

Abstract
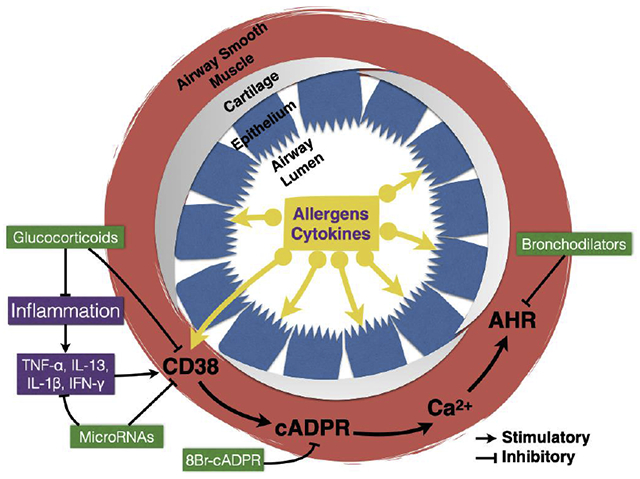
The worldwide socioeconomical burden associated with chronic respiratory diseases is substantial. Enzymes involved in the metabolism of nicotinamide adenine dinucleotide (NAD) are increasingly been implicated in chronic airway diseases. One such enzyme, CD38, utilizes NAD to produce several metabolites, including cyclic ADP ribose (cADPR), which is involved in calcium signaling in airway smooth muscle (ASM). Up-regulation of CD38 in ASM caused by exposure to cytokines or allergens lead to enhanced calcium mobilization by agonists and the development of airway hyperresponsiveness (AHR) to contractile agonists. Glucocorticoids and microRNAs can suppress CD38 expression in ASM, whereas cADPR antagonists such as 8Br-cADPR can directly antagonize intracellular calcium mobilization. Bronchodilators act via CD38-independent mechanisms. CD38-dependent mechanisms could be developed for chronic airway diseases therapy.
Keywords: Asthma, COPD, calcium, bronchoconstriction
Graphical abstract
Introduction
Chronic obstructive pulmonary disease (COPD) and asthma are the most common chronic respiratory diseases and are among the leading causes of morbidity and mortality worldwide. In 2015, it was estimated that 358 million people across all ages and ethnicities were affected by asthma and 174 million by COPD, although the death burden from COPD was eight times higher than from asthma [1]. In the United States, chronic obstructive lung diseases (e.g., asthma and COPD) are the 3rd leading cause of death. Among Americans of all age groups, the estimated lifetime asthma prevalence is 8.4% [2] with an associated economic burden of $81.9 billion dollars [3]. Whereas the risk of COPD is largely related to modifiable behaviors (e.g., smoking), risk factors for asthma appear to be substantially more heterogeneous and less understood. Use of symptomatic therapy (e.g., corticosteroids, bronchodilators) has significantly decreased asthma mortality but the overall asthma prevalence is on the rise [1]. A better understanding of underlying causes and mechanisms is necessary to improve disease prevalence. This brief review intends to summarize current knowledge regarding the role of CD38 in the pathogenesis of chronic obstructive pulmonary diseases. The reader is kindly referred to recent, more in depth reviews if interested in more details regarding regulatory mechanisms [4] and therapeutic potential [5].
Nicotinamide adenine dinucleotide (NAD) in health and disease
The importance of nicotinamide adenine dinucleotide (NAD) and related metabolic enzymes in health and disease is becoming increasingly appreciated [6]. For example, sirtuins (SIRT) are a family of NAD-dependent enzymes with an increasingly recognized role in metabolism, ageing [7], as well as in airway inflammation during allergic airway diseases [8; 9]. Another NAD-consuming enzyme, poly(ADP-ribose) polymerase (PARP), appears to be involved in airway inflammation, hyperresponsiveness and remodeling [10–13]. Finally, CD38 is another NAD-consuming enzyme that has emerged as an important determinant of airway hyperresponsiveness (AHR) associated with allergic airway diseases [14–31]. This body of literature suggests that NAD metabolism may be an important determinant of altered airway function in chronic obstructive pulmonary diseases and constitute a feasible therapeutic target.
CD38 protein
CD38 is a type II, 45 kDa glycoprotein with 300 amino acids distributed in three domains comprised by a short N-terminal cytoplasmic tail (21 amino acids), a single chain transmembrane portion (23 amino acids) and a large C-terminal extracellular domain (256 amino acids) [32]. The short cytoplasmic tail does not appear to contain any known motifs (Src homology domain 2 or 3 – SH2 or SH3, antigen receptor activation – ARAM, or pleckstrin homology – PH) that could mediate interactions with other signaling molecules and no enzymatic activity has been demonstrated [32; 33]. The enzymatic activities reside in the extracellular domain whereby CD38 catalyzes the cleavage of the nicotinamide group from NAD (NADase activity) leading to the formation of an enzyme-ADP-ribosyl intermediary complex, which then partitions between two possible pathways. An intramolecular reaction between the ribosyl carbon-1 and the nitrogen-1 of the adenine ring (ADP-ribosyl cyclase activity) yields cyclic adenosine diphosphate ribose (cADPR; ~3% final product) and a macroscopically irreversible hydrolysis reaction (cADPR hydrolase or NAD glycohydrolase) produces adenosine diphosphate ribose (ADPR; ~97% final product). Thus, CD38 is a more efficient NAD glycohydrolase (i.e., direct conversion of NAD to ADPR) than ADP-ribosyl cyclase [34–36]. CD38 is also responsible for the generation of nicotinic acid adenine dinucleotide phosphate (NAADP) from NADP. The cADPR and NAADP products regulate Ca2+ signaling and contractility in smooth muscle cells [22].
CD38 and immunity
CD38 is a pleiotropic cell surface molecule within the immune system, acting as a multifunctional enzyme and receptor. CD38 generates transmembrane signals upon engagement with its counter-receptor, CD31, with agonistic monoclonal antibodies and during G-protein coupled receptor activation. The effects mediated include production of pro-inflammatory and regulatory cytokines by monocytes, NK cells, activated B and T lymphocytes, proliferation of T lymphocytes and protection of mature B lymphocytes from apoptosis. Defective CD38 signaling in neutrophils impairs innate immunity to bacterial infection, and inadequate dendritic cell priming of B- and T-cells affect humoral immune responses to antigens [37].
CD38 and airway function
Our laboratory was the first to demonstrate the contribution of CD38 to the regulation of airway hyperresponsiveness (AHR). We first determined the airway phenotype of CD38 knockout mice as compared to that of the wild-type mice. Airway challenge with inhaled methacholine resulted in significantly attenuated changes in resistance to airflow and dynamic compliance in the CD38 knockout mice compared to wild-type controls [27]. Additionally, ASM cells isolated from CD38 knockout mice developed significantly lower intracellular calcium responses to acetylcholine and endothelin-1 than wild-type controls. In ASM cells, CD38 utilizes NAD to form cADPR and ADPR, but the release of calcium in response to agonist activation is cADPR-dependent as it can be antagonized by the competitive cADPR antagonist 8Br-cADPR [27]. These evidences suggested that the attenuated airway responsiveness to inhaled methacholine likely stemmed from decreased intracellular calcium responses in ASM cells due to the loss of CD38 activity and cADPR production in these cells.
Since asthma is an inflammatory disease in which pro-inflammatory cytokines have a role in inducing abnormal ASM function and in the development of AHR, we determined the methacholine responsiveness of CD38 knockout and wild-type mice exposed to the asthma-relevant cytokines IL-13 and TNF-α [25; 26]. It was previously known that the pro-inflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-1β, interferon (IFN)-γ or the T helper type 2 (TFI2) cytokine IL-13 significantly up-regulate CD38 expression, cADPR production and intracellular calcium responsiveness in ASM cells [23; 24; 38–41]. Intranasal challenge with IL-13 induced robust AHR to inhaled methacholine in wild-type mice whereas AHR was significantly lower in the CD38 knockout mice. The ASM reactivity to inhaled methacholine was significantly lower in the CD38 knockout mice than in wild-type controls. Using tracheal ring preparations, we showed that CD38 within ASM mediated the IL-13-associated increase in isometric force generation to carbachol stimulation whereas absence of CD38 did not affect relaxation of ASM to the β-adrenergic receptor agonist isoproterenol [25]. A single intranasal challenge with TNF-α caused AHR to inhaled methacholine in wild-type but not in CD38 knockout mice whereas the differences in airway phenotype were no longer present following more prolonged exposure to TNF-α [26]. Together, these results provided evidence that CD38 expressed in ASM cells plays a significant role in AHR to two asthma-relevant cytokines. More recent findings showed that the CD38 signaling pathway mediated AHR due to high-fat diet in mice, thus extending its possible role to obese asthma phenotype [30].
Earlier studies suggested that CD38 knockout mice developed attenuated T cell-dependent humoral immune response to protein antigens [42]. The underlying mechanisms for this attenuated allergen-induced antibody response were complex and included inefficient migration of CD38-deficient dendritic cells from sites of inflammation to the draining lymph nodes and defective priming of T cells [37]. Interestingly, airway inflammation in response to IL-13 was similarly robust in both wild-type and CD38 knockout mice, yet AHR was significantly lower in the former [25]. The role of CD38 in the responses to intranasal challenge with TNF-α also seemed disconnected from the development of airway inflammation [26]. Since asthma is triggered by allergen exposure leading to airway inflammation and release of cytokines and chemokines, we asked whether CD38 knockout mice would develop an attenuated airway phenotype following allergen sensitization and challenge [14]. We compared the airway responsiveness of CD38 knockout and wild-type mice following intranasal sensitization and intranasal challenge with fungal antigens as well as following intraperitoneal sensitization followed by intranasal challenge with ovalbumin antigen. Regardless of the route of allergen sensitization, the CD38 knockout mice developed significantly attenuated AHR to inhaled methacholine compared to wild-type mice. Once again, the airway inflammatory response characterized by the number and types of inflammatory cells in the bronchoalveolar lavage (BAL) fluid was comparable in the two groups of mice. Furthermore, the changes in BAL fluid content of IL-5 and IL-13 were similar between CD38 knockout and wild-type mice in both allergen models. However, the magnitude of increase in eotaxin-2 levels in BAL fluid was significantly less in the CD38 knockout mice following fungal allergen challenge but not following ovalbumin. These findings indicated that CD38 expression is relevant for the development of heightened AHR following allergen sensitization and challenge, but appears to be a less important determinant of airway inflammation.
To further explore this mechanism, we developed bone marrow chimeras to assess if reciprocal transfer of bone marrow-derived cells from wild-type and CD38 knockout mice would restore the airway phenotype in the hosts. The airway phenotype under naive conditions was unchanged by reciprocal bone marrow transfer. Following ovalbumin challenge, the airway phenotype in CD38 knockout mice was partially reversed by bone marrow transfer from either source whereas the airway phenotype of WT hosts was preserved [14]. These results thus confirm that loss of CD38 in hematopoietic cells is insufficient to prevent AHR to allergen and thus the contribution of CD38 to AHR stems from its presence and activity in resident airway cells. Altogether, the above findings in the cytokine and allergen models are consistent with the role of CD38 in ASM calcium responses and contractility to agonists and suggest that this CD38 mechanism is an important contributor to heightened bronchoconstriction in allergic airway diseases. Interestingly, injection of dendritic cells overexpressing CD38 into ovalbumin-immunized female mice prior to ovalbumin challenge led to lower airway inflammation compared to controls as well as increased in the levels of the Th1 cytokine IFN-γ concurrently with a decrease in the Th2 cytokine IL-4 [43]. Thus, it is possible that CD38 expression in dendritic cells and ASM cells has opposing roles in terms of inflammation and AHR during allergic airway disease.
The presence of increased CD38 expression in the lungs of asthmatic human patients was first reported in a study analyzing its distribution in human tissues [44]. Since then, the role of CD38 in human ASM cell function has been extensively investigated in cells obtained from patients with asthma and healthy controls [15–21; 23; 28; 29]. In human ASM from asthmatics, CD38 amplifies calcium signaling and inflammatory response to pro-inflammatory cytokines via mechanisms involving both transcriptional and post-transcriptional regulation [16–21; 24]. Glucocorticoids, which are commonly used in both asthma and COPD therapy [45; 46], significantly suppress TNF-α-induced CD38 expression in human ASM from asthmatics [15; 17; 20]. Treatment of human ASM with microRNAs with (miRNAs) miR-140-3p and miR-708 negatively regulate CD38 expression by mechanisms that involve translational repression as well as indirectly by transcriptional mechanisms [19]. This results in profound anti-inflammatory effect as evidenced by significant decreases in the expression and release of several chemokines and asthma related genes [47]. Thus, there is good understanding of the role of CD38 in ASM from human asthmatics.
The role of CD38 on COPD is less clear. In COPD patients, expression of CD38 occurs in both alveolar and interstitial macrophages (small and large), although expression is greater in the small macrophages [48]. “Small” and “large” macrophage subpopulations have been identified in the sputum of COPD patients based on pattern of forward scatter during flow cytometry analysis. Compared to large macrophages, COPD small macrophages show higher levels of TNF-α, both constitutively and in response to lipopolysaccharide challenge [49]. Current smoking on COPD patients is associated with lower CD38 expression in large interstitial macrophages compared to ex-smokers [48]. Levels of a soluble form of CD38 (sCD38) in the serum and exhaled breath condensate of patients with moderate COPD were higher than in healthy non-smoker controls [50], but the significance of this finding is unclear. In addition, patients with significant smoking history (> 25 pack years) had significantly higher CD38 expression in epithelium-denuded bronchial smooth muscle when compared to lifetime non-smokers. In vitro, cigarette smoke extract concentration-dependently increased CD38 expression and calcium responsiveness in human ASM, as well as enhanced TNF-α release and proliferative capacity [51]. Lastly, in vitro evidence suggests that CD38 might play a role in the response to respiratory viral infections [52], which are known to worsen the symptoms of asthma and COPD patients. In that study, the inflammatory response of human monocyte-derived dendritic cells infected with respiratory syncytial virus was significantly reduced by inhibiting CD38 enzymatic activity with the flavonoid kuromanin, as well as by inhibiting CD38 downstream signaling with the cADPR antagonist 8Br-cADPR [52].
Conclusions and future directions
This review outlines the current understanding of the role of CD38/cADPR signaling in chronic obstructive pulmonary diseases. Cytokines, glucocorticoids and miRNAs modulate CD38 expression in human ASM, especially in asthmatics. Furthermore, CD38 significantly contributes to the asthmatic phenotype in mice challenged with inhaled allergens. The role CD38 in the pathophysiology of COPD appears to be emerging, but additional studies are necessary to elucidate this possibility. Studies are warranted to better understand the epigenetic interactions between the CD38 signaling system and factors such as viral infections, cigarette smoking and ozone, which contribute to or exacerbate obstructive pulmonary diseases such as asthma and COPD.
Acknowledgments
Mathur S. Kannan and Deepak A. Deshpande received support through grants from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
References:
- [1].Collaborators, G.B.D.C.R.D. (2017) Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med 5, 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA and Liu X (2012) Trends in asthma prevalence, health care use, and mortality in the United States, 2001-2010. NCHS Data Brief, 1–8. [PubMed] [Google Scholar]
- [3].Nurmagambetov T, Kuwahara R and Garbe P (2018) The Economic Burden of Asthma in the United States, 2008-2013. Ann Am Thorac Soc 15, 348–356. [DOI] [PubMed] [Google Scholar]
- [4]*.Deshpande DA, Guedes AGP, Graeff R, Dogan S, Subramanian S, Walseth TF and Kannan MS (2018) CD38/cADPR Signaling Pathway in Airway Disease: Regulatory Mechanisms. Mediators Inflamm 2018, 8942042. [DOI] [PMC free article] [PubMed] [Google Scholar]; In an in depth review, the authors outline how the CD38 signaling in airway smooth muscle is regulated in conditions relevant to allergic airway diseases.
- [5]*.Deshpande DA, Guedes AGP, Lund FE, Subramanian S, Walseth TF and Kannan MS (2017) CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacol Ther 172, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a comprehensive review in which the authors describe using text and diagrams the feasibility of targeting CD38 for therapy of allergic airway diseases.
- [6].Connell NJ, Houtkooper RH and Schrauwen P (2019) NAD(+) metabolism as a target for metabolic health: have we found the silver bullet? Diabetologia 62, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Houtkooper RH, Pirinen E and Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nature Reviews Molecular Cell Biology 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ma K, Lu N, Zou F and Meng FZ (2019) Sirtuins as novel targets in the pathogenesis of airway inflammation in bronchial asthma. Eur J Pharmacol 865, 172670. [DOI] [PubMed] [Google Scholar]
- [9].Colley T, Mercado N, Kunori Y, Brightling C, Bhavsar PK, Barnes PJ and Ito K (2016) Defective sirtuin-1 increases IL-4 expression through acetylation of GATA-3 in patients with severe asthma. J Allergy Clin Immunol 137, 1595–1597.e1597. [DOI] [PubMed] [Google Scholar]
- [10].Sethi GS, Sharma S and Naura AS (2019) PARP inhibition by olaparib alleviates chronic asthma-associated remodeling features via modulating inflammasome signaling in mice. IUBMB life 71, 1003–1013. [DOI] [PubMed] [Google Scholar]
- [11].Oumouna M, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, Fallon K and Boulares H (2006) Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol 177, 6489–6496. [DOI] [PubMed] [Google Scholar]
- [12].Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al-Khami AA, Lammi MR, Kim H, Zea AH, Davis C, Okpechi S, Wyczechowska D, Al-Ghareeb K, Mansy MS, Ochoa A, Naura AS and Boulares AH (2015) PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite-induced disease in mice. Clin Sci (Lond) 129, 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ghonim MA, Pyakurel K, Ibba SV, Al-Khami AA, Wang J, Rodriguez P, Rady HF, El-Bahrawy AH, Lammi MR, Mansy MS, Al-Ghareeb K, Ramsay A, Ochoa A, Naura AS and Boulares AH (2015) PARP inhibition by olaparib or gene knockout blocks asthma-like manifestation in mice by modulating CD4(+) T cell function. J Transl Med 13, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guedes AG, Jude JA, Paulin J, Rivero-Nava L, Kita H, Lund FE and Kannan MS (2015) Airway responsiveness in CD38-deficient mice in allergic airway disease: studies with bone marrow chimeras. Am J Physiol Lung Cell Mol Physiol 308, L485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kang BN, Jude JA, Panettieri RA Jr., Walseth TF and Kannan MS (2008) Glucocorticoid regulation of CD38 expression in human airway smooth muscle cells: role of dual specificity phosphatase 1. Am J Physiol Lung Cell Mol Physiol 295, L186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jude JA, Tirumurugaan KG, Kang BN, Panettieri RA, Walseth TF and Kannan MS (2012) Regulation of CD38 expression in human airway smooth muscle cells: role of class I phosphatidylinositol 3 kinases. Am J Respir Cell Mol Biol 47, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tirumurugaan KG, Kang BN, Panettieri RA, Foster DN, Walseth TF and Kannan MS (2008) Regulation of the cd38 promoter in human airway smooth muscle cells by TNF-alpha and dexamethasone. Respiratory research 9, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jude JA, Dileepan M, Subramanian S, Solway J, Panettieri RA Jr., Walseth TF and Kannan MS (2012) miR-140-3p regulation of TNF-alpha-induced CD38 expression in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 303, L460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dileepan M, Jude JA, Rao SP, Walseth TF, Panettieri RA, Subramanian S and Kannan MS (2014) MicroRNA-708 regulates CD38 expression through signaling pathways JNK MAP kinase and PTEN/AKT in human airway smooth muscle cells. Respiratory research 15, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF and Kannan MS (2006) Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. FASEB J 20, 1000–1002. [DOI] [PubMed] [Google Scholar]
- [21].Kang BN, Deshpande DA, Tirumurugaan KG, Panettieri RA, Walseth TF and Kannan MS (2005) Adenoviral mediated anti-sense CD38 attenuates TNF-alpha-induced changes in calcium homeostasis of human airway smooth muscle cells. Can J Physiol Pharmacol 83, 799–804. [DOI] [PubMed] [Google Scholar]
- [22].Deshpande DA, White TA, Dogan S, Walseth TF, Panettieri RA and Kannan MS (2005) CD38/cyclic ADP-ribose signaling: role in the regulation of calcium homeostasis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 288, L773–788. [DOI] [PubMed] [Google Scholar]
- [23].Deshpande DA, Walseth TF, Panettieri RA and Kannan MS (2003) CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyperresponsiveness. FASEB J 17, 452–454. [DOI] [PubMed] [Google Scholar]
- [24].Deshpande DA, Dogan S, Walseth TF, Miller SM, Amrani Y, Panettieri RA and Kannan MS (2004) Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: role of CD38/cyclic adenosine diphosphate ribose pathway. Am J Respir Cell Mol Biol 31, 36–42. [DOI] [PubMed] [Google Scholar]
- [25].Guedes AG, Paulin J, Rivero-Nava L, Kita H, Lund FE and Kannan MS (2006) CD38-deficient mice have reduced airway hyperresponsiveness following IL-13 challenge. Am J Physiol Lung Cell Mol Physiol 291, L1286–1293. [DOI] [PubMed] [Google Scholar]
- [26].Guedes AG, Jude JA, Paulin J, Kita H, Lund FE and Kannan MS (2008) Role of CD38 in TNF-alpha-induced airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol 294, L290–299. [DOI] [PubMed] [Google Scholar]
- [27].Deshpande DA, White TA, Guedes AG, Milla C, Walseth TF, Lund FE and Kannan MS (2005) Altered airway responsiveness in CD38-deficient mice. Am J Respir Cell Mol Biol 32, 149–156. [DOI] [PubMed] [Google Scholar]
- [28].Tirumurugaan KG, Jude JA, Kang BN, Panettieri RA, Walseth TF and Kannan MS (2007) TNF-alpha induced CD38 expression in human airway smooth muscle cells: role of MAP kinases and transcription factors NF-kappaB and AP-1. Am J Physiol Lung Cell Mol Physiol 292, L1385–1395. [DOI] [PubMed] [Google Scholar]
- [29].Jude JA, Solway J, Panettieri RA Jr., Walseth TF and Kannan MS (2010) Differential induction of CD38 expression by TNF-{alpha} in asthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 299, L879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]*.Chong L, Zhang W, Yu G, Zhang H, Zhu L, Li H, Shao Y and Li C (2018) High-fat-diet induces airway hyperresponsiveness partly through activating CD38 signaling pathway. Int Immunopharmacol 56, 197–204. [DOI] [PubMed] [Google Scholar]; Using a mouse model of obesity, the authors showed upregulation of the CD38 signaling in lungs and airway smooth muscle, along with heightened responsiveness to allergen and cytokine challenges.
- [31].Jain D, Keslacy S, Tliba O, Cao Y, Kierstein S, Amin K, Panettieri RA Jr., Haczku A and Amrani Y (2008) Essential role of IFNbeta and CD38 in TNFalpha-induced airway smooth muscle hyper-responsiveness. Immunobiology 213, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jackson DG and Bell JI (1990) Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J Immunol 144, 2811–2815. [PubMed] [Google Scholar]
- [33].Shubinsky G and Schlesinger M (1997) The CD38 lymphocyte differentiation marker: new insight into its ectoenzymatic activity and its role as a signal transducer. Immunity 7, 315–324. [DOI] [PubMed] [Google Scholar]
- [34].Zocchi E, Franco L, Guida L, Benatti U, Bargellesi A, Malavasi F, Lee HC and De Flora A (1993) A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem Biophys Res Commun 196, 1459–1465. [DOI] [PubMed] [Google Scholar]
- [35].Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF and Lee HC (1993) Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262, 1056–1059. [DOI] [PubMed] [Google Scholar]
- [36].Lee HC (2006) Structure and enzymatic functions of human CD38. Mol Med 12, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Partida-Sanchez S, Rivero-Nava L, Shi G and Lund FE (2007) CD38: an ectoenzyme at the crossroads of innate and adaptive immune responses. Adv Exp Med Biol 590, 171–183. [DOI] [PubMed] [Google Scholar]
- [38].Tliba O, Panettieri RA Jr., Tliba S, Walseth TF and Amrani Y (2004) Tumor necrosis factor-alpha differentially regulates the expression of proinflammatory genes in human airway smooth muscle cells by activation of interferon-beta-dependent CD38 pathway. Mol Pharmacol 66, 322–329. [DOI] [PubMed] [Google Scholar]
- [39].Sathish V, Thompson MA, Sinha S, Sieck GC, Prakash YS and Pabelick CM (2014) Inflammation, caveolae and CD38-mediated calcium regulation in human airway smooth muscle. 1843, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sieck GC, White TA, Thompson MA, Pabelick CM, Wylam ME and Prakash YS (2008) Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. American Journal of Physiology-Lung Cellular and Molecular Physiology 294, L378–L385. [DOI] [PubMed] [Google Scholar]
- [41].Wu Y, Zou F, Lu Y, Li X, Li F, Feng X, Sun X and Liu Y (2019) SETD7 promotes TNF-alpha-induced proliferation and migration of airway smooth muscle cells in vitro through enhancing NF-kappaB/CD38 signaling. Int Immunopharmacol 72, 459–466. [DOI] [PubMed] [Google Scholar]
- [42].Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, Murray R, Schuber F and Howard MC (1998) Mice deficient for the ectonicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 92, 1324–1333. [PubMed] [Google Scholar]
- [43]*.Wang J, Zhu W, Chen Y, Lin Z and Ma S (2016) CD38 gene-modified dendritic cells inhibit murine asthma development by increasing IL-12 production and promoting Th1 cell differentiation. Mol Med Rep 14, 4374–4382. [DOI] [PubMed] [Google Scholar]; Using bone marrow-derived dendritic cells overexpressing CD38, the authors provided evidence that CD38 activity in dendritic cells function to alleviate asthma by restoring Th1/Th2 balance during airway inflammation.
- [44].Fernandez JE, Deaglio S, Donati D, Beusan IS, Corno F, Aranega A, Forni M, Falini B and Malavasi F (1998) Analysis of the distribution of human CD38 and of its ligand CD31 in normal tissues. J Biol Regul Homeost Agents 12, 81–91. [PubMed] [Google Scholar]
- [45].Sobieraj DM, Weeda ER, Nguyen E, Coleman CI, White CM, Lazarus SC, Blake KV, Lang JE and Baker WL (2018) Association of Inhaled Corticosteroids and Long-Acting beta-Agonists as Controller and Quick Relief Therapy With Exacerbations and Symptom Control in Persistent Asthma: A Systematic Review and Meta-analysis. Jama 319, 1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cazzola M, Rogliani P, Stolz D and Matera MG (2019) Pharmacological treatment and current controversies in COPD. F1000Res 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dileepan M, Sarver AE, Rao SP, Panettieri RA Jr., Subramanian S and Kannan MS (2016) MicroRNA Mediated Chemokine Responses in Human Airway Smooth Muscle Cells. PLoS One 11, e0150842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48]*.Dewhurst JA, Lea S, Hardaker E, Dungwa JV, Ravi AK and Singh D (2017) Characterisation of lung macrophage subpopulations in COPD patients and controls. Scientific reports 7, 7143. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using human patients with COPD, the authors demonstrated for the first time a pattern of high CD38 expression in small macrophages, which appeared primed towards a pro-inflammatory phenotype.
- [49].Frankenberger M, Menzel M, Betz R, Kassner G, Weber N, Kohlhaufl M, Haussinger K and Ziegler-Heitbrock L (2004) Characterization of a population of small macrophages in induced sputum of patients with chronic obstructive pulmonary disease and healthy volunteers. 138, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kubysheva N, Postnikova L, Soodaeva S, Novikov V, Eliseeva T, Batyrshin I, Li T, Klimanov I and Chuchalin A (2017) Relationship of the Content of Systemic and Endobronchial Soluble Molecules of CD25, CD38, CD8, and HLA-I-CD8 and Lung Function Parameters in COPD Patients. Dis Markers 2017, 8216723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wylam ME, Sathish V, Vanoosten SK, Freeman M, Burkholder D, Thompson MA, Pabelick CM and Prakash YS (2015) Mechanisms of Cigarette Smoke Effects on Human Airway Smooth Muscle. PLOS ONE 10, e0128778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schiavoni I, Scagnolari C, Horenstein AL, Leone P, Pierangeli A, Malavasi F, Ausiello CM and Fedele G (2018) CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology 154, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]