

Abstract
Background
Asiatic Citrus Canker, caused by Xanthomonas citri pv. citri, severely impacts citrus production worldwide and hampers international trade. Considerable regulatory procedures have been implemented to prevent the introduction and establishment of X. citri pv. citri into areas where it is not present. The effectiveness of this surveillance largely relies on the availability of specific and sensitive detection protocols. Although several PCR- or real-time PCR-based methods are available, most of them showed analytical specificity issues. Therefore, we developed new conventional and real-time quantitative PCR assays, which target a region identified by comparative genomic analyses, and compared them to existing protocols.
Results
Our assays target the X. citri pv. citri XAC1051 gene that encodes for a putative transmembrane protein. The real-time PCR assay includes an internal plant control (5.8S rDNA) for validating the assay in the absence of target amplification. A receiver-operating characteristic approach was used in order to determine a reliable cycle cut-off for providing accurate qualitative results. Repeatability, reproducibility and transferability between real-time devices were demonstrated for this duplex qPCR assay (XAC1051-2qPCR). When challenged with an extensive collection of target and non-target strains, both assays displayed a high analytical sensitivity and specificity performance: LOD95% = 754 CFU ml− 1 (15 cells per reaction), 100% inclusivity, 97.2% exclusivity for XAC1051-2qPCR; LOD95% = 5234 CFU ml− 1 (105 cells per reaction), 100% exclusivity and inclusivity for the conventional PCR. Both assays can detect the target from naturally infected citrus fruit. Interestingly, XAC1051-2qPCR detected X. citri pv. citri from herbarium citrus samples. The new PCR-based assays displayed enhanced analytical sensitivity and specificity when compared with previously published PCR and real-time qPCR assays.
Conclusions
We developed new valuable detection assays useful for routine diagnostics and surveillance of X. citri pv. citri in citrus material. Their reliability was evidenced through numerous trials on a wide range of bacterial strains and plant samples. Successful detection of the pathogen was achieved from both artificially and naturally infected plants, as well as from citrus herbarium samples, suggesting that these assays will have positive impact both for future applied and academic research on this bacterium.
Keywords: Asiatic Citrus canker, Surveillance, Real-time quantitative PCR, Diagnostics, Cycle cut-off, Ancient DNA
Background
Over the last half century, there has been a dramatic increase in biological invasions worldwide as a result of globalization and increased international trade and travel [1]. To limit the threat posed by the introduction of exotic plant pathogens and pests through trade and human transport, many countries have tightened border biosecurity surveillance, as well as phytosanitary inspection, and quarantine measures. This biosecurity effort has slowed down the introduction and establishment of pathogens despite the increase in trade and the international movement of people. However, biosecurity measures have adopted to differing degrees across the agricultural sector. Measures to protect annual crop and pasture species have had a positive impact. In contrast, the biosecurity effort should be enhanced for perennial crops such as forest and fruit tree species, which remain vulnerable [2]. When a plant disease outbreak is observed in a new area, quick and appropriate management measures, such as containment or eradication, are necessary to avoid the establishment and further spread of the pathogen. For efficient biosecurity surveillance and plant disease management, identifying regulated pathogens fast and accurately is essential. Misdiagnosis can have a severe economic impact, for example, if unwanted pathogens are introduced or inappropriate management options applied [3–5].
Xanthomonas citri pv. citri, the causal agent of Asiatic citrus canker (ACC), is an example of a high-concern regulated pathogen that threatens an economically important fruit crop (i.e., collectively, citrus fruit rank #1 fruit crop worldwide). This bacterial pathogen causes serious direct and indirect economic losses due to reduced crop yield and quality, the cost of eradication, containment (including the destruction of nursery, grove or backyard trees) or integrated management measures, as well as embargos on the movement of fruit. In addition, it raises concerns about environmental issues linked to increased pesticide use and the development of resistance to antimicrobials [4, 6]. For example, more than one billion US dollars was spent over a decade in Florida in an attempt to eradicate the pathogen [7]. Australia organized several successful ACC eradication campaigns in the past [8, 9], and is currently conducting a response plan in the Northern Territory at a cost of millions of A$.
Although at least four distinct xanthomonads are pathogenic to citrus, only two of them, X. citri pv. citri and X. citri pv. aurantifolii, cause visually indistinguishable canker-like symptoms. They are the causal agents of Asiatic and South American citrus canker, respectively. However, only the former bacterium has a major agricultural significance, because it is the only one associated with serious canker outbreaks even in countries where both pathovars occur concomitantly. Within X. citri pv. citri, strains differ in host range among citrus lines and can be classified into three distinct pathotypes. Pathotype A strains have the greatest global economic impact on the citrus industry. They are widely distributed and induce canker on a broad range of rutaceous hosts, including many Citrus species, hybrids or related genera such as trifoliate orange (Poncirus trifoliata) [4]. Pathotype A* strains are pathogenic to a restricted range of citrus hosts. Most outbreaks occur on Mexican lime (Citrus x aurantiifolia) in Asia, the Arabian Peninsula and Eastern Africa [10–12]. Pathotype Aw has been reported to date on the Indian subcontinent, in the Arabian Peninsula and the USA. Natural infections are restricted to Mexican lime and alemow (C. x macrophylla). It has the unique feature of causing a hypersensitive response when inoculated into some non-host citrus lines such as C. x paradisi and C. x sinensis [13, 14]. Pathotypes represent phylogenetically-coherent lineages based on whole-genome sequencing (WGS) data or genotyping data [15–18]. These techniques have made it possible to identify previously unreported sublineages within pathotypes A and A*, which are responsible for outbreaks both in the pathogen’s area of origin or in regions where it has recently emerged [11, 16, 18].
Long-distance dissemination of X. citri pv. citri occurs primarily when humans transport diseased citrus plant material [19]. Since the early 2000s, there have been reports of several cases of geographical expansion and successful pathogen establishment in some Western and Eastern African countries, and the Caribbean [10, 20, 21]. X. citri pv. citri is a major threat to disease-free areas (e.g. New Zealand, Australia, and countries in Southern Africa and the Mediterranean) where it is listed as a quarantine organism. Preventing the establishment of X. citri pv. citri in new areas very much depends on the availability of specific detection protocols and the implementation of surveillance and quarantine measures. Given the significance of ACC, numerous molecular detection methods have been developed, (i) conventional PCR assays [22–27], (ii) real-time quantitative qPCR assays [28, 29]; and (iii) Lamp assay [30]. Issues of inclusivity (i.e., the ability of the assay to detect all strains of the target organism) and/or exclusivity (i.e., the capacity to generate negative responses from non-target strains) occur with most of the previous assays developed for X. citri pv. citri [31]. Therefore, we evaluated recently published diagnostic tools and developed a new system.
Many microbial genomes that are publicly available constitute a valuable resource for identifying new, specific molecular markers. In this study, we describe the development of highly specific conventional and real-time PCR assays from a DNA marker selected using in silico comparative genomic analysis of X. citri pv. citri genomes and non-target Xanthomonas genomes. The real-time PCR assay amplified an endogenous plant DNA sequence present in the sample (5.8S rDNA) as a co-extracted and co-amplified internal control. It reveals any flaws in DNA extraction and the presence of PCR inhibitors [32–34]. These protocols were further validated using naturally infected citrus material collected in the field. These new molecular tools were independently evaluated and compared to existing protocols by the French Agency for Food, Environmental and Occupational Health & Safety (ANSES).
Results
Selection of a specific DNA fragment for PCR and qPCR assay design
The comparative genomic analysis of 30 X. citri pv. citri genomes against 30 other Xanthomonas genomes using the MicroScope platform [35] resulted in the selection of 33 coding DNA sequences (CDS), which were present in all the X. citri pv. citri genomes and absent in the non-target genomes used (Table S1). CDS with sizes < 100 bp (n = 2) and CDS corresponding to mobile elements (integrases, transposases, phage- and plasmid-borne genes) were not considered further (n = 5). The CDS XAC1051 (564 pb) encoding for a putative transmembrane protein displayed the best in silico specificity among the remaining candidates. A homologous sequence was only found in the X. citri pv. cajani strain LMG 558. It was split into two fragments located on two different contigs of its draft sequence. Thus, XAC1051 was used to design the qPCR Taqman® assay and conventional PCR assays (Table 1). The Primer Express® software used to design systems could not generate an efficient probe/primer system to allow the lack of amplification of X. citri pv. cajani strains. Conversely, primers were successfully designed to prevent the target amplification in X. citri pv. cajani for the conventional PCR assay.
Table 1.
Primers and probes used in this study
| Primers/probes | Sequence 5′ > 3’ | Amplicon size |
|---|---|---|
| XAC1051-2qPCR | ||
| P-XAC1051-MGB (6-Fam™) | CGGTGAGAAGCTGTAC | 58 bp |
| qPCR-XAC1051-F | AGAGGCGCACTATGGCTTTC | |
| qPCR-XAC1051-R | CAACCCAGGACCTGCAAGAA | |
| P-citrus5.8S- MGB (Vic™) | ATCCCGTGAACCATCG | 94 bp |
| citrus5.8S -F | GCGAAATGCGATACTTGGTGTGA | |
| citrus5.8S-R | CGTGCCCTCGGCCTAATG | |
| XAC1051-F/R PCR | ||
| XAC1051-F | AAATTCTTGTCGATCTGCTGGCT | 499 bp |
| XAC1051-R | GCCGCCGCATAATTCTTCTCAC | |
Analytical specificity of XAC1051-2qPCR
The 58 bp targeted DNA region of X. citri pv. citri strain IAPAR 306, including the primers and probes perfectly matched (100% identity and 100% query coverage) with sequences belonging to all the 91 X. citri pv. citri genomes available on NCBI, three X. citri pv. citri historical genomes from herbarium samples used in this study and the X. citri pv. cajani strain LMG 558. Conversely, no significant similarity was found with sequences from the other 2790 non-target xanthomonads.
All X. citri pv. citri strains showed a FAM-positive signal when tested with the real-time PCR assay. The typical exponential amplification curves and Ct values ranged from 27.7 to 31.8 (mean of 29.7 and standard deviation of 1.0). Among the 101 non-target strains, only three strains of X. citri pv. cajani tested positive with the real-time PCR assay with Ct values ranging from 22.9 to 23.7.
Dynamic range
The dynamic range of the duplex quantitative real-time PCR was assayed with three independent 10-fold dilution series of strain IAPAR 306 in each of the five different plant matrices. Calibration curves were constructed for each plant matrix by plotting the obtained Ct values against the Log10 of the standard concentrations (Fig. 1 and Fig. S1). A very strong linear relationship was observed between Ct values and the logarithm of bacterial concentration down to 1 × 103 CFU ml-1 for all plant matrices with 0.988 > r2 > 0.992. The PCR efficiency (E) ranged from 90 to 102% according to the different plant matrices. As target concentration decreased, some of the replicate samples appeared to be negative (undetermined results): 22 negative signals out of 45 and 39 out of 45 registered for the concentrations of 102 CFU ml− 1 and 101 CFU ml− 1, respectively. Nevertheless, when the calibration curves included data from the 102 CFU ml− 1 concentration, the coefficient and efficiency values were still acceptable (0.976 > r2 > 0.991 and 92% < E < 105%). In other words, the Ct values obtained for low target concentrations could be valid and may be considered as positive signals in routine diagnosis.
Fig. 1.
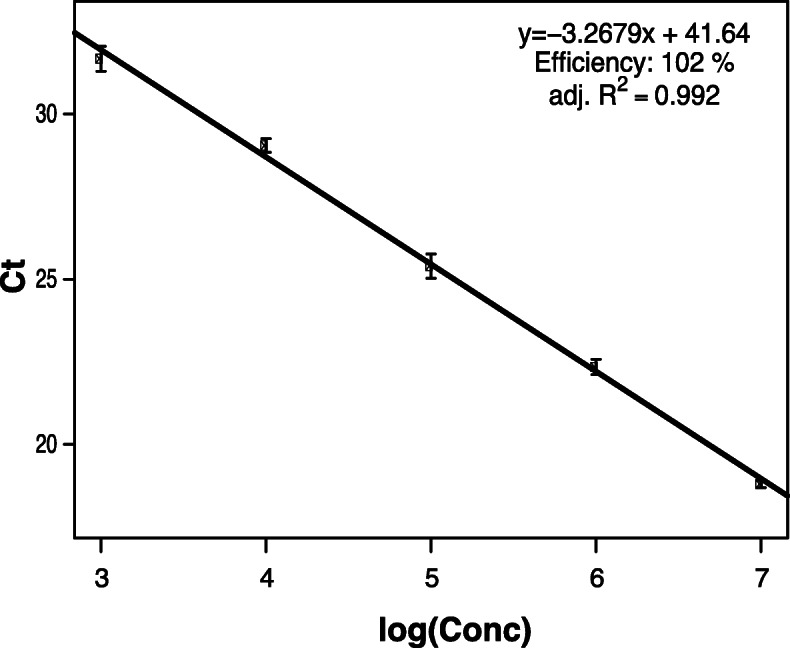
Standard curve obtained for a dilution series of the X. citri pv. citri strain IAPAR 306 in a sweet orange matrix. XAC1051-2qPCR was run on total DNA extracted from sweet orange leaves spiked with serially suspensions obtained from 10-fold serial dilution (107 to 102 CFU ml− 1). The standard curve was constructed using linear regression analysis of the threshold cycle (Ct) values for the serial dilutions over the Log10 of the initial target concentrations (compilation of all series and runs, corresponding to 9 replicates at each concentration level). The linear regression equation, the efficiency value and the adjusted R2 are indicated
Plant internal control
A VIC-positive signal, with Ct values ranging from 22.6 to 30.9, was detected for all symptomless (n = 90) and spiked plant samples with bacterial concentrations ranging from 1 × 101 to 1 × 104 CFU ml− 1 (n = 180). For higher bacterial concentrations, the number of VIC-positive signals decreased proportionally to the increase in bacterial populations: 15/36, 3/36 and 0/36 positive signals were registered for the concentrations of 1 × 105 CFU ml− 1, 1 × 106 CFU ml− 1 and 1 × 107 CFU ml− 1, respectively.
Analytical sensitivity in plant extracts
Before determining the analytical sensitivity, the first step was to implement a Ct cut-off, i.e., the value of Ct beyond which the real-time PCR signal was no longer considered as positive. Based on Youden’s index, the cut-off was estimated at 35.4 (Fig. 2). This cycle cut-off value was used to convert the quantitative data into binary data.
Fig. 2.
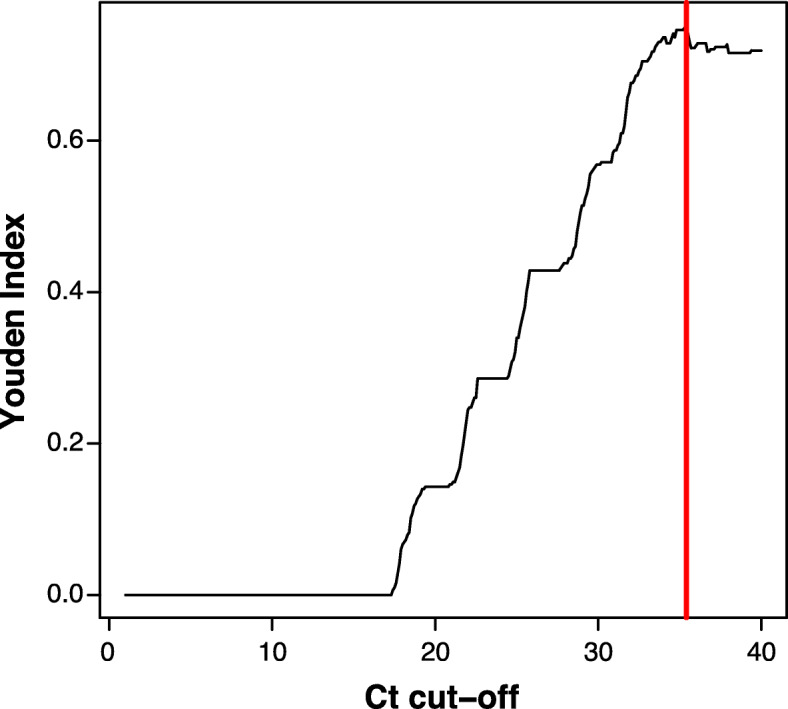
Determining the Ct cut-off according to the Youden index J. The optimal cut-off point is the PCR cycle with the highest value of the Youden index value, which represents a trade-off between sensitivity and specificity. The Ct cut-off was estimated to be 35.4
The limit of detection (LOD95%) was then determined for the whole dataset by plotting the numbers of positive responses obtained at the different concentrations against the Log10 of bacterial concentrations (Fig. 3). Log-Log was the best fitting curve with an estimated LOD95% of 754 CFU ml− 1 (95% CI 600–949), which corresponds to 15 bacteria per reaction. The same samples tested with the XAC1051-PCR assay yielded to an estimated LOD95% of 5234 CFU ml− 1 (95% CI 3656–7482), which corresponds to 105 bacteria per test sample.
Fig. 3.
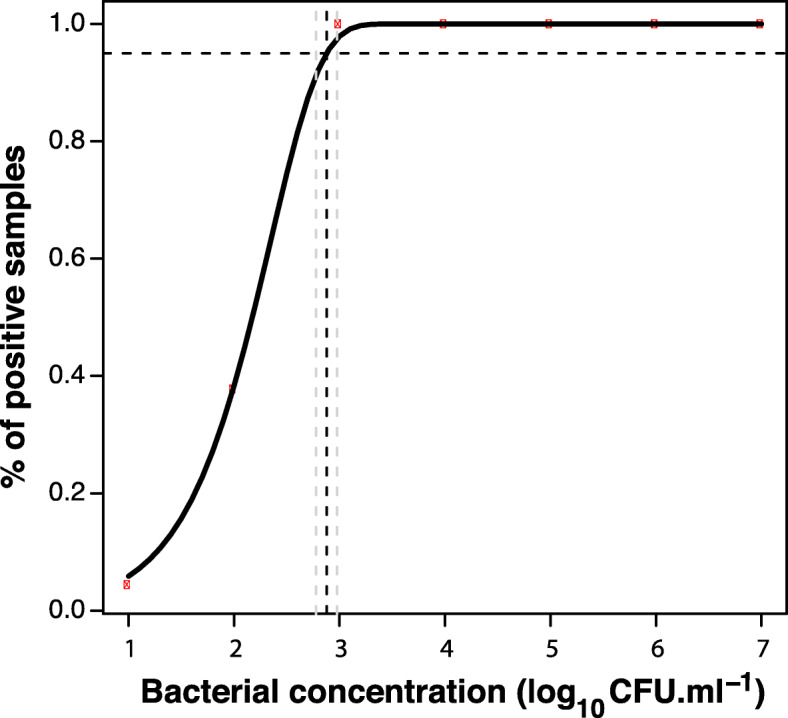
Determining the limit of detection (LOD95%) of the real-time qPCR assay. The x-axis represents the log of the bacterial concentrations, and the y-axis represents the probability of detection (POD) of replicate samples with a Ct value below 35.4 (cut-off). Each red point on the graph corresponds to means of 45 data samples. The smooth fitting line represents the best fitting model to the data points, based on a least squares approach using a probit model. The dark dotted vertical line indicates the bacterial concentration corresponding to LOD 95% (754 CFU ml− 1) and the two clear dotted lines indicate the corresponding 95% confidence interval
Repeatability, reproducibility and transferability
The intra-assay coefficients of variation obtained for all Ct value triplicates ranged from 0.035 to 2.482% with a median of 0.429%. The inter-assay coefficients of variations were calculated using the Ct values obtained for the three independent dilution series in each plant matrix and ranged from 0. 492 to 2.775% with a median of 1.558%. These low values reflected the repeatability and the reproducibility of the assay.
The qPCR efficiency values collected during the transferability assessment were all included between accepted range (90–110%) and all correlation coefficients were superior to 0.98 (Table 2). Low intra- and inter-assay Cv values were obtained for all experiments. Cut-off values were determined for each experiment involving a different qPCR device. These cut-off values were used to convert the quantitative data into qualitative data and to estimate the LOD95% values. LOD95% values were not statistically significant when the XAC1051-2qPCR protocol was tested on QS and LC480 devices.
Table 2.
Characteristics of the XAC1051-2qPCR assay performed using three different devices
| Plant matrix | StepOnePlus | QS | LC480 | |
|---|---|---|---|---|
| qPCR Efficiency | lemon | 93% | 94% | 97% |
| orange | 102% | 103% | 106% | |
| R2 | lemon | 0.992 | 0.981 | 0.984 |
| orange | 0.988 | 0.986 | 0.987 | |
| Intra-assay variation Cv range (median) |
0.07–2.48% (0.34) |
0.13–4.6% (0.60) |
0.14–3.6% (0.81) |
|
| Inter-assay variation Cv range (median) |
0.49–2.5% (1.50) |
0.55–3% (1.30) |
0.84–2.4% (1.50) |
|
| Cut-off values | 35.9 | 38.43 | 38.89 | |
|
LOD95% (CI) |
2.90 (2.76–3.04) |
3.04 (2.96–3.11) |
3.09 (2.92–3.25) |
|
| Vic Ct range | 25.8–31.2 | 22.3–27.9 | 23.4–29.7 |
Detection from naturally infected fruit
In order to detect X. citri pv. citri from symptomatic fruits collected in the field, we used XAC1051-PCR, XAC1051-2qPCR and enumeration of Xanthomonas-like colonies on semi-selective agar medium (Table S2). All samples showing canker lesions were tested positive using these methods for all four assayed citrus lines, while all the healthy citrus control samples tested negative. Non-repeatable low qPCR signals (Ct > 35.4), interpreted as negative results, were obtained for some replicates of a few symptomless samples. Some doubtful Xanthomonas colonies were observed on KC medium for some tangor samples. Nevertheless, suspensions from these colonies were tested negative by XAC1051-2qPCR. The plant signal was detected for all symptomless samples, which confirmed that the failure to detect X. citri pv. citri was not due to a technical problem during the step of DNA extraction and/or PCR amplification.
Detection from herbarium citrus samples
DNA extracted from the three herbarium specimens displayed substantial fragmentation (between 70 and 90 nt on average, see Table S3), as expected for DNA obtained from this type of material [36]. Nevertheless, they all tested positive with the Xac-qPCR assay. The presence of X. citri pv. citri in these samples was further confirmed by analyzing the next generation sequencing data (Table S3) obtained from the same samples during the course of another study (Rieux, unpublished data).
Comparison of XAC1051-based conventional and real-time PCR assays with existing molecular tests
All X. citri pv. citri strains, irrespective of pathotype, tested positive with all five PCR assays and the three qPCR assays, with the exception of strain NCPPB 211 for J-Taqpth-qPCR (Table 3 and Table S4). The X. citri pv. aurantifolii B and C strains only tested positive only with the conventional Jpth1/2 and VM3/VM4 PCR assays and the VM-Syb-qPCR assay, which is consistent with previously published data [31].
Table 3.
Comparison of the specificity of several PCR and real-time qPCR protocols
| Assay | X. citri pv. citri | X. citri pv. aurantifolii (n = 5) | X. euvesicatoria pv. citrumelonis (n = 2) | Other X. citria pathovars (n = 9) | Other Xanthomonasa species (n = 11) | Saprophytic xanthomonadsb (n = 15) | |||
|---|---|---|---|---|---|---|---|---|---|
| A (n = 63) | A* (n = 11) | Aw (n = 4) | |||||||
| PCR | Jpth1/2 | 62c | 11 | 4 | 5 | 0 | 7 | 1 | 0 |
| PCR | VM3/4 | 63 | 11 | 4 | 5 | 0 | 7 | 1 | 0 |
| PCR | XACF/R | 63 | 11 | 4 | 0 | 0 | 4 | 0 | 0 |
| PCR | XCF/R | 63 | 11 | 4 | 0 | 0 | 9 | 0 | 0 |
| PCR | XAC1051-F/R | 63 | 11 | 4 | 0 | 0 | 0 | 0 | 0 |
| qPCR | XAC1051-2qPCR | 63 | 11 | 4 | 0 | 0 | 1 | 0 | 0 |
| qPCR | J-Taqpth-qPCR | 63 | 11 | 4 | 0 | 0 | 3 | 0 | 0 |
| qPCR | VM-Syb-qPCR | 63 | 11 | 4 | 5 | 0 | 7 | 1 | 0 |
a Isolated from Citrus spp. but not pathogenic to Citrus spp.
b Isolated from Citrus spp.
c Number of positive samples
In terms of exclusivity, the XAC1051-F/R PCR assay displayed 100% exclusivity whereas the other PCR assays picked up some non-target strains, with exclusivity values of 77.8% for both Jpth1/2 and VM 3/4 primers, 88.8% for XACF/R primers and 75.0% for XCF/R, respectively. Most of the PCR-positive non-target strains were phylogenetically close to X. citri [37]. Among the qPCR assays, XAC1051-2qPCR displayed the best exclusivity because only one non-target strain of X. citri pv. cajani was amplified, as seen above (97.2% exclusivity). J-Taqpth-qPCR showed an acceptable specificity (91.7% exclusivity) whereas VM-Syb-qPCR assay displayed only 77.8% exclusivity (i.e., the same value as for the conventional primer pairs).
The sensitivity of the different molecular assays was compared using different combinations of X. citri pv. citri strains and plant matrices (Table S5). Of the different conventional tests, the XAC1051-F/R assay was the most sensitive, with a detection threshold of 3 × 103 ml− 1 in most of the plant matrices. A few samples were positive at 3 × 104 ml− 1. The Miyoshi XCF/R protocol yielded similar results whereas all other conventional PCR assays were found less sensitive (3 × 104 to 3 × 107 CFU ml− 1, depending on plant matrices and strains tested). Cut-off thresholds were determined for each real-time protocol using the ROC method. As expected, real-time PCR assays displayed a higher level of sensitivity than the conventional PCRs with almost identical sensitivity (a majority of detection limits at 3 × 103 CFU ml− 1).
Discussion
Xanthomonas citri pv. citri is a major threat to global citrus production. The success of surveillance strategies and quarantine measures to control the international movement of X. citri pv. citri is highly dependent on the availability of rapid and reliable in planta detection tools. Previous studies have shown that a number of existing diagnostic protocols developed for X. citri pv. citri display insufficient exclusivity or in some cases inclusivity [31]. In this study, we developed new, highly specific and sensitive molecular assays to diagnose, detect and quantify X. citri pv. citri in citrus tissues. We compared the new assays to existing diagnostic tools using a broad collection of target and non-target strains and in different citrus matrices. Importantly, the present study evaluated how PCR protocols reacted to an extensive X. citri pv. citri strain collection, a feature that most earlier studies failed to achieve. Indeed, we assayed representative samples of all the lineages/sublineages of this bacterium, which have been reported to date throughout the world [11, 16–18, 38].
The selected target gene, XAC1051, encodes for a hypothetical transmembrane protein and is present on the chromosome of strains for which a complete genome sequence is available. This gene is part of a genomic region previously considered to be specific to X. citri pv. citri, when compared to X. citri pv aurantifolii B and C strains [39].
When a conventional PCR format was used, our assay appeared perfectly specific with 100% inclusivity and exclusivity, values that outcompete other conventional PCR assays.
Real-time quantitative PCR assays have significant benefits compared to conventional PCR, including shorter turnaround time, reproducibility and sensitivity. In addition, this method can be used for both qualitative and quantitative assessments (for recent examples, see [40, 41]). We therefore developed a duplex real-time PCR assay, targeting the same bacterial gene, and including a plant internal control. This control targets a plant 5.8S rDNA sequence conserved among Citrus species. We demonstrated successful amplification of the plant internal control for X. citri pv. citri concentrations ≤1 × 104 CFU ml− 1. The plant signal was inhibited when higher bacterial concentrations were present in the extracts, which is consistent with previously published data [42]. Importantly, the plant control always yielded positive reactions in the absence of a bacterial signal. Therefore, it shows that a negative response for the bacterium is not due to a failure in the DNA extraction or PCR amplification process.
The XAC-1051-2qPCR assay was shown to be highly specific (100% inclusivity and 97.2% exclusivity). It displayed the best specificity when compared to the other real-time PCR assays available to date. Only strains of X. citri pv. cajani, responsible for bacterial leaf spot disease of pigeon pea (Cajanus cajan, Fabaceae) [43] tested positive with this molecular assay. This pathogen seems geographically restricted to India where pigeon pea is its sole known host species. X. citri pv. cajani was identified as the closest relative to X. citri pv. citri in a recent phylogenomic analysis [38], suggesting that this DNA region may have been present in their most recent common ancestor. If necessary, suspect samples can be confirmed by performing the XAC1051-F/R PCR assay.
When real-time quantitative PCR is used as a qualitative method, a Ct cut-off, i.e., the PCR cycle number above which any sample response value (Ct) is considered to be a false positive, must be set. Indeed, depending on experimental conditions, some false positives with high Ct values may be registered. These values result from a spontaneous increase in fluorescence background emissions and/or low-level DNA cross-contaminations [44, 45]. The cut-off varies depending on the experiment context: inherent characteristics of the real-time PCR system, qPCR instrument, qPCR mix and DNA template. A statistical approach based on ROC analysis, where both false positive and false negative qPCR signals are considered, allowed us to establish an optimal Ct cut-off. The estimated Ct cut-off was applied to determine the level of sensitivity of our assay in different plant matrices. The XAC-1051-2qPCR assay was shown to be highly sensitive with the capacity to detect approximatively 15 target cells per reaction. It demonstrated high repeatability and reproducibility. It also proved to be transferable between PCR cyclers with an optimization step, without compromising sensitivity and specificity. Importantly, the XAC-1051-2qPCR assay showed a good ability to detect the target from naturally infected citrus fruits. Interestingly, as predicted in silico, the XAC-1051-2qPCR assay was able to detect X. citri pv. citri from some herbarium samples dating back to 1911 (Table S3), despite the small quantities, the high fragmentation and the chemical modifications expected when ancient DNA (aDNA) is obtained from samples of this type [36]. The success of this assay is probably due to its high sensitivity and the small size of the target DNA (58pb). Interestingly, this result suggests that molecular tools, such as our XAC-1051-2qPCR assay, are useful for screening herbaria for plant pathogens. Screening is a prerequisite for further investigations such as metagenomics or population genetic analyses geared to reconstructing the evolutionary history of plant pathogens [46, 47].
Conclusions
Herein, we conducted a thorough comparative analysis of several conventional and quantitative PCR protocols using the same strain collection and plant samples. Thus, we hope to provide end-users with precise information with regard to the respective advantages and limitations of the different protocols in order to help them select one or more complementary methods for testing plant material or microbial cultures.
In agreement with previous studies [48–50], we conclude that genome-informed identification of targets is a powerful aid when it comes to developing highly specific diagnostic techniques for plant pathogens.
Methods
Bacterial strains and culture conditions
Ninety-eight strains of X. citri pv. citri, representing the currently known genetic diversity of this pathovar were used in this study (Table S6). This collection included the strain LMG 696. This X. citri pv. citri pathotype A* strain was recently authenticated by WGS data, after initially being mislabeled as X. campestris pv. durantae [38].
The present study also examined 101 non-target strains representing other bacterial genera, other pathovars of X. citri, other Xanthomonas strains pathogenic to rutaceous species and saprophytic Xanthomonas strains isolated from citrus (Table S7).
To compare the different PCR and real-time quantitative PCR protocols (see 2.10 and 3.4), a specific collection of strains was used, including some of the strains of Tables S6 and S7. This collection is listed separately in Table S4 to facilitate comprehension.
Strains were stored at − 80 °C on beads in cryovials (Microbank Prolab Diagnostics) or freeze-dried for long-term storage. All strains were streaked on yeast-peptone-glucose agar (YPGA; yeast extract 7 g l− 1, peptone 7 g l− 1, glucose 7 g l− 1, and agar 18 g l− 1; pH 7.2) plates at 28 °C for 3–4 days to check for purity. Subcultures were produced from single colonies on YPGA plates incubated at 28 °C for 48 h. Bacterial suspensions were prepared and diluted in 0.01 M Tris buffer pH 7.2 (Sigma 7–9 Sigma-Aldrich, Saint-Quentin Fallavier, France) unless otherwise stated. Plant or canker lesion homogenates were prepared in the same buffer supplemented with 2% polyvinylpyrrolidone (PVP) with an average mol wt of 40,000 (Sigma-Aldrich).
Selection of a DNA target specific to Xanthomonas citri pv. citri and a plant internal control
A preliminary bioinformatics screening of candidate CDSs was performed using the “gene phyloprofile” tool in the MicroScope platform (Genoscope, Evry, France) [35] on 30 X. citri pv. citri genomes, including pathotype A, A* and AW strains against 30 non-target genomes of Xanthomonas (other species and pathovars). The aim was to select CDSs conserved in all X. citri pv. citri genomes that had limited or no identity to CDSs from non-target genomes present in the database. Then, using the selected nucleotide sequences as query, we performed BLASTn and Megablast searches against NCBI databases (February 2020): nr/nt, draft (n = 2225) and complete genomes (n = 565) of Xanthomonas group (taxid = 32,033), complete plasmids (n = 17,302) and complete bacteriophages (n = 2717). In silico presence and identity of chosen target regions was further confirmed in the three ancient genomes used in this study (see above, detection from herbarium citrus samples, and Table S3).
The 5.8S rDNA sequence from C. x aurantiifolia (MF797954) was selected to develop a endogenous plant internal control. This multicopy DNA region is conserved in the Rutaceae family, particularly among Citrus species.
Duplex real-time quantitative PCR (XAC1051-2qPCR) and PCR assays
Taqman® probe and primers were designed from the X. citri pv. citri XAC1051 gene and the plant 5.8S rDNA sequence using Primer Express® software Version 3.0 (Applied Biosystems, Courtaboeuf, France) and were provided by Applied Biosystems (Courtaboeuf, France). The different Taqman® probe and primer systems were checked in Oligo 7.6 (Molecular Biology Insights, Inc., Cascade, CO, USA) in order to minimize interactions between the different oligonucleotides. The selected primers and probes are listed in Table 1.
Amplifications were carried out in 15-μl reaction volumes (in HPLC grade water) containing 7.5 μl of 2× Mastermix (Applied Biosystems), 600 nM of qPCR-XAC1051-F and qPCR -XAC1051-R primers, 425 nM of 5′FAM-labeled XAC-1051 MGB probe (P-XAC1051-MGB), 50 nM of citrus5.8SF and citrus5.8SR primers, 50 nM of 5′VIC-labeled citrus5.8S MGB probe (P-citrus5.8S- MGB) and 2 μl (pure bacterial suspensions) or 5 μl (total plant DNA extract) of template DNA.
The real-time PCR cycling conditions included a step at 50 °C for 2 min, an initial denaturation step at 95 °C for 2 min followed by 40 cycles of denaturation and annealing/elongation for 15 s at 95 °C and 1 min at 60 °C, respectively. Analyses were performed using the StepOnePlus software version v2.2.2. Each sample was at least duplicated.
Conventional primers were also designed from the XAC1051 gene using Oligo 7.6 (Table 1). Amplifications were carried out in 25-μl reaction volumes containing 5 μl of Green GoTaq® Reaction Buffer, 2 mM MgCl2, 0.5 μM of XAC1051-F and XAC1051-R, 0.2 mM each dNTP, 1.25 U of GoTaq® DNA Polymerase (Promega,) and 2 μl of template DNA. PCR amplifications were performed using a Veriti™ Thermal Cycler (Applied Biosystems, Courtaboeuf, France). The amplification program included denaturation at 95 °C for 2 min, 35 cycles consisting of denaturation at 95 °C for 45 s, annealing at 65 °C for 45 s, and extension at 72 °C for 1 min, and a final extension step at 72 °C for 5 min.
Specificity of XAC1051-2qPCR assay
The in silico-determined specificity of the 58 bp target region from X. citri pv. citri was subject to an addition experimental check following the guidelines in the EPPO PM 7/98 (4) standard protocol [51]. The real time PCR protocol was assayed on pure cultures of target (n = 98) and non-target (n = 101) strains (Tables S6 and S7). Spectrophotometrically adjusted suspensions containing approx. 1 × 108 CFU ml− 1 were diluted 100 or 10,000-fold for non-target and target strain assays, respectively. The suspensions were heated at 95 °C for 2 min and chilled on ice.
Dynamic range in the plant matrix
The dynamic range of the real-time PCR assay, i.e., the range of initial template concentrations for which accurate Ct values are obtained, was determined on the dilution series of the strain IAPAR 306 in different citrus matrices: sweet orange (C. x sinensis), clementine mandarin (C. reticulata), grapefruit (C. x paradisi), lemon (C. x limon) and makrut lime (C. hystrix). Overnight cultures of IAPAR 306 were adjusted spectrophotometrically to a concentration of approx. 1 × 108 CFU ml− 1 and serially 10-fold diluted. Fruit peel (0.1 g) was homogenized in 10 ml buffer using a grinder (Homex 6, Bioreba, Reinach, Switzerland) and spiked with bacterial suspensions at final concentrations ranging from 1 × 101 to 1 × 107 CFU ml− 1. Three replicated dilution series were performed in each citrus matrix. Total DNA was extracted from 2 ml homogenates using DNeasy Plant Mini kit (Qiagen, Courtaboeuf, France). Three qPCR replicates were carried out at each contamination level (nine Ct values were thus registered for each plant matrix and contamination level). Non-template controls (NTC) consisting of plant matrix and mix without DNA were included as negative samples (n = 18). Standard curves were generated for each citrus matrix by plotting Ct values against the logarithm of initial DNA concentrations. The reaction efficiency E was calculated according to the slope of the standard curves as follows: . The XAC1051-F/R PCR assay was also performed in duplicate using the same samples.
Cut-off Ct value and limit of detection (LOD)
A ROC (Receiver operating characteristic) was used in order to determine the Ct cut-off value, i.e., the PCR cycle number above which signals are no longer interpreted as positive [52, 53]. This analysis, based on the determination of the Youden J index, which considers false positives and negatives [42, 54], was performed on Ct values obtained (see § 2.5. above) for samples with a priori positive status, i.e., citrus spiked samples with different bacterial concentrations (n = 135) and for samples with a priori negative status, i.e., NTC (n = 110). Samples with Ct values higher than the Ct cut-off value were then considered negative.
The analytical sensitivity of the XAC1051-2qPCR and the XAC1051-F/R PCR were estimated by determining the 95% limit of detection (LOD95%), i.e., the concentration at which a detection probability of 95% is expected [55–57] as explained previously [42].
Repeatability, reproducibility and transferability
Repeatability (i.e., the level of agreement between replicates of a sample tested under the same conditions) and reproducibility (i.e., the ability of a test to provide consistent results when applied to aliquots of the same sample tested under different conditions (time, personnel, equipment, location, etc.)) were estimated according to the EPPO standard PM 7/76 (5) [58]. Repeatability was evaluated by computing intra-assay coefficients of variation ( based on Ct mean values of qPCR triplicates obtained for the concentrations ranging from 1 × 103 CFU ml− 1 to 1 × 107 CFU ml− 1 (70 Cv values). Reproducibility was evaluated on three qPCR runs independently performed for each plant matrix at different times. Inter-assay Cv values were calculated from the Ct values (PCR triplicate means) obtained for concentrations ranging from 1 × 103 CFU ml− 1 to 1 × 107 CFU ml− 1 (25 Cv values).
The protocol’s transferability was evaluated by reproducibility experiments performed by different operators, at different periods and in different laboratories (Cirad and ANSES). Dilution series of suspensions prepared from the IAPAR 306 strain in lemon and orange matrices already tested on the StepOnePlus device (Applied Biosystems) were also assayed using the Light Cycler LC 480 (Roche Life Science, Meylan, France) and the Quantstudio5 (QS5) (Applied Biosystems) real-time PCR systems. The application of the StepOnePlus master mix and real time cycling conditions for other devices gave poor results and required optimization (data not shown). Successful amplifications were obtained for both LC480 and QS5 devices when using the GoTaq® probe qPCR master mix kit (Promega) and the following cycling conditions: a step at 95 °C for 2 min followed by 45 cycles of denaturation and annealing/elongation for 15 s at 95 °C and 1 min at 60 °C, respectively. The concentrations of the different primers and probes remained the same as for the StepOnePlus. Efficiency and correlation coefficients were calculated for each fruit/real-time device data set. Cut-off values and LOD95% were estimated for each real-time qPCR device data set. Intra-assay and inter coefficients of variation were also calculated for each real-time qPCR device data set.
Detection from naturally infected fruit
Fifteen fruit (several species) showing typical ACC symptoms were collected in citrus groves in Reunion (Table S2). Three lesions per fruit (0.1 g each) were sampled and independently homogenized in 10 ml buffer (45 samples). Fifty microliters were plated in duplicates on KC medium to estimate target concentrations [59]. Total DNA was extracted from 2 ml homogenates using DNeasy Plant Mini kit (Qiagen, Courtaboeuf, France). Fifteen citrus fruits showing no canker symptoms were analyzed as well, with two or three samples (same size as diseased samples) collected independently per fruit and processed as described above (43 samples). Conventional and real time quantitative PCR assays were performed on the different samples as described in § 2.3.
Detection from herbarium citrus samples
The duplex PCR assay was also used to screen three herbarium citrus samples bearing typical citrus canker lesions, and collected between 1911 to 1992 in different areas (Table S3). They were provided by the Royal Mauritius Herbarium (acronym: MAU) and the National Herbarium of the Muséum National d’Histoire Naturelle, France (acronym: P). DNA extraction was performed in a bleach-cleaned facility room according to the protocol described in §2.5 using 0.01 g of leaf fragments (instead of fruit peel) as starting material. DNA concentration and fragment size were measured with Qubit (Invitrogen life Technologies) and TapeStation (Agilent Technologies) high sensitivity assays, respectively, according to the manufacturers’ instructions. The XAC1051-dqPCR assay was performed as described in § 2.3.
Comparison of XAC1051-based conventional and real-time PCR assays with existing molecular tests
This comparison was performed by a laboratory (ANSES), which is different to the one where the qPCR XAC1051-based conventional and real-time PCR assays were developed (Cirad). We considered a selection of published PCR and real-time PCR protocols based on previously published data [31] or preliminary experimental and/or in silico data analyses (in the case of the most recent protocols). This excluded a recently published multiplex protocol designed to detect and differentiate between several citrus-associated xanthomonads [23], because the primers selected for X. citri pv. citri pathotype A perfectly matched in silico and reacted in preliminary assays with five other Xanthomonas citri pathovars. Table S8 presents the published protocols (and associated experimental conditions), which passed the first screen and were compared to the XAC1051-based conventional and real-time PCR assays in terms of analytical specificity and sensitivity.
In the first assay, bacterial suspensions in sterile distilled water (containing approx. 1 × 104 or 1 × 106 CFU ml− 1 for target and non-target strains, respectively) were used. A set of X. citri pv. citri strains (n = 78) and other xanthomonads (n = 42) (Table S4) was assayed in duplicate to compare the analytical specificity following the guidelines in EPPO PM 7/98 (4) standard protocol [51].
Then, the comparison of analytical sensitivity was carried out on different plant matrices spiked with tenfold-diluted bacterial suspensions. Plant matrices included the leaf or fruit peel of sweet orange grapefruit, lemon, Tahiti lime (C. x latifolia), clementine (C. x clementina) and makrut lime. They were spiked with 10-fold dilutions of the X. citri pv. citri strain CFBP 2525 with final concentrations ranging from 3 × 102 to 3 × 107 CFU ml− 1. In addition, leaves and fruit peels of Mexican lime (i.e., a host species susceptible to all X. citri pv. citri pathotypes) were spiked with 10-fold dilutions of the following strains: pathotype A strains JJ238–29 and LH001–1 (lineage 1 and 2, respectively), pathotype Aw strain LG115 (lineage 3), pathotype A* strain CFBP 2911 (lineage 4), the X. citri pv. aurantifolii pathotype B strain CFBP 2902 or the X. citri pv. aurantifolii pathotype C strain CFBP 2866 (same final concentrations as CFBP 2525). Homogenate production (0.1 mg plant matrix 5 ml buffer) and DNA extractions were performed as described above. PCR or real-time PCR assays were performed in duplicate.
Supplementary information
Acknowledgements
We would like to express our thanks to the Plant Protection Platform (3P, IBISA) and to C. Boyer for her helpful contribution.
Authors’ contributions
Conceived and designed the experiments: IR, AC, NB, AR, LG and OP. Provided biological material, performed the experiments and acquired the data: VML, AMor, NB, AMou, AR, PC, LG, MG and CB. Run bioinformatic analyses: IR, AR and PC. Analysed and interpreted the data: IR, VML, AC, NB, AMou, AR, PC, LG, CV, FC and OP. Wrote the manuscript: IR and OP. Revised and approved the manuscript: all authors. All authors read and approved the final manuscript.
Funding
The European Union Agricultural Fund for Rural Development (FEAFRD contract 2014FR06RDRP004), the European Regional Development Fund (ERDF contract GURDT I2016–1731-0006632), the Région Réunion, the French Agropolis Foundation (Labex Agro – Montpellier, E-SPACE project number 1504–004), the Agence Nationale pour la Recherche (JCJC MUSEOBACT contract ANR-17-CE35–0009-01), ANSES and CIRAD provided financial support.
Availability of data and materials
Available as Supplementary Material. R scripts available upon request.
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12866-020-01972-8.
References
- 1.Paini DR, Sheppard AW, Cook DC, De Barro PJ, Worner SP, Thomas MB. Global threat to agriculture from invasive species. Proc Natl Acad Sci U S A. 2016;113(27):7575–7579. doi: 10.1073/pnas.1602205113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sikes BA, Bufford JL, Hulme PE, Cooper JA, Johnston PR, Duncan RP. Import volumes and biosecurity interventions shape the arrival rate of fungal pathogens. PLoS Biol. 2018;16(5):e2006025. doi: 10.1371/journal.pbio.2006025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bull CT, Koike ST. Practical benefits of knowing the enemy: modern molecular tools for diagnosing the etiology of bacterial diseases and understanding the taxonomy and diversity of plant-pathogenic bacteria. Annu Rev Phytopathol. 2015;53:157–180. doi: 10.1146/annurev-phyto-080614-120122. [DOI] [PubMed] [Google Scholar]
- 4.Graham JH, Gottwald TR, Cubero J, Achor DS. Xanthomonas axonopodis pv. citri: factors affecting successful eradication of citrus canker. Mol Plant Pathol. 2004;5(1):1–15. doi: 10.1046/j.1364-3703.2004.00197.x. [DOI] [PubMed] [Google Scholar]
- 5.Miller SA, Beed FD, Harmon CL. Plant disease diagnostic capabilities and networks. Annu Rev Phytopathol. 2009;47:15–38. doi: 10.1146/annurev-phyto-080508-081743. [DOI] [PubMed] [Google Scholar]
- 6.Richard D, Ravigné V, Rieux A, Facon B, Boyer C, Boyer K, et al. Adaptation of genetically monomorphic bacteria: evolution of copper resistance through multiple horizontal gene transfers of complex and versatile mobile genetic elements. Mol Ecol. 2017;26(7):2131–2149. doi: 10.1111/mec.14007. [DOI] [PubMed] [Google Scholar]
- 7.Gottwald TR, Irey M. Post-hurricane analysis of citrus canker II: Predictive model estimation of disease spread and area potentially impacted by various eradication protocols following catastrophic weather events. Plant Health Prog. 2007. 10.1094/PHP-2007-0405-01-RS.
- 8.Broadbent P, Fahy PC, Gillings MR, Bradley JK, Barnes D. Asiatic citrus canker detected in a pummelo orchard in northern Australia. Plant Dis. 1992;76(8):824–829. [Google Scholar]
- 9.Gambley CF, Miles AK, Ramsden M, Doogan V, Thomas JE, Parmenter K, et al. The distribution and spread of citrus canker in emerald, Australia. Australas Plant Pathol. 2009;38:547–557. [Google Scholar]
- 10.Derso E, Vernière C, Pruvost O. First report of Xanthomonas citri pv. citri-A* causing citrus canker on lime in Ethiopia. Plant Dis. 2009;93(2):203. doi: 10.1094/PDIS-93-2-0203B. [DOI] [PubMed] [Google Scholar]
- 11.Pruvost O, Goodarzi T, Boyer K, Soltaninejad H, Escalon A, Alavi SM, et al. Genetic structure analysis of strains causing citrus canker in Iran reveals the presence of two different lineages of Xanthomonas citri pv. citri pathotype A*. Plant Pathol. 2015;64(4):776–84. 10.1111/ppa.12324.
- 12.Vernière C, Hartung JS, Pruvost OP, Civerolo EL, Alvarez AM, Maestri P, et al. Characterization of phenotypically distinct strains of Xanthomonas axonopodis pv. citri from Southwest Asia. Eur J Plant Pathol. 1998;104(5):477–487. [Google Scholar]
- 13.Rybak M, Minsavage GV, Stall RE, Jones JB. Identification of Xanthomonas citri ssp. citri host specificity genes in a heterologous expression host. Mol Plant Pathol. 2009;10(2):249–262. doi: 10.1111/j.1364-3703.2008.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun XA, Stall RE, Jones JB, Cubero J, Gottwald TR, Graham JH, et al. Detection and characterization of a new strain of citrus canker bacteria from key Mexican lime and Alemow in South Florida. Plant Dis. 2004;88(11):1179–1188. doi: 10.1094/PDIS.2004.88.11.1179. [DOI] [PubMed] [Google Scholar]
- 15.Bui Thi Ngoc L, Vernière C, Jarne P, Brisse S, Guérin F, Boutry S, et al. From local surveys to global surveillance: three high throughput genotyping methods for the epidemiological monitoring of Xanthomonas citri pv. citri pathotypes. Appl Environ Microbiol. 2009;75(4):1173–1184. doi: 10.1128/AEM.02245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon JL, Lefeuvre P, Escalon A, Barbe V, Cruveiller S, Gagnevin L, et al. Comparative genomics of 43 strains of Xanthomonas citri pv. citri reveals the evolutionary events giving rise to pathotypes with different host ranges. BMC Genomics. 2015;16(1):1098. doi: 10.1186/s12864-015-2310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong K, Munoz-Bodnar A, Arias Rojas N, Poulin L, Rodriguez RL, Gagnevin L, et al. CRISPR elements provide a new framework for the genealogy of the citrus canker pathogen Xanthomonas citri pv. citri. BMC Genomics. 2019;20(1):917. doi: 10.1186/s12864-019-6267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pruvost O, Magne M, Boyer K, Leduc A, Tourterel C, Drevet C, et al. A MLVA genotyping scheme for global surveillance of the citrus pathogen Xanthomonas citri pv. citri suggests a worldwide geographical expansion of a single genetic lineage. PloS One. 2014;9(6):e98129. doi: 10.1371/journal.pone.0098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schubert T, Rizvi S, Sun X, Gottwald T, Graham J, Dixon W. Meeting the challenge of eradicating citrus canker in Florida - again. Plant Dis. 2001;85(4):341–356. doi: 10.1094/PDIS.2001.85.4.340. [DOI] [PubMed] [Google Scholar]
- 20.Leduc A, Traoré YN, Boyer K, Magne M, Grygiel P, Juhasz C, et al. Bridgehead invasion of a monomorphic plant pathogenic bacterium: Xanthomonas citri pv. citri, an emerging citrus pathogen in Mali and Burkina Faso. Environ Microbiol. 2015;17(11):4429–4442. doi: 10.1111/1462-2920.12876. [DOI] [PubMed] [Google Scholar]
- 21.Richard D, Boyer C, Javegny S, Boyer K, Grygiel P, Pruvost O, et al. First report of Xanthomonas citri pv. citri pathotype A causing Asiatic citrus canker in Martinique, France. Plant Dis. 2016;100(9):1946. [Google Scholar]
- 22.Cubero J, Graham JH. Genetic relationship among worldwide strains of Xanthomonas causing canker in citrus species and design of new primers for their identification by PCR. Appl Environ Microbiol. 2002;68(3):1257–1264. doi: 10.1128/AEM.68.3.1257-1264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fonseca NP, Felestrino EB, Caneschi WL, Sanchez AB, Cordeiro IF, Lemes CGC, et al. Detection and identification of Xanthomonas pathotypes associated with citrus diseases using comparative genomics and multiplex PCR. PeerJ. 2019;7:e7676. doi: 10.7717/peerj.7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartung JS, Pruvost OP, Villemot I, Alvarez A. Rapid and sensitive colorimetric detection of Xanthomonas axonopodis pv. citri by immunocapture and a nested-polymerase chain reaction assay. Phytopathology. 1996;86(1):95–101. [Google Scholar]
- 25.Kingsley MT, Fritz LK. Identification of the citrus canker pathogen Xanthomonas axonopodis pv citri A by fluorescent PCR assays. Phytopathology. 2000;90:S42. [Google Scholar]
- 26.Miyoshi T, Sawada H, Tachibana Y, Matsuda I. Detection of Xanthomonas campestris pv. citri by PCR using primers from the spacer region between the 16S and 23S rRNA genes. Ann Phytopath Soc Jpn. 1998;64(4):249–254. [Google Scholar]
- 27.Park DS, Hyun JW, Park YJ, Kim JS, Kang HW, Hahn JH, et al. Sensitive and specific detection of Xanthomonas axonopodis pv. Citri by PCR using pathovar specific primers based on hrpW gene sequences. Microbiol Res. 2006;161:145–149. doi: 10.1016/j.micres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Cubero J, Graham JH. Quantitative real-time polymerase chain reaction for bacterial enumeration and allelic discrimination to differentiate Xanthomonas strains on citrus. Phytopathology. 2005;95(11):1333–1340. doi: 10.1094/PHYTO-95-1333. [DOI] [PubMed] [Google Scholar]
- 29.Mavrodieva V, Levy L, Gabriel DW. Improved sampling methods for real-time polymerase chain reaction diagnosis of citrus canker from field samples. Phytopathology. 2004;94(1):61–68. doi: 10.1094/PHYTO.2004.94.1.61. [DOI] [PubMed] [Google Scholar]
- 30.Rigano LA, Marano MR, Castagnaro AP, Do Amaral AM, Vojnov AA. Rapid and sensitive detection of citrus bacterial canker by loop-mediated isothermal amplification combined with simple visual evaluation methods. BMC Microbiol. 2010;10(1):176. doi: 10.1186/1471-2180-10-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delcourt S, Vernière C, Boyer C, Pruvost O, Hostachy B, Robène-Soustrade I. Revisiting the specificity of PCR primers for diagnostics of Xanthomonas citri pv. citri by experimental and in silico analyses. Plant Dis. 2013;97(3):373–378. doi: 10.1094/PDIS-04-12-0351-RE. [DOI] [PubMed] [Google Scholar]
- 32.Muska A, Peck E, Palmer S. Standards and controls: concepts for preparation and use in real-time PCR application. In: Mackay IM, editor. Real-time PCR in microbiology: from diagnosis to characterization. Norfolk, UK: Caister Academic Press; 2007. pp. 101–131. [Google Scholar]
- 33.Ioos R, Fabre B, Saurat C, Fourrier C, Frey P, Marçais B. Development, comparison, and validation of real-time and conventional PCR tools for the detection of the fungal pathogens causing brown spot and red band needle blights of pine. Phytopathology. 2010;100(1):105–114. doi: 10.1094/PHYTO-100-1-0105. [DOI] [PubMed] [Google Scholar]
- 34.Schenck N, Fourrier-Jeandel C, Ioos R. A robust and specific real-time PCR tool for the detection of Phytophthora lateralis in plant tissues. Eur J Plant Pathol. 2016;146(2):231–244. [Google Scholar]
- 35.Vallenet D, Calteau A, Dubois M, Amours P, Bazin A, Beuvin M, et al. MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 2020;48(D1):D579–DD89. doi: 10.1093/nar/gkz926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss CL, Schuenemann VJ, Devos J, Shirsekar G, Reiter E, Gould BA, et al. Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R Soc Open Sci. 2016;3(6):160239. doi: 10.1098/rsos.160239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Constantin EC, Cleenwerck I, Maes M, Baeyen S, Van Malderghem C, De Vos P, et al. Genetic characterisation of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016;65(5):792–806. [Google Scholar]
- 38.Patane JSL, Martins J, Jr, Rangel LT, Belasque J, Digiampietri LA, Facincani AP, et al. Origin and diversification of Xanthomonas citri subsp. citri pathotypes revealed by inclusive phylogenomic, dating, and biogeographic analyses. BMC Genomics. 2019;20(1):700. doi: 10.1186/s12864-019-6007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreira AN, Laia ML, De Souza RF, Zaini PA, Da Silva AC, Da Silva AM, et al. Development and validation of a Xanthomonas axonopodis pv. citri DNA microarray platform (XACarray) generated from the shotgun libraries previously used in the sequencing of this bacterial genome. BMC Res Notes. 2010;3:150. doi: 10.1186/1756-0500-3-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdulai M, Basim H, Saba CKS. Rapid identification and detection of Xanthomonas phaseoli pv. manihotis, causing bacterial blight disease in cassava by real-rime PCR using LNA probe. Int J Agric Biol. 2020;23(2):259–268. [Google Scholar]
- 41.Villela JGA, Ritschel P, Barbosa MAG, Baccin KMS, Rossato M, Maia JDG, et al. Detection of Xanthomonas citri pv. viticola on grapevine by real-time PCR and BIO-PCR using primers designed from pathogenicity and xanthomonadin gene sequences. Eur J Plant Pathol. 2019;155(2):445–459. [Google Scholar]
- 42.Jouen E, Chiroleu F, Maillot-Lebon V, Chabirand A, Merion S, Boyer C, et al. A duplex quantitative real-time PCR assay for the detection and quantification of Xanthomonas phaseoli pv. dieffenbachiae from diseased and latently infected anthurium tissue. J Microbiol Meth. 2019;161:74–83. doi: 10.1016/j.mimet.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Kulkarni YS, Patel MK, Abhyankar SG. A new bacterial leaf-spot and stem canker of pigeon pea. Curr Sci. 1950;19(12):384. [PubMed] [Google Scholar]
- 44.Burns DL. Biochemistry of type IV secretion. Curr Opinion Microbiol. 1999;2(1):25–29. doi: 10.1016/s1369-5274(99)80004-6. [DOI] [PubMed] [Google Scholar]
- 45.Caraguel CG, Stryhn H, Gagne N, Dohoo IR, Hammell KL. Selection of a cutoff value for real-time polymerase chain reaction results to fit a diagnostic purpose: analytical and epidemiologic approaches. J Vet Diagn Investig. 2011;23(1):2–15. doi: 10.1177/104063871102300102. [DOI] [PubMed] [Google Scholar]
- 46.Li W, Song Q, Brlansky RH, Hartung JS. Genetic diversity of citrus bacterial canker pathogens preserved in herbarium specimens. Proc Natl Acad Sci U S A. 2007;104(47):18427–18432. doi: 10.1073/pnas.0705590104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida K, Burbano HA, Krause J, Thines M, Weigel D, Kamoun S. Mining herbaria for plant pathogen genomes: back to the future. PLoS Pathog. 2014;10(4):e1004028. doi: 10.1371/journal.ppat.1004028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang JM, Hamilton JP, Diaz MGQ, Van Sluys MA, Burgos MRG, Vera Cruz CM, et al. Genomics-based diagnostic marker development for Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola. Plant Dis. 2010;94(3):311–319. doi: 10.1094/PDIS-94-3-0311. [DOI] [PubMed] [Google Scholar]
- 49.Langlois PA, Snelling J, Hamilton JP, Bragard C, Koebnik R, Verdier V, et al. Characterization of the Xanthomonas translucens complex using draft genomes, comparative genomics, phylogenetic analysis, and diagnostic LAMP assays. Phytopathology. 2017;107(5):519–527. doi: 10.1094/PHYTO-08-16-0286-R. [DOI] [PubMed] [Google Scholar]
- 50.Triplett LR, Hamilton JP, Buell CR, Tisserat NA, Verdier V, Zink F, et al. Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl Environ Microbiol. 2011;77(12):3930–3937. doi: 10.1128/AEM.00028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anonymous PM 7/98 (2) specific requirements for laboratories preparing accreditation for a plant pest diagnostic activity. EPPO Bull. 2014;44(2):117–147. [Google Scholar]
- 52.Grosdidier M, Aguayo J, Marcais B, Ioos R. Detection of plant pathogens using real-time PCR: how reliable are late C-t values? Plant Pathol. 2017;66(3):359–367. [Google Scholar]
- 53.Nutz S, Doll K, Karlovsky P. Determination of the LOQ in real-time PCR by receiver operating characteristic curve analysis: application to qPCR assays for Fusarium verticillioides and F. proliferatum. Anal Bioanal Chem. 2011;401(2):717–726. doi: 10.1007/s00216-011-5089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3(1):32–35. doi: 10.1002/1097-0142(1950)3:1<32::aid-cncr2820030106>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 55.Broeders S, Huber I, Grohmann L, Berben G, Taverniers I, Mazzara M, et al. Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci Tech. 2014;37(2):115–126. [Google Scholar]
- 56.Burns M, Valvidia H. Modelling the limit of detection in real-time quantitative PCR. Eur Food Res Technol. 2008;226:1513–1524. [Google Scholar]
- 57.Kolm C, Martzy R, Brunner K, Mach RL, Krska R, Heinze G, et al. A complementary isothermal amplification method to the U.S. EPA quantitative polymerase chain reaction approach for the detection of enterococci in environmental waters. Environ Sci Technol. 2017;51(12):7028–7035. doi: 10.1021/acs.est.7b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anonymous PM7/76(5) use of EPPO diagnostic protocols. EPPO Bull. 2018;48(3):373–377. [Google Scholar]
- 59.Pruvost O, Roumagnac P, Gaube C, Chiroleu F, Gagnevin L. New media for the semi-selective isolation and enumeration of Xanthomonas campestris pv. mangiferaeindicae, the causal agent of mango bacterial black spot. J Appl Microbiol. 2005;99(4):803–815. doi: 10.1111/j.1365-2672.2005.02681.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Available as Supplementary Material. R scripts available upon request.