

Abstract
Aims
Natriuretic peptides are useful for diagnosis and prognostication of heart failure of any cause. Now, research aims to discover novel biomarkers that will more specifically define the heart failure phenotype. DNA methylation plays a critical role in the development of cardiovascular disease with the potential to predict fundamental pathogenic processes. There is a lack of data relating DNA methylation in heart failure that specifically focuses on patients with severe multi‐vessel coronary artery disease. To begin to address this, we conducted a pilot study uniquely exploring the utility of powerful whole‐genome methyl‐binding domain‐capture sequencing in a cohort of cardiac surgery patients, matched for the severity of their coronary artery disease, aiming to identify candidate peripheral blood DNA methylation markers of ischaemic cardiomyopathy and heart failure.
Methods and results
We recruited a cohort of 20 male patients presenting for coronary artery bypass graft surgery with phenotypic extremes of heart failure but who otherwise share a similar coronary ischaemic burden, age, sex, and ethnicity. Methylation profiling in patient blood samples was performed using methyl‐binding domain‐capture sequencing. Differentially methylated regions were validated using targeted bisulfite sequencing. Gene set enrichment analysis was performed to identify differences in methylation at or near gene promoters in certain known Reactome pathways. We detected 567 188 methylation peaks of which our general linear model identified 68 significantly differentially methylated regions in heart failure with a false discovery rate <0.05. Of these regions, 48 occurred within gene bodies and 25 were located near enhancer elements, some within coding genes and some in non‐coding genes. Gene set enrichment analyses identified 103 significantly enriched gene sets (false discovery rate <0.05) in heart failure. Validation analysis of regions with the strongest differential methylation data was performed for two genes: HDAC9 and the uncharacterized miRNA gene MIR3675. Genes of particular interest as novel candidate markers of the heart failure phenotype with reduced methylation were HDAC9, JARID2, and GREM1 and with increased methylation PDSS2.
Conclusions
We demonstrate the utility of methyl‐binding domain‐capture sequencing to evaluate peripheral blood DNA methylation markers in a cohort of cardiac surgical patients with severe multi‐vessel coronary artery disease and phenotypic extremes of heart failure. The differential methylation status of specific coding genes identified are candidates for larger longitudinal studies. We have further demonstrated the value and feasibility of examining DNA methylation during the perioperative period to highlight biological pathways and processes contributing to complex phenotypes.
Keywords: Ischaemic cardiomyopathy, Heart failure, Coronary artery bypass graft surgery, DNA methylation, Epigenome‐wide association study, Male
Introduction
Coronary artery disease (CAD) and heart failure (HF) are leading global causes of morbidity and mortality. 1 Natriuretic peptides, such as B‐type natriuretic peptide, and the prohormone, N‐terminal pro‐B‐type natriuretic peptide, are valuable biomarkers utilized for HF diagnosis and prognostication. 2 However, a future goal is to apply an array of biomarkers to detect and guide therapy of a broader spectrum of HF, including in its asymptomatic early stages. Research to discover novel genetic and epigenetic biomarkers associated with myocardial ischaemia, myocardial wall tension/stretch, myocardial inflammation, and fibrosis therefore represents a key area of investigation. 3
Part of the reparative process following myocardial infarction (MI) requires synthesis of new extracellular matrix that, in some patients, may adversely remodel the left ventricle reducing the ejection fraction during systole. 4 Ischaemic cardiomyopathy does not occur in isolation. Ischaemia and infarction are potent inflammatory triggers associated with local and varying degrees of systemic innate immune activation. A phase of chronic extracellular matrix remodelling involving monocytes and macrophages becomes apparent. 5 The roles of inflammatory cellular subsets in this process are of increasing interest as it becomes apparent that HF may represent a state of dysregulation of inflammatory and fibrotic responses to chronic cardiomyocyte stress. 6 These processes are fundamentally susceptible to regulation at the genomic level, and epigenetic factors, such as DNA methylation, which ultimately determine cellular phenotypes, are likely to play a critical role.
DNA methylation is an epigenetic mark that is increasingly recognized to play a critical role in cardiovascular disease. Notably, methylation variation associated with the development of aortic atheroma is detectable in peripheral blood leucocytes prior to the development of vascular lesions. 7 Furthermore, distinct patterns of peripheral blood DNA methylation have been related to the risk of ischaemic heart disease 8 and coronary events. 9 Variation in cardiomyocyte DNA methylation and gene expression has been demonstrated in end‐stage cardiomyopathy 10 and dilated cardiomyopathy (DCM). 11 , 12 Importantly, significant overlap with the cardiomyocyte DNA methylation changes potentially contributing to cardiomyopathy has been related to changes in peripheral blood DNA methylation. Indeed, these methylation markers of early systolic dysfunction were shown to outperform N‐terminal pro‐B‐type natriuretic peptide. 13 These findings provide strong justification for further studies to identify DNA methylation markers of HF that may precede current clinical predictors and highlight cellular pathways that contribute to the pathogenesis of HF.
There is currently a lack of data focusing specifically on patients with severe multi‐vessel CAD, HF, and peripheral blood DNA methylation. Given the wide‐ranging impact of diabetic and atheromatous processes within the body, it is highly likely that the status of the peripheral blood methylome may indicate important elements that determine the myocardial response to hypoxic, hypertensive, and hyperglycaemic stress. We therefore conducted a pilot methylome‐wide study, as a planned substudy of a large clinical trial, 14 enrolling patients with severe multi‐vessel CAD and angina requiring coronary artery bypass graft surgery. The Aspirin and Tranexamic Acid for Coronary Artery Surgery trial was a large multicentre randomized controlled trial investigating the impact of aspirin and tranexamic acid on bleeding and thrombotic complications following surgery. For this pilot study, we specifically aimed to demonstrate the explorative utility of methyl‐binding domain (MBD)‐capture sequencing to profile the differences in preoperative peripheral blood DNA methylation in a cohort of patients who present with phenotypic extremes of HF but who otherwise share a similar coronary ischaemic burden, age, sex, and ethnicity.
Methods
Patient inclusion
Following ethical approval (The Alfred Hospital Ethics Committee, HREC Number 11/05), we identified 10 men (55–83 years) with multi‐vessel CAD and HF and having elective or semi‐elective coronary artery bypass graft surgery (Table 1 ). These patients were generally (80%) type 2 diabetic. Cases were matched with 10 male diabetic controls (57–81 years) without HF (Table 1 ). Any information suggesting a patient had impaired left ventricular (LV) function or had experienced symptoms of HF resulted in exclusion from the control group. Patients with contributing cardiac valvular pathology (severe mitral regurgitation) were also excluded. Because of the potentially important role of the inflammatory response in HF, patients on corticosteroids or other anti‐inflammatory medications (e.g. COX‐II inhibitors and non‐steroidal anti‐inflammatory medications excluding aspirin) were excluded. Semi‐elective denoted patients who presented with progressive symptoms and signs of HF and/or angina (small ischaemic events such as non‐ST‐elevation MI) and required surgery prior to discharge from hospital. Patients whose HF and/or angina was acutely related to critical coronary events (preoperative ST‐elevation MI, percutaneous coronary intervention on that admission, inotropic support, and intra‐aortic balloon pump support) were excluded. Elective patients were admitted on the day of surgery with stable HF and/or angina.
Table 1.
Cases (HF) and controls
| Variable | HF (range) | Controls (range) |
|---|---|---|
| Number | 10 | 10 |
| Age | 67 (55–83) | 64 (50–81) |
| Diabetes | 80% | 100% |
| Hypertension | 90% | 80% |
| Angina | 80% | 100% |
| Ejection fraction (%) | 27% (15‐35) | 62% (57–71) |
| HF symptoms | 100% | 0% |
| NYHA (1–4) | 2.5 (3–4) | 1.4 (1–2) |
| Surgical LV grade (1–4) | 3.2 (3–4) | 1 (1) |
| No. of distal coronary grafts | 3.7 (3–5) | 3.8 (2–6) |
| % elective vs. semi‐elective | 60% | 60% |
HF, heart failure; LV, left ventricle; NYHA, New York Heart Association.
LV grade: surgical assessment of the left ventricular systolic function. Ejection fraction reported by preoperative transthoracic echocardiography.
Methylation profiling by methyl‐binding domain‐capture sequencing
Whole blood was collected into EDTA tubes, and DNA was extracted using a by DNeasy Blood & Tissue Kit (Qiagen Pty Ltd, Hilden, Germany) and quantified by Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA was fragmented by sonication. The MethylMiner Methylated DNA Enrichment Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to enrich methylated DNA fragments from 500 ng of input material. DNA was eluted from beads with 2 M NaCl solution and was purified by NucleoSpin columns to concentrate DNA and remove salts. Five nanograms of methyl‐enriched DNA underwent library construction using the NEBNext Ultra II DNA Library Prep (New England Biolabs, Ipswich, MA, USA), and the barcoded libraries underwent equimolar pooling and were sent to the Australian Genome Research Facility (AGRF, Melbourne, Australia) for 100 cycles, single end sequencing on HiSeq 2500 (Illumina, San Diego, CA, USA) using version four reagents.
Methyl‐binding domain‐capture sequencing data analysis
Reference human genome (GRCh38 primary assembly) was downloaded from Ensembl alongside Ensembl v84 gene annotation in GTF format. 15 Methyl‐binding domain‐capture sequencing (MBD‐seq) reads were quality trimmed using FastX Toolkit (FastX Toolkit 0.0.14) with a minimum base quality threshold of 20 and minimum read length of 20 nt. Reads were mapped to the human genome using BWA‐MEM (Version 0.7.10). 16 CpG dinucleotide content was determined with a custom BASH script. Peak calling was performed using MACS v1.4d 17 software comparing each patient dataset to a pool of inputs we generated previously. Peak size, GC, and CpG proportions were determined with a custom BASH script. Reads mapped to peaks were counted using featureCounts (v1.4.2) with mapping quality ≥10 and otherwise default settings. 18 In parallel, reads were counted at promoters, as defined as regions ±1 kb of transcriptional start sites, generated using BEDTools software. 19 If a gene possessed multiple transcriptional start sites, the read counts were aggregated to gene‐wise counts with featureCounts. Both methylation matrices underwent filtering to discard regions/genes with fewer than 10 reads per sample on average. General linear model feature of edgeR (Version 3.14.0) 20 was used to identify regions/genes with altered methylation in CCF as compared with control after correcting for patient age. Patient age was scaled in R prior to inclusion in the model. CpG island coordinates were downloaded from the UCSC table browser (http://genome.ucsc.edu/cgi‐bin/hgTables). CpG shore coordinates were derived using BEDTools by extracting regions flanking (up to 2 kb) CpG islands. Genome compartments intron, exon, intergenic, gene body, and coding sequence were obtained from the Ensembl v84 gene annotation file or derived with BEDTools. Open chromatin regions were defined by DNase hypersensitivity experiments from the ENCODE project (wgEncodeRegDnaseClusteredV3.bed.gz). Distal regulatory element coordinates were obtained from a previous study, 21 and coordinates were converted to GRCh38 using the liftOver tool (https://genome.ucsc.edu/util.html). Repeat element coordinates were obtained from RepeatMasker data in the UCSC table browser. For pathway analyses, genes were ranked from most up‐methylated to most down‐methylated using the product of negative log2 of the adjusted P‐value and the sign of the fold change as determined by edgeR. Annotation of differentially methylated regions (DMRs) was performed using GREAT 22 for Supporting Information, Table S2, after coordinate conversion from GRCh38 to GRCh19/Hg19 using liftOver. GSEAPreranked (v2.2.2) 23 was used with the ‘classic’ algorithm to determine differential pathways based on promoter methylation from MSigDB v5. 24
Validation of differentially methylated CpGs with targeted bisulfite sequencing
Profile plots of methylation signal across DMRs were generated using a custom R script. The BiSearch tool was used to design bisulfite PCR primers to amplify DMRs of interest. 25 Primer sequences used are as follows: MIR3675_F:GATTGAAAAAAGTTTAATAAG, MIR3675_R:TCCTAAAAAAACRAAAAAAACC, HDAC9_F:AAATGATTTAAGAGTTAATGAG, and HDAC9_R:AATCCCACCATTAAAATCAAA. Bisulfite conversion of 100 ng DNA was performed using EpiTect Bisulfite Kit (Qiagen). HotStar Tag (Qiagen) was used to amplify regions from bisulfite converted DNA using with 38 cycles of PCR with the recommended thermal cycling. Amplicons were quantified using MultiNA bioanalyzer (Shimadzu Scientific Instruments, Kyoto, Kyoto Japan). Amplicons from the same patient were pooled to equimolar amounts. NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) was used to generate MiSeq compatible barcoded sequencing libraries. No size selection was performed, and library was amplified with 6 cycles of PCR. Barcoded libraries were combined in equimolar amounts to make a single pooled library. Sequencing was conducted on Illumina MiSeq with the MiSeq Reagent Kit v3 (600 cycles). PEAR was used to assemble forward and reverse reads into a single sequence, 26 which was subsequently mapped to the regions of interest using Bismark. 27 After methylation calling with Bismark methylation extractor, differential methylation analysis was performed using methylKit that uses logistic regression to determine significance using replicate information. 28 Patient age was not considered in the determination of P‐values for differential methylation. Boxplots were generated using the ggplot2 package in the R programming language.
Results
Methyl‐binding domain capture sequencing
Methyl‐binding domain capture was performed on isolated sheared genomic DNA, and these were sequenced to produce an average of 73 million reads per sample [standard deviation (SD) = 8.7 million] (Supporting Information, Table S1 ). Over 99% of these reads mapped to the human genome, and 76.1% (SD = 2.2%) were mapped uniquely (mapQ ≥ 10). As expected, the CpG dinucleotide content in MBD‐seq datasets was elevated (6.54%, SD = 0.65%) when compared with background samples (2.45%). This result indicates that the MBD capture enriched for methylated DNA.
Methylation analysis
Peak calling identified 548k (SD = 55k) enriched regions per sample, occupying 562 Mbp (SD = 65 Mbp) of the genome. Individual peak sets were merged into a list of 643k peaks, which occupied 890 Mbp and had a median size of 973 bp. The CpG dinucleotide content was 3.0% compared with the genome‐wide level of 1.9%. The peak set median GC content was 44.1% compared with the genome‐wide level of 39.3%. We next counted uniquely mapped reads aligned to these regions and omitted peaks below the detection threshold (10 reads per sample on average). This resulted in a matrix of 567 188 detected methylation peaks, accounting for on average 64.3% of MBD‐seq reads (SD = 2.2%). In contrast, only 23.3% of input reads were assigned to peak regions. This result demonstrates that the MBD‐seq identified methylated regions in patients with ischaemic cardiomyopathy with severe multi‐vessel coronary disease requiring coronary artery bypass graft surgery.
Differentially methylated positions in ischaemic cardiomyopathy with heart failure
We implemented a general linear model statistical approach to identify DMRs in HF while correcting for variation in age. We identified 68 DMRs (false discovery rate ≤0.05; Figure 1 A ) when compared with 63 DMRs without adjustment for age. As shown in Figure 1 B , 48 DMRs occurred within gene bodies and 25 were located near enhancer elements (and full list in Supporting Information, Table S2). Of the DMRs that intersect gene bodies with reduced methylation in HF, 19 were protein‐coding genes (ABCB1, ARID5B, CAV2, DIS3, DOPEY2, GREM1, HDAC9, HS6ST3, IQCA1, JARID2, MYO1H, NFIA, OPA1, PACRG, RPS6KA5, RUNDC3B, SOS2, SOX6, and TC2N) and four non‐coding RNA genes. From DMRs with increased methylation in HF, 19 protein‐coding genes were identified (ACSS2, DAAM2, GPA33/DUSP27, EDEM1, FAM212B, FRMD4A, KCNK10, MICALCL, MYO5A, PARP4, PDSS2, PLA2G4E, RALGAPA1, RGS9, SCAMP1, and TRPM6) and 12 non‐coding RNA genes were identified. To identify broad trends of altered methylation sites across different genomic compartments, we intersected these compartments with DMRs passing a looser significance threshold (nominal P ≤ 0.01). When compared with the genome‐wide background, gene bodies, protein‐coding regions, CpG shores, and regions immediately flanking transcriptional start sites were overrepresented, while DMRs were less frequently identified in CpG islands and intergenic regions (Supporting Information, Figure S1 A). We also observed differential methylation of Alu repeat elements, with Alu Y elements strongly overrepresented in DMRs with elevated methylation in HF, while Alu S elements were overrepresented in DMRs with lower methylation in HF (Supporting Information, Figure S1 B).
Figure 1.
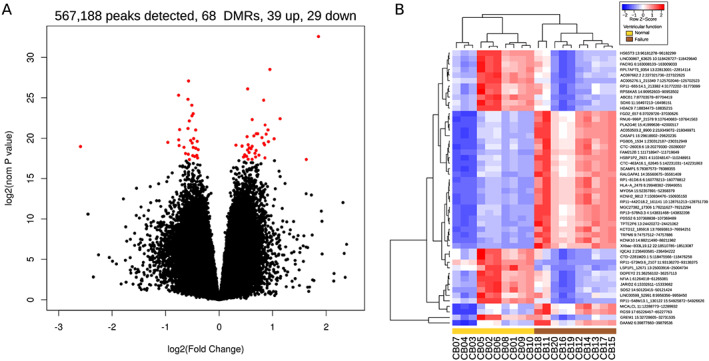
Differential methylation among coronary artery disease patients with and without heart failure. (A) Methyl‐binding domain‐capture sequencing identified 567, 188 peaks, of which 68 were differentially methylated (false discovery rate <0.05) between normal and heart failure groups after correction for age, as shown in the volcano plot in red points. (B) Heatmap of the top 50 differentially methylated regions (DMRs) selected by low P‐value.
Gene set enrichment analysis
To further understand the potential functional implications of differential methylation, we performed gene set enrichment analysis (GSEA) for methylation differences at or near known gene promoters in particular known Reactome pathways. 29 We revealed 103 significantly (false discovery rate <0.05) enriched gene sets in HF with promoter down‐methylation occurring more than up‐methylation (87 vs. 16). Table 2 demonstrates top‐ranked selected gene sets with high positive and negative enrichment scores (Table 2 ).
Table 2.
Top 10 selected significantly enriched gene sets in coronary artery disease patients with heart failure
| ES | Reactome ID (R‐HSA) | Reactome description | n | P‐value | FDR q‐value | Key gene pathway area and function |
|---|---|---|---|---|---|---|
| −0.39 | 72187 | mRNA processing | 50 | <0.0001 | <0.0001 | Metabolism of RNA: mRNA splicing and transport |
| −0.35 | 159236 | mRNA transport | 68 | <0.0001 | <0.0001 | Metabolism of RNA: mature RNA transport via nuclear pore complex |
| −0.31 | 3214841 | Methylation of histones | 42 | <0.0001 | 0.0005 | Chromatin organization: methylation of lysine 5 of histone H3 |
| −0.59 | 390666 | Serotonin receptors | 10 | 0.002 | 0.01 | Signal transduction: 5‐HT1 and 5‐HT5A receptors that bind serotonin |
| −0.38 | 264876 | Insulin processing | 24 | <0.0001 | 0.01 | Metabolism of proteins: translocation of insulin secretory granules |
| 0.30 | 381070 | Activates chaperones | 50 | <0.0001 | 0.008 | Metabolism of proteins: unfolded protein response in the endoplasmic reticulum |
| 0.42 | 449836 | Other interleukins | 23 | <0.0001 | 0.011 | Immune system: interleukin signalling: IL‐34, IL‐32, and CD4 |
| 0.33 | 2029481 | FC gamma receptor activation | 38 | <0.0001 | 0.015 | Innate immunity: FCGR‐dependent phagocytosis: immunoglobulin light chains |
| 0.39 | 5576893 | Plateau phase | 25 | <0.0001 | 0.018 | Cardiac conduction: calcium transport |
| 0.33 | 445355 | Smooth muscle contraction | 33 | 0.02 | 0.025 | Muscle contraction: tropomysin 2 and myosin light chains |
ES, enrichment score (negative: reduced promoter methylation; positive: increased promoter methylation); FDR: false discovery rate; n, number of genes in the gene set.
Targeted validation of differential methylation using bisulfite sequencing
Validation of the MBD‐seq data was performed using bisulfite sequencing to determine methylation at two DMRs of the same DNA samples. As the average DMR size was larger than what can be routinely amplified by bisulfite PCR, we visualized methylation signals across DMRs to target groups of CpG nucleotides with the strongest differential methylation. The DMRs selected were located within or nearby to the gene HDAC9 and the uncharacterized miRNA gene MIR3675 (Figure 2 A ). While not all targets could be successfully amplified in all samples, at least five individuals per group were obtained for these two regions of interest. High‐throughput 300 bp sequencing of both termini gave coverage of 112k‐fold on average (SD = 148k). The HDAC9 DMR portion analysed, within intron 19 (chr7:18 834 681–18 834 888), had high methylation in the control group (91.8%), and the HF group had lower methylation at two of the four CpG sites analysed. Sites c34 and c49 were lower in HF by 0.62% and 0.99%, respectively (Figure 2 B ). The MIR3675 DMR portion analysed (chr1:16 547 840–16 548 225) contained 30 CpG sites, 15 of that had intermediate methylation levels (30–70%) and the remainder were highly methylated (>70%). Overall, MIR3675 methylation levels were 3.7% higher in HF patients, but there were 15 CpG sites where HF patients had methylation 5% higher than the control group (Figure 2 B ). These results confirm two regions of altered methylation in HF and validate the use of MBD‐seq for the detection of altered methylation in the perioperative samples.
Figure 2.
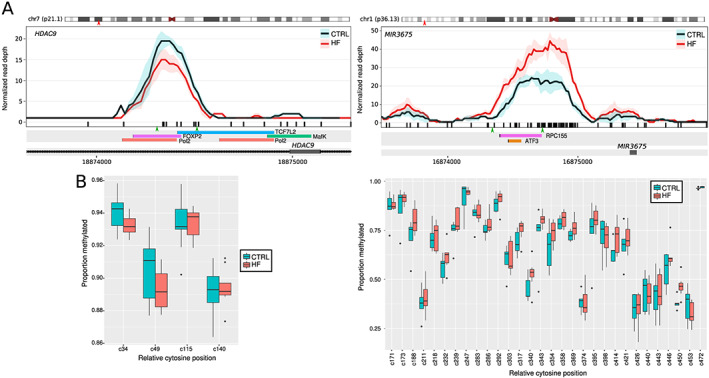
Targeted bisulfite sequencing‐based validation of two heart failure (HF)‐associated differentially methylated regions (DMRs). (A) Methylation landscape of methyl‐binding domain‐capture sequencing signal across two selected DMRs, HDAC9 and MIR3675. Red arrowheads indicate approximate location on the chromosome ideogram. Black bars indicate CpG sites. Green arrows indicate locations of primers for bisulfite sequencing. Below, tracks of transcription factor binding sites from the ENCODE project data are indicated. (B) Bisulfite sequencing of HDAC9 and MIR3675 that reveal single‐nucleotide‐resolution cytosine methylation at these DMRs depicted in boxplots confirms genome‐wide findings in the same samples.
Discussion
This is the first study utilizing the perioperative environment to create a genomics biorepository to investigate the relationship between peripheral blood DNA methylation and a phenotypic extreme identified within a clinical trial. We demonstrate the utility of MBD‐seq to identify differential methylation at multiple sites throughout the genome in ischaemic cardiomyopathy and HF and to further reveal where altered methylation occurs within different genomic contexts and biological pathways.
Cardiac remodelling and HDAC9
The most well‐described protein‐coding gene identified and validated in this study with links to cardiomyopathy and hypertrophic remodelling is the class IIa histone deacetylase enzyme, HDAC9 (HDAC9; Ch7). HDAC9 is well known in cardiovascular medicine for its action to repress myocyte enhancer factor‐2, a transcription factor that reprograms cardiac gene expression causing cardiac hypertrophy. 30 HDAC9 acts to suppress cardiac hypertrophy, 31 , 32 and our data highlights the methylation status of HDAC9 as a potential marker of a propensity for pathological cardiac remodelling in severe CAD. The data also provide further evidence in support of the therapeutic potential of HDAC inhibitors to treat and potentially prevent the progression to HF. 33 While still at a preclinical stage, HDAC inhibition paradoxically has been shown to be antihypertrophic, 34 the net effect of modified histone acetylation of genes 35 that results in antifibrotic, anti‐inflammatory, and anti‐apoptotic actions on an array of cell types in addition to cardiomyocytes.
Inflammation and HDAC9
Numerous clinical studies have demonstrated an association between reduced vascular events, lipid‐lowering therapies (statins/ezetimibe), and improved outcomes when the level of systemic inflammation is reduced. 39 This is further supported by the observation that lowering systemic inflammation with inhibition of the pro‐inflammatory cytokine, interleukin 1β using canakinumab, in patients with stable CAD reduced cardiovascular mortality by 31%. 40 Recent data for HF in mice implicate a change in the role of myocardial regulatory T lymphocytes (Tregs), becoming pro‐inflammatory, exacerbating inflammation and adverse remodelling. 41 This suggests that Tregs may underlie immune activation in HF and raises another potential effect of HDAC9 activity in this process. By means of its interaction with the transcriptional regulator, forkhead family protein FOXP3 in human Tregs, 42 , 43 , 44 HDAC9 activity and inhibition thereof is a target with widespread immunomodulatory effects that may be of benefit in HF. 45 This hypothesis is supported by the observation that in patients with HF, FOXP3 expression decreases and HDAC9 expression increases 46 as B‐type natriuretic peptide levels rise. The reduced methylation of HDAC9 observed in this study may be an epigenetic marker of increased inflammation mediated by altered Treg activity in HF.
Coenzyme Q10 synthesis and PDSS2
Another highlighted cellular process of interest is the coenzyme Q10 (CoQ10) biosynthetic pathway. This pathway is also well known in HF therapy with dietary supplementation being shown to improve cardiac tolerance to hypoxia, reduce HF symptoms, and reduce major adverse cardiovascular events. 36 , 37 In this study, we identified an increased methylation of the decaprenyl diphosphate synthase subunit 2 (PDSS2; Ch 6) gene. This gene is critical to CoQ10 activity and myocardial mitochondrial function, 38 and the methylation status may prove to be a marker of the capacity of the myocardium to recover from hypoxic stress and potentially indicate therapeutic efficacy of CoQ10 supplementation.
Additional candidate novel biomarkers of heart failure
JARID2 (Ch6) demonstrates reduced methylation. Jumonji (Jumonji and AT‐rich interaction domain containing 2) protein is a transcriptional repressor required for normal murine cardiac development 47 and, with reduced methylation of jarid2, has been associated with greater progression of isoproterenol induced HF. 48 The impact of reduced methylation of JARID2 is unclear; however, in humans, myocardial JARID2 protein is decreased by 60% in HF and altered methylation may actually be an early marker of ventricular decompensation. 49
GREM1 (Ch15) demonstrates reduced methylation. Gremlin‐1 is an antagonist of bone morphogenic proteins, and its expression has been significantly correlated with myocardial fibrosis and impaired LV ejection fraction in nonischaemic HF. 50 Furthermore, Gremlin‐1 levels have been shown to be elevated in patients with CAD. Its action as an inhibitor of macrophage migration inhibitory factor, leading to increased risk of atherosclerotic plaque instability has led to the proposal that gremlin‐1/macrophage migration inhibitory factor ratio may act as a marker of the severity and instability of CAD.
Gene set enrichment analysis
The enrichment analysis revealed highly ranked genes with decreased promoter methylation involved with mRNA processing (THOC1: Rank 30384), mRNA transport (TPR: Rank 30180), and the nuclear pore complex (NUP54: Rank 30085). Others included diverse areas such as histone methylation (KMT2A: Rank 30305), serotonin receptors (HTR1D: Rank 28393), and myosin VA (MYO5A: Rank 29307), an actin‐based motor protein involved in vesicular transport, insulin processing, and cardiomyocyte potassium channel transport. 51 In contrast, enriched gene sets with increased methylation contained highly ranked genes involved in protein metabolism (TPP1: Rank 421) and components of the nuclear lamina previously associated with DCM (LMNA: Rank 1368). 52 One gene (HYOU1: Rank 1943) is thought to have a cytoprotective role in hypoxia and has been highlighted in DCM. 53 Within the immune system, interleukin signalling (IL32: Rank 284; IL34: Rank 668) and immunoglobulin light chains (IGLV1‐47: Rank 152) were highly ranked. IL34 expression has recently been associated with post‐operative atrial fibrillation. 54 Some genes have been directly related to cardiac conduction (CACNB1: Rank 392, voltage‐gated L‐type calcium channels), contractility (TPM2: Rank 191, tropomyosin 2 slow‐type muscle fibre 55 ; MYL7: Rank 400, myosin light chain 7), and cardiomyopathy. 56
Overlap with previous studies of DNA methylation and heart failure
There have been previous microarray studies of HF including one by Hass and colleagues 11 using the Illumina 27k Infinium array of LV tissues comprising eight controls and nine patients with DCM, which identified three differentially methylated genes LY75, ERBB3, and ADORA2A. In a larger study of LV (n = 19) and right ventricular (n = 9) tissues in DCM using the 450k Infinium array, 1555 differentially methylated genes were identified, 57 including ARID5B and NFIA, which were also identified here. Although these regions do not overlap, the ARID5B DMR identified here is located nearby three differentially methylated probes (ARID5B +1.2 kb; NFIA +79 and +182 kb). Another microarray study using MeDIP‐chip profiling of a small number of LV tissues identified three DMRs, 10 although these were not significant in the present study. The 2017 study by Meder et al. 13 described mRNA (RNA‐seq) and DNA methylation (Infinium 450k array) in both LV tissue and peripheral blood in DCM, highlighting several probes with robust differences that were replicated in independent cohorts. The Infinium microarray platform measures methylation at 450k and more recently 850k CpG sites, which represents 1.6% and 3.0% of the 28.3 million CpG sites in the genome. 58 This means that 98.4% of genomic CpG sites are yet to be probed in HF. Even with the most comprehensive array available, 97% of CpG sites remain unassessed.
In contrast to previous studies, we selected MBD‐seq over microarrays or reduced representation bisulfite sequencing as this approach has more comprehensive genome coverage. With sufficient read depth, MBD‐seq is able to profile virtually all CpG sites within mappable parts of the genome, containing ~24.3 M CpG sites (86%) albeit without the single base resolution of bisulfite sequencing. To demonstrate, only 13.4% of detected peaks (76 834/570 961) and only 10.1% of DMRs (7/69) overlap a probe with the 450k Infinium array. Similarly, only 5.3% of detected peaks (30 562/570 961) and only 1.4% of DMRs (1/69) overlap a CpG site with ≥10‐fold coverage by ENCODE RRBS (HAIB Methyl RRBS GM12878 Rep1). While MBD‐seq lacks the ability of whole‐genome bisulfite sequencing to call absolute levels of methylation at single‐nucleotide resolution, at the level of genes and DMRs, concordance between the methods is above 90%. 59 Moreover, as MBD‐seq requires fewer reads, it is considerably cheaper than whole‐genome bisulfite sequencing, making it applicable for larger studies. Targeted bisulfite sequencing of a small number of DMRs in the same specimens confirmed the ability of MBD‐seq to identify DMRs, as well as providing absolute methylation quantification of these loci at single‐nucleotide resolution.
Limitations of the current study
This is a pilot study and is not powered or specifically designed to discover HF biomarkers. The study size, male sex, and lack of validation in all patients at all genomic sites preclude conclusions of that nature. However, these preliminary observations demonstrate the utility of this method in perioperative genomic research and provide justification for further, candidate‐based, approaches in a larger cohort of patients, with ongoing co‐measurement of established biomarkers. The complexity of HF, coronary atheroma, and diabetic phenotypes combined with variation in medication administration further limits the generalizability of these findings; however, these data, when combined with other validation cohorts, will contribute to a greater understanding of the relationship between ischaemic cardiomyopathy and peripheral markers of systolic dysfunction.
Conclusions
We have demonstrated the utility of MBD‐seq combined with subsequent targeted bisulphite sequencing to evaluate peripheral blood DNA methylation markers in a cohort of cardiac surgical patients with severe multi‐vessel CAD and phenotypic extremes of HF. The differential methylation status of multiple specific coding genes identified, such as HDAC9, is a candidates for larger longitudinal studies. We have further demonstrated the value and feasibility of examining DNA methylation as part of perioperative medical research to highlight cellular pathways and processes contributing to complex phenotypes within a clinical trial.
Ethics approval and consent to participate
All studies were approved by The Alfred Hospital Ethics Committee (HREC Number: 11/05), and all participants provided informed written consent.
Consent for publication
Not applicable.
Availability of data and material
The dataset supporting the conclusion of this article is available in the NCBI GEO repository (https://www.ncbi.nlm.nih.gov/geo/) under Accession Number GSE134766.
Conflict of interest
None declared.
Author contributions
C.R.B. was responsible for the concept, design, and assembly of the patient cohort. C.R.B., P.S.M., S.W., and K.B. were responsible for the biobank creation. A.W.K., R.T., and H.T. were responsible for the tissue processing and DNA preparation. A.K., I.K., H.K., A.E.‐O., and M.Z. were responsible for next‐generation sequencing and validation analyses. M.Z. was responsible for bioinformatics and data repository creation. C.R.B., M.Z., and K.B. drafted the manuscript. All authors contributed to the critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript.
Funding
This research was funded by a services support grant through the Baker IDI Heart and Diabetes Institute. C.R.B. is supported by the Australian and New Zealand College of Anaesthetists (ANZCA). K.B. is supported by an E. H. Flack Fellowship. Additional infrastructure funding to the Baker Heart and Diabetes Institute and Murdoch Children's Research Institute was provided by the Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme and the Victorian Government's Operational Infrastructure Support Program. A.E.‐O. is a National Health and Medical Research Council (NHMRC) Senior Research Fellow (1154650 and 0526681). The funding bodies had no role in the design, analysis, interpretation, or reporting of results.
Supporting information
Figure S1. Differential methylation enriched in genomic compartments. Regions with differential methylation (nom p‐val<0.01) were intersected with various annotated functional compartments (A), major repeat element classes (B), and Alu subclasses (C).
Table S1. Pre‐operative history: Control (GRP 1 CB no 1‐10) Case (GRP 2 CB no 11‐20)
Table S2. Pre‐operative cardiac data: Controls (GRP 1) and Case (GRP 2)
Table S3. Cardiac medications: Control (GRP 1) and Case (GRP 2)
Table S4. Blood tests control: (GRP 1) and Cases (GRP 2)
Table S5. MBD‐seq data metrics
Table S6. Heart failure DMRs after adjusting for age. DMRs (all FDR≤0.05) are sorted by nominal p‐value. Gene annotations were generated using GREAT. Enhancer annotations were obtained from GeneCards.
Acknowledgements
We would like to express our gratitude to our study participants for their time. The authors acknowledge the use of the Australian Genome Research Facility and the support it receives from the Commonwealth of Australia.
Bain, C. R. , Ziemann, M. , Kaspi, A. , Khan, A. W. , Taylor, R. , Trahair, H. , Khurana, I. , Kaipananickal, H. , Wallace, S. , El‐Osta, A. , Myles, P. S. , and Bozaoglu, K. (2020) DNA methylation patterns from peripheral blood separate coronary artery disease patients with and without heart failure. ESC Heart Failure, 7: 2468–2478. 10.1002/ehf2.12810.
References
- 1. Roth GA, Johnson C, Abajobir A, Abd‐Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, Alla F, Alvis‐Guzman N, Amrock S, Ansari H, Ärnlöv J, Asayesh H, Atey TM, Avila‐Burgos L, Awasthi A, Banerjee A, Barac A, Bärnighausen T, Barregard L, Bedi N, Ketema EB, Bennett D, Berhe G, Bhutta Z, Bitew S, Carapetis J, Carrero JJ, Malta DC, Castañeda‐Orjuela CA, Castillo‐Rivas J, Catalá‐López F, Choi J‐Y, Christensen H, Cirillo M, Cooper L, Criqui M, Cundiff D, Damasceno A, Dandona L, Dandona R, Davletov K, Dharmaratne S, Dorairaj P, Dubey M, Ehrenkranz R, Zaki MES, Farao EJA, Esteghamati A, Farid T, Farvid M, Feigin V, Ding EL, Fowkes G, Gebrehiwot T, Gillum R, Gold A, Gona P, Gupta R, Habtewold TD, Hafezi‐Nejad N, Hailu T, Hailu GB, Hankey G, Hassen HY, Abate KH, Havmoeller R, Hay SI, Horino M, Hotez PJ, Jacobsen K, James S, Javanbakht M, Jeemon P, John D, Jonas J, Kalkonde Y, Karimkhani C, Kasaeian A, Khader Y, Khan A, Khang Y‐H, Khera S, Khoja AT, Khubchandani J, Kim D, Kolte D, Kosen S, Krohn KJ, Kumar GA, Kwan GF, Lal DK, Larsson A, Linn S, Lopez A, Lotufo PA, Razek HMAE, Malekzadeh R, Mazidi M, Meier T, Meles KG, Mensah G, Meretoja A, Mezgebe H, Miller T, Mirrakhimov E, Mohammed S, Moran AE, Musa KI, Narula J, Neal B, Ngalesoni F, Nguyen G, Obermeyer CM, Owolabi M, Patton G, Pedro J, Qato D, Qorbani M, Rahimi K, Rai RK, Rawaf S, Ribeiro A, Safiri S, Salomon JA, Santos I, Milicevic MS, Sartorius B, Schutte A, Sepanlou S, Shaikh MA, Shin M‐J, Shishehbo M, Shore H, Silva DAS, Sobngwi E, Stranges S, Swaminathan S, Tabarés‐Seisdedos R, Atnafu NT, Tesfay F, Thakur JS, Thrift A, Topor‐Madry R, Truelsen T, Tyrovolas S, Ukwaja KN, Uthman O, Vasankari T, Vlassov V, Vollset SE, Wakayo T, Watkins D, Weintraub R, Werdecker A, Westerman R, Wiysonge CS, Wolfe C, Workicho A, Xu G, Yano Y, Yip P, Yonemoto N, Younis M, Yu C, Vos T, Naghavi M, Murray C. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol 2017; 70: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yancy, C. W. , Jessup, M. , Bozkurt, B. , Butler, J. , Casey, D. E. , Colvin, M. M. , Drazner, M. H. , Filippatos, G. S. , Fonarow, G. C. , Givertz, M. M. , Hollenberg, S. M. , Lindenfeld, J. , Masoudi, F. A. , McBride, P. E. , Peterson, P. N. , Stevenson, L. W. & Westlake, C. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017; 136: e137‐e161. [DOI] [PubMed] [Google Scholar]
- 3. Nadar MMSSK. Biomarkers in routine heart failure clinical care. Card Fail Rev 2019; 5: 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larose E, Rodés‐Cabau J, Pibarot P, Rinfret S, Proulx G, Nguyen CM, Déry J‐P, Gleeton O, Roy L, Noël B, Barbeau G, Rouleau J, Boudreault J‐R, Amyot M, Larochellière RD, Bertrand OF. Predicting late myocardial recovery and outcomes in the early hours of ST‐segment elevation myocardial infarction traditional measures compared with microvascular obstruction, salvaged myocardium, and necrosis characteristics by cardiovascular magnetic resonance. J Am Coll Cardiol 2010; 55: 2459–2469. [DOI] [PubMed] [Google Scholar]
- 5. Frantz S, Falcao‐Pires I, Balligand J‐L, Bauersachs J, Brutsaert D, Ciccarelli M, Dawson D, de Windt LJ, Giacca M, Hamdani N, Hilfiker‐Kleiner D, Hirsch E, Leite‐Moreira A, Mayr M, Thum T, Tocchetti CG, van der Velden J, Varricchi G, Heymans S. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail 2018; 20(3): 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 2016; 119: 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar‐Garea A, Ballestar E, Esteller M, Zaina S. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 2004; 279: 29147–29154. [DOI] [PubMed] [Google Scholar]
- 8. Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, Sparrow D, Vokonas P, Schwartz J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010; 21: 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aslibekyan S, Agha G, Colicino E, Do AN, Lahti J, Ligthart S, Marioni RE, Marzi C, Mendelson MM, Tanaka T, Wielscher M, Absher DM, Ferrucci L, Franco OH, Gieger C, Grallert H, Hernandez D, Huan T, Iurato S, Joehanes R, Just AC, Kunze S, Lin H, Liu C, Meigs JB, van Meurs JBJ, Moore AZ, Peters A, Prokisch H, Räikkönen K, Rathmann W, Roden M, Schramm K, Schwartz JD, Starr JM, Uitterlinden AG, Vokonas P, Waldenberger M, Yao C, Zhi D, Baccarelli AA, Bandinelli S, Deary IJ, Dehghan A, Eriksson J, Herders C, Jarvelin M‐R, Levy D, Arnett DK. Association of methylation signals with incident coronary heart disease in an epigenome‐wide assessment of circulating tumor necrosis factor α. JAMA Cardiol 2018; 3: 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Movassagh M, Choy M‐K, Goddard M, Bennett MR, Down TA, Foo RS‐Y. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. Plos One 2010; 5: e8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, Marquart S, Sedaghat‐Hamedani F, Kayvanpour E, Köhler D, Wolf NM, Hassel S, Nietsch R, Wieland T, Ehlermann P, Schultz J‐H, Dösch A, Mereles D, Hardt S, Backs J, Hoheisel JD, Plass C, Katus HA, Meder B. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med 2013; 5: 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe T, Okada H, Kanamori H, Miyazaki N, Tsujimoto A, Takada C, Suzuki K, Naruse G, Yoshida A, Nawa T, Tanaka T, Kawasaki M, Ito H, Ogura S, Okura H, Fujiwara T, Fujiwara H, Takemura G. In situ nuclear DNA methylation in dilated cardiomyopathy: an endomyocardial biopsy study. Esc Hear Fail 2020; 7: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meder B, Haas J, Sedaghat‐Hamedani F, Kayvanpour E, Frese K, Lai A, Nietsch R, Scheiner C, Mester S, Bordalo DM, Amr A, Dietrich C, Pils D, Siede D, Hund H, Bauer A, Holzer DB, Ruhparwar A, Mueller‐Hennessen M, Weichenhan D, Plass C, Weis T, Backs J, Wuerstle M, Keller A, Katus HA, Posch AE. Epigenome‐wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation 2017; 136: 1528–1544. [DOI] [PubMed] [Google Scholar]
- 14. Myles PS, Smith JA, Forbes A, Silbert B, Jayarajah M, Painter T, Cooper DJ, Marasco S, McNeil J, Bussières JS, Wallace S, Network, A. I. of the A. C. T . Stopping vs. continuing aspirin before coronary artery surgery. New Engl J Medicine 2016; 374: 728–737. [DOI] [PubMed] [Google Scholar]
- 15. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho‐Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Keenan S, Lavidas I, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Nuhn M, Parker A, Patricio M, Pignatelli M, Rahtz M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Birney E, Harrow J, Muffato M, Perry E, Ruffier M, Spudich G, Trevanion SJ, Cunningham F, Aken BL, Zerbino DR, Flicek P. Ensembl 2016. Nucleic Acids Res 2016; 44: D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. (2013).
- 17. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 2008; 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 19. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee B‐K, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature 2012; 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 2010; 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA‐P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007; 23: 3251–3253. [DOI] [PubMed] [Google Scholar]
- 24. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tusnády, G. E. , Simon, I. , Váradi, A. & Arányi, T. BiSearch: primer‐design and search tool for PCR on bisulfite‐treated genomes. Nucleic Acids Res 2005; 33: e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics 2014; 30: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 2011; 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Akalin A, Kormaksson M, Li S, Garrett‐Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: a comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biol 2012; 13: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D'Eustachio P. The Reactome pathway knowledgebase. Nucleic Acids Res 2018; 46: D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium‐dependent regulator of cell division, differentiation and death. Trends Biochem Sci 2002; 27: 40–47. [DOI] [PubMed] [Google Scholar]
- 31. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal‐responsive repressors of cardiac hypertrophy. Cell 2002; 110: 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 2004; 24: 8467–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Pharmacol Toxicol 2012; 52: 303–319. [DOI] [PubMed] [Google Scholar]
- 34. Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang C‐L, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose‐dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 2003; 278: 28930–28937. [DOI] [PubMed] [Google Scholar]
- 35. Ooi JYY, Tuano NK, Rafehi H, Gao X‐M, Ziemann M, Du X‐J, El‐Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 2015; 10: 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenfeldt F, Marasco S, Lyon W, Wowk M, Sheeran F, Bailey M, Esmore D, Davis B, Pick A, Rabinov M, Smith J, Nagley P, Pepe S. Coenzyme Q10 therapy before cardiac surgery improves mitochondrial function and in vitro contractility of myocardial tissue. J Thorac Cardiovasc Surg 2005; 129: 25–32. [DOI] [PubMed] [Google Scholar]
- 37. Mortensen, S. A. , Rosenfeldt, F. , Heart, A. K. J. & 2014. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q‐SYMBIO: a randomized double‐blind trial. Jacc Hear Fail 2014; 2: 641‐649. [DOI] [PubMed] [Google Scholar]
- 38. Quinzii, C. M. , Hirano, M. & DiMauro, S. CoQ10 deficiency diseases in adults. Mitochondrion 2007; 7 Suppl: S122–S126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E, Investigators, P. or A. E. and I. T.‐T. in M. I. 22 (PROVE I.‐T. 22) . C‐reactive protein levels and outcomes after statin therapy. New Engl J Medicine 2005; 352: 20–28. [DOI] [PubMed] [Google Scholar]
- 40. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group, C. T . Relationship of C‐reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 2018; 391: 319–328. [DOI] [PubMed] [Google Scholar]
- 41. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory T‐lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 2019; 139: 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, Basu S, Riley JL, Hancock WW, Shen Y, Saouaf SJ, Greene MI. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc National Acad Sci 2007; 104: 4571–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4: 330–336. [DOI] [PubMed] [Google Scholar]
- 44. Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol 2010; 22: 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L, de Zoeten EF, Greene MI, Hancock WW. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nast Rev Drug Discov 2009; 8: 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liao P‐P, Liu L‐H, Wang B, Fang X, Zhou S‐Q, Li W, Li W, Zhang Y‐Q, Guan S‐M. Correlation between histone deacetylase 9 and regulatory T cell in patients with chronic heart failure. Curr Medical Sci 2018; 38: 199–203. [DOI] [PubMed] [Google Scholar]
- 47. Mysliwiec MR, Carlson CD, Tietjen J, Hung H, Ansari AZ, Lee Y. Jarid2 (Jumonji, AT rich interactive domain 2) regulates NOTCH1 expression via histone modification in the developing heart. J Biol Chem 2012; 287: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen H, Orozco LD, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM. DNA methylation indicates susceptibility to isoproterenol‐induced cardiac pathology and is associated with chromatin states: novelty and significance. Circ Res 2016; 118: 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bovill E, Westaby S, Reji S, Sayeed R, Crisp A, Shaw T. Induction by left ventricular overload and left ventricular failure of the human Jumonji gene (JARID2) encoding a protein that regulates transcription and reexpression of a protective fetal program. J Thorac Cardiovasc Surg 2008; 136: 709–716. [DOI] [PubMed] [Google Scholar]
- 50. Mueller KAL, Tavlaki E, Schneider M, Jorbenadze R, Geisler T, Kandolf R, Gawaz M, Mueller II, Zuern CS. Gremlin‐1 identifies fibrosis and predicts adverse outcome in patients with heart failure undergoing endomyocardial biopsy. J Card Fail 2013; 19: 678–684. [DOI] [PubMed] [Google Scholar]
- 51. Schumacher‐Bass SM, Vesely ED, Zhang L, Ryland KE, McEwen DP, Chan PJ, Frasier CR, McIntyre JC, Shaw RM, Martens JR. Role for myosin‐V motor proteins in the selective delivery of Kv channel isoforms to the membrane surface of cardiac myocytes. Circ Res 2014; 114: 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, Edvardsen T, Haugaa KH. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J 2017; 39: 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hodatsu A, Fujino N, Uyama Y, Tsukamoto O, Okazaki AI, Yamazaki S, Seguchi O, Konno T, Hayashi K, Kawashiri M a, Asano Y, Kitakaze M, Takashima S, Yamagishi M. Impact of cardiac myosin light chain kinase gene mutation on development of dilated cardiomyopathy. Esc Hear Fail 2019; 6: 406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sigurdsson MI, Saddic L, Heydarpour M, Chang T‐W, Shekar P, Aranki S, Couper GS, Shernan SK, Muehlschlegel JD, Body SC. Post‐operative atrial fibrillation examined using whole‐genome RNA sequencing in human left atrial tissue. BMC Med Genomics 2017; 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Purcell IF, Bing W, Marston SB. Functional analysis of human cardiac troponin by the in vitro motility assay: comparison of adult, foetal and failing hearts. Cardiovasc Res 1999; 43: 884–891. [DOI] [PubMed] [Google Scholar]
- 56. England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci 2012; 70: 1221–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jo B‐S, Koh I‐U, Bae J‐B, Yu H‐Y, Jeon E‐S, Lee H‐Y, Kim J‐J, Choi M, Choi SS. Data of methylome and transcriptome derived from human dilated cardiomyopathy. Data Brief 2016; 9: 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luo, Y. , Lu, X. & Xie, H. Dynamic Alu methylation during normal development, aging, and tumorigenesis. Biomed Res Int 2014; 784706‐784712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, Olshen A, Ballinger T, Zhou X, Forsberg KJ, Gu J, Echipare L, O'Geen H, Lister R, Pelizzola M, Xi Y, Epstein CB, Bernstein BE, Hawkins RD, Ren B, Chung W‐Y, Gu H, Bock C, Gnirke A, Zhang MQ, Haussler D, Ecker JR, Li W, Farnham PJ, Waterland RA, Meissner A, Marra MA, Hirst M, Milosavljevic A, Costello JF. Comparison of sequencing‐based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 2010; 28: 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Differential methylation enriched in genomic compartments. Regions with differential methylation (nom p‐val<0.01) were intersected with various annotated functional compartments (A), major repeat element classes (B), and Alu subclasses (C).
Table S1. Pre‐operative history: Control (GRP 1 CB no 1‐10) Case (GRP 2 CB no 11‐20)
Table S2. Pre‐operative cardiac data: Controls (GRP 1) and Case (GRP 2)
Table S3. Cardiac medications: Control (GRP 1) and Case (GRP 2)
Table S4. Blood tests control: (GRP 1) and Cases (GRP 2)
Table S5. MBD‐seq data metrics
Table S6. Heart failure DMRs after adjusting for age. DMRs (all FDR≤0.05) are sorted by nominal p‐value. Gene annotations were generated using GREAT. Enhancer annotations were obtained from GeneCards.
Data Availability Statement
The dataset supporting the conclusion of this article is available in the NCBI GEO repository (https://www.ncbi.nlm.nih.gov/geo/) under Accession Number GSE134766.