

Abstract
Immunomodulatory drugs (IMiDs) lenalidomide and pomalidomide show remarkable anti-tumor activity in multiple myeloma (MM) via directly inhibiting MM cell growth in the bone marrow (BM) microenvironment and promoting immune effector cell function. They are known to bind to the ubiquitin 3 ligase CRBN complex and thereby trigger degradation of IKZF1/3. In this study, we demonstrate that IMiDs also directly bind and activate zeta-chain-associated protein kinase-70 (Zap-70) via its tyrosine residue phosphorylation in T cells. IMiDs also triggered phosphorylation of Zap-70 in NK cells. Importantly, increased granzyme-B (GZM-B) expression and NK cell activity triggered by IMiDs is associated with Zap-70 activation and inhibited by Zap-70 knockdown, independent of CRBN. We also demonstrate a second mechanism whereby IMiDs trigger GZM-B and NK cytotoxicity which is CRBN- and IKZF3-mediated and inhibited by knockdown of CRBN or IKZF-3, independent of Zap-70. Our studies therefore show that IMiDs can enhance NK and T cell cytotoxicity in (1) ZAP-70-mediated CRBN independent, as well as (2) CRBN-mediated ZAP-70 independent mechanisms; and provide the framework for developing novel therapeutics to activate Zap-70 and thereby enhance T and NK anti-MM cytotoxicity.
Introduction
Among the most important treatment advances in multiple myeloma (MM) is the development of immunomodulatory drugs (IMiDs) thalidomide (Thal), lenalidomide (Len), and pomalidomide (Pom). Their multiple anti-MM effects include: induction of growth arrest and apoptosis in tumor cells; downregulation of adhesion molecules and MM cell binding to cellular components and extracellular matrix proteins in the bone marrow (BM); anti-angiogenesis; modulation of cytokines; and immunomodulation associated with enhanced T cell, NK cell, and NK-T cell activity, along with decreased regulatory T cell activity 1–3. Multiple groups have shown that Thal , Len, and Pom directly bind to cereblon (CRBN), forming an E3 ubiquitin ligase complex with damaged DNA binding protein 1 (DDB1), cullin-4A, and regulator of cullins1 4, 5, thereby triggering proteasomal degradation of IKZF1 and IKZF3 followed by downregulation of interferon regulatory factor 4 and MM cell growth 6, 7. Recently, we have also shown that Pom directly binds to TP53 regulating kinase and inhibits its activity, which is associated with significant MM cell growth inhibition via both p53-dependent and -independent pathways 8.
Studies have also begun to delineate the molecular mechanisms whereby IMiDs mediate their immune effects. For example, Len triggers CD28 tyrosine phosphorylation in T cells, followed by NF-κB activation 9. IMiDs induce IL-2 and γ-interferon, while inhibiting suppressor of cytokine signaling, in CD4+ T-cells, CD8+ T-cells, and NK-T cells from both BM and peripheral blood (PB) of MM patients 10. This upregulation of immune activity by Pom and Len is, at least in part, mediated by their binding to CRBN and triggering degradation of IKZF1 and IKZF3 in T cells, thereby allowing for increased transcription and secretion of cytokines including IL-2 11. We have demonstrated that IL-2-primed PB mononuclear cells (PBMCs) treated with IMiDs showed significantly increased lysis of MM cell lines, which was not major histocompatibility complex-class restricted 12. We and others have also reported that IMiDs enhance both NK cell and NK-T cell cytotoxicity and antibody-dependent cellular cytotoxicity (ADCC), at least in part due to triggering IL-2 production from T cells 13–18. Moreover, a recent study has shown that Len can enhance secretion of IFN-γ and GZM-B from antigen-specific T-cells 19. To date, however, the molecular mechanisms whereby IMiDs induce NK cell cytotoxicity have not been elucidated.
In this study, we characterized the role of zeta-chain-associated protein kinase 70 (Zap-70), a 70 kDa cytoplasmic protein tyrosine kinase composed of two SH2 domains and a carboxy-terminal kinase domain initiating T-cell responses by the antigen receptor 20, in mediating the increased NK cell-cytotoxicity triggered by IMiDs. We show that IMiDs directly bind and activate Zap-70. Importantly, increased GZM-B expression and NK cell activity triggered by IMiDs is associated with Zap-70 activation and inhibited by Zap-70 knockdown, independent of CRBN. A second mechanism whereby IMiDs trigger GZM-B and NK cytotoxicity is CRBN- and IKZF3-mediated and can be inhibited by knockdown of CRBN or IKZF-3, independent of Zap-70. Our studies therefore show that IMiDs can enhance NK and T cell cytotoxicity in 1) ZAP-70-mediated CRBN independent, as well as 2) CRBN-mediated Zap-70 independent mechanisms. They further validate the potential of developing novel therapeutics to activate Zap-70 and thereby enhance T and NK MM cytotoxicity.
Materials and Methods
Cells
U266 myeloma cell line and Jurkat T-cell leukemia cell line were obtained from American Type Culture Collection (ATCC, Manassas, MD) and cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 100U/mL of penicillin, and 100ug/mL of streptomycin. KHYG-1 natural killer (NK) cell leukemia line was purchased from German Collection of Microorganism and Cell Cultures GmbH (DSMZ, Germany), and cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 100U/mL of penicillin, 100ug/mL of streptomycin, and 10 ng/ml IL-2. NK-92 NK cell line was obtained from ATCC and cultured in αMEM supplemented with 12.5% fetal bovine serum, 12.5% horse serum, 2mM L-glutamine, 1.5g/L sodium bicarbonate, 0.2mM inositol, 0.1mM 2-mercaptoethanol and 200U IL-2. Cell lines have been tested and authenticated by STR DNA fingerprinting analysis (Molecular Diagnostic Laboratory, DFCI). They were also regularly tested for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland) and were used within three months after thawing.
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers using Ficoll-Paque PLUS density gradient media (GE Healthcare, Uppsala, Sweden). T-cells and NK cells were further purified from PBMCs by negative selection procedure using RosetteSep Separation System® (StemCell Technologies, Vancouver, Canada). Purity (> 90%) of T and NK cells was confirmed by flow cytometric analysis. All experiments with healthy volunteer’s samples were performed under auspices of an DFCI Institutional Review Board approved protocol after informed consent was obtained.
Reagents and antibodies
Len) and Pom were purchased from Sigma-Aldrich (St. Louis, MO) and Selleck Chemicals (Houston, TX), respectively. CC-220 (Iberdomide) was purchased from Cayman Chemicals (Ann Arbor, MI). Dexamethasone was purchased form Sigma-Aldrich (St Louis, MO). Anti-actin and -perforin Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); and anti-IKZF1 Ab was obtained from R&D Systems (Minneapolis, MN). All other Abs were purchased from Cell Signaling Technologies (Danvers, MA). The information of Abs are also listed in Supplementary Table S1.
Growth inhibition assay
Cell growth was assessed by measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide (MTT, Chemicon International, Temecula, CA) dye absorbance. Cells were pulsed with 10 μl of 5 mg/ml MTT to each well for the last 4h of 48h and/or 72h cultures, followed by 100 μl isopropanol containing 0.04N HCl. Absorbance was measured at 570/630 nm using a spectrophotometer (Molecular Devices Corp., Sunnyvale CA).
Pom-based reagent and immunoblotting
Affi-Gel 10 (Bio-Rad, Hercules, CA)-linked Pom was generated as described in our previous study 8 and provided by TAIHO PHARMACEUTICAL CO., LTD. (Tsukuba, Ibaraki, Japan). After pre-cleaning, the Jurkat cell lysates with a control reagent (ethanolamine-affigel) to reduce non-specific binding, the lysates were incubated with either the ethanolamine-affigel or pomalidomide-based affinity reagent (80 uL of each, 50% slurry) for 1 hour at 4°C to identify candidate binding proteins. In competition assays, 10-fold excess concentration (1 mM) Pom was added to lysate from Pom-based affinity reagents. Affinity reagents were then washed three times with NP-40 lysis buffer, and binding proteins were then eluted with 2X SDS sample buffer at 50°C. The eluted samples were subjected to immunoblotting.
Zap-70 kinase assay
Zap-70 kinase activity was measured using ADP-Glo kinase assay system (Promega, Cat # V8311), according to manufacturer’s protocol.
NK cell assay
U266 target cells were stained with 10 μM of calcein-AM (Thermo Fisher Scientific) for 30 min at 37°C, and then washed 3 times with RPMI media. The cells were then seeded into a 96-well plate at 15,000 cells/well. Effector NK cells were added at various E:T ratios and incubated for 4h. After centrifugation, supernatants were collected for measurement of fluorescence. The spontaneous release and the maximum release were obtained from target cells without effector cells and from target cells with 1% Triton X-100, respectively. Specific target cell lysis was calculated as [(test release - spontaneous release)/(maximum release - spontaneous release)] x 100.
siRNA transfection
For siRNA transfection, non-targeted, Zap-70, CRBN, IKZF1, and IKZF3 ON-Targetplus siRNA SMARTpools were purchased from Thermo Scientific (Lafayette, CO). siRNA transfection was carried out by Amaxa electroporation system using the “Cell Line Nucleofector® Kit T solution (Lonza, Koln, Germany). Sequences of small interference RNAs are listed in Supplementary Table S2.
Immunoblotting and densitometric analysis
Cells were harvested and lysed using RIPA lysis buffer (Cell Signaling Technology) containing 1 mM PMSF. Whole cell lysates were subjected to SDS-PAGE, transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA), and immunoblotted with indicated Abs. Densitometric analysis of immunoblotting was carried out using ImageJ software version 1.48 (National Institute of Health).
RNA extraction and reverse transcription polymerase chain reaction
RNA was extracted using Trizol (Invitrogen) and quantified by a Nanodrop spectrophotometer (Labtech). Specifically, 5x106 cells were pelleted, washed with cold PBS, and resuspended in 1 mL Trizol. Cells were then incubated with 1-bromo-3-chloropropane (Sigma), washed first with Isopropyl alcohol and then with 75% Ethanol, and then resuspended in Nuclease Free-water (Invitrogen). After quantification, 2000 ng of RNA was used to synthesize cDNA using the SuperScript II First-Strand Synthesis Kit (Invitrogen), according to the manufacturer’s instructions. To evaluate the expression levels of GZM-B and GAPDH (internal control), quantitative real time PCR (QRT-PCR) was performed using SYBR GREEN PCR Master Mix (Applied Biosystem), after optimization of the primer conditions as in our prior studies 8. Primers used for quantitative RT-PCR are listed in the Supplementary Table S3.
Results
Pom induces phosphorylation of Zap-70 in T cells
To characterize the effect of IMiDs on Zap-70 function in immune effector cells, we first evaluated whether Pom triggered phosphorylation of Zap-70 and its known downstream target linker of activated T-cells (LAT) in PBMCs from healthy volunteers. As shown in Figure 1A (upper panel), Pom induced phosphorylation of both Zap-70 and LAT in PBMCs in a dose-dependent fashion. ImageJ densitometric analysis confirmed 42% increased p-Zap-70 after Pom (1 μM) treatment (Fig. 1a, lower panel). The increased p-Zap-70, p-LAT, as well as downstream p-ERK in PBMCs triggered by Pom is also time-dependent (Fig. 1b). Of note, Len similarly triggered p-Zap-70 and p-LAT in PBMCs (Fig. 1c). Since Zap-70 is a mediator of T-cell receptor signaling, we next examined whether IMiDs triggered p-Zap-70 in T cells from healthy volunteers. As in PBMCs, Pom treatment induced p-Zap-70 and p-LAT in primary normal donor T cells (Fig. 1d). Pom similarly induced p-Zap-70 in Jurkat cells in a dose-dependent fashion (Fig. 1e), without altering their proliferation (Fig. 1f).
Figure 1. IMiDs induce phosphorylation of Zap-70 in PBMCs and Jurkat cells.
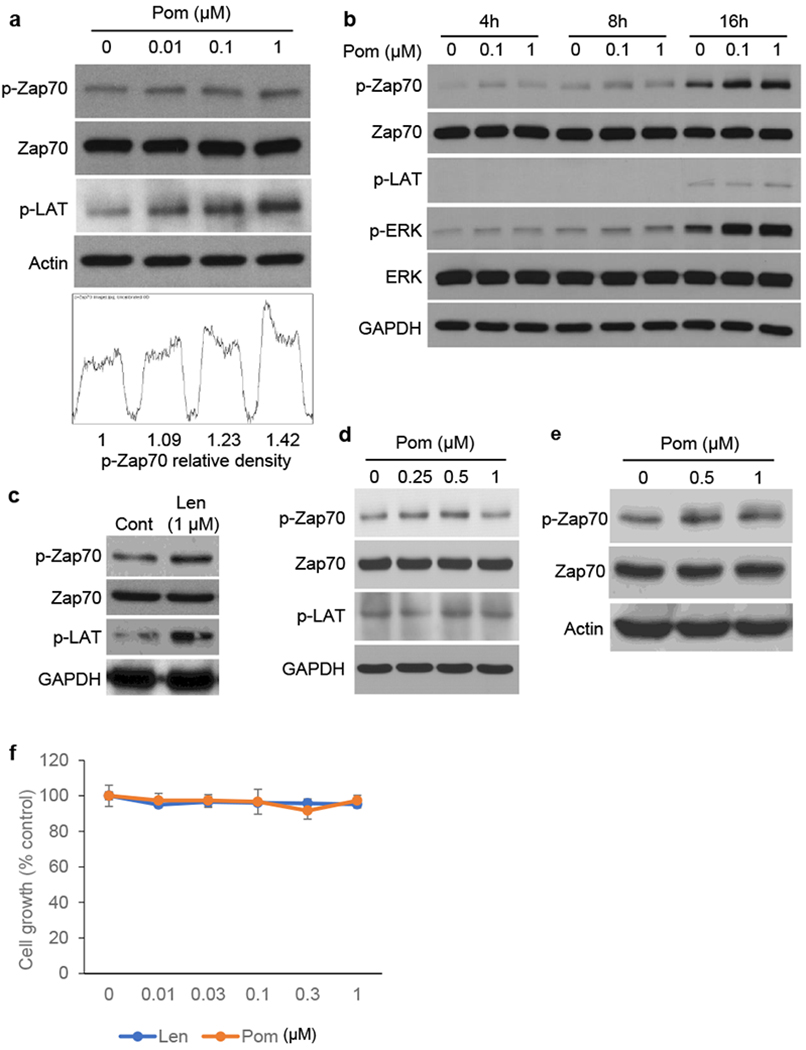
(a) PBMCs were cultured with Pom (0.01 – 1 μM) for 16h. Upper panel shows immunoblotting for Zap 70, p-Zap70 and p-LAT. Lower panel shows densitometric analysis of Zap-70. (b) PBMCs were cultured with Pom (0.1 and 1 μM) for the indicated time periods. (c) PBMCs were cultured with Len (1uM) for 16h. (d) Primary T-cells from healthy volunteer were cultured with Pom (0.25 – 1 μM) for 16h. (e) Jurkat cells were cultured with Pom (0.5 and 1 μM) for 16h. Whole cell lysates were subjected to immunoblotting (a - e) using indicated Abs. (f) Jurkat cells were cultured with Pom (0.01 – 1 μM) for 72h. Cell growth was assessed by MTT assay. Data are representative of two independent experiments and values are expressed in mean ± SD.
IMiDs directly bind and activate Zap-70
Since the Ab used for evaluation of p-Zap-70 (Cell Signaling Technology, catalogue # 2704) also recognizes p-Syk (spleen tyrosine kinase), we next validated that the increased phosphorylation observed by immunoblotting after Pom treatment was p-Zap-70. Specifically, we knocked down Zap-70 in Jurkat cells, and then immunoblotted cell lysates with p-Zap-70 and Zap-70 Abs; control cells were transfected with scrambled (Sc) siRNA and similarly immunoblotted. The control blot showed 2 bands (upper p-Syk and more prominent lower p-Zap-70), and the lower band was significantly downregulated in Zap-70 knock down cells (Fig. 2a). We also performed ELISA assay to specifically detect p-Zap-70 (Tyr319) in Jurkat cells. As expected, this assay also showed that both Len and Pom (Len < Pom) increased p-Zap-70 in a dose-dependent fashion (Fig. 2b).
Figure 2. IMiDs bind and activate Zap-70.
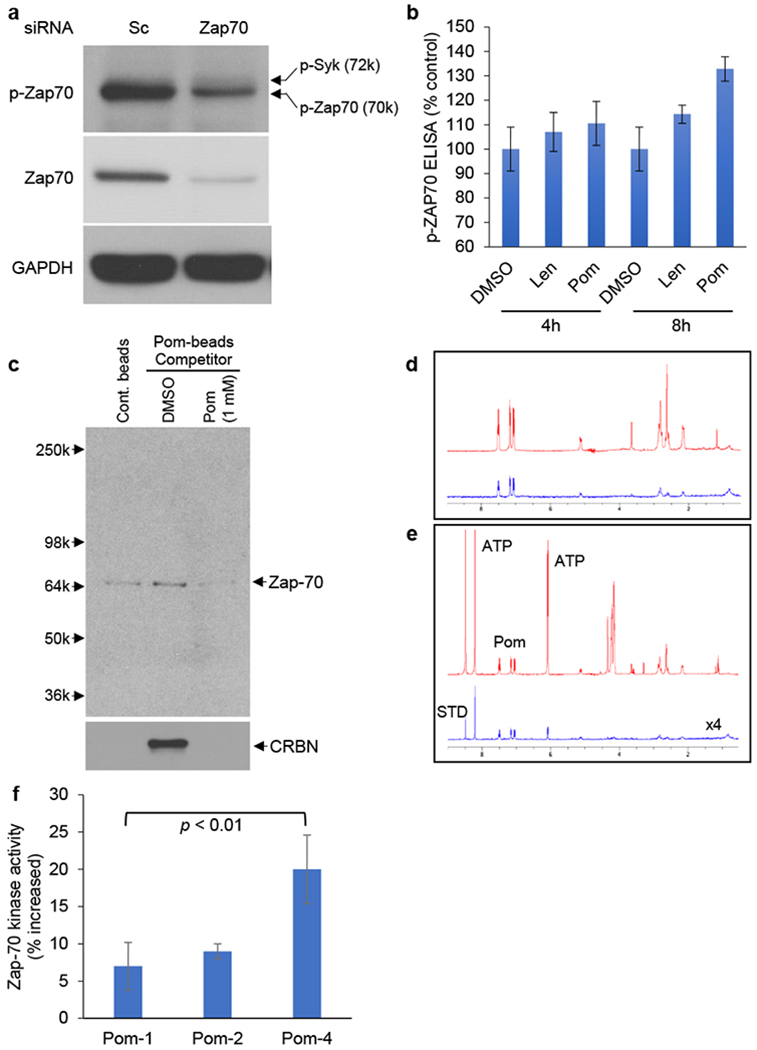
(a) Jurkat cells were transfected with scrambled (Sc) siRNA or Zap-70 siRNA. Whole cell lysates were subjected to immunoblotting using indicated Abs. (b) Jurkat cells were treated with Len (1 and 3 μM) or Pom (1 and 3 μM) for 4h and 8h. Whole cell lysates were subjected to p-Zap ELISA assay. (c) Jurkat whole cell lysates were incubated with Pom-beads in the presence or absence of competitor (1 mM free Pom) for 1h. After elution, samples were subjected to immunoblotting using anti-Zap-70 and CRBN Abs. (d) Saturation-transfer difference resulting from the binding of Pom to Za-70. Pom is at 320uM and Zap-70 is approximately 2uM in deuterated PBS solution. The top (red) spectrum shows the normal 1D spectrum for Pom plus protein, and the bottom (blue) spectrum shows the STD. (e) Saturation-transfer difference resulting from the binding of Pom to Zap-70 in the presence of 2.56mMATP. Pom is at 320uM and Zap70 is approximately 2uM in deuterated PBS solution. The top (red) spectrum shows the normal 1D spectrum for ATP plus Pom plus protein, and the bottom (blue) spectrum shows the STD. Note that both ATP and Pom show binding to Zap-70. (f) Non cell-based Zap-70 kinase assay was carried out, according to manufacturer’s protocol. For b and f, data are representative of three independent experiments and values are expressed in mean ± SD.
Our previous studies have shown that Pom binds not only to CRBN, but also to TP53RK, thereby inhibiting its function 8. By immunoblotting using Jurkat cell lysates, we here demonstrated that Pom-immobilized beads (Pom-beads) pulled down Zap-70, which was inhibited by free Pom. CRBN served as a positive control of pulled down protein by Pom-beads (Fig. 2c). As in our prior studies 8, we next carried out nuclear magnetic resonance (NMR) spectroscopy to confirm that Pom directly binds to Zap-70 (Fig. 2d and 2e). Len similarly binds to Zap-70 (Supplementary Fig. S1a and S1b). In vitro Zap-70 kinase assay confirmed that IMiDs induce activation of Zap-70 function via phosphorylation (Fig. 2f), consistent with upregulation of downstream p-LAT observed by immunoblotting (Figure 1a–1d). Taken together, these data show that IMIDs directly bind to Zap-70 and stimulate its activity.
Pom induced p-Zap-70 independent of CRBN
Since IMiDs are known to bind CRBN and trigger proteasomal degradation of IKZF1 and IKZF3, we next examined whether CRBN affects expression of Zap-70 or p-Zap-70 in Jurkat cells. We observed no significant change in constitutive Zap-70 (Fig. 3a) and p-Zap-70, or in p-Zap-70 induced by Pom (Fig. 3b), in CRBN-knockdown (KD) versus control Sc KD Jurkat cells. To evaluate the biologic role of Zap-70, we next knocked down Zap-70 in Jurkat cells (Fig. 3c, left panel), and observed significant inhibition of their cell growth. (Fig. 3c, right panel). These results suggest that Zap-70 is a growth factor and independent of CRBN in Jurkat cells.
Figure 3. CRBN expression does not regulate phosphorylation or protein expression of Zap-70.
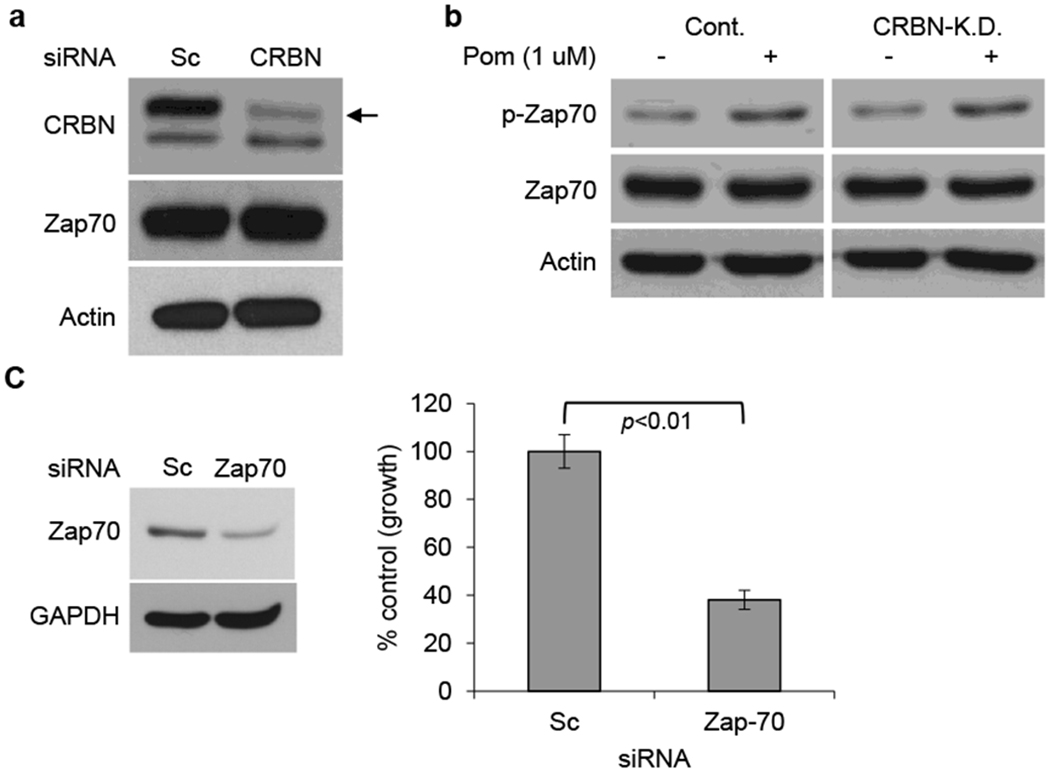
(a) Jurkat cells were transfected with scrambled siRNA (Scsi) or CRBN siRNA (CRBNsi). Whole cell lysates were subjected to immunoblotting using indicated Abs. (b) Jurkat cells transfected with Scsi or CRBNsi were cultured with Pom (1 μM) for 16h. Whole cell lysates were subjected to immunoblotting using indicated Abs. (c) Jurkat cells were transfected with Scsi or Zap-70 siRNA (Zap-70si) (left panel). The transfectants were further cultured for 72h, and cell growth was assessed by MTT assay (right panel). Data are representative of three independent experiments and values are expressed in mean ± SD.
Zap-70 mediates Pom-induced upregulation of NK cell activity
Zap-70 is a crucial mediator of T-cell receptor signaling 20; however, its role in NK cells has not yet been delineated. We therefore next validated the biologic impact of Zap-70 in NK cells using KHYG-1 NK cell line. As in PBMCs, Jurkat, or primary T-cells, Pom similarly enhanced p-Zap-70 in KHYG-1 cells (Fig. 4a), associated with increased cytotoxicity against U266 cells in a dose-dependent fashion (Fig. 4b). Importantly, Zap-70 KD significantly reduced cytotoxic activity of both Pom-treated (Fig. 4c) and Len-treated KHYG-1 cells (Supplementary Fig. S2a), without significantly impacting growth (Fig. 4d and Supplementary Fig. S2b). Finally, as in Jurkat cells (Fig 3a and 3b), CRBN KD in KHYG-1 cells did not alter constitutive Zap-70 protein and p-Zap-70, or Pom-induced p-Zap-70, expression (Fig. 4e). Consistent with KHYG-1 cells, Pom induced increased p-Zap70 and upregulated NK activity. Of note, neither Pom nor Zap-70 KD altered growth in NK-92 cells (Supplementary Fig S3a, S3b, S3c and S3d).
Figure 4. Zap-70 mediates Pom-induced upregulation of NK cell activity.
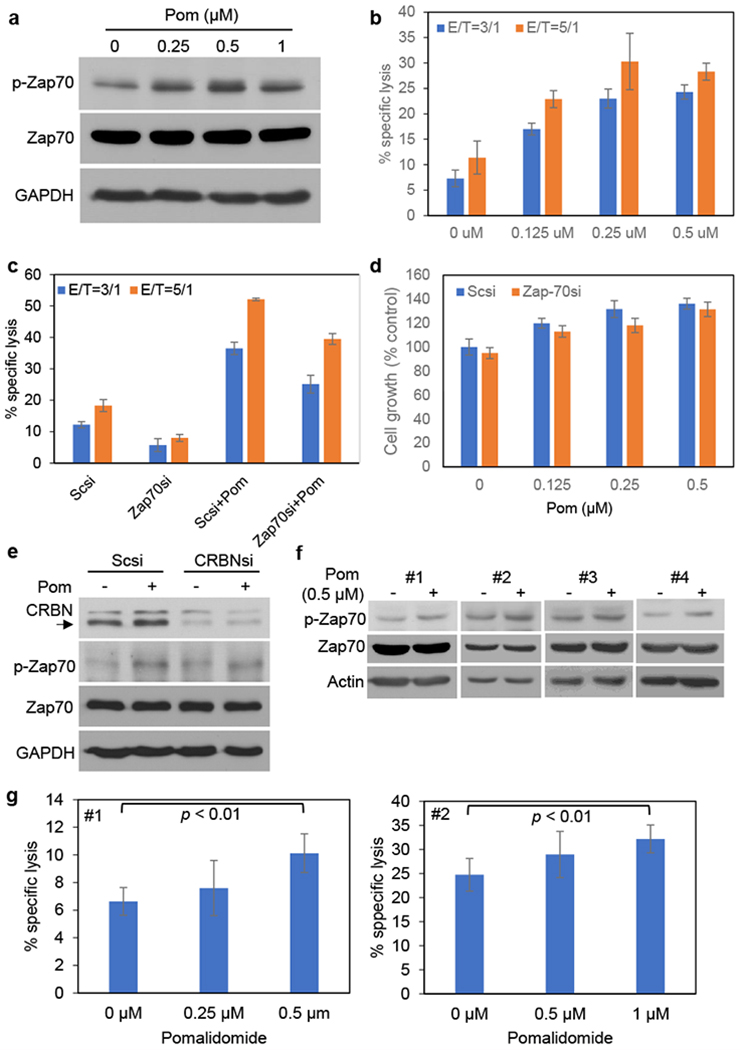
KHYG-1 cells were cultured with Pom (0.25 - 1 μM) for 24h. (a) Whole cell lysates were subjected to immunoblotting using indicated Abs. (b) KHYG-1 cells were incubated with calcein-AM-stained U266 cells for 4h at the indicated effector/target (E/T) ratios. Percent specific lysis was calculated as described previously. (c) KHYG-1 cells were transfected with Scsi or Zap-70si, and then cultured with Pom (0.25 μM) for 72h in the absence of IL-2. Viable cell number was determined, and cells were then incubated with calcein AM-labeled U266 target cells for 4h at indicated effector/target (E/T) ratios. Percent specific lysis was calculated as described previously. (d) After transfection with Scsi or Zap-70si, cells were cultured with Len or Pom for 72h. Cell growth was assessed by MTT assay. (e) KHYG-1 cells were transfected with scrambled (Scsi) or CRBNsi. The transfectants were then cultured with Pom (0.5 μM) for 24h, and whole cell lysates were subjected to immunoblotting using indicated Abs. The arrow indicates CRBN expression. (f) Primary NK cells (#1, #2, #3, #4) were isolated from healthy volunteer donor PBMCs, as described in Materials and Methods. NK cells were cultured with Pom (0.5 μM) for 24h, and whole cell lysates were subjected to immunoblotting using indicated Abs. (g) Isolated primary NK cells (#1, #2) were cultured with Pom (left panel: 0.25 and 0.5 μM, right panel: 0.5 and 1 μM) for 24h, and were then incubated with calcein AM-labeled U266 for 4h at E/T ratio of 5/1 (left panel) and 10/1 (right panel). Percent specific lysis was calculated as previously described. For b, c, d, data are representative at least two independent experiments and value are expressed in mean ± SD.
We similarly examined the effect of Pom on p-Zap-70 and NK cell activity in primary NK cells isolated from healthy volunteer donors (#1, #2, #3, #4). Importantly and as in KHYG-1 NK cell line, Pom upregulated p-Zap-70 in primary NK cells (Fig. 4f, #1, #2, #3, #4) and in a dose dependent fashion (0 – 1uM) significantly enhanced their NK cytolytic activity (Fig. 4g, #1, #2), without significantly effecting NK cell growth. (Supplementary Fig. S4, #1, #2). These results indicate that Zap-70 mediates, at least in part, constitutive and IMiDs-induced upregulation of NK cell activity.
Pom upregulates GZM-B expression via Zap-70
We next examined the molecular mechanism whereby IMiDs enhance NK cell activity. A previous study has demonstrated that Len upregulates GZM-B expression in MM patient T-cells 19. Here we showed that Pom (0-1uM) in a dose-dependent fashion upregulates GZM-B expression in both KHYG-1 cells (Fig. 5a) and primary NK cells (Fig. 5b, #1, #2). Similar results were observed in NK-92 cells treated with Pom (Supplementary Fig S3a). Since Zap-70 KD inhibited Pom-induced upregulation of KHYG-1 cell killing activity (Fig. 4c), we next examined whether Zap-70 KD also altered GZM-B expression. As expected, Zap-70 KD decreased both baseline and Pom-induced GZM-B upregulation in KHYG-1 cells (Fig. 5c). Consistent with KHYG-1 cells, we also observed downregulation of GZM-B in NK-92 cells after Zap70 KD (Supplementary Figure S5). Taken together, these results suggest that Zap-70 plays an important role regulating GZM-B expression in NK cells.
Figure 5. Pom upregulates granzyme-B expression via Zap-70.
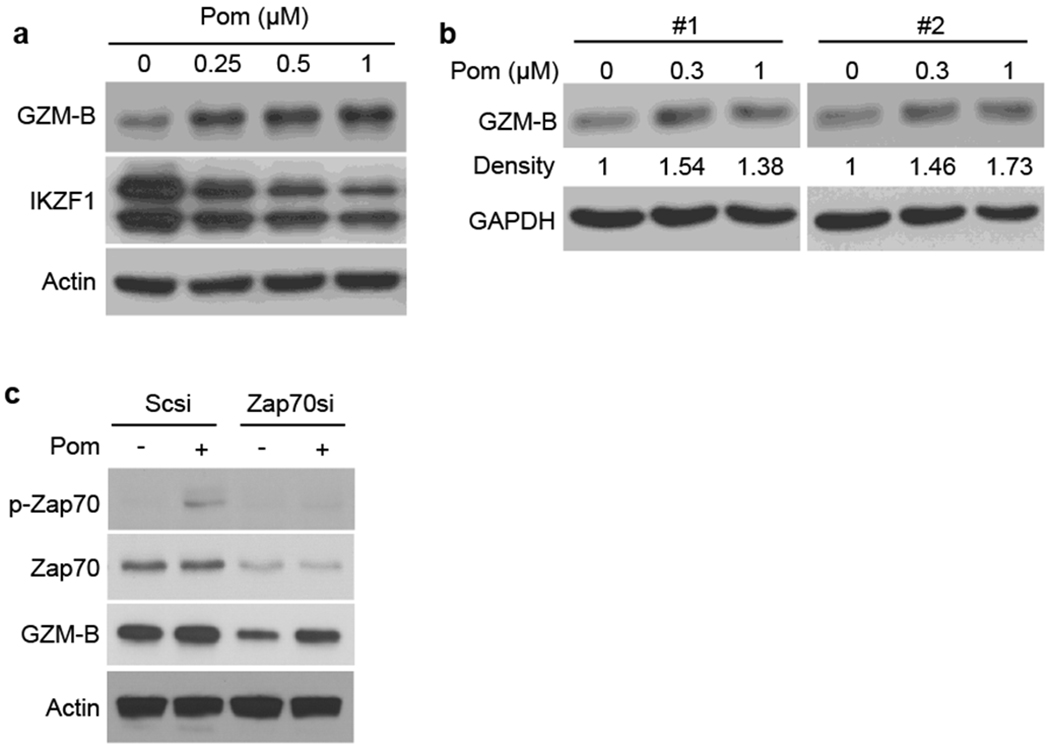
(a) KHYG1 cells were cultured with Pom (0.25 – 1 μM) for 24h. (b) Isolated primary NK cells from healthy volunteers (#1, #2) were cultured with Pom (0.3 – 1 μM) for 24h. (c) KHYG-1 cells were transfected with Scsi or Zap-70si. After 48h, cells were cultured for 24h in the absence or presence (0.5 μM) of Pom. (A-C) Whole cell lysates and RNAs were subjected to immunoblotting using indicated Abs.
Pom upregulates GZM-B expression via CRBN
We next examined whether CRBN also mediates Pom-induced GZM-B upregulation in KHYG-1 cells. Although CRBN KD minimally downregulated constitutive GZM-B expression, it significantly inhibited upregulation of GZM-B triggered by Pom (Fig. 6a). Real-time qPCR confirmed that CRBN transcriptionally regulates GZM-B expression (Supplementary Figure S6). Consistent with downregulation of GZM-B, both constitutive and Pom-induced cell killing activity was significantly inhibited in CRBN KD KHYG-1 cells (Fig. 6b). Taken together, these results indicate that Pom-induced enhanced GZM-B and NK cell activity is also mediated, at least in part, by CRBN.
Figure 6. Pom upregulates granzyme-B expression via CRBN.
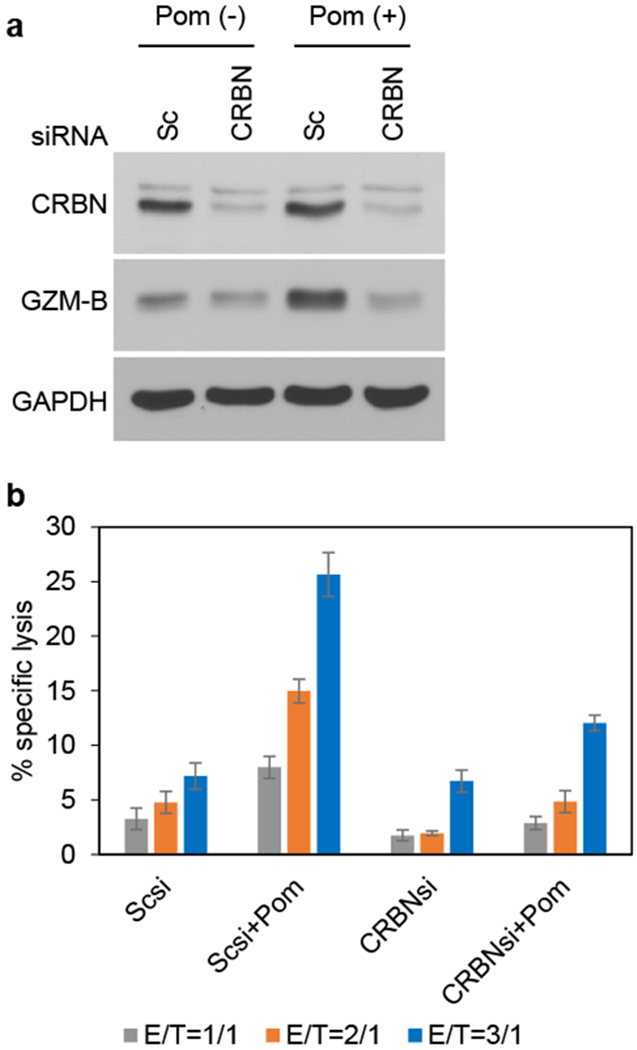
(a) KHYG-1 cells were transfected with Scsi or CRBNsi. After 48h, cells were cultured in the absence or presence of Pom (0.5 μM) for 24h, and cell lysates immunoblotted with indicated Abs. (b) KHYG-1 cells were transfected with Scsi or Zap-70si, and then cultured for 72h with Pom (0.25 μM), in the absence of IL-2. Cells were counted and incubated with calcein AM-labeled U266 for 4h at indicated effector/target (E/T) ratios. Data are representative two independent experiments and value are expressed in mean ± SD.
Pom upregulates granzyme-B expression via IKZF3
Since IKZF1 and/or IKZF3 are downstream degradation targets of CRBN, we next defined their roles in modulating constitutive and Pom-induced GZM-B expression. IKZF3 KD, but not IKZF1 KD, enhanced both baseline and Pom-induced GZM-B expression (Fig. 7a). These results indicate that IKZF3 serves a transcriptional repressor of GZM-B; and conversely, that Pom activation of CRBN E3 ligase and proteasomal degradation of IKZF3 leads to GZM-B upregulation in KHYG-1 cells. We further confirmed that IKZF3 KD significantly upregulated NK cell activity, which is further enhanced in the presence of Pom (Supplementary Fig. S7). Consistent with this view, proteasome inhibitor bortezomib downregulated Pom-induced GZM-B expression in a dose-dependent fashion, associated with upregulation of IKZF3 (Fig. 7b). CC-220 (iberdomide) is a more potent IMiD with enhanced binding affinity to CRBN relative to Len or Pom, and is now under evaluation in phase 1-2 clinical trials in multiple myeloma. We showed that CC-220 induced p-Zap-70 in a dose-dependent fashion (Supplementary Fig. S8a), which was associated with enhanced NK cell activity (Supplementary Fig. S8b). We next compared potency of Len, Pom, and CC-220 in triggering GZM-B in KHYG-1 cells. Interestingly, CC-220 more potently upregulated GZM-B than Len or Pom, which was associated with downregulation of IKZF3 (Fig. 7c). Real-time qPCR of GZM-B further supported this result (Fig. 7d). Of note, none of these IMiDs altered perforin expression, indicating that IMiDs-induced upregulation of NK cell activity is predominantly mediated by GZM-B (Fig. 7c). Taken together, our results show that IMiDs-induced GZM-B upregulation is differentially mediated in NK cells via Zap-70 and via CRBN/IKZF3 pathways.
Figure 7. IKZF3 plays a critical role in Pom-induced GZM-B expression.
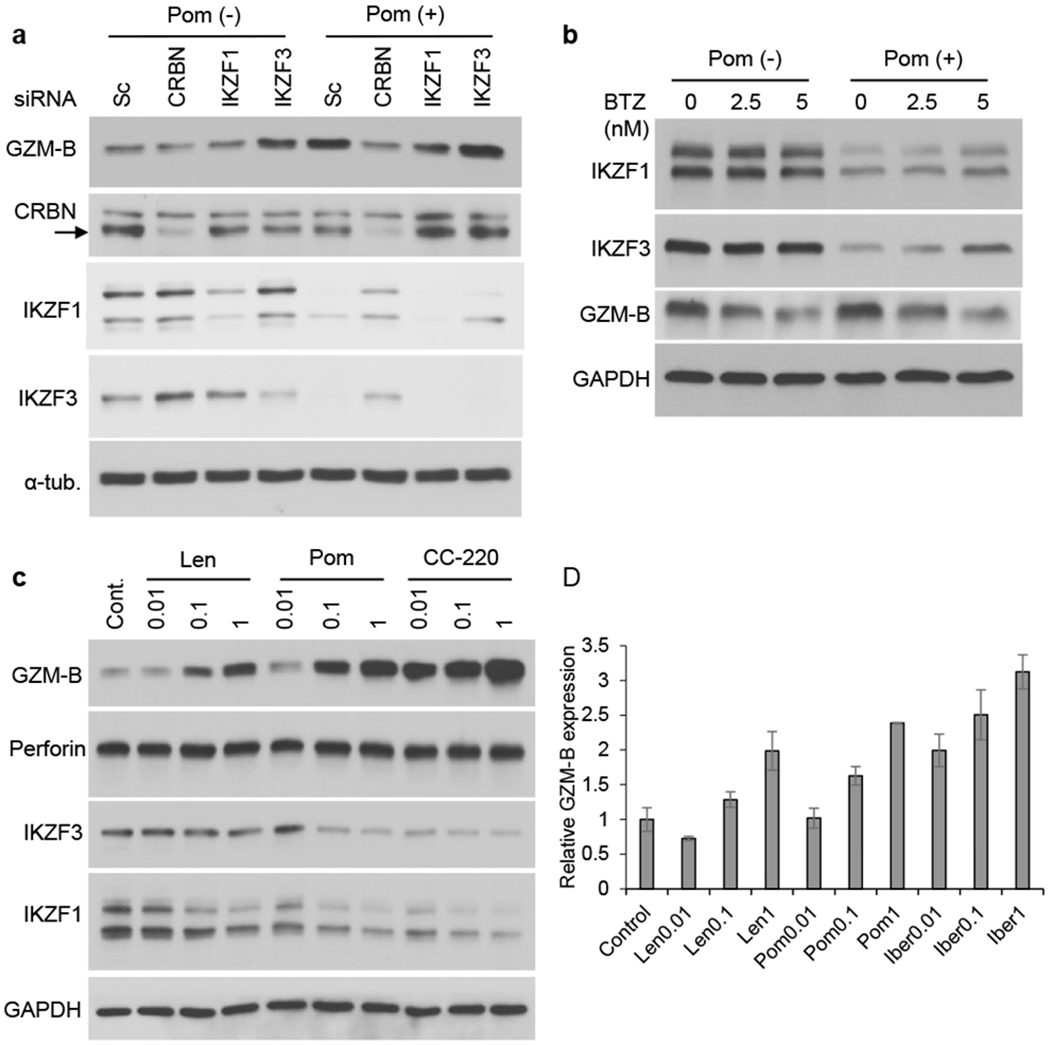
(a) KHYG-1 cells were transfected with CRBN, IKZF1, or IKZF3 siRNA. The transfectants were then cultured for 24h with Pom (0.5 μM). The arrow indicates CRBN. (b) KHYG-1 cells were cultured with Pom for 24h (0.5 μM), in the presence or absence of bortezomib (BTZ; 2.5 and 5 nM). (c, d) KHYG-1 cells were cultured for 24h with Len, Pom or CC-220 (0.01 – 1 μM). Whole cell lysates and RNAs were subjected to immunoblotting using indicated Abs (c) and real-time qPCR (d), respectively. Data are representative three independent experiments and value are expressed in mean ± SD.
Discussion
IMiDs trigger CRBN E3 ligase activity and proteasomal degradation of IKZF1 (Ikaros) and IKZF3 (Aiolos) 6, 7, followed by downregulation of c-Myc and IRF4 as well as inhibition of MM cell growth. We and others have also shown that IMiDs activate T-cells 9 and NK cells 12, 13. Since IKZF1/3 are repressors of IL-2 transcription in T cells, IMiDs activation of T cells by IMiDs is, at least in part, due to CRBN-mediated proteasomal degradation of IKZF1/3, thereby upregulating IL-2 transcription and secretion by T-cells 11, 21. More recent studies have shown that Len enhances the antigen-specific secretion of IFN-γ and GZM-B by MM patient T cells 19. Multiple studies have also shown that IMiDs enhance antibody-dependent cellular cytotoxicity (ADCC) mediated by anti-CD38 22 and anti-CD 20 23, 24 monoclonal Abs. To date, however, the molecular mechanism of IMiDs-induced NK cell activation has not been fully elucidated.
Zap-70 is a Syk family protein tyrosine kinase expressed in T cells, which plays a critical role mediating T cell activation in response to T cell receptor (TCR) engagement. Following TCR engagement, Zap-70 is rapidly phosphorylated on several tyrosine residues through autophosphorylation and transphosphorylation by the Src family lymphocyte-specific protein tyrosine kinase (Lck) 20. In turn, tyrosine phosphorylation of Zap-70 is associated with increased kinase activity and downstream signaling events in T cells. Importantly, Zap-70 expression has also been correlated with disease progression and identified as a prognostic factor in chronic lymphocytic leukemia 25. However, the biologic role of Zap-70 in IMiDs-induced T-cell and/or NK cell activation is not yet known. In this study, we show that Zap-70 is phosphorylated by IMiDs treatment in both T-cells and NK cells. We next demonstrated that IMiDs directly phosphorylate and activate Zap-70, as validated by in vitro non-cell-based Zap-70 kinase assays, Zap-70 pull-down experiments using Pom-immobilized beads, and NMR spectroscopy. Our Zap-70 kinase assay showed that IMiDs directly activate Zap-70 without any other co-factors except ATP; and both Pom-beads pull-down and NMR experiments also demonstrate direct binding of IMiDs to Zap-70. Taken together, these results indicate that IMiDs directly bind to Zap-70 and trigger its activation. Finally, previous studies show that IMiDs bind to CRBN to facilitate proteasomal degradation of target substrate proteins including IKZF1/3 6, 7 and CK1α 26. It has been also reported that Zap-70 is a proteasome substrate 27, 28. We therefore next asked whether CRBN could modulate constitutive or IMiDs-induced Zap-70 expression, and showed that neither phosphorylation nor protein expression of Zap-70 were altered by CRBN KD. Taken together, these results indicate that regulation of Zap-70 expression and its function are independent of CRBN E3 ligase activity.
The functional significance of Zap-70 activity in NK cells has not yet been reported. We therefore next studied the biologic impact of Zap-70 in mediating IMiDs-induced enhanced NK cell activity. Zap-70 downregulation did not significantly alter constitutive or IMiDs-induced proliferation, suggesting that it is not a growth or survival factor. Since Len enhances GZM-B release from antigen-specific T-cells 19, we next asked whether Len and/or Pom could enhance GZM-B expression in NK cells. Importantly, we confirmed that both Len and Pom (Pom > Len) upregulated GZM-B expression in a dose- and time-dependent fashion, associated with enhanced NK cell activity. Conversely, Zap-70 KD significantly inhibits IMiDs-induced GZM-upregulation, resulting in reduced NK cell activity. These results suggest that Zap-70 plays a role in maintaining constitutive and IMiDs induced killing activity in NK cells. Our ongoing studies are delineating Zap-70 downstream signaling modulating GZM-B expression.
Importantly, we here also observed that CRBN KD inhibits both constitutive and IMiDs-induced NK cell activity. To delineate the molecular mechanism whereby CRBN modulates NK cell activity, we examined whether CRBN KD, in the presence or absence of Pom, alters GZM-B expression in NK cells. As in Zap-70 KD, GZM-B expression was also downregulated in CRBN KD cells, both before and after Pom treatment. These results suggest that CRBN negatively regulates GZM-B expression. Previous studies show that IKZF1/3 are transcriptional repressors of IL-2; and that IMiDs-induced CRBN-mediated proteasomal degradation of IKZF1/3 relieves this repression, thereby enhancing IL-2 secretion from T-cells 11, 21. These data suggest that IKZF1/3 may be also a repressor of GZM-B gene expression in NK cells; and conversely, that CRBN KD may abrogate proteasomal degradation of IKZF1/3 triggered by IMiDs. We therefore individually knocked down CRBN, IKZF1 or IKZF3, and then assessed the impact on GZM-B expression in the presence or absence of IMiDs. Although IKZF3 protein expression was not completely downregulated in our KD cells, both constitutive and Pom-induced GZM-B were upregulated by IKZF3 KD. A previous study has demonstrated that IKZF1 binds to the promoter of GZM-B 29, but our results do not show significant GZM-B changes in IKZF1 KD cells. Rather they suggest that IKZF3, but not IKZF1, regulates GZM-M expression in NK cells.
Recently, more potent CRBN modulating agents including CC-220 (iberdomide) are being evaluated in MM clinical trials, with early evidence of activity even in the setting of Len and Pom resistance. These agents bind CRBN with a higher affinity and more potently trigger degradation of IKZF1/3 than Pom 30, 31. We therefore next examined the ability of CC-220 to trigger degradation of IKZF1/3 and upregulation of GZM-B. Importantly, CC-220 more potently degrades IKZF1/3 and upregulates GZM-B than either Len or Pom. In contrast, expression of perforin was not altered by Len, Pom, or CC-220, suggesting that IMiDs-induced enhanced cytotoxic activity is associated with upregulation of GZM-B, but not perforin, in NK cells.
Since IMiDs are often combined with dexamethasone (Dex) in MM therapies, we also examined the impact of Dex on Pom-induced NK cell activity in KHYG-1 cells and observed that Dex significantly downregulated NK cell activity, even in the presence of Pom (Supplementary Fig. S9). Although this should be evaluated in the clinical setting, our results suggest that Dex may have a negative impact on cytotoxic effector cells.
In conclusion, our results show that IMiDs: directly bind and activate Zap-70 associated with increased GZM-B expression and NK cell activity, which can be inhibited by Zap-70 knockdown independent of CRBN; and trigger GZM-B and NK cytotoxicity via CRBN-mediated IKZF3 degradation, which can be inhibited by KD of CRBN or IKZF-3 independent of Zap-70. IMiDs can therefore enhance NK and T cell cytotoxicity in 1) ZAP-70-mediated CRBN independent, as well as 2) CRBN-mediated ZAP-70 independent mechanisms. These studies provide the basis for developing novel therapeutics to activate Zap-70 and thereby enhance T and NK cell anti-MM cytotoxicity.
Supplementary Material
Acknowledgement
We thank TAIHO PHARMACEUTICALS for providing Pom-immobilized beads.
This study was supported by the National Institute of Health Grant; SPORE-P50100707 (KCA), R01-CA050947 (K.C.A.) and R01-CA178264 (TH and KCA); and the Sheldon and Miriam Medical Research Foundation (K.C.A). K.C.A. is an American Cancer Society Clinical Research Professor.
Footnotes
Compliance with ethical standards
Conflict of Interest
KC Anderson serves on advisory boards to Celgene, Millennium, Janssen, Sanofi, Bristol Myers Squibb, Gilead, Precision Biosciences, and Tolero, and is a Scientific Founder of OncoPep and C4 Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Hideshima T, Chauhan D, Shima Y, Raje N, Davies FE, Tai Y-T, et al. Thalidomide and its analogues overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 2000; 96: 2943–50. [PubMed] [Google Scholar]
- 2.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood 2002; 99: 4525–30. [DOI] [PubMed] [Google Scholar]
- 3.Anderson KC. The Rapid Evolution of Novel Therapies in Multiple Myeloma. J Natl Compr Canc Netw 2016; 14: 493–96. [DOI] [PubMed] [Google Scholar]
- 4.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science 2010; 327: 1345–50. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012; 26: 2326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014; 343: 301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014; 343: 305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hideshima T, Cottini F, Nozawa Y, Seo HS, Ohguchi H, Samur MK, et al. p53-related protein kinase confers poor prognosis and represents a novel therapeutic target in multiple myeloma. Blood 2017; 129: 1308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004; 103: 1787–90. [DOI] [PubMed] [Google Scholar]
- 10.Gorgun G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010; 116: 3227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4 (CRBN). Br J Haematol 2014; 164: 811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001; 98: 210–16. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol 2005; 128: 192–203. [DOI] [PubMed] [Google Scholar]
- 14.Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, et al. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood 2006; 108: 618–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol 2008; 140: 36–45. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res 2008; 14: 4650–57. [DOI] [PubMed] [Google Scholar]
- 17.Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, et al. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood 2013; 121: 423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pittari G, Vago L, Festuccia M, Bonini C, Mudawi D, Giaccone L, et al. Restoring natural killer cell immunity against multiple myeloma in the era of new drugs. Front Immunol 2017; 8: 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuber B, Dai J, Waraich WA, Awwad MHS, Engelhardt M, Schmitt M, et al. Lenalidomide overcomes the immunosuppression of regulatory CD8(+)CD28(−) T-cells. Oncotarget 2017; 8: 98200–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, et al. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol 2010; 2: a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagner PR, Chiu H, Ortiz M, Apollonio B, Wang M, Couto S, et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br J Haematol 2017; 179: 399–409. [DOI] [PubMed] [Google Scholar]
- 22.van de Donk N Immunomodulatory effects of CD38-targeting antibodies. Immunol Lett 2018; 199: 16–22. [DOI] [PubMed] [Google Scholar]
- 23.Leonard JP, Jung SH, Johnson J, Pitcher BN, Bartlett NL, Blum KA, et al. Randomized trial of lenalidomide alone versus lenalidomide plus rituximab in patients with recurrent follicular lymphoma: CALGB 50401 (Alliance). J Clin Oncol 2015; 33: 3635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leonard JP, Trneny M, Izutsu K, Fowler NH, Hong X, Zhu J, et al. AUGMENT: A phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol 2019; 37: 1188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Wang Y, Yang J, Bi Y, Wang H. ZAP-70 in chronic lymphocytic leukemia: A meta-analysis. Clin Chim Acta 2018; 483: 82–8. [DOI] [PubMed] [Google Scholar]
- 26.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015; 523: 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paolini R, Molfetta R, Piccoli M, Frati L, Santoni A. Ubiquitination and degradation of Syk and ZAP-70 protein tyrosine kinases in human NK cells upon CD16 engagement. Proc Natl Acad Sci U S A 2001; 98: 9611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang M, Chen T, Li X, Yu Z, Tang S, Wang C, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nat Immunol 2015; 16: 1253–62. [DOI] [PubMed] [Google Scholar]
- 29.Wargnier A, Legros-Maida S, Bosselut R, Bourge JF, Lafaurie C, Ghysdael CJ, et al. Identification of human granzyme B promoter regulatory elements interacting with activated T-cell-specific proteins: implication of Ikaros and CBF binding sites in promoter activation. Proc Natl Acad Sci U S A 1995; 92: 6930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito T, Handa H. Recent topics in IMiDs and cereblon. Rinsho Ketsueki 2017; 58: 2067–73. [DOI] [PubMed] [Google Scholar]
- 31.Matyskiela ME, Zhang W, Man HW, Muller G, Khambatta G, Baculi F, et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem 2018; 61: 535–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.