

Abstract
A 74‐year‐old man had abnormal left ventricular (LV) function according to a perioperative test at a local hospital and was transferred to our institution for further evaluation and treatment. His electrocardiogram demonstrated the presence of premature ventricular contraction with a QRS complex of the right bundle branch block type and superior axis. His echocardiography showed systolic dysfunction of the LV (LV ejection fraction, 44.6%). Cardiac computed tomography imaging revealed banded and patchy densities observed frequently from the middle to epicardial layer of the LV wall. Cardiac magnetic resonance imaging showed fat signals on fat‐selective images and late gadolinium enhancement in the mid‐wall to subepicardial layers in the LV myocardium. Endomyocardial biopsy revealed the histological presence of fibrofatty replacement. A genetic analysis revealed a nonsense mutation in the desmoplakin gene. Thus, he was diagnosed with left‐dominant arrhythmogenic cardiomyopathy. To prevent fatal ventricular arrhythmias, an implantable cardioverter defibrillator was successfully implanted.
Keywords: Left‐dominant arrhythmogenic cardiomyopathy, Desmoplakin gene (DSP), Ventricular tachycardia
Introduction
Many cases of sudden cardiac death are considered arrhythmic death, and ventricular tachyarrhythmias, including ventricular fibrillation, play an important role in the development of arrhythmic death. Because the benefits of an implantable cardioverter defibrillator (ICD) have been demonstrated for preventing sudden death due to tachycardia have been demonstrated, it is important how to indicate ICDs for the treatment of fatal ventricular tachyarrhythmias. Sustained ventricular tachycardia associated with organic heart disease is often fatal, and the causes include ischaemic heart disease, cardiomyopathy [arrhythmogenic right ventricular cardiomyopathy (ARVC), hypertrophic cardiomyopathy, and dilated cardiomyopathy (DCM)], postcardiac surgery and cardiac sarcoidosis as well as Brugada syndrome, long QT syndrome, and catecholaminergic polymorphic ventricular tachycardia. Therefore, it is important to stratify risks based on clinical and laboratory findings associated with each condition and disease and to make a differential diagnosis to prevent sudden death.
Description
Case report
A 74‐year‐old man had abnormal left ventricular (LV) function according to a perioperative test for a patellar fracture at a local hospital and was referred to our university hospital for further evaluation and treatment. He had hypertension and dyslipidaemia. His medications included losartan potassium plus hydrochlorothiazide (Preminent® Tablet LD) daily, 2.5 mg bisoprolol fumarate daily, and 80 mg fenofibrate (Lipidil® Tablet) daily. He had no symptoms suggestive heart failure such as dyspnoea on exertion. An electrocardiogram showed a normal sinus rhythm with a heart rate of 68, low QRS amplitude in the limb leads, and flat or inverted T waves in the inferolateral leads. There was a premature ventricular contraction with a QRS complex of the right bundle branch block type and superior axis (Figure 1 ). In addition, the prolonged intrisicond deflection (widening of the initial part of the QRS complex) in V1 suggested that the epicardial basal left ventricle may be the origin of premature ventricular contraction. 1 Echocardiography showed global and regional systolic dysfunction in the LV (hypokinesis in the anterior, anteroseptal and inferoseptal walls as well as the apex, mild hypokinesis in inferior, inferolateral and anterolateral walls, and scar in mid anteroseptal and mid inferoseptal walls; LV ejection fraction, 44.6%) and preserved right ventricular volume and contraction (Supporting Information, Movie S1 ). No family history of cardiovascular disease or sudden cardiac death was recorded.
FIGURE 1.
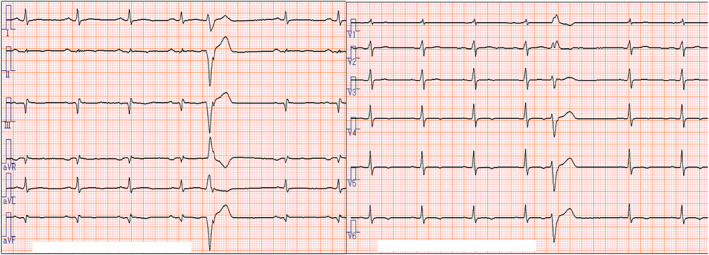
Electrocardiogram on admission.
Cardiac computed tomography (CT) imaging revealed banded and patchy fat densities observed frequently from the middle to epicardial layer of the LV wall (Figure 2 C A– 2 and Supporting Information, Movie S2 ). Cardiac magnetic resonance (CMR) imaging showed fat signals on fat‐selective images and late gadolinium enhancement (LGE) in the mid‐wall to subepicardial layers in the LV myocardium (Figure 2 F D– 2 ). No evidence of coronary stenosis was found according to coronary angiography. Endomyocardial biopsy from the right side of the interventricular septum revealed moderate fibrofatty replacement and mild hypertrophy (Figure 3 ).
FIGURE 2.
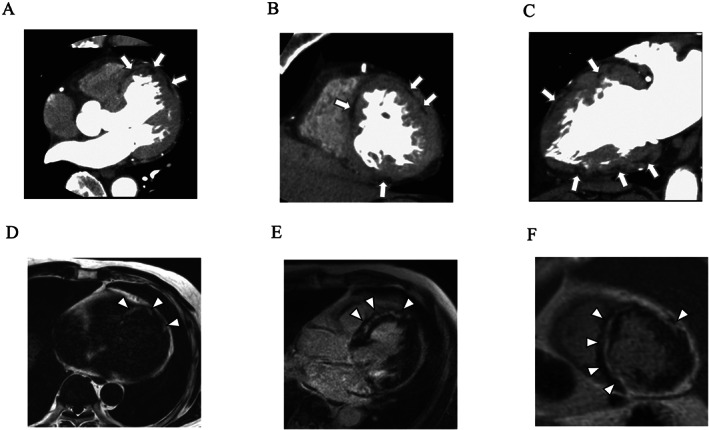
Cardiac computed tomography images: (A) axial image, (B) short‐axis image, and (C) vertical long‐axis image. Arrows indicate fat densities. Cardiac magnetic resonance imaging images: (D) fat‐selective image with mDIXON sequence (axial image), (E) late gadolinium enhancement (LGE) image (horizontal long‐axis image), and (F) LGE image (short‐axis image). Arrowheads indicate fat signals in (D) and LGEs in (E) and (F).
FIGURE 3.
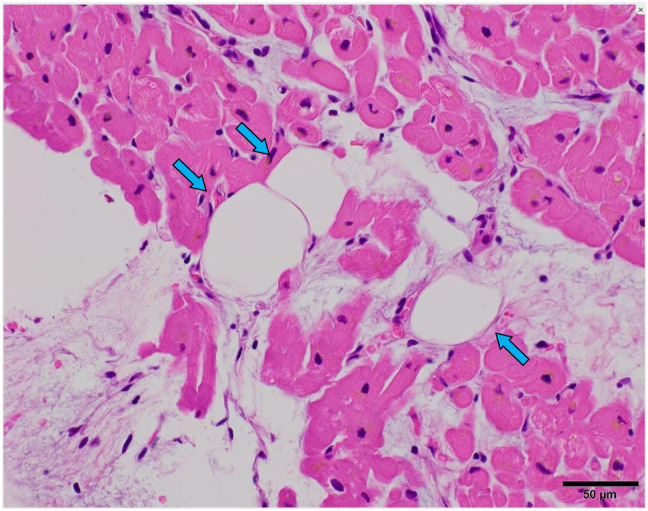
Histological findings from endomyocardial biopsy. Blue arrows indicate adipose tissue.
Left‐dominant arrhythmogenic cardiomyopathy (LDAC) was diagnosed based on the findings of arrhythmia derived from the LV, LV systolic dysfunction, presence of intramyocardial fat on CT and CMR, LGE in the LV, and histological presence of fibrofatty replacement. 2
To search for the genetic background of LDAC, we performed a genetic analysis as described previously. 3 Figure 4 shows the family tree (A) and electropherogram of the sequencing analysis of exon 23 in DSP (B). The genetic analysis revealed a nonsense mutation in DSP (c.5212C > T, p.R1738*). Because there are 24 exons in DSP, nonsense‐mediated decay should be involved in degrading abnormal mRNA and decreasing the expression level of desmoplakin encoded by DSP, which may lead to cardiomyopathy. Two sons of the proband were asymptomatic and carried no mutations.
FIGURE 4.
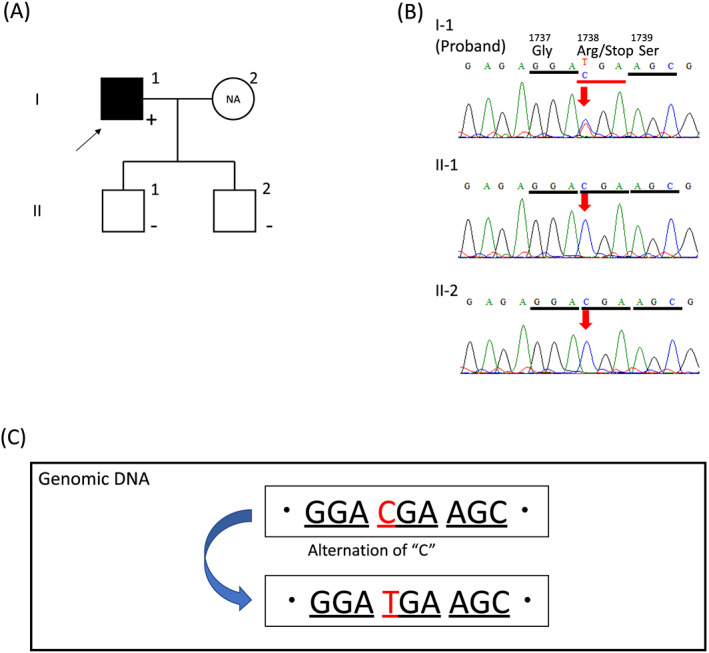
Genetic analyses of the family. (A) Family tree shows an affected proband (filled square) and asymptomatic sons (open squares). Plus sign (+), mutation positive; negative sign (−), mutation negative; NA, not available. (B) Electropherogram of the sequencing analysis. A T to C substitution at nucleotide 5212 caused a nonsense mutation at codon 1738 in the sequence of the proband (I‐1), with no substitution in those of II‐1 and II‐2. Panel (C) indicates the c.5212C > T, p.R1738* mutation. (···GGACGAAGC··) instead of the normal subject data (···GGATGAAGC···).
He was confirmed to have nonsustained ventricular tachycardia (Supporting Information, Figure S1 ). Desmosomal gene mutation is recognized as one of the independent risk factors for adverse events in ARVC patients. 4 The Japanese Circulation Society (JCS) recommends the implantation of an ICD in individuals with ARVC as the primary prevention of sudden cardiac death as a class IIa (as judged from the available findings and opinions, a method of evaluation or treatment is likely to be beneficial and effective) indication in patients with nonsustained ventricular tachycardia and desmosomal gene mutations. 5 Hence, we decided to implant an ICD in this patient.
A regular medical check‐up policy was initiated. The shock results of the ICD have not been confirmed.
Discussion
Sen‐Chowdhry et al. 2 identified the clinical features of LDAC (Table 1 ). We have already reported a case of LDAC with heterozygous mutations in DSP and MYBPC3. 3 DSP is the desmosomal gene associated with arrhythmogenic cardiomyopathy. Differentiation between LDAC and DCM is made according to the clinical characteristics, and the principal way to distinguish LDAC from DCM is a predisposition to ventricular arrhythmia that exceeds the degree of morphological abnormality and impairment of systolic function of the left ventricle. 2 Moreover, the differences in the distinctive imaging phonotypes are comprehensively reviewed, 6 and a genetic search might be very useful for distinguishing between LDAC and DCM. Furthermore, recently, the Heart Rhythm Society (HRS) released an expert consensus statement that included an algorithm for considering the presence of an arrhythmogenic cardiomyopathy. 7 Recently, Augusto et al. reported that subepicardial LGE in CMR with a ring‐like pattern (at least three contiguous segments in the same short‐axis slice) was observed in 78.1% of DSP/FLNC genotypes. 6 The present case is consistent with these algorithms.
TABLE 1.
Clinical features of left‐dominant arrhythmogenic cardiomyopathy
| 1. Unexplained ventricular arrhythmia of right bundle block configuration. |
| 2. Unexplained T‐wave inversion in inferior or lateral leads. |
| 3. Mild left ventricular dilation and/or systolic impairment. |
| 4. Myocyte loss with fibrofatty or fibrotic replacement confirmed by biopsy or late gadolinium enhancement in the left ventricle on cardiac magnetic resonance. |
Arrhythmogenic right ventricular cardiomyopathy is listed as a differential diagnosis, but right ventricular volume and contraction were preserved (Supporting Information, Movie S1), and no findings meeting the recent Task Force Criteria 8 were found. An alternative diagnosis in this particular patient would be triglyceride deposit cardiomyovasculopathy (TGCV). Hirano et al. reported an intractable disease called TGCV that caused severe heart failure, arrhythmia, and coronary artery disease as a result of the accumulation of triglycerides in the myocardium and coronary arteries. 9 TGCV is associated with coronary stenosis 10 and a decrease in the wash‐out rate (wash‐out rate < 10) of iodine‐123‐15‐(p‐iodophenyl)‐3‐(R,S)‐methylpentadecanoic acid (123I‐BMIPP) scintigraphy. 11 Moreover, TGCV has been reported in peripheral blood (smear), polymorphonuclear leukocytes and monocytes with vacuolar degeneration (Jordans' anomaly), 12 , 13 and no findings were confirmed in the present case. In addition, no abnormal fluoroglucose (FDG) accumulation was observed on positron emission tomography (figure not shown), and no findings such as granuloma formation were observed in the pathological tissue (Figure 3 ). Rather, this case is consistent with the clinical features of LDAC. 2 In the present case, the fact that fibrofatty replacement was clearly delineated by both cardiac CT and CMR is a highly novel finding.
Pinamonti et al. suggested that LDAC can be considered one of the three possible patterns of the spectrum of ARVC, in consideration of the histopathologic and genetic similarities. 14 Hence, we have decided to comply with ARVC guidelines regarding the adaptation of ICD implantation in this patient. The JCS Guideline (2018 JCS/JHRS Guideline on Non‐Pharmacotherapy of Cardiac Arrhythmias), 5 which was published in March 2019, was prepared based on the consensus statement issued in 2015 4 and the reports that Japanese patients with high scoring based on ARVC diagnostic criteria. 8 However, the 2019 HRS expert consensus statement, 7 which was published in November 2019, the LDAC has been proposed to recognize arrhythmogenic cardiomyopathy of LV origin as distinct from ARVC. The ICD implantation of this patient was performed in July 2019; the decision to implant the ICD in the case report patient may have been made between 2018 JCS guideline 5 and 2019 HRS expert consensus statement. 7
To our knowledge, this is the first report of a DSP mutation (c.5212C > T, p.R1738*) accompanying by the results of imaging and histological investigations.
Conflict of interest
None declared.
Funding
None.
Supporting information
Figure S1. Monitored electrocardiogram findings obtained during hospitalization
Movie S1. Transthoracic ultrasound cardiogram
Movie S2. Cardiac computed tomography images (short‐axis image)
Acknowledgements
The patient provided consent for the publication of this study. We thank all paramedical staff and clinical secretaries for their kind support during this work.
Tsuruta, Y. , Sueta, D. , Takashio, S. , Oda, S. , Sakamoto, K. , Kaikita, K. , Kato, K. , Ohno, S. , Horie, M. , and Tsujita, K. (2020) Left‐dominant arrhythmogenic cardiomyopathy with a nonsense mutation in DSP . ESC Heart Failure, 7: 3174–3178. 10.1002/ehf2.12790.
References
- 1. Rosenbaum MB. Classification of ventricular extrasystoles according to form. J Electrocardiol 1969; 2: 289–297. [DOI] [PubMed] [Google Scholar]
- 2. Sen‐Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol 2008; 52: 2175–2187. [DOI] [PubMed] [Google Scholar]
- 3. Sakamoto N, Natori S, Hosoguchi S, Minoshima A, Noro T, Akasaka K, Sato N, Ohno S, Ikeda Y, Ishibashi‐Ueda H, Horie M, Hasebe N. Left‐dominant arrhythmogenic cardiomyopathy with heterozygous mutations in DSP and MYBPC3. Circ Cardiovasc Imaging 2019; 12: e008913. [DOI] [PubMed] [Google Scholar]
- 4. Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Pelliccia A, Duru F, Protonotarios N, Estes NM 3rd, McKenna WJ, Thiene G, Marcus FI, Calkins H. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an International Task Force Consensus Statement. Circulation 2015; 132: 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The Japanese Circulation Society [Internet]. [cited 2020 Mar. 20]. Available from: http://www.j-circ.or.jp/guideline/pdf/JCS2018_kurita_nogami191120.pdf (in Japanese).
- 6. Augusto JB, Eiros R, Nakou E, Moura‐Ferreira S, Treibel TA, Captur G, Akhtar MA, Protonotarios A, Gossios TD, Savvatis K, Syrris P, Mohiddin S, Moon JC, Elliott PM, Lopes LR. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype‐imaging phenotype study. Eur Heart J Cardiovasc Imaging 2019. [DOI] [PubMed] [Google Scholar]
- 7. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, Estes NAM 3rd, Hua W, Indik JH, Ingles J, James CA, John RM, Judge DP, Keegan R, Krahn AD, Link MS, Marcus FI, McLeod CJ, Mestroni L, Priori SG, Saffitz JE, Sanatani S, Shimizu W, van Tintelen JP, Wilde AAM, Zareba W. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: executive summary. Heart Rhythm 2019; 16: e373–e407. [DOI] [PubMed] [Google Scholar]
- 8. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31: 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirano K, Ikeda Y, Zaima N, Sakata Y, Matsumiya G. Triglyceride deposit cardiomyovasculopathy. N Engl J Med 2008; 359: 2396–2398. [DOI] [PubMed] [Google Scholar]
- 10. Ikeda Y, Hirano K, Fukushima N, Sawa Y. A novel type of human spontaneous coronary atherosclerosis with triglyceride deposition. Eur Heart J 2014; 35: 875. [DOI] [PubMed] [Google Scholar]
- 11. Hirano K, Ikeda Y, Sugimura K, Sakata Y. Cardiomyocyte steatosis and defective washout of iodine‐123‐beta‐methyl iodophenyl‐pentadecanoic acid in genetic deficiency of adipose triglyceride lipase. Eur Heart J 2015; 36: 580. [DOI] [PubMed] [Google Scholar]
- 12. Inaba T, Nomura N, Takahashi M, Ishizuka K, Yoshika K, Yuasa S, Nakanishi M, Fujita N. Characteristic scattergram of white blood cells obtained using the Pentra MS CRP hematology analyzer in a patient with neutral lipid storage disease. Lab Hematol 2013; 19: 22–24. [DOI] [PubMed] [Google Scholar]
- 13. Suzuki A, Nagasaka H, Ochi Y, Kobayashi K, Nakamura H, Nakatani D, Yamaguchi S, Yamaki S, Wada A, Shirata Y, Hui SP, Toda T, Kuroda H, Chiba H, Hirano KI. Peripheral leukocyte anomaly detected with routine automated hematology analyzer sensitive to adipose triglyceride lipase deficiency manifesting neutral lipid storage disease with myopathy/triglyceride deposit cardiomyovasculopathy. Mol Genet Metab Rep 2014; 1: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pinamonti B, Brun F, Mestroni L, Sinagra G. Arrhythmogenic right ventricular cardiomyopathy: From genetics to diagnostic and therapeutic challenges. World J Cardiol 2014; 6: 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Monitored electrocardiogram findings obtained during hospitalization
Movie S1. Transthoracic ultrasound cardiogram
Movie S2. Cardiac computed tomography images (short‐axis image)