

Abstract
Brainstem gliomas are molecularly heterogeneous diseases, many of which are difficult to safely surgically resect and have limited treatment options due to their eloquent location. These constraints pose challenges to biopsy, which limits the use of routine molecular profiling and identification of personalized therapies. Here, we explored the potential of sequencing of circulating tumor DNA (ctDNA) isolated from the cerebrospinal fluid (CSF) of brainstem glioma patients as a less invasive approach for tumor molecular profiling. CSF was obtained from patients either intraoperatively (91.2%, 52/57), from ventricular-peritoneal shunt (3.5%, 2/57), or by lumbar puncture (5.3%, 3/57), all prior to surgical manipulation of the tumor. Deep sequencing of glioma-associated genes was performed on CSF-derived ctDNA and, where available, matched blood and tumor DNA from 57 patients, including nine medullary and 23 diffuse intrinsic pontine gliomas (DIPG). At least one tumor-specific mutation was detected in over 82.5% of CSF ctDNA samples (47/57). In cases with primary tumors harboring at least one mutation, alterations were identified in the CSF ctDNA of 97.3% of cases (36/37). In over 83% (31/37) of cases, all primary tumor alterations were detected in the CSF, and in 91.9% (34/37) of cases, at least half of the alterations were identified. Among ten patients found to have primary tumors negative for mutations, 30% (3/10) had detectable somatic alterations in the CSF. Finally, mutation detection using plasma ctDNA was less sensitive than sequencing the CSF ctDNA (38% vs. 100%, respectively). Our study indicates that deep sequencing of CSF ctDNA is a reliable technique for detecting tumor-specific alterations in brainstem tumors. This approach may offer an alternative approach to stereotactic biopsy for molecular profiling of brainstem tumors.
Keywords: Liquid biopsy, ctDNA, DIPG, Brainstem, H3K27M mutation
Introduction
Brainstem gliomas are heterogeneous diseases, many of which are difficult to safely surgically resect due to their location in highly eloquent regions of the brain. In particular, those tumors affecting the pons, known as diffuse intrinsic pontine gliomas (DIPG), which are more frequent in children, have a dismal survival of less than 1 year [18]. Despite numerous clinical trials, chemotherapy has proven to be ineffective in prospective studies, while radiation therapy, the standard of care, only prolongs survival by a few months [11, 13, 24].
In an effort to better understand the pathogenesis of human brainstem gliomas and to identify new therapeutic targets, recent collaborative efforts have leveraged the latest next-generation sequencing technologies on tissues obtained by stereotactic biopsy or post-mortem for comprehensive molecular characterization. These studies have identified the major genetic alterations and potential therapeutic targets of brainstem gliomas in adult and pediatric populations, including frequent alterations in H3F3A, HIST1H3B/C, ACVR1, PPM1D, IDH1, TP53, and ATRX [2, 10, 23, 33, 34, 38–40].
Based on these findings, brainstem gliomas are made up of multiple genetic subtypes and are distinct from supratentorial tumors [2, 4, 10, 31, 33, 34, 39, 40]. These results highlight the need for molecularly guided treatment and rational clinical trial design to ensure personalized treatment of homogeneous populations (Suppl. Table 1, Online Resource 1). Stereotactic biopsy is becoming increasingly accepted as a safe approach for tumor profiling. However, traditional biopsies still pose some risk to patients, in particular in eloquent regions of the brain, and these approaches do not provide opportunities for dynamic monitoring [20, 30]. Reports of intratumoral heterogeneity in brainstem tumors such as DIPG make adequate sampling for effective therapy selection challenging [3, 14, 26].
Liquid biopsy, or sampling of tumor-derived DNA released into the circulation, offers an opportunity to profile tumors less invasively, while also enabling dynamic disease monitoring and capturing disease heterogeneity [1, 7]. Although plasma is a common source for circulating tumor DNA (ctDNA), brain tumors often lack detectable plasma ctDNA due to the blood brain barrier. However, for brain tumors, sampling the cerebrospinal fluid (CSF) has proven to be an adequate source for ctDNA [6, 25, 29, 37]. For brainstem gliomas, due to their anatomic location, the potential risks of stereotactic biopsy, liquid biopsy approaches may offer an alternative for obtaining critical genetic information for these tumors and also may enable tracking of tumor dynamics using molecular endpoints throughout a patient’s clinical course.
Here, we investigated the use of deep sequencing of ctDNA from the CSF of brainstem glioma patients to identify tumor-specific genetic alterations. Our results indicate that CSF ctDNA is a potent resource to genetically characterize and identify potential therapeutic targets for these heterogeneous and difficult to sample tumors.
Methods
Ethics statement
This research was approved by the Institutional Review Board (IRB) and Ethics Committee of Beijing Tiantan Hospital (Beijing, China), which has full accreditation of the Association for the Accreditation of Human Research Protection Program (AAHPP). All methods were carried out in accordance with the approved guidelines. Written informed consent was obtained from all patients.
Patients and sample collection
Patients with primary tumor in the brainstem or fourth ventricle (n = 57) were enrolled in this retrospective study between August 2015 and September 2017 in the Department of Neurosurgery at Beijing Tiantan Hospital. The median age at diagnosis was 12.0 y (range 3.7 y to 56.0 y). All patients underwent MRI scans. CSF (5–10 ml) was collected at the time of surgery or ventricular-peritoneal shunt prior to any surgical manipulation of the tumor in 53 patients and from preoperative lumbar puncture in 4 patients (Fig. 1a–d). Fresh tumor tissue was obtained from surgical resection or open biopsy for 47 patients in the study and confirmed by a neuropathologist to contain greater than 70% tumor cells by H&E staining. Additionally, blood samples for germline DNA controls from each patient were obtained.
Fig. 1.
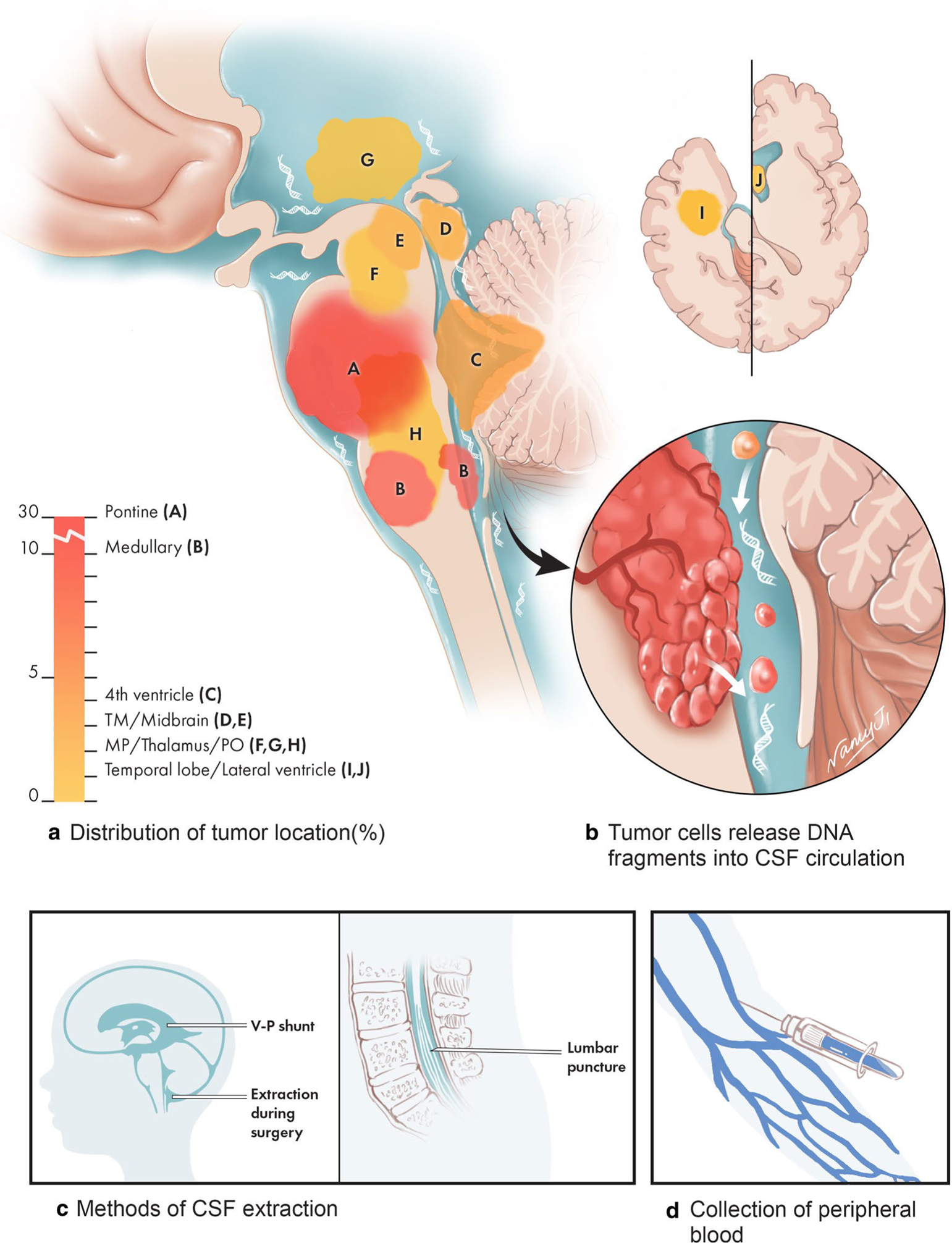
The anatomic distribution of brainstem tumors that underwent CSF-derived ctDNA sequencing and methods used to collect samples. The majority of patient tumors in this study are diffuse intrinsic pontine glioma (DIPG), or medullary tumors, while the remaining are other brainstem tumors also located in eloquent regions of the brain. Due to their anatomic locations, procedures such as surgical resection and stereotactic biopsy are challenging and may harbor risks for patients. Circulating tumor DNA is shed into the CSF and was isolated in our study by either VP shunt, during surgery, or by lumbar puncture
Tumor volume calculation on MRI scans
A conventional MRI scan was performed on all patients preoperatively. Tumor volumes were determined through manual segmentation of the corresponding MRI scans using a 3D slicer (version 4.8.0) [8]. Contrast-enhanced T1WI and T2WI/T2Flair images were used as the primary reference for determining tumor boundaries. When patchy enhancement was observed, the hyperintensity on the T2WI/T2Flair images was used to delineate the tumor boundaries. This process was performed by a single neurosurgeon. The senior author then independently verified all results.
Extraction of genomic DNA and cell-free DNA
EDTA tubes containing whole blood (5 ml) samples or CSF (5–10 ml) were centrifuged for 10 min at 1900 g, and the supernatants from these samples were further centrifuged for 10 min at 16,000 g. Samples were then collected and stored at − 80 °C until extraction. Genomic DNA was extracted from fresh tumor tissue using the QIAamp DNA Tissue & Blood Kit (Qiagen; Germantown, MD, USA) [12, 19]. Cell-free DNA was extracted from plasma (3 ml) and CSF using the MagMAX™ CellFree DNA Isolation Kit (ThermoFisher Scientific; Waltham, MA, USA). Finally, all isolated DNAs were quantified with the Qubit 2.0 Fluorometer using the Qubit dsDNA HS Assay kit (Life Technologies; Carlsbad, CA, USA) according to the recommended protocol.
NGS library preparation and sequencing data analysis
Genomic DNA was sheared into 150–200 bp fragments with the Covaris M220 Focused-ultrasonicator™ Instrument (Covaris; Woburn, MA, USA). Fragmented DNA and ctDNA libraries were constructed with the KAPA HTP Library Preparation Kit (Illumina platforms; KAPA Biosystems; Wilmington, MA, USA) following the manufacturer’s instructions [5, 17]. DNA libraries were captured with a designed brain cancer panel of 68 genes (GenetronHealth; Beijing, China) that included major brain tumor related genes. The captured samples were subjected to Illumina HiSeq X-Ten for paired end sequencing.
Sequencing reads from the HiSeq X-Ten platform were demultiplexed allowing zero mismatches in barcodes, and the read quality statistics were calculated by FastQC. Sequence adapters and low-quality regions were removed with Trimmomatic (v0.36), and then mapped to the hg19 reference genome with BWA (v0.7.10). PCR duplicates were marked using Picard. Local realignment was run using GATK. Pileup files that were converted from bam files were generated for the genomic regions targeted by exome enrichment. Using the pileup file as input, SNV or indel was called with SAMtools (v0.1.1722) and Pindel; structural variation was detected with Crest. The criteria we adopted for retaining a mutation in CSF and plasma ctDNA were that it had an allele fraction of ≥ 0.1% and a total of ≥ 4 reads. Additionally, known recurrent loci were manually reviewed using Integrative Genomics Viewer (IGV v2.3.34) in the target tissue as compared to the normal blood DNA. All mutations were annotated for genes and function as well as repeated genomic regions using ANNOVAR, Oncotator and Vep. The dbNSFP and the Exome Aggregation Consortium (ExAC) database were used to filter out either the benign mutations with pp2_hdiv score < 0.452 or polymorphic nonsynonymous mutations sites. Finally, all mutations were annotated for genes using ANNOVAR, Oncotator and Vep.
Results
Clinical and pathological features affecting the detection of CSF ctDNA
We isolated CSF ctDNA from 57 patients diagnosed with primary CNS tumors of the brainstem (Suppl. Table 2, Online Resource 2). The median age was 12.0 years old (range 3.7–56 years old) and tumors were classified as WHO grades I–IV. The majority of tumors were located in the pons (n = 30) and medulla (n = 9), while the remaining tumors were located in the fourth ventricle (n = 4), the thalamus (n = 2), or had intermediate locations between two regions of the brainstem (Fig. 1). The median concentration of cell-free DNA fragments from the 57 patients was 3.2 ng/mL for CSF, which was lower than the median concentration of 8.4 ng/mL for plasma. All patients in the study had detectable cell-free DNA fragments in their CSF.
We investigated the relationship between clinical features and the detectability of CSF ctDNA, (Suppl. Table 3, Online Resource 3; Suppl. Figure 1, Online Resource 4), meaning tumor-specific alterations (identified both in tumor sample and CSF ctDNA) or new compared to the germline DNA controls, in particular for cases without available tumor tissue. The results demonstrated that there were significant differences in tumor grade (p = 0.001), location abutting the CSF space (p = 0.028), and symptom duration (p = 0.05) between samples with detectable alterations in the CSF versus those with undetected alterations. Additionally, samples with detectable CSF alterations generally also had increased amounts of DNA in the CSF (p = 0.022). Other features such as tumor location in the brainstem, sex and age did not significantly vary between both groups, while CSF-detectable alterations trended towards being present in larger tumors (p = 0.097), although CSF ctDNA was still successfully identified in tumors as small as 0.95 cm3. Although tumor location was not a significant variable affecting the detectability of CSF ctDNA, both of the two tumors (one pilocytic astrocytoma and one glioblastoma) located in regions away from CSF reservoirs had undetectable tumor-specific alterations in the cell-free DNA fragments from the CSF. On univariate logistic regression analysis, tumor grade, in particular lower grades (I, p = 0.001; I and II, p = 0.016) vs. higher grades had significant effects on CSF ctDNA detectability (Suppl. Table 4, Online Resource 5).
Tumor-specific alterations detected in CSF-derived ctDNA
We used a panel that captures the coding regions of the 68 most frequently mutated genes (Suppl. Table 5, Online Resource 6) in brain tumors and performed next generation sequencing on CSF ctDNA isolated from 57 patients (Suppl. Table 6, Online Resource 7). We sequenced CSF ctDNA to an average depth of 1581× (range 102–1864×). The mutant allele fraction (MAF) of variants detected in the CSF-derived ctDNA was 12.1% (range 0.15–100%). At least one tumor-specific mutation among 68 brain tumor-associated genes was detected in over 82.5% of patients (47/57) (Fig. 2). The average number of different mutations identified in the CSF ctDNA was 2.07 (range 0–5). The most frequently altered genes were H3F3A (47.7%), TP53 (43.86%), ATRX (12.28), PDGFRA (10.53), FAT1 (8.77%), HIST1H3B (8.77%), PPM1D (8.77%), IDH1 (7.02%), NF1 (7.02%), PIK3CA (7.02%) and ACVR1 (7.02%). The average MAF for the most frequently mutated genes in CSF were the following: H3F3A (26.18%), TP53 (20.81%), ATRX (27.68%), PDGFRA (20.76%), FAT1 (19.69%), HIST1H3B (12.54%), PPM1D (22.01%), IDH1 (1.67%), NF1 (3.50%), PIK3CA (22.50%) and ACVR1 (13.25%).
Fig. 2.
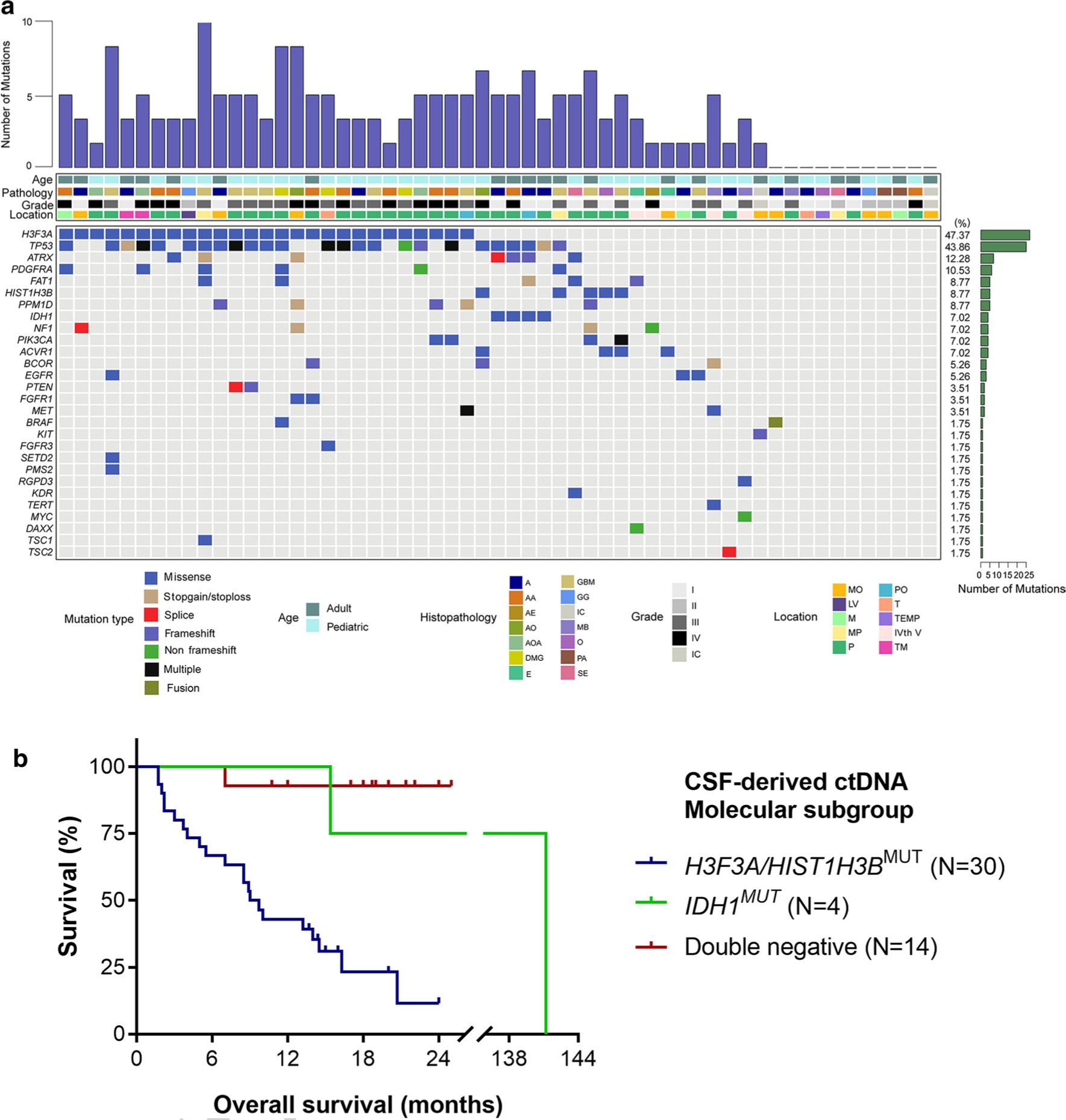
Mutational characteristics for cerebrospinal fluid cell-free DNA. a CSF ctDNA was successfully isolated from patients with brainstem tumors (N = 57). We used a panel-based next generation sequencing approach to identify recurrent tumor-specific alterations in the majority of CSF ctDNA samples. The most frequently mutated genes included H3F3A, TP53, ATRX and PDGFRA. Tumors were located in the following locations: MO medullary oblongata, P pontine, TM thalamus and midbrain, M midbrain, MP midbrain and pontine, PO pontine and medullary oblongata, TEMP temporal lobe, LV lateral ventricle, T thalamus, IVth V the fourth ventricle, IC inconclusive. The following histologies were included in our study: A astrocytoma AA anaplastic astrocytoma, DMG diffuse midline glioma, GBM glioblastoma, AOA anaplastic oligoastrocytoma, PA pilocytic astrocytoma, GG ganglioglioma, E ependymoma, AO anaplastic oligodendroglioma, SE subependymoma, O oligodendroglioma, AE anaplastic ependymoma, MB medulloblastoma. b Kaplan–Meier estimates of overall survival in brainstem and thalamic tumors based on molecular subgroups from CSF-derived ctDNA. The H3F3A/HIST1H3B mutant subgroup has a significantly worse survival of 9.35 months as compared to the IDH mutant group (141.2 months, p = 0.0318) and the double negative group (only one death in this subgroup, undefined median OS, p < 0.0001)
For patients with tumors of the brainstem and thalamus, we examined the survival trends in overall survival based on the most frequent genomic alterations detected in the CSF ctDNA, namely those in the histone H3.3 and H3.1 genes (H3F3A or HIST1H3B), IDH mutation, and tumors negative for both markers (“double-negative”). The patients with detectable CSF-derived ctDNA alterations in H3.3 and H3.1 exhibited significantly worse survival of 9.35 months as compared to the IDH mutant group (141.2 months, p = 0.0318) and the double negative group (only one death in this subgroup, undefined median OS, p < 0.0001, Fig. 2b). The two-year survival of the H3-mutant group was only 11.6%, as compared to the IDH-mutant (75%) and double negative (92.9%) groups.
The sensitivity and specificity of CSF liquid biopsy
Matched DNA from the primary tumor biopsy and germline DNA from the patient’s blood (reference DNA) were available for 47 cases and were sequenced to determine the specificity and sensitivity of sequencing CSF ctDNA (Fig. 3a). In 97.3% of cases (36/37) with detectable mutations in the primary tumor, matched tumor-specific alterations were identified in the CSF ctDNA. In 83.8% (31/37) of cases, all primary tumor alterations were detected in the CSF, and in 91.9% (34/37) of cases, at least half of the alterations were identified in the CSF (Suppl. Table 2, Online Resource 2; Fig. 3b, c).
Fig. 3.
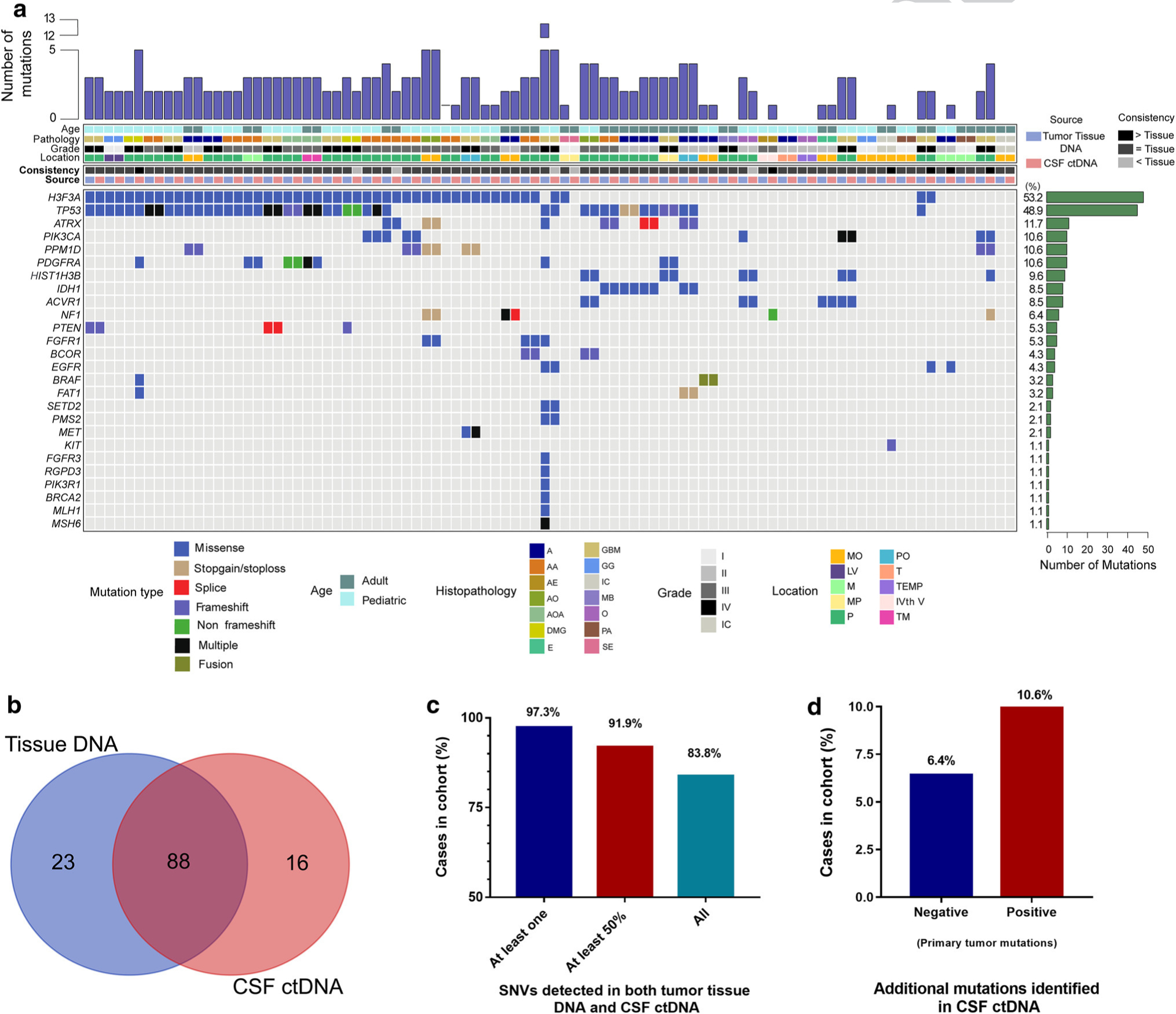
Sequencing of CSF-derived ctDNA accurately detects tumor-specific alterations found in the primary tumor tissue (N = 47). a Using parallel sequencing of the primary tumor DNA as well as CSF-derived ctDNA, tumor-specific alterations were identified and compared to one another. b, c The majority of cases show a high level of consistency between the tissue DNA detected variants and those identified in the CSF ctDNA. d In 17.0% of cases overall, mutations were detected in the CSF ctDNA that were undetectable in the primary tumor. In half of these cases, the primary tumor had undetectable mutations from the tissue specimen
We also found that among cases in which alterations were undetected in the primary tumor DNA, mutations were readily detected in the CSF-derived ctDNA in 30% (3/10) of these cases (6.4% of entire cohort, Suppl. Table 2, Online Resource 2; Fig. 3d). The alterations exclusively detected in the CSF ctDNA were mutations in NF1 (p.Ala2035_Ala2037del), KIT (p.Gln775ArgfsTer39), and EGFR (p.Ser768Ile). We also found that among primary tumors sequenced that had detectable mutations, 10.6% of these tumors had additional somatic alterations detected exclusively in the CSF-derived ctDNA, not originally identified in the primary tumor (Suppl. Table 2, Online Resource 2; Fig. 3d). These additional alterations were PDGFRA, BRAF, FAT1 in case RD993, EGFR in case RE964, HIST1H3B, NF1 in case RF007, and TP53 (p.Arg273Cys) in case RF182.
CSF ctDNA offers improved detection of brain tumor mutations compared to plasma
We detected tumor-specific alterations in the CSF ctDNA in the majority of brainstem glioma patients using next-generation sequencing, indicating that CSF was a reliable source for liquid biopsy. However, we sought to examine if the plasma could also be used as a source of ctDNA for less invasive profiling of brainstem tumors. We therefore isolated ctDNA from the plasma of eight patients with matching CSF ctDNA, primary tumor DNA and germline DNA, and performed targeted capture sequencing on these samples. The genetic profiles generated from CSF ctDNA, primary tumor tissue, and plasma were compared (Fig. 4a–c). Tumor-specific mutations were detected in the CSF ctDNA in 100% (8/8) of cases, but in the plasma ctDNA in only 37.5% (3/8) of cases (Fig. 4c). Furthermore, for the four mutations detected in both CSF and plasma, the median MAF was higher in all CSF ctDNA than in plasma ctDNA (Fig. 4d, e).
Fig. 4.
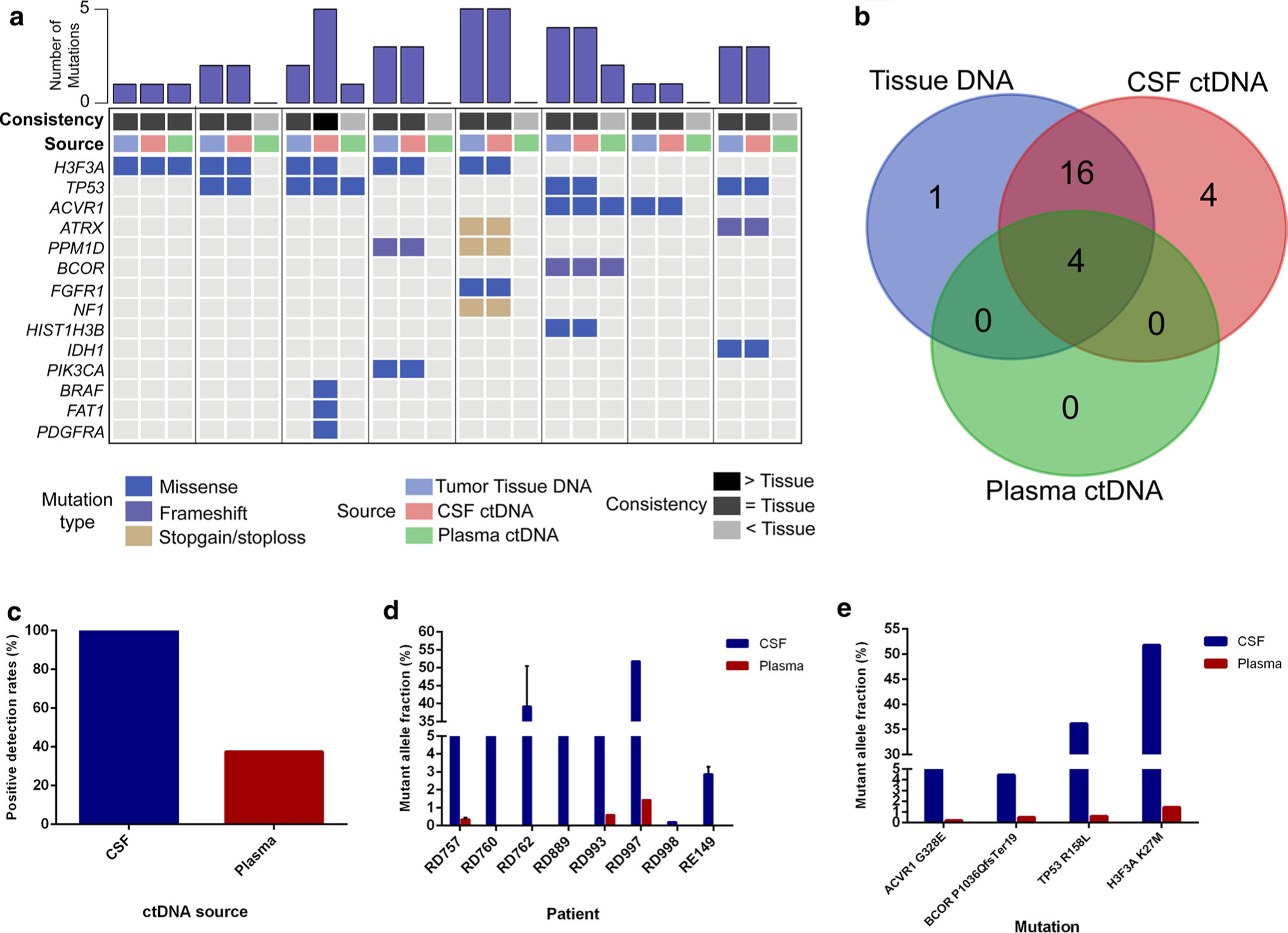
CSF-ctDNA provides robust detection of tumor-specific alterations for brainstem tumors as compared to plasma ctDNA. a For eight cases with available primary tumor tissue DNA, both the CSF ctDNA and plasma ctDNA were isolated and sequenced by NGS for genes frequently mutated in brain tumors. b CSF identified the majority of alterations in the primary tumor, but also identified additional alterations undetected in the tumor. c Positive detection rates were higher for CSF ctDNA as compared to plasma ctDNA, as were the d, e mutant allele fractions of the variants identified
Discussion
Recent genetic profiling studies of brainstem tumors, in particular DIPG, have identified the most frequent molecular alterations in these tumors and highlighted the existence of multiple distinct molecular subtypes of these tumors. Considering the limited treatment options available, these molecular markers will be critical for personalized therapy selection and patient stratification. However, the significant risks associated with biopsy of these tumors pose challenges to truly harnessing these molecular markers for treating patients.
Here, we have shown, through comparison of mutational analyses from deep sequencing of ctDNA isolated from the CSF with the matched primary tumor samples that the CSF represents an attractive source for liquid biopsy of tumors located in the eloquent brain areas, such as brainstem and thalamus. The major genetic alterations associated with brainstem tumors were readily detected in the CSF-derived ctDNA and these mutation patterns reflected distinct survival trends consistent with more aggressive H3F3A/HIST1H3B-mutant tumors and less aggressive tumors with IDH1 mutation or lacking both of these mutations. Sampling of the CSF for this analysis may assist in determining the expected clinical course for patients and need for immediate therapeutic intervention. H3F3A/HIST1H3B-mutated tumors may be candidates for early intervention with radiotherapy and targeted therapies including epigenetic modifiers, such as HDAC and BET inhibitors, which are currently in clinical trials for DIPG patients [22]. Similar to previous studies, the IDH-mutated brainstem gliomas had co-occurring mutations in TP53 and ATRX and were present in adults with a more favorable prognosis (median OS = 141.2 months) [40]. These tumors resemble their cerebral counterparts in terms of their genetic signature and less aggressive clinical course. IDH1-mutated brainstem gliomas may therefore benefit from treatment such as concomitant radiotherapy (RT) and chemotherapy followed by adjuvant therapy [9, 27]. Patients with double negative tumors (H3F3A/HIST1H3B and IDH wildtype) demonstrated even better prognosis with only one death during follow-up and unreached median OS. These tumors were largely Grade I and II tumors and in our clinical experience, these tumors are often optimal candidates for surgical resection. For those undergoing total or near-total resection, they may be able to expect long-term tumor free of stable disease on serial observation, with RT and/or chemotherapy reserved for tumor recurrence [21, 28]. Other alterations were identified by sequencing of the CSF-derived ctDNA for which targeted therapies are in development or currently available, including mutations in PDGFRA, EGFR, IDH1, BRAF, MET and PPM1D. Based on the location of these tumors, the challenges of surgical resection and biopsy, and limited treatment options currently available, molecular profiling from the CSF may offer an alternative approach to identify patients that are candidates for new targeted therapies. Here, we show for the first time that a panel-based approach can accurately be used to sensitively detect multiple tumor-specific mutations from brainstem tumors by sampling the CSF, and that this approach is superior to plasma ctDNA in these tumor types. In addition, we show that this approach can be accurately used in a variety brainstem tumor types (including DIPG) of both adult and pediatric populations. Prior studies have focused on detection of single variants (such as H3 variants) in DIPG [15].
In several cases, the primary tumor was sequenced with no mutations being identified, but alterations were detectable in the CSF ctDNA. These results may be due to the sampling bias inherent in traditional biopsy or due to tumor heterogeneity known to be present in brainstem tumors such as DIPG, highlighting an additional potential benefit of CSF ctDNA over primary tumor DNA. However, this analysis is difficult to accurately assess in our study as the primary tumor samples were sequenced to lower depth than the CSF and plasma (either WGS to ~ 60× or 68-gene panel sequencing to 500×).
Recent studies of supratentorial brain tumors have also shown a similar pattern in which CSF ctDNA was unable to consistently detect alterations in tumors unless they abutted the CSF space. In our study, both tumors with undetectable CSF ctDNA alterations were non-brainstem tumors that were not directly adjacent to CSF reservoir (one oligodendroglioma of the temporal lobe and one pilocytic astrocytoma of the thalamus) [37]. For this reason, brainstem tumors are being bathed by the CSF and therefore may be ideal targets for CSF ctDNA sequencing. Patients with supratentorial gliomas often exhibit increased intracranial pressure (ICP), which is a strong contraindication for CSF sampling by lumbar puncture. However, patients with brainstem gliomas less commonly demonstrate symptoms of hydrocephalus or elevated intracranial pressure, making them much better candidates for safe use of lumbar puncture for CSF sampling.
We found that ctDNA isolated from the plasma was not a reliable DNA source for the detection of tumor-specific mutations, with the majority of patients having undetectable mutations in the plasma ctDNA. Additionally, plasma ctDNA-detected mutations exhibited lower overall MAF than from the CSF ctDNA. Thus, direct CSF sampling might be superior for sensitive detection of tumor-specific alterations in brainstem glioma.
Despite our encouraging findings of detectable and consistent mutation profiles in the ctDNA of brainstem glioma patients compared to tumor tissue, there were several cases in our study that lacked any detectable alterations by both approaches. For more comprehensive annotation of these cases, the 68 gene panels could be expanded to encompass additional alterations. Additionally, several of these cases with undetectable mutations were Grade I tumors, indicating this liquid biopsy approach may be more appropriate for aggressive tumors. In addition, in our study, the majority of CSF samples was obtained during open biopsy or V-P shunt. Although tumor-specific mutations were detected by sequencing the ctDNA from all the three CSF samples collected by traditional lumbar puncture, the majority of samples in this study were obtained from intraoperative sampling prior to tumor manipulation. Larger studies should build of these promising findings to ensure the utility and sensitivity of lumbar puncture-based CSF ctDNA detection for primary tumor profiling. Additionally, the incidence of hydrocephalus in pediatric DIPG varies from < 10% to approximately 25% [16, 32, 35, 36]. We believe lumbar puncture as an approach to CSF sampling for ctDNA should be avoided in patients with signs of hydrocephalus. Finally, our results could be expanded to determine if serial sampling of the CSF could be used to monitor tumor dynamics, including tracking minimal residual disease, recurrence, and response to therapy.
In conclusion, our study shows that deep sequencing of CSF-derived ctDNA from patients with tumors of the brainstem can accurately detect tumor-specific mutations. The molecular signature of tumors is becoming increasingly important for designing personalized therapy for cancer patients, ensuring homogeneous treatment populations and advancing clinical management of devastating cancers such as DIPG. This approach can potentially facilitate these goals in brainstem glioma patients and ultimately help to make significant advances in prolonging the lives of affected patients.
Supplementary Material
Acknowledgements
We thank Lin Qiao and Xuefeng Guo for their help in collecting samples. We would like to thank Honglin Zhu and Yufei Yang for their helpful advices in data analysis. The authors thank Nancy Chu Ji, who was hired to assist in the illustration for Fig. 1, which was inspired by Wang et al. [37].
Funding Financial support was provided by the National Key Technology Research and Development Program of the Ministry of Science and Technology of China (Grant nos. 2014BAI04B01 and 2015BAI12B04), Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (Grant no. ZYLX201608), and Grant for C.P from Beijing Municipal Bureau of Human Resources and Social Security (Grant no. 2017-ZZ-117).
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00401-018-1936-6) contains supplementary material, which is available to authorized users.
Conflict of interest HY is a founder of Genetron Health and receives royalties from Personal Genome Diagnostics (PGDX) and Agios. YJ is a co-founder of and Scientific Advisor for Genetron Health. BHD serves as a scientific consultant for Genetron Health.
References
- 1.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al. (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Boufet E, Bartels U et al. (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46:451–456. 10.1038/ng.2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bugiani M, Veldhuijzen van Zanten SEM, Caretti V, Schellen P, Aronica E, Noske DP, Vandertop WP, Kaspers GJL, van Vuurden DG, Wesseling P et al. (2017) Deceptive morphologic and epigenetic heterogeneity in diffuse intrinsic pontine glioma. Oncotarget 8:60447–60452. 10.18632/oncotarget.19726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castel D, Philippe C, Calmon R, Le Dret L, Trufaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L (2015) Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cracolici V, Mujacic I, Kadri S, Alikhan M, Niu N, Segal JP, Rosen LE, Sarne DH, Morgan A, Desouky S et al. (2018) Synchronous and metastatic papillary and follicular thyroid carcinomas with unique molecular signatures. Endocr Pathol 29:9–14. 10.1007/s12022-017-9491-6 [DOI] [PubMed] [Google Scholar]
- 6.De Mattos-Arruda L, Mayor R, Ng CK, Weigelt B, Martinez-Ricarte F, Torrejon D, Oliveira M, Arias A, Raventos C, Tang J et al. (2015) Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 6:8839 10.1038/ncomms9839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz LA Jr, Bardelli A (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32:579–586. 10.1200/JCO.2012.45.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fedorov A, Beichel R, Kalpathy-Cramer J, Finet J, Fillion-Robin JC, Pujol S, Bauer C, Jennings D, Fennessy F, Sonka M et al. (2012) 3D Slicer as an image computing platform for the quantitative imaging network. Magn Reson Imaging 30:1323–1341. 10.1016/j.mri.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher BJ, Hu C, Macdonald DR, Lesser GJ, Coons SW, Brachman DG, Ryu S, Werner-Wasik M, Bahary JP, Liu J et al. (2015) Phase 2 study of temozolomide-based chemoradiation therapy for high-risk low-grade gliomas: preliminary results of Radiation Therapy Oncology Group 0424. Int J Radiat Oncol Biol Phys 91:497–504. 10.1016/j.ijrobp.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA et al. (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46:462–466. 10.1038/ng.2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazier JL, Lee J, Thomale UW, Noggle JC, Cohen KJ, Jallo GI (2009) Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies: a review. J Neurosurg Pediatr 3:259–269. 10.3171/2008.11.peds08281 [DOI] [PubMed] [Google Scholar]
- 12.Gerasimidis K, Bertz M, Quince C, Brunner K, Bruce A, Combet E, Calus S, Loman N, Ijaz UZ (2016) The effect of DNA extraction methodology on gut microbiota research applications. BMC Res Notes 9:365 10.1186/s13104-016-2171-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hargrove D, Bartels U, Boufet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7:241–248. 10.1016/s1470-2045(06)70615-5 [DOI] [PubMed] [Google Scholar]
- 14.Hofman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, Ramani A, Brudno M, Kumar SS, Drissi R et al. (2016) Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4:1 10.1186/s40478-015-0269-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, James CD, Shilatifard A, Saratsis AM (2017) Detection of histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun 5:28 10.1186/s40478-017-0436-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jansen MH, van Zanten SEV, Aliaga ES, Heymans MW, Warmuth-Metz M, Hargrave D, van der Hoeven EJ, Gidding CE, de Bont ES, Eshghi OS (2015) Survival prediction model of children with diffuse intrinsic pontine glioma based on clinical and radio-logical criteria. Neuro Oncol 17:160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang T, Li X, Wang J, Su C, Han W, Zhao C, Wu F, Gao G, Li W, Chen X et al. (2017) Mutational landscape of cfDNA identifies distinct molecular features associated with therapeutic response to first-line platinum-based doublet chemotherapy in patients with advanced NSCLC. Theranostics 7:4753–4762. 10.7150/thno.21687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johung TB, Monje M (2017) Diffuse intrinsic pontine glioma: new pathophysiological insights and emerging therapeutic targets. Curr Neuropharmacol 15:88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadri S, Long BC, Mujacic I, Zhen CJ, Wurst MN, Sharma S, McDonald N, Niu N, Benhamed S, Tuteja JH et al. (2017) Clinical validation of a next-generation sequencing genomic oncology panel via cross-platform benchmarking against established amplicon sequencing assays. J Mol Diagn 19:43–56. 10.1016/j.jmoldx.2016.07.012 [DOI] [PubMed] [Google Scholar]
- 20.Kieran MW, Goumnerova LC, Prados M, Gupta N (2016) Biopsy for diffuse intrinsic pontine glioma: a reappraisal. J Neurosurg Pediatr 18:390–391. 10.3171/2015.6.PEDS15374 [DOI] [PubMed] [Google Scholar]
- 21.Klimo P, Panandiker ASP, Thompson CJ, Boop FA, Qaddoumi I, Gajjar A, Armstrong GT, Ellison DW, Kun LE, Ogg RJ et al. (2013) Management and outcome of focal low-grade brainstem tumors in pediatric patients: the St. Jude experience. J Neurosurg Pediatr 11:274–281. 10.3171/2012.11.peds12317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long W, Yi Y, Chen S, Cao Q, Zhao W, Liu Q (2017) Potential new therapies for pediatric diffuse intrinsic pontine glioma. Front Pharmacol 8:495 10.3389/fphar.2017.00495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M et al. (2017) Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32(520–537):e525 10.1016/j.ccell.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandell LR, Kadota R, Freeman C, Douglass EC, Fontanesi J, Cohen ME, Kovnar E, Burger P, Sanford RA, Kepner J (1999) There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: results of a Pediatric Oncology Group phase III trial comparing conventional vs. hyperfractionated radiotherapy. Int J Radiat Oncol Biol Phys 43:959–964 [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Ricarte F, Mayor R, Martinez-Saez E, Rubio-Perez C, Pineda E, Cordero E, Cicuendez M, Poca MA, Lopez-Bigas N, Ramon YCS et al. (2018) Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumour DNA from cerebrospinal fluid. Clin Cancer Res. 10.1158/1078-0432.ccr-17-3800 [DOI] [PubMed] [Google Scholar]
- 26.Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho CY, Kambhampati M, Hwang EI, Faury D et al. (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7:11185 10.1038/ncomms11185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olar A, Wani KM, Alfaro-Munoz KD, Heathcock LE, van Thuijl HF, Gilbert MR, Armstrong TS, Sulman EP, Cahill DP, Vera-Bolanos E et al. (2015) IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol 129:585–596. 10.1007/s00401-015-1398-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan CC, Chen X, Xu C, Wu WH, Zhang P, Wang Y, Wu T, Tang J, Xiao XR, Wu Z et al. (2016) Brainstem gangliogliomas: prognostic factors, surgical indications and functional outcomes. J Neurooncol 128:445–453. 10.1007/s11060-016-2131-z [DOI] [PubMed] [Google Scholar]
- 29.Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, Omuro A, Lin X, Fleisher M, Grommes C et al. (2016) Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol 34:2404–2415. 10.1200/JCO.2016.66.6487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puget S, Beccaria K, Blauwblomme T, Roujeau T, James S, Grill J, Zerah M, Varlet P, Sainte-Rose C (2015) Biopsy in a series of 130 pediatric diffuse intrinsic pontine gliomas. Childs Nerv Syst 31:1773–1780. 10.1007/s00381-015-2832-1 [DOI] [PubMed] [Google Scholar]
- 31.Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L et al. (2012) Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One 7:e30313 10.1371/journal.pone.0030313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roujeau T, Di Rocco F, Dufour C, Bourdeaut F, Puget S, Sainte Rose C, Zerah M (2011) Shall we treat hydrocephalus associated to brain stem glioma in children? Childs Nerv Syst 27:1735–1739. 10.1007/s00381-011-1538-2 [DOI] [PubMed] [Google Scholar]
- 33.Saratsis AM, Kambhampati M, Snyder K, Yadavilli S, Devaney JM, Harmon B, Hall J, Raabe EH, An P, Weingart M et al. (2014) Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol 127:881–895. 10.1007/s00401-013-1218-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor KR, Mackay A, Trufaux N, Butterield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D et al. (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. 10.1038/ng.2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanan MI, Eisenstat DD (2015) DIPG in children—what can we learn from the past? Front Oncol 5:237 10.3389/fonc.2015.00237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veldhuijzen van Zanten SEM, Lane A, Heymans MW, Baugh J, Chaney B, Hofman LM, Doughman R, Jansen MHA, Sanchez E, Vandertop WP et al. (2017) External validation of the diffuse intrinsic pontine glioma survival prediction model: a collaborative report from the International DIPG Registry and the SIOPE DIPG Registry. J Neurooncol 10.1007/s11060-017-2514-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, Ptak J, Brem H, Chaichana K, Gallia GL et al. (2015) Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA 112:9704–9709. 10.1073/pnas.1511694112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253. 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al. (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444–450. 10.1038/ng.2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, Yang S, Jones S, Wang S, Zhou W et al. (2014) Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 46:726–730. 10.1038/ng.2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.