

Summary
Studies revealing molecular mechanisms underlying neural specification have majorly focused on the role played by different transcription factors, but less on non-nuclear components. Earlier, we reported mitochondrial superoxide dismutase (SOD2) to be essential for self-renewal and pluripotency of mouse embryonic stem cells (mESCs). In the present study, we found SOD2 to be specifically required for neural lineage, but not the meso- or endoderm specification. Temporally, SOD2 regulated early neural genes, but not the matured genes, by modulating mitochondrial dynamics—specifically by enhancing the mitochondrial fusion protein Mitofusin 2 (MFN2). Bio-complementation strategy further confirmed SOD2 to enhance mitochondrial fusion process independent of its antioxidant activity. Over-expression of SOD2 along with OCT4, but neither alone, transdifferentiated mouse fibroblasts to neural progenitor-like colonies, conclusively proving the neurogenic potential of SOD2. In conclusion, our findings accredit a novel role for SOD2 in early neural lineage specification.
Subject Areas: Developmental Genetics, Molecular Genetics, Developmental Neuroscience
Graphical Abstract
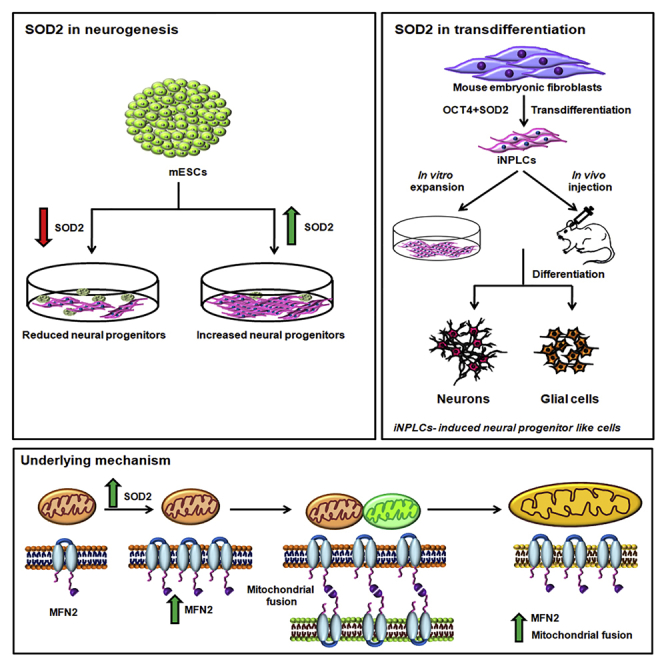
Highlights
-
•
SOD2 is essential for early neural differentiation of mESCs
-
•
Up-regulation of MFN2, but not MFN1, underlies SOD2-mediated neurogenesis
-
•
Antioxidant enzymatic activity of SOD2 is dispensable for mitochondrial fusion
-
•
Overexpression of SOD2 with OCT4 transdifferentiate MEFs to iNPLCs
Developmental Genetics; Molecular Genetics; Developmental Neuroscience
Introduction
Neurogenesis is an intricate developmental process that occurs as early as E10 in mice (Martynoga et al., 2012). The process includes a series of events including the formation of neural progenitor cells and their proliferation, migration, further differentiation, and commitment to form a functional circuitry of neurons across the brain (Stiles and Jernigan, 2010). Although the existence of neural stem cells (NSCs) in humans is highly debated, there is abundant evidence to support the existence of NSCs and neurogenesis in adult rodents (Eriksson et al., 1998; Toni et al., 2007). NSCs in adult brain are predominantly present in the sub-ventricular zone (SVZ) of the lateral ventricles and in the hippocampal dentate gyrus region (Alvarez-buylla and Garcı, 2002; Bond et al., 2015). Various intrinsic and extrinsic factors including transcription factors (TFs), epigenetic modulators, neurotropic factors, and neurogenic niche govern the generation and commitment of neurons in the developing brain.
Being a highly organized and energy-demanding process, neurogenesis requires active involvement of dynamic organelles like mitochondria to provide unlimited supply of energy (Agostini et al., 2016; Arrázola et al., 2019; Beckervordersandforth et al., 2017). During the course of energy production, mitochondria release superoxide radicals, which are further converted to hydrogen peroxide (Turrens, 2003). Mitochondrial SOD2, also known as manganese superoxide dismutase, is the major superoxide scavenging enzyme residing in the mitochondrial matrix (Wang et al., 2018). Upon selective knockdown of SOD2 in adult mice, although liver and other endocrine organs did not show any abnormality, the animal developed cardiomyopathy and severe damage in the brain leading to death within 3 weeks (Oh et al., 2012). Specific knockout of SOD2 in the brain also leads to encephalopathy and consequent death of the mice perinatally (Izuo et al., 2015). SOD2 level is highest in the central nervous system and is profusely expressed during early neural development, at as early as E7.5–8.5 in mice (Yon et al., 2011). Neural protection obtained by mitochondrial deacetylase SIRT3 has also been reported to be due to increased expression of SOD2 (Cheng et al., 2016). Increased SOD2 levels have also relieved the symptoms of Alzheimer's (Massaad et al., 2009) and Parkinson's disease (Klivenyi et al., 1998) in mouse models. Although the neuroprotective role of SOD2 is well appreciated, its role in neural lineage determination remains elusive.
Recent studies revealed mitochondrial dynamics to be one of the key regulators of neural fate specification. Variation in mitochondrial mass and structure, changes in fission and fusion events, and mitochondrial bioenergetics are important for maintaining neural tissue homeostasis (Van Laar and Barmen, 2013). Involvement of mitochondrial dynamics in neural fate decision is well appreciated during embryonic cortical neurogenesis. Although NSCs in the ventricular zone feature elongated mitochondria, mitochondrial fission dictates progenitor fate in the SVZ and further maturation of neural progenitors to neurons re-exhibits mitochondrial fusion process (Khacho and Slack, 2018; Khacho et al., 2016, 2019; Laaper and Jahani-Asl, 2018). Enhanced mitochondrial fragmentation drives NSCs to neuronal differentiation by mitochondria-to-nuclear retrograde signaling and regulation of nuclear gene expression (Khacho and Slack, 2018; Khacho et al., 2016; Laaper and Jahani-Asl, 2018). Loss of function of mitochondrial fusion or fission proteins has been shown to result in abnormal neuronal development with compromised synapse formation. For instance, mitochondrial fusion protein Mitofusin (MFN2) plays a significant role in neuronal maturation and synapse formation (Fang et al., 2016). Conditional knockout of MFN2 has been shown to result in smaller cerebella and motor deficits (Chen et al., 2007). Similarly, fission protein (DRP1)-deficient mice showed impaired forebrain development (Ishihara et al., 2009). Together, these studies illustrate the importance of mitochondrial dynamics in neuronal development. Despite the growing curiosity to unravel the role of mitochondrial parameters in neural commitment, very little is known about the mitochondrial resident protein SOD2 in neural fate specification.
In the present study we show SOD2 to be essential for early NPC generation, but not for mesoderm or endoderm specification. The neurogenic potential of SOD2 is further established by its ability to override the BMP4-mediated neural inhibition. Attempt to understand the underlying molecular mechanism showed increased mitochondrial fusion by SOD2, mediated by enhanced mitochondrial fusion protein MFN2, but not MFN1. Finally, over-expression of SOD2 along with OCT4 in mouse embryonic fibroblast (MEF) is sufficient to generate induced neural progenitor like cells (iNPLCs), conclusively indicating the neural propensity conferred by SOD2.
Results
SOD2 Expression Is Essential for Neural Fate Specification of mESCs
In our previous study we had reported the role of SOD2 in maintaining STAT3-mediated pluripotency of mouse embryonic stem cells (mESCs) (Sheshadri et al., 2015). In order to understand the role of SOD2 in lineage specification, we analyzed the expression of SOD2 in tri-lineage differentiation of mESCs and found SOD2 to be specifically up-regulated in neural differentiation but not in endo- and mesodermal differentiation of mESCs (Figures 1A and 1B). The observation was further substantiated by the expression of SOD2 in the adult brain tissue of 3-week-old mice (Figure S1A). Directed differentiation of mESCs to neurons and oligodendrocytes showed SOD2 to be specifically detected in neural differentiation, but not in oligodendrocyte differentiation (Figures 1C and S1B), indicating that SOD2 is probably not crucial in other ectodermal sub lineages. Forced expression of SOD2 during neural differentiation demonstrated enhanced expression of neural specific markers—Pax6, Sox1, Zic1 and Foxg1 at transcript level (Figure 1D) and FOXG1 at protein level (Figure 1E). Addition of BMP4, a potent inhibitor of neural differentiation, to neural induction media reduced the expression of neural genes and Sod2S, but forced expression of SOD2 in this condition, induced the expression of early neural markers, suggesting that SOD2 facilitates neural differentiation from mESCs even under non-permissive culture condition (Figure 1F).
Figure 1.

SOD2 Is Essential for the Expression of Neuroectodermal Genes
(A and B) mRNA (A) and protein (B) levels of SOD2 in endoderm, mesoderm, and neural differentiation of mESCs. mRNA levels are plotted as mean ± SE of biological triplicates and statistical significance has been calculated using paired Student's t test, ∗p < 0.05, ∗∗p < 0.01. (C) mRNA expression of SOD2 and specific markers in neural (i) and oligodendrocyte (ii) differentiation of mESCs. Comparison of mRNA (D) and protein (E) levels of neural markers in SOD2 over-expression against the vector control. (F) Comparison of mRNA levels of neural markers in the vector control, with and without BMP4 treatment, and SOD2 over-expression with BMP4 treatment. (G) Transcript analysis of germ layer markers in endodermal (i), mesodermal (ii), and ectodermal (iii) differentiation from mESCs upon SOD2 knockdown. Expression analysis of early neural marker SOX1 upon SOD2 knockdown in early neural differentiation of mESCs by immunoblotting (H) and immunofluorescence (I). Scale bar represents 50 μm. Un-cropped full western blot images are available in Data S1. List of primers used for transcript analysis and antibodies used for protein detection are available in Tables S1 and S2, respectively.
To further examine the essentiality of SOD2 in lineage specification and neural induction, we introduced short hairpin RNA (shRNA) specifically targeting SOD2 into mESCs and differentiated them into three derm lineages. Knockdown of SOD2 specifically compromised neural differentiation as evidenced by the down-regulation in the expression of neural lineage markers—Zic1, Sox1 and Foxg1 (Figure 1G) at transcript and SOX1 at protein levels (Figures 1H and 1I). However, the genes representing the expression of endo- and mesodermal differentiation remained unaltered, suggesting the dispensability of SOD2 in these conditions. To assess whether the effect is specific to SOD2, we tested the role of another SOD family member SOD1 in neural commitment. Neural differentiation of mESCs transduced with SOD1 shRNA (Figure S2A) showed no reduction in transcript levels of Pax6, Sox1, and Nestin compared with scramble control (Figure S2B). Immunofluorescence for neural marker SOX1 also failed to show any modulation upon Sod1 shRNA (Figure S2C). We thus ruled out the involvement of SOD1 in mESC neural differentiation. Taken together, these results show that SOD2 is essential for neural commitment of mESCs.
SOD2 Specifically Modulates Early Neural Differentiation
To ascertain the stage-specific role of SOD2 during neural differentiation, we segregated the differentiation process into two stages—early neural differentiation comprising neural progenitors and late neural differentiation majorly consisting of matured neurons (Figure S1C). During the course of differentiation, biphasic expression pattern of SOD2 was observed wherein lower expression of SOD2 was observed during early neural differentiation compared with mESCs and higher expression during late neural differentiation (Figure 2A). The observed biphasic expression was further supported by a similar trend in superoxide dismutase activity of SOD2 (Figure S1D). Estimation of ROS levels using DCFHDA staining during differentiation showed lower ROS levels in cells of early neural differentiation compared with mESCs and cells of late neural differentiation (Figure 2B), similar to biphasic SOD2 expression. Over-expression of SOD2 using doxycycline-inducible system (Figure 2C) during early neural differentiation showed enhanced expression of early neural markers Foxg1, Zic1, Pax6, and Sox1 (Figure 2Di), but the over-expression of SOD2 during late neural differentiation resulted in unaltered expression of matured neural marker Map2 (Figure 2Dii). However, persistent expression of early neural marker was observed even during the late-stage differentiation when doxycycline-induced SOD2 expression was elicited during late-stage neural differentiation (Figure 2Dii). This suggested SOD2 to specifically modulate early neural differentiation. To further probe into the essentiality of SOD2 in the two stages of neural differentiation, we generated inducible shRNA and knocked down SOD2 during early and late neural differentiation (Figure 2E) and analyzed the expression of respective neural markers. We found the expression of early neural markers—Foxg1, Zic1, Pax6, and Sox1—to be compromised upon SOD2 knockdown during early neural differentiation (Figure 2Fi), but the expression of matured neural markers—Tau, Nurr1, Map2 and NeuN—remained unchanged upon SOD2 knockdown during late neural differentiation (Figure 2Fii). Altogether, these findings indicate that SOD2 promotes neural differentiation by activating the expression of early neuroectodermal genes.
Figure 2.

SOD2 Specifically Modulates Early Neural Differentiation
(A) Modulation of protein expression of SOD2 across neural differentiation of mESCs. (B) Analysis of ROS levels using DCFHDA staining during early and late neural differentiation of mESCs. (C) mESCs were transfected with inducible SOD2 over-expression construct and 48 h after addition of doxycycline, cells were harvested and expression of SOD2 analyzed by western blotting. (D) Gene expression analysis of specific neural markers upon SOD2 over-expression induced during (i) early neural differentiation (days 0–3) and (ii) late neural differentiation (days 3–7). (E) mESCs were transfected with inducible SOD2 shRNA construct and 48 h after addition of doxycycline, cells were harvested and expression of SOD2 analyzed by western blotting. (F) Transcript analysis of specific neural markers upon SOD2 knockdown induced during (i) early neural differentiation (days 0–3) and (ii) late neural differentiation (days 3–7). Un-cropped full western blot images are available in data S1. List of primers used for transcript analysis and antibodies used for protein detection are available in Tables S1 and S2, respectively.
SOD2 along with OCT4 Is Sufficient to Transdifferentiate MEFs to Neural Progenitor-like Cells
In our previous study we showed a neural transcription factor ZIC3 along with Yamanaka factors to directly transdifferentiate human fibroblast to neural progenitor-like cells (Kumar et al., 2012). Since our results indicated the requirement of SOD2 for neural commitment of mESCs, we wanted to understand whether SOD2 has a similar ability to transdifferentiate MEFs. To address this, we transduced MEFs with various combinations of reprogramming factors OCT4 (O), SOX2 (S), and KLF4 (K) along with SOD2. Except for OSK combination, colonies appeared in all other combinations tested at as early as 12 days (Figure S3A). Interestingly, a minimal combination of OCT4 and SOD2 was sufficient to generate the colonies (Figure 3A), but not individually (data not shown). We continued our further characterization with clones obtained by transducing minimal cocktail of OCT4 and SOD2 transgenes. Expression of endogenous Oct4 was not detected in these cells, although transgene expression of Oct4 and Sod2 was witnessed (Figures S3B and S3C). Lack of fibroblast genes Col1A1, Col3A1, and Dkk1 indicated that the cells had indeed lost their fibroblast identity (Figure S3D). Continuing the characterization, we expanded these colonies on feeder-free conditions and performed transcript analysis of the pluripotency markers—Oct4, Sox2, Nanog, and Rex1—and found that they lacked the expression of pluripotent genes compared with an established iPSC line (Figure 3B). Also, epiblast specific gene Fgf5 was not expressed in the colonies (Figure 3C).
Figure 3.

SOD2 along with OCT4 Transdifferentiates MEFs to iNPLCs
(A) Phase contrast images of mouse embryonic fibroblasts (MEF) and colonies obtained from OCT4 and SOD2-mediated transdifferentiation.
(B–K) Characterization of three independent clones obtained by OCT4 and SOD2-transduced cells. Scale bar represents 50 μm. (B) Gene expression analysis of pluripotency markers—Oct3/4, Nanog, and Rex1. (C) Transcript analysis of Epiblast specific marker Fgf5. Gene expression analysis of (D) Endodermal markers—Sox17, Cxcr4, and Gata6; (E) Mesodermal markers—Mixl1, Fkl1, and Vegf; and (F) Neural markers—Pax6, Foxg1, and Zic1 in iNPLC colonies. (G) Flow cytometric analysis of neural specific marker ZIC1 in MEF and i-NPLCs. (H) Protein levels as analyzed by western blot of early neural markers—ZIC1, SOX1, PAX6, and FOXG1, oligodendrocyte progenitor marker—OLIG2, and fibroblast marker—VIMENTIN in i-NPLCs. (I) Representative immunofluorescence images of neural markers—SOX1, ZIC1, PAX6—and oligodendrocyte progenitor marker—OLIG2—in i-NPLCs. (J) Microarray analysis showing the global gene expression analysis of mouse brain derived neural progenitor cells and i-NPLCs. (K) Comparison of mRNA levels of early neural markers between endogenous mouse neural progenitors obtained P2 infant brain and i-NPLCs with respect to MEF control.
(L–N) Transcript analysis of neural genes at different time points in MEFs transduced with either Oct4 and WT SOD2 (L) or OCT4 and mutSOD2 (M). Analysis of neural transcripts in cells cultured in NSC medium (N). (O) Immunofluorescence analysis of NESTIN in MEFs transduced with either OCT4 and WT or OCT4 and mutSOD2. Scale bar represents 100 μm. Mean ± SE of biological triplicates, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control MEF. Un-cropped full western blot images are available in data S1. List of primers used for transcript analysis and antibodies used for protein detection are available in Tables S1 and S2, respectively.
Furthermore, to establish the identity of these cells, we performed transcript analysis of tri-lineage specific genes in three individual clones. Although there was minimal expression of endodermal markers (Figure 3D) and mesodermal markers (Figure 3E) in these cells, there was a high expression of neural lineage genes Pax6, Foxg1, and Zic1 (Figure 3F). Flow cytometric analysis for neural specific marker ZIC1 showed that 94.4% of these cells expressed ZIC1 as against 5.47% in MEF (Figure 3G). Immunoblotting revealed expression of neural specific proteins—SOX1, PAX6, FOXG1, and ZIC1—and the oligodendrocyte marker, OLIG2, with a concomitant down-regulation of the fibroblast marker, VIMENTIN (Figure 3H), suggesting the loss of fibroblast memory. Nuclear expression of SOX1, ZIC1, OLIG2, and PAX6 was also observed in these colonies by immunofluorescence (Figure 3I). This suggested that over-expression of OCT4 and SOD2 conferred neural identity to the fibroblast cells and therefore, henceforth these colonies will be designated as induced neural progenitor like cells (iNPLCs). iNPLCs exhibited high morphological homogeneity and could be stably expanded for more than 20 passages without compromise in the expression of neural progenitor genes. With passages, there was decrease in the expression of SOD2 transgene (Figure S3E). In order to determine how similar iNPLCs were to mouse brain derived neural progenitor cells, we performed a global gene expression analysis of iNPLCs and neural progenitor cells (NPCs) derived from P2 mouse brain. Analysis showed that, of 35,715 genes analyzed, expression of 22,910 genes from iNPLCs and P2 mouse NPCs overlapped, accounting for 64% similarity between the two populations (Figure 3J). PCR analysis of a few neural progenitor genes further confirmed a similar expression pattern between iNPLCs and P2 mouse NPCs (Figure 3K). These results show the ability of SOD2 along with OCT4 to transdifferentiate MEFs to iNPLCs.
A hallmark of a bona fide NPLC is its ability to differentiate to matured neurons and glia in in vitro and in vivo conditions. On providing appropriate cues, iNPLCs could efficiently differentiate to neurons that expressed Map2, Tau, and Nurr1 in vitro (Figure 4A). Also, in glial differentiation conditions, the cells expressed Gfap and S100β (Figure 4B). Differentiated iNPLCs also stained positive for matured neural markers—SYNAPTOPHYSIN, NF200kDa—and glial protein—GFAP (Figure 4C). These results validated the in vitro differentiation ability of the iNPLCs. Injection of GFP-tagged iNPLCs in the cortex of the neonatal mouse brain and subsequent analysis after 4 weeks showed GFP-positive cells in mouse brain co-expressing matured neural markers MAP2, TAU, TUJ1 (Figure 4D). In vivo maturation of iNPLCs thereby conclusively established their neural identity. Thus, the transdifferentiation of MEFs to iNPLCs further reiterated the neurogenic potential of SOD2.
Figure 4.

iNPLCs Differentiate to Mature Neurons In Vitro and In Vivo
(A and B) mRNA levels of matured neural markers (A) and glial markers (B) in iNPLC clones differentiated in vitro. (C) Representative immunofluorescence image of matured neuronal and glial markers in iNPLC clones differentiated in vitro. (D) Representative immunohistochemistry image showing in vivo maturation of injected iNPLC cells harboring GFP plasmid. Mean ± SE, n = 3 independent experiments. Scale bar represents 50 μm. List of primers used for transcript analysis and antibodies used for protein detection are available in Tables S1 and S2, respectively.
SOD2 Regulates Neurogenesis through Mitochondrial Remodeling
The combined results from mESC differentiation and the transdifferentiation indicate that SOD2 strongly favors the neural lineage over other derm layers during differentiation. To understand the mechanism behind SOD2-mediated neural commitment, we analyzed the conventional role of SOD2 such as altering cell proliferation and quenching of ROS. There was no significant change in cell proliferation upon SOD2 knockdown during neural differentiation as evidenced by Ki67 staining, BrdU incorporation assay, and cell cycle analysis (Figures S4A–S4C). We then speculated that the abrogation of neural differentiation of mESCs upon SOD2 knockdown could be due to increased ROS insult. To resolve this, we knocked down SOD2 and treated the cells with N-acetyl cysteine (NAC), a ROS quencher, during the neural differentiation of mESCs. Gene expression analysis showed no appreciable rescue in the expression of neural markers upon NAC administration in SOD2 knockdown background (Figure S4D). This suggested the dispensability of antioxidant defense in SOD2-mediated neural commitment. In all, these results indicated that the conventional role of SOD2 in regulating cell proliferation and ROS fails to address the mechanism of SOD2-mediated neurogenesis.
As SOD2 is a nuclear-encoded mitochondrial resident enzyme, we sought to observe any modulation in mitochondrial architecture and dynamics as the underlying mechanism. In our initial attempt we transfected MEFs with plasmid encoding DsRed that localizes to mitochondria (mito-DsRed). Fluorescence imaging clearly showed an increase in mitochondrial length upon over-expression of SOD2 (Figure S5A). We quantified the mitochondrial length and categorized them into short: < 0.02 μm; medium: 0.02–0.04 μm; and long: > 0.04 μm. Results clearly indicated an increase in the number of long mitochondria upon SOD2 over-expression, with a concomitant decrease in the short mitochondria (Figure S5B). Quantifying the number of mitochondrial contacts in a time frame showed SOD2 over-expression to increase the number of contacts between mitochondria at 40-, 80-, and 120-s time intervals compared with control (Figure S5C).To further prove this in a cell of neuronal origin, we inhibited SOD2 expression in P40H1, an immortal cell line derived from the hippocampus of a 40-day-old adult immortomouse (Demaegdt et al., 2009) and labeled mitochondria with mito-DsRed. After 72 h of SOD2 inhibition, we observed mitochondria were more fragmented when compared with the scrambled control (Figure 5A). We quantified the length of these mitochondria and found a very significant increase in the number of short mitochondria and a distinct decrease in the number of long mitochondria upon knockdown of SOD2 expression (Figure 5B). On the other hand, ectopic expression of SOD2 showed a significant decrease in the number of short mitochondria and increase in the number of long mitochondria (Figures 5C and 5D). To evaluate the contribution of conventional role of SOD2 in mitochondrial fusion, we generated SOD2 mutant (mutSOD2) constructs wherein the active site residues were mutated to D183N and W185F. Over-expression of mutSOD2 failed to quench the mitochondrial superoxide radicals compared with WT SOD2, as seen by the MitoSox staining (Figure 5E). We further validated the mutSOD2 construct by overexpressing WT and mutSOD2 in the background of SOD2-shRNA that targets the endogenous 3′ UTR and not the overexpressed SOD2 (Figures S6A and S6B). Surprisingly, over-expression of mutSOD2 also enhanced mitochondrial length similar to that of WT wherein significant decrease in the number of short mitochondria and a very significant increase in the number of long mitochondria were observed (Figures 5C and 5D). This observation supported the unconventional role of SOD2—independent of its antioxidant activity—in regulating mitochondrial morphology.
Figure 5.

SOD2 Enhances Mitochondrial Fusion Process
(A) Representative confocal images showing modulation in mitochondrial architecture upon SOD2 knockdown in P40H1 cell line. The enlarged image of the boxed region is represented beside each main image. (B) Quantification of mitochondrial length upon SOD2 knockdown in P40H1 cell line. (C) Representative confocal images showing modulation in mitochondrial architecture upon over-expression of wild-type and redox activity mutant constructs of SOD2 in P40H1 cell line. (D) Change in mitochondrial length upon the over-expression of WT and mutant SOD2 in P40H1 cell line. (E) Mitochondrial superoxide levels upon over-expression of SOD2 wild-type and mutant SOD2 constructs as quantified using Mitosox Red staining. (F) Quantification of percentage of mitochondrial contacts upon SOD2 over-expression. (G) (i) Schematic representation depicting the bio-complementation assays used to quantify the mitochondrial fusion process and (ii) the relative fold change in mitochondrial fusion upon SOD2 over-expression as assayed using bio-complementation assay. Data representative of mean ± SE, n = 3 independent experiments, ∗∗p < 0.01, ∗∗∗p < 0.001. Scale bar represents 100 pixels. Twelve images per condition were analyzed and length of ∼400 mitochondria was calculated. See also Videos S1 and S2.
To understand transient fusion events that predict enhanced mitochondrial fusion, we quantified mitochondrial contact events and found that SOD2 over-expression significantly enhanced the transient mitochondrial fusion (∼4.5-fold increase in mitochondrial contact compared with control) (Figure 5F, Videos S1 and S2 related to Figure 5). Videos S1 and S2 show the frequency of contacts between mitochondria in control and pMIG SOD2 over-expressing P40H1 cells, respectively. As a direct evidence for mitochondrial fusion, we performed a Venus bio-complementation-based mitochondrial fusion assay where the fusion of mitochondria would lead to Venus fluorescence by the bio-complementation of N terminus (NVZL) and C terminus (CVZL) of Venus localized in two different mitochondria (Figure 5Gi). As a proof of experiment, combined expression of NVZL and CVZL in HEK 293Ts generated venous fluorescence, but not transfected individually (Figure S5D). The specificity of mitochondrial isolation, used for bio-complementation assay, was shown by enrichment of mitochondrial-specific marker COX-6A (Figure S5E). Initial experimental validation in MEF showed increased mitochondrial fusion upon SOD2 expression (Figure S5F). Upon over-expression of SOD2 in neuronal cell line P40H1, we observed a highly significant increase in Venus fluorescence implying enhanced mitochondrial fusion compared with vector alone control (Figure 5Gii). These results suggest that SOD2 positively regulates mitochondrial fusion. These mitochondrial changes upon SOD2 manipulation led us to hypothesize a role for SOD2 in enhancing neural commitment via mitochondrial fusion.
SOD2 Regulates Neurogenesis by MFN2-Mediated Mitochondrial Fusion
Having deciphered the effect of SOD2 on mitochondrial fusion, we wanted to understand the changes in mitochondrial fusion and fission proteins upon SOD2 over-expression. We found up-regulation in the expression of fusion protein MFN2, but no modulation in MFN1 and fission protein FIS1, upon SOD2 over-expression (Figure 6A). This up-regulation of Mfn2 was evident upon over-expression of WT SOD2 and mutSOD2 at the mRNA and protein levels (Figure S7A and S7C) and SOD2-deficient cells expressed reduced amounts of Mfn2 (Figure S7B). This led us to hypothesize that MFN2 is an intermediate player in SOD2-mediated neural commitment. To address this, we initially knocked down MFN1 and MFN2 (Figure S7D) individually and simultaneously and looked at the mitochondrial architecture. Mitochondria exhibited fragmentation in each of these conditions compared with the controls, which corroborated the previous results (Chen et al., 2003). Interestingly, SOD2 over-expression rescued the mitochondrial length when MFN2 was inhibited individually or together with MFN1. However, the rescue in mitochondrial length upon SOD2 over-expression was not observed when MFN1 alone was knocked down. These results suggested that SOD2 exerts its effect on mitochondrial fusion by regulating MFN2 specifically and not MFN1 (Figure 6B). To further determine whether the antioxidant activity of SOD2 is required for MFN2-mediated fusion process, WT and mutSOD2 were over-expressed in cells where MFN2 is either inhibited individually or combined with MFN1. Surprisingly, similar to SOD2 WT, we found mutSOD2 to rescue the mitochondrial length only in those cells where MFN2 was inhibited but not in the cells where MFN1 was knocked down (Figure 6B). The observation was further supported by the quantitative assessment of mitochondrial length using mito DsRed transfected images under all the genetic manipulation conditions in P40H1 cells (Figure 6C). These results demonstrated the dispensability of the antioxidant activity of SOD2 in mitochondrial fusion process. To validate this observation, we sought after transdifferentiation approach wherein we overexpressed mutSOD2 along with OCT4 to transdifferentiate MEFs. Post over-expression, we observed generation of colonies resembling those generated with WT SOD2 (Figure S3F). Upon transcript analysis, cells overexpressing mutSOD2 and OCT4 showed expression of neural markers Pax6, Foxg1, and Zic1 similar to those transduced with OCT4 and WT SOD2 (Figures 3L and 3M). Culturing these cells in NSC medium further induced neural signatures that were undetected in MEF control (Figure 3N). Transduction of SOD2 WT and mutSOD2 individually along with OCT4 resulted in generation of NESTIN-positive cells, thus confirming their NPLC identity (Figure 3O) and thus reiterated the antioxidant activity of SOD2 to be trivial in regulating neural commitment.
Figure 6.

SOD2-Induced Neurogenesis Is by Enhancing Mitochondrial Fusion Process
(A) Protein levels of mitochondrial fission (FIS1) and fusion (MFN1 and MFN2) markers upon SOD2 over-expression. (B) Representative images of mitochondria upon WT or mutSOD2 over-expression in cells where Mfn1, Mfn2, or both are knocked down. (C) Quantification of mitochondrial length in the conditions as mentioned in the (B). (D) mRNA levels of early neural markers (Foxg1 and Nestin) in P40H1 cells with conditions wherein SOD2 is over-expressed in cells with Mfn2 knockdown. (E) Representative immunofluorescence images of NESTIN in neural cells with Mfn 1, Mfn 2 individual or simultaneous knockdown and subsequent SOD2 over-expression. Scale bar represents 50 μm. (F) Relative fold change in the percentage of NESTIN-positive cells upon knockdown of Mfn 1, Mfn2, or both, and subsequent SOD2 over-expression. (G) Transcript analysis showing rescue in neural markers Sox1, Nestin, and Zic1 of mESCs differentiated to neural progenitors under Mfn2 shRNA and over-expression of SOD2 conditions. (H) Immunofluorescence images of NESTIN and SOX1 showing rescue in neural differentiation upon over-expression of SOD2 in Mfn2 knockdown background. (I) Transcript analysis of Nestin and (J) immunofluorescence for NESTIN and SOX1 showing rescue in neural differentiation of mESCs upon over-expression of antioxidant activity mutant SOD2 under Mfn2 shRNA condition. Scale bar represents 100 μm. Data representative of mean ± SE, n = 3 independent experiments, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Number of mitochondria counted per condition ∼400. Un-cropped full western blot images are available in data S1. List of primers used for transcript analysis and antibodies used for protein detection are available in Tables S1 and S2, respectively.
To convincingly demonstrate that the role of SOD2 in neurogenesis is via specific modulation of MFN2 expression, we analyzed the expression of neural genes in P40H1 over-expressing SOD2 in MFN1/MFN2 knockdown conditions. There was no detectable change in the expression of neural marker NESTIN in the conditions where either MFN1 was knocked down or SOD2 over-expressed in MFN1 knockdown condition. On the other hand, there was a clear decrease in the expression of Foxg1 and Nestin at the transcript level (Figure 6D) and number of NESTIN-positive cells upon MFN2 knockdown, which was significantly rescued upon over-expression of SOD2 (Figures 6E and 6F). To authenticate the observation, we choose an independent model system of differentiating mESCs to NPCs. Similar inhibition of MFN2 during differentiation of mESCs to NPCs led to compromised expression of neural progenitor markers Sox1, Nestin, and Zic1 (Figure 6G) at the transcript level and SOX1 and NESTIN at the protein level (Figure 6H). Over-expression of SOD2 in MFN2 knockdown background led to rescue in the expression of these markers similar to the controls (Figures 6G and 6H). To test whether the SOD's antioxidant activity is required for the rescue in the expression of these genes, we over-expressed mutSOD2 in place of WT SOD2 in cells lacking MFN2 expression and observed a similar rescue in the transcript level of Nestin (Figure 6I) and SOX1 and NESTIN at the protein level to that of WT SOD2 (Figure 6J). These results implicate a potential role for SOD2 in neurogenesis by modulating the mitochondrial length and dynamics via the regulation of MFN2, independent of its antioxidant activity.
Discussion
Mitochondrial resident SOD2 is traditionally viewed as an enzyme involved in dismutation of superoxide anion to hydrogen peroxide. The role played by SOD2 beyond its antioxidant activity, especially in terms of derm layer specification during development, remains largely unexplored. In 2012, Hou et al. reported that SOD2 knockout mice have significantly lesser proliferating NPCs compared with WT. This difference was attributed to increased superoxide flashes, in cells lacking SOD2, ultimately resulting in abrogation of neural progenitors in embryonic mouse cortex (Hou et al., 2012). It has also been shown that increased expression of SOD2 attenuates apoptosis of mesodermal cardiomyocytes and ameliorates myocardial infarction (Long et al., 2017). Despite these initial clues, a comprehensive understanding of SOD2's involvement in lineage specification remained elusive.
Previous studies from our laboratory and others had elucidated the role of SOD2 in maintaining stem cell pluripotency (Sheshadri et al., 2015; Solari et al., 2015). Following this observation, we expected that its inhibition would enhance tri-lineage differentiation of mESCs. However, upon knockdown of SOD2, mESCs failed to acquire neural fate without impeding the expression of meso- and endoderm lineage genes. These observations are well corroborated with previous in vivo studies wherein mice lacking SOD2 specifically in brain, but not other antioxidant enzymes, showed perinatal death and exhibited spongiform neurodegeneration in motor cortex, hippocampus, and brainstem (Izuo et al., 2015). Surprisingly, although knockdown and over-expression of SOD2 during differentiation of mESCs showed a modulation in neural differentiation at early stage, the results from normal differentiation condition showed a reduction in SOD2 during early stage of differentiation in contrast to anticipated up-regulation. The probable reason for this conflicting observation is that, as SOD2 is also involved in maintenance of pluripotency of mESCs, down-regulation of SOD2 is necessary to exit from the pluripotent state to get differentiated. Therefore, a critical level of SOD2 is essential for conferring neural fate and expressing SOD2 over and above this level resulted in persistent maintenance of enhanced expression of neural progenitor genes during maturation with concomitant decrease in matured genes. So, during normal neural differentiation decreased expression of SOD2 is probably implemented to favor the transition of NPCs to matured neurons. To test the neurogenic potential of SOD2, we sought a more stringent approach, i.e., transdifferentiation, using SOD2. In the present study, a minimal cocktail of SOD2 and OCT4 was sufficient to transdifferentiate MEF to iNPLC, advocating the neurogenic potential of SOD2. As reported earlier, the probable role of OCT4 in this cocktail is to shape chromatin accessibility and pave way for other transcription factors to bind to its promoter (King and Klose, 2017). Further differentiation of these cells to neurons in vitro and in vivo conclusively established the ability of SOD2 to confer neural identity to terminally differentiated MEFs. These observations conclusively advocated a pivotal role for SOD2 in conferring neural proclivity.
Having convincingly precluded cell proliferation and antioxidant defense as plausible mechanisms, we contemplated on modulation in mitochondrial architecture and dynamics as an alternative mechanism underlying SOD2-mediated neural differentiation. Liu et al., demonstrated that, by increasing the mitochondrial membrane potential, transdifferentiation of human dermal stem cells to functional neurons could be achieved (Liu et al., 2019). Previous studies also reported mitochondrial dynamics established through regulated fission and fusion to dictate the fate decisions of neural stem cells (Khacho and Slack, 2018; Khacho et al., 2016, 2019). These observations compelled us to investigate the role of mitochondrial dynamics as a probable mechanism underlying SOD2-mediated neurogenesis. An independent report by Zhao et al. showed SOD2 to increase neurite growth in primary cortical neurons by decreasing fission protein DRP1R and in turn reducing mitochondrial fragmentation (Zhao et al., 2019). However, in our present study, by multiple approaches, we found that SOD2 enhances mitochondrial fusion by specifically up-regulating MFN2, but not MFN1. Probably, this up-regulation of MFN2 might be a chosen mechanism to favor neurogenesis by SOD2.
MFN2 is a mitochondrial outer membrane protein that mediates outer mitochondrial membrane (OMM) fusion. Several studies have shown the importance of MFN2 in neuronal maturation (Fang et al., 2016), cerebellar development (Chen et al., 2007), and synapse formation (Fang et al., 2016). Studies by Pham et al. showed specific loss of Mfn2, but not Mfn1, to impair dopaminergic neuron function and result in motor deficit (Pham et al., 2012). Compared with Mfn2, levels of Mfn1 are very low in the nervous system. Probably owing to this, conditional knockout of Mfn2, but not Mfn1, results specifically in neurodegeneration. In our study also, we observed knockdown of Mfn2, but not Mfn1, to result in reduced neural gene expression. Also, in corroboration with previous reports, knockdown of either Mfn1 or Mfn2 resulted in mitochondrial fragmentation, but SOD2 had the ability to rescue mitochondrial fragmentation only in Mfn2 knockdown condition, reiterating SOD2 to specifically regulate Mfn2-mediated effects and this turns out to be one of the underlying mechanisms in SOD2-mediated neurogenesis. However, our attempts to understand how SOD2 influences the MFN2 activity neither showed SOD2's physical interaction with MFN2 and modulation of its fusion activity nor did it prevent MFN2 from undergoing protein degradation (data not shown).
In conclusion, our work defines an important antioxidant-independent role for SOD2 in specifically up-regulating the fusion protein MFN2 and promoting neural specification. This property of SOD2 has assisted in generating a novel cocktail for transdifferentiating mouse fibroblasts to iNPLCs. Collectively, our work provides a new functional implication for SOD2 in neural lineage specification.
Limitations of the Study
Our study reveals a crucial role of SOD2 in the regulation of early neural fate commitment using different model systems. We also show the ability of SOD2 to switch the fate of MEFs to neural progenitor-like cells in vitro. And these were independent of antioxidant activity of SOD2. Along the process we found SOD2 to influence mitochondrial dynamics where in it facilitates fusion process by up-regulating MFN2. A major limitation of the study is that the mechanism by which SOD2 regulates MFN2 expression remains elusive. We attempted to understand the relation between SOD2 and MFN2 by performing co-immunoprecipitation experiments. The results indicated the absence of any interaction between SOD2 and MFNs. As an alternative, we carried out experiments to see if SOD2 shielded MFNs from undergoing degradation. Although MFNs were degraded with time in presence of Cycloheximide, their stability was not maintained even under SOD2 over-expression. Despite attempts, the study leaves behind an unanswered query of mechanism underlying SOD2-mediated up-regulation of MFN2.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anujith Kumar: anujith.kumar@manipal.edu.
Materials Availability
All the in-house generated plasmid constructs as mentioned in Table S2, Transparent Methods supplemental file, are available upon request.
Data and Code Availability
The data that support the findings of this study are available from the Lead Contact on reasonable request. Microarray data have been deposited in Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE154756.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by grants from Department of Biotechnology, India (BT/PR 6165/GBD/27/369/2012, BT/PR8508/MED/30/1019/2013 and BT/PR15508/MED/31/316/2015) to A.K. We thank Prof. Panicker for kindly providing the cell line P40H1. We thank Prof. Heidi Mcbride for the plasmids C-mito LZV and N-mito VZL. pLV-mitoDsRed was a gift from Pantelis Tsoulfas (Addgene plasmid #44386). We thank Central Imaging and Flow Cytometry Facility, National Center for Biological Sciences, Bangalore, for helping in confocal imaging.
Author Contributions
Conceptualization, S.B., P.S., J.P., and A.K.; Methodology and Formal Analysis, S.B., P.S., and A.K.; Investigation, S.B., P.S., J.P.J., and C.P.; Writing – Original Draft, S.B., J.P.J., and A.K.; Writing – Review & Editing: S.B., P.S., J.P., and A.K.; Supervision, A.K.; Funding Acquisition, A.K.
Declaration of Interests
The authors declare no competing interest.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101564.
Supplemental Information
References
- Agostini M., Romeo F., Inoue S., Niklison-Chirou M.V., Elia A.J., Dinsdale D., Morone N., Knight R.A., Mak T.W., Melino G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016;23:1502–1514. doi: 10.1038/cdd.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-buylla A., Garcı J.M. Neurogenesis in adult subventricular zone. J. Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrázola M.S., Andraini T., Szelechowski M., Mouledous L., Arnauné-Pelloquin L., Davezac N., Belenguer P., Rampon C., Miquel M.C. Mitochondria in developmental and adult neurogenesis. Neurotox. Res. 2019;36:257–267. doi: 10.1007/s12640-018-9942-y. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R., Ebert B., Schäffner I., Moss J., Fiebig C., Shin J., Moore D.L., Ghosh L., Trinchero M.F., Stockburger C. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron. 2017;93:560–573.e6. doi: 10.1016/j.neuron.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A.M., Ming G., Song H. Adult mammalian neural stem cells and neurogenesis: Five Decades Later. Cell Stem Cell. 2015;17:385–395. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 co-ordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cheng A., Yang Y., Zhou Y., Maharana C., Lu D., Peng W., Liu Y., Wan R., Marosi K., Misiak M. Mitochondrial SIRT3 mediates adaptive responses to neurons to exercise and metabolic and excitatory challenges. Cell Metabol. 2016;23:128–142. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaegdt H., Lukaszuk A., De Buyser E., De Backer J.P., Szemenyei E., Tóth G., Chakravarthy S., Panicker M., Michotte Y., Tourwé D., Vauquelin G. Selective labeling of IRAP by the tritiated AT4 receptor ligand [3H] Angiotensin IV and its stable analog [3H] AL-11. Mol. Cell Endocrinol. 2009;13:77–86. doi: 10.1016/j.mce.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Eriksson P.S., Perfilieva E., Bjork- Eriksson T., Alborn A.M., Nordborg C., Peterson D.A., Gage F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998;4:1313. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Fang D., Yan S., Yu Q., Chen D., Yan S.S. Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci. Rep. 2016;6:31462. doi: 10.1038/srep31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Ouyang X., Wan R., Cheng H., Mattson M.P., Cheng A. Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells. 2012;30:2535–2547. doi: 10.1002/stem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuo N., Nojiri H., Uchiyama S., Noda Y., Kawakami S., Kojima S., Sasaki T., Shirasawa T., Shimizu T. Brain-specific superoxide dismutase 2 deficiency causes perinatal death with spongiform encephalopathy in mice. Oxid. Med. Cell. Longev. 2015;2015:1–10. doi: 10.1155/2015/238914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Taguchi N. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell. Biol. 2009;11:958. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Khacho M., Slack R.S. Mitochondrial dynamics in the regulation of neurogenesis: from development to the adult brain. Dev. Dyn. 2018;247:47–53. doi: 10.1002/dvdy.24538. [DOI] [PubMed] [Google Scholar]
- Khacho M., Clark A., Svoboda D.S., Harper M., Park D.S., Slack R.S. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:1–16. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Khacho M., Harris R., Slack R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019;20:34–48. doi: 10.1038/s41583-018-0091-3. [DOI] [PubMed] [Google Scholar]
- King H.W., Klose R.J. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife. 2017;6:1–24. doi: 10.7554/eLife.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P., St. Clair D., Wermer M., Yen H.C., Oberley T., Yang L., Beal M.F. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol. Dis. 1998;5:253–258. doi: 10.1006/nbdi.1998.0191. [DOI] [PubMed] [Google Scholar]
- Kumar A., Declercq J., Eggermont K., Agirre X., Prosper F., Verfaille C.M. Zic3 induces conversion of human fibroblasts to stable neural progenitor- like cells. J. Mol. Cell Biol. 2012;4:252–255. doi: 10.1093/jmcb/mjs015. [DOI] [PubMed] [Google Scholar]
- Laaper M., Jahani-Asl A. Regulation of neural stem cell fate decisions by mitochondrial dynamics. Neural Regen. Res. 2018;13:1548–1549. doi: 10.4103/1673-5374.237115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., He Z., April S.L., Trefny M.P., Rougier J.S., Salemi S., Olariu R., Widmer H.R., Simon H.U. Biochemical re-programming of human dermal stem cells to neurons by increasing mitochondrial membrane potential. Cell Death Differ. 2019;26:1048–1061. doi: 10.1038/s41418-018-0182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long B., Gan T.Y., Zhang R.C., Zhang Y.H. miR-23a regulates cardiomyocyte apoptosis by targeting manganese superoxide dismutase. Mol. Cell. 2017;40:542–549. doi: 10.14348/molcells.2017.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B., Drechsel D., Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb. Perspect. Biol. 2012;4:a008359. doi: 10.1101/cshperspect.a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad C.A., Washington T.M., Pautler R.G., Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.S., Sullivan K.A., Wilkinson J.E., Backus C., Hayes J.M., Sakowski S.A., Feldman E.L. Neurodegeneration and early lethality in superoxide dismutase 2-deficient mice: a comprehensive analysis of the central and peripheral nervous systems. Neuroscience. 2012;212:201–213. doi: 10.1016/j.neuroscience.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham A.H., Meng S., Chu Q.N., Chan D.C. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheshadri P., Ashwini A., Jahnavi S., Bhonde R., Prasanna J., Kumar A. Novel role of mitochondrial manganese superoxide dismutase in STAT3 dependent pluripotency of mouse embryonic stem cells. Sci. Rep. 2015;5:1–12. doi: 10.1038/srep09516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solari C., Echegaray C.V., Cosentino M.S., Petrone M.V., Waisman A., Luzzani C., Francia M., Villodre E., Lenz G., Miriuka S. Manganese superoxide dismutase gene expression is induced by Nanog and Oct4, essential pluripotent stem cells’ transcription factors. PLoS One. 2015;10:1–12. doi: 10.1371/journal.pone.0144336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J., Jernigan T.L. The basics of brain development. Neuropsychol. Rev. 2010;20:327–348. doi: 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N., Teng E.M., Bushong E.A., Aimone J.B., Zhao C., Consiglio A., Van Praag H., Martone M.E., Ellisman M.H., Gage F.H. Synapse formation on neurons born in the adult hippocampus. Nat. Neurosci. 2007;10:727–734. doi: 10.1038/nn1908. [DOI] [PubMed] [Google Scholar]
- Turrens J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar V.S., Berman S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson's disease. Neurobiol. Dis. 2013;51:43–55. doi: 10.1016/j.nbd.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Branicky R., Noë A., Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018;217:1915–1928. doi: 10.1083/jcb.201708007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yon J.M., Baek I.J., Lee B.J., Yun Y.W., Nam S.Y. Dynamic expression of manganese superoxide dismutase during mouse embryonic organogenesis. Int. J. Dev. Biol. 2011;55:327–334. doi: 10.1387/ijdb.103270jy. [DOI] [PubMed] [Google Scholar]
- Zhao Q., Lu D., Wang J., Liu B., Cheng H., Mattson M.P., Cheng A. Calcium dysregulation mediates mitochondrial and neurite outgrowth abnormalities in SOD2 deficient embryonic cerebral cortical neurons. Cell Death Differ. 2019;26:1600–1614. doi: 10.1038/s41418-018-0230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the Lead Contact on reasonable request. Microarray data have been deposited in Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE154756.