP's biology does not lend itself to enzymatic or activity assays, we chose to apply several non-activity-based screening modalities 19F-observed and saturation transfer difference (STD) NMR fragment screening, differential scanning fluorimetry (DSF), DNA-encoded library (DEL) selection, and an in silico screen. We selected a fragment-based drug discovery paradigm as a starting point based on PrP's small size and lack of obvious binding pockets (49) and the success of this method in identifying ligands for targets refractory to other approaches (50). Our campaign utilized 19F NMR and STD NMR, because these approaches are sensitive to weak affinity binders (51), have low false-positive rates (52), and allow searching a large swath of chemical space through small, highly soluble fragments that can later be optimized into larger, higher-affinity molecules (53). DSF was employed to find compounds that directly influence the thermal stability of PrP. This technique quantifies protein thermal stability by measuring fluorescence of a solvatochromic dye (SYPRO Orange) as a function of temperature as it binds to unfolded regions of a protein (54). In principle, DSF hits should have the desired property of stabilizing the target protein. DEL selection uses pooled, individually DNA-barcoded molecules added to an immobilized recombinant protein, enabling single-pot panning of thousands to trillions of compounds. Nonspecific molecules are washed away, and putative binders are eluted, PCR amplified, and subjected to next-generation DNA sequencing for identification. We used a DEL of peptide macrocycles (55) diversified through incorporation of many nonnatural amino acids, some of which are acyclic, aromatic, or spirogenic and tailored to conform to Lipinski's Rule of Five (56) and Kihlberg (57) rules. Macrocycles display better stability and have a lower entropic cost of binding than linear peptides, making them suitable for targeting surfaces of proteins (58, 59). Finally, we employed the artificial intelligence-based in silico screening method, AtomNet® (60), which uses a protein structure-based, convolutional neural network to predict molecular binding affinities. This technique was recently used to discover a selective binder and degrader of Miro1 (61).
Results
Fragment-based drug discovery through NMR screening
From five commercial and internal sources (Fig. 1A), we selected 6,630 low molecular weight, high solubility fragments for a fragment-based drug discovery campaign. The compounds in these libraries were mostly small (<300 Da) and had a range of AlogP values and hydrogen bond donor and acceptor sites that mostly fell within the Rule of Three (62) for fragment-based screening (Fig. 1, B–D). Fragments were screened against either HuPrP23-231 or HuPrP90-231 using pooled, 19F ligand-observed or STD NMR methods as a primary screen, singleton STD NMR for retesting, and protein-observed 1H-15N TROSY NMR for validation (Table 1).
Figure 1.

Physicochemical properties of fragment libraries screened. A, composition of fragment libraries. B, scatter plot of AlogP versus molecular weight (MW) for all fragments screened against PrP using 19F or STD NMR. C and D, number of hydrogen bond donors (HBD) or hydrogen bond acceptors (HBA). Orange denotes compliance with the “Rule of Three” (62), and black denotes noncompliance.
Table 1.
Summary of NMR fragment screeninga
| Library name | Compounds screened | Pools with hits | Retested by STD | Retested by TROSY | Validated hits |
|---|---|---|---|---|---|
| Broad Institute 19F library | 785 | 14 | 14 | 5 | 0 |
| Broad Institute 1st gen STD library | 1,116 | 16 | 15 | 4 | 0 |
| Broad Institute 2nd gen STD library | 1,823 | 43 | 55 | 34 | 1 |
| Schreiber chiral collection | 381 | 1 | 5 | 0 | 0 |
| ChemBridge high-solubility subset | 2,525 | 31 | 149 | 37 | 0 |
| Total | 6,630 | 105 | 238 | 80 | 1 |
a “Compounds screened” lists the total number of compounds in a given collection of molecules. “Pools with hits” indicates the number compound pools with observed hits. “Retested by STD” indicates the number of individual compounds that were retested for STD signal from each hit pool. “Retested by TROSY” shows the number of compounds that were advanced to 2D TROSY NMR.
Of 6,630 compounds tested, 238 initial hits were retested as singletons by STD NMR, of which 80 were further tested by TROSY NMR. A single compound, 5,6-dichloro-2-methyl-1H-benzimidazole (1), gave a reproducible STD signal in the presence of PrP (Fig. 2A) and induced chemical shift perturbations (CSPs) in the TROSY spectrum of HuPrP90-231 (Fig. 2, B–C, Fig. S1 and S2A). Mapping these residues onto an NMR structure of HuPrP (PDB entry 1HJM) (63) revealed no discernable pocket, with shifts scattered across the structure (Fig. 2D). Nonetheless, we observed similar resonance shifts in the full-length protein HuPrP23-231, suggesting that this binding is not an artifact of using a truncated construct (Fig. S2A). CSPs were confirmed to be dose-responsive for several residues (Fig. 2E and Fig. S2B), and the compound caused a small (∼0.2 °C) but significant decrease in melting temperature by DSF (Fig. S2D).
Figure 2.
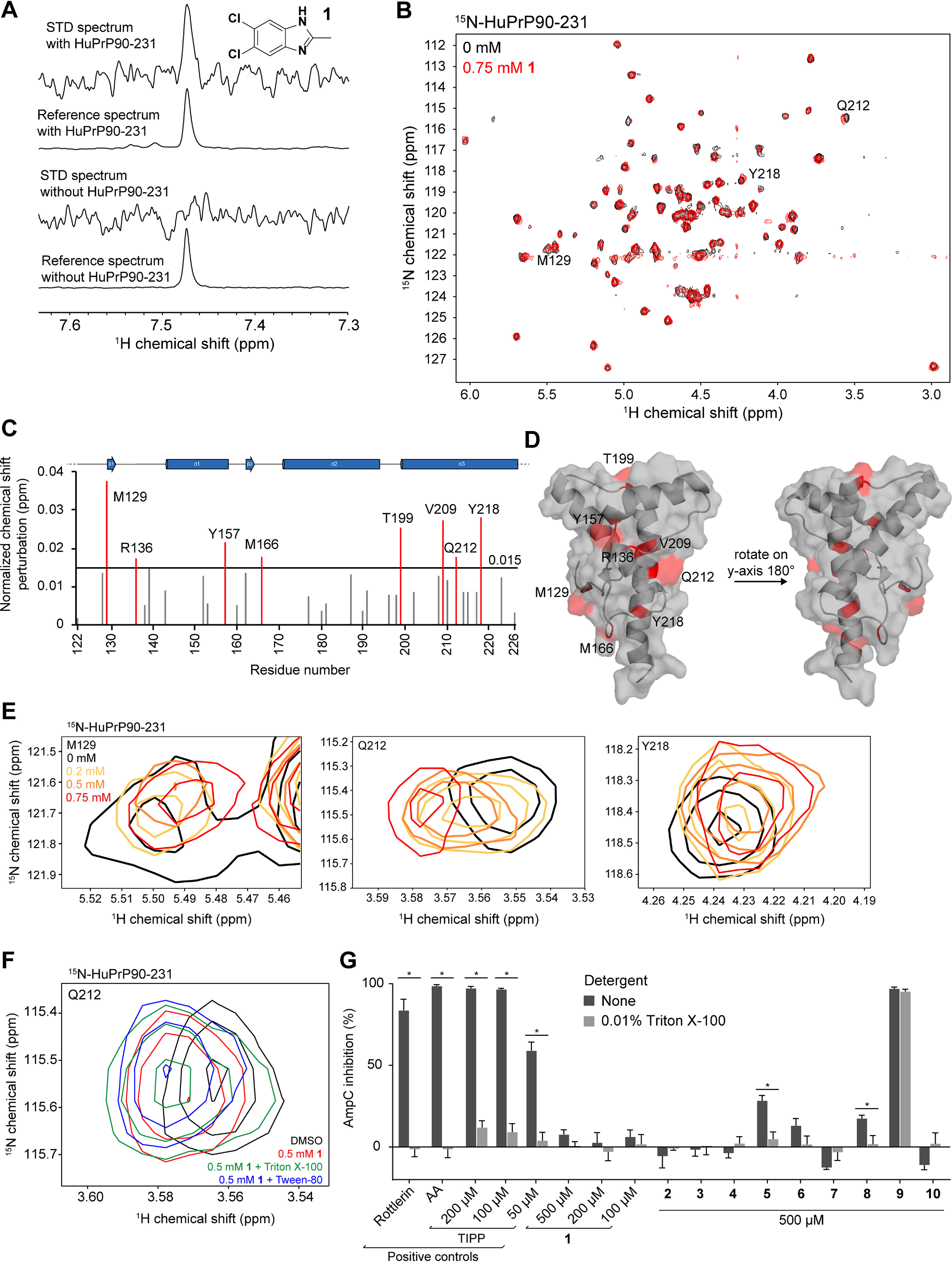
Validation and characterization of a benzimidazole fragment hit. A, STD NMR spectra of 1 (5,6-dichloro-2-methyl-1H-benzimidazole) with and without HuPrP90-231. STD spectra are scaled to 16× the reference spectra. B, TROSY spectrum of 15N-HuPrP90-231 with DMSO (black) or 0.75 mm 1 (red). Peaks that shift greater than 0.015 ppm are denoted with the residue number. C, normalized chemical shift perturbations upon addition of 0.75 mm compound 1. D, residues that shift more than 0.015 ppm were mapped onto the NMR structure of PrP (PDB entry 1HJM) (63). E, concentration-dependent CSPs of residues Q212, M129, and Y218 upon addition of 1. F, 1H-15N TROSY chemical shifts in the presence of 0.75 mm 1 with and without detergents. G, AmpC inhibition assay of 1 and its analogs. Rottlerin (10 μm), anacardic acid (AA, 10 μm), and tetraiodophenolphthalein (TIPP) are prototypical aggregators. Adding detergent to small-molecule aggregates dissociates them and attenuates inhibition of AmpC. *, significance cutoff between detergent and nondetergent tests was p < 0.01 after correction for multiple comparisons. Data are means ± S.D. of four intrarun technical replicates performed on the same microplate.
Because the CSPs caused by 1 are so small, we wanted to be certain that this compound was not perturbing HuPrP because of nonspecific colloidal aggregation. To test whether 1 is causing CSPs because of aggregation, 15N-HuPrP90-231 was incubated with 1 in the presence or absence of nonionic detergent Triton X-100 or Tween-20. The CSPs resulting from 1 were preserved in the presence of detergent, suggesting that 1 is not an aggregator (Fig. 2F). To assess compound aggregation by an orthogonal method, we used the well-established AmpC β-lactamase inhibition assay (64). AmpC is inhibited by small molecules that form aggregates, and these aggregates can be disrupted by addition of detergent. No significant inhibition of AmpC was observed with 1 even at 500 μm concentrations, although the positive control compounds rottlerin and anacardic acid (AA) showed inhibition at 10 μm that could be relieved upon detergent addition (Fig. 2G). Analogs of 1 (compounds 2–10) were also tested; the majority of them did not inhibit AmpC, and none inhibited AmpC as well as the positive controls. Collectively, these data argue that 1 is not an aggregator and does not cause PrP CSPs via aggregation.
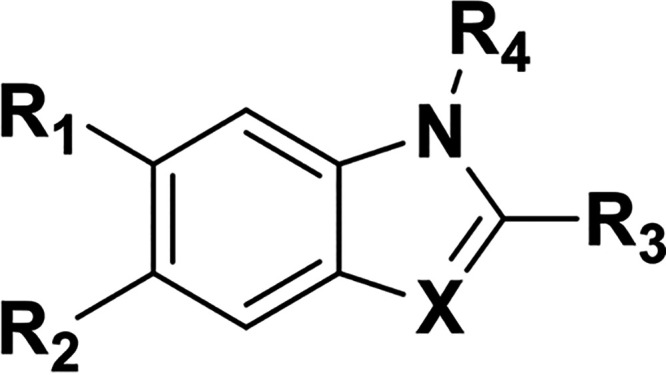
The small magnitude of CSPs and lack of saturation at concentrations up to 0.75 mm suggested that 1 has a Kd for PrP in the millimolar range, too weak to interrogate by many non-NMR orthogonal biophysical assays. In an attempt to find a stronger binder, we tested 54 analogs of 1 from commercial sources and the Broad Institute's internal library by STD NMR and TROSY (Table 2, Table S1, and Fig. S3). Of the 20 compounds most similar to 1, 11 demonstrated positive STD and TROSY signal (Table 2); however, none of the analogs had TROSY CSPs larger than 1 by visual inspection; thus, they were not subjected to further biophysical assays. Despite efforts to soak unliganded PrP crystals with 1 and 20 of its analogs, no electron density attributable to a compound was identified (Table S1).
Table 2.
Analogs of compound 1 tested by STD and TROSY NMRa
| Compound |
 |
STD | TROSY | ||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | X | |||
| 1 | Cl | Cl | Me | H | N | + | + |
| 2 | Me | Me | Me | H | N | + | − |
| 3 | F | F | Me | H | N | − | ± |
| 4 | H | CN | Me | H | N | − | − |
| 5 | Cl | Cl | OH | H | N | − | ± |
| 6 | Cl | Cl | H | H | N | + | + |
| 7 | Cl | H | Me | H | N | − | − |
| 8 | Cl | Cl | Et | H | N | + | + |
| 9 | Cl | Cl | Me | Et | N+-Et | − | ± |
| 10 | Cl | Cl | Me | Me | N | + | + |
| 11 | Br | Cl | H | H | N | + | ± |
| 12 | Br | Br | H | H | N | + | ± |
| 13 | Cl | Cl | CH2OH | H | CH | + | ± |
| 14 | Cl | Cl | H | H | CH | + | ± |
| 15 | Cl | Br | H | H | CH | + | ± |
| 16 | Cl | OMe | H | H | CH | ± | |
| 17 | OMe | Cl | H | H | CH | − | |
| 18 | Cl | F | H | H | CH | − | |
| 19 | F | Cl | H | H | CH | ± | |
| 20 | Br | Cl | H | H | CH | + | ± |
a Analogs of 1 were initially tested by STD NMR, and positive STD hits were assayed by TROSY. + indicates positive STD or TROSY signal; − indicates no STD or TROSY signal; ± indicates borderline positive signal. Blank cells indicate that the analog was not tested. All spectra were assessed by visual inspection.
Thermal shift screening
We tested a library of 30,013 compounds from the Novartis screening set for external collaborations (SSEC) in singleton for thermal stabilization of PrP using DSF (Fig. 3, Table 3 and Fig. S5, A–C). An ideal DSF screen would possess a high signal-to-baseline fluorescence ratio, tightly distributed melting temperatures in the apo condition, and a ≥10:1 molar ratio of soluble ligand to protein (54). We varied a range of assay parameters, including protein concentration, dye concentration, assay volume, use of HuPrP90-231 or HuPrP23-231, and buffer conditions, including buffering agent, metals, and DMSO concentration (Fig. S4). We obtained acceptable melt curves only at high protein concentrations (Fig. S4). Our final screening conditions achieved a ∼5:1 signal-to-baseline fluorescence and a 0.06 °C median absolute deviation (MAD) with 30 μm HuPrP90-231; median Tm was 68.5 °C (Fig. 3, A and B). Compounds were screened at 100 μm for a >3:1 ligand-to-protein ratio, although we lack empirical data on their solubility over the temperature ramp. We chose hit compounds that either positively or negatively affected the melting temperature (Tm) of PrP based on separate criteria (see Experimental procedures). An internally developed pipeline was used to perform Boltzmann fitting of the fluorescence data and call Tm values. Irregular melt curves were automatically flagged and discarded. Two hit criteria were chosen for positive Tm shifters: (1) a statistical cutoff of greater than 3× MAD of the DMSO control wells (0.17 °C) and (2) an initial fluorescence of less than 6 to eliminate compounds that have intrinsic fluorescence or distort the melt curve. Because there were so many negative shifters, stricter hit cutoffs of −0.7 °C > ΔTm > −9 °C were applied. Even though negative shifters are predicted to destabilize PrP, one hypothesis is that such compounds bind a partially folded or destabilized form of PrP and could exhibit antiprion properties (65). Compounds that passed our hit criteria were retested by DSF in triplicate (Fig. 3C). Here, we applied stricter hit cutoffs (see Experimental procedures) because of throughput limitations of our orthogonal heteronuclear single-quantum coherence spectroscopy (HSQC) NMR assay. The 183 reproducible positive hits were passed through frequent hitter and PAINS filtering (66), narrowing the list to 117 compounds, of which we were able to test 93 for PrP binding by HSQC using 15N-HuPrP90-231. Even though the melt curves of PrP with these compounds were often very robust and reproducible (Fig. 3D), none of the compounds tested by HSQC led to PrP CSPs at 100 μm concentration (Fig. 3, E and F). This suggested that the observed thermal shifts were not mediated by binding PrP and were likely artifacts. In support of this interpretation, when we tested eight of the validation screen hits by an orthogonal thermal shift method, differential scanning calorimetry, we were unable to reproduce the change in melting temperature (ΔTm) observed in DSF (Fig. S5, D and E).
Figure 3.

Thermal shift screening. A, a box plot of the HuPrP90-231 median Tm (blue dash) for the DMSO controls from each of the 86 384-well plates screened. These values are relative to the median from all plates combined. Light blue rectangles around each median represent ±3× MAD. B, scatter plot of ΔTm data from the initial screen with red dots indicating hits that were screened in triplicate. Red dots were compounds called as hits (see Experimental procedures for details). Gray dots were compounds that were flagged for having melt curve analysis errors. Black dots are compounds that did not meet our hit calling threshold but were not flagged. Compounds that resulted in PrP shifts of greater than or less than one degree were plotted at +1 or −1, respectively. C, scatter plot of ΔTm values from primary screening versus triplicate screening data. D, fluorescence melt curves of HuPrP90-231 with DMSO and a positive ΔTm shifter (fluorobenzamide shown in panel F). n = 128 for DMSO and n = 4 for test compound. E, HSQC spectrum of 15N-HuPrP90-231 with hit (fluorobenzamide shown in panel F; 100 μm). F, chemical structure of selected fluorobenzamide hit, boxed in red in panels B and C.
Table 3.
Summary of thermal shift screening resultsa
| Screen | Compounds | Total hits | Positive hits | Negative hits |
|---|---|---|---|---|
| Primary (singleton) | 30,013 | 1,129 | 492 | 637 |
| Validation (triplicate) | 1,129 | 282 | 176 | 106 |
| HSQC | 84 | 0 |
a “Compounds” provides the total number of compounds tested in that screening step. “Positive hits” and “negative hits” list the number of molecules that shifted the Tm of HuPrP90-231 positively or negatively, respectively.
DNA-encoded library selection
We performed a selection using HuPrP90-231 with a DEL library of 256,000 macrocycles. Barcode rank abundance in the unenriched library was plotted against enrichment observed in the PrP condition versus a no-protein control condition, revealing enriched compounds across three structural scaffolds (Fig. 4A). The *KRD scaffold was judged to be a likely covalent binder and was not pursued further. Representative compounds from the CC*S and *CJS series were resynthesized off-DNA (Figs. 4, B and C), as both cis and trans isomers, for validation. None of these compounds produced appreciable CSPs against 15N-HuPrP90-231 using TROSY at 200 μm, suggesting that these hits were either false positives or have affinities too weak to be detected by NMR (Fig. 4, D and E).
Figure 4.
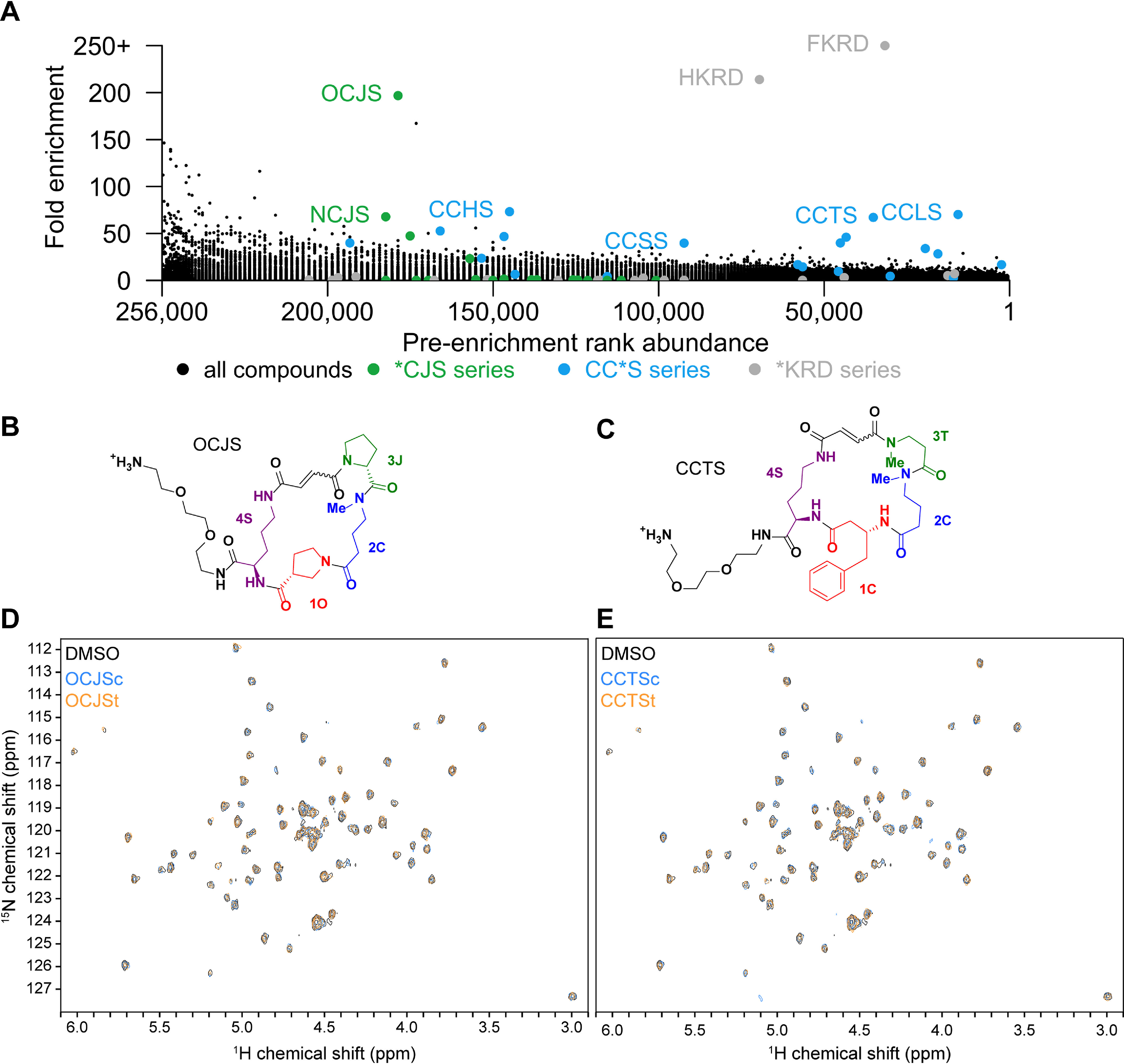
Selection of PrP binders from a DNA-encoded macrocycle library. A, enrichment plot of macrocycle DEL compounds versus input rank. *KRD series compounds (gray) are frequent hitters. B and C, structures of the DEL hits OCJS and CCTS synthesized off-DNA. DEL macrocycles are synthesized as stereoisomers on-DNA, so each cis/trans isomer pair was synthesized off-DNA for testing (c, cis; t, trans). (D and E) TROSY spectra of 15N-HuPrP90-231 with the cis- and trans-isomers (200 μm) of OJCS and CCTS.
In silico screening
We used Atomwise's AtomNet® convolutional neural network method (60) to search for compounds that bind PrP at a specific site. Since there are no reported structures of human PrP bound to a lead-like ligand, we instead used the reported structure of mouse PrP bound to promazine (PDB entry 4MA7) (67) to create a homology model of human PrP also bound to promazine between helix 2 (α2) and the two beta strands (β1 and β2) (Fig. 5A). The regions that were modeled share a high degree of sequence identity with only 12 amino acid differences over residues A117-R230 (mouse PrP numbering). The promazine binding site was screened against 6,922,894 molecules. After additional filtering, the top 81 compounds (Table S2) were selected as predicted binders and assayed for binding using both DSF and STD NMR. By DSF, none of the compounds (90 μm) increased the Tm of PrP more than three standard deviations (0.79 °C), and none of the compounds (100 μm) showed an appreciable STD signal in the presence of HuPrP90-231 (Fig. 5, B and C).
Figure 5.
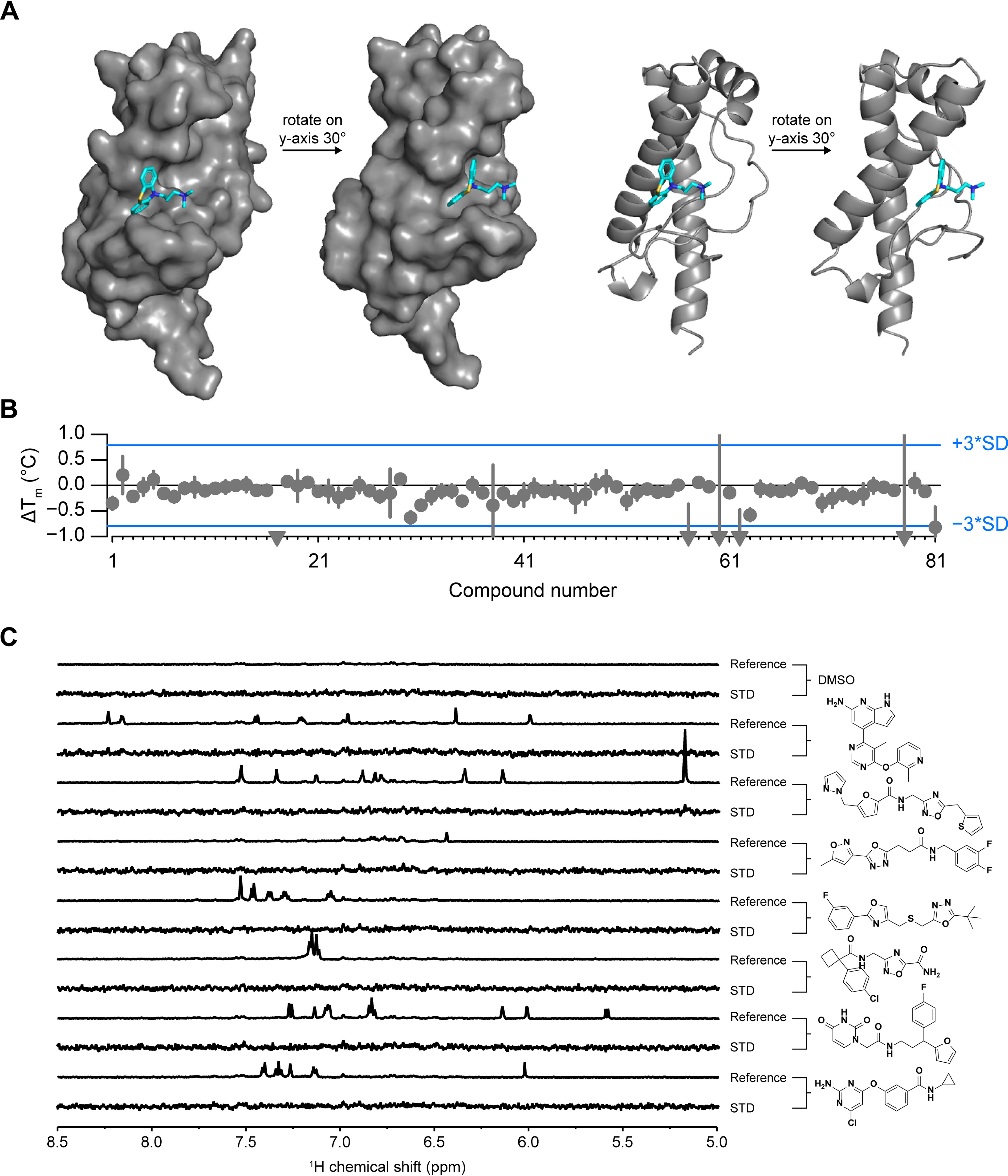
In silico screening. A, homology model of HuPrP with ligand bound (promazine) that was used for neural network in silico screening. B, DSF ΔTm values for all 81 Atomwise compounds with HuPrP90-231. Compounds that resulted in PrP shifts of less than one degree were plotted at −1. Error bars represent standard deviations for three measurements made on the same day using the same batch of protein-dye mix. S.D. of the DMSO conditions (n = 32 technical replicates per plate) was 0.26 °C. C, STD NMR of selected compounds that had a ΔTm of >0.05 °C by DSF. STD spectra are scaled to 5× the reference spectrum intensity.
Discussion
We pursued four different screening modalities aimed at discovering binders of the human prion protein. Despite the large number of molecules tested and complementary approaches used, we were unable to identify any hits suitable for advancement into medicinal chemistry. Our fragment screening campaign identified compound 1 (5,6-dichloro-2-methyl-1H-benzimidazole) and several analogs that weakly bind PrP and were validated with orthogonal NMR assays. However, the poor affinity of these compounds (>1 mm) coupled with the absence of improved binding of chemical analogs effectively precluded their validation through non-NMR methods, and none were pursued further. Meanwhile, our thermal shift, DNA-encoded library, and in silico screening approaches yielded no validated hits at all.
A variety of target-specific technical challenges may have contributed to our inability to identify binders by the approaches employed here. Some reports indicate that the transfer of NMR saturation is weak for smaller proteins, which may have produced false negatives in our STD NMR screens (68, 69). SYPRO Orange dye fluorescence in the presence of unfolded PrP was weak, which necessitated DSF screening at 30 μm protein concentration; compounds were accordingly screened at 100 μm, but solubility limitations may have prevented saturable binding with a maximum thermal shift. Whereas our DEL comprised 256,000 compounds, its size and chemical diversity may not have been sufficient to encompass PrP binders strong enough to survive our stringent selection procedure. Our in silico screen utilized a homology model based on a crystal structure of promazine bound to mouse PrP, but promazine has not been shown to bind human PrP in solution, and promazine analogs that exert antiprion activity in cells appear to do so through an orthogonal mechanism (48). In general, without a positive control available, it is difficult to guide the optimization of screening assays. Taken as a whole, our experimental screens cannot be considered definitive given their modest scale, but considering the diverse methods and compound sets employed, our results hint toward relative rarity of PrP binders in chemical space.
Alternative screening approaches might also improve the probability of discovering a high-affinity PrP binder. We used recombinantly expressed PrP from Escherichia coli for our experiments, which lacks posttranslational modifications (PTMs), including two N-linked glycosylations and a GPI anchor, and many of our experiments used a truncated construct lacking most of the unstructured N terminus and were performed under chelating buffer conditions. Coordination of divalent cations like copper(II) and zinc(II) by full-length PrP can give rise to interdomain interactions (70–72) that might create new binding sites. Alternatively, purification of PrP from mammalian cells (73) and insertion into nanodiscs (74) or micelles might more faithfully recapitulate PrP's PTMs and endogenous membrane environment, potentially yielding binding sites not present on recombinant PrP. Encouragingly, DEL screening has been used successfully with nanodisc immobilized proteins (75). We could also extend our fragment-based drug design strategy by using chemoproteomics (76) to directly assess PrP ligandability on the cell surface. Multiple approaches may be necessary, because targets with low NMR and thermal shift hit rates are reported to, on average, also have lower hit-to-lead development success rates (77).
Overall, despite various technical limitations, our inability to identify even weak binders through multiple orthogonal screening modalities is striking. The absence of obvious binding pockets on PrP's structure, together with the predominance of indirect mechanisms of action revealed in phenotypic screening campaigns, have led to the perception that PrP is a difficult target for small-molecule discovery (31). Our data may provide some support for this conclusion. On balance, our results motivate an emphasis on non-small-molecule technologies, such as oligonucleotide therapeutics, as a means for targeting PrP but do not rule out the possibility that small-molecule binders could be discovered through an expanded screening effort.
Experimental procedures
AlogP and H-bond donor/acceptor calculations
SMILES strings were parsed to yield molecular weight, AlogP, and hydrogen bond donor and acceptor counts using RCDK (78).
Purification of HuPrP90-231 and HuPrP23-231
Recombinant PrP glycerol stocks were a generous gift from Byron Caughey and Andrew Hughson (NIAID Rocky Mountain Laboratory). The purification protocol was adapted from published procedures (79). Two 4-ml cultures of E. coli were started from a glycerol stock in Terrific Broth (TB) with kanamycin (25 μg/ml) and chloramphenicol (25 μg/ml) and incubated (6 h, 37 °C, 220 rpm). Those cultures were used to inoculate 1 liter of autoinduction medium (AIM) (Millipore, 71300) culture made with TB plus kanamycin (25 μg/ml) and chloramphenicol (25 μg/ml) in a 4-liter baffled flask and incubated (22 h, 37 °C, 180 rpm). E. coli was harvested by centrifugation (4,300 × g, 12 min, 4 °C) into four bottles (250 ml each), and the pellets were frozen at −80 °C. The following amounts of reagents used are based on a single pellet from 250 ml of media. The cell pellet was thawed at room temperature, resuspended in lysis buffer (14 ml) (Millipore, 71456-4) by vortexing, and homogenized with a tissue homogenizer (60 s, 50% speed) using disposable tips. The homogenate was incubated (20 min, room temperature [r.t.], end-over-end agitation), and 20 μl of the whole-cell lysate was saved and diluted 1:20 (fraction L). The lysate was clarified by centrifugation (16,000 × g, 20 min, 4 °C), and 20 μl of the supernatant was saved and diluted 1:20 (fraction S). Lysis buffer (14 ml) was added to the pellet, which was homogenized with the tissue homogenizer (50% speed, 60 s) and incubated (20 min, r.t., end-over-end agitation). 0.1× lysis buffer (∼20 ml) was added to a total volume of 34 ml, and the homogenate was centrifuged (16,000 × g, 15 min, 4 °C). 30 μl of the supernatant was saved (fraction W). The pellet was resuspended in 30 ml 0.1× lysis buffer using a tissue homogenizer (50% speed, 1 min, r.t.) and centrifuged (16,000 × g, 15 min, 4 °C). 30 μl of the supernatant (fraction W2) was saved. 10.5 ml of unfolding buffer (8 M guanidine HCl in 100 mm NaPO4, pH 8.0) was added to the inclusion body pellet and homogenized with the tissue homogenizer (50–100% power, 1 min, r.t.). The homogenate was incubated (50 min, r.t., end-over-end agitation) and centrifuged (8,000 × g, 5 min, 4 °C). 30 μl of the supernatant was saved (fraction D). All four supernatants were combined into a 50-ml conical tube and stored (7 days, 4 °C). 15 g of semi-dry nickel-nitrilotriacetic acid resin (Qiagen, 30450) was weighed out into each of three conical tubes, denaturing buffer was added to 30-ml total volume, and the solution was incubated (10 min, end-over-end incubation, r.t.). Denatured PrP was evenly added to each of the three conical tubes and incubated (40 min, r.t.). PrP-bound resin was added to a column (Cytiva, 28988948), and 30 μl of the unbound was saved (fraction UB). ÄKTA pure (Cytiva) lines were equilibrated with denaturing buffer (6 M guanidinium HCl, 100 mm sodium phosphate, pH 8.0) in line A and refolding buffer (100 mm sodium phosphate, 10 mm Tris, pH 8.0) in line B and then 100% denaturing buffer (line A only). A gradient was run from 0–100% refolding buffer (2.25 ml/min, 240 min, 4 °C) and then 100% refolding buffer (2.25 ml/min, 30 min, 4 °C). The protein was eluted with a gradient from 0–100% elution buffer (500 mm imidazole, 100 mm sodium phosphate, pH 6.0) (6 ml/min, 45 min, 4 °C) and then 100% elution buffer (6 ml/min, 15 min, 4 °C). 30 μl of each fraction (fraction #) was saved. A 100-μl sample of the resin slurry was saved (fraction B). The fractions containing PrP were dialyzed (7-kDa MWCO, Thermo Fisher, 68700) against 6 liters of dialysis buffer (10 mm sodium phosphate, pH 5.8) (overnight, 4 °C) and 4 liters of dialysis buffer (4–6 h, 4 °C). The dialyzed elution was centrifuged (4,300 × g, 10 min, 4 °C) to pellet precipitated protein. A 30-μl sample of the final protein (fraction F) was saved. [HuPrP90-231] was measured by its absorbance at 280 nm (ε = 22,015 M−1 cm−1) (MW = 16.145 kDa). [HuPrP23-231] was measured by its absorbance at 280 nm (ε = 57,995 M−1 cm−1) (MW = 22.963 kDa). Protein was aliquotted, frozen in N2(l), and stored at −80 °C. For SDS-PAGE analysis, protein samples (30 μl) were mixed with 10 μl 4× loading buffer (4× LDS buffer [Thermo Fisher, NP0007] with 10 mm TCEP [Thermo Fisher, 77720]). For fraction B, 25 μl of 4× loading buffer was added. Fractions D and UB were ethanol precipitated before SDS-PAGE by adding 270 μl of ethanol, vortexing, and incubating on dry ice (5 min). D and UB were centrifuged (21,000 × g, 5 min, 4 °C), and the supernatant was discarded. 300 μl 90% ethanol (−80 °C) was added, vortexed, and centrifuged (21,000 × g, 5 min, 4 °C). The supernatant was discarded and the pellet was allowed to dry. 600 μl of 1× loading buffer was added to sample D, and 40 μl 1× loading buffer was added to sample UB. All gel samples were incubated (90 °C, 5 min). Fractions were analyzed by SDS-PAGE with Coomassie staining (10 μl of each sample into a 15-well Bis-Tris NuPAGE gel [Thermo Fisher, NP0321BOX], 180 V, 40 min). The full amino acid sequences of HuPrP constructs are as follows. The N-terminal methionine of HuPrP90-231 was mostly removed by endogenous proteases as expected based on the second residue being glycine (80); this was verified by intact protein LC-MS (Fig. S5C). HuPrP23-231, however, retains the N-terminal methionine (81), again as expected given the second residue is lysine (80). HuPrP90-231, MGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSS; HuPrP23-231, MKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSS.
Purification of 15N-HuPrP90-231 and 15N-HuPrP23-231
The same procedure was used to purify 15N-HuPrP with the following modifications. 15N AIM was composed of 1× BioExpress 15N cell growth medium (Cambridge Isotope Laboratory, CGM-1000-N) in an EMD Millipore Overnight Express induction system (Sigma-Aldrich, 71300-M) with kanamycin (25 μg/ml) and chloramphenicol (25 μg/ml). Dialysis buffer was 20 mm HEPES, pH 7.4, 50 mm NaCl.
NMR data acquisition and analysis
Spectra were acquired on a 600-MHz Bruker Avance III NMR spectrometer equipped with a 5-mm QCI cryoprobe using 3-mm sample tubes (Bruker, Z112272) and a SampleJet for sample handling. Spectra were analyzed in TopSpin version 4.0.2 and MestreNova version 10.0.1. Hit identification was performed by visual inspection of the data.
19F NMR screening
HuPrP23-231 (final concentration, 9 μm) or matched dialysis buffer alone (20 mm HEPES, pH 6.8, no-protein control condition) was combined with 10% (v/v) D2O and 4.5% (v/v) DMSO containing a pool of 10 19F fragments per NMR tube (45 μm each) with a total volume of 200 μl for each sample. A 19F NMR spectrum was obtained for each sample using a standard 1H-decoupled one-pulse experiment with 64 scans and a spectral width of 237 ppm with the carrier frequency at −100 ppm. The sample temperature was 280 K. Fragment hits were identified by comparison of both the peak position and peak width between the control (no protein) sample and the protein-containing sample. For this screen, minimal line-broadening was observed; fragment hits were identified by visual review of chemical shifts, with perturbation of ≥0.005 (3 Hz) as an approximate threshold. Hit peaks from pools were compared with reference spectra to identify the likely hit fragment. Resupplied fragments were tested by STD NMR and/or 1H-15N TROSY NMR as described below.
STD NMR screening
HuPrP90-231 was buffer exchanged into 20 mm HEPES-d18, pH 7.4, 150 mm NaCl in ∼99% D2O using a 5-kDa MWCO centrifugal concentrator (Millipore, C7715). Preplated fragment pools were thawed in a desiccator at room temperature. For the Broad Institute 1st and 2nd generation STD libraries, 180 μl of HuPrP90-231 (11 μm) in deuterated buffer was added to each well, mixed with the fragments (1.6%, v/v, DMSO-d6), and transferred immediately to a 3-mm NMR tube. The ChemBridge high-solubility subset and Schreiber chiral fragment collection screens used HuPrP90-231 at 10 μm, 2% (v/v) DMSO-d6, in 20 mm HEPES-d18, pH 6.8, 25 mm NaCl in ∼99% D2O. Fragments were pooled with eight (Broad 1st and 2nd Gen STD and ChemBridge high-solubility subset) or five (Schreiber chiral fragment collection) fragments per tube, always at a final concentration of 200 μm each. The Schreiber chiral fragment collection consisted of n = 381 compounds synthesized in-house, many of which have been described previously (82–84). In silico screening hits (100 μm) were mixed with HuPrP90-231 (11 μm) in 20 mm HEPES-d18, pH 7.4, 150 mm NaCl in ∼99% D2O with 1% (v/v) DMSO-d6. Ligand-observed screening was done using STD NMR. On-resonance irradiation of the protein was done at −0.25 ppm and off-resonance irradiation at 30 ppm. To saturate the protein, a 2-s train of 50-ms Gaussian pulses separated by 1-ms delays was used. A 27-ms spin-lock pulse was used to suppress protein signals, and water suppression was accomplished using excitation sculpting with the gradient pulse scheme. The sample temperature was 280 K. Hit pools were identified by visual inspection, and fragment hits were confirmed as singletons using the same experimental conditions as those described above. Compound 1 (5,6-dichloro-2-methyl-1H-benzimidazole) was purchased from two different vendors (Combi-Blocks, HC-3145, and Key Organics, PS-4319) and retested for an STD signal, which was reproducible across vendors. The compound from Key Organics was deemed more pure than those of other sources by NMR, TLC, and LC-MS and was used for the majority of the experiments presented here.
1H-15N TROSY NMR of 15N-HuPrP90-231 and 15N-HuPrP23-231
15N-HuPrP90-231 (50-60 μm) in 20 mm HEPES, pH 7.4, 50 mm NaCl was combined with D2O (10%, v/v) and ligand (0-1 mm) or DMSO (2%, v/v), mixed, and added to a 3-mm NMR tube. 15N-HuPrP23-231 in 20 mm HEPES, pH 7.4, 150 mm NaCl was combined with D2O (10%, v/v) and compound 1 (0.3-1 mm) or DMSO (3%, v/v), mixed, and added to a 3-mm NMR tube. All concentrations are final. 1H-15N TROSY spectra were acquired at 298 K with 64 scans and 128 increments. Chemical shift perturbations (CSPs) were identified by visual inspection. Quantification of dose-response CSPs was performed with MestReNova, version 10.0.1. Compound 1 was reproducible for CSPs across two different vendor sources used for retesting (described above).
1H-15N HSQC NMR of 15N-HuPrP90-231
15N-HuPrP90-231 (50 μm, 160 μl) in 20 mm HEPES, pH 7.4, 50 mm NaCl was combined with 18 μl D2O (10%, v/v) and ligand (100 μm final concentration, 1.8 μl) or DMSO (1%, v/v), mixed, and added to a 3-mm NMR tube. 1H-15N HSQC spectra were acquired using a 600-MHz Bruker Avance II spectrometer at 298 K. 2D data were processed and analyzed by using TopSpin version 4.0.2 software. CSPs were identified by visual inspection.
AmpC aggregation counterscreen
Compounds were tested for colloidal aggregation using an AmpC β-lactamase counterscreen (64). Recombinant E. coli AmpC was expressed in Rosetta cells and purified using a published protocol (85). The enzymatic assay was performed in 50 mm sodium phosphate, pH 7.0, ±0.01% (v/v) Triton X-100 in clear UV-transparent 96-well half-area microplates (Corning, 3679) in 150-μl final reaction volumes. The final concentration of DMSO was 1.0% (v/v). Compounds were incubated with 5 nM AmpC in a 143.5-μl reaction solution for 5 min at r.t., followed by the addition of 1.5 μl of nitrocefin substrate (Cayman, 15424) dissolved in DMSO (100 μm initial substrate concentration). Reaction solutions were gently mixed by multichannel pipette. Reaction progress was continuously monitored by absorbance at 482 nm for 5 min at r.t. on a SpectraMax M3 plate reader. Percent activity was calculated from reaction rates (slope) and normalized to DMSO-only controls after background subtraction with an enzyme-free reaction. AA, rottlerin, and 3′,3′',5′,5′'-tetraiodophenolphthalein (TIPP) (Combi-Blocks, QE-5474) were used as positive controls. The significance difference between detergent and nondetergent tests was defined as p < 0.01 (after correction for multiple comparisons). Four intrarun technical replicates were performed on the same microplate.
DSF screening
All concentrations are final assay concentrations unless otherwise indicated. All reagents were diluted in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA. Protein was thawed and centrifuged (4500 × g, 10 min, 4 °C) to pellet precipitate. HuPrP90-231 (30 μm) was mixed with SYPRO Orange dye (10×) (Thermo Fisher, S6651), centrifuged (4500 × g, 10 min, 4 °C), and decanted into a new bottle and covered in foil. Compound stocks dissolved in DMSO:H2O (90:10, v/v) were dispensed into 384-well barcoded plates (200 nl, 5 mm stock concentration, 100 μm final concentration) (4titude, 4Ti-0381). 10 μl of protein-dye mix was added to each well with a Multidrop™ Combi Reagent Dispenser (Thermo Fisher), shaken (2 min, r.t.), and centrifuged (1 min, r.t.). All Combi lines were covered to block ambient light. A Roche LightCycler II was used for fluorescence measurements with filter set 465/580. A temperature ramp from 30–90 °C, rate of 0.07 °C/s, and 6 acquisitions per second (6.5-min run) were used to collect the data. At the beginning of each day, 4 plates with no compounds were run to equilibrate the system. Melting temperatures (Tm) were calculated by fitting the fluorescence data to a Boltzmann curve. ΔTm values were calculated by taking the Tm with compound and subtracting it from the average of the DMSO control wells on each plate (n = 32 DMSO wells per plate). After removing error code-flagged wells, positive ΔTm hits were picked using the following criteria: ΔTm > 3× MAD (0.17 °C), initial fluorescence intensity of <6. Negative ΔTm hits were picked using the following criteria: −0.7 °C > ΔTm > −9 °C, initial fluorescence intensity of <6. 1,129 hit compounds were tested in triplicate, all wells flagged with error codes 3 and 4 were removed, and hits were chosen based on the following criteria: ΔTm sign must be the same as the primary screen, 19.93 > Tm > 0.3, initial fluorescence of <4, change in fluorescence of 2–17.1. Hits from the triplicate validation screen were filtered for frequent hitters and PAINS, and 84 compounds were tested by HSQC NMR. DSF data were analyzed using Tibco Spotfire and RStudio.
Differential scanning calorimetry
HuPrP90-231 (30 μm) was mixed with buffer or compound (100 μm) in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, 2% (v/v) DMSO (400 μl total volume per well). All concentrations are final assay concentrations. Data were acquired using a MicroCal VP-Capillary DSC instrument from Malvern Panalytical, and data were analyzed using Origin software, provided by the vendor. A temperature ramp from 20–100 °C was conducted at a rate of 200 °C/h. Four buffer-only runs preceded the sample runs to equilibrate the system. n = 4 for DMSO controls and n = 1 for each compound. 3× S.D. was used as a hit cutoff.
Macrocycle DEL screening
DNA-encoded library selection was performed as described previously using a 256,000-compound macrocycle library (55). Briefly, 40 μg of HuPrP90-231, purified as described above, was loaded onto His Dynabeads (Invitrogen, 10103D), relying on the intrinsic metal-binding properties of untagged PrP. Beads were washed, blocked, incubated with 50 μl of DNA-encoded library (60 min, 4 °C), and washed three times, and protein was eluted with 300 mm imidazole. Barcodes were sequenced on an Illumina MiSeq, and enrichment was calculated against a no-protein condition run in parallel.
In silico screening
The homology model of human PrP with bound ligand promazine was built on the template of the mouse PrP structure (67) (PDB entry 4MA7, chain A) using SWISS-MODEL (86). The binding site is surrounded by residues V122, G124, L125, G126, Y128, Y162, I182, Q186, V189, and T190 on the human PrP homology model.
The virtual screen was carried out using the AtomNet neural network, the first deep convolutional neural network for structure-based drug design (60, 61). A single global AtomNet model was employed to predict binding affinity of small molecules to a target protein. The model was trained with experimental Ki, Kd, and IC50 values of several million small molecules and protein structures spanning several thousand different proteins, curated from both public databases and proprietary sources. Because AtomNet is a global model, it can be applied to novel binding sites with no known ligands, a prerequisite to most target-specific machine-learning models. Another advantage of using a single global model in prospective predictions is that it helped prevent the so-called model overfitting. The following three-step procedure was applied to train the AtomNet model. The first step is to define the binding site on a given protein structure using a flooding algorithm (87) based on an initial seed. The initial starting point of the flooding algorithm may be determined using either a bound ligand annotated in the PDB database, crucial residues as revealed by mutagenesis studies, or identification of catalytic motifs previously reported. The second step is to shift the coordinates of the protein-ligand cocomplex to a three-dimensional Cartesian system with an origin at the center-of-mass of the binding site. Data augmentation was performed by randomly rotating and translating the protein structure around the center-of-mass of the binding site to prevent the neural network from memorizing a preferred orientation of the protein structure. The third step is to sample the conformations or poses of a small-molecule ligand within the binding site pocket. For a given ligand, an ensemble of poses was generated, and each of these poses represented a putative cocomplex with the protein. Each generated cocomplex was then rasterized into a fixed-size regular three-dimensional grid, where the values at each grid point represent the structural features that are present at each point. Similar to a photo pixel containing three separate channels representing the presence of red, green, and blue colors, our grid points represent the presence of different atom types. These grids serve as the input to a convolutional neural network and define the receptive field of the network. A network architecture of a 30 × 30 × 30 grid with 1 Å spacing for the input layer, followed by five convolutional layers of 32 × 33, 64 × 33, 64 × 33, 64 × 33, and 64 × 23 (number of filters × filter-dimension), and a fully connected layer with 256 ReLU hidden units was used. The scores for each pose in the ensemble were combined through a weighted Boltzmann averaging to produce a final score. These scores were compared against the experimentally measured pKi or pIC50 (converted from Ki or IC50) of the protein and ligand pair, and the weights of the neural network were adjusted to reduce the error between the predicted and experimentally measured affinity using a mean-square-error loss function. Training was done using the ADAM adaptive learning method (88), the backpropagation algorithm, and minibatches with 64 examples per gradient step.
The Mcule small-molecule library, purchasable from the chemical vendor Mcule, was used for the in silico screen. The original Mcule library version v20180817, containing ∼10 million compounds in SMILES format, was downloaded from Mcule's website (SCR_018921). Every compound in the library was pushed through a standardization process, including the removal of salts, isotopes, and ions, and conversion to neutral form, namely, conversion of functional groups and aromatic rings to consistent representations. Additional filters were applied on some molecular properties, including molecular weight between 100 and 700 Da, total number of chiral centers in a molecule of ≤6, total number of atoms in a molecule of ≤60, total number of rotatable bonds of ≤15, and only molecules containing C, N, S, H, O, P, B, and halogens. Other filters, such as toxicophores, Eli Lilly's MedChem Rules (89), and PAINS, were also applied to remove compounds with undesirable substructures, resulting in the final library of 6,922,894 unique compounds.
For each small molecule, we generated a set of 64 poses within the binding site. Each of these poses was scored by the trained model, and the molecules were ranked by their scores. Because of a lack of a well-defined small molecule binding pocket on the human prion protein structure, there was low confidence in the predicted binders. Regardless, the top 50,000 ranking compounds were clustered based on chemical similarity and filtered for CNS drug-like properties using the Lipinski's CNS rules (90) with MW of ≤400, clogP of ≤5, number of hydrogen bond donors of ≤3, and number of hydrogen bond acceptors of ≤7. The final set of 81 compounds containing diverse chemical scaffolds were selected and sourced from Mcule.
DSF of in silico hits and compound 1
Compounds were diluted to 1 mm in DMSO-HBS (20 mm HEPES, pH 7.4, 150 mm NaCl) (20:80, v/v). 1 μl of compound was added to each well of a 384-well plate (90 μm final concentration). Mixtures of HuPrP90-231 (30 μm) were prepared with SYPRO Orange dye (10×) in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA buffer and then centrifuged (4000 × g, 10 min, 4 °C). 10 μl of protein-dye mix supernatant was added to each compound and mixed (2%, v/v, DMSO final concentration). A Roche LightCycler II was used for fluorescence measurements with filter set 465/580. A temperature ramp from 30–90 °C, rate of 0.07 °C/s, and 8 acquisitions per second (∼18 min per plate) were used to collect the data. Three independent plates (using the same protein-dye mix) were measured with n = 32 for DMSO controls per plate and n = 1 for each compound per plate. ΔTm values were calculated by subtracting the average apo Tm from the Tm with compound. Three ΔTm values per compound were averaged and plotted. Error bars represent standard deviations (0.26 °C). For DSF of compound 1 with HuPrP90-231, the same experimental parameters were used as described above, except compound 1 was used at 500 μm concentration and n = 8 intrarun technical replicates were performed on the same assay plate.
Intact protein LC-MS
Purified HuPrP90-231 was diluted to 2 μm in 20 mm HEPES, pH 7.4, 50 mm NaCl. 1 μl of diluted protein was injected onto a Waters BioAccord LC-ToF (composed of an ACQUITY I-Class UPLC and RDa detector with ESI source). Mobile phase A consisted of 0.1% formic acid (Millipore LiChroPur) in LC-MS-grade water (JTBaker), and mobile phase B consisted of 0.1% formic acid in LC-MS-grade acetonitrile (JTBaker). Protein was trapped on a C4 column (ACQUITY UPLC Protein BEH, 300 Å, 1.7 μm, 2.1 × 50 mm) held at 80 °C for the entire analysis. The protein was desalted for 1 min before elution with a gradient of 5% to 85% mobile phase B in 2.5 min. Ionization was performed with a 55-V cone voltage and 550 °C ionization temperature. The instrument scan rate was 0.2 scans/s over 50 to 2000 m/z. PrP eluted at an observed retention time of 2 min. The PrP charge envelope was deconvoluted into the intact mass with the MaxEnt1 function using UNIFI software (Waters).
Data availability
Raw data and source code will be made available in a public GitHub repository: http://github.com/ericminikel/binder_screening.
Supplementary Material
Acknowledgments
Novartis provided reagents, expertise in screening sciences, and efforts to perform the work as part of a research collaboration agreement. We thank Dr. Kirk Clark (Novartis) for guidance on the DSF screen and related NMR validation experiments.
This article contains supporting information.
Author contributions—A. G. R., M. F. M., S. M. V., A. J. L., K. T. N., and E. V. M. conceptualization; A. G. R., M. F. M., D. C., S. M. V., J. L. D., A. J. L., A. I. C., D. L. U., J. B. Y., R. N. S., O. K. S., J. R. S., V. L. R., J. A. M., M. S., M. C. N., K. T. N., and E. V. M. data curation; A. G. R., J. B. Y., J. A. M., M. S., K. T. N., D. S. A., and E. V. M. formal analysis; A. G. R. and E. V. M. writing-original draft; A. G. R., M. F. M., D. C., S. M. V., J. L. D., A. J. L., A. I. C., D. L. U., J. B. Y., C. T. L., A. J. C., R. N. S., O. K. S., J. R. S., V. L. R., J. A. M., M. S., M. C. N., K. T. N., S. K. W., D. R. L., F. F. W., V. K. K., D. S. A., S. L. S., and E. V. M. writing-review and editing; M. F. M., S. M. V., A. J. L., C. T. L., A. J. C., M. C. N., S. K. W., D. R. L., F. F. W., V. K. K., D. S. A., S. L. S., and E. V. M. supervision; S. M. V., D. R. L., and E. V. M. funding acquisition; D. S. A. resources.
Funding and additional information—This work was supported by the National Institutes of Health (F31 AI122592 to E. V. M., R35 GM118062 to D. R. L., supporting A. I. C. and D. L. U., and T32 HL007627 to J. L. D.), Prion Alliance, BroadIgnite, and an anonymous organization. Atomwise provided effort and reagents in kind as part of an Artificial Intelligence Molecular Screen (AIMS) Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—E. V. M. has received consulting fees from Deerfield Management and Guidepoint and has received research support in the form of unrestricted charitable contributions from Charles River Laboratories and Ionis Pharmaceuticals. S. L. S. serves on the Board of Directors of the Genomics Institute of the Novartis Research Foundation (“GNF”); is a shareholder and serves on the Board of Directors of Jnana Therapeutics; is a shareholder of Forma Therapeutics; is a shareholder and advises Kojin Therapeutics, Kisbee Therapeutics, Decibel Therapeutics, and Eikonizo Therapeutics; serves on the Scientific Advisory Boards of Eisai Co., Ltd., Ono Pharma Foundation, Exo Therapeutics, and F-Prime Capital Partners; and is a Novartis Faculty Scholar. S. M. V. has received speaking fees from Illumina and Biogen and has received research support in the form of unrestricted charitable contributions from Charles River Laboratories and Ionis Pharmaceuticals. D. R. L. is a consultant for and cofounder of Exo Therapeutics, which uses DNA-encoded libraries for drug development. D. C., D. S. A., O. K. S., and S. K. W. are employees of Novartis. K. T. N. is an employee of Atomwise.
- STD
- saturation transfer difference
- DSF
- differential scanning fluorimetry
- ASOs
- antisense oligonucleotides
- DEL
- DNA-encoded library
- CSPs
- chemical shift perturbations
- AA
- anacardic acid
- HSQC
- heteronuclear single-quantum coherence spectroscopy
- PTMs
- posttranslational modifications.
References
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vallabh S. M., Minikel E. V., Schreiber S. L., and Lander E. S. (2020) Towards a treatment for genetic prion disease: trials and biomarkers. Lancet Neurol. 19, 361–368 10.1016/S1474-4422(19)30403-X [DOI] [PubMed] [Google Scholar]
- 3. Nazor Friberg K., Hung G., Wancewicz E., Giles K., Black C., Freier S., Bennett F., Dearmond S. J., Freyman Y., Lessard P., Ghaemmaghami S., and Prusiner S. B. (2012) Intracerebral infusion of antisense oligonucleotides into prion-infected mice. Mol Ther. Nucleic Acids 1, e9 10.1038/mtna.2011.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raymond G. J., Zhao H. T., Race B., Raymond L. D., Williams K., Swayze E. E., Graffam S., Le J., Caron T., Stathopoulos J., O'Keefe R., Lubke L. L., Reidenbach A. G., Kraus A., Schreiber S. L., Mazur C., et al. (2019) Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight 4, 5 10.1172/jci.insight.131175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Minikel E. V., Zhao H. T., Le J., O'Moore J., Pitstick R., Graffam S., Carlson G. A., Kavanaugh M. P., Kriz J., Kim J. B., Ma J., Wille H., Aiken J., McKenzie D., Doh-Ura K., et al. (2020) Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 10.1093/nar/gkaa616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aguzzi A., Lakkaraju A. K. K., and Frontzek K. (2018) Toward therapy of human prion diseases. Annu. Rev. Pharmacol. Toxicol. 58, 331–351 10.1146/annurev-pharmtox-010617-052745 [DOI] [PubMed] [Google Scholar]
- 7. Dyer C. (2018) British man with CJD gets experimental treatment in world first. BMJ 363, k4608 10.1136/bmj.k4608 [DOI] [PubMed] [Google Scholar]
- 8. Bulawa C. E., Connelly S., Devit M., Wang L., Weigel C., Fleming J. A., Packman J., Powers E. T., Wiseman R. L., Foss T. R., Wilson I. A., Kelly J. W., and Labaudinière R. (2012) Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 109, 9629–9634 10.1073/pnas.1121005109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurer M. S., Schwartz J. H., Gundapaneni B., Elliott P. M., Merlini G., Waddington-Cruz M., Kristen A. V., Grogan M., Witteles R., Damy T., Drachman B. M., Shah S. J., Hanna M., Judge D. P., Barsdorf A. I., Huber P., et al. (2018) ATTR-ACT study investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379, 1007–1016, 10.1056/NEJMoa1805689 [DOI] [PubMed] [Google Scholar]
- 10. Van Goor F., Hadida S., Grootenhuis P. D. J., Burton B., Stack J. H., Straley K. S., Decker C. J., Miller M., McCartney J., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., and Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wainwright C. E., Elborn J. S., Ramsey B. W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J. C., De Boeck K., Flume P. A., Konstan M. W., McColley S. A., McCoy K., McKone E. F., Munck A., et al., TRAFFIC Study Group, TRANSPORT Study Group (2015) Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 373, 220–231 10.1056/NEJMoa1409547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peretz D., Williamson R. A., Kaneko K., Vergara J., Leclerc E., Schmitt-Ulms G., Mehlhorn I. R., Legname G., Wormald M. R., Rudd P. M., Dwek R. A., Burton D. R., and Prusiner S. B. (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739–743 10.1038/35089090 [DOI] [PubMed] [Google Scholar]
- 13. Enari M., Flechsig E., and Weissmann C. (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. U S A 98, 9295–9299 10.1073/pnas.151242598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. White A. R., Enever P., Tayebi M., Mushens R., Linehan J., Brandner S., Anstee D., Collinge J., and Hawke S. (2003) Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422, 80–83 10.1038/nature01457 [DOI] [PubMed] [Google Scholar]
- 15. Hafner-Bratkovic I., Bester R., Pristovsek P., Gaedtke L., Veranic P., Gaspersic J., Mancek-Keber M., Avbelj M., Polymenidou M., Julius C., Aguzzi A., Vorberg I., and Jerala R. (2011) Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J. Biol. Chem. 286, 12149–12156 10.1074/jbc.M110.213926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu Y.-L., Yang X., Ren Z., McDonnell D. P., Norris J. D., Willson T. M., and Greene G. L. (2005) Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 18, 413–424 10.1016/j.molcel.2005.04.014 [DOI] [PubMed] [Google Scholar]
- 17. Hanan E. J., Liang J., Wang X., Blake R., Blaquiere N., and Staben S. T. (2020) Monomeric targeted protein degraders. J. Med. Chem. 10.1021/acs.jmedchem.0c00093 [DOI] [PubMed] [Google Scholar]
- 18. Lai A. C., and Crews C. M. (2017) Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 10.1038/nrd.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chamberlain P. P., and Hamann L. G. (2019) Development of targeted protein degradation therapeutics. Nat. Chem. Biol 15, 937–944 10.1038/s41589-019-0362-y [DOI] [PubMed] [Google Scholar]
- 20. Banik S., Pedram K., Wisnovsky S., Riley N., and Bertozzi C. (2019) Lysosome targeting chimeras (LYTACs) for the degradation of secreted and membrane proteins. Nature 584, 291–297 10.1038/s41586-020-2545-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barreca M. L., Iraci N., Biggi S., Cecchetti V., and Biasini E. (2018) Pharmacological agents targeting the cellular prion protein. Pathogens 7, 27 10.3390/pathogens7010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kocisko D. A., Baron G. S., Rubenstein R., Chen J., Kuizon S., and Caughey B. (2003) New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 77, 10288–10294 10.1128/JVI.77.19.10288-10294.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawasaki Y., Kawagoe K., Chen C., Teruya K., Sakasegawa Y., and Doh-Ura K. (2007) Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J. Virol. 81, 12889–12898 10.1128/JVI.01563-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghaemmaghami S., May B. C. H., Renslo A. R., and Prusiner S. B. (2010) Discovery of 2-aminothiazoles as potent antiprion compounds. J. Virol. 84, 3408–3412 10.1128/JVI.02145-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wagner J., Ryazanov S., Leonov A., Levin J., Shi S., Schmidt F., Prix C., Pan-Montojo F., Bertsch U., Mitteregger-Kretzschmar G., Geissen M., Eiden M., Leidel F., Hirschberger T., Deeg A. A., et al. (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol. 125, 795–813 10.1007/s00401-013-1114-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giles K., Berry D. B., Condello C., Dugger B. N., Li Z., Oehler A., Bhardwaj S., Elepano M., Guan S., Silber B. M., Olson S. H., and Prusiner S. B. (2016) Optimization of aryl amides that extend survival in prion-infected mice. J Pharmacol. Exp. Ther. 358, 537–547 10.1124/jpet.116.235556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berry D. B., Lu D., Geva M., Watts J. C., Bhardwaj S., Oehler A., Renslo A. R., DeArmond S. J., Prusiner S. B., and Giles K. (2013) Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. U.S.A. 110, E4160–E4169 10.1073/pnas.1317164110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu D., Giles K., Li Z., Rao S., Dolghih E., Gever J. R., Geva M., Elepano M. L., Oehler A., Bryant C., Renslo A. R., Jacobson M. P., Dearmond S. J., Silber B. M., and Prusiner S. B. (2013) Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol. Exp. Ther. 347, 325–338 10.1124/jpet.113.205799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giles K., Berry D. B., Condello C., Hawley R. C., Gallardo-Godoy A., Bryant C., Oehler A., Elepano M., Bhardwaj S., Patel S., Silber B. M., Guan S., DeArmond S. J., Renslo A. R., and Prusiner S. B. (2015) Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J. Pharmacol. Exp. Ther. 355, 2–12 10.1124/jpet.115.224659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawatake S., Nishimura Y., Sakaguchi S., Iwaki T., and Doh-Ura K. (2006) Surface plasmon resonance analysis for the screening of anti-prion compounds. Biol. Pharm. Bull. 29, 927–932 10.1248/bpb.29.927 [DOI] [PubMed] [Google Scholar]
- 31. Poncet-Montange G., St Martin S. J., Bogatova O. V., Prusiner S. B., Shoichet B. K., and Ghaemmaghami S. (2011) A survey of antiprion compounds reveals the prevalence of non-PrP molecular targets. J. Biol. Chem. 286, 27718–27728 10.1074/jbc.M111.234393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferreira N. C., Ascari L. M., Hughson A. G., Cavalheiro G. R., Góes C. F., Fernandes P. N., Hollister J. R., da Conceição R. A., Silva D. S., Souza A. M. T., Barbosa M. L. C., Lara F. A., Martins R. A P., Caughey B., and Cordeiro Y. (2018) A promising antiprion trimethoxychalcone binds to the globular domain of the cellular prion protein and changes its cellular location. Antimicrob. Agents Chemother. 62, e01441–17 10.1128/AAC.01441-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caughey B., Brown K., Raymond G. J., Katzenstein G. E., and Thresher W. (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red [corrected]. J. Virol. 68, 2135–2141 10.1128/JVI.68.4.2135-2141.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hafner-Bratkovic I., Gaspersic J., Smid L. M., Bresjanac M., and Jerala R. (2008) Curcumin binds to the alpha-helical intermediate and to the amyloid form of prion protein–a new mechanism for the inhibition of PrP(Sc) accumulation. J Neurochem 104, 1553–1564 10.1111/j.1471-4159.2007.05105.x [DOI] [PubMed] [Google Scholar]
- 35. McGovern S. L., Caselli E., Grigorieff N., and Shoichet B. K. (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med. Chem. Am. Chem. Soc. 45, 1712–1722 10.1021/jm010533y [DOI] [PubMed] [Google Scholar]
- 36. Nicoll A. J., Trevitt C. R., Tattum M. H., Risse E., Quarterman E., Ibarra A. A., Wright C., Jackson G. S., Sessions R. B., Farrow M., Waltho J. P., Clarke A. R., and Collinge J. (2010) Pharmacological chaperone for the structured domain of human prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 17610–17615 10.1073/pnas.1009062107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Caughey W. S., Raymond L. D., Horiuchi M., and Caughey B. (1998) Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. U.S.A. 95, 12117–12122 10.1073/pnas.95.21.12117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kocisko D. A., Caughey W. S., Race R. E., Roper G., Caughey B., and Morrey J. A. (2006) Porphyrin increases survival time of mice after intracerebral prion infection. Antimicrob. Agents Chemother. 50, 759–761 10.1128/AAC.50.2.759-761.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caughey B., and Raymond G. J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67, 643–650 10.1128/JVI.67.2.643-650.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gabizon R., Meiner Z., Halimi M., and Ben-Sasson S. A. (1993) Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol. 157, 319–325 10.1002/jcp.1041570215 [DOI] [PubMed] [Google Scholar]
- 41. Doh-Ura K., Ishikawa K., Murakami-Kubo I., Sasaki K., Mohri S., Race R., and Iwaki T. (2004) Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J. Virol. 78, 4999–5006 10.1128/JVI.78.10.4999-5006.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Macedo B., and Cordeiro Y. (2017) Unraveling prion protein interactions with aptamers and other PrP-binding nucleic acids. Int J Mol. Sci. 18, 1023 10.3390/ijms18051023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kocisko D. A., Vaillant A., Lee K. S., Arnold K. M., Bertholet N., Race R. E., Olsen E. A., Juteau J.-M., and Caughey B. (2006) Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 50, 1034–1044 10.1128/AAC.50.3.1034-1044.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gunther E. C., Smith L. M., Kostylev M. A., Cox T. O., Kaufman A. C., Lee S., Folta-Stogniew E., Maynard G. D., Um J. W., Stagi M., Heiss J. K., Stoner A., Noble G. P., Takahashi H., Haas L. T., Schneekloth J. S., Merkel J., et al. (2019) Rescue of transgenic Alzheimer's pathophysiology by polymeric cellular prion protein antagonists. Cell Rep. 26, 145–158 10.1016/j.celrep.2018.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reidenbach A. G., Minikel E. V., Zhao H. T., Guzman S. G., Leed A. J., Mesleh M. F., Kordasiewicz H. B., Schreiber S. L., and Vallabh S. M. (2019) Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides. Biomolecules 10, 1 10.3390/biom10010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Massignan T., Cimini S., Stincardini C., Cerovic M., Vanni I., Elezgarai S. R., Moreno J., Stravalaci M., Negro A., Sangiovanni V., Restelli E., Riccardi G., Gobbi M., Castilla J., Borsello T., et al. (2016) A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein. Sci. Rep. 6, 23180 10.1038/srep23180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vogtherr M., Grimme S., Elshorst B., Jacobs D. M., Fiebig K., Griesinger C., and Zahn R. (2003) Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J. Med. Chem. 46, 3563–3564 10.1021/jm034093h [DOI] [PubMed] [Google Scholar]
- 48. Stincardini C., Massignan T., Biggi S., Elezgarai S. R., Sangiovanni V., Vanni I., Pancher M., Adami V., Moreno J., Stravalaci M., Maietta G., Gobbi M., Negro A., Requena J. R., Castilla J., et al. (2017) An antipsychotic drug exerts anti-prion effects by altering the localization of the cellular prion protein. PLoS One 12, e0182589 10.1371/journal.pone.0182589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zahn R., Liu A., Lührs T., Riek R., von Schroetter C., López García F., Billeter M., Calzolai L., Wider G., and Wüthrich K. (2000) NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 145–150 10.1073/pnas.97.1.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scott J. D., Li S. W., Brunskill A. P. J., Chen X., Cox K., Cumming J. N., Forman M., Gilbert E. J., Hodgson R. A., Hyde L. A., Jiang Q., Iserloh U., Kazakevich I., Kuvelkar R., Mei H., Meredith J., et al. (2016) Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer's disease. J. Med. Chem. 59, 10435–10450 10.1021/acs.jmedchem.6b00307 [DOI] [PubMed] [Google Scholar]
- 51. Harner M. J., Frank A. O., and Fesik S. W. (2013) Fragment-based drug discovery using NMR spectroscopy. J Biomol. NMR 56, 65–75 10.1007/s10858-013-9740-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gossert A. D., and Jahnke W. (2016) NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 97, 82–125 10.1016/j.pnmrs.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 53. Hajduk P. J., and Greer J. (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 6, 211–219 10.1038/nrd2220 [DOI] [PubMed] [Google Scholar]
- 54. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 55. Usanov D. L., Chan A. I., Maianti J. P., and Liu D. R. (2018) Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem. 10, 704–714 10.1038/s41557-018-0033-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lipinski C. A., Lombardo F., Dominy B. W., and Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 10.1016/S0169-409X(00)00129-0 [DOI] [PubMed] [Google Scholar]
- 57. Doak B. C., Over B., Giordanetto F., and Kihlberg J. (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 21, 1115–1142 10.1016/j.chembiol.2014.08.013 [DOI] [PubMed] [Google Scholar]
- 58. Driggers E. M., Hale S. P., Lee J., and Terrett N. K. (2008) The exploration of macrocycles for drug discovery–an underexploited structural class. Nat. Rev. Drug Discov. 7, 608–624 10.1038/nrd2590 [DOI] [PubMed] [Google Scholar]
- 59. Villar E. A., Beglov D., Chennamadhavuni S., Porco J. A., Kozakov D., Vajda S., and Whitty A. (2014) How proteins bind macrocycles. Nat. Chem. Biol. 10, 723–731 10.1038/nchembio.1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wallach I., Dzamba M., and Heifets A. (2015) AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. CoRR. [Google Scholar]
- 61. Hsieh C.-H., Li L., Vanhauwaert R., Nguyen K. T., Davis M. D., Bu G., Wszolek Z. K., and Wang X. (2019) Miro1 marks Parkinson's disease subset and Miro1 reducer rescues neuron loss in Parkinson's models. Cell Metab. 30, 1131–1140 10.1016/j.cmet.2019.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Congreve M., Carr R., Murray C., and Jhoti H. (2003) A “rule of three” for fragment-based lead discovery? Drug Discov Today 8, 876–877 10.1016/S1359-6446(03)02831-9 [DOI] [PubMed] [Google Scholar]
- 63. Calzolai L., and Zahn R. (2003) Influence of pH on NMR structure and stability of the human prion protein globular domain. J. Biol. Chem. 278, 35592–35596 10.1074/jbc.M303005200 [DOI] [PubMed] [Google Scholar]
- 64. Feng B. Y., and Shoichet B. K. (2006) A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 1, 550–553 10.1038/nprot.2006.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spagnolli G., Massignan T., Astolfi A., Biggi S., Brunelli P., Libergoli M., Ianeselli A., Orioli S., Boldrini A., Terruzzi L., Maietta G., Rigoli M., Lorenzo N. L., Fernandez L. C., Tosatto L., et al. (2020) Pharmacological protein inactivation by targeting folding intermediates. bioRxiv 10.1101/2020.03.31.018069 [DOI] [PMC free article] [PubMed]
- 66. Baell J. B., and Nissink J. W. M. (2018) Seven year itch: pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem Biol. 13, 36–44 10.1021/acschembio.7b00903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baral P. K., Swayampakula M., Rout M. K., Kav N. N. V., Spyracopoulos L., Aguzzi A., and James M. N. G. (2014) Structural basis of prion inhibition by phenothiazine compounds. Structure 22, 291–303 10.1016/j.str.2013.11.009 [DOI] [PubMed] [Google Scholar]
- 68. Mayer M., and Meyer B. (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem. Int. Ed. Engl. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 69. Chessari G., Buck I. M., Day J. E. H., Day P. J., Iqbal A., Johnson C. N., Lewis E. J., Martins V., Miller D., Reader M., Rees D. C., Rich S. J., Tamanini E., Vitorino M., Ward G. A., et al. (2015) Fragment-based drug discovery targeting inhibitor of apoptosis proteins: discovery of a non-alanine lead series with dual activity against cIAP1 and XIAP. J. Med. Chem. 58, 6574–6588 10.1021/acs.jmedchem.5b00706 [DOI] [PubMed] [Google Scholar]
- 70. Spevacek A. R., Evans E. G. B., Miller J. L., Meyer H. C., Pelton J. G., and Millhauser G. L. (2013) Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 21, 236–246 10.1016/j.str.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Evans E. G. B., Pushie M. J., Markham K. A., Lee H.-W., and Millhauser G. L. (2016) Interaction between prion protein's copper-bound octarepeat domain and a charged C-terminal pocket suggests a mechanism for N-terminal regulation. Structure 24, 1057–1067 10.1016/j.str.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McDonald A. J., Leon D. R., Markham K. A., Wu B., Heckendorf C. F., Schilling K., Showalter H. D., Andrews P. C., McComb M. E., Pushie M. J., Costello C. E., Millhauser G. L., and Harris D. A. (2019) Altered domain structure of the prion protein caused by Cu2+ binding and functionally relevant mutations: analysis by cross-linking, MS/MS, and NMR. Structure 27, 907–922 10.1016/j.str.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Blochberger T. C., Cooper C., Peretz D., Tatzelt J., Griffith O. H., Baldwin M. A., and Prusiner S. B. (1997) Prion protein expression in Chinese hamster ovary cells using a glutamine synthetase selection and amplification system. Protein Eng. 10, 1465–1473 10.1093/protein/10.12.1465 [DOI] [PubMed] [Google Scholar]
- 74. Denisov I. G., and Sligar S. G. (2017) Nanodiscs in membrane biochemistry and biophysics. Chem. Rev. 117, 4669–4713 10.1021/acs.chemrev.6b00690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ahn S., Pani B., Kahsai A. W., Olsen E. K., Husemoen G., Vestergaard M., Jin L., Zhao S., Wingler L. M., Rambarat P. K., Simhal R. K., Xu T. T., Sun L. D., Shim P. J., Staus D. P., et al. (2018) Small-molecule positive allosteric modulators of the β2-adrenoceptor isolated from DNA-encoded libraries. Mol. Pharmacol. 94, 850–861 10.1124/mol.118.111948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Parker C. G., Galmozzi A., Wang Y., Correia B. E., Sasaki K., Joslyn C. M., Kim A. S., Cavallaro C. L., Lawrence R. M., Johnson S. R., Narvaiza I., Saez E., and Cravatt B. F. (2017) Ligand and target discovery by fragment-based screening in human cells. Cell 168, 527–541 10.1016/j.cell.2016.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chilton M., Clennell B., Edfeldt F., and Geschwindner S. (2017) Hot-spotting with thermal scanning: a ligand- and structure-independent assessment of target ligandability. J. Med. Chem. 60, 4923–4931 10.1021/acs.jmedchem.7b00208 [DOI] [PubMed] [Google Scholar]
- 78. Guha R. (2007) Chemical informatics functionality in R. J. Stat. Softw. 18, 1–16 10.1360/jos180001 [DOI] [Google Scholar]
- 79. Orrù C. D., Groveman B. R., Hughson A. G., Manca M., Raymond L. D., Raymond G. J., Campbell K. J., Anson K. J., Kraus A., and Caughey B. (2017) RT-QuIC assays for prion disease detection and diagnostics. Methods Mol. Biol. 1658, 185–203 10.1007/978-1-4939-7244-9_14 [DOI] [PubMed] [Google Scholar]
- 80. Frottin F., Martinez A., Peynot P., Mitra S., Holz R. C., Giglione C., and Meinnel T. (2006) The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteomics 5, 2336–2349 10.1074/mcp.M600225-MCP200 [DOI] [PubMed] [Google Scholar]
- 81. Minikel E. V., Kuhn E., Cocco A. R., Vallabh S. M., Hartigan C. R., Reidenbach A. G., Safar J. G., Raymond G. J., McCarthy M. D., O'Keefe R., Llorens F., Zerr I., Capellari S., Parchi P., Schreiber S. L., et al. (2019) Domain-specific quantification of prion protein in cerebrospinal fluid by targeted mass spectrometry. Mol. Cell. Proteomics 18, 2388–2400, 10.1074/mcp.RA119.001702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haftchenary S., Nelson S. D., Furst L., Dandapani S., Ferrara S. J., Bošković ŽV., Figueroa Lazú S., Guerrero A. M., Serrano J. C., Crews D. K., Brackeen C., Mowat J., Brumby T., Bauser M., Schreiber S. L., et al. (2016) Efficient routes to a diverse array of amino alcohol-derived chiral fragments. ACS Comb. Sci. 18, 569–574 10.1021/acscombsci.6b00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nelson S. D., Wawer M. J., and Schreiber S. L. (2016) Divergent synthesis and real-time biological annotation of optically active tetrahydrocyclopenta[c]pyranone derivatives. Org. Lett. 18, 6280–6283 10.1021/acs.orglett.6b03118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gerry C. J., Hua B. K., Wawer M. J., Knowles J. P., Nelson S. D., Verho O., Dandapani S., Wagner B. K., Clemons P. A., Booker-Milburn K. I., Boskovic Z. V., and Schreiber S. L. (2016) Real-time biological annotation of synthetic compounds. J. Am. Chem. Soc. 138, 8920–8927 10.1021/jacs.6b04614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Usher K. C., Blaszczak L. C., Weston G. S., Shoichet B. K., and Remington S. J. (1998) Three-dimensional structure of AmpC beta-lactamase from Escherichia coli bound to a transition-state analogue: possible implications for the oxyanion hypothesis and for inhibitor design. Biochemistry 37, 16082–16092 10.1021/bi981210f [DOI] [PubMed] [Google Scholar]
- 86. Bordoli L., and Schwede T. (2012) Automated protein structure modeling with SWISS-MODEL workspace and the protein model portal. Methods Mol. Biol. 857, 107–136 10.1007/978-1-61779-588-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hendlich M., Rippmann F., and Barnickel G. (1997) LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol. Graph Model. 15, 359–363 10.1016/S1093-3263(98)00002-3 [DOI] [PubMed] [Google Scholar]
- 88. Kingma D. P., Ba J., and Adam A. (2017) Method for stochastic optimization. [Google Scholar]
- 89. Bruns R. F., and Watson I. A. (2012) Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 55, 9763–9772 10.1021/jm301008n [DOI] [PubMed] [Google Scholar]
- 90. Pajouhesh H., and Lenz G. R. (2005) Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2, 541–553 10.1602/neurorx.2.4.541 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and source code will be made available in a public GitHub repository: http://github.com/ericminikel/binder_screening.