

In this study, Yang et al. investigated how lagging-strand telomeres lacking TRF1 or BLM form fragile telomeres, structures that resemble common fragile sites (CFSs). They found that fragile telomeres in BLM-deficient cells involved double-strand break (DSB) formation by the SLX4/SLX1 nuclease, and the DSBs were repaired by POLD3/POLD4-dependent break-induced replication (BIR), resulting in fragile telomeres containing conservatively replicated DNA.
Keywords: break-induced replication, telomere, BLM, G4, SLX1, SLX4, POLD3, POLD4, fragile telomere, TRF1
Abstract
TRF1 facilitates the replication of telomeric DNA in part by recruiting the BLM helicase, which can resolve G-quadruplexes on the lagging-strand template. Lagging-strand telomeres lacking TRF1 or BLM form fragile telomeres—structures that resemble common fragile sites (CFSs)—but how they are formed is not known. We report that analogous to CFSs, fragile telomeres in BLM-deficient cells involved double-strand break (DSB) formation, in this case by the SLX4/SLX1 nuclease. The DSBs were repaired by POLD3/POLD4-dependent break-induced replication (BIR), resulting in fragile telomeres containing conservatively replicated DNA. BIR also promoted fragile telomere formation in cells with FokI-induced telomeric DSBs and in alternative lengthening of telomeres (ALT) cells, which have spontaneous telomeric damage. BIR of telomeric DSBs competed with PARP1-, LIG3-, and XPF-dependent alternative nonhomologous end joining (alt-NHEJ), which did not generate fragile telomeres. Collectively, these findings indicate that fragile telomeres can arise from BIR-mediated repair of telomeric DSBs.
Mammalian telomeres are composed of long arrays of duplex TTAGGG repeats that end in a 3′ overhang. Telomeric DNA is bound by the shelterin complex, which protects the chromosome ends from DNA damage response and double-strand break (DSB) repair pathways (de Lange 2018). In addition, shelterin promotes the semiconservative replication of telomeric DNA. Deletion of the TRF1 subunit of shelterin causes fork stalling in the telomeric DNA, as shown by DNA combing experiments, and results in S-phase-dependent activation of ATR signaling (Sfeir et al. 2009). A prominent phenotype of TRF1 loss is the formation of fragile telomeres, which are detected in metaphase as multiple and/or highly extended telomeric fluorescence in situ hybridization (FISH) signals at individual chromatid ends (Martinez et al. 2009; Sfeir et al. 2009). Since fragile telomeres also occur when cells are treated with the DNA polymerase inhibitor aphidicolin, they are thought to reflect telomeric replication stress (Sfeir et al. 2009). In addition to TRF1, several other factors (e.g., the BLM and RTEL1 helicases and AKTIP) are implicated in repressing fragile telomere formation (Sfeir et al. 2009; Vannier et al. 2012; Zimmermann et al. 2014; Burla et al. 2015).
TRF1 facilitates telomere replication, at least in part, by recruiting BLM (Sfeir et al. 2009; Zimmermann et al. 2014). BLM localizes to telomeres in late S/G2 and its role in promoting telomere replication requires its recruitment by TRF1 (Barefield and Karlseder 2012; Zimmermann et al. 2014). Because BLM can resolve G-quadruplex (G4) structures in vitro (Sun et al. 1998; Mohaghegh et al. 2001; Huber et al. 2002), it was proposed that BLM might promote telomere replication by removing these obstacles from the lagging-strand ([TTAGGG]n) telomeric template. Consistent with this hypothesis, BLM depletion induces a fragile telomere phenotype predominantly at lagging-strand telomeres and increases telomeric G4 signals (Zimmermann et al. 2014). However, unlike loss of TRF1, deletion of BLM does not induce ATR signaling at telomeres, and does not create fragile leading strand telomeres or formation of sister telomere associations, indicating that other aspects of TRF1 contribute to its replication function (Martinez et al. 2009; Sfeir et al. 2009; Zimmermann et al. 2014). It remains to be determined how persistent G4 structures affect lagging-strand telomeric DNA replication and exacerbate telomere fragility.
The term “fragile telomere” was first coined because the interrupted telomeric FISH signals resemble common fragile sites (CFSs), which appear as gaps or breaks in metaphase chromosomes of aphidicolin-treated cells (Durkin and Glover 2007; Sfeir et al. 2009). CFS expression is thought to involve cleavage of stalled replication forks by MUS81/EME1, followed by mitotic DNA synthesis (MiDAS) (Minocherhomji et al. 2015). MiDAS is a form of break-induced replication (BIR), which is a homology-directed repair pathway that can heal one-ended DNA breaks using a homologous template. BIR requires the POLD3 and POLD4 subunits of Polδ and can be mediated by either RAD51 or RAD52 (Anand et al. 2013). Both MiDAS and other forms of BIR have been documented at telomeres (Dilley et al. 2016; Roumelioti et al. 2016; Min et al. 2017; Özer et al. 2018; Lu et al. 2019), but the role of BIR in fragile telomere formation remains to be determined.
Here, we aimed to understand the molecular mechanism underlying fragile telomere formation in cells lacking BLM. We found that fragile telomeres in BLM-deficient cells appeared to be independent of MUS81/EME1 and did not involve factors involved in replication fork reversal. Instead, the induction of fragile telomeres required the structure-specific endonuclease SLX1 and its associated SLX4 scaffold, which generated transient DSBs at telomeres. Importantly, fragile telomere formation involved the Polδ subunits POLD3 and POLD4—two key components of the BIR machinery—and both strands of the fragile telomeres were newly replicated, consistent with the conservative replication mode of BIR. Cells with induced telomeric DSBs showed BIR-dependent fragile telomeres, as did ALT cells, which have spontaneous telomeric damage. BIR-dependent repair of telomeric DSBs was in competition with alternative nonhomologous end joining (alt-NHEJ), resulting in fewer fragile telomeres when alt-NHEJ was active. Together, the data implicate both DSB formation and BIR in fragile telomere formation.
Results
Depletion of fork remodelers does not affect fragile telomeres in BLM-deficient cells
We previously speculated that persistent G4 structures could induce fork stalling in telomeres lacking BLM (Zimmermann et al. 2014). Since reversal of stalled forks at telomeres has been implicated in the fragile telomere phenotype of RTEL1-deficient cells (Margalef et al. 2018), we tested whether the fragile telomere phenotype in BLM-deficient cells involves fork reversal. We used SV40-immortalized conditional BlmF/F mouse embryonic fibroblasts (MEFs), which showed the expected loss of BLM protein and formation of fragile telomeres at 96 h after infection with Hit&Run Cre retrovirus (Fig. 1A,B). In this setting, short-hairpin RNAs (shRNAs) were used to deplete ZRANB3, SMARCAL1, or HLTF, each of which is required for fork remodeling (Bansbach et al. 2009; Ciccia et al. 2009, 2012; Weston et al. 2012; Yuan et al. 2012; Kile et al. 2015; Cortez 2019; Rickman and Smogorzewska 2019). Despite efficient depletion of the fork remodelers (Fig. 1C), the frequency of fragile telomeres was unchanged (Fig. 1D). Although this negative result does not exclude replication fork arrest/reversal in response to persistent G4 structures, it is consistent with the finding that a block in lagging-strand replication does not result in fork arrest in vitro (Taylor and Yeeles 2018). Rather, repriming by Polα/Primase allows forks to progress, creating unreplicated gaps on the lagging-strand product (Taylor and Yeeles 2018). Together, these findings raised the possibility that BLM loss results in telomeres harboring unreplicated ss gaps rather than stalled forks.
Figure 1.

SLX1/SLX4 contribute to fragile telomere formation in Blm-deficient cells. (A) Western blot analysis of BLM in BlmF/F MEFs ± Cre (96 h). γ-Tubulin serves as the loading control. (B) Telomere FISH on metaphase spreads of BlmF/F MEFs ± Cre (96 h) with Cy3-[CCCTAA]3 probes (green) and DAPI staining (red). Fragile telomeres are marked by an asterisk. (C) Knockdown of ZRANB3, SMARCAL1, and HTLF with shRNAs (6 d) in BlmF/F MEFs verified by Western blotting. Cells infected with an shRNA targeting Luciferase (shLuc) were used as the control. γ-Tubulin serves as the loading control and an asterisk marks a nonspecific band detected by the HLTF antibody. (D) Quantification of fragile telomeres detected by FISH (q arms only) in BlmF/F MEFs ± Cre (96 h) with shRNAs targeting Luc, ZRANB3, SMARCAL1, or HTLF as described in C. (E) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs ± Cre (96 h) after CRISPR/Cas9 targeting of Slx4 with three different sgRNAs. Control cells were infected with an sgRNA targeting Luciferase (sgLuc). The relative level of SLX4 mRNA normalized to GAPDH was determined by RT-qPCR and compared with the sgLuc sample (set to 100). (F) Western blot analysis of SLX1 after CRISPR/Cas9 targeting of Slx1 with three different sgRNAs. γ-Tubulin serves as the loading control. (G) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs ± Cre (96 h) after CRISPR/Cas9 targeting of Slx1 with three different sgRNAs as in F. (H) Western blot analysis of the expression of FLAG-SLX4 and various mutants in BlmF/F MEFs with γ-Tubulin as the loading control. (I) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs + Cre (96 h) expressing empty vector (−), sgRNA-resistant WT FLAG-SLX4 or various mutants with CRISPR/Cas9 targeting of Luc or Slx4. (J) PLA foci (red) of TRF1 and γH2AX detected in BlmF/F MEFs ± Cre (96 h). (K) Quantification of PLA foci as in J in BlmF/F MEFs ± Cre (96 h) with CRISPR/Cas9 targeting of Luc, Slx4, or Slx1. Data are means ± SD of four independent experiments of >100 nuclei each. P-values were from paired two-tailed t-tests. (*) P ≤ 0.05. (L) PLA foci (red) of FLAG-TRF1 and 53BP1 detected in BlmF/F MEFs ± Cre (96 h). (M) Quantification of FLAG-TRF1/53BP1 PLA foci in BlmF/F MEFs ± Cre (96 h) with CRISPR/Cas9 targeting of Luc, Slx4, or Slx1. Data are means ± SD of three independent experiments of >100 nuclei each. For the fragile telomere analyses in D, E, G, and I, data are means ± SD from three independent experiments with ∼2000 telomeres analyzed per experiment. All P-values except for the ones in K were derived from unpaired two-tailed t-tests. (***) P ≤ 0.001, (**) P ≤ 0.01, (*) P ≤ 0.05, (n.s.) P > 0.05.
SLX4 and SLX1 promote fragile telomere formation
Because fragile telomeres resemble CFSs, we asked whether their formation involved the MUS81/EME1 nuclease, which is implicated in CFS expression (Minocherhomji et al. 2015). Three independent single guide RNAs (sgRNAs) were used in CRISPR/Cas9 targeting of Mus81 in pools of BlmF/F MEFs (Supplemental Fig. S1A,B). Despite substantial reduction of MUS81 protein levels (Supplemental Fig. S1A), indicating successful CRISPR/Cas9 targeting in a majority of the cells, depletion of MUS81 had no discernible effect on the fragile telomere phenotype induced by Blm deletion (Supplemental Fig. S1B).
In contrast, three independent sgRNAs targeting Slx4 significantly diminished the fragile telomere phenotype associated with Blm deletion, even though the targeting was inefficient with as much as 30% of SLX4 mRNA remaining (Fig. 1E). The phenotype was rescued by the expression of an sgRNA resistant version of Slx4 (see Fig. 1H,I, discussed below). SLX4 is a scaffold protein that interacts with three nucleases: MUS81/EME1, XPF/ERCC1, and SLX1 (Wyatt and West 2014). As MUS81 did not appear to be a major contributor to the fragile telomere phenotype, we tested XPF and SLX1. Unexpectedly, each of three independent sgRNAs to Xpf increased the frequency of fragile telomeres in BLM-deficient cells but not in BLM-proficient cells (Supplemental Fig. S1C). How XPF depletion exacerbates the fragile telomere phenotype in BLM-deficient cells is addressed below (see Fig. 6, below). In contrast to XPF and MUS81, CRISPR/Cas9 targeting of Slx1 with three different sgRNAs significantly decreased the frequency of fragile telomeres in BLM-deficient cells (Fig. 1F,G) and this phenotype was rescued by an sgRNA resistant version of Slx1 (Supplemental Fig. S1D,E). SLX4 depletion also reduced the expression level of SLX1 (Fig. 1H) and depletion of both SLX1 and SLX4 did not further lower the frequency of telomere fragility, suggesting that SLX1 functions with SLX4 (Supplemental Fig. S1F,G). Consistent with the involvement of SLX1 and SLX4 in the formation of fragile telomeres, a version of SLX4 that does not interact with SLX1 (SLX4ΔSLX1) (Kim et al. 2013) did not rescue targeting of the endogenous SLX4 (Fig. 1H,I). The finding that SLX4ΔSLX1 does not restore SLX1 levels in SLX4-depleted cells is consistent with the loss of the SLX1 interaction (Fig. 1H). In contrast to SLX4ΔSLX1, an SLX4 mutant that lacked the ability to bind MUS81 (SLX4ΔMUS81 [Kim et al. 2013])—expressed at the same level as SLX4ΔSLX1—promoted fragile telomere formation to the same level as wild-type SLX4 (Fig. 1H,I). These data indicate that SLX4-bound SLX1 promotes fragile telomere formation.
Figure 6.

Alt-NHEJ competes with BIR at telomeric DSBs. (A) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs ± Cre (96 h) treated with or without 2 μM olaparib for 20 h. All P-values were derived from two-tailed paired t-test. (*) P ≤ 0.05. (B) Western blot analysis of Ligase 3 in BlmF/F MEFs with γTubulin as the loading control. (C) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs ± Cre (96 h) with CRISPR/Cas9 targeting of Luc or Xpf and/or shRNAs targeting Luc or LIG3. (D) Quantification of leading- and lagging-end q arm fragile telomeres detected by CO-FISH in BlmF/F MEFs ± Cre (96 h) treated with or without 2 μM olaparib for 20 h. (E) Quantification of leading- and lagging-end q arm fragile telomeres detected by CO-FISH in BlmF/F MEFs ± Cre (96 h) with CRISPR/Cas9 targeting of Luc or Xpf and/or shRNAs targeting Luc or LIG3. For all fragile telomere analyses in this figure, the data are means ± SD of three independent experiments of ∼2000 telomeres analyzed per experiment. All P-values in this figure except for panel A were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (**) P ≤ 0.01, (*) P ≤ 0.05. (F) Model showing how alt-NHEJ and BIR act on telomeric DSBs in Blm-deficient cells and the effect of alt-NHEJ on the detection of fragile telomeres.
The involvement of SLX4/SLX1 in the formation of fragile telomeres documented above predicts that DSBs are formed in telomeres that are replicating in the absence of BLM. We therefore monitored the appearance of γH2AX at telomeres using TRF1 and γH2AX antibodies in a proximity ligation assay (PLA). Initial control experiments indicated that the formation of PLA foci was dependent on the presence of both antibodies and increased upon deletion of BLM (Supplemental Fig. S1H). In four independent experiments (Fig. 1J,K), deletion of Blm led to an increase of the PLA foci and these telomere damage foci were diminished by depletion of SLX4 and SLX1, consistent with SLX4/SLX1-mediated DSB formation. Similarly, a PLA assay for colocalization of 53BP1 with FLAG-tagged TRF1 showed the presence of DNA damage foci at telomeres in Blm-deficient cells and the formation of these foci was again dependent on SLX4/SLX1 (Fig. 1L,M). These data suggest that SLX4/SLX1 create DSBs at telomeres lacking BLM.
POLD3 and POLD4 promote fragile telomere formation in Blm-deficient cells
Since BIR underlies CFS expression and since the data on SLX4/SLX1 suggested formation of DSBs that can be repaired by BIR, we tested whether the fragile telomere phenotype of Blm-deficient cells is promoted by POLD3 and POLD4, the two Polδ subunits required for BIR (Anand et al. 2013). Knockdown of POLD3 in BlmF/F MEFs reduced the POLD3 protein level (Fig. 2A) and diminished the occurrence of fragile telomeres after Blm deletion (Fig. 2B). The effect of the POLD3 shRNA was repressed by an shRNA-resistant form of wild-type POLD3 but not by a BIR-deficient version of POLD3 lacking the PCNA interacting motif (ΔPIP) (Fig. 2C,D; Ducoux et al. 2001; Dilley et al. 2016). Similar results with two independent shRNAs to POLD4 further implicated BIR in the fragile telomere phenotype associated with BLM deficiency (Fig. 2B).
Figure 2.
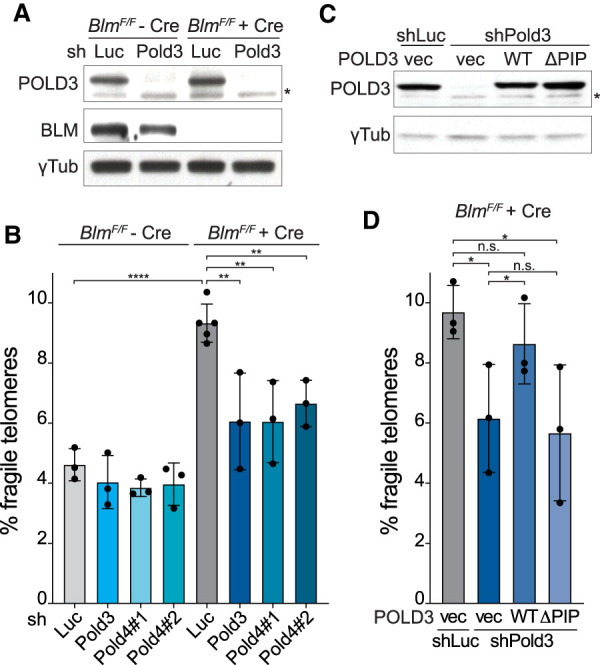
POLD3 and POLD4 promote fragile telomere formation in Blm-deficient cells. (A) Western blot to monitor POLD3 knockdown with shRNA (96 h) and Blm deletion with Cre (96 h) in BlmF/F MEFs. γ-Tubulin serves as the loading control, and an asterisk marks a nonspecific band detected by the POLD3 antibody. (B) Quantification of q arm fragile telomeres detected by FISH in BlmF/F MEFs ± Cre (96 h) after the knockdown of Luc or POLD3 as in A or POLD4 with two different shRNAs. The relative POLD4 mRNA levels determined by RT-qPCR, normalized to GAPDH and compared with the shLuc sample (set at 100), were 42 for sh#1 and 15 for sh#2. Data are means ± SD of three or more independent experiments of ∼2000 telomeres analyzed per experiment. All P-values were derived from two-tailed unpaired t-test. (****) P ≤ 0.0001, (**) P ≤ 0.01. (C) Western blot analysis of BlmF/F MEFs expressing empty vector, shRNA-resistant WT, or ΔPIP POLD3. All cells were treated with Cre and shLuc or shPold3 as in A. γ-Tubulin serves as the loading control. An asterisk indicates a nonspecific band detected by the POLD3 antibody. (D) Quantification of q arm fragile telomeres detected by FISH in cells as described in C. Data are means ± SD of three independent experiments of ∼2000 telomeres analyzed per experiment. All P-values were derived from two-tailed paired t-test. (*) P ≤ 0.05, (n.s.) P > 0.05.
We next queried the involvement of RAD52, which contributes to CFS expression (Bhowmick et al. 2016) and to the BIR-like telomere maintenance mechanism in ALT cells (Min et al. 2017; Verma et al. 2019; Zhang et al. 2019). CRISPR/Cas9 targeting of Rad52 with two different sgRNAs did not result in a significant change in fragile telomere frequency in BLM-deficient cells (Supplemental Fig. S2A), suggesting that the BIR pathway is not solely dependent on RAD52. Similarly, in ALT cells, some of the telomeric BIR is not dependent on RAD52 (Verma et al. 2019; Zhang et al. 2019).
Since it is possible that BIR engages only one end of the DSB induced by SLX4/SLX1, we considered the fate of the other end. There was no evidence for homologous recombination at telomeres lacking BLM, since no telomere sister chromatid exchanges (T-SCEs) were induced by deletion of Blm and/or depletion of POLD3 (Supplemental Fig. S2B,C). We also did not detect shortened telomeric DNA fragments in Blm-deficient cells or changes in telomere length that depended on the presence of SLX4/SLX1 (Supplemental Fig. S2D). The low frequency of the SLX4/SLX1 cleavage event likely prevents detection of the fate of the released telomeric DNA fragment. Furthermore, as shown below (Fig. 6, below), the cleaved telomeres can be repaired by alt-NHEJ, preventing detection of cleavage products.
CO-FISH confirms BIR-mediated fragile telomere formation
To corroborate the role of BIR in the formation of fragile telomeres, we determined whether fragile telomeres are formed by conservative DNA replication. As BIR involves synthesis of both DNA strands, fragile telomeres that are BIR products should not be detectable in chromosome orientation FISH (CO-FISH) experiments (Fig. 3A; Roumelioti et al. 2016). In CO-FISH, newly synthesized DNA strands labeled with BrdU and BrdC are removed upon Hoechst/UV nicking and exonuclease III treatment so that only the remaining parental DNA strands are detected (Fig. 3B). To test whether telomeres with both strands containing BrdU and BrdC escape detection in CO-FISH as predicted, we incubated wild-type MEFs with BrdU and BrdC for sufficient time to allow passage through one or two rounds of DNA replication (16 or 26 h, respectively) before performing CO-FISH (Fig. 3B). Analogous to the classic Meselson and Stahl experiment (Meselson and Stahl 1958), one round of labeling allowed detection of two telomeres at each chromosome end in CO-FISH, whereas cells labeled for two rounds of DNA replication showed only one telomeric signal per chromosome end (Fig. 3B), implying that telomeres with both strands containing BrdU and BrdC were not detectable. Applying this approach, we compared the frequency of fragile telomeres in Blm-deficient cells detected by FISH versus CO-FISH. Consistent with BIR, the frequency of fragile telomeres detected with CO-FISH was significantly diminished compared with FISH (Fig. 3C,D), implying absence of the parental DNA strand and synthesis of both strands of the fragile telomeres. As expected, the few fragile telomeres that remained detectable by CO-FISH preferentially involved the lagging-strand telomeres (Fig. 3E) and were formed in a manner that depended on SLX4/SLX1 and POLD3 (Fig. 3F,G). As shown below (Fig. 6, below), these fragile telomeres are only detected in CO-FISH because they contain a segment of telomeric DNA replicated by semiconservative DNA replication that has been fused to the BIR product by alt-NHEJ.
Figure 3.

Fragile telomeres of Blm-deficient cells arise from conservative replication. (A) Model for BIR-mediated fragile telomere formation and their removal after CO-FISH. (B) Schematic and images of CO-FISH on cells cultured in the presence of BrdU and BrdC for 16 or 26 h. The substituted DNA strands are removed by treatment with Hoechst 33258, UV, and exonuclease III. Telomeres replicated by leading-strand DNA synthesis were hybridized [TTAGGG]3 (green) and lagging-strand telomeres with [CCCTAA]3 (red). Cells labeled for 16 h (one S phase) show two signals per chromosome end, whereas cells labeled for 26 h (two S phases) show one signal per chromosome end, indicating that telomeres lacking a parental strand are poorly detected by CO-FISH. (C) Comparison of telomere FISH and CO-FISH performed on parallel metaphase spreads of BlmF/F MEFs + Cre (96 h). FISH was performed with [CCCTAA]3 (green), CO-FISH was done with [TTAGGG]3 (green) and [CCCTAA]3 (red), and DNA was stained with DAPI (blue). Fragile telomeres are marked by an asterisk. (D) Quantification of q arm fragile telomeres in BlmF/F ± Cre cells (96 h) detected by FISH and CO-FISH on the same samples derived from BrdU/BrdC-labeled cells. (E) Quantification of leading- and lagging-end q arm telomeres using CO-FISH as in D. Note that a sample with 6% lagging fragile telomeres and 2% leading fragile telomeres will show an average of 4% fragile telomeres when both sisters are scored (as is the case in D). (F) Quantification of leading- and lagging-end q arm fragile telomeres using CO-FISH in BlmF/F MEFs ± Cre (96 h) with CRISPR/Cas9 targeting of Luc, Slx4, or Slx1. (G) Quantification of leading- and lagging-end q arm fragile telomeres using CO-FISH in BlmF/F MEFs ± Cre (96 h) with shRNAs targeting Luc or Pold3. For all fragile telomere analyses in this figure, data are means ± SD of three independent experiments of ∼2000 telomeres analyzed per experiment. All P-values in this figure were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (**) P ≤ 0.01, (n.s.) P > 0.05.
Fragile telomere formation in Blm-deficient cells is likely to take place in S phase
BIR at CFSs takes place in mitosis, as demonstrated by incorporation of EdU at CSFs after nuclear envelope breakdown (Minocherhomji et al. 2015). To test whether BIR at telomeres lacking BLM similarly takes place in mitosis, cells were labeled with EdU for 2 h before metaphase cells harvested, thereby allowing detection of EdU incorporation in metaphase spreads from cells that had already completed S phase at the time of EdU addition. MEFs treated with aphidicolin showed interstitial EdU incorporation in ∼40% of the metaphase chromosomes (Supplemental Fig. S3A), consistent with the previously reported MiDAS at CFSs (Minocherhomji et al. 2015). In contrast, no EdU incorporation was detected at fragile telomeres in BLM-deficient cells (Supplemental Fig. S3A). Although the lack of EdU labeling of fragile telomeres could be explained if the BIR tracts are shorter at fragile telomeres than at CFSs, it is possible that the telomeric BIR takes place before entry into mitosis. In agreement with the fragile telomeres being formed in S phase, telomere dysfunction-induced foci (TIFs) (Takai et al. 2003) formed after Blm deletion were only observed in S phase (as detected based on pan-nuclear EdU incorporation) (Supplemental Fig S3B,C). Together these data suggest that the formation of DSBs at telomeres lacking BLM and the repair of these DSBs by BIR most likely occurs in S phase rather than in mitosis.
BIR promotes fragile telomere formation upon FokI-induced telomeric DSBs
To further interrogate the role of DSBs and BIR in the formation of fragile telomeres, telomeric DSBs were induced by targeting the FokI endonuclease to telomeres through fusion to TRF1 (Myc-FokI-ERT2-TRF1) (Tang et al. 2013; Cho et al. 2014; Doksani and de Lange 2016). Previous work showed that this FokI-TRF1 fusion does not interfere with the function of the endogenous TRF1 and does not induce a fragile telomere phenotype if the fused FokI is not catalytically active (Doksani and de Lange 2016). Consistent with prior data, TRF1 fused to the wild-type (WT) or nuclease-dead (DA) FokI localized to telomeres (Fig. 4A,B). As expected, induction of WT but not the DA version of FokI resulted in TIFs, visualized based on colocalization of 53BP1 IF signals with telomeres (Fig. 4B). As previously reported (Doksani and de Lange 2016), induction of telomeric DSBs with WT FokI-ERT2-TRF1 resulted in fragile telomeres (Fig. 4C). Consistent with BIR, depletion of POLD3 ameliorated the fragile telomere phenotype while not affecting the induction of DSBs by FokI (Fig. 4C–E). Furthermore, the fragile telomeres formed upon FokI induction largely escaped detection in CO-FISH, indicating a conservative mode of replication (Fig. 4F). As was the case for telomeres lacking BLM, the residual fragile telomeres that escaped degradation during CO-FISH were diminished upon Pold3 knockdown (Fig. 4G), again indicating the involvement of BIR in the formation of fragile telomeres in cells with telomeric DSBs. The origin of the fragile telomeres that resist degradation in CO-FISH is addressed below.
Figure 4.

BIR mediates fragile telomeres formation in cells with telomeric DSBs. (A) Western blot analysis of Myc-FokI-ERT2-TRF1 expression in wild-type MEFs infected with empty vector, nuclease-dead (DA), or wild-type (WT) Myc-FokI-ERT2-TRF1. γ-Tubulin serves as the loading control. (B) Immunofluorescence (IF)-FISH analysis of MEFs expressing DA or WT Myc-FokI-ERT2-TRF1. Telomeres were detected by FISH with an Alexa 488-[TTAGGG]3 probe. The FokI-ERT2-TRF1 alleles were detected with anti-Myc antibodies and Alexa 647 secondary antibodies and 53BP1 with anti-53BP1 antibodies and Alexa 555 secondary antibodies. DNA was stained with DAPI. Images were false colored for presentation purposes. (C) Quantification of q arm fragile telomeres detected by FISH in cells expressing DA or WT Myc-FokI-ERT2-TRF1 after shRNA knockdown of Luc or POLD3 (96 h). (D) Western blot to monitor POLD3 knockdown with shRNA (96 h) in cells expressing DA or WT Myc-FokI-ERT2-TRF1. γ-Tubulin serves as the loading control. (E) Quantification of the TIF response in cells expressing DA or WT Myc-FokI-ERT2-TRF1 after shRNA knockdown of POLD3. Cells were induced with 4-OHT for 24 h before fixation. Data are means ± SD of three independent experiments of >50 nuclei each. (F) Quantification of q arm fragile telomere frequency in cells expressing DA or WT Myc-FokI-ERT2-TRF1 by FISH and CO-FISH. Cells were treated with 4-OHT for 24 h before harvesting for metaphase analysis. (G) Quantification of q arm fragile telomeres detected by CO-FISH in cells expressing DA or WT Myc-FokI-ERT2-TRF1 after shRNA knockdown of Luc or POLD3 (96 h). For all fragile telomere analyses, data are means ± SD of three independent experiments of ∼2000 telomeres analyzed per experiment. All P-values in this figure were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (**) P ≤ 0.01, (*) P ≤ 0.05, (n.s.) P > 0.05.
BIR contributes to the fragile telomere phenotype eof ALT cells
BIR has been implicated in the telomere maintenance in ALT cells and it is thought that DSBs in the telomeric DNA are frequent in these cells (Dilley et al. 2016; Roumelioti et al. 2016; Min et al. 2017). Fragile telomeres have been reported in several ALT cell lines (Arora et al. 2014; Min et al. 2017). Consistent with this report, five ALT cell lines showed a high frequency of fragile telomeres (Fig. 5A). Because ALT lines have very long telomeres, their fragile telomere phenotypes were compared with three cell lines with very long telomeres that do not use ALT for telomere maintenance (Fig. 5A). In addition, we included the JFCF6/T.1F cell line in the comparison, which is similar to the precursor of two of the queried ALT lines (JFCF6/T.1M and JFCF6/T.1R) (Fig. 5A).
Figure 5.

BIR contributes to fragile telomere formation in ALT cells. (A) Quantification of the frequency of fragile telomeres by FISH in non-ALT cell lines and a panel of ALT cell lines. Asterisks indicate P-values from pairwise comparisons between HeLa1.3 and the ALT cell lines. The HeLaST and HT1080ST (super telomerase) (Cristofari and Lingner 2006) cells with very long telomeres were treated with 10 μM BIBR1532 to inhibit telomerase for better comparison with telomerase-negative ALT cells. (B) Western blot analysis of CRISPR/Cas9 targeting of POLD3 with two different sgRNAs (5 d) in JFCF-6/T.1R cells. (C) Quantification of fragile telomeres detected by FISH in JFCF-6/T.1R cells after POLD3 targeting in bulk by CRISPR/Cas9 as in B. (D) Western blot analysis of CRISPR/Cas9 targeting of POLD3 with two different sgRNAs (5 d) in U2OS cells. (E) Quantification of fragile telomeres detected by FISH in U2OS cells after POLD3 targeting in bulk by CRISPR/Cas9 as in D. (F) Western blot analysis of CRISPR/Cas9 targeting of POLD3 with two different sgRNAs (5 d) in WI38.VA12-2RA cells. (G) Quantification of fragile telomeres detected by FISH in WI38.VA12-2RA cells after POLD3 targeting in bulk by CRISPR/Cas9 as in F. (H) Quantification of fragile telomeres in two ALT cell lines detected by FISH and CO-FISH. (I) Western blot analysis of CRISPR/Cas9 targeting of RAD51AP1 (5 d) in U2OS cells. An asterisk marks a nonspecific band detected by the RAD51AP1 antibody. (J) Quantification of fragile telomeres detected by FISH in U2OS cells after RAD51AP1 targeting in bulk by CRISPR/Cas9 as in I. For all fragile telomere analyses, data are means ± SD of three independent experiments of ∼2000 telomeres analyzed per experiment. All P-values in this figure were derived from two-tailed unpaired t-test. (****) P ≤ 0.0001, (***) P ≤ 0.001, (**) P ≤ 0.01, (*) P ≤ 0.05, (n.s.) P > 0.05.
In support of a role for BIR in the ALT fragile telomere phenotype, CRISPR/Cas9 targeting of POLD3 using two different sgRNAs significantly reduced the frequency of fragile telomeres in three ALT cell lines (Fig. 5B–G). Furthermore, the detection of fragile telomeres in ALT cell lines was significantly reduced in CO-FISH versus FISH (Fig. 5H). Finally, targeting of RAD51AP1 in U2OS cells with CRISPR/Cas9 significantly decreased telomere fragility (Fig. 5I,J). RAD51AP1 has recently been shown to be a key player in the ALT pathway, contributing to BIR-like and RAD52/RAD51-dependent telomere maintenance mechanisms in U2OS cells (Barroso-González et al. 2019). We did not detect an effect of RAD52 depletion on the fragile telomere phenotype in ALT cells (Supplemental Fig. S4A,B), perhaps due to redundancy with RAD51. Similarly, prior work showed that RAD52 deletion does not affect the BIR process following the induction of telomeric breaks by FokI-TRF1 in ALT cells (Verma et al. 2019). Collectively, the results point to a role of BIR in fragile telomeres formation in ALT cells.
PARP1-, LIG3-, and XPF-dependent alt-NHEJ competes with BIR
Since DSBs within the telomeric repeat array are often repaired by alt-NHEJ (Doksani and de Lange 2016), we determined whether this repair pathway competes with BIR and affects the formation of fragile telomeres. Treatment with the PARP inhibitor (PARPi) olaparib significantly increased the frequency of fragile telomeres induced by Blm deletion but did not affect the level of fragile telomeres in Blm-proficient cells (Fig. 6A). Similarly, depletion of ligase 3 (LIG3) resulted in an increase of the fragile telomere phenotype in Blm-deficient cells (Fig. 6B,C). Finally, depletion of XPF, which has been implicated in DNA flap cleavage during alt-NHEJ (Ma et al. 2003; Bennardo et al. 2008), promoted the fragile telomere phenotype in Blm-deficient cells (Fig. 6C; Supplemental Fig. S1C). XPF appeared to be epistatic with LIG3 (Fig. 6B,C). These results indicate that alt-NHEJ can compete with BIR in the repair of telomeric DSBs.
Interestingly, when the telomeres were examined using CO-FISH, inhibition of alt-NHEJ with PARPi or by depleting LIG3 or XPF reduced the frequency of lagging-strand fragile telomeres in Blm-deficient cells to background levels (Fig. 6D,E). This result indicates that CO-FISH detection of fragile lagging-strand telomeres in Blm-deficient cells relies on an alt-NHEJ dependent process. The same phenomenon was observed in cells with FokI-induced telomeric DSBs (Supplemental Fig. S5A–C). When alt-NHEJ was inhibited, the frequency of fragile telomeres detected by FISH increased (Supplemental Fig. S5A,B). However, when fragile telomeres were monitored using CO-FISH, inhibition of alt-NHEJ led to a reduction in the fragile telomeres that remained (Supplemental Fig. S5C).
The simplest explanation of these data is that the fragile telomeres in Blm-deficient cells that are detected by CO-FISH represent BIR products that are fused (by alt-NHEJ) to the distal segment of the cleaved telomere (Fig. 6F).
Discussion
These data indicate that BIR can contribute to the fragile telomere phenotype of cells with telomeric DSBs, including in Blm-deficient cells where SLX4/SLX1 cleavage of replication intermediates creates a break, in cells with FokI-induced telomeric DSBs, and in ALT cells, which harbor spontaneous DNA damage. The involvement of BIR in fragile telomere formation was evident based on its dependence on DSB formation, the POLD3 and POLD4 subunits of Polδ, and the conservative mechanism of DNA replication. Interestingly, this BIR-dependent fragile telomere formation differed from the BIR involved in generating the CFSs with regard to the nucleases involved and its timing. Whereas CFS-associated BIR occurs during mitosis and relies on MUS81/EME1, BIR at fragile telomeres occurs before mitosis and depends on SLX4/SLX1. Consistent with fragile telomeres arising from BIR acting on induced or constitutive telomeric DSBs, this process was in competition with alt-NHEJ. Although these data show that BIR-dependent repair of DSBs is one of the pathways that generate fragile telomeres, additional processes that create this telomere anomaly remain to be deciphered.
The role of SLX4/SLX1 in telomere processing in Blm-deficient cells
It has long been anticipated that G4 formation in the lagging-strand telomeric template could pose a problem. We considered the possibility that lagging-strand replication problems may lead to fork stalling and generate reversed forks that could be processed by SLX4/SLX1 into the DSB used by BIR. However, cleavage of reversed fork seems an unlikely source of DSBs in Blm-deficient cells, since the fragile telomere phenotype was unaffected by depletion of fork reversal factors, such as ZRANB3, SMARCAL1, and HTLF. Second, cleavage of reversed forks is unlikely to be orientation-specific, and therefore fails to explain the lagging-strand specificity of fragile telomere formation in Blm-deficient cells. Third, in vitro data argue that a block in lagging-strand DNA synthesis does not result in fork stalling (Taylor and Yeeles 2018). Therefore, we propose that in Blm-deficient cells, the lagging-end telomeres contain unreplicated gaps, most likely at sites where G4 structures have persisted, and that the ssDNA in these gaps is cleaved by SLX4/SLX1 to form the DSBs used by BIR (Fig. 6F). Importantly, this model accounts for the lagging-strand prevalence of fragile telomere formation in Blm-deficient cells.
The role of alt-NHEJ in repairing telomeric DSBs
It is known that telomeric DSBs are often repaired by alt-NHEJ (Doksani and de Lange 2016). The data indicate that this repair pathway is in competition with BIR. When DSBs are created by FokI or when they result from SLX1/SLX4 cleavage of telomeres replicated in the absence of BLM, alt-NHEJ diminishes the formation of fragile telomeres by BIR. In addition, the data show that alt-NHEJ can act on the DNA end after it has been extended by BIR, leading to ligation of the distal segment of the cleaved telomere (Fig. 6F). During BIR, the DNA end is thought to be frequently released from the template (Smith et al. 2007). Our results suggest that whenever the BIR product is disengaged from the template, alt-NHEJ might be able to terminate BIR by ligating the end to the distal telomeric fragment. The distal end of the cleaved telomere is expected to remain in proximity to the BIR product since it is associated with the sister telomere through cohesion.
Telomeric BIR and ALT
ALT cells have the ability to use BIR to extend their telomeres (Dilley et al. 2016; Roumelioti et al. 2016). Our results with non-ALT cells, including Blm-deficient cells and cells with FokI-induced telomeric DSBs, suggest that telomeric BIR is not unique to ALT. Telomeric BIR in non-ALT cells can explain how telomerase-positive cells can copy an inserted molecular marker from one telomere to another (Neumann et al. 2013). In non-ALT cells, the telomeric damage needed to initiate BIR may be infrequent, in part explaining why spreading of a marker from one telomere to others is rare. Given that BIR is active at telomeres in non-ALT cells with telomeric DSBs, the unique aspect of ALT cells may be their propensity to form DSBs in the telomeric DNA (Cesare et al. 2009). In addition, ALT cells and other cells that lack the ATRX chromatin remodeler show prominent loss of telomere cohesion and disengagement of the distal telomeric fragment generated by a DSB (Lovejoy et al. 2020). The high level of BIR-dependent processes in ALT, including the formation of fragile telomeres may therefore also be due the loss of the distal telomeric fragment that could have been used by alt-NHEJ to prevent or terminate BIR.
Materials and methods
Cell culture, cloning, and viral infections
SV40-LT-immortalized BlmF/F MEFs were described previously (Chester et al. 2006; Wu et al. 2012). MEFs were cultured in DMEM (Cellgro) supplemented with 100 U/mL penicillin (Gibco), 0.1 mg/mL streptomycin (Gibco), 0.2 mM L-glutamine (Gibco), 0.1 mM nonessential amino acids (Gibco), and 10% bovine calf serum (HyClone). ALT and non-ALT human cell lines were described previously (Cristofari and Lingner 2006; Takai et al. 2010; Lovejoy et al. 2012) and cultured with 10% fetal bovine serum (Gibco) with the same supplements. Cre recombinase was introduced by two retroviral infections with Hit&Run Cre in pMMP at 12-h intervals and cells were harvested 96 h after the second infection in all experiments. Mouse Pold3 cDNA was cloned by RT-PCR into pLPC-N-Myc, and shRNA-resistant (TATACAAAAAGCTATGCTAAA in place of the shRNA target site CATCCAGAAAGCTATGCTAAA) WT and ΔPIP mutant (deletion of amino acids 451–461) were generated by site-directed mutagenesis using PCR. Mouse Slx4 and Slx1 cDNAs were purchased from Origene and cloned into pWZL-FLAG-Hygro vector. sgRNA-resistant Slx4 (ATCTGAAACAATGCGCCGTC in place of sgRNA#2 ACTTGAAGCAGTGTGCGGTG) and Slx1 (CGGAAAAAGGGAGGTGCCTGG in place of sgRNA#1 GCAAGAAAGGTGGAGCATGG) were generated by site-directed mutagenesis using PCR. The following mutants of Slx4 were made by site-directed mutagenesis using PCR: ΔXPF, Δ233–380; ΔMUS81, Δ1314–1397; and ΔSLX1, Δ1407–1565 (Kim et al. 2013). Myc-FokI-ERT2-TRF1 constructs were described previously (Doksani and de Lange 2016). MEFs infected with retroviral vectors were selected with 2.5 μg/mL puromycin or 135 μg/mL hygromycin for 3 d. Cells expressing Myc-FokI-ERT2-TRF1 were treated with 0.5 μM 4-hydroxytamoxifen (Sigma-Aldrich) for 24 h before harvesting. The following shRNAs were obtained from Sigma-Aldrich: shZranb3 (TRCN0000239044), shSmarcal1 (TRCN0000317573), shHltf (TRCN0000349351), shPold3 (TRCN0000279480), shPold4#1 (TRCN0000111341), shPold4#2 (TRCN0000111344), and shLig3 (TRCN0000070978). These shRNAs and a control shLuc (CGCTGAGTACTTCGAAATGTC) were cloned into the pLKO.1 vector. sgRNAs were designed using the designer from the Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) and top hits were picked. The following sgRNAs were cloned into the pLentiCRISPRv2 vector (Addgene 52961): sgLuc (ACAACTTTACCGACCGCGCC), sgSlx4 (#1: CTCACACCATGACAGCCTGG; #2: ACTTGAAGCAGTGTGCGGTG; #3: TGTGCGGTGAGGATGGAAGT), sgSlx1 (#1: GCAAGAAAGGTGGAGCATGG; #2: CGGGTCCGGCAACACAACGC; #3: TCGGGTCCGGCAACACAACG), sgMus81 (#1: TACCCGCTACCATTGCGCAG; #2: AAGCAGCACCTAGCATCAGG; #3: CCACAGCTTCCTAACCAAAG), sgXpf (#1: GAAATGGTAGAGGAGCCGGT; #2: TATGAAGTCTACACGCAGGG; #3: ACAAAGGGTCCAAGTAATGG), and sgRAD52 (#1: GACTTCCCACCAACAAAGGG; #2: GATGGTTGTCACACCCTCTG). MEFs were infected with the lentiviral vectors and selected with 2.5 μg/mL puromycin. For human cells, the following sgRNAs were cloned into the pLentisgRNA vector (Addgene 71409): sgPOLD3 (#1: AAAGCCATGCTAAAGGACAG; #2: CAACAAGGAAACGAAAACAG), sgRAD52 (#1: AGGCCATCCAGAAGGCCCTG; #2: GGGAGTCTGTGCATTTGTGA), and sgRAD51AP1 (#1: GAAATCCAGAACAGCACCAA; #2: TGCACATTAGTGGTGACTGT). Cells were infected, selected in 2 μg/mL puromycin, infected with adenoviral Cas9 (Vector Biolabs 1900), and harvested after 5 d.
Western blotting
Harvested cells were lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS, 50 mM Tris-HCl at pH 8.0, supplemented with protease inhibitor cocktail tablets [Roche] and 0.5 mM PMSF) for 30 min on ice and centrifuged at maximum speed in a microfuge for 20 min at 4°C. Protein concentrations in the supernatant were determined using the Pierce BCA protein assay kit (Thermo Fisher 23227) and 10–20 μg of protein in 20–30 μL of Laemmli buffer (10% glycerol, 60 mM Tris-HCl at pH 6.8, 2% SDS, 0.001% bromophenol blue, 1.25% 2-mercaptoethanol) was resolved on precast 4%–12% SDS–polyacrylamide gels and transferred to nitrocellulose membranes after electrophoresis. The membranes were blocked in 5% milk and incubated with primary antibodies in 1% milk for 2 h, followed by three washes with TBST. The membranes were then incubated with horseradish peroxidase-conjugated sheep antimouse or donkey antirabbit secondary antibodies (GE Healthcare) for 1 h, followed by three washes with TBST, and then developed using the SuperSignal ECL kit (Thermo Fisher 34580) and XAR films (LabScientific) according to the manufacturer's protocol. The following primary antibodies were used: mBLM (Abcam ab2179), mZRANB3 (Abclonal A9555), mSMARCAL1 (Millipore ABE1836), mHLTF (Proteintech 14286-1-AP), mSLX1 (Proteintech 21158-1-AP), mMUS81 (Abclonal A6818), mPOLD3 and hPOLD3 (Proteintech 21935-1-AP), mLIG3 (BD Biosciences 611876), mTRF1 (1449), hRAD52 (Santa Cruz Biotechnology sc-365341), hRAD51AP1 (a gift from R. O'Sullivan [Barroso-González et al. 2019]), and γ-tubulin (Sigma-Aldrich GTU88).
RT-qPCR
Total RNA was extracted with the RNeasy Plus minikit (Qiagen 74134), and cDNA synthesis was performed with the SuperScript IV first strand synthesis kit (Thermo Fisher 18091050) as per the manufacturer's instructions. The following gene-specific primers were used for first strand synthesis: mSlx4 (TGGTGACTCCTTTCCGTTTC), mPold4 (TCTGTCCTGAAGTTCTTGGTG), mXpf (TCCGAAACTCACGCATGTC), mRad52 (CCAAGTGCATTCCCAAAACTC), and mGapdh (ATTGTCATACCAGGAAATGAGCTT). qPCR was done with SYBR Green PCR master mix (Thermo Fisher 4309155). The following primer pairs were used for qPCR: mSlx4 (For: AGGGAGCAACATGTGAACAG; Rev: AGTGACTGATTCGGCTCTTG), mPold4 (For: CATCACTGACTCCTATCCTGTTG; Rev: CCAGGTCAAACTGCCTCAG), mXpf (For: CCCAGATACGTGGTTCTGTATG; Rev: GCAGAGCAGTCAGGTAGC), mRad52 (For: AATACCAGGCCATCCAGAAAG; Rev: CCCAGCCATTGTAACCAAAC), and mGapdh (For: GTGTTCCTACCCCCAATGTGT; Rev: ATTGTCATACCAGGAAATGAGCTT). qPCR was performed on a QuantStudio 12K Flex real-time PCR system (Life Technologies) at The Rockefeller University Genomics Resource Center.
FISH and CO-FISH
Telomere FISH on metaphase spreads was conducted as described previously (Celli and de Lange 2005). Briefly, cells were treated with 0.2 μg/mL colcemid for 1 h before harvesting, suspended in 0.075 M KCl, swollen for 30 min at 37°C, and fixed in 3:1 methanol:glacial acetic acid overnight. Fixed cells were dropped onto glass slides and dried overnight. Hybridization was performed with 20 nM Cy3-[CCCTAA]3 PNA probes (PNA Bio) in 100 μL of hybridization mix (10 mM Tris-HCl at pH 7.2, 70% deionized formamide, 0.5% blocking reagent [Roche 11096176001]) placed onto the slides and heated for 10 min at 80°C. Slides were kept in a dark humidified chamber for 2 h, washed twice with wash buffer I (70% formamide, 10 mM Tris-HCl at pH 7.2) for 15 min each, and then washed three times for 5 min with wash buffer II (100 mM Tris-HCl at pH 7.2, 150 mM NaCl, 0.08% Tween-20) with DAPI added to the second wash. Slides were dehydrated with 70%, 95%, and 100% ethanol before being mounted with ProLong Gold Antifade reagent (Life Technologies P36934). For EdU detection experiments, cells were incubated with 10 μM EdU for 2 h before harvesting with colcemid added during the second hour. Click chemistry was performed after the wash with FISH wash buffer I using the Click-iT EdU Alexa fluor 647 imaging kit (Invitrogen C10340).
The procedure for telomere CO-FISH was described previously (Bailey et al. 2001; Celli et al. 2006). Briefly, cells were incubated with 7.5 μM BrdU and 2.5 μM BrdC for 16 h before harvesting and metaphase spreads were prepared as above. To degrade the newly synthesized strands, slides were rehydrated with PBS, treated with 0.5 mg/mL RNase A for 15 min at 37°C, stained with 0.5 μg/mL Hoechst 33258 in 2× SSC for 15 min at room temperature, and exposed to 365-nm UV light for 5.4 × 103 J/m2, followed by treatment with 100 μL of 10 U/μL exonuclease III (Promega M1811) for 30 min at 37°C. Slides were then washed with PBS and serially dehydrated as above. Cells were hybridized with 2.5 nM Cy3-[TTAGGG]3 (PNA Bio) probes for 2 h at room temperature, rinsed with wash buffer I, and hybridized with 20 nM Alexa 488-[CCCTAA]3 (PNA Bio) probes for 2 h at room temperature. Washing and mounting steps were as described above. Images were obtained on a DeltaVision microscope.
IF-FISH
IF-FISH was conducted as described previously (Takai et al. 2003). Cells were plated onto coverslips 24 h before fixation with 3% paraformaldehyde in PBS for 10 min. Coverslips were then incubated with blocking solution (0.1% BSA, 3% goat serum, 0.1% Triton X-100, 2 mM EDTA in PBS) for 30 min, followed by primary antibody incubation in blocking solution for 1 h. After three washes with PBST, cells were incubated with secondary antibodies in blocking solution for 30 min, followed by three washes with PBST. Cells were fixed again with 3% paraformaldehyde for 5 min before washing twice with PBS and dehydrating with 70%, 95%, and 100% ethanol. The FISH procedure was the same as described above, using the Alexa 488-[TTAGGG]3 (PNA Bio) probes. After FISH, coverslips were washed twice with wash buffer I for 15 min each, and three times with PBST with DAPI added to the second PBST wash. Coverslips were dehydrated again before mounting onto slides. Cells were imaged using the DeltaVision microscope, and images were deconvoluted. TIF analysis was done by automated focus counting as described previously (Doksani and de Lange 2016). The following antibodies were used for IF: Myc (Cell Signaling 9B11), 53BP1 (Abcam ab175933), goat antirabbit A555 (Thermo Fisher A32732), and goat antimouse A647 (Thermo Fisher A21237).
Telomere restriction fragment (TRF) analysis
TRF analysis was conducted as described previously (Celli and de Lange 2005). Briefly, 1 million cells were embedded in 1% agarose plugs and treated with 1 μg/mL proteinase K overnight, followed by extensive wash with TE, after which the plugs were digested with 60 U of AluI and MboI overnight. Plugs were run out in a 1% agarose gel using pulse field gel electrophoresis and the gel was dried. [γ-32P]ATP-labeled TelC probe ([CCCTAA]4) was used for probing telomeric overhang under native condition overnight. After washing and imaging, the gel was denatured, reprobed, and imaged again. Imaging was performed with a GE Typhoon system.
Proximity ligation assay (PLA)
PLA assay was performed using a Duolink PLA kit following the manufacturer's protocol. The following antibodies were used: TRF1 (1449) and γH2AX (Millipore 05636), FLAG (Sigma M2), and 53BP1 (Abcam ab175933).
Statistics and reproducibility
Data sets were analyzed using GraphPad Prism 8 and differences between samples were determined using a two-tailed, unpaired t-test unless stated otherwise. Error bars represent SD unless noted otherwise. Significance levels are given as follows: P ≤ 0.0001 (****), P ≤ 0.001 (***), P ≤ 0.01 (**), P ≤ 0.05 (*), and P > 0.05 (n.s. [not significant]). Experiments were repeated at least three times.
Data availability
All data supporting the findings of this study are available here and in the Supplemental Material.
Competing interest statement
T.d.L is a member of the Scientific Advisory Board of Calico. The remaining authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank members of the de Lange laboratory for helpful comments on this manuscript. Z.Y. is grateful for Dr. Nanda Sasi's help in troubleshooting experiments. We thank Roddy O'Sullivan for providing the RAD51AP1 antibody and advice, and Joachim Lingner for providing the supertelomerase cell lines. Research reported here was supported by a grant from the National Institutes of Health (AG016642) to T.d.L. T.d.L. is an American Cancer Society Rose Zarucki Trust Research Professor.
Author contributions: Z.Y. and T.d.L. designed the experiments. Z.Y. performed all experiments with the exception of the experiments involving ALT cells, which were performed by C.A.L., K.K.T., and Z.Y. Z.Y. and T.d.L. wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.328575.119.
References
- Anand RP, Lovett ST, Haber JE. 2013. Break-induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397 10.1101/cshperspect.a010397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. 2014. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 5: 5220 10.1038/ncomms6220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SM, Cornforth MN, Kurimasa A, Chen DJ, Goodwin EH. 2001. Strand-specific postreplicative processing of mammalian telomeres. Science 293: 2462–2465. 10.1126/science.1062560 [DOI] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. 2009. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23: 2405–2414. 10.1101/gad.1839909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield C, Karlseder J. 2012. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 40: 7358–7367. 10.1093/nar/gks407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso-González J, García-Expósito L, Hoang SM, Lynskey ML, Roncaioli JL, Ghosh A, Wallace CT, Modesti M, Bernstein KA, Sarkar SN, et al. 2019. RAD51AP1 is an essential mediator of alternative lengthening of telomeres. Mol Cell 76: 11–26.e7. 10.1016/j.molcel.2019.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. 2008. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4: e1000110 10.1371/journal.pgen.1000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID. 2016. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126. 10.1016/j.molcel.2016.10.037 [DOI] [PubMed] [Google Scholar]
- Burla R, Carcuro M, Raffa GD, Galati A, Raimondo D, Rizzo A, La Torre M, Micheli E, Ciapponi L, Cenci G, et al. 2015. AKTIP/Ft1, a new Shelterin-interacting factor required for telomere maintenance. PLoS Genet 11: e1005167 10.1371/journal.pgen.1005167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7: 712–718. 10.1038/ncb1275 [DOI] [PubMed] [Google Scholar]
- Celli GB, Lazzerini Denchi E, de Lange T. 2006. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol 8: 885–890. 10.1038/ncb1444 [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR. 2009. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol 16: 1244–1251. 10.1038/nsmb.1725 [DOI] [PubMed] [Google Scholar]
- Chester N, Babbe H, Pinkas J, Manning C, Leder P. 2006. Mutation of the murine Bloom's syndrome gene produces global genome destabilization. Mol Cell Biol 26: 6713–6726. 10.1128/MCB.00296-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, Greenberg RA. 2014. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159: 108–121. 10.1016/j.cell.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. 2009. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev 23: 2415–2425. 10.1101/gad.1832309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. 2012. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47: 396–409. 10.1016/j.molcel.2012.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. 2019. Replication-Coupled DNA repair. Mol Cell 74: 866–876. 10.1016/j.molcel.2019.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofari G, Lingner J. 2006. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J 25: 565–574. 10.1038/sj.emboj.7600952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. 2018. Shelterin-mediated telomere protection. Annu Rev Genet 52: 223–247. 10.1146/annurev-genet-032918-021921 [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. 2016. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58. 10.1038/nature20099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, de Lange T. 2016. Telomere-internal double-strand breaks are repaired by homologous recombination and PARP1/Lig3-Dependent end-joining. Cell Rep 17: 1646–1656. 10.1016/j.celrep.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducoux M, Urbach S, Baldacci G, Hübscher U, Koundrioukoff S, Christensen J, Hughes P. 2001. Mediation of proliferating cell nuclear antigen (PCNA)-dependent DNA replication through a conserved p21Cip1-like PCNA-binding motif present in the third subunit of human DNA polymerase δ. J Biol Chem 276: 49258–49266. 10.1074/jbc.M106990200 [DOI] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. 2007. Chromosome fragile sites. Annu Rev Genet 41: 169–192. 10.1146/annurev.genet.41.042007.165900 [DOI] [PubMed] [Google Scholar]
- Huber MD, Lee DC, Maizels NP. 2002. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res 30: 3954–3961. 10.1093/nar/gkf530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA. 2015. HLTF's ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol Cell 58: 1090–1100. 10.1016/j.molcel.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Spitz GS, Veturi U, Lach FP, Auerbach AD, Smogorzewska A. 2013. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121: 54–63. 10.1182/blood-2012-07-441212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JHJ, Sung PA, Jasin M, et al. 2012. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8: e1002772 10.1371/journal.pgen.1002772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Takai K, Huh MS, Picketts DJ, de Lange T. 2020. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol 18: e3000594 10.1371/journal.pbio.3000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, O'Rourke JJ, Sobinoff AP, Allen JAM, Nelson CB, Tomlinson CG, Lee M, Reddel RR, Deans AJ, Pickett HA. 2019. The FANCM–BLM–TOP3A–RMI complex suppresses alternative lengthening of telomeres (ALT). Nat Commun 10: 2252 10.1038/s41467-019-10180-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JL, Kim EM, Haber JE, Lee SE. 2003. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol 23: 8820–8828. 10.1128/MCB.23.23.8820-8828.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, Boulton SJ. 2018. Stabilization of reversed replication forks by telomerase drives telomere catastrophe. Cell 172: 439–453.e14. 10.1016/j.cell.2017.11.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. 2009. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23: 2060–2075. 10.1101/gad.543509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M, Stahl FW. 1958. The replication of DNA in Escherichia coli. Proc Natl Acad Sci 44: 671–682. 10.1073/pnas.44.7.671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Wright WE, Shay JW. 2017. Alternative lengthening of telomeres mediated by mitotic DNA synthesis engages break-induced replication processes. Mol Cell Biol 37: e00226-17 10.1128/MCB.00226-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. 2015. Replication stress activates DNA repair synthesis in mitosis. Nature 528: 286–290. 10.1038/nature16139 [DOI] [PubMed] [Google Scholar]
- Mohaghegh P, Karow JK, Brosh RM Jr, Bohr VA, Hickson ID. 2001. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res 29: 2843–2849. 10.1093/nar/29.13.2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann AA, Watson CM, Noble JR, Pickett HA, Tam PP, Reddel RR. 2013. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev 27: 18–23. 10.1101/gad.205062.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özer Ö, Bhowmick R, Liu Y, Hickson ID. 2018. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 9: 15836–15846. 10.18632/oncotarget.24745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, Smogorzewska A. 2019. Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol 218: 1096–1107. 10.1083/jcb.201809012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, Gagos S. 2016. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep 17: 1731–1737. 10.15252/embr.201643169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103. 10.1016/j.cell.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS. 2007. Template switching during break-induced replication. Nature 447: 102–105. 10.1038/nature05723 [DOI] [PubMed] [Google Scholar]
- Sun H, Karow JK, Hickson ID, Maizels NP. 1998. The Bloom's syndrome helicase unwinds G4 DNA. J Biol Chem 273: 27587–27592. 10.1074/jbc.273.42.27587 [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556. 10.1016/S0960-9822(03)00542-6 [DOI] [PubMed] [Google Scholar]
- Takai KK, Hooper S, Blackwood S, Gandhi R, de Lange T. 2010. In vivo stoichiometry of shelterin components. J Biol Chem 285: 1457–1467. 10.1074/jbc.M109.038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. 2013. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20: 317–325. 10.1038/nsmb.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MRG, Yeeles JTP. 2018. The initial response of a eukaryotic replisome to DNA damage. Mol Cell 70: 1067–1080.e12. 10.1016/j.molcel.2018.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. 2012. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149: 795–806. 10.1016/j.cell.2012.03.030 [DOI] [PubMed] [Google Scholar]
- Verma P, Dilley RL, Zhang T, Gyparaki MT, Li Y, Greenberg RA. 2019. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev 33: 221–235. 10.1101/gad.319723.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston R, Peeters H, Ahel D. 2012. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev 26: 1558–1572. 10.1101/gad.193516.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Takai H, de Lange T. 2012. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-In by POT1b-associated CST. Cell 150: 39–52. 10.1016/j.cell.2012.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, West SC. 2014. Holliday junction resolvases. Cold Spring Harb Perspect Biol 6: a023192 10.1101/cshperspect.a023192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, Chen J. 2012. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell 47: 410–421. 10.1016/j.molcel.2012.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Yadav T, Ouyang J, Lan L, Zou L. 2019. Alternative lengthening of telomeres through two distinct break-induced replication pathways. Cell Rep 26: 955–968.e3. 10.1016/j.celrep.2018.12.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Kibe T, Kabir S, de Lange T. 2014. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev 28: 2477–2491. 10.1101/gad.251611.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available here and in the Supplemental Material.