

Abstract
Rationale: Two distinct phenotypes of acute respiratory distress syndrome (ARDS) with differential clinical outcomes and responses to randomly assigned treatment have consistently been identified in randomized controlled trial cohorts using latent class analysis. Plasma biomarkers, key components in phenotype identification, currently lack point-of-care assays and represent a barrier to the clinical implementation of phenotypes.
Objectives: The objective of this study was to develop models to classify ARDS phenotypes using readily available clinical data only.
Methods: Three randomized controlled trial cohorts served as the training data set (ARMA [High vs. Low Vt], ALVEOLI [Assessment of Low Vt and Elevated End-Expiratory Pressure to Obviate Lung Injury], and FACTT [Fluids and Catheter Treatment Trial]; n = 2,022), and a fourth served as the validation data set (SAILS [Statins for Acutely Injured Lungs from Sepsis]; n = 745). A gradient-boosted machine algorithm was used to develop classifier models using 24 variables (demographics, vital signs, laboratory, and respiratory variables) at enrollment. In two secondary analyses, the ALVEOLI and FACTT cohorts each, individually, served as the validation data set, and the remaining combined cohorts formed the training data set for each analysis. Model performance was evaluated against the latent class analysis–derived phenotype.
Measurements and Main Results: For the primary analysis, the model accurately classified the phenotypes in the validation cohort (area under the receiver operating characteristic curve [AUC], 0.95; 95% confidence interval [CI], 0.94–0.96). Using a probability cutoff of 0.5 to assign class, inflammatory biomarkers (IL-6, IL-8, and sTNFR-1; P < 0.0001) and 90-day mortality (38% vs. 24%; P = 0.0002) were significantly higher in the hyperinflammatory phenotype as classified by the model. Model accuracy was similar when ALVEOLI (AUC, 0.94; 95% CI, 0.92–0.96) and FACTT (AUC, 0.94; 95% CI, 0.92–0.95) were used as the validation cohorts. Significant treatment interactions were observed with the clinical classifier model–assigned phenotypes in both ALVEOLI (P = 0.0113) and FACTT (P = 0.0072) cohorts.
Conclusions: ARDS phenotypes can be accurately identified using machine learning models based on readily available clinical data and may enable rapid phenotype identification at the bedside.
Keywords: ARDS phenotypes, machine learning, classifier models
At a Glance Commentary
Scientific Knowledge on the Subject
Using latent class analysis, two phenotypes of acute respiratory distress syndrome (ARDS) with divergent characteristics and clinical outcomes have consistently been identified in secondary analysis of randomized controlled trials. To date, however, the identification of these phenotypes has been contingent on quantification of research biomarkers. The lack of point-of-care testing for these biomarkers has limited their implementation at the bedside.
What This Study Adds to the Field
We have developed and validated models that only use readily available clinical data and can classify ARDS phenotypes with high accuracy. Importantly, the identified phenotypes shared clinical characteristics and differential treatment responses observed in the original latent class analysis–derived phenotypes. Contingent on prospective validation, these models can facilitate the implementation of ARDS phenotypes at the bedside.
In critical care medicine, clinical syndromes such as acute respiratory distress syndrome (ARDS) and sepsis have come to define the specialty. Both sepsis and ARDS are highly prevalent clinical disorders associated with high mortality and morbidity (1, 2). Despite decades of preclinical and clinical studies, no disease-altering interventions have been successfully tested in either of these syndromes (3, 4). In recent years, a proposed explanation for the near ubiquitous failures of these clinical trials implicates the broad defining criteria for these syndromes, which inevitably lead to clinical and biological heterogeneity.
To address the issue of heterogeneity, investigators are increasingly using unsupervised learning methods to seek distinct phenotypes nested within these syndromes. Clustering algorithms using experimental biomarkers, including plasma proteins and transcriptomic data, have most consistently identified distinct phenotypes that offer novel biological insights (5, 6). More pertinently, many of these studies have identified phenotypes that potentially offer routes to both prognostic and predictive enrichment of clinical trials (5, 6). Although the potential of their clinical applicability may be tantalizing, practically, the lack of point-of-care testing for many of the key defining biomarkers limits the applicability of phenotypes in the clinical setting. The clinical implementation of these phenotypes, therefore, represents one of the foremost challenges facing the field.
Specifically pertaining to ARDS, our group has performed latent class analysis (LCA) in five randomized controlled trial (RCT) cohorts of ARDS using 30–40 clinical and biological variables and has consistently identified two distinct phenotypes of ARDS (7–10). These phenotypes have been termed hypoinflammatory and hyperinflammatory, with the latter characterized by high plasma levels of inflammatory biomarkers. In addition, the phenotypes have widely divergent clinical outcomes, and differential treatment responses have been identified to positive end-expiratory pressure (PEEP) strategy (7), fluid therapy (8), and simvastatin (9).
Bedside identification of these phenotypes is seemingly dependent on the rapid quantification of plasma biomarkers such as IL-8, protein C, IL-6, and sTNFR-1 (soluble tumor necrosis factor receptor 1) (8, 11). Currently, point-of-care or clinically validated assays are unavailable for most of these biomarkers, thereby limiting real-time prospective identification of the phenotypes.
The advent of modern machine learning algorithms could potentially allow the development of highly accurate phenotype classification models that are solely reliant on readily available data. One such algorithm, namely gradient-boosted machines (GBMs), is increasingly being applied for prediction in the data science industry and is known to outperform simpler models, such as logistic regression, in many clinical research fields, including critical care (12–14). The primary objective of this study was to use GBMs to develop ARDS phenotype classifier models using only clinical variables that are readily available on admission to the emergency room or ICU. Secondary objectives of the study were 1) to test whether the differential treatment responses observed in prior LCA studies can be identified using these clinical classifier models and 2) to develop models that use a more limited set of readily available clinical data. A portion of the work contained in this manuscript has been previously published in abstract form (15). The data have been updated since then.
Methods
Study Population
Clinical data were obtained from patients enrolled in four NHLBI ARDS Network RCTs: namely, ARMA (High vs. Low Vt) (16), ALVEOLI (Assessment of Low Vt and Elevated End-Expiratory Pressure to Obviate Lung Injury) trial (17), FACTT (Fluids and Catheter Treatment Trial) (18), and SAILS (Statins for Acutely Injured Lungs from Sepsis) (19). Patients were enrolled in these trials within 48 hours of the onset of ARDS. All patients in the trials were mechanically ventilated and admitted to ICUs. Data from before or at the time of enrollment were used for the purposes of this study. Full details of the trials can be found in the original published studies (16–19). Subjects were assigned into hypoinflammatory or hyperinflammatory phenotypes using LCA-derived phenotypes from a prior study (11). In this study, ARMA (study arm only), ALVEOLI, and FACTT were combined into a single cohort for LCA, and SAILS was analyzed individually (20). These phenotypes served as the reference standard for model development and model performance evaluation. The severity of disease scores, clinical outcomes, and treatment assignments were not incorporated during the model development.
Data Synthesis and Analysis
Figure 1 provides an outline of the analysis plan. For each phase of analysis, separate training and validation data sets were created. For the primary analysis, data from ARMA, ALVEOLI, and FACTT cohorts were merged to form the training data set, and the most contemporaneous cohort, SAILS, served as the validation data set. For secondary analyses, two models were created; in secondary model 1, the ARMA, ALVEOLI, and SAILS cohorts were merged to form the training data set, and the FACTT cohort served as the validation data set. In secondary model 2, the ARMA, FACTT, and SAILS cohorts were combined to form the training data set, and the ALVEOLI cohort served as the validation data set. The rationale for the resampling analyses in secondary models 1 and 2 were twofold. First, these analyses served as a form of cross-validation of the approach to estimate the external generalizability of the proposed algorithms and phenotypes. Second, they allowed the testing of differential treatment responses in the identified phenotypes, as seen in the original LCA-derived phenotypes. The ARMA cohort was not used as a validation data set because it constituted the smallest and oldest data set that excluded half the study population (those receiving high Vts). Consequently, the heterogeneous treatment effect in phenotypes was not tested in the ARMA cohort in the original LCA study (7).
Figure 1.
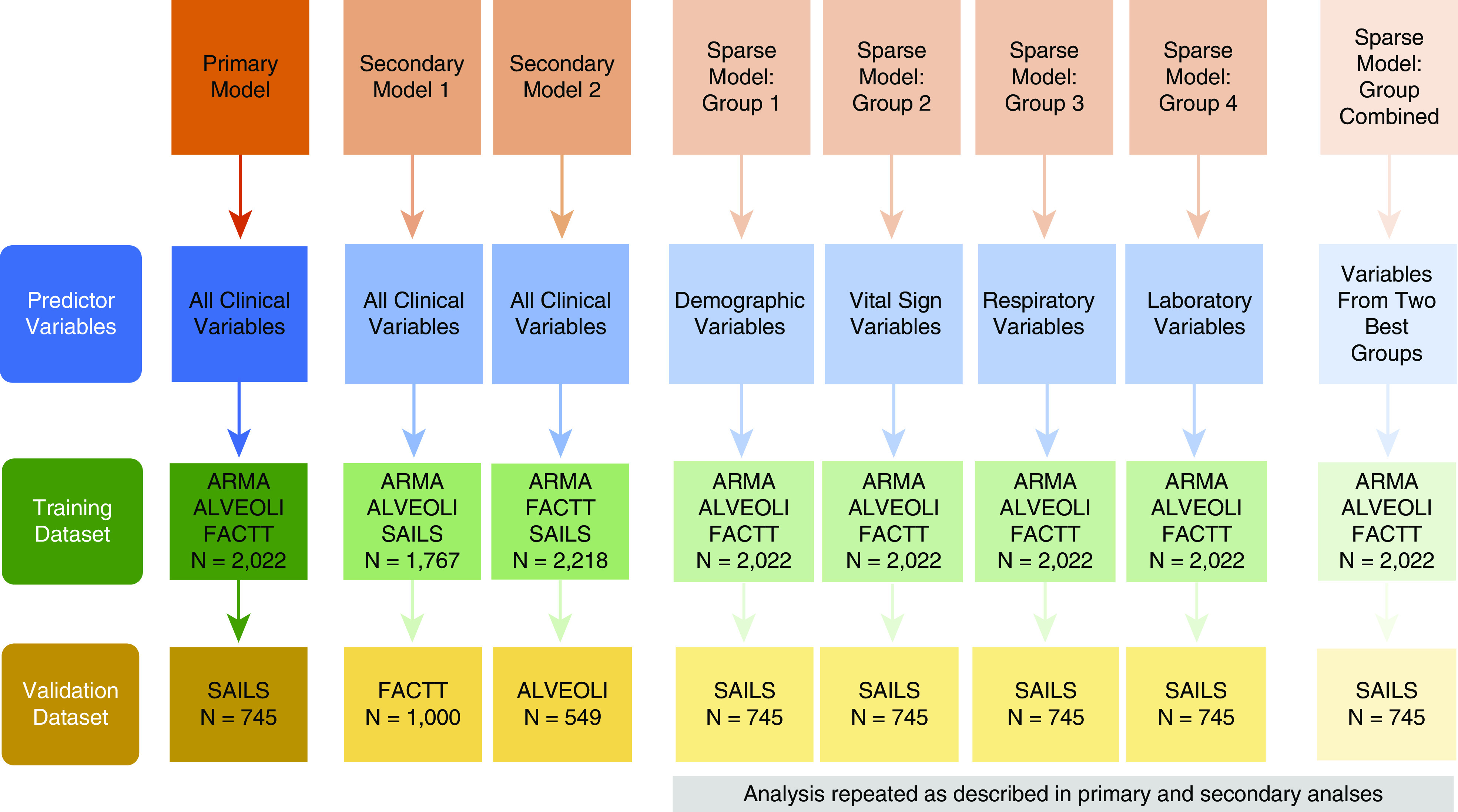
A schematic of the analysis plan and the data sets used in the primary, secondary, and sparse variable set analyses. ALVEOLI = Assessment of Low Vt and Elevated End-Expiratory Pressure to Obviate Lung Injury; ARMA = High vs. Low Vt; FACTT = Fluids and Catheter Treatment Trial; SAILS = Statins for Acutely Injured Lungs from Sepsis.
Next, classifier models were developed using sparse sets of variables that were grouped according to variable type. The four types were demographics, vital signs, respiratory data, and laboratory data (Table 1). The data sets from the primary analysis were used to train and test model performance. A priori, a decision was made to use the variables from the two best performing groups in the validation data set to develop a new (“combined”) model, and its accuracy was evaluated in the validation data set. The rationale for this approach was to develop a model comprised of a more parsimonious set of variables.
Table 1.
Variables Used for the Clinical Classifier Model
| Variables | Groups |
|---|---|
| Age, yr | Demographic (group 1) |
| Sex, F | |
| Race, white | |
| Body mass index | |
| ARDS risk factor: pneumonia | |
| ARDS risk factor: sepsis | |
| ARDS risk factor: aspiration | |
| ARDS risk factor: trauma | |
| ARDS risk factor: other | |
| PaO2/FiO2 ratio, mm Hg | Respiratory (group 2) |
| PaCO2, mm Hg | |
| e, ml/min | |
| Vt, ml | |
| Peak end-expiratory pressure, cm H2O | |
| Temperature, °C; high | Vital signs (group 3) |
| Heart rate, beats/min; high | |
| Systolic blood pressure, mm Hg; low | |
| Respiratory rate, breaths/min−1; high | |
| Vasopressor use at baseline, yes/no | |
| Hematocrit, % | Laboratory (group 4) |
| White cell count, 103/μl; high | |
| Platelets, 103/μl; low | |
| Sodium, mmol/L; high | |
| Glucose, mg/dl; high | |
| Creatinine, mg/dl; high | |
| Bicarbonate, mmol/L; low | |
| Albumin, g/dl; low | |
| Bilirubin, mg/dl; high |
Definition of abbreviation: ARDS = acute respiratory distress syndrome.
Predictor Variables
The list of predictor variables used in the classifier models are presented in Table 1. Only data that were used in the original LCA-modeling studies and that were deemed to be readily available in routine clinical workflow at the point of trial enrollment were considered for predictor variables. Urine output over the prior 24 hours was excluded because it may not consistently be available at the bedside, and plateau pressure was excluded because of high missingness (>25%).
Model Training and Testing
A variant of the gradient-boosted trees algorithm known as XGBoost (extreme gradient boosting) was used to develop the clinical classifier models (21). GBM is an ensemble- or decision tree–based method whereby each new tree in the model aims to correct the classification errors of previous trees in the ensemble (22). This learning procedure enables the iterative refinement of the model, and residual errors are minimized in new trees until the model is maximally optimized, leading to more accurate predictions. Hyperparameters were tuned using a grid search strategy in the training data set using 10-fold cross-validation. Additional details of the XGBoost analyses are available in the online supplement.
The performance of the final tuned model was evaluated in the validation data set, which was kept isolated from the model training process throughout the analysis. The area under the receiver operating characteristic curve (AUC) was used to evaluate model performance. In the validation data set, patients were assigned the phenotypes based on their highest probability (≥0.5). Sensitivity, specificity, and accuracy of phenotype assignments were computed. Clinical outcomes and treatment interactions were determined based on phenotype assignments, and these were compared with LCA-derived phenotypes. Furthermore, to test the model performance over a range a probability cutoffs, the analyses were repeated for all models by assigning classes using cutoffs of 0.3, 0.4, 0.6, and 0.7.
Between-group differences were tested using Student’s t tests and Wilcoxon-rank sum tests depending on the distribution of the variable. Differences in outcomes between phenotypes were tested using Pearson’s χ2 test. To evaluate differential treatment responses, logistic regression models were constructed by introducing interaction terms of phenotype assignment and treatment, with mortality at Day 90 as the outcome variable. XGBoost models were developed using the R package XGBoost. All analyses were performed using R version 3.4.1.
Results
Values for variables at baseline for the training data set and validation data set in the primary analysis are summarized in Table E1 in the online supplement. For model performance, only data generated in the validation data sets are presented throughout.
Primary Analysis
Data were available for 2,022 patients in the training data set and 745 patients in the validation data set. For the primary analysis, the final tuned (“clinical classifier”) model, when tested in the validation data set (SAILS), had an AUC of 0.95 (95% confidence interval [CI], 0.94–0.96). Using a probability cutoff of 0.5 or more to assign the hyperinflammatory phenotype, the model specificity was 0.98, sensitivity was 0.63 (Table 2), and accuracy was 0.85. The model performance over a range of probability cutoffs is presented in Table 3. There was a strong positive correlation between the probabilities generated by the clinical classifier model and those generated by the LCA (r = 0.81; P < 0.0001).
Table 2.
Confusion Matrix Comparing Phenotype Classification Derived by the Gradient-Boosting Machine (Clinical Classifier) Model with Original LCA-derived Classification in the Primary Validation Data Set (SAILS)*
| LCA-assigned Hyperinflammatory Class | LCA-assigned Hypoinflammatory Class | Total | |
|---|---|---|---|
| Clinical classifier–derived hyperinflammatory class | 175 (sensitivity 0.63) | 8 | 183 |
| Clinical classifier–derived hypoinflammatory class | 102 | 460 (specificity 0.98) | 562 |
| Total | 277 | 468 | — |
Definition of abbreviations: LCA = latent class analysis; SAILS = Statins for Acutely Injured Lungs from Sepsis.
Probability cutoff of ≥0.5 to assign phenotype.
Table 3.
Model Performance and Accuracy of Clinical Classifier Model over a Range of Probability Cutoffs in the Validation Data Set (SAILS) in the Primary Analysis*
| Probability Cutoff | Sensitivity | Specificity | Accuracy | Total Patients [n (%)] |
Mortality at Day 90 [n (%)] |
P Value for Treatment Interaction | ||
|---|---|---|---|---|---|---|---|---|
| Hypoinflammatory | Hyperinflammatory | Hypoinflammatory | Hyperinflammatory | |||||
| ≥0.3 | 0.78 | 0.93 | 0.87 | 493 (66) | 252 (34) | 117 (24) | 87 (35) | 0.7960 |
| ≥0.4 | 0.71 | 0.97 | 0.87 | 533 (72) | 212 (28) | 124 (23) | 80 (38) | 0.3300 |
| ≥0.6 | 0.54 | 0.99 | 0.82 | 593 (80) | 152 (20) | 145 (24) | 59 (39) | 0.3897 |
| ≥0.7 | 0.45 | 0.99 | 0.79 | 618 (83) | 127 (17) | 154 (25) | 50 (39) | 0.2650 |
Definition of abbreviation: SAILS = Statins for Acutely Injured Lungs from Sepsis.
For each phenotype, proportions of patients, mortality at Day 90, and P values for interaction term of phenotypes with randomized intervention (with mortality as outcome) are also presented.
As with LCA-derived phenotypes, the hyperinflammatory phenotype identified using the clinical classifier model had significantly higher levels of plasma IL-6, IL-8, soluble tumor necrosis factor receptor 1 (Figure 2), intercellular adhesion molecule 1, and plasminogen activator factor 1 (see Figure E1). As with the LCA-derived phenotypes, plasma protein C levels were significantly lower in the hyperinflammatory group (Figure 2). The absolute median values and interquartile ranges of the biomarkers in the phenotypes of the clinical classifier models were remarkably similar to those in the LCA-derived phenotypes (data not shown). When phenotype was assigned by the clinical classifier model, mortality at Day 90 was significantly higher in the hyperinflammatory phenotype compared with the hypoinflammatory phenotype (38% vs. 24%; P = 0.0002). Ventilator-free days were also significantly fewer in the hyperinflammatory phenotype (median 13 vs. 21 d; P < 0.0001). As with the LCA-derived phenotypes, no treatment interaction was observed with treatment groups (rosuvastatin vs. placebo) and clinical classifier–derived phenotypes (P = 0.359).
Figure 2.

Differences in the plasma biomarker levels in the validation data set (SAILS [Statins for Acutely Injured Lungs from Sepsis] trial) at baseline in the hypoinflammatory and hyperinflammatory phenotypes as identified by the clinical classifier model developed in the primary analysis. P values represent the Wilcoxon rank sum test. (A) IL-6; y-axis upper limit is restricted to 10,000 with 19 observations censored (15 hyperinflammatory and 4 hypoinflammatory). (B) IL-8; y-axis upper limit is restricted to 1,000 with 31 observations censored (24 hyperinflammatory and 7 hypoinflammatory). (C) Protein C. (D) Soluble tumor necrosis factor receptor 1; y-axis upper limit is restricted to 40,000 with three observations censored (all hyperinflammatory). TNF = tumor necrosis factor.
The three most important predictor variables in the clinical classifier model were bicarbonate, vasopressor use, and creatinine. The top 10 variables of importance in the clinical classifier model are presented in Figure 3.
Figure 3.

Top 10 most important variables in the training data set in the primary analysis. Importance was scaled to 100; 100 represents the most important predictor variable, and a decreasing value represents diminishing importance. BP = blood pressure.
Secondary Analysis
FACTT as the validation data set (secondary model 1)
In this analysis, the training data set comprised 1,767 patients (ARMA, ALVEOLI, and SAILS) and the validation data set comprised 1,000 patients (FACTT). The AUC for the secondary model 1 in the validation data set was 0.94 (95% CI, 0.92–0.96). Table E2 has a summary of model accuracy, sensitivity, and specificity using a probability cutoff 0.5 or more to assign class. Clinical outcomes were significantly worse in the hyperinflammatory phenotype compared with the hypoinflammatory phenotype (Tables 4 and 5). In line with LCA-assigned phenotypes, a significant interaction was identified between the phenotype and fluid treatment strategy for mortality at Day 90 (P = 0.0072) in the validation (FACTT) data set.
Table 4.
Mortality at Day 90 in Phenotypes Identified in the Secondary Model 1 Using the Classifier Models (Probability Cutoff ≥ 0.5)*
| Model | Mortality at Day 90 in the Hypoinflammatory Phenotype |
Mortality at Day 90 in the Hyperinflammatory Phenotype |
P Value | ||||
|---|---|---|---|---|---|---|---|
| Total [n (%)] | Liberal Fluid [n (%)] | Conservative Fluid [n (%)] | Total [n (%)] | Liberal Fluid [n (%)] | Conservative Fluid [n (%)] | ||
| Clinical classifier | 145/678 (21) | 81/321 (25) | 64/357 (18) | 139/322 (43) | 69/176 (39) | 70/146 (48) | 0.0072 |
| Sparse combined | 153/693 (22) | 86/333 (26) | 67/360 (19) | 131/307 (43) | 64/164 (42) | 67/143 (51) | 0.0124 |
| LCA (8) | 161/727 (22) | 93/355 (26) | 68/372 (18) | 123/273 (45) | 57/142 (40) | 66/131 (50) | 0.004 |
Definition of abbreviations: ALVEOLI = Assessment of Low Vt and Elevated End-Expiratory Pressure to Obviate Lung Injury; ARMA = High vs. Low Vt; FACTT = Fluids and Catheter Treatment Trial; LCA = latent class analysis; SAILS = Statins for Acutely Injured Lungs from Sepsis.
Training data sets: ARMA, ALVEOLI, and SAILS; validation data set: FACTT (n = 1,000). P value represents the interaction between phenotype assignment and randomly assigned treatment strategy for mortality at Day 90. LCA-derived classes were extracted from the original LCA study (8) and not from the derivation data set. Outcomes are shown in phenotypes derived by three different models: clinical-classifier model composed of all the predictor variables, the sparse-combined model composed of only laboratory values and vital signs variables, and the original latent class model (8). Outcomes were substratified by randomized treatment strategy in the original trials.
Table 5.
Mortality at Day 90 in Phenotypes Identified in the Secondary Model 2 Using the Classifier Models (Probability Cutoff ≥ 0.5)*
| Model | Mortality at Day 90 in the Hypoinflammatory Phenotype |
Mortality at Day 90 in the Hyperinflammatory Phenotype |
P Value | |||||
|---|---|---|---|---|---|---|---|---|
| Total [n (%)] | Low PEEP [n (%)] | High PEEP [n (%)] | Total [n (%)] | Low PEEP [n (%)] | High PEEP [n (%)] | |||
| Clinical classifier | 73/372 (20) | 27/184 (15) | 46/188 (25) | 75/177 (42) | 42/89 (47) | 33/88 (38) |
0.0113 | |
| Sparse combined | 85/402 (21) | 35/200 (18) | 50/202 (25) | 63/147 (43) | 34/73 (47) | 29/74 (39) |
0.0748 | |
| LCA (7) | 81/404 (20) | 33/202 (16) | 48/202 (24) | 67/145 (46) | 36/71 (51) | 31/74 (42) | 0.049 | |
Definition of abbreviations: ALVEOLI = Assessment of Low Vt and Elevated End-Expiratory Pressure to Obviate Lung Injury; ARMA = High vs. Low Vt; FACTT = Fluids and Catheter Treatment Trial; LCA = latent class analysis; PEEP = positive end-expiratory pressure; SAILS = Statins for Acutely Injured Lungs from Sepsis.
Training data sets: ARMA, FACTT, and SAILS; validation data set: ALVEOLI trial (n = 549). P value represents the interaction between phenotype assignment and randomly assigned treatment-strategy for mortality at Day 90. LCA-derived classes were extracted from the original LCA study (7) and not from the derivation data set. Outcomes are shown in phenotypes derived by three different models: clinical-classifier model comprising all the predictor variables, the sparse-combined model comprising only laboratory values and vital signs variables, and the original latent class model (7). Outcomes were substratified by randomized treatment strategy in the original trials.
ALVEOLI as the validation data set (secondary model 2)
For this analysis, the training data set comprised 2,218 patients (ARMA, FACTT, and SAILS) and the validation data set comprised 549 patients (ALVEOLI). The AUC for secondary model 2 was 0.94 (95% CI, 0.92–0.95). Table E3 has a summary of model accuracy, sensitivity, and specificity using a probability cutoff of 0.5 or more to assign class. Clinical outcomes were significantly worse in the hyperinflammatory group, and significant treatment interaction was observed with model-derived phenotypes and PEEP strategy for mortality at Day 90 in the validation data set (P = 0.0113; Tables 4 and 5).
Sparse variable-set modeling
Of the sparse set of variables, the laboratory variables model had the highest AUC (0.92; 95% CI, 0.90–0.94) followed by the vital signs model (AUC, 0.79; 95% CI, 0.76–0.82) in the validation data set (SAILS). Demographic variables had the lowest AUC (0.58; 95% CI, 0.53–0.62; Figure 4).
Figure 4.

Receiver operating characteristic curves of the four grouped sparse variable set classifier models in the validation cohort (SAILS [Statins for Acutely Injured Lungs from Sepsis] trial) of the primary analysis. For each model, the area under the curve is presented in the legend box. AUC = area under the curve; ROC = receiver operating characteristic.
A further model was created combining the laboratory and vital signs variables (groups 3 and 4; Table 1). In the primary analysis data set, this combined model had an AUC of 0.94 (95% CI, 0.93–0.96) in the validation data set (SAILS). The addition of further groups to the model had no notable improvements in model performance (see Table E4). Using a probability cutoff of 0.5 or more to assign class, clinical outcomes were again worse in the hyperinflammatory phenotype (90-d mortality 38% vs. 23% [P ≤ 0.0001]; ventilator-free days median 14 vs. 21 d [P < 0.0001]).
When the combined group model was developed and evaluated using data sets described in the secondary analyses, the findings of clinical outcomes in the phenotypes identified were also significantly divergent, in keeping with the prior models (Tables 4 and 5). In addition, significant treatment interactions were observed with phenotype assignment with fluid management strategy (P = 0.0124) when the FACTT served as the validation cohort (Table 4). With ALVEOLI as the validation cohort, a similar pattern of significant divergent clinical outcomes were also observed (Table 5). The observed differential treatment response was similar to the original LCA study but failed to reach statistical significance (P = 0.0748; Table 5).
The proportional composition of phenotypes and their divergent clinical outcomes remained consistent across a range of probabilities in all models (Tables E5–E7). When detected in the original LCA studies, significant treatment interaction with phenotypes assignment were also observed with most probability cutoffs in all models (Tables E5–E7).
Discussion
The clinical implementation of biologically derived phenotypes has been limited because of a lack of point-of-care or clinical grade laboratory tests for the defining biomarkers (23, 24). In the presented study, a novel machine learning–based approach is described that uses readily available clinical data to accurately identify phenotypes derived from complex composite biological and clinical data. The findings of this study and the models it describes offer a potential pipeline for the clinical translation of biomarker-derived phenotypes and their imminent clinical application. The presented study not only describes highly accurate bedside-amenable classifier models, but more importantly, the differential treatment responses that were identified in prior LCA-derived phenotypes were also observed here. Differences in levels of biomarkers between these phenotypes were also remarkably similar to those in the original LCA-derived phenotypes. Collectively, the findings of this study suggest that the hyperinflammatory and hypoinflammatory phenotypes of ARDS can be identified based on clinical variables alone, at least in selected RCT-based populations with ARDS.
Prior efforts to develop accurate parsimonious models to identify ARDS phenotypes have mostly been reliant on the plasma quantification of research biomarkers. In addition to these biomarkers, bicarbonate and vasopressor use were the two most consistently identified clinical components of such parsimonious models (8, 11). It was, therefore, anticipated that these two were the most important clinical variables in the clinical classifier models developed here. The top 10 variables of importance for the classifier models further identify the factors separating the two phenotypes. Given that the LCA-derived hyperinflammatory phenotype is known to be associated with increased incidence of shock, acidosis, and organ dysfunction (7), it is unsurprising that vasopressor use, bicarbonate, creatinine, platelets, and bilirubin were among the most important variables in the model. The observed differences in top 10 variables between the identified phenotypes would, in part, explain the differences in mortality between the two phenotypes. It is worth noting, however, that although on one level these phenotypes represent the severity of a pathophysiological process, they also seem to capture information that is unique. For example, when the same populations were stratified by other measures of disease severity, such as APACHE score or PaO2/FiO2, differential treatment responses were not observed (9, 11).
Prior logistic regression–based classifier models developed exclusively using clinical variables have only demonstrated modest accuracy during validation (AUCs of 0.75–0.80) (8). There are several factors that may have resulted in the models presented in this study performing better than the models developed in prior studies. The current analysis represents a larger data set of patients with ARDS, which allowed enhanced model development and tuning. In addition, the models presented in this study likely benefited from the enhanced predictive accuracy afforded by using GBM. For this reason, GBM is widely applied in big data analytics and consistently used by the top performers of machine learning predictive modeling competitions (e.g., Kaggle) (21). In addition, compared with other machine learning ensemble algorithms such as random forest, GBM models have a built-in functionality to deal with missing values. This gives GBM the distinct advantage of using data from, and assigning class to, all observations in the cohort without the need to impute data. Consequently, the GBM models may be more likely to detect heterogeneous treatment effects should they exist because all the observations used in the LCA analysis are being used to test interactions. Furthermore, the ability to classify phenotypes in the face of missing data makes these models more viable in the real-life clinical setting and offsets some of the disadvantages inherent to GBM model complexity. Another advantage of GBM models is that the data can be used in its original scale, whereas other methods, such as support vector machines, require data transformation (e.g., Z scale standardization). Such transformations require prior knowledge of variable distribution, which limits their value in the prospective setting. The ability to handle missing data and use original scale predictor variables makes GBM models uniquely attractive for prospective implementation.
The findings from using sparse categories of variables indicate that laboratory values, followed by vital signs, were the most important variables for phenotype classification. Put differently, these variables were the best at predicting the biological signature described by plasma biomarkers in LCA-derived ARDS phenotypes. The significant differences in levels of IL-6, IL-8, and sTNFR-1 between the identified phenotypes substantiate this finding. Unsurprisingly, given the independent association of these biomarkers with adverse outcomes in ARDS (25), mortality at Day 90 and ventilator-free days were significantly worse in the hyperinflammatory phenotype. Of great interest, however, was the ability to detect differential treatment response in both the ALVEOLI and FACTT trials in the GBM model–identified phenotypes. Taken together, these findings suggest that the clinical classifier models closely mimic phenotypes identified by LCA and have potential clinical utility for personalizing care for patients with ARDS.
This study, necessarily, has limitations. All presented data are retrospective secondary analyses of previously conducted RCTs. Further, all the RCTs analyzed were conducted by the same network (the NHLBI ARDS Network), and the type and timing of data collection were reasonably uniform. The generalizability of these and prior LCA-derived phenotypes in unselected populations with ARDS are also unknown. The presented models must, therefore, be interpreted with caution. The assumption of the linkage between phenotypes described by the clinical classifier models to underlying biological signatures may not be valid outside these RCT populations. Prospective validation of the models in both non–ARDS Network RCTs and in observational cohorts is required before their use in clinical settings. Furthermore, the variables used to generate the model were carefully curated for RCTs. Model performance in the context of data extracted directly from electronic medical records and/or real-time data is also currently unknown and likely represents a major challenge in the bedside implementation of these models.
The best probability cutoff to use to classify phenotypes remains unknown. It is interesting to note that in SAILS, a sepsis-associated ARDS cohort, a lower probability cutoff led to higher classification accuracy, whereas when ALVEOLI and FACTT, which were composed of nonspecific ARDS risk factors, served as validation cohorts, higher cutoffs led to more accurate classification. Imbalance in outcome prevalence can often lead to the default cutoff of 0.5 not being the most accurate at classification nor the most representative of the probability distribution (26). To that end, for the primary analysis, the prevalence of the hyperinflammatory phenotype in SAILS (validation data set) was 37%, whereas in the training data set it was 29%; this difference may explain the modest accuracy with the default cutoff. The cross-validation across permutations of the data sets is reassuring that the observed high AUCs in the validation data sets are robust over a range of prevalence of the hyperinflammatory phenotype. Nonetheless, when translating the models to the prospective setting, in which prior population data would not be available, selecting the optimal cutoff threshold at an individual level may be challenging. To an extent, the objective of identifying the phenotypes would determine the optimal threshold. For example, if the objective were to use the classifier model as a screening tool, then a lower cutoff would be desirable. Conversely, for studies in which specificity is more important, higher cutoffs could be used. Whether the optimal probability cutoff requires risk factor–dependent adjustment on an individual level also requires further evaluation. To determine the optimal cutoff, the models require validation in more generalized population with ARDS, and this reiterates the necessity for further studies.
As a consequence of the complexity of the underlying algorithms, it is not possible to decipher the precise mechanics of the XGBoost models, so they are sometimes referred to as concealed in a “black box.” The variables with the highest predictive power in these models, such as bicarbonate, vasopressor use, and creatinine, were also the most influential nonprotein biomarker variables in the original LCA studies, lending validity to the algorithms and the phenotypes they identify. Furthermore, the strong paired correlation between the probabilities generated by LCA models and the XGBoost models also indicate local (per sample) validity of the presented models.
Potential pathways of investigation to evaluate these models prospectively include either developing a mobile interface or a web-based application. Alternatively, the models could be incorporated into existing electronic health record systems, in which they could serve as a screening tool for patients with ARDS to recruit in future clinical trials based on phenotypes, with the caveats mentioned above. Another important use of the models could be to interrogate previously conducted ARDS trials that have been inaccessible because of the lack of biological specimens. Finally, once the phenotypes have been identified, these models may offer an inexpensive approach to studying the phenotypes and their trajectory longitudinally over time. All these avenues of implementation are technically relatively straightforward and could be developed imminently; however, before their widespread use, these models need validating in observational cohorts using real-life data. This represents the main rate-limiting step in the bedside usage of these models because of the limited availability of cohorts with biomarker-derived LCA phenotypes to serve as the comparative gold standard.
In summary, this study presents machine learning models that can accurately identify ARDS phenotypes exclusively using clinically available data. The ability to identify phenotypes independent of measuring plasma biomarkers could accelerate our understanding and application of ARDS inflammatory phenotypes. Moreover, the study represents the culmination of a body of work from phenotype identification using a composite of biological and clinical data to the development of models that can identify these phenotypes using clinical data at the bedside. The presented study offers an algorithmic pipeline to other investigators seeking to implement biologically derived phenotypes in the clinical setting. The authors make a commitment to sharing the model with investigators seeking to validate it against LCA-derived phenotypes or seeking to use the model in prior or prospective RCT cohorts.
Supplementary Material
Footnotes
Supported by NIH grants HL140026 (C.S.C.), T32-GM008440 (P.S.), and R01-GM123193 (M.M.C.).
Author Contributions: P.S., M.M.C., and C.S.C. were all involved in study conception, design, analysis, and writing the manuscript.
The authors make a commitment to sharing the model with investigators seeking to validate it against latent class analysis–derived phenotypes or seeking to use the model in prior or prospective randomized controlled trial cohorts.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202002-0347OC on June 18, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. LUNG SAFE Investigators; ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315:788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, et al. Milwaukee Initiative in Critical Care Outcomes Research (MICCOR) Group of Investigators. Nationwide trends of severe sepsis in the 21st century (2000-2007) Chest. 2011;140:1223–1231. doi: 10.1378/chest.11-0352. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med. 2017;5:524–534. doi: 10.1016/S2213-2600(17)30188-1. [DOI] [PubMed] [Google Scholar]
- 4.Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20:195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Sinha P, Calfee CS. Phenotypes in acute respiratory distress syndrome: moving towards precision medicine. Curr Opin Crit Care. 2019;25:12–20. doi: 10.1097/MCC.0000000000000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarma A, Calfee CS, Ware LB. Biomarkers and precision medicine: state of the art. Crit Care Clin. 2020;36:155–165. doi: 10.1016/j.ccc.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA NHLBI ARDS Network. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2:611–620. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. ARDS Network. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. 2017;195:331–338. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Irish Critical Care Trials Group. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. 2018;6:691–698. doi: 10.1016/S2213-2600(18)30177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS NHLBI ARDS Network. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med. 2018;44:1859–1869. doi: 10.1007/s00134-018-5378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med. 2020;8:247–257. doi: 10.1016/S2213-2600(19)30369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Ho KM, Hong Y. Machine learning for the prediction of volume responsiveness in patients with oliguric acute kidney injury in critical care. Crit Care. 2019;23:112. doi: 10.1186/s13054-019-2411-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayaru L, Ypsilantis PP, Nanapragasam A, Choi RC, Thillanathan A, Min-Ho L, et al. Prediction of outcome in acute lower gastrointestinal bleeding using gradient boosting. PLoS One. 2015;10:e0132485. doi: 10.1371/journal.pone.0132485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Churpek MM, Yuen TC, Winslow C, Meltzer DO, Kattan MW, Edelson DP. Multicenter comparison of machine learning methods and conventional regression for predicting clinical deterioration on the wards. Crit Care Med. 2016;44:368–374. doi: 10.1097/CCM.0000000000001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinha P, Churpek MM, Calfee CS. Machine learning classifier models can identify ARDS phenotypes using readily available clinical data [abstract] Am J Respir Crit Care Med. 2019;199:A1014. doi: 10.1164/rccm.202002-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 17.Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, et al. National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 18.Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354:2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 19.Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, et al. National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med. 2014;370:2191–2200. doi: 10.1056/NEJMoa1401520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analyses of randomised controlled trials. Lancet Respir Med. 2020;8:247–257. doi: 10.1016/S2213-2600(19)30369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen T, Guestrin C. San Francisco, CA: 2016. XGBoost: a scalable tree boosting system. Presented at the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; pp. 785–794. [Google Scholar]
- 22.Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29:1189–1232. [Google Scholar]
- 23.Ginsburg GS, Phillips KA. Precision medicine: from science to value. Health Aff (Millwood) 2018;37:694–701. doi: 10.1377/hlthaff.2017.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed MU, Saaem I, Wu PC, Brown AS. Personalized diagnostics and biosensors: a review of the biology and technology needed for personalized medicine. Crit Rev Biotechnol. 2014;34:180–196. doi: 10.3109/07388551.2013.778228. [DOI] [PubMed] [Google Scholar]
- 25.Binnie A, Tsang JL, dos Santos CC. Biomarkers in acute respiratory distress syndrome. Curr Opin Crit Care. 2014;20:47–55. doi: 10.1097/MCC.0000000000000048. [DOI] [PubMed] [Google Scholar]
- 26.Freeman EA, Moisen GG. A comparison of the performance of threshold criteria for binary classification in terms of predicted prevalence and kappa. Ecol Modell. 2008;217:48–58. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.