

ABSTRACT
Inflammatory bowel disease (IBD) pathogenesis involves significant contributions from genetic and environmental factors. Loss-of-function single-nucleotide polymorphisms (SNPs) in the protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene increase IBD risk and are associated with altered microbiome population dynamics in IBD. Expansion of intestinal pathobionts, such as adherent-invasive E. coli (AIEC), is strongly implicated in IBD pathogenesis as AIEC increases pro-inflammatory cytokine production and alters tight junction protein regulation – suggesting a potential mechanism of pathogen-induced barrier dysfunction and inflammation. We aimed to determine if PTPN2 deficiency alters intestinal microbiome composition to promote expansion of specific bacteria with pathogenic properties. In mice constitutively lacking Ptpn2, we identified increased abundance of a novel mouse AIEC (mAIEC) that showed similar adherence and invasion of intestinal epithelial cells, but greater survival in macrophages, to the IBD-associated AIEC, LF82. Furthermore, mAIEC caused disease when administered to mice lacking segmented-filamentous bacteria (SFB), and in germ-free mice but only when reconstituted with a microbiome, thus supporting its classification as a pathobiont, not a pathogen. Moreover, mAIEC infection increased the severity of, and prevented recovery from, induced colitis. Although mAIEC genome sequence analysis showed >90% similarity to LF82, mAIEC contained putative virulence genes with >50% difference in gene/protein identities from LF82 indicating potentially distinct genetic features of mAIEC. We show for the first time that an IBD susceptibility gene, PTPN2, modulates the gut microbiome to protect against a novel pathobiont. This study generates new insights into gene-environment-microbiome interactions in IBD and identifies a new model to study AIEC-host interactions.
KEYWORDS: AIEC, Caco-2, colitis, epithelial, inflammatory bowel disease, LF82, macrophage, microbiome
Introduction
Genome-wide association studies (GWAS) have identified an association between loss-of-function single-nucleotide polymorphisms (SNPs) in the protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene, that encodes the T-cell protein tyrosine phosphatase (TCPTP), and several autoimmune diseases including Crohn’s disease, ulcerative colitis, celiac disease, type 1 diabetes and rheumatoid arthritis.1-5 Crohn’s disease (CD) and ulcerative colitis (UC), collectively known as inflammatory bowel disease (IBD), are chronic intestinal inflammatory conditions whose etiology is unclear. Several factors such as genetics and alterations in the intestinal microbiome are critical determinants of IBD pathogenesis.6 TCPTP has an essential role in restricting inflammation as homozygous Ptpn2 knockout mice exhibit substantially increased expression of pro-inflammatory cytokines and uncontrolled systemic inflammation.7,8 TCPTP regulation of inflammation is due, at least in part, to restriction of pro-inflammatory signaling pathways mediated by members of the Janus kinases (JAKs) and signal transducer and activator of transcription (STAT) families of signaling molecules (JAK-STATs).
JAK-STAT signaling can be activated by inflammatory cytokines such as interferon-gamma (IFN-γ) that is involved in several autoimmune diseases including IBD and celiac disease.8,9 TCPTP can restrict intestinal epithelial cell (IEC) barrier defects and tight junction remodeling induced by IFN-γ.8-11 The intestinal epithelium forms a selectively permeable barrier between the lumen and the submucosa through the formation of multiprotein complexes of desmosomes, adherens junctions and tight junctions (TJ) that regulate paracellular permeability.12,13 The intestinal epithelial barrier is essential to maintain appropriate compartmentalization of tissue versus lumenal factors in the gut. Specifically, the epithelium ensures that lumenal microbes and their products are restricted from accessing lamina propria immune cells or gaining access to the underlying vasculature. Indeed, when intestinal epithelial integrity is compromised, translocation of bacteria and bacterial products such as lipopolysaccharide can occur and trigger inflammatory responses that in severe cases lead to sepsis.14-16
Approximately 1012–1013 bacteria exist in the adult human gastrointestinal tract with more than 35,000 bacterial species that play a major role in maintaining intestinal homeostasis.17-19 Alterations in the intestinal microbiome are a major environmental factor in the pathogenesis of IBD.20-22 Expansion of pathobionts, such as adherent-invasive Escherichia coli (AIEC), is associated with IBD pathogenesis. This is likely due to a combination of pathological activities of AIEC which include induction of pro-inflammatory cytokine (IFN-γ, tumor necrosis factor alpha [TNF-α], interleukin 13 [IL-13]) production; increasing susceptibility to intestinal inflammation in genetically susceptible hosts; and disrupting expression and distribution of epithelial TJ proteins leading to increased intestinal permeability.23-31
A major gap in our understanding of how complex inflammatory diseases such as IBD arise, relates to how genetic susceptibility alters the intestinal environment to favor the expansion of commensal microbes with pathogenic potential (i.e., pathobionts) prior to the manifestation of clinical disease.32 Clinical genetic studies have provided some clues in this regard. A role for PTPN2 as a clinically relevant modulator of the gut microbiome was first identified in IBD patients where PTPN2 SNPs influenced the population dynamics of intestinal microbes, while a separate study also identified dysbiosis and increased disease severity in patients harboring PTPN2 SNPs.33,34 16S ribosomal RNA (rRNA) gene analysis showed that Ptpn2 deficiency in mouse CD4 T-lymphocytes aggravated adoptive T-cell transfer colitis and promoted a broad dysbiosis featuring typical colitis-associated increases in Bacteroidetes and Proteobacteria, and decreased Firmicutes, in stool samples.35 In this study we aimed to use a more rigorous approach of sequencing the internal transcribed spacer (ITS) variable region of bacteria to (i) identify if constitutive Ptpn2 loss alters intestinal microbiome composition; and (ii) identify specific bacterial species modulated by Ptpn2.36
Here, we report that Ptpn2-deficient mice exhibit a highly significant increase in abundance of a novel mouse adherent-invasive E. coli that has significant, but also distinct, genetic overlap with the human LF82 originally isolated from a Crohn’s disease patient.37 This mouse AIEC (mAIEC) caused weight loss in mice housed in specific pathogen-free (SPF) conditions and lacking segmented-filamentous bacteria (SFB-free), and in germ-free mice reconstituted with a microbiome, thus supporting its classification as a pathobiont rather than a pathogen. Furthermore, mAIEC exacerbated and delayed recovery from dextran sulfate sodium (DSS) colitis in SPF-SFB-free mice, thus confirming its pathogenic properties. Our data identify a novel mouse AIEC that may prove to be a valuable tool for the study of pathobiont-induced disease and for the interactions of genetic and environmental factors contributing to autoimmune diseases such as IBD.
Methods
Animal procedures
Ethical Statement on Mouse Studies – All animal care and procedures were performed in accordance with institutional guidelines and approved by the University of California, Riverside Institutional Animal Care and Use Committee under Protocol #A20190032E.
Housing and husbandry of experimental animals
– Constitutive Ptpn2 knockout (KO) male and female mice were generated by breeding of heterozygous (Het) mice on a BALB/c background and genotyped as previously described.7 Wild-type (WT) and Ptpn2-Het littermate male and female mice were used as controls. All mice used for microbiome analysis were approximately 3 weeks old (19–23 d of age) at time of sacrifice and were housed in specific pathogen-free (SPF) conditions at the University of California, Riverside.
Wild-type 10-week old confirmed SFB-free C57Bl/6 female mice were purchased from JAX labs (Stock# 000664) and housed in SPF conditions.
Germ-free ~12-week old C57Bl/6 male and female mice were generated from within the germ-free facility at the University of California, Riverside.
Microbiome studies
– Lumenal contents (distal ileum, cecum, proximal and distal colon) and mucosal-associated microbes (small and large intestine) were isolated as previously published.38 Intestinal epithelial cells (IEC) were isolated from small and large intestines using the dithiothreitol (DTT)-based (ThermoFisher Scientific, Waltham, MA) intraepithelial lymphocyte release method and the Percoll-based (GE Healthcare Bio-Sciences, Pittsburg, PA) density gradient purification method as previously described.39,40 DNA was isolated from the samples using the DNeasy PowerSoil Kit (Qiagen, Germantown, MD) and a 30-s bead-beating step using a Mini-Beadbeater-16 (BioSpec Products, Bartlesville, OK, USA). Illumina bacterial rRNA internal transcribed spacer (ITS) region libraries were constructed as previously described.36,41 DNA sequencing (single-end 250 base) was performed using an Illumina MiSeq (Illumina Inc., San Diego, CA).
Bacterial rRNA ITS data processing
– The UPARSE pipeline was used for de-multiplexing, length trimming, quality filtering and operational taxonomic unit (OTU) picking using default parameters or recommended guidelines as initially described and which have been updated at https://www.drive5.com/usearch/manual/uparse_pipeline.html.42 Briefly, after demultiplexing and using the recommended 1.0 expected error threshold, sequences were trimmed to a uniform length of 245bp, which kept 65.1% of reads. De-replicated sequences were subjected to error-correction (denoised) and chimera filtering to generate zero-radius operational taxonomic units (ZOTUs) using UNOISE3.43 An OTU table was then generated using the otutab command. ZOTUs having non-bacterial DNA were identified by performing a local BLAST search of their seed sequences against the nucleotide database.44 ZOTUs were removed if any of their highest scoring BLAST hits contained taxonomic IDs within the Rodent family, Fungal or Viridiplantae kingdoms, or PhiX. Taxonomic assignments to bacterial ZOTUs were made by finding the lowest common taxonomic level of the highest BLAST hits excluding unclassified designations. The bacterial rRNA ITS sequences have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the SRA BioProject Accession Number PRJNA609140.
Bacterial rRNA ITS data analysis
– QIIME was used to calculate Hellinger beta diversity distance matrices, which were depicted using principle coordinates analysis (PCoA), and statistically assessed by performing Adonis tests.45,46 Statistical differences among the phylotypes were determined using edgeR and the false discovery rate (FDR) method.47-49 Prism (GraphPad, La Jolla, CA) was used to create taxonomic plots.
Isolating the E. coli phylotype bacterium (mAIEC)
– The mouse AIEC (mAIEC) used in this study was isolated using standard microbiological procedures. Briefly, mAIEC was isolated from distal colon luminal contents – a region where a significant increase in relative abundance of mAIEC was found (see Figure 3(a)) – from a constitutive Ptpn2-knockout (KO) mouse using BBA plates – Brucella Agar (BD, Franklin Lakes, NJ) + 5% defribrinated sheep blood (Colorado Serum Company, Denver, CO) – and incubated aerobically at 37°C overnight. We performed a three-step procedure to identify and purify the mAIEC. First, Escherichia coli colonies were tentatively identified using the PCR-based, Ribosomal Intergenic Spacer Analysis (RISA) method and small portions of individual colonies from the BBA plates as the templates, and the PCR primers: 1507 F, GGTGAAGTCGTAACAAGGTA and 23SR, GGGTTBCCCCATTCRG (see Suppl. Figure 3 for additional information on this method and PCR amplicon results).50 Second, colonies with an E. coli banding pattern from the RISA were purified by performing two successive streak-plating procedures to obtain single colonies. The resulting colonies were grown overnight in LB broth, and stored in aliquots in Brucella Broth + 20% glycerol at –80°C. Finally, the strain used in this study – mAIEC strain UCR-PP2 – was confirmed to have the identical rRNA ITS nucleotide sequence as the E. coli phylotype identified by the Illumina sequence analysis by (i) PCR amplifying the rRNA ITS region of individual colonies using the RISA primers described above, and (ii) using the Sanger method to obtain the nucleotide sequence of the PCR amplicons.36
Figure 3.
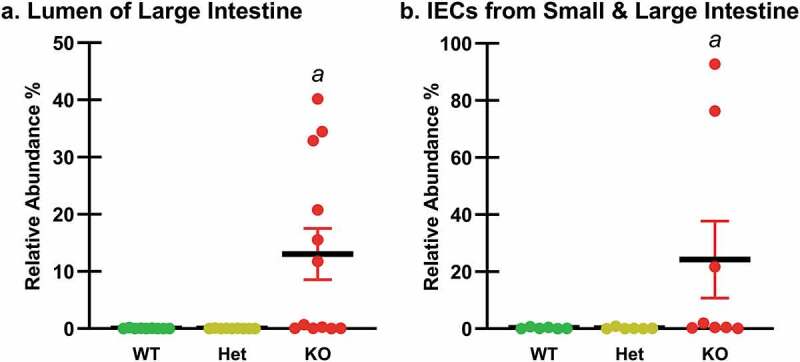
Relative Abundances of the E. coli Phylotype by Intestinal Region and Ptpn2 Genotype. The relative abundances of the E. coli phylotype were examined in the regions identified by the beta diversity analysis (Figure 1(a)); (a) the lumen of the large intestine (cecum, proximal and distal colon) (n = 9–12), and (b) intestinal epithelial cells (IECs) from the small and large intestine (n = 6–8). Differences were determined using edgeR analyses at the species level. Data are shown as mean relative abundances (thick horizontal lines) ± standard error. Ptpn2-KO mice showed higher relative abundance of mAIEC in the lumen of the large intestine (aP ≤ 0.003) and in IECs from the small and large intestine (aP < 0.001) compared with WT and Het mice.
Bacterial constructs
– To generate a constitutive red fluorescent mAIEC (mAIECred), pKB985 which encodes for mCherry was transformed into chemically competent mAIEC.51,52 Plasmid maintenance was achieved during subculture using chloramphenicol (20 µg/mL) (Sigma-Aldrich, St. Louis, MO).
Bacterial infection studies
– Bacteria from stocks frozen at –80°C in 1:1 vol/vol glycerol:LB were cultured overnight in Luria–Bertani (LB) broth at 37°C, 250–300rpm, and regrown the next day in fresh LB to exponential phase growth. Culture was pelleted, washed with phosphate buffered saline (PBS), and resuspended in PBS. The bacteria used were the LF82 human AIEC (kindly provided by the late Dr. Arlette Darfeuille-Michaud), mAIEC, and K12 (a noninvasive E. coli, ATCC 25404). Caco-2 brush border (Caco-2BBe) cells were seeded at 2 × 105 cells per well in 24-well plates, cultured until confluent, and infected at a multiplicity of infection (MOI) of 10 bacteria/cell as previously described.53,54 Media was changed to antibiotic and serum-free media 24 h prior to infection. For adherence and invasion studies, Caco-2BBe cells were infected with bacteria for 3 h followed by 1 h with fresh media with 100 µg/mL Gentamicin (Sigma Aldrich, St. Louis, MO) for invasion studies. Bacterial survival was performed using murine J774 macrophages (Mϕ) (seeded similarly as Caco-2BBe cells above) (ATCC) infected with bacteria at a MOI of 20 for 2 h followed by a PBS wash, incubation with fresh media containing 100 µg/mL Gentamicin for 1 h followed by incubation with fresh media containing 20 µg/mL Gentamicin for 24 h. After bacterial infection, cells were washed with PBS and lysed with 1% Triton-X for 5 min. Lysates were plated onto Luria Bertani agar plates and cultured overnight at 37°C.
For imaging of mAIECred in epithelial cells by immunofluorescence, Caco-2BBe cells were seeded at 5 × 105 cells per round coverslip, cultured until 70% confluent, and infected with mAIECred following the invasion studies described above. Cells were washed with PBS (x3) followed by fixation with 4% paraformaldehyde for 20 min at room temperature. Cells were then permeabilized with 0.3% Triton X-100 (ThermoFisher Scientific, Waltham, MA) for 5 min followed by blocking with 5% Bovine Serum Albumin (BSA) for 10 min at room temperature. Cells were incubated with Alexa Fluor 488-Phalloidin antibody (1:1000) (abcam, Cambridge, United Kingdom) for 90 min at room temperature following by nuclei staining with 4,6-diamididino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA). Confocal analysis was performed using a CSU-X-1 spinning-disk confocal imager (Yokogawa, Japan) attached to a Zeiss 130 Axio Observer inverted microscope. Hardware was controlled by Micro-Manager imaging software. Phalloidin was visualized using a 488 nm excitation laser and FITC filter set. DAPI was visualized using a 405 nm excitation laser and DAPI filter set. mCherry was visualized using a 561 nm excitation laser and mCherry filter set. Images were analyzed using ImageJ software.
Mice were infected by oral gavage with 106 colony forming units (CFU)/mL of mAIECred, or 109 CFU/mL of mAIEC, or K12 in 100 µL PBS/mouse. Body weight was monitored daily. Colonization and bacterial burden were measured by overnight culture of homogenized samples suspended in 500 µL of PBS.
Induction and assessment of colitis severity and histological score
– Colitis was induced by supplementation of drinking water with dextran-sodium sulfate (DSS) (molecular weight 36,000–50,000) (MP Biomedicals, Irvine, CA) at 3% for 7 d as described.55 Mice were randomized to three groups: 1) H2O group did not receive DSS but received daily single gavages of PBS (H2O-PBS) or bacteria (H2O-K12, H2O-mAIEC) for the first four consecutive days (dosing previously determined, unpublished data (Spalinger et al.)); 2) acute DSS group received DSS for 7 d with daily single gavages of PBS (DSS-PBS) or bacteria (DSS-K12, DSS-mAIEC) for the first four consecutive days of DSS treatment followed by 4 d of recovery post DSS treatment; and 3) recovery from colitis group received DSS for 7 d with daily single gavages of PBS (rec-PBS) or bacteria (rec-K12, rec-mAIEC) for four consecutive days post DSS treatment.
To assess disease severity of colitis in animals, disease activity index (DAI) was monitored daily as follows: Appearance (0 = smooth, shiny fur; 1 = dull coat, 2 = dull, ruffled fur), Activity (0 = normal, active, 1 = reduced activity, moves when cage is opened, 2 = reluctant to move when touched), Interest (0 = active interest, 1 = reduced interest when cage is opened, 2 = self-isolation, does not interact), Weight (0 = weight gain or no change, 0.5 = weight loss up to 3%, 1 = weight loss up to 5%, 1.5 = weight loss up to 7%, 2 = weight loss over 7%), and Stool consistency (0 = solid, 1 = soft, 2 = very soft but in shape, 3 = liquid) for a total score range of 0–11.56,57 Myeloperoxidase, an indicator of infiltration/activation of neutrophilic granulocytes was measured as previously described.57,58 Histological scoring for inflammatory infiltration and epithelial cell damage was performed on H&E-stained sections of the most distal 1 cm of the mouse colon.35,56,57 Briefly, sections from the most distal 1 cm of the mouse colon were deparaffinized in Citrisolv and rehydrated using a series of alcohol washes with decreasing concentration, before staining with Hematoxylin for 10 min. The sections were then rinsed with tap water for 10 mins and stained in Eosin Y solution for 15 sec, briefly rinsed in tap water and dehydrated in ethanol with ascending concentrations followed by Citrisolv, and finally mounted with Permount (ThermoFisher Scientific, Waltham, MA). Microscopic assessment was performed using a Leica DM5500 microscope attached to a DFC365 FX camera using a 63× oil immersion objective with an additional 2× digital zoom. The individual images were converted to tiff files with the LAS-V4.12 lite software, and Photoshop (Adobe) was used to create the final figures (Suppl. Figure 2).
In-vivo barrier permeability
– Mice were gavaged with 80 mg/mL of fluorescein isothiocyanate (FITC)-dextran (4 kDa) (FD4) and 20 mg/mL of rhodamine B-dextran (70 kDa) (RD70). After 5 h, blood was collected by tail bleed (germ-free mice) or retro-orbital bleed into serum collection tubes. Blood was centrifuged at 4°C, 1,500 g, for 15 min, and serum analyzed for FITC-dextran and rhodamine B-dextran concentration with the Veritas Microplate luminometer (Turner Biosystems, Sunnyvale, CA), GloMax software (Promega, Madison, WI), using excitation/emission wavelengths of 490/510-570 nm and 525/580-640 nm, respectively.59,60 Standard curves for calculating fluorophore concentration in the samples were obtained by diluting the fluorophore stock in water.
Escherichia coli genome sequencing, assembly, annotation and analysis
– Genome sequences of our mAIEC (strain UCR-PP2) and E. coli K12 (ATCC 25404) were obtained using shotgun DNA sequencing (Novogene, Sacramento, CA). Genomes were assembled using Spades 3.11.1 and annotated using Prokka 1.13.3.61,62 The genome of mAIEC was compared with the genome sequence of the human LF82 isolate.37 Putative virulence genes found in E. coli’s and other AIECs including LF82 were identified by comparing the known sequence of each gene from K12. Putative virulence genes common between mAIEC; another opportunistic mouse E. coli with some AIEC features, NC101; and the human LF82 AIEC; or genes unique to each isolate, were compared and the percent identity of each gene and predicted protein between the isolates was determined using EMBOSS Water.63,64
Statistical analyses
– We set critical significance level α = 0.05 and analyzed our data using parametric statistics. Data are expressed as mean ± SD for n independent observations per group unless stated otherwise. Between-group inferences were made by using 1-way or 2-way analysis of variance (ANOVA) and controlled by the false discovery rate (FDR) procedure where applicable.65
Results
Constitutive PTPN2-deficient mice have an altered intestinal microbiome and expansion of proteobacteria
Since PTPN2 genotyped human IBD patients show altered intestinal microbiomes, we analyzed the microbiomes of constitutive Ptpn2-deficient mice.34 At the community level, analysis of the bacterial rRNA ITS region revealed three distinct regional groups (Figure 1(a)) (Adonis tests, P = 0.001). One group was samples from the lumen of the large intestine, comprised of the cecum, proximal colon and distal colon. Another group contained the intestinal epithelial cell (IEC) samples from the small and large intestine, while the third group was samples from the ileal lumen.
Figure 1.
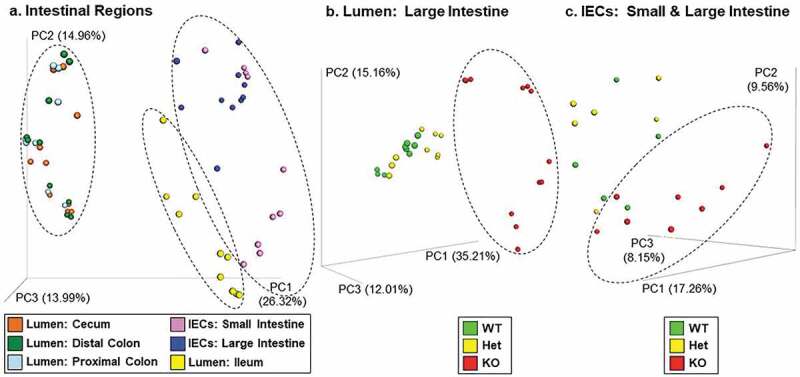
Bacterial Community Analysis by Region and Ptpn2 Genotype. Beta diversity distance values were generated from bacterial rRNA ITS sequences and displayed using principal-coordinates analysis (PCoA). Adonis tests determined that (a) the three regional groups in the ellipses were different (P = 0.001; n = 18–24), (b) KO was different than WT and Het (P < 0.03; n = 9–12) in the lumen of the large intestine (cecum, proximal and distal colon), and (c) KO was different than WT (P = 0.022; n = 6–8) in the intestinal epithelial cells (IECs) from the small and large intestine.
An examination of these three regional groups by Ptpn2 genotype showed that constitutive Ptpn2-knockout (KO) mice had different bacterial communities in the lumen of the large intestine compared with heterozygous (Het) and wild-type (WT) mice (Adonis tests, P < 0.030) (Figure 1(b)). In the group comprised of the IECs from the small and large intestine, the communities in Ptpn2-KO mice were different than those in the WT mice (Adonis tests, P = 0.022) (Figure 1(c)). An analysis of the bacterial communities in the mesenteric lymph nodes and spleen showed that bacterial composition in Ptpn2-KO mice was different than in WT mice (P = 0.005) (Suppl. Figure 1).
To further investigate the taxa in these regional groups, phyla and species plots were constructed (Figure 2). The most abundant gut bacterial phyla have been reported to be Bacteroidetes, Firmicutes and Actinobacteria.66 In our study, the most abundant phylum in the lumen of the large intestine was the Bacteroidetes (Figure 2(a)). However, the most abundant phylum in the IECs of the small and large intestine was the Proteobacteria (Figure 2(c)). We posit that this difference is caused by the distinct environmental conditions associated with the IECs and the lumen of the gut.
Figure 2.
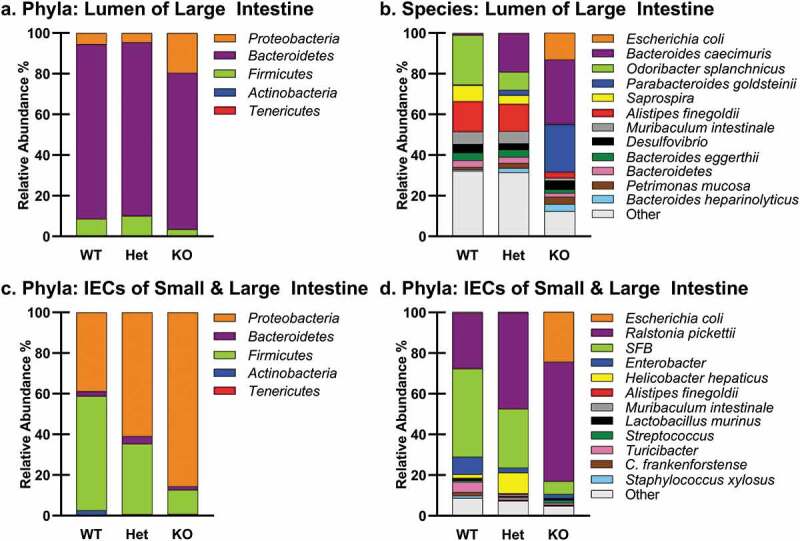
Bacterial Taxa by Region and Ptpn2 Genotype. Bacterial phyla and species plots of (a,b) the lumen of the large intestine (n = 9–12), and (c,d) intestinal epithelial cells (IECs) from the small and large intestine (n = 6–8).
An analysis of the taxa by Ptpn2 genotype showed more Proteobacteria in Ptpn2-KO mice than in the WT and Het mice in samples from the lumen of the large intestine and small and large intestinal IEC samples (Figure 2(a,c)). An examination at a finer taxonomic level showed that the proteobacterial species that increased most dramatically in Ptpn2-KO mice in these regional groups was an Escherichia coli phylotype (Figure 2(b,d)).
PTPN2-deficient mice harbor significant expansion of a novel E. coli phylotype with high sequence identity to AIECs
To examine the relative abundance of this E. coli phylotype in these regional groups, edgeR analyses were performed. In the group comprised of the three regions of the large intestine lumen, the E. coli phylotype was more abundant in Ptpn2-KO mice compared with WT and Het mice (Figure 3(a)). The relative abundance of this E. coli phylotype was also significantly higher in Ptpn2-KO mice than in WT and Het mice in the group comprised of the IEC samples from the small and large intestine (Figure 3(b)).
Using NCBI BLAST, we determined that the rRNA ITS region (250 nucleotides) of the E. coli phylotype had 100% sequence identity with the first described AIEC – the human clinical isolate, LF82, identified in Crohn’s disease patients.26,44 The key factor enabling us to identify this E. coli phylotype as a potential AIEC was our analysis of the bacterial rRNA ITS region.36 When this sequence was first analyzed in 2015, there were only five exact matches in NCBI’s nucleotide (nt) database and one of them was LF82, suggesting that our E. coli phylotype could be an AIEC. Subsequent BLAST analyses of this E. coli rRNA ITS sequence in 2019 using NCBI’s nt database (updated on 2019/10/03 and excluding “Uncultured/environmental sample sequences”) identified 91 exact matches. Conversely, when we performed a similar analysis using a portion of the 16S rRNA gene (primers 515 F and 806 R, 253 nucleotides) of our isolated mouse E. coli, we obtained 12,352 identical matches. These data demonstrated the value of examining the rRNA ITS region of bacteria to identify our bacterium as a putative E. coli, and further provided the impetus for isolating this bacterium.36 Since there is no genetic marker to conclusively distinguish AIEC from other E. coli, we next proceeded to confirm the phenotypic properties of our novel mouse E. coli.67,68
Confirmation of phenotypic properties of a novel mouse AIEC
Since AIEC have defined criteria to establish their adherent and invasive properties, we next determined if our E. coli isolate met these criteria in comparison to the human AIEC LF82 and the noninvasive E. coli K12.23 Since, we did not observe any difference in bacterial adherence between human Caco-2BBe IECs and young-adult mouse colonic (YAMC) IECs (data not shown), we continued our studies using the better characterized Caco-2 cell line. Our E.coli isolate (mAIEC) showed greater adherence to and invasion of IECs than the control E. coli K12 (Figure 4(a–c)). Of note, the human LF82 AIEC adhered to and invaded human IECs at a much greater level than mAIEC. However, mAIEC showed greater infection of murine Mϕ (J774A.1) than the human LF82 AIEC and K12 (Figure 4(d–f)). These data confirm that our novel mouse E. coli is phenotypically an AIEC.
Figure 4.
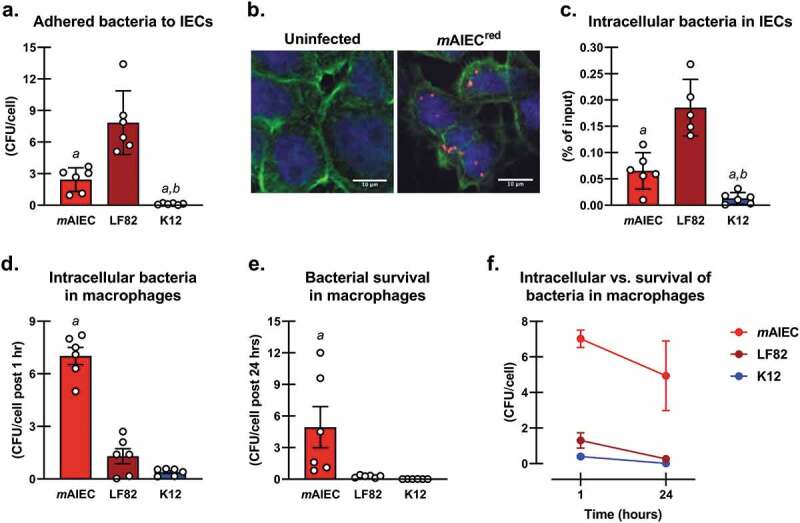
Confirmation of Phenotypic Properties of a Novel Mouse AIEC. Phenotypic characterization of a novel putative mAIEC compared with the human AIEC LF82 and the noninvasive E. coli K12. Caco-2BBe IECs (a,b,c) and J774A.1 murine macrophages (d,e,f) were seeded at 2 × 105 cells/well in a 24-well plate until confluent, or 5 × 105 cells/well on round coverslips until 70% confluent for (b), and then exposed for 3 h to individual bacteria (MOI of 10 bacteria/cell). 1-way ANOVA revealed interactions for all variables (P ≤ 0.004; n = 6 from three independent experiments). (a) Bacteria adhered to Caco-2BBe cells as colony forming units (CFU)/cell (aP < 0.001 cf. LF82, bP ≤ 0.049 cf. mAIEC); (b) Immunofluorescence of uninfected and mAIECred invaded Caco-2BBe cells (n = 2–5); (c) Intracellular bacteria as % of input in Caco-2BBe cells (aP < 0.001 cf. LF82, bP ≤ 0.03 cf. mAIEC); (d) Intracellular bacteria as CFU/cell post 1 h in murine macrophages (aP < 0.001 cf. all groups); (e) Survival of bacteria in murine macrophages as CFU/cell post 24 h (aP < 0.001 cf. all groups); and (f) Relationship of intracellular bacteria and survival of bacteria in murine macrophages (no difference between mAIEC levels at 1 h vs. 24 h, P = 0.49). These data show that mAIEC attach to IECs, and can survive within macrophages, thus confirming AIEC properties.
mAIEC colonizes and causes mild disease in SFB-free mice
We next tested whether our novel mAIEC colonizes mouse intestine and causes disease in vivo in the absence of SFB, since SFB is known to elicit a Th17 response which prevents AIEC colonization.69 Whereas H2O-PBS and H2O-K12 mice showed stable body weights, H2O-mAIEC mice showed significant body weight loss at 4 and 7 days post infection compared with H2O-PBS mice (Figure 5(a)). Interestingly, the drop in body weight coincided with a mild increase in disease activity index (DAI) with significantly higher DAI at day 4 and 5 post infection in H2O-mAIEC mice compared with H2O-PBS and H2O-K12 mice (Figure 5(b)). H2O-mAIEC mice showed no change in FD4 permeability (Figure 5(e)) or distal colon bacterial burden (see Figure 9(b)) on day 7 post-infection coinciding with normal DAI scores at day 7. However, H2O-mAIEC mice showed increased spleen weight (Figure 5(c)), and colon shortening (Figure 5(d)) 7 d post infection compared with H2O-PBS and H2O-K12 mice. This observation could potentially be explained by the sustained fecal bacterial burden of mAIEC up to at least 15 d post infection (Suppl. Figure 2(a)). These data demonstrate that mAIEC caused mild and transient disease activity.
Figure 5.
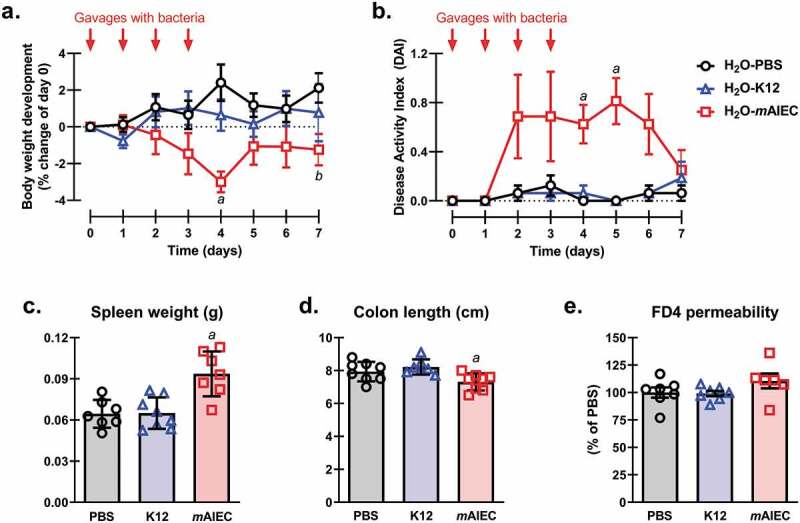
mAIEC Colonizes and Causes Mild Disease in SFB-free Mice. Oral gavage of mAIEC (109 bacteria/mouse in 100 µL PBS; day 0–3) caused (a) weight loss (n = 8; also see Figure 7(a)); (b) mild disease (n = 8; also see Figure 8(b)); (c) splenomegaly (n = 7–8; also see Figure 7(c)); (d) colon shortening (n = 7–8; also see Figure 7(d)); and (e) intestinal FD4 permeability (n = 6–7; also see Figure 8(a)) in confirmed SFB-free C57Bl/6 mice (10 weeks old; 20–22 g; JAX labs) compared with control E. coli (K12) or PBS after 7 d. 2-way repeated measures (RM) ANOVA of body weight and DAI revealed an interaction (mean ± SEM from two independent experiments; P ≤ 0.002). mAIEC reduced body weight at day 4 compared with all groups (aP ≤ 0.01) and day 7 compared with PBS (aP = 0.03), and DAI at days 4 and 5 compared with all groups (aP ≤ 0.02). 1-way ANOVA of spleen weight and colon length, but not FD4 permeability revealed an interaction (mean ± SD from two independent experiments; P ≤ 0.01). mAIEC increased spleen weight compared with all groups (aP ≤ 0.001) and reduced colon length compared with K12 (aP ≤ 0.01).
Figure 9.

mAIEC Infection During DSS Worsens Barrier Permeability and Increases Bacterial Burden. Co-administration of mAIEC and DSS (3%; 7 d) in drinking water exacerbated (a) FD4 permeability (as % of H2O-PBS; n = 6–8); and bacterial burden of mAIEC in the (b) distal colon (n = 5–8); (c) spleen (n = 6–8); and (d) liver (n = 6–8) in confirmed SFB-free C57Bl/6 mice (10 weeks old; 20–22 g) compared with control E. coli (K12) or PBS. 2-way ANOVA revealed an interaction (mean ± SD from two independent experiments; P ≤ 0.03). DSS induced an increase in permeability independent of infection (aP = 0.01 cf. H2O mice) and there was no difference between the DSS mice (P ≥ 0.13). DSS-mAIEC mice had greater bacterial burden in their distal colon (aP ≤ 0.04 cf. DSS-K12 mice), spleen (aP ≤ 0.03 cf. all mice), and liver (aP ≤ 0.02 cf. all mice).
mAIEC requires a microbiome to invade germ-free mice
Having shown that mAIEC only caused transient disease in the presence of other gut bacteria, we next tested whether mAIEC alone caused disease by colonizing germ-free (GF) male and female mice with mAIEC or K12. Although mAIEC-infected mice showed fluctuations in body weight as early as 5 d post-infection compared with K12-colonized mice, this did not reach significance despite sustained mAIEC fecal bacterial burden (Figure 6, Suppl. Figure 2(b)). Interestingly, fecal microbiota transplant (FMT) of cecal content from a Ptpn2-KO mouse caused sustained, statistically significant, weight loss in mice compared with GF mice colonized with K12, and intriguingly, mAIEC-dependent weight loss was restricted to male GF mice (Figure 6). These data demonstrate that mAIEC is capable of causing disease only in the presence of other gastrointestinal microbes.
Figure 6.
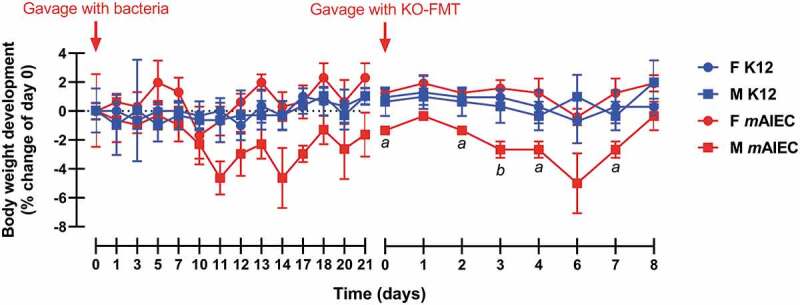
mAIEC Does Not Cause Disease in Germ-free Mice Without a Microbiome. Oral gavage of mAIEC (109 bacteria/mouse in 100 µL PBS) to germ-free mice (C57Bl/6; 12–13 weeks old; n = 3, two experiments). Whereas mAIEC infection had no significant effect on body weight, fecal microbiota transplant (FMT) of cecal content from a Ptpn2-KO mouse (KO-FMT) by oral gavage after 21 d of mAIEC colonization caused a selective decrease in body weight in male vs. female mAIEC-infected mice while KO-FMT into control K12 E. coli infected mice was without effect (n = 3). 2-way RM ANOVA revealed an interaction (mean ± SD; P ≤ 0.02). Body weight of male mAIEC infected mice was significantly lower at days 0, 2, 4, and 7 post-KO-FMT compared with female mAIEC infected mice (aP ≤ 0.02) and at day 3 compared with female mice (bP ≤ 0.005).
mAIEC worsens DSS colitis
We next wanted to determine if mAIEC modifies inflammation by worsening the response to an acute inflammatory episode. Whereas DSS control mice (DSS-PBS or DSS-K12) did not show a significant loss in body weight, mice treated with DSS and infected with mAIEC (DSS-mAIEC) showed a significant and sustained drop in body weight beginning at 4 d compared with H2O control mice (H2O-PBS & H2O-K12) and DSS-PBS mice, and 5–7 d compared with H2O control mice and DSS treated (DSS-PBS, DSS-K12, DSS-mAIEC) mice (Figure 7(a)). Consequently, DSS-mAIEC mice showed severe disease indicated by a significant increase in DAI at 7 d, induction of splenomegaly, and a further shortening in colon length compared with H2O control and DSS-treated mice (Figure 7(b–d)). Additionally, DSS-mAIEC mice showed a higher DAI, increased histological score, and MPO activity compared with DSS-PBS and DSS-K12 mice (Figures 7(e–f), 8).
Figure 7.

mAIEC Worsens Disease During Acute DSS Colitis. Co-administration of mAIEC and DSS (3%; 7 d) in drinking water exacerbated (a) weight loss (n = 8); (b) disease activity (n = 8); (c) splenomegaly (n = 7–8); (d) colon shortening (n = 7–8); (e) mean histology score (n = 5); and (f) MPO activity (n = 5) in confirmed SFB-free C57Bl/6 mice (10 weeks old; 20–22 g; JAX labs) compared with control E. coli (K12) or PBS. 2-way RM ANOVA of body weight and 1-way RM ANOVA of DAI revealed an interaction (mean ± SEM from two independent experiments, P < 0.001). mAIEC alone in the absence of DSS caused a mild decrease in body weight (4 d, aP ≤ 0.04 cf. H2O groups) and increase in DAI (4 d, cP ≤ 0.04 cf. H2O-PBS and DSS-PBS; 5 d, dP ≤ 0.04 cf. H2O groups). Although DSS-PBS mice showed a very mild decrease in body weight (7 d, dP ≤ 0.03 cf. H2O-PBS) and increase in DAI (7 d, fP ≤ 0.02 cf. H2O groups), and DSS-K12 mice showed an increase in DAI (6 d, eP ≤ 0.03 cf. H2O-PBS and H2O-K12; 7 d, fP ≤ 0.04 cf. H2O groups), mAIEC potentiated the loss in body weight (4 d, bP = 0.02 cf. H2O-PBS and K12 and DSS-PBS; 5–7 d, aP ≤ 0.04 cf. all groups), and increased DAI (3 d, aP ≤ 0.005 cf. H2O-PBS and K12 and DSS groups; 4–7 d, bP ≤ 0.04 cf. all groups). 1-way ANOVA of all other parameters revealed an interaction (mean ± SD from two independent experiments except for mean histology score and MPO activity; P ≤ 0.001). mAIEC alone in the absence of DSS and DSS-PBS and K12 equally caused splenomegaly (aP ≤ 0.02 cf. H2O-PBS and K12). Additionally, mAIEC alone in the absence of DSS caused colon shortening (aP = 0.006 cf. H2O-K12). However, DSS-PBS and K12 showed more pronounced shortening of the colon (bP ≤ 0.03 cf. H2O groups and cP ≤ 0.005 cf. H2O-PBS and K12, respectively). mAIEC further potentiated the increase in spleen weight (bP ≤ 0.04 cf. all groups) and colon shortening (dP ≤ 0.001 cf. all groups). All DSS-treated mice showed an increase in mean histology score (aP ≤ 0.02 cf. H2O groups) and this effect was worsened when DSS mice were infected with mAIEC (bP ≤ 0.001 cf. DSS groups). MPO activity was increased in DSS-PBS but not DSS-K12 mice, and this effect was not worsened when DSS mice were infected with mAIEC (aP ≤ 0.008 cf. all groups).
Figure 8.

Histology of Distal Colons from Colitis Mice Infected with E. coli. Hematoxylin and Eosin stained sections (5 µM) of the most distal 1 cm of the mouse colon from water treated or DSS-colitis mice gavaged with PBS, control E. coli K12, or mAIEC.
As an additional determination of intestinal epithelial integrity and function, we assessed in vivo permeability in the same mice used in Figures 7 and 8 and found that DSS-PBS, DSS-K12 and DSS-mAIEC mice showed increased permeability to FD4 compared with H2O-PBS, H2O-K12 or H2O-mAIEC mice, and there was no difference in FD4 permeability between DSS-mAIEC mice and DSS-PBS or DSS-K12 mice (Figure 9(a)). However, only DSS-mAIEC mice showed higher levels of bacterial burden in distal colon, spleen and liver compared with H2O-PBS, H2O-K12 and H2O-mAIEC mice and DSS-PBS and DSS-K12 mice (Figure 9(b–d)). Interestingly, there was no significant bacterial burden found in mesenteric lymph nodes (mLN) (data not shown). Overall, these data confirm that mAIEC: (i) invades SFB-free mouse intestine, (ii) causes disease and weight loss; (iii) increases intestinal permeability; and (iv) exacerbates disease in the setting of inflammation.
mAIEC prevents recovery from DSS colitis
Since mAIEC worsened acute colitis, we wanted to determine if it also impaired recovery from colitis. DSS-induced colitis in all of the mice in the recovery group, regardless of bacterial infection, as evidenced by reduced body weight; however, mice infected with mAIEC during the recovery phase (rec-mAIEC) showed a more severe body weight loss and slower recovery at 13 and 14 d compared with mice treated with PBS (rec-PBS) and mice infected with K12 (rec-K12) during the recovery phase (Figure 10(a)). Consequently, rec-mAIEC mice showed a higher and more sustained DAI from day 9 through 14 compared with rec-PBS and rec-K12 mice (Figure 10(b)). This coincided with splenomegaly and bacterial burden in distal colon but no change in colon length or FD4 permeability in rec-mAIEC mice compared with rec-PBS and rec-K12 mice (Figure 10(c–f)). Overall, these data show that mAIEC prevents recovery of acute colitis.
Figure 10.

mAIEC Impairs Recovery from Colitis. Administration of mAIEC post DSS (3%; 7 d) in drinking water impaired recovery of (a) body weight (n = 7–8); (b) disease (n = 7–8); and (d) splenomegaly (n = 7–8); with no effect on (c) colon length (n = 6–8); (e) FD4 permeability (as % of rec-PBS; n = 7–8); or (f) bacterial burden (n = 5–8) of mAIEC to the distal colon in confirmed SFB-free C57Bl/6 mice (10 weeks old; 20–22 g; JAX labs) compared with control E. coli (K12) or PBS. 2-way RM ANOVA of body weight and DAI revealed an interaction (mean ± SEM from two independent experiments; P < 0.001). mAIEC prevented recovery of body weight loss (13–14 d, aP ≤ 0.03 cf. rec-PBS and K12), and decrease in DAI (9–14 d, aP ≤ 0.03 cf. rec-PBS and K12). One-way ANOVA of spleen weight and distal colon bacterial burden revealed an interaction (mean ± SEM from two independent experiments; P < 0.001) with no effect on colon length or FD4 permeability (aP ≥ 0.59). mAIEC increased spleen weight during the recovery phase (aP ≤ 0.002 cf. rec-PBS and K12) and was detected in distal colon tissue (aP ≤ 0.001 cf. rec-PBS and K12).
Genome sequence analysis of mAIEC
Since the rRNA ITS region of mAIEC and the human AIEC LF82 had 100% sequence identity, we next wanted to identify how similar these bacteria are across their genomes. We sequenced the genome of our novel mouse AIEC and compared it with the published genome sequence of LF82 (GenBank CU651637.1). mAIEC showed approximately 90.3% sequence identity to the genome of the human AIEC LF82. We further probed for the presence in our mAIEC of putative virulence genes found in E. coli’s and other AIECs including LF82. Putative virulence gene sequences and their predicted protein products were compared using EMBOSS Water (Table 1).63,64 There were 14 total putative virulence genes that were used in this analysis. In particular, mAIEC showed two distinct differences: mAIEC yfcU and tnpB had very low % gene and protein identities when compared to the same gene and protein sequences from LF82 and NC101 (Table 1). Since analyses using BLAST determined that the closest matches to these genes and proteins were yfcU or tnpB for all three bacteria, this suggests that these proteins all have similar functions but that the sequences from our mAIEC are very different from those from LF82 and NC101.
Table 1.
Summary of Putative Virulence Genes Between the Novel mAIEC, LF82 AIEC and NC101 E. coli.
| Gene | Reference | Function | % gene identity with NC101 | % query covered | % putative protein identity with NC101 | % query covered | % gene identity with LF82 (Accession CU651637.1) | % query covered | % putative protein identity with LF82 | % query covered |
|---|---|---|---|---|---|---|---|---|---|---|
| fimH | 70 | Terminal subunit of Type 1 pilus | *99.1 | 100 | 99 | 100 | 99.4 | 100 | 99.5 | 100 |
| yfcU | 71 | Outer membrane usher protein | 55.5 | 98 | 44.6 | 97 | 54.7 | 87 | 44.7 | 97 |
| yadC | 72 | Fimbrial-like protein | *92.8 | 100 | 91.2 | 100 | *93.2 | 100 | 91.2 | 100 |
| csgE | 73 | Curli production assembly/transport component | 99.7 | 100 | 100 | 100 | 99.7 | 100 | 100 | 100 |
| yehA | 74 | Putative fimbrial-like protein | 99.3 | 100 | 99.4 | 100 | 99.4 | 100 | 99.4 | 100 |
| hcpA | 73 | Major exported protein | 98.6 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| fimA | 73 | Type-1 fimbrial protein A chain | 90.2 | 100 | 90.7 | 100 | *91.4 | 100 | 91.3 | 100 |
| ygiL | 75 | Fimbrial family protein | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| ftsh | 76 | ATP-dependent zinc metalloprotease | 99.3 | 100 | 100 | 100 | 99.3 | 100 | 100 | 100 |
| vat | 77 | Vacuolating autotransporter toxin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| malX | 73 | PTS system maltose- and glucose- specific EIICB component | 99.7 | 99 | 100 | 100 | 99.7 | 100 | 100 | 100 |
| tnpB | 78 | IS200/IS605 family element transposase accessory protein | 43.5 | 99 | 34.1 | 93 | 47.6 | 99 | 34.1 | 94 |
| dsbA | 73 | Thiol:disulfide interchange protein | 99.4 | 100 | 100 | 100 | 99.7 | 100 | 100 | 100 |
| fliA | 79 | RNA polymerase sigma factor | 98.9 | 100 | 100 | 100 | 99.8 | 100 | 100 | 100 |
*For the indicated gene comparisons, when the % gene identity was greater than % protein identity, there was a higher percentage of nucleotide differences in codon positions 1 + 2 (32%) than when % gene identity was less than % protein identity (9%). Similar comparisons of the genes with equal % identities and the two genes with much greater dissimilarities (yfcU and tnpB) were not included in this analysis.
Discussion
One of the contributing factors to IBD development and progression is alteration of intestinal microbiome composition that favors expansion of commensal bacteria with potential to cause disease. In mice with loss of expression of the IBD risk gene, Ptpn2, we found significant alterations in intestinal bacteria compared with wild-type littermates. Moreover, Ptpn2-deficient mice showed a selective increase in abundance of Proteobacteria, specifically E. coli. Intriguingly, the highest increase in abundance of lumenal and mucosal-associated bacteria in Ptpn2-KO mice was of an E. coli that shared >90% genome sequence similarity to the human IBD-associated AIEC LF82. Furthermore, IBD patients that have been genotyped for the PTPN2 IBD risk allele rs1893217 show altered microbiomes and increased presence of Proteobacteria, thus further validating our in vivo system as a model of IBD.23,33,34 Although LF82 was originally isolated from the ileum of a Crohn’s disease patient, additional AIEC have since been identified in ulcerative colitis patients and isolated from other intestinal regions.37,80-82 Whereas mAIEC was detected albeit at low levels in lumenal and mucosal-associated samples from wild-type mice indicating that this bacterium is part of the resident microbiota, Ptpn2-KO mice showed higher abundance of mAIEC in both the lumen of the gut and the epithelium of the small and large intestine. This was a notable finding because IBD-associated AIEC are able to adhere to and invade IECs. Moreover, the finding that our novel mAIEC was present in IECs in vivo further supports its identity as an AIEC. Overall, reduced expression of the IBD candidate gene, Ptpn2, alters intestinal microbial communities in mice to favor expansion of this novel mouse AIEC.
We confirmed the adherent, invasive and survival properties of mAIEC relative to the well-studied human AIEC LF82.23 We found that mAIEC adhere to and invade intestinal epithelial cells, although to a lesser extent than LF82. This was expected as we used human IECs with a mouse bacterium to comply with the criteria for identification as AIEC that were established for LF82.23 However, the absolute values of mAIEC adherence and invasion are not significantly different from the published adherence and invasive properties for an AIEC, and there was no difference in adherence of mAIEC to young adult mouse colonic (YAMC) IEC lines compared with human IECs.23,26 Intriguingly, survival of mAIEC in J774A.1 mouse macrophages was significantly higher than LF82 and K12, and this was not solely a host-specific effect since mAIEC invaded human THP-1 macrophages similar to LF82 (Spalinger et al., unpublished manuscript; under review). Given that our current studies were performed with mouse macrophages, this may suggest that our novel mAIEC represents a better tool to study AIEC-induced disease in a mouse host than the human LF82 isolate. Overall, these data confirm the epithelial adherent and invasive properties and survival in macrophages of our novel mAIEC.
We also validated that our novel mouse AIEC can cause disease in SFB-free mice. These mice were housed under SPF conditions reflecting the housing conditions of the Ptpn2-KO mice in which we found a high abundance of mAIEC (SPF mice with low abundance of SFB, data not shown). We confirmed that infection of mAIEC, but not K12, results in mild disease evident by reduced body weight and slightly increased disease activity. The lack of an overt response to infection, and that these mice appear to recover by day 7 despite delayed recovery of spleen weight and colon length due to pathologic changes to the tissue, may be explained by the fact that these mice already have a microbiome that could partially outcompete or restrict mAIEC pathogenicity.83 Therefore, we infected germ-free mice and found that mono-colonization with mAIEC had no significant effect on body weight despite sustained levels of bacterial burden. Interestingly, introduction of a microbiome by FMT in mAIEC-infected germ-free mice caused a significant and sustained reduction in body weight, and intriguingly, this effect seemed to be specific to male mice despite similar mAIEC and K12 bacterial burden levels in male and female germ-free mice pre-FMT. However, female mice also showed large fluctuations in body weight indicating that mAIEC resulted in a response in both male and female germ-free mice albeit with qualitative differences in the magnitude of the effect that appears to be sex-dependent. We showed for the first time that our mAIEC (a pathobiont) can cause transient disease in mice with no known genetic alterations or additional stressors such as DSS-induced colitis.
Since we isolated mAIEC from Ptpn2-KO mice that develop systemic inflammation, and since AIEC have been shown to be increased in CD and UC patients, we investigated the effects of mAIEC in an inflammatory setting. Mice exposed to DSS to induce mild colitis and simultaneously infected with mAIEC displayed more severe disease (greater body weight loss, increased disease scores, bacterial burden) compared with control mice treated with DSS and water treated mice; and increased macromolecular intestinal permeability compared with water-treated mice. We speculate that inflammation is required in the host to create an ideal environment for AIEC to expand and cause a more severe and sustained disease, consistent with it being a pathobiont (such as the human LF82 AIEC) rather than a strict pathogen.84 In fact, AIEC have been shown to worsen the response to an inflammatory flare, and impede a susceptible host from recovery against acute colitis in IBD patients, thus potentiating IBD progression.84,85 Of note, mice infected with mAIEC fail to recover (body weight loss and increased disease activity) from DSS-induced colitis.
To identify possible factors that contribute to mAIEC colonization in mice, we compared the genome sequence of the novel mouse AIEC to the human LF82 AIEC and another opportunistic mouse E. coli with some AIEC features, NC101. The genome of mAIEC showed >90% sequence identity to LF82. Intriguingly, two putative virulence genes, yfcU and tnpB, from our mAIEC were compared to those from LF82 and NC101. The mAIEC gene and putative protein sequences had very low percent identities when compared to the other two bacteria providing further evidence for the novelty of our strain and indicating potentially different gene variants or protein isoforms that may give rise to differences in pathogenicity (e.g., adherence/invasion) between mAIEC, LF82 and NC101.
While a number of studies have investigated potential contributions of commensal bacteria that possess or acquire pathogenic potential, such as the LF82 AIEC in the pathogenesis of IBD, it still remains elusive as to how IBD susceptibility genes modulate the intestinal microbiome to maintain intestinal homeostasis and how disruption of this interaction precipitates changes in the microbiome that promote disease. We show here for the first time how loss of an IBD-associated gene, Ptpn2, in mice (Ptpn2-deficient), independent of an external stressor, alters the intestinal microbiome and favors expansion of a novel mouse AIEC that is capable of both initiating and exacerbating disease. Thus, PTPN2 plays a key role as a “microbial modulator” of the microbiome to protect against pathobiont expansion and colonization. We propose that whole-body Ptpn2-deficiency in mice may serve as a useful model to investigate how host genetics modulate the balance of intestinal microbes, and that this novel mouse AIEC can be utilized to interrogate mechanisms of pathobiont-induced intestinal inflammation.
Supplementary Material
Acknowledgments
We are grateful to Dr. Jason Stajich (UCR) for valuable discussions. We are grateful to Dr. David Lo (UCR) for providing access to the CSU-X-1 spinning-disk confocal imager.
Funding Statement
This work was supported by a Crohn’s and Colitis Foundation Research Fellowship Award (A.S.); Crohn’s and Colitis Foundation Senior Research Award (D.F.M.); NIH-2R01-DK091281 (D.F.M.); American Gastroenterological Association IBD Research Award (D.F.M.); UCR Office of Research & Economic Development Pilot Award (D.F.M. & J.B.); National Institute of General Medical Sciences grant R35GM124724 (to A.H.).
Disclosure of Potential Conflicts of Interest
The authors report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M, et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40(6):713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Study C . Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spalinger MR, Voegelin M, Biedermann L, Zeitz J, Rossel JB, Sulz MC, Frei P, Scharl S, Vavricka SR, Fried M, et al. The clinical relevance of the IBD-associated variation within the risk gene locus encoding protein tyrosine phosphatase non-receptor type 2 in patients of the Swiss IBD Cohort. Digestion. 2016;93(3):182–192. doi: 10.1159/000444479. [DOI] [PubMed] [Google Scholar]
- 5.Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JHM, Howson JMM, Stevens H, McManus R, Wijmenga C, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359(26):2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khor B, Gardet A, Xavier RJ.. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.You-Ten KE, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, Lapp WS, Tremblay ML. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J Exp Med. 1997;186(5):683–693. doi: 10.1084/jem.186.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, Lapp WS. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood. 2004;103(9):3457–3464. doi: 10.1182/blood-2003-09-3153. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, Matsuda T. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem Biophys Res Commun. 2002;297(4):811–817. doi: 10.1016/S0006-291X(02)02291-X. [DOI] [PubMed] [Google Scholar]
- 10.Krishnan M, McCole DF. T cell protein tyrosine phosphatase prevents STAT1 induction of claudin-2 expression in intestinal epithelial cells. Ann N Y Acad Sci. 2017;1405(1):116–130. doi: 10.1111/nyas.13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scharl M, Paul G, Weber A, Jung BC, Docherty MJ, Hausmann M, Rogler G, Barrett KE, McCole DF. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology. 2009;137(6):2030–2040. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 2003;4(3):225–236. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 13.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Assimakopoulos SF, Triantos C, Thomopoulos K, Fligou F, Maroulis I, Marangos M, Gogos CA. Gut-origin sepsis in the critically ill patient: pathophysiology and treatment. Infection. 2018;46(6):751–760. doi: 10.1007/s15010-018-1178-5. [DOI] [PubMed] [Google Scholar]
- 15.Guerville M, Boudry G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointest Liver Physiol. 2016;311(1):G1–G15. doi: 10.1152/ajpgi.00098.2016. [DOI] [PubMed] [Google Scholar]
- 16.Bein A, Zilbershtein A, Golosovsky M, Davidov D, Schwartz B. LPS induces hyper-permeability of intestinal epithelial cells. J Cell Physiol. 2017;232(2):381–390. doi: 10.1002/jcp.25435. [DOI] [PubMed] [Google Scholar]
- 17.Chowdhury SR, King DE, Willing BP, Band MR, Beever JE, Lane AB, Loor JJ, Marini JC, Rund LA, Schook LB, et al. Transcriptome profiling of the small intestinal epithelium in germfree versus conventional piglets. BMC Genomics. 2007;8:215. doi: 10.1186/1471-2164-8-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghoshal UC, Shukla R, Ghoshal U, Gwee KA, Ng SC, Quigley EMM. The gut microbiota and irritable bowel syndrome: friend or foe? Int J Inflam. 2012;2012:151085. doi: 10.1155/2012/151085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stecher B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. In: Conway T, Cohen PS, editors. Metabolism and bacterial pathogenesis; Washington (DC): SM Press; 2015. p. 297–320. doi:10.1128/microbiolspec.MBP-0008-2014. [DOI] [PubMed] [Google Scholar]
- 21.Rogler G, Luc B, Scharl M. New insights into the pathophysiology of inflammatory bowel disease: microbiota, epigenetics and common signalling pathways. Swiss Med Wkly. 2018;148:w14599. doi: 10.4414/smw.2018.14575. [DOI] [PubMed] [Google Scholar]
- 22.Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, Bowman D, Scherl EJ, Simpson KW. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PLoS One. 2012;7(7):e41594. doi: 10.1371/journal.pone.0041594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Medina M, Garcia-Gil LJ. Escherichia coli in chronic inflammatory bowel diseases: an update on adherent invasive Escherichia coli pathogenicity. World J Gastrointest Pathophysiol. 2014;5(3):213–227. doi: 10.4291/wjgp.v5.i3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut. 2014;63(7):1069–1080. doi: 10.1136/gutjnl-2013-304909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, Gonzalez A, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12(2):139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 27.Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Jezek GE, Islas-Islas M, Torres AG. Escherichia coli isolated from a Crohn’s disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol. 2008;298(5–6):397–409. doi: 10.1016/j.ijmm.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 28.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52(3):439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shawki A, McCole DF. Mechanisms of intestinal epithelial barrier dysfunction by adherent-invasive Escherichia coli. Cell Mol Gastroenterol Hepatol. 2017;3(1):41–50. doi: 10.1016/j.jcmgh.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao AP, Petrof EO, Kuppireddi S, Zhao Y, Xia Y, Claud EC, Sun J. Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLoS One. 2008;3(6):e2369. doi: 10.1371/journal.pone.0002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. 2011;141(5):769–776. doi: 10.3945/jn.110.135657. [DOI] [PubMed] [Google Scholar]
- 32.Molodecky NA, Kaplan GG. Environmental risk factors for inflammatory bowel disease. Gastroenterol Hepatol (N Y). 2010;6:339–346. [PMC free article] [PubMed] [Google Scholar]
- 33.Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommeren S, Imhann F, Stempak JM, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6(12):107. doi: 10.1186/s13073-014-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yilmaz B, Spalinger MR, Biedermann L, Franc Y, Fournier N, Rossel JB, Juillerat P, Rogler G, Macpherson AJ, Scharl M. The presence of genetic risk variants within PTPN2 and PTPN22 is associated with intestinal microbiota alterations in Swiss IBD cohort patients. PLoS One. 2018;13(7):e0199664. doi: 10.1371/journal.pone.0199664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spalinger MR, Kasper S, Chassard C, Raselli T, Frey-Wagner I, Gottier C, Lang S, Atrott K, Vavricka SR, Mair F, et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 2015;8(4):918–929. doi: 10.1038/mi.2014.122. [DOI] [PubMed] [Google Scholar]
- 36.Ruegger PM, Clark RT, Weger JR, Braun J, Borneman J. Improved resolution of bacteria by high throughput sequence analysis of the rRNA internal transcribed spacer. J Microbiol Methods. 2014;105:82–87. doi: 10.1016/j.mimet.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–1413. doi: 10.1016/S0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 38.Presley LL, Wei B, Braun J, Borneman J. Bacteria associated with immunoregulatory cells in mice. Appl Environ Microbiol. 2010;76(3):936–941. doi: 10.1128/AEM.01561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujiwara D, Chen L, Wei B, Braun J. Small intestine CD11c+ CD8+ T cells suppress CD4+ T cell-induced immune colitis. Am J Physiol Gastrointest Liver Physiol. 2011;300(6):G939–G947. doi: 10.1152/ajpgi.00032.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannock GW, Crichton CM, Savage DC. A method for harvesting non-cultivable filamentous segmented microbes inhabiting the ileum of mice. FEMS Microbiol Lett. 1987;45(6):329–332. doi: 10.1111/j.1574-6968.1987.tb02409.x. [DOI] [Google Scholar]
- 41.Ginnan NA, Dang T, Bodaghi S, Ruegger PM, Peacock BB, McCollum G, England G, Vidalakis G, Roper C, Rolshausen P, et al. Bacterial and fungal next generation sequencing datasets and metadata from citrus infected with ‘candidatus liberibacter asiaticus’. Phytobiomes. 2018;2:64–70. doi: 10.1094/PBIOMES-08-17-0032-A. [DOI] [Google Scholar]
- 42.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 43.Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016;081257. [Google Scholar]
- 44.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 45.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73(5):1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 50.Borneman J, Triplett EW. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl Environ Microbiol. 1997;63(7):2647–2653. doi: 10.1128/AEM.63.7.2647-2653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mills E, Baruch K, Aviv G, Nitzan M, Rosenshine I. Dynamics of the type III secretion system activity of enteropathogenic Escherichia coli. mBio. 2013;4(4). doi: 10.1128/mBio.00303-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandel M, Higa A. Calcium-dependent bacteriophage DNA infection. J Mol Biol. 1970;53(1):159–162. doi: 10.1016/0022-2836(70)90051-3. [DOI] [PubMed] [Google Scholar]
- 53.Martinez-Medina M, Aldeguer X, Lopez-Siles M, Gonzalez-Huix F, Lopez-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15(6):872–882. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 54.Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. Isme J. 2007;1(5):403–418. doi: 10.1038/ismej.2007.52. [DOI] [PubMed] [Google Scholar]
- 55.Kasper SH, Spalinger MR, Leonardi I, Gerstgrasser A, Raselli T, Gottier C, Atrott K, Frey-Wagner I, Fischbeck-Terhalle A, Rogler G, et al. Deficiency of protein tyrosine phosphatase non-receptor type 2 in intestinal epithelial Cells has no appreciable impact on dextran sulphate sodium colitis severity but promotes wound healing. Digestion. 2016;93(4):249–259. doi: 10.1159/000445289. [DOI] [PubMed] [Google Scholar]
- 56.Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nat Protoc. 2006;1(6):2900–2904. doi: 10.1038/nprot.2006.446. [DOI] [PubMed] [Google Scholar]
- 57.Friedman DJ, Kunzli BM, Rahim YI, Sevigny J, Berberat PO, Enjyoji K, Csizmadia E, Friess H, Robson SC. CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. 2009;106(39):16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Raselli T, Frey-Wagner I, Gutte PM, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. 2016;126(5):1783–1800. doi: 10.1172/JCI83669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai PY, Zhang B, He WQ, Zha JM, Odenwald MA, Singh G, Tamura A, Shen L, Sailer A, Yeruva S, et al. IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe. 2017;21(6):671–681. doi: 10.1016/j.chom.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edelblum KL, Sharon G, Singh G, Odenwald MA, Sailer A, Cao S, Ravens S, Thomsen I, El Bissati K, McLeod R, et al. The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell Mol Gastroenterol Hepatol. 2017;4(2):285–297. doi: 10.1016/j.jcmgh.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 63.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 64.Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol. 2000;279(1):R1–R8. doi: 10.1152/ajpregu.2000.279.1.R1. [DOI] [PubMed] [Google Scholar]
- 66.Marchesi JR. Human distal gut microbiome. Environ Microbiol. 2011;13(12):3088–3102. doi: 10.1111/j.1462-2920.2011.02574.x. [DOI] [PubMed] [Google Scholar]
- 67.Camprubi-Font C, Lopez-Siles M, Ferrer-Guixeras M, Niubo-Carulla L, Abella-Ametller C, Garcia-Gil LJ, Martinez-Medina M. Comparative genomics reveals new single-nucleotide polymorphisms that can assist in identification of adherent-invasive Escherichia coli. Sci Rep. 2018;8(1):2695. doi: 10.1038/s41598-018-20843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martinez-Medina M, Mora A, Blanco M, Lopez C, Alonso MP, Bonacorsi S, Nicolas-Chanoine MH, Darfeuille-Michaud A, Garcia-Gil J, Blanco J. Similarity and divergence among adherent-invasive Escherichia coli and extraintestinal pathogenic E. coli strains. J Clin Microbiol. 2009;47(12):3968–3979. doi: 10.1128/JCM.01484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang HJ, Xu B, Wang H, Xu B, Wang GD, Jiang MZ, Lei C, Ding ML, Yu PF, Nie YZ, et al. IL-17 is a protection effector against the adherent-invasive Escherichia coli in murine colitis. Mol Immunol. 2018;93:166–172. doi: 10.1016/j.molimm.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 70.Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007;117(6):1566–1574. doi: 10.1172/JCI30504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boisen N, Scheutz F, Rasko DA, Redman JC, Persson S, Simon J, Kotloff KL, Levine MM, Sow S, Tamboura B, et al. Genomic characterization of enteroaggregative Escherichia coli from children in Mali. J Infect Dis. 2012;205(3):431–444. doi: 10.1093/infdis/jir757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verma R, Rojas TCG, Maluta RP, Leite JL, da Silva LPM, Nakazato G, Dias da Silveira W, McCormick BA. Fimbria-encoding gene yadC has a pleiotropic effect on several biological characteristics and plays a role in avian pathogenic Escherichia coli pathogenicity. Infect Immun. 2016;84(1):187–193. doi: 10.1128/IAI.01138-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, Ziebell K, Torres AG, Karmali MA, Coombes BK. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics. 2010;11:667. doi: 10.1186/1471-2164-11-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ravan H, Amandadi M. Analysis of yeh fimbrial gene cluster in Escherichia coli O157: h7in order to find a genetic marker for this serotype. Curr Microbiol. 2015;71(2):274–282. doi: 10.1007/s00284-015-0842-6. [DOI] [PubMed] [Google Scholar]
- 75.Spurbeck RR, Stapleton AE, Johnson JR, Walk ST, Hooton TM, Mobley HLT. Fimbrial profiles predict virulence of uropathogenic Escherichia coli strains: contribution of ygi and yad fimbriae. Infect Immun. 2011;79(12):4753–4763. doi: 10.1128/IAI.05621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allen KJ, Lepp D, McKellar RC, Griffiths MW. Examination of stress and virulence gene expression in Escherichia coli O157: h7using targeted microarray analysis. Foodborne Pathog Dis. 2008;5(4):437–447. doi: 10.1089/fpd.2008.0100. [DOI] [PubMed] [Google Scholar]
- 77.Gibold L, Garenaux E, Dalmasso G, Gallucci C, Cia D, Mottet-Auselo B, Fais T, Darfeuille-Michaud A, Nguyen HTT, Barnich N, et al. The Vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn’s disease-associated Escherichia coli. Cell Microbiol. 2016;18(5):617–631. doi: 10.1111/cmi.12539. [DOI] [PubMed] [Google Scholar]
- 78.Mattar R, Marques SB, Monteiro M, Dos Santos AF, Iriya K, Carrilho FJ. Helicobacter pylori cag pathogenicity island genes: clinical relevance for peptic ulcer disease development in Brazil. J Med Microbiol. 2007;56(Pt 1):9–14. doi: 10.1099/jmm.0.46824-0. [DOI] [PubMed] [Google Scholar]
- 79.Claret L, Miquel S, Vieille N, Ryjenkov DA, Gomelsky M, Darfeuille-Michaud A. The flagellar sigma factor FliA regulates adhesion and invasion of Crohn disease-associated Escherichia coli via a cyclic dimeric GMP-dependent pathway. J Biol Chem. 2007;282(46):33275–33283. doi: 10.1074/jbc.M702800200. [DOI] [PubMed] [Google Scholar]
- 80.O’Brien CL, Bringer MA, Holt KE, Gordon DM, Dubois AL, Barnich N, Darfeuille-Michaud A, Pavli P. Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut. 2017;66(8):1382–1389. doi:10.1136/gutjnl-2015-311059. [DOI] [PubMed] [Google Scholar]
- 81.Lee JG, Han DS, Jo SV, Lee AR, Park CH, Eun CS, Lee Y. Characteristics and pathogenic role of adherent-invasive Escherichia coli in inflammatory bowel disease: potential impact on clinical outcomes. PLoS One. 2019;14(4):e0216165. doi: 10.1371/journal.pone.0216165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kotlowski R, Bernstein CN, Sepehri S, Krause DO. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut. 2007;56(5):669–675. doi: 10.1136/gut.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hausmann M, Rechsteiner T, Caj M, Benden C, Fried M, Boehler A, Rogler G. A new heterotopic transplant animal model of intestinal fibrosis. Inflamm Bowel Dis. 2013;19(11):2302–2314. doi: 10.1097/MIB.0b013e3182a6a0f3. [DOI] [PubMed] [Google Scholar]
- 84.Mirsepasi-Lauridsen HC, Vallance BA, Krogfelt KA, Petersen AM. Escherichia coli pathobionts associated with inflammatory bowel disease. Clin Microbiol Rev. 2019;32:2. doi: 10.1128/CMR.00060-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Small CL, Xing L, McPhee JB, Law HT, Coombes BK. Acute infectious gastroenteritis potentiates a Crohn’s disease pathobiont to fuel ongoing inflammation in the post-infectious period. PLoS Pathog. 2016;12(10):e1005907. doi: 10.1371/journal.ppat.1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.