

Abstract
Viruses promote infection by hijacking host ubiquitin machinery to counteract or redirect cellular processes. Adenovirus encodes two early proteins, E1B55K and E4orf6, that together co-opt a cellular ubiquitin ligase complex to overcome host defenses and promote virus production. Adenovirus mutants lacking E1B55K or E4orf6 display defects in viral RNA processing and protein production, but previously identified substrates of the redirected ligase do not explain these phenotypes. Here we used a quantitative proteomics approach to identify substrates of E1B55K/E4orf6-mediated ubiquitination that facilitate RNA processing. While all currently known cellular substrates of E1B55K/E4orf6 are degraded by the proteasome, we uncovered RNA-binding proteins (RBPs) as high-confidence substrates that are not decreased in overall abundance. We focused on two RBPs, RALY and hnRNP-C, which we confirm are ubiquitinated without degradation. Knockdown of RALY and hnRNP-C increased levels of viral RNA splicing, protein abundance, and progeny production during infection with E1B55K-deleted virus. Furthermore, infection with E1B55K-deleted virus resulted in increased interaction of hnRNP-C with viral RNA, and attenuation of viral RNA processing. These data suggest viral-mediated ubiquitination of RALY and hnRNP-C relieves a restriction on viral RNA processing, revealing an unexpected role for non-degradative ubiquitination in manipulation of cellular processes during virus infection.
Keywords: Adenovirus, ubiquitin ligase, proteomics, RNA processing
Viruses have evolved mechanisms to alter cellular pathways to promote infection and inactivate host defenses. This can be achieved through viral factors that redirect host post-translational protein modifications (PTMs) such as ubiquitination to regulate protein function and turnover. Viruses interface with the host ubiquitin system by encoding ubiquitin ligases, redirecting host ligases, or regulating enzymatic deubiquitination1–3. Ubiquitination leads to diverse outcomes, including proteasome-mediated degradation, protein relocalization, and altered protein or nucleic acid interactions4–7. This functional diversity makes ubiquitin regulation a potent target for viral manipulation.
Adenovirus (Ad) encodes two early proteins (E1B55K and E4orf6) which integrate into an existing host ubiquitin ligase complex containing Elongin-B/C, Cullin5, and RBX18,9. E4orf6 hijacks the host ligase through binding to Elongin-B/C, and E1B55K recruits substrates for ubiquitination9. Deleting these Ad proteins or expressing a dominant-negative Cullin5 reduces virus production, demonstrating the importance of redirecting the ubiquitin complex10–18. Several host proteins have been identified as targets for proteasomal degradation mediated by the Ad serotype 5 (Ad5) E1B55K/E4orf6 complex, including MRE11, RAD50, NBS1, DNA Ligase IV, BLM, and p538,19–23. Degradation of these proteins represses DNA damage signaling and apoptosis during infection24–26. Viral mutants lacking either E1B55K or E4orf6 are also defective for late RNA processing, and exhibit reduced late RNA, protein, and progeny production, with little impact on early stages of infection11–16. These deficiencies are not explained by known substrates for E1B55K/E4orf610–16.
In this study, we employed proteomics to identify substrates ubiquitinated in the presence of Ad5 E1B55K/E4orf6. We combined antibody-based di-glycine remnant enrichment and mass spectrometry (K-ɛ-GG)27,28 to quantify ubiquitinome changes, together with whole cell proteomics (WCP) to quantify protein abundance changes. Our analysis suggests that E1B55K/E4orf6 facilitate different outcomes of ubiquitination, with the majority of ubiquitinated substrates being unchanged in abundance. RNA-binding proteins (RBPs) were enriched among ubiquitinated substrates. We validated RALY and hnRNP-C as two host RBPs that are modified to overcome restrictions on viral late transcript production. These substrates provide a potential mechanistic link between E1B55K/E4orf6-redirected ubiquitination and viral RNA processing, highlighting viral exploitation of non-degradative ubiquitination as a strategy to manipulate host pathways.
RESULTS
Functional E1B55K/E4orf6 complex is required for viral late RNA splicing.
Ubiquitination promoted by E1B55K/E4orf6 targets proteins for proteasomal degradation or may impact function without affecting protein abundance (Fig. 1a). We assessed the role of E1B55K/E4orf6-mediated ubiquitination on viral RNA-processing and protein expression by inactivating the complex through E1B55K deletion or Cullin5 inhibition29. Compared to wild-type (WT) Ad5, infection with E1B55K mutant virus (ΔE1B) resulted in decreased viral late proteins (hexon, penton, fiber and protein VII) but minimal impact on early protein (DBP) production (Fig. 1b; Extended Data Fig. 1a). We hypothesized that Cullin5 inhibition would mimic ΔE1B infection. Cullin ligases are activated by NEDD8 modification30,31, which can be inhibited by the small molecule MLN492429. Inhibition of NEDDylation (NEDDi) was confirmed by decreased slower-migrating modified Cullin5, and reduced degradation of MRE11 and BLM (Fig. 1b). NEDDi substantially decreased viral late protein levels for WT Ad5 infection but only marginally decreased early protein DBP (Fig. 1b; Extended Data Fig. 1a), and did not further alter the late protein defect observed with ΔE1B infection (Fig. 1b). Increased E1B55K levels observed upon NEDDi in WT likely reflect inhibition of auto-ubiquitination, commonly observed for substrate recognition components of ubiquitin ligases32. We assessed viral RNA-processing during NEDDi or E1B55K deletion. Levels of late transcripts from the major late promoter (MLP) and fiber regions were reduced during NEDDi in WT infection, similar to decreases with ΔE1B (Fig. 1c). We labelled nascent RNA with 4sU to quantitate transcription and RNA turnover during infection (Extended Data Fig. 1b). E1B55K deletion did not significantly decrease transcription or RNA decay of early (E1A and E4) or late (MLP) viral RNAs. We blocked transcription with Actinomycin D to measure decay of viral transcripts, and observed that E1B55K-deletion did not alter mRNA turnover (Extended Data Fig. 1c). We used quantitative reverse-transcription PCR (RT-qPCR) to determine the ratio of spliced:unspliced transcripts (Extended Data Fig. 1d). Both NEDDi and E1B55K-deletion reduced splicing efficiency of viral late transcripts (MLP and fiber), without negatively impacting early E1A (Fig. 1d; Extended Data Fig. 1e,f). Since incorrect splicing causes nuclear retention and degradation of unspliced RNA33,34, we hypothesized that splicing failures could explain the export defect and decrease in viral late RNA. Detection of RNA by fluorescence in situ hybridization (FISH) for fiber transcripts showed less fiber RNA reaches the cytoplasm upon NEDDi and E1B55K-deletion (Fig. 1e; Extended Data Fig. 1g). These data highlight that E1B55K/E4orf6-directed ubiquitination is important for Ad late RNA splicing, export, and protein production. We sought to identify substrates of E1B55K/E4orf6 that could explain the RNA-processing phenotypes.
Figure 1 |. E1B55K deletion or inhibition of Cullin-mediated ubiquitination decreases adenovirus late RNA splicing and RNA processing.
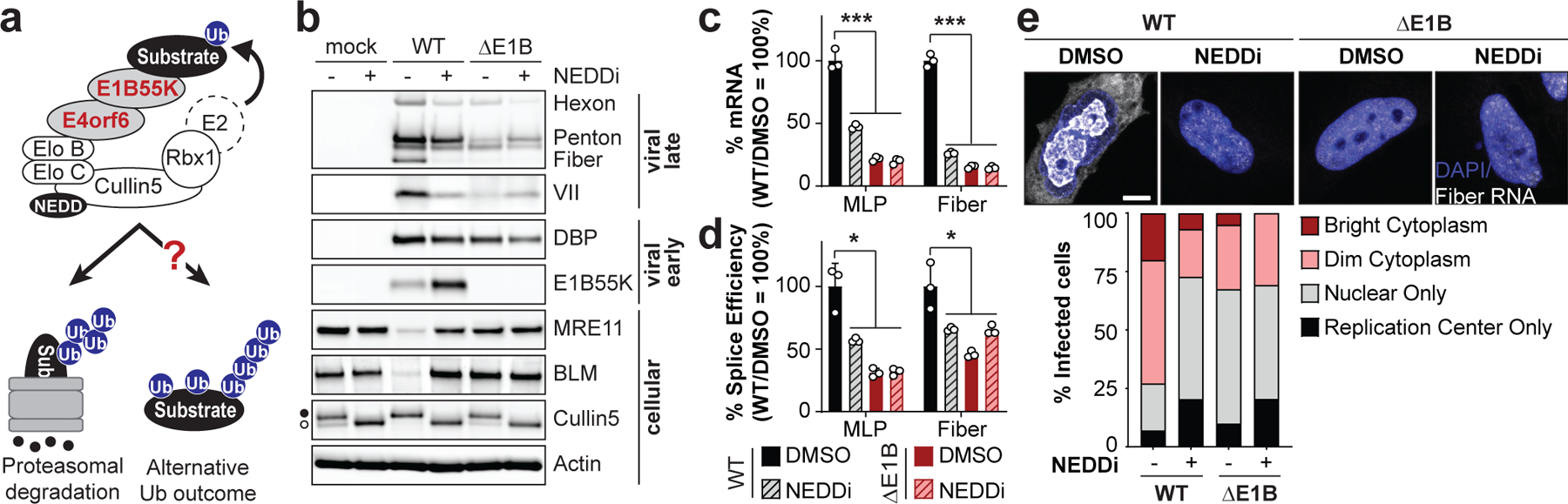
a, Model of E1B55K/E4orf6 association with the host Cullin5 complex and possible functional outcomes. b-e, HeLa cells infected with wild-type (WT) or E1B55K-deleted (ΔE1B) Ad5 at multiplicity of infection (MOI) of 10. Cells were treated with either DMSO or NEDDi (neddylation inhibitor MLN4924) at 8 hours post-infection (hpi) and assayed at 24 hpi. b, Immunoblot analysis of viral and cellular protein abundance. The neddylated (●) and unmodified (○) forms of Cullin5 are indicated. c, Bar graph representing spliced RNA levels of viral late transcripts from the major late promoter (MLP) and fiber by quantitative reverse transcription PCR (RT-qPCR). d, Bar graph representing splicing efficiency (ratio of spliced to unspliced transcripts) of MLP and fiber relative to WT DMSO control by RT-qPCR. e, Representative RNA FISH visualizing localization of fiber transcripts (white) in relation to nuclear DNA stained with DAPI (blue) and quantification of observed pattern for >50 HeLa cells. Scale bar=10 μm. All data are representative of two (e) or three (others) biologically independent experiments. All graphs show mean+SD. Significance was calculated using an unpaired, two-tailed Student’s t-test, * p <0.05, *** p <0.005.
Proteomics reveals ubiquitination of RNA-binding proteins in the presence of E1B55K/E4orf6.
To identify cellular substrates ubiquitinated in the presence of E1B55K/E4orf6 complex, we used proteomics approaches to profile changes in ubiquitination and associated WCP (Fig. 2a; Extended Data Fig. 2). We expressed E1B55K and E4orf6 from non-replicating viral vectors19,35, and monitored degradation kinetics for known substrates to determine optimal conditions (Extended Data Fig. 2a). Tryptic cleavage leaves two C-terminal glycine residues from ubiquitin on modified lysines (K-ɛ-GG), which can be immuno-enriched27, allowing identification and quantification by mass spectrometry. The number of enriched peptides and proteins identified, as well as proteins quantitated in WCP, were reproducible across timepoints (Extended Data Fig. 2b,c; Supplementary Table 1). Changes in peptide modification were normalized to changes in total protein abundance, for the 10 h timepoint. E1B55K/E4orf6 expression induced significant ubiquitination increases for 38 peptides (Fig. 2b). Additionally, 51 peptides were ubiquitinated only upon E1B55K/E4orf6 expression. Substrates displaying increased or unique ubiquitination upon E1B55K/E4orf6 expression included known targets MRE11 and RAD50 (Fig. 2b). There were 46 proteins significantly decreased in total protein abundance by E1B55K/E4orf6 expression, including known E1B55K/E4orf6 substrates (Fig. 2b). Peptide-level ubiquitinome data were transformed into protein-level data and plotted against their associated WCP fold-changes (Fig. 2c). We identified 120 host proteins as putative substrates of ubiquitination directed by the viral complex (protein-based K-ɛ-GG increase >2-fold). Among these, 25 proteins were ubiquitinated and decreased in abundance, including known targets MRE11 and RAD50 (Fig. 2c,d, red; Supplementary Table 2). An additional 91 proteins showed increased ubiquitination but were not significantly changed in overall abundance, and are predicted as non-degraded substrates of E1B55K/E4orf6 (Fig. 2c,d, blue, Supplementary Table 2). These data provide evidence that E1B55K/E4orf6 can facilitate non-degradative ubiquitination, and suggest the majority of potential substrates fall into this category.
Figure 2 |. Unbiased proteomics reveals RNA-binding proteins among putative non-degraded substrates of Ad E1B55K/E4orf6.
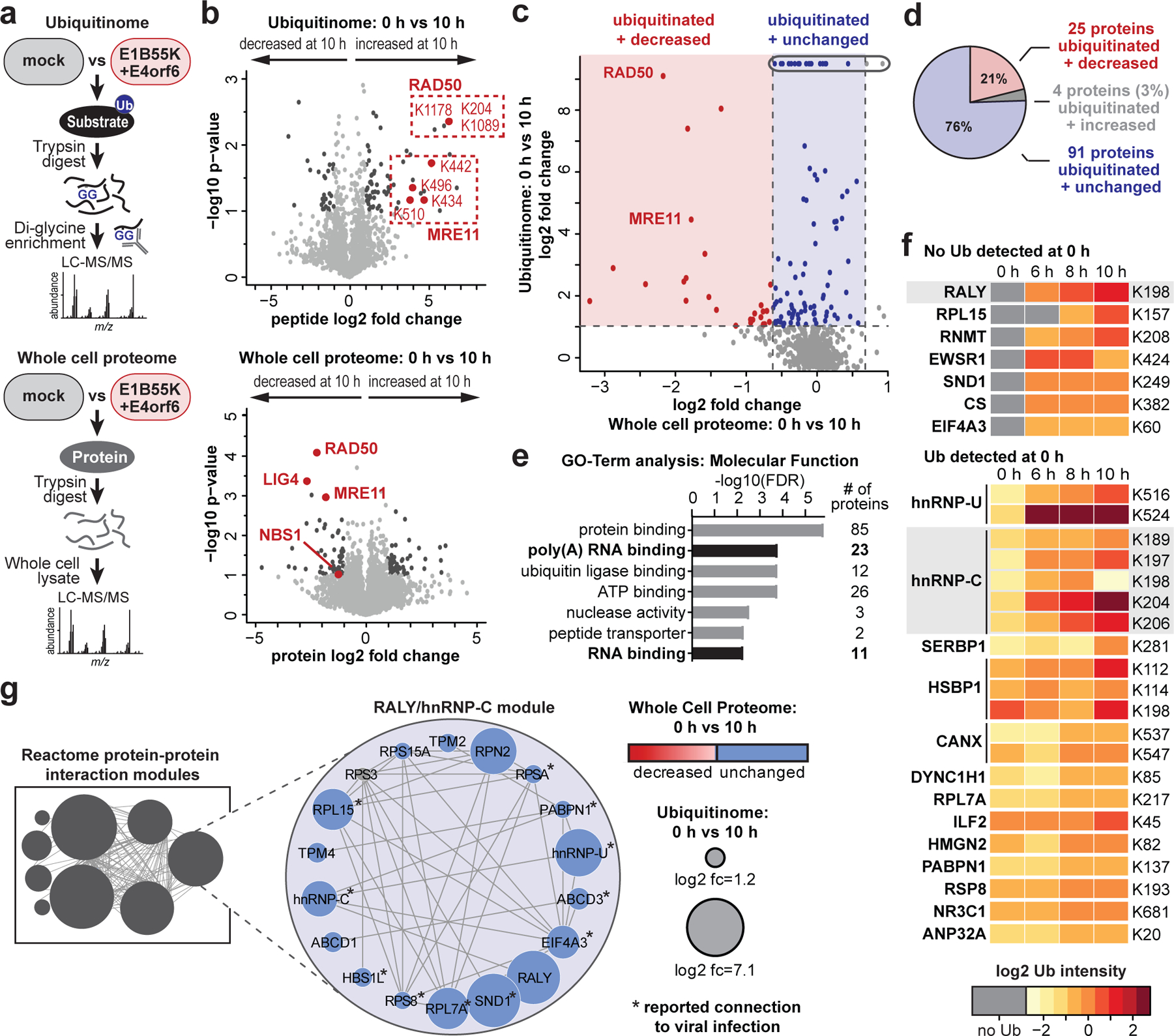
a, Proteomics workflow for identification of E1B55K/E4orf6 substrates. HeLa cells were transduced with recombinant Ad vectors expressing E1B55K and E4orf6 (MOI=10), and subjected to di-glycine remnant profiling (K-ɛ-GG), and whole cell proteomics. b, Volcano plots showing log2 fold-changes (fc) between 0 h and 10 h for ubiquitination (above) and protein abundance (below). Ubiquitinated peptides are normalized to protein abundance. Significance was calculated using an unpaired, two-tailed (whole cell proteome) or one-tailed (normalized K-ɛ-GG fc) Student’s t-test, without correction for multiple comparisons. Dark grey: K-ɛ-GG peptides and whole cell proteins with log2 fc >1 or <−1 and p <0.10. Red: ubiquitinated K-ɛ-GG peptides or whole cell proteins of known substrates of E1B55K/E4orf6. c, Scatter plot showing changes in protein abundance (X-axis) plotted against calculated protein-level ubiquitination (Y-axis). Red: putative degraded substrates - ubiquitination increased (log2 fc >1), abundance decreased (log2 fc <−1SD). Known degraded targets MRE11 and RAD50 are indicated. Blue: putative non-degraded substrates - ubiquitination increased (log2 fc >1), abundance unchanged (−1SD <log2 fc <+1SD). Circled dots: ubiquitinated during infection, undetectable ubiquitination in mock. d, Pie chart indicating proportion of predicted substrates (n=120) that decrease (red), increase (grey), or remain unchanged (blue) in protein abundance upon E1B55K/E4orf6 expression. e, Bar graph representing Gene Ontology (GO) Molecular Function analysis of predicted substrates (n=120) generated by ReactomeFI (FDR is Benjamini-Hochberg method). Categories containing RNA-binding proteins are highlighted. f, Heat map of ubiquitinated lysine residues within RNA-binding proteins with normalized log2 abundance z-score >−0.5 and maximum log2 fc >1 at any time-point of E1B55K/E4orf6 transduction. Colors correspond to average z-score of ubiquitination. Highly ubiquitinated proteins RALY and hnRNP-C are highlighted (grey). g, RALY and hnRNP-C protein-protein interaction module (expanded, right) from interaction network generated with Reactome-Fl in Cytoscape. Nodes represent proteins and edges represent Reactome-based protein-protein interactions. Node size: relative protein-based ubiquitination log2 fc. Node color: protein abundance log2 fc. * denotes proteins with reported roles for viral infections. All data are representative of three biologically independent experiments.
Gene ontology (GO) analysis of predicted E1B55K/E4orf6 substrates revealed enrichment of “poly(A) RNA binding” and “RNA-binding” annotations (Fig. 2e; Supplementary Table 3). Since E1B55K-deletion has been shown to induce RNA-processing defects, we focused on the 26 proteins included within these RNA-related categories. Seven RBPs were predicted to be ubiquitinated only upon E1B55K/E4orf6 expression (Fig. 2f; Extended Data Fig. 3a), of which RALY had the highest ubiquitination. Among the other RBPs, hnRNP-C is a RALY-interacting protein that had the most sites with increased ubiquitination (Fig. 2f; Extended Data Fig. 3b). Reactome36 protein-protein interaction database analysis found RALY and hnRNP-C together in an interaction module with other RBPs (Fig. 2g; Extended Data Fig. 4 and Supplementary Table 3). Our data also reveal potential ubiquitination substrates in other cellular processes, including deubiquitination (USP5, USP16, USP25), folding chaperones (multiple heat shock proteins), intracellular transport (RAB family members), metabolism (citrate synthase), and antigen presentation (TAP1, TAP2) (Extended Data Fig 4a; Supplementary Table 3). The RALY/hnRNP-C module included proteins with reported association with viruses (Fig. 2g; Supplementary Information 1). Both RALY and hnRNP-C are expressed at high levels in all tissues37 and are implicated in multiple steps of RNA-processing, including splicing and export38–43. Additionally, hnRNP-C is reported to bind Ad late transcripts44. We therefore chose to validate RALY and hnRNP-C as substrates of E1B55K/E4orf6-directed ubiquitination.
RALY and hnRNP-C are ubiquitinated but not degraded upon E1B55K/E4orf6 expression.
RALY and hnRNP-C share ~43% homology, and are 63% homologous in the coiled-coil domain that contains all lysine residues identified with increased ubiquitination (Fig. 3a; Extended Data Fig. 4). The single detected ubiquitination site in RALY (K198) is conserved with the most strongly-modified site in hnRNP-C (K204). Since E1B55K recruits substrates for ubiquitination, we examined interaction of E1B55K with host RBPs (Fig. 3b). Immunoprecipitation (IP) of E1B55K isolated RALY and hnRNP-C during WT but not ΔE1B infection (Fig. 3b). In reciprocal experiments, E1B55K was detected upon IP of RALY and hnRNP-C during WT infection, confirming interaction of the E1B55K/E4orf6 complex with these RBPs (Fig. 3b). hnRNP-C and RALY interact in reciprocal IPs, as reported previously45, and this association was maintained during infection. To confirm E1B55K/E4orf6-induced ubiquitination, we transfected RALY-Flag or hnRNP-C-Flag, together with HA-ubiquitin and E4orf6 into HEK293 cells (this cell line expresses E1B55K46). IP for HA followed by Flag immunoblotting revealed high-molecular-weight ubiquitin complexes of RALY and hnRNP-C upon E4orf6 expression (Fig. 3c, Extended Data Fig. 6a). Alternative isoform hnRNP-C2 was also ubiquitinated upon E4orf6 expression (Extended Data Fig. 6b). The overall stoichiometry for ubiquitination is low relative to total protein abundance of RALY and hnRNP-C, as is the case with many PTMs. We demonstrated that NEDDi decreased E4orf6-induced RALY and hnRNP-C ubiquitination (Fig. 3d, Extended Data Fig. 6c). We also verified that hnRNP-C was ubiquitinated during WT but not ΔE1B infection (RALY antibody quality proved insufficient for this assay) (Fig. 3e). WCP analysis showed RALY and hnRNP-C abundances were stable during E1B55K/E4orf6 expression (Extended Data Fig. 3a) and was confirmed during WT infection (Fig. 3f, Extended Data Fig. 6d). RALY and hnRNP-C abundances were also stable during transduction of A549 and U2OS cells with E1B55K/E4orf6 vectors, as well as E4orf6 expression in HEK293 cells (Extended Data Fig. 6e). RALY and hnRNP-C abundances also remained stable upon cycloheximide treatment during infection (Fig. 3g). Finally, mRNA levels for RALY and hnRNP-C are stable during WT infection (Extended Data Fig. 6f). We hypothesize that E1B55K/E4orf6 can facilitate either degradative or non-degradative ubiquitination for distinct substrates. We therefore examined ubiquitination differences between MRE11, RAD50, RALY, and hnRNP-C. Ubiquitination assays were performed by transfection of HEK293 cells with and without proteasome inhibitor MG132 (Fig. 3h). E4orf6 expression increased MRE11 ubiquitination, which further increased upon proteasome inhibition, consistent with MRE11 degradation upon E1B55/E4orf6-mediated ubiquitination. In contrast, E4orf6 expression increased ubiquitination of RALY and hnRNP-C without further increase upon MG132 treatment. This suggests that ubiquitination of RALY and hnRNP-C likely does not result in proteasomal degradation. We next examined ubiquitin chains attached to each substrate. The ubiquitin linkage most commonly associated with proteasomal degradation is K48. To probe for K48 linkages, we performed native HA-ubiquitin IPs and compared ubiquitination levels after treatment with DUBs47 that are linkage-indiscriminate (DUBPan) or K48-linkage-specific (DUBK48) (Fig. 3i, Extended Data Fig. 6g). MRE11 and RAD50 showed a clear decrease in ubiquitination upon treatment with both DUBs, indicating attachment of K48-linked ubiquitin. In contrast, RALY and hnRNP-C ubiquitination decreased with DUBPan but not DUBK48 treatment. This suggests that RALY and hnRNP-C are substrates for non-K48 ubiquitination, distinct from degraded substrates MRE11 and RAD50. Our data support non-degradative outcomes for hnRNP-C/RALY ubiquitination, although it is possible that degradation occurs within a sub-population too small to distinguish by these assays. Together, these data validate that RALY and hnRNP-C are ubiquitinated by the E1B55K/E4orf6 complex without subsequent proteasomal degradation.
Figure 3 |. RALY and hnRNP-C are non-degraded substrates of Ad E1B55K/E4orf6-mediated ubiquitination.
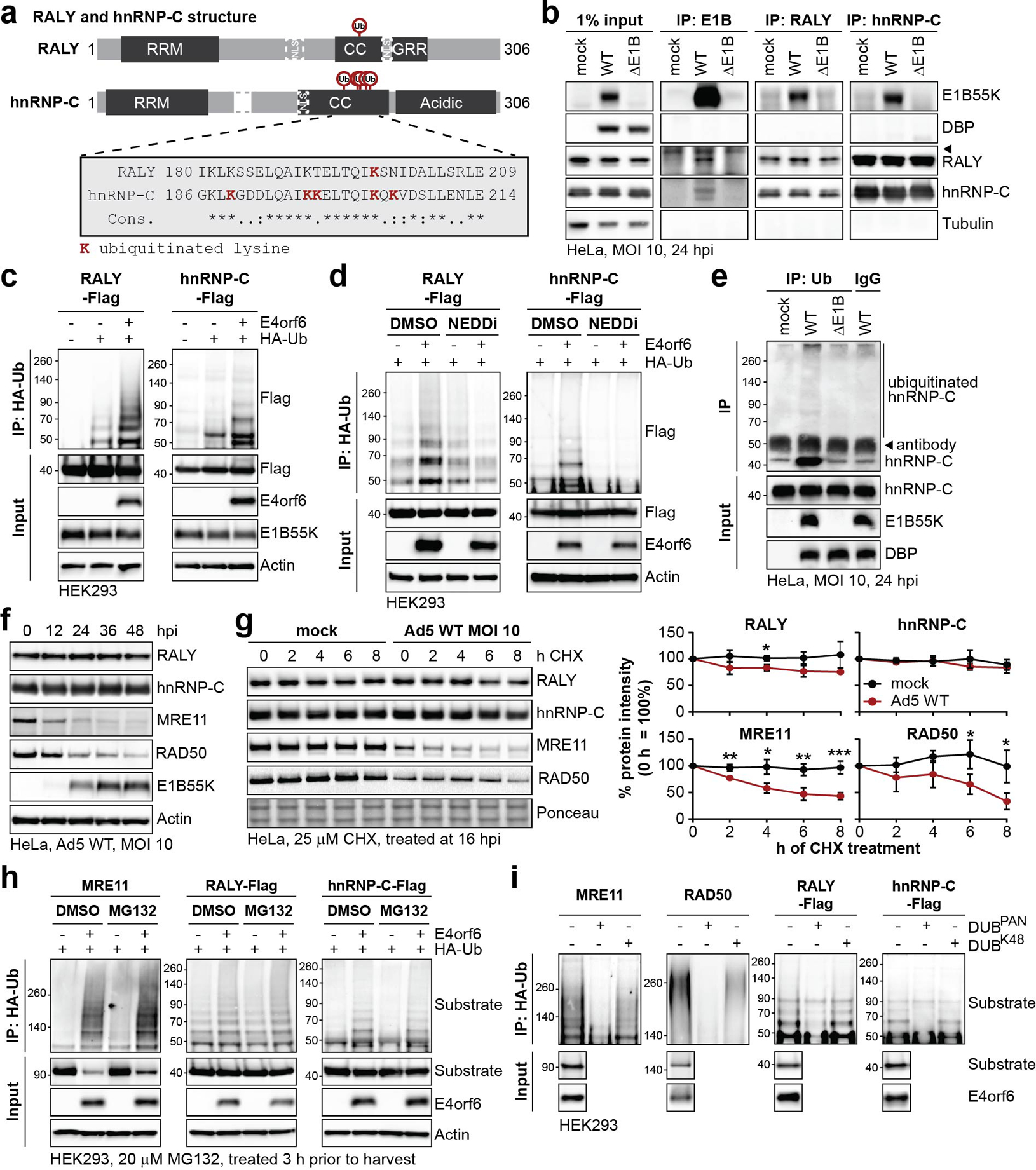
a, Domain structure of RALY and hnRNP-C. RRM=RNA recognition motif, NLS = nuclear localization sequence, CC=coiled-coil, GRR=glycine rich region. CC region shown below contains all detected lysine residues with increased ubiquitination upon E1B55K/E4orf6 expression highlighted in red. b, Immunoblot analysis of E1B55K, RALY, and hnRNP-C immunoprecipitations (IP) probing for pull-down of viral and cellular proteins during mock, WT and ΔE1B infection of HeLa cells at MOI of 10 for 24 h. DBP and Tubulin served as negative controls.◂ denotes antibody heavy chain. c, HEK293 cells transfected with indicated constructs for 24 h followed by denaturing IP with HA antibody and immunoblot analysis of RALY-Flag or hnRNP-C-Flag. d, HEK293 cells transfected with the indicated constructs for 24 h and treated with DMSO or NEDDi at 6 h prior to harvest, followed by denaturing IP with HA antibody and immunoblot analysis of RALY-Flag or hnRNP-C-Flag. e, Immunoblot of denaturing IP of hnRNP-C probing for ubiquitin during mock, WT and ΔE1B infections in HeLa cells at MOI of 10 for 24 h. ◂ indicates antibody heavy chain. f, Immunoblot analysis of protein levels over a time course of Ad5 WT infection (MOI=10) of HeLa cells. g, Immunoblot analysis and quantification of RALY, hnRNP-C, MRE11, and RAD50 over a time course of cycloheximide (CHX) treatment of mock or Ad5 WT infected HeLa cells. Shown is mean+SD. Statistical significance was calculated using an unpaired, two-tailed Student’s t-test, * p< 0.05, ** p <0.01, *** p <0.005. h, HEK293 cells transfected with the indicated constructs for 24 h and treated with DMSO or proteasome inhibitor MG132 at 3 h prior to harvest followed by denaturing IP with HA antibody and immunoblot analysis of MRE11, RALY-Flag, and hnRNP-C-Flag. i, HEK293 cells transfected with indicated constructs for 24 h followed by denaturing IP with HA antibody, treatment with the indicated deubiquitinating enzymes (DUBs) and immunoblot analysis of MRE11, RAD50, RALY-Flag, and hnRNP-C-Flag. All data are representative of three (b, e, g), four (d, h), or five (c) biologically independent experiments. Size markers in kDa are shown for ubiquitination immunoblots.
RALY and hnRNP-C are detrimental for viral late RNA processing.
To determine whether RALY and hnRNP-C impact Ad infection, we used siRNA knockdown in HeLa and HBEC3-KT cells infected with WT or ΔE1B virus. RALY/hnRNP-C knockdown did not affect viral protein levels during WT infection, suggesting that during infection with a fully-competent virus their presence has no significant impact (Fig. 4a, Extended Data Fig. 7a,b). RALY/hnRNP-C depletion rescued the viral late protein defect of ΔE1B virus almost to WT levels. We examined whether knockdown also affects progeny production of ΔE1B virus. ΔE1B virus produced >100-fold fewer viral particles than WT, in infection of HeLa or HBEC3-KT cells, which was significantly rescued by RALY/hnRNP-C knockdown (Fig. 4b). These data suggest that RALY and hnRNP-C are detrimental to Ad infection, and that E1B55K/E4orf6-mediated ubiquitination relieves this restriction. Since RALY and hnRNP-C are involved in RNA splicing and export, we hypothesized that depletion selectively increases late RNA-processing without affecting earlier stages of infection. We therefore examined viral DNA replication by quantifying genome accumulation using qPCR (Fig. 4c). There was a modest decrease (2-fold) in DNA replication for ΔE1B virus compared to WT, in agreement with prior reports48. Viral DNA accumulation was not significantly affected upon RALY/hnRNP-C depletion (Fig. 4c), confirming effects occur after viral genome replication. We then quantified RNA levels of both viral early (E1A) and late (MLP, fiber) transcripts (Fig. 4d). RALY and hnRNP-C depletion rescued mRNA levels for MLP and fiber to WT levels during ΔE1B infection (Fig. 4d) without impacting E1A transcript (Extended Data Fig. 7c). MLP and fiber splicing efficiency was reduced for ΔE1B virus, and rescued to WT levels upon RALY/hnRNP-C knockdown (Fig. 4e, Extended Data Fig. 7c). siRNA treatment also increased cytoplasmic fiber RNA staining in ΔE1B infection, while not impacting WT (Fig. 4f). Depletion of RALY or hnRNP-C individually increased viral late protein, RNA, and splicing efficiency of ΔE1B virus, with a greater effect for hnRNP-C than RALY knockdown (Extended Data Fig. 7e–g). We combined siRNA-mediated knockdown with NEDDi during WT infection. RALY and hnRNP-C knockdown completely rescued the NEDDi defect without impacting viral early proteins or RNA (Fig. 4g–i, Extended Data Fig. 7h–j). These data suggest that RALY and hnRNP-C are detrimental to late stages of Ad infection and that ubiquitination or depletion can overcome these defects.
Figure 4 |. Knockdown of RALY and hnRNP-C rescues the RNA processing defect caused by absence of a functional E1B55K/E4orf6 complex.
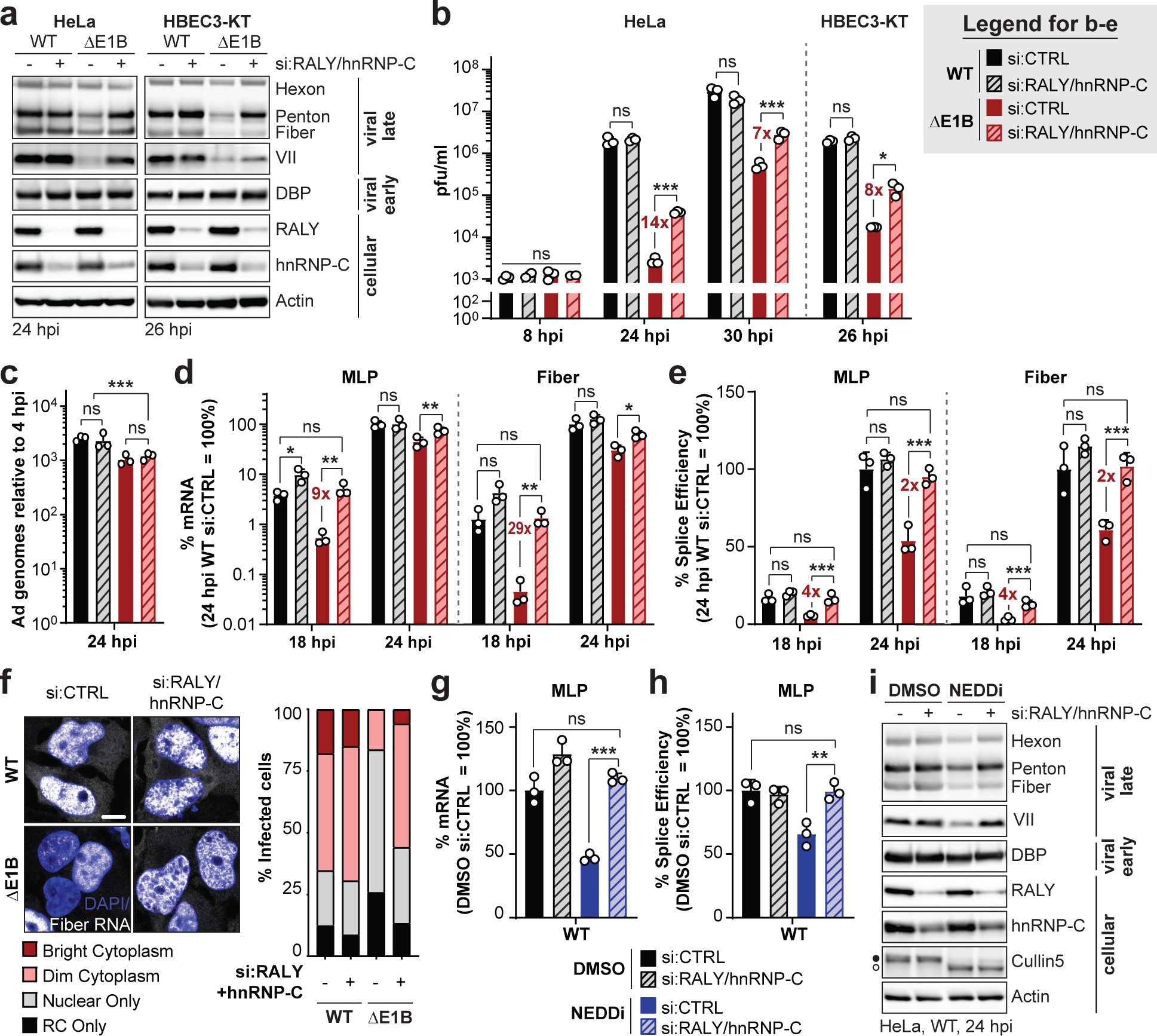
a-f, HeLa cells or HBEC3-KT (only a,b) transfected with control (si:CTRL) or RALY and hnRNP-C (si:RALY/hnRNP-C) siRNA 24 h prior to infection with Ad5 WT or ΔE1B (MOI 10), harvested at respective time points. a, Immunoblot analysis of viral and cellular protein levels. b, Bar graph representing plaque assays for viral progeny. pfu=plaque forming units. c, Bar graph representing qPCR of viral genomes normalized to input. d, Bar graph representing spliced RNA levels of viral late transcripts MLP and fiber measured by RT-qPCR. e, Bar graph representing splicing efficiency (ratio of spliced to unspliced transcripts) of MLP and fiber measured by RT-qPCR. f, Representative RNA FISH visualizing localization of fiber transcripts (white) in relation to nuclear DNA stained with DAPI (blue) and quantification of observed pattern for >100 HeLa cells. RC=replication center. Scale bar=10 μm. g-i. HeLa cells transfected with control (si:CTRL) or RALY and hnRNP-C (si:RALY/hnRNP-C) siRNA for 24 h prior to infection with Ad5 WT (MOI=10), treated with either DMSO or NEDDi at 8 hpi and processed at 24 hpi. g, Bar graph representing spliced RNA levels of MLP measured by RT-qPCR. h, Bar graph representing splicing efficiency of MLP measured by RT-qPCR. i, Immunoblot analysis of viral and cellular protein levels, with neddylated (●) and unmodified (○) forms of Cullin5 indicated. All data are representative of two (f) or three (others) independent experiments. All graphs show mean+SD. Statistical significance was calculated using an unpaired, two-tailed Student’s t-test, * p <005, ** p <0.01, *** p <0.005.
Infection causes global changes to hnRNP-C RNA-binding.
Our data suggest that viral-mediated non-degradative ubiquitination of RALY and hnRNP-C relieves a restriction on viral late RNA-processing. Non-degradative ubiquitination can alter protein localization, e.g. by obscuring nuclear localization sequences49. We examined RALY and hnRNP-C localization by immunofluorescence (IF) in control and Ad-infected cells (Extended Data Fig. 8a). RALY and hnRNP-C showed nucleoplasmic but not nucleolar localization in uninfected cells, as reported37. Upon infection, both proteins were excluded from viral replication centers marked by DBP or USP7 in a pattern matching viral RNA and other RBPs50,51. However, there was no obvious localization difference between WT and ΔE1B, suggesting viral-induced ubiquitination does not alter localization. RALY and hnRNP-C ubiquitination was within coiled-coil domains (Fig. 3a), which are involved in multimerization and protein-RNA interaction52. Therefore, we examined whether protein-complex formation is affected during infection by treating cells with disuccinimidyl suberate (DSS) crosslinker (Extended Data Fig. 8b). Crosslinking caused a mobility shift of hnRNP-C and RALY, consistent with multimerization52. During WT or ΔE1B infection these patterns were similar, suggesting viral-induced ubiquitination does not significantly affect protein-complex formation. Next, we employed targeted proteomic identification of RNA-binding regions (RBR-ID)53,54 for hnRNP-C (Extended Data Fig. 8c, Supplementary Table 4). We compared mock, WT, and ΔE1B infections in SILAC-labeled55 cells for control (light) or 4sU-pulsed (heavy) conditions. Protein-RNA photo-crosslinking at 24 hpi was followed by hnRNP-C IP, nuclease treatment, protein digestion, and mass spectrometry analysis. RNA-crosslinked peptides retain RNA adducts, causing a mass shift and loss of the unmodified signal compared to control. Decreased heavy:light ratios thus identify RNA-binding regions in proteins (Fig. 5a). In uninfected cells the strongest binding region of hnRNP-C was the RNA-recognition motif (RRM), which provides specificity for poly-U motifs56. Surprisingly, our data indicate that upon both WT and ΔE1B infection RNA-binding of the RRM dramatically decreased, while coiled-coil domain RNA-binding increased (Fig. 5a). Approximately 20 amino acids downstream of the hnRNP-C ubiquitination sites, we detected an RNA-binding peak in mock samples that decreased in WT but increased in ΔE1B (Fig. 5a), highlighting a region potentially impacted by Ad-mediated ubiquitination. In co-immunoprecipitated RALY, we also detected infection-mediated changes in RRM interactions, and potential ubiquitin-mediated differences between WT and ΔE1B near ubiquitination sites (Extended Data Fig. 8d). We observed only minimal differences in the hnRNP-C interactome during infections (Extended Data Fig. 8e,f, Supplementary Table 5). In summary, RBR-ID revealed major changes to hnRNP-C RNA-binding during infection and potential ubiquitin-mediated differences between Ad5 WT and ΔE1B.
Figure 5 |. Interaction of hnRNP-C with viral late RNA increases in the absence of functional E1B55K/E4orf6 complex.
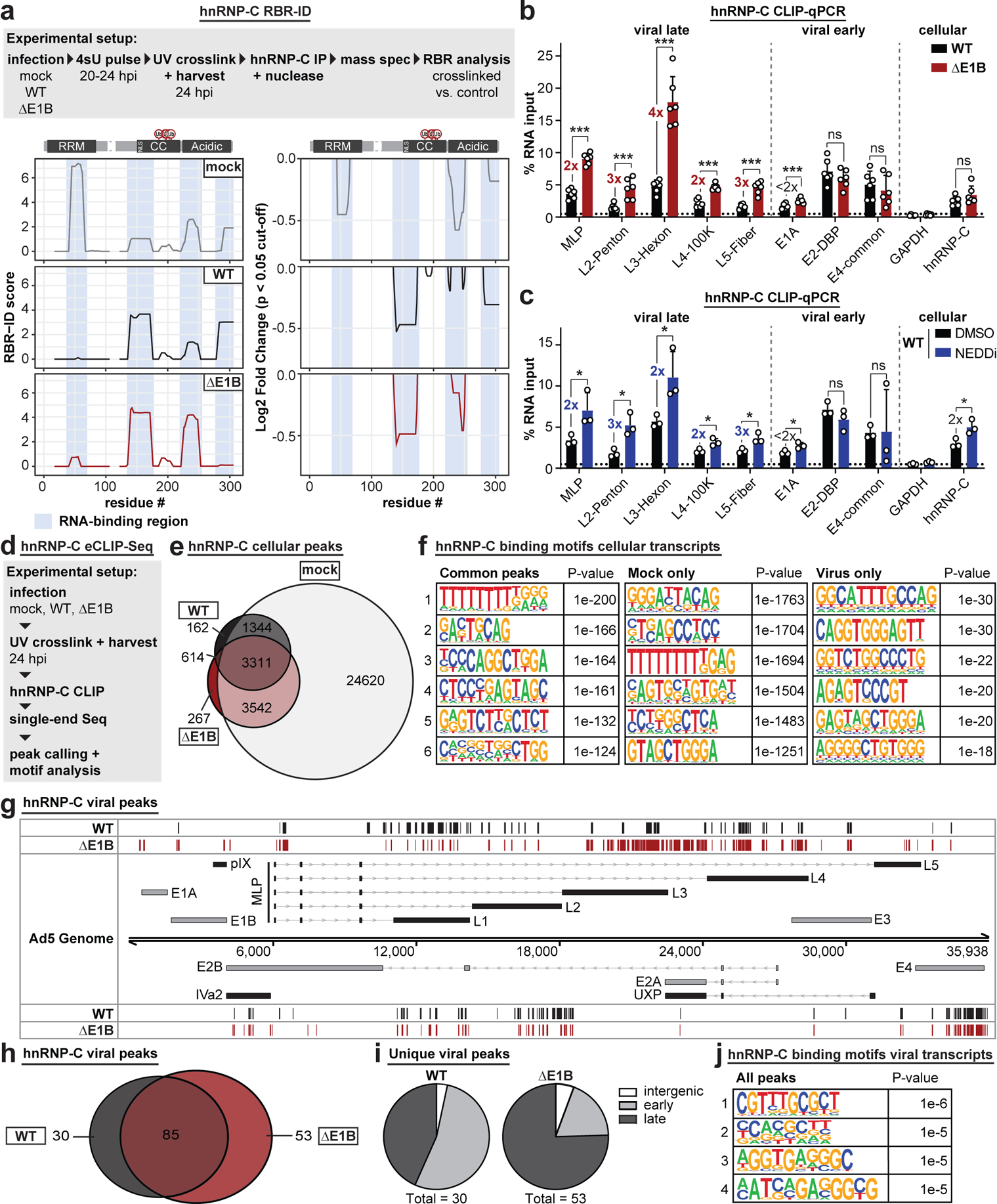
a, RBR-ID (RNA-binding region identification) for hnRNP-C comparing mock (grey), Ad5 WT (black), and ΔE1B (red) conditions at 24 hpi (MOI=10). Experimental setup (above), smoothed residue-level RBR-ID score plotted along primary sequence (left), and smoothed residue-level fold-change of crosslinked/control conditions with significance threshold p < 0.05 (right). hnRNP-C domain structure with ubiquitination sites shown above graphs. RNA-binding regions shaded in blue. n=3 biologically independent experiments. b-c, Bar graphs showing results of hnRNP-C CLIP (cross-linking immunoprecipitation) and RT-qPCR for viral transcripts. GAPDH: negative control. hnRNP-C: positive control. Graphs show mean+SD, n=6 (b) or 3 (c) biologically independent experiments. Statistical significance was calculated using an unpaired, two-tailed Student’s t-test, * p <0.05, ** p <0.01, *** p <0.005. b, HeLa cells infected with WT Ad5 or ΔE1B (MOI=10), UV-crosslinked, and harvested at 24 hpi. c, HeLa cells infected with WT Ad5 (MOI=10), treated with DMSO or NEDDi at 8 hpi, UV-crosslinked, and harvested at 24 hpi. d, Experimental setup for hnRNP-C eCLIP-Seq. e, Venn diagram for hnRNP-C peaks in host transcripts for mock (grey), Ad5 WT (black), and ΔE1B (red). f, Top six motifs for hnRNP-C binding sites in host transcripts in all 3 conditions (left), mock only (middle), or virus only (right, WT only + ΔE1B only + WT and ΔE1B). n=3,311, 24,620, and 614 peaks, respectively. g, hnRNP-C peaks in Ad transcripts for Ad5 WT (black) and ΔE1B (red). Simplified schematic of the Ad transcriptome with forward-facing (above) genome and reverse-facing (below) transcription units. Shown are exons (bars) and introns (lines with arrowheads) of early (grey) and late (black) viral genes. h, Venn diagram of hnRNP-C peaks called in viral transcripts for Ad5 WT (black) and ΔE1B (red). i, Pie charts of unique peaks for WT and ΔE1B showing the location in intergenic, early, or late transcription units. j, Top four motifs for hnRNP-C binding sites in viral transcripts for all conditions. n=168 peaks. Enriched motifs in f and j were identified with HOMER Software Package using hypergeometric enrichment calculations and adjustments for multiple comparisons. Data from e-j are representative of two biologically independent experiments.
Increased interaction of hnRNP-C and RALY with viral late RNA in the absence of ubiquitination.
To determine the impact of Ad-mediated ubiquitination on hnRNP-C interaction with viral RNA, we performed crosslinking-immunoprecipitation (CLIP) followed by RT-qPCR (Extended Data Fig. 9a,b). The hnRNP-C transcript served as positive control and GAPDH as negative control for the assay57. All viral transcripts were detected above background under WT conditions, however, there was a 2–4-fold increase specifically in late but not early RNAs during ΔE1B infection. This suggests viral-induced ubiquitination of hnRNP-C specifically decreases interaction with viral late transcripts. Since overall ubiquitination stoichiometry is low relative to total protein abundance, these observations may represent a localized effect or a dominant-negative impact. These results scaled linearly over a ten-fold dilution of input material (Extended Data Fig. 9c). In contrast, non-UV-crosslinked hnRNP-C CLIP-qPCR or IgG control CLIP precipitated minimal RNA (Extended Data Fig. 9d,e). Available RALY antibodies were not suitable for this technique; therefore, we created an inducible RALY-Flag cell line and performed Flag CLIP-qPCR. RALY interactions with viral late RNA were also increased in ΔE1B infections, while early RNA-binding was unchanged or decreased (Extended Data Fig. 9f). We also repeated hnRNP-C CLIP-qPCR with NEDDi during WT infection. Similar to ΔE1B infection, hnRNP-C interaction with viral late transcripts increased ≥2-fold with NEDDi (Fig. 5c, Extended Data Fig. 9g). This further supports that hnRNP-C interaction with viral late RNAs increases without a functional E1B55K/E4orf6 complex.
To determine whether ubiquitination during infection alters hnRNP-C RNA-binding, we globally analyzed binding sites using enhanced CLIP followed by sequencing (eCLIP-Seq58) (Fig. 5d; Extended Data Fig. 9h,i). We observed dramatic reduction in hnRNP-C binding to host RNAs upon infection, with >24,000 peaks unique to mock, and only ~3,000 common to all 3 conditions (Fig. 5e, Extended Data Fig. 9j). Binding motif analysis revealed the known hnRNP-C poly-U/poly-T binding motif among common and mock-specific peaks (Fig. 5f). We also detected hnRNP-C peaks in host transcripts that were specific to infection, suggesting potential roles for ubiquitination in manipulating hnRNP-C RNA-binding (Extended Data Fig. 9k). The hnRNP-C poly-U motif was decreased in virus-specific peaks on host transcripts (15th most-enriched motif in viral unique transcripts), supporting the RBR-ID that shows decreased RNA-binding of the hnRNP-C RRM (Fig. 5a). The number and location of hnRNP-C peaks on viral transcripts differed between WT and ΔE1B infection (Fig. 5g,h). Analyzing peaks unique to WT or ΔE1B suggested that E1B55K deletion increased hnRNP-C binding mainly in viral late transcripts (Fig. 5i). Differences were especially pronounced in the L3-L5 region of the major late transcription unit (Fig. 5g). These results were consistent with the CLIP-qPCR data (Fig. 5b). Additionally, several hnRNP-C binding sites in viral late RNA regions such as MLP and fiber are unique to ΔE1B (Fig. 5g). We did not observe a poly-U preference of hnRNP-C in viral transcripts (Fig. 5j). However, the most prominent motif for infection-specific hnRNP-C binding sites within both host and viral transcripts is similar. While this motif was present in mock, it was not among top RNA-binding motifs identified (Fig. 5f). This suggests infection alters the equilibrium of major and minor RNA-binding sites for hnRNP-C. In summary, RBR-ID and eCLIP-Seq data highlight changes in hnRNP-C RNA-binding during infection. These results support a model in which E1B55K/E4orf6-induced ubiquitination overcomes detrimental effects on viral RNA-processing by reducing hnRNP-C/RALY interaction with viral late RNA (Extended Data Fig. 10).
DISCUSSION
Viruses commonly redirect cellular pathways towards efficient viral production. E1B55K/E4orf6 hijack the host Cullin5 ubiquitin ligase to redirect ubiquitination and stimulate viral late RNA nuclear export and late protein synthesis. While prior studies identified binding partners and a limited number of E1B55K/E4orf6 substrates8,9,19–23,59–62, they did not explicitly link potential cellular substrates to viral RNA-processing. Here we combined ubiquitinome analysis with global proteomic quantification to identify substrates of the E1B55K/E4orf6 complex. We predicted 120 substrates, with enrichment of RBPs that are potentially involved in viral RNA-processing. We focused on two highly ubiquitinated RBPs, RALY and hnRNP-C, identified as non-degraded substrates of the E1B55K/E4orf6 complex. We demonstrated differential interaction of hnRNP-C and RALY with viral late transcripts in the presence of an active E1B55K/E4orf6 complex, supporting a model in which ubiquitination promotes efficient RNA-processing by altering RNA-binding (Extended Data Fig. 10). Since hnRNP-C has reported roles in alternative splicing38,40,41, we propose that Ad-directed ubiquitination results in exclusion from RNP complexes on late viral RNA. Ubiquitin and related proteins have emerging roles in regulating splicing and spliceosomal complex dynamics through altered protein-protein interactions63. Both virus and host enzymes are likely to regulate RBPs via PTM pathways such as ubiquitin and SUMO that can alter RNA-binding64. Analyzing host splicing changes during Ad infection may provide insights into host pathways altered by RBP ubiquitination. In addition to ubiquitin-mediated changes in interaction between hnRNP-C/RALY and viral RNA, we observed global changes in hnRNP-C RNA-binding during infection, independent of the E1B55K/E4orf6 complex. Understanding how Ad induces RRM RNA-binding decreases and associated changes in binding motif may provide novel insights into RBP functional regulation.
Manipulation of host ubiquitin machinery during virus infection has traditionally been studied in the context of proteasomal degradation, with few known examples of non-degradative ubiquitination1–3. Our data suggest that most predicted substrates of E1B55K/E4orf6 are ubiquitinated without significant abundance changes, suggesting a major role for non-degradative ubiquitination. This finding highlights the need to analyze both ubiquitinome and whole cell proteomes when predicting substrates. We propose that harnessing ubiquitination without proteasome-mediated degradation provides increased flexibility and more rapid approaches to alter cellular pathways and host responses. Viral redirection of ubiquitination may present good model systems to study how ubiquitin ligases in general can facilitate both degradative and non-degradative ubiquitination of distinct substrates. Given increasing appreciation that cellular ubiquitin ligases (such as Cullin complexes) can facilitate formation of multiple ubiquitin chain linkages65–67, viral infections also provide systems to probe the ubiquitin code.
In addition to contributing to fundamental knowledge of cellular biology, Ad has been developed as a vector for gene delivery and oncolytic cancer treatment. Mutant viruses lacking E1B55K have been shown to replicate conditionally in cancer cells, with selectivity that is likely due to preferential viral late mRNA export (rather than p53 inactivation)68–70. Since many cancers have altered RNA-processing, Ad ΔE1B use in oncolytic therapies may be complemented by defects in substrates of the E1B55K/E4orf6 complex. Our work suggests that alterations in these substrates, such as RBPs RALY and hnRNP-C, may make tumor cells more susceptible to ΔE1B-based oncolytic viruses.
Materials and Methods
Cell culture
All cell lines were obtained from the American Type Culture Collection (ATCC) and cultured at 37°C and 5% CO2. HeLa (Cat#: ATCC CCL-2), HEK293 (Cat#: ATCC CRL-1573), and U2OS cells (Cat#: ATCC HTB-96) were grown in DMEM (Corning, Cat#: 10–013-CV) supplemented with 10% v/v fetal bovine serum (FBS) (VWR, Cat#: 89510–186) and 1% v/v Pen/Strep (100 U/ml of penicillin, 100 μg/ml of streptomycin, Gibco, Cat#: 15140–122). A549 cells (Cat#: ATCC CCL-185) were maintained in Ham’s F-12K medium (Gibco, Cat#: 21127–022) supplemented with 10% v/v FBS and 1% v/v Pen/Strep. Primary like HBEC3-KT (Cat#: ATCC CRL-4051) were grown in Airway Epithelial Cell Basal Medium (Cat#: ATCC PCS-300–030) supplemented with Bronchial Epithelial Cell Growth Kit (Cat#: ATCC PCS-300–040) and 1% v/v Pen/Strep. The RALY-Flag inducible cell line was generated using a HeLa acceptor cell line kindly provided by E. Makeyev71 and used as previously reported. RALY-Flag was cloned from the pcDNA3.1 plasmid described below and inserted into the inducible plasmid cassette using restriction enzymes BsrGI and AgeI. Sequence confirmed clones were transfected into the HeLa acceptor cells along with plasmid encoding the Cre recombinase. Clones were selected by puromycin (1 μg/mL) and induced with doxycycline (0.5 μg/mL) to express RALY-Flag. Protein expression was verified by immunoblot. All cell lines tested negative for mycoplasma using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich). The cell lines were not authenticated.
Viruses and infection
Ad5 wild-type (WT) was purchased from ATCC. The Ad5 E1B55K-deletion mutant dl110 has been described previously10 and was a gift from G. Ketner. The E1 deletion mutant recombinant adenovirus vectors expressing E1B55K (rAd E1B55K)35 and E4orf6 (rAd E4orf6)19 were obtained from P. Branton. All viruses were propagated on HEK293 cells, purified using two sequential rounds of ultracentrifugation in CsCl gradient and stored in 40% v/v glycerol at −20°C. Viral titers were determined by plaque assay on HEK293 cells for all but rAd E4orf6. For this virus we assumed a plaque forming unit-to-particle ratio of 1:50. All infections were carried out using a multiplicity of infection (MOI) of 10 and harvested at indicated hours post infection (hpi). Infections were performed on monolayers of cells by dilution of the virus in respective low serum growth medium. After 2 h at 37°C additional full serum growth medium was added. For plaque assays, the virus infection media was removed after 2 h and cells were washed 1x in PBS before addition of full serum growth medium.
Plasmids, siRNA and transfection
Full-length RALY with a carboxyl-terminal Flag-tag (cDNA obtained from Dharmacon, Cat#: MHS6278–202857995) and hnRNP-C isoforms 1 and 2 with a carboxyl-terminal Flag-tag (cDNA containing plasmids were a gift from K. Lynch) and RFP were cloned into the pcDNA3.1 vector using the BamHI and XbaI restriction sites. The pRK5 vector encoding Ad5 E4orf6 was generated by subcloning from purified Ad5 DNA as previously described72. The expression vector for HA-tagged tetra-ubiquitin as previously described73 was a gift from R. Greenberg DNA transfections were performed using the standard protocol for Lipofectamine2000 (Invitrogen).
The following siRNAs were obtained from Dharmacon: non-targeting control (Cat#: D-001206-13-05), RALY (Cat#: M-012392-00-0005) and hnRNP-C (Cat#: M-011869-01-0005; Cat#: L-011869-03-0005 only used for hnRNP-C single knockdown in Extended Data Fig. 7). siRNA transfections were performed using the standard protocol for Lipofectamine RNAiMAX (Invitrogen).
Antibodies and inhibitors
The following primary antibodies for viral proteins were obtained: Adenovirus late protein antibody staining Hexon, Penton and Fiber (gift from J. Wilson74, species: rabbit, WB 1:10,000), Protein VII (gift from H. Wodrich75, Clone: Chimera 2–14, WB 1:200), DBP (gift from A. Levine76, Clone: B6–8, WB 1:1000, IF 1:400), E1B55K (gift from A. Levine77, Clone: 58K2A6, WB 1:500) and E4orf6 (gift from D. Ornelles78, Clone: RSA#3, WB 1:500).
The following primary antibodies were used for cellular proteins: MRE11 (Novus Biologicals, Catalog#: NB100–142, WB 1:1000), BLM (Abcam, Catalog#: ab476, WB 1:1000), Cullin5 (Bethyl Laboratories, Catalog#: A302–173A, WB 1:200), Actin (Sigma-Aldrich, Catalog#: A5441–100UL, WB 1:5000), RALY (Bethyl Laboratories, Catalog#: A302–070A, WB 1:1000; Bethyl Laboratories, Catalog#: A302–069A, IF 1:500, IP 5 μl = 5 μg), hnRNP-C (Santa Cruz Biotechnology, Catalog#: sc-32308, WB 1:1000, IF 1:1000, IP 25 μl=5 μg), Tubulin (Santa Cruz Biotechnology, Catalog#: sc-69969, WB 1:1000), Flag (Sigma-Aldrich, Catalog#: F7425-.2MG, WB 1:1000; Sigma-Aldrich, Catalog#: F3165–1MG, IP 5 μg), Ubiquitin (Santa Cruz, Catalog#: sc-9133, IP 10 μl=2 μg; Abcam, Catalog#: ab7780, IP 5 μl), NBS1 (Novus Biologicals, Catalog#: NB100–143, WB 1:1000), RAD50 (GeneTex, Catalog#: GTX70228, WB 1:1000) and USP7 (Bethyl Laboratories, Catalog#: A300–033A, IF 1:500).
Horseradish peroxidase-conjugated (HRP) secondary antibodies for immunoblot were purchased from Jackson Laboratories. Anti-mouse IgG conjugated to HRP for immunoblot of immunoprecipitation samples (used in Fig. 3b) was purchased from Abcam (Cat#: ab131368). Fluorophore-conjugated secondaries for immunofluorescence were purchased from Life Technologies.
Cycloheximide (CHX) was purchased from Calbiochem (Cat#: 293764), dissolved in DMSO to a stock concentration of 25 mM and used at a final concentration of 25 μM. NEDDylation inhibitor MLN4924 was purchased from Sigma-Aldrich (Cat#: 505477), dissolved in DMSO to a stock concentration of 1 mM and used at a final concentration of 3 μM. Proteasome inhibitor MG132 was purchased from Sigma-Aldrich (Cat#: 474791) at a concentration of 10 mM in DMSO and used at a final concentration of 20 μM.
Immunoblotting
Protein samples were prepared using lithium dodecyl sulfate (LDS) loading buffer (NuPage) supplemented with 25 mM dithiothreitol (DTT) and boiled at 95°C for 10 min. Equal amounts of protein lysate were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Millipore) at 30 V for at least 60 min (overnight for ubiquitination assays). Membranes were stained with Ponceau to confirm equal loading and blocked in 5% w/v milk in Tris-buffered saline with Tween-20 (TBST) supplemented with 0.05% w/v sodium azide. Membranes were incubated with primary antibodies overnight, washed for 30 min in TBST, incubated with HRP-conjugated secondary for 1 h and washed again for 30 min in TBST. Proteins were visualized with Pierce ECL Western Blotting Substrate (Thermo Scientific) and detected using a Syngene G-Box. Images were processed and assembled in Adobe CS6. Immunoblots were quantified by pixel densitometry using the Syngene GeneTools software.
Immunofluorescence
HeLa cells were grown on coverslips in 24-well plates, infected with indicated viruses and fixed at 24 hpi in 4% w/v paraformaldehyde in PBS for 10 mins. Cells were permeabilized with 0.5% v/v Triton-X in PBS for 10 mins. The samples were blocked in 3% w/v BSA in PBS (+ 0.05% w/v sodium azide) for 30 mins, incubated with primary antibodies in 3% w/v BSA in PBS (+ 0.05% w/v sodium azide) for 1 h, followed by secondary antibodies and 4,6-diamidino-2-phenylindole (DAPI) for 2 h. Secondary antibodies used were Alexa Fluor α-rabbit 488 and α-mouse 555. Coverslips were mounted onto glass slides using ProLong Gold Antifade Reagent (Cell Signaling Technologies). Immunofluorescence was visualized using a Zeiss LSM 710 Confocal microscope (Cell and Developmental Microscopy Core at UPenn) and ZEN 2011 software. Images were processed in ImageJ and assembled in Adobe CS6.
RNA fluorescence in situ hybridization
RNA FISH was performed following previously established protocols79, with the following modifications. Thirty-two singly labeled DNA oligonucleotides targeting the Fiber open reading frame were designed using the Stellaris smFISH probe designer and ordered with a 3’ mdC-TEG-Amino label from LGC Biosearch. Fiber FISH probes were pooled and labeled with ATTO 647N NHS-Ester (ATTO-TEC, Cat#: AD 647N-31), isopropanol precipitated and purified by HPLC as previously described79. GAPDH probes labeled with Cy3 were used as a counterstain to demarcate cytoplasmic boundaries and were a kind gift from Sydney Schaffer, University of Pennsylvania80. All probe sequences can be found in Supplementary Information 2. HeLa cells were grown on coverslips, harvested, fixed, and permeabilized as described for conventional immunofluorescence above. After permeabilization, cells on coverslips were equilibrated in Wash Buffer (2X SSC, 10% formamide) before being inverted over 30 μl Hybridization Buffer (2X SSC, 10% formamide, 10% dextran sulphate) containing 500 nM Fiber and GAPDH FISH probes and incubated at 37°C in a humidified chamber overnight. The following day coverslips were washed twice with Wash Buffer for 30 minutes at 37°C with DAPI added to the second wash, briefly washed three times at room temperature with 2X SSC, and then affixed to glass slides using clear nail polish. Images were acquired on a Zeiss LSM 710 microscope with ten z-stacks of 0.7 μm each in the z-direction. Images were deconvoluted by maximum intensity projection in the z-direction using ImageJ. Fiber RNA localization was scored as described in Extended Data Fig. 1 over 41–160 individual cells. Representative images were further processed in ImageJ and assembled in Adobe CS6.
RNA isolation and RT-qPCR
Total RNA was isolated from infected cells at the indicated timepoints using the RNeasy Micro Kit (Qiagen). Complementary DNA (cDNA) was synthesized using 1 μg of input RNA and the High Capacity RNA-to-cDNA Kit (Thermo Fisher). Quantitative PCR was performed by standard protocol using diluted cDNA, primers for different viral and cellular transcripts (see Supplementary Information 2 for complete list of primers) and SYBR Green (Thermo Scientific) using the QuantStudio 7 Flex Real-Time PCR System (Thermo Scientific). The relative values for each transcript were normalized to a control RNA (actin or HPRT).
RNA transcription and stability profiling
To assess relative RNA transcription rate and RNA half-life, cells were treated with 200 μM 4-thiouridine (4sU; Sigma T4509) for exactly 30 min. Infection was stopped and RNA harvested using 1 ml TRIzol (Thermo Fisher Scientific), following manufacturer’s instructions. A fraction of the total RNA was reserved as input, and the remaining 4sU-labeled nascent RNA was biotinylated using MTSEA-Biotin-XX (Biotium; 90066) as previously described81,82. Nascent RNA was separated from unlabeled RNA using MyOne C1 Streptavidin Dynabeads (Thermo Fisher Scientific; 65–001), biotin was removed from nascent RNA using 100 mM dithiothreitol (DTT), and RNA was isopropanol precipitated. Total RNA (1 μg) and an equivalent volume of nascent RNA were converted to cDNA and qPCR was performed as described above. Relative transcription rates were determined by the ΔΔCt method to compare nascent transcript levels between control and siRNA treated cells normalized to nascent GAPDH RNA. RNA half-life was determined using the previously described formula t1/2 = -t × [ln(2)/DR] where t is the 4sU labeling time (0.5 h) and DR is the decay rate defined as Nascent/Total RNA83. Half-lives were normalized to the half-life of GAPDH set at 8 h as previously determined84.
RNA decay measurement using Actinomycin D
To determine the decay of viral mRNA species, HeLa cells infected with either Ad5 WT or ΔE1B were treated with 1 μg/ml Actinomycin D (Cayman Chemical, Cat#: 11421) at 24 hpi. RNA harvested using RLT buffer (from Qiagen RNA isolation kit) at 0, 1, 2, 4, 6, and 8 hours after treatment. RNA levels were quantified using RT-qPCR and normalized to 0 hours of Actinomycin D to determine RNA decay.
Viral genome accumulation by qPCR
Infected cells were harvested by trypsinization at 4 and 24 hpi and total DNA was isolated using the PureLink Genomic DNA kit (Invitrogen). qPCR was performed using primers for the Ad5 DBP and cellular tubulin (see Supplementary Information 2 for primers). Values for DBP were normalized internally to tubulin and to the 4 hpi timepoint to control for any variations in virus input. qPCR was performed using the standard protocol for SYBR Green and analyzed with the QuantStudio 7 Flex Real-Time PCR System.
Plaque assay
Infected cells seeded in 12-well plates were harvested by scraping at the indicated timepoints and lysed by three cycles of freeze-thawing. Cell debris was removed from lysates by centrifugation at max speed (21,130 g), 4°C, 5 min. Lysates were diluted serially in DMEM supplemented with 2% v/v FBS and 1% v/v Pen/Strep to infect HEK293 cells seeded in 12-well plates. After incubation for 2 h at 37°C, the infection media was removed, and cells were overlaid with DMEM containing 0.45% w/v SeaPlaque agarose (Lonza) in addition to 2% v/v FBS and 1% v/v Pen/Strep. Plaques were stained using 1% w/v crystal violet in 50% v/v ethanol between 6 to 7 days post-infection.
Immunoprecipitation
Approximately 2×107 cells were harvested, washed, pelleted and flash frozen for each immunoprecipitation. For IP of hnRNP-C and RALY 50 μl of Protein G Dynabeads (Thermo Fisher) per sample were washed 3x in IP buffer (50 mM HEPES pH 7.4, 150 mM KCl, 2 mM EDTA, 0.5% v/v NP-40, 0.5 mM DTT, 1x cOmplete Protease Inhibitor Cocktail (Roche)) and incubated with 5 μg of antibody (α-hnRNP-C or α-RALY) rotating at 4°C for 2h. Cell pellets were resuspended in 1 ml IP buffer and incubated for 1 h on ice. Samples were sonicated with a Diagenode Biorupter on low setting for 30 s on and 30 s off for ten rounds at 4°C and spun at max speed (21,130 g) for 10 min at 4°C. 300 μl of sample were added to washed beads and incubated rotating at 4°C for 2h. Beads were washed 4x in IP wash (same as above but with only 0.05% v/v NP-40). Samples were eluted in 50 μl 1x LDS sample buffer with 25 mM DTT by boiling for 10 min at 95°C and further processed for analysis by immunoblot.
The following changes were made to the protocol for IP of E1B55K: IP buffer contained 50 mM Tris-HCl pH 7.4, 0.1% v/v Triton X-100, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1x cOmplete Protease Inhibitor Cocktail.
Denaturing in vivo ubiquitination assay
Approximately 1×107 cells were washed, pelleted and stored at −80°C for each immunoprecipitation. For HEK293 cells, the pellets were thawed on ice and resuspended in 100 μl of Lysis buffer (1% w/v SDS, 5 mM EDTA, 10 mM DTT, 1x cOmplete Protease Inhibitor Cocktail) with 1 μl Benzonase (Sigma-Aldrich) by vortexing. Samples were incubated on ice for 5 min and then further denatured by heating to 95°C for 5 min. 900 μl of Wash buffer (10 mM Tris-HCl pH 7.4, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 1% v/v Triton X-100, 0.2 mM Na3VO4, 1x cOmplete Protease Inhibitor Cocktail), passed 10 times through a 23G syringe and spun at max speed (21,130 g) for 5 min at 4°C. A minimum of 800 μl of sample was added to 50 μl washed Pierce Anti-HA Magnetic beads (Thermo Fisher). Sample was incubated with beads rotating for 1 h at 4°C, washed 3x in Wash buffer and eluted in 1x LDS sample buffer with 25 mM DTT for further processing by immunoblot.
The following changes were made to the protocol for HeLa cells: the Lysis buffer contained 1% w/v SDS in PBS, TBST was used as wash buffer, Protein G Dynabeads incubated for 1 h with a mix of both α-ubiquitin antibodies listed above were used for the IP.
De-ubiquitination assay
Approximately 1×107 HEK293 cells were washed, pelleted and stored at −80°C for each immunoprecipitation. The pellets were resuspended in 1 ml IP buffer B (20 mM HEPES-KOH pH 7.4, 110 mM potassium acetate, 2 mM MgCl2, 0.1% v/v Tween-20, 0.1% v/v Triton X-100, 150 mM NaCl, 1 mM DTT, 0.1 mM PTSF) containing 1x cOmplete Protease Inhibitor Cocktail, 20 μM PR-619 (LifeSensors, Cat#: SI9619–5X5MG), 5 mM 1,10-phenanthroline (LifeSensors, Cat#: SI9649), and 1 μl/ml Benzonase (Sigma-Alrich). Samples were incubated on ice for 30 min, sonicated with a Diagenode Biorupter on low setting for 30 s on and 30 s off for five rounds at 4°C and spun at max speed (21,130 g) for 5 min at 4°C. 925 μl of sample was added to 100 μl washed Pierce Anti-HA Magnetic beads (Thermo Fisher). Sample was incubated with beads rotating for 2 h at 4°C, washed 3x in IP buffer B, resuspended in 100 μl of IP buffer B and split into three 30 μl aliquots. 1 μl of 20 mM PR-619 was added to sample 1 (untreated), 1 μl of USP2 (LifeSensors, Cat#: DB501) was added to sample 2 (DUBPAN) and 2 μl of OTUB1 (LifeSensors, Cat#: DB201) was added to sample 3 (DUBK48). Samples were incubated at 30°C for a minimum of 1 h. Samples were eluted by addition of 10 μl 4x LDS sample buffer with 100 mM DTT and boiling at 95°C for 10 min for further processing by immunoblotting.
CLIP-qPCR
The CLIP protocol was adapted from existing protocols58. In short, approximately 2×107 cells were crosslinked on ice with 0.8 J/cm2 UV 254 nm in a UV Stratalinker 2400 (Stratagene), washed in PBS with 2 mM EDTA and 0.2 mM PMSF, flash frozen in liquid nitrogen and stored at −80°C. 50 μl of Protein G Dynabeads per sample were washed 3x in iCLIP lysis buffer A (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.2% v/v NP-40, 0.1% w/v SDS, 0.5% w/v Sodium deoxycholate, 1x cOmplete Protease Inhibitor Cocktail), resuspended in 100 μl iCLIP lysis buffer A and incubated with 5 μg of α-hnRNP-C antibody, 5 μg of α-Flag antibody (mouse), or 5 μl of Normal Mouse Serum Control (Thermo Fisher) rotating 1 h at 4°C. Cell pellets were resuspended in 1 ml of iCLIP lysis buffer B (same as buffer A but with 1% v/v NP-40 and 11 μl of Murine RNase inhibitor (NEB) per 1 ml) and incubated on ice for 15 min. Samples were sonicated with a Diagenode Biorupter on low setting for 30 s on and 30 s off for five rounds at 4°C. 2 μl of TURBO DNase (Thermo Fisher) were added and samples incubated at 37°C for 6 min. Lysates were cleared by centrifugation at max speed (21,130 g) for 15 min at 4°C and supernatants transferred to a new tube. 300 μl of lysate were added to washed beads and incubated rotating at 4°C for 2 h. Beads were washed 2x in High Salt buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 0.2% v/v NP-40, 0.1% w/v SDS, 0.5% w/v Sodium deoxycholate), 2x in Wash buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% v/v Tween-20) and 2x in Proteinase K buffer (100mM Tris-HCl pH 7.4, 50 mM NaCl, 10 mM EDTA, 0.2% w/v SDS). Beads were resuspended in 50 μl Proteinase K buffer and 10 μl removed and processed for immunoblot analysis. 10 μl of Proteinase K (NEB) and 2 μl Murine RNase Inhibitor were added to the remaining beads or to 30 μl of input (10%) and incubated at 50°C for 1 h. The RNA was extracted using a standard protocol for TRIzol (Thermo Fisher) and further processed for RT-qPCR.
seCLIP-Seq
Sample preparation
The CLIP protocol was adapted from existing protocols85. In short, approximately 2×107 HeLa cells were crosslinked on ice with 0.8 J/cm2 UV 254 nm in a UV Stratalinker 2400 (Stratagene), washed in PBS with 2 mM EDTA and 0.2 mM PMSF, flash frozen in liquid nitrogen and stored at −80°C. Protein G Dynabeads (100 μl per sample) were washed 3x in iCLIP lysis buffer A (see CLIP-qPCR), resuspended in 100 μl iCLIP lysis buffer A and incubated with 10 μg of α-hnRNP-C antibody rotating 1 h at RT. Cell pellets were resuspended in 1 ml of iCLIP lysis buffer B and incubated on ice for 15 min. Samples were sonicated with a Diagenode Biorupter on low setting for 30 s on and 30 s off for five rounds at 4°C. Samples were incubated with 2 μl of TURBO DNase (Thermo Fisher) and 10 μl of 1:10 diluted RNase I (Thermo Fisher) in Thermomixer at 1200 rpm at 37°C for 5 min, samples placed on ice and 22 μl SUPERase·In RNase Inhibitor was added. Cleared lysate (500 μl) was added to washed beads and incubated rotating at 4°C for 2 h. Beads were washed 2x in High Salt buffer, 2x in Wash buffer and 2x FastAP buffer. FastAP master mix (100 μl) and FastAP enzyme (8 μl) was added and samples were incubated with a Thermomixer at 1200 rpm at 37°C for 15 min. T4 PNK enzyme (7 μl) and 300 PNK master mix were added and samples were incubated with a Thermomixer at 1200 rpm at 37 °C for 20 min. Beads were washed and resuspended in Ligase buffer with 2.5 μl RNA Ligase high conc., 2.5 μl of RNA adapters (3SR_RNA), and incubated at RT for 75 min. Beads were washed and a fraction saved for immunoblotting. For the remaining fraction, beads were resuspended in lysis buffer with DTT, eluted by incubation in Thermomixer, 1200 rpm, 70 °C, run on SDS-PAGE and transferred onto Nitrocellulose o/n, 30V. Lanes for the RBP band (plus 75 kDa) and size-matched input were cut from the membranes RNA was eluted with 20 μl of Proteinase K (Thermo Fisher) in a Thermomixer at 1200 rpm at 50 °C for 1 h. RNA was extracted with acid phenol/chloroform/isoamyl alcohol (pH 4.5), and concentrated using RNA Clean and Concentrator (Zymo). Size-matched inputs were ligated to 3SR_RNA. All RNA samples were reverse transcribed with 0.9 μl AffinityScript Enzyme at 55 °C for 45 min with RT primer SR_RTv2. RNA and excess primers were removed with 3.5 μl ExoSAP-IT 1 M NaOH. cDNA was purified using 10 μl MyONE Silane beads and ligated to 5’ linker SR_DNA o/n at RT. After clean up, cDNA was quantified by qPCR using NEBNext universal and index primers (NEB E7335S). Libraries were indexed using NEBNext High-Fidelity PCR Master Mix (NEB M0541S) for 11 cycles (size-matched input) or 15 cycles (hnRNP-C IP). Libraries were size selected by 1.0x AmpureXP beads (Beckman Coulter A63880), quantified by QuBit HS DNA and Bioanalyzer High Sensitivity DNA assay, and pooled for sequencing. Sequencing was performed using the Illumina NextSeq 500 Sequencing System with 75bp single-end reads.
Data analysis
Preprocessing involved adapter cutting using cutadapt (v. 1.18)86 and extracting the UMIs using umi_tools (v 1.0.0)87. Alignment was achieved using GSNAP (v 2019-09-12)88. Reads were aligned to the human and adenovirus 5 genome simultaneously. After the alignment we used umi_tools to deduplicate reads based on the UMIs, which was followed by removing all non-unique reads. We then used clipper (v0.1.4)89 to find significant enriched IP peaks over the input on the human genome. To identify enriched peaks on the virus genome we employed a sliding window approach, by counting fragments overlapping 10bp wide windows along both the forward and reverse strand on the genome. If two consecutive windows were significantly enriched over input, they were merged into one peak. Motif analysis was conducted using the Homer suite90.
Analyzing protein complexes by crosslinking
Cells were crosslinked using disuccinimidyl suberate (DSS, Thermo Fisher) dissolved to 100 mM in DMSO and further diluted to 0.1 mM, 0.3 mM and 1 mM in PBS. Cells seeded as a monolayer in 6-well plates were washed once with PBS, overlaid with 500 μl with PBS or the different DSS dilutions and incubated at room temperature for 30 min. The reaction was quenched by addition of 500 μl of 20 mM Tris-HCl pH 7.4, washed twice with PBS and further processed for immunoblot analysis.
Di-glycine remnant profiling by mass spectrometry
Cell lysis and initial desalting
Approximately 10 mg of input was generated from 5×15 cm plates of HeLa cells transduced with rAd E1B55K and rAd E4orf6 constructs for 0 h (mock), 6 h, 8 h, and 10 h. Each timepoint was produced in biological triplicate. Cell were harvested with 0.25% Trypsin (Gibco), washed 1x in PBS, flash frozen in liquid nitrogen and stored at −80°C. Pellets were thawed, resuspended in 1 ml of lysis buffer (6 M urea, 2 M thiourea, in 50 mM ammonium bicarbonate pH 8.0) with 1x Halt Protease Cocktail inhibitor solution, and incubated for ~5 min on ice. Samples were then diluted 10-fold in 50 mM ammonium bicarbonate, reduced with 10 mM DTT, alkylated with 20 mM iodoacetamide, and digested with trypsin protease overnight. Digestion was quenched by acidification to pH 2 with trifluoroacetic acid (TFA) and samples were desalted over Waters tC18 SepPak cartridges (Cat#: 036805). A 10% aliquot was set aside for global proteomic analysis and all samples were dried to completion.
Di-glycine (K-ε-GG) enrichment, fractionation, and desalting
A Cell Signaling PTMScan ubiquitin remnant motif kit (Cat#: 5562) was used to enrich for peptides that had been ubiquitinated. Aliquoted beads were cross-linked for 30 minutes in 100 mM sodium borate and 20 mM dimethyl pimelimidate (Thermo Scientific), following the protocol outlined by Udeshi et al.28. Tryptic peptides were resuspended in IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and immunoprecipitated with the provided antibody for 2 h at 4°C. Samples were eluted in LC-MS grade water (Thermo Fisher) with 0.15% v/v TFA and separated into either 3 high-pH fractions (enriched ubiquitinated peptides) or 7 high-pH fractions (global proteome) over C18 columns (The Nest Group, MicroSpin column C18 silica, part#: SEM SS18V, lot#: 091317). Fractionated samples were desalted a final time over Oligo R3 reverse-phase resin (Thermo Scientific, Cat#:1-1339-03).
Data acquisition and search parameters
All solvents used in analysis of MS samples were LC-MS grade. Samples were analyzed with an Easy-nLC system (Thermo Fisher) running 0.1% v/v formic acid (Buffer A) and 80% v/v acetonitrile with 0.1% v/v formic acid (Buffer B), coupled to an Orbitrap Fusion Tribrid mass spectrometer. Peptides were separated using a 75 μm i.d. silica capillary column packed in-house with Repro-Sil Pur C18-AQ 3 μm resin and eluted with a gradient of 3–38% Buffer B over 85 minutes. Full MS scans from 300–1500 m/z were analyzed in the Orbitrap at 120,000 FWHM resolution and 5×105 AGC target value, for 50 ms maximum injection time. Ions were selected for MS2 analysis with an isolation window of 2 m/z, for a maximum injection time of 50 ms, to a target AGC of 5×104.
MS raw files were analyzed by MaxQuant software version 1.6.0.16, and MS2 spectra were searched against a target + reverse database with the Andromeda search engine using the Human UniProt FASTA database [9606] (reviewed, canonical entries; downloaded November 2017) and adenovirus serotype 5 UniProt FASTA database (reviewed, canonical entries; downloaded February 2018). Ad5 proteins detected in our proteomics data likely reflect the ability of E1B55K and E4orf6 to complement an E1A-deleted vector91. The search included variable modifications of methionine oxidation, N-terminal acetylation, and GlyGly on lysine residues, with a fixed modification of carbamidomethyl cysteine. For global proteome samples, iBAQ quantification was performed on unique+razor peptides using unmodified, oxidized methionine, and N-terminally acetylated forms. Trypsin cleavage was specified with up to 2 missed cleavages allowed. Match between runs was enabled, but restricted to matches within a single biological replicate by separating replicates into independent searches. Match between runs parameters included a retention time alignment window of 20 min and a match time window of 0.7 min. False discovery rate (FDR) was set to 0.01.
Proteomics and bioinformatics analysis
Data normalization and filtering
MaxQuant output was filtered to remove identified contaminant and reverse proteins. MaxQuant “Intensity” and “iBAQ”92 label-free quantification values were used to measure abundances for the K-ε-GG and WCP data, respectively. Abundances were transformed to log2 values, with unidentified values assigned as “NA”. K-ε-GG and WCP data were normalized separately. Data were normalized by subtracting the sample medians from log2 transformed abundances within replicates. Both the KεGG and the WCP datasets were filtered at each timepoint to require quantification in at least 2 of 3 replicates to be included in the calculations of fold change, z-score, or for hypothesis testing. Data that contained less than 2 replicate quantifications in each timepoint was removed entirely from the analysis. Peptides or proteins are considered uniquely identified in one timepoint compared to another if there were at least 2 replicate quantifications for one timepoint and 0 replicate quantifications for the compared timepoint. Peptides uniquely ubiquitinated during transduction are defined as those not quantified in any mock cell samples but found in 2–3 replicates from transduced cells. Since these unique peptides were not identified in mock conditions, they therefore do not have quantification values. The lack of quantification values precludes calculation of associated fold-changes or p-values since both of these calculations require numerical values for both compared conditions. Therefore, in these cases, we used z-scores to assess abundance of ubiquitination during expression of E1B55K/E4orf6, and for downstream analysis to define the most highly ubiquitinated proteins.
Fold change, p-value, and z-score calculations
The fold change across timepoints was calculated by comparing the log2 transformed, normalized peptide or protein abundances for compared timepoints. Fold changes were calculated both on a per-replicate basis and by comparing averaged abundances across timepoints. Hypothesis testing was performed using unpaired, two-tailed Students t-tests, when comparing log2 transformed, normalized replicate abundances across timepoints. Hypothesis testing using one-sided t-tests, with null hypothesis of fold change equal 0, was implemented when evaluating log2 fold changes. Multiple testing correction was not performed. Peptide ubiquitin intensity Z-scores were used to measure relative ubiquitin abundances for a peptide at the respective timepoint. Z-scores were calculated by averaging the peptide intensities for each replicate identification within the timepoint and comparing to the mean and standard deviation of averaged values within that timepoint.
Protein ubiquitin abundance calculation
The di-glycine technique quantifies peptide-based abundance of the K-ε-GG modification. In order to quantify protein-based K-ε-GG abundance changes, we implemented a calculation to combine the peptide-based fold changes for cases in which multiple K-ε-GG peptides comprise a modified protein. If a single K-ε-GG peptide was identified for a modified protein, that K-ε-GG peptide abundance fold change represented the protein-based K-ε-GG fold change. For cases in which multiple K-ε-GG peptides were quantified for a single protein, the fold changes of each peptide were weighted by the abundance of that peptide and the weighted fold changes were averaged to calculate the protein-based K-ε-GG fold change. In cases for which a peptide was uniquely identified in the mock or 10 h transduction timepoint, a log2 fold change of plus or minus 7, respectively, representing the largest fold changes identified in the dataset, was assigned to this peptide. The K-ε-GG abundance log2 fold changes, for each identified replicate, were normalized by the total protein abundance log2 fold change of the corresponding replicate of the same protein in the corresponding whole cell proteome. The normalization of the K-ε-GG data by WCP data was performed for the 10 h timepoint, which was the time at which the WCP infection data were generated. The replicate-based normalized log2 fold changes were averaged and hypothesis testing was performed for the log2 fold changes using onesided t-tests. The normalization of the K-ε-GG fold change by total protein fold change was performed to identify differentially increased or decreased ubiquitination, beyond what would be expected if modification abundance was driven solely by changes in total protein abundance.
K-ε-GG and whole cell proteome comparison
The protein-based K-ε-GG and corresponding whole cell proteome data were compared to identify proteins that exhibited an increase in K-ε-GG abundance and to predict the effect of ubiquitination on total protein abundance. Proteins that exhibited a protein-based, normalized K-ε-GG log2 fold change > 1 were classified as being increased in ubiquitination in response to E1B55K/E4orf6 expression. Proteins that exhibited whole cell proteome log2 fold change greater than the mean fold change +/− 1 standard deviation, or which were uniquely identified in the 0 or 10 hour timepoint, were classified as increased or decreased in total protein abundance. Proteins for which total protein expression did not deviate more than +/− 1 standard deviation from the mean fold change were classified as unchanged in protein abundance in response to E1B55K/E4orf6 expression. Proteins that were ubiquitinated and decreased in total protein abundance were predicted to be potential substrates of E1B55K/E4orf6 ubiquitin-mediated degradation. Proteins that were ubiquitinated and unchanged in total protein abundance were predicted to be non-degraded substrates of E1B55K/E4orf6.
Gene ontology and protein-protein interaction network analysis
The proteins that exhibited increased protein-based ubiquitination were analyzed using the ReactomeFI plug-in (6.1.0)36 within the Cytoscape network visualization software (3.4.0)93. The protein-protein interaction network was generated using the Gene Set analysis within the ”2016” ReactomeFI network version with “linker genes” included. The network was clustered using the in-built ReactomeFI clustering algorithm. Gene ontology “Molecular Function”, “Biological Processes” and Reactome Pathway analysis was performed within the ReactomeFI application for the entire network as well as for each clustered module. Network node attributes included size, which corresponded to degree of increased ubiquitination, and color, which corresponded to total protein increase or decrease. Network edges were set to non-directed, solid lines for all types of Reactome protein-protein interactions.
Targeted hnRNP-C RBR-ID
Cell growth
Heavy and light media were prepared by supplementing SILAC DMEM (Thermo #88364) with 800 μM of Lysine (Sigma #L8662–25G) and 400 μM Arginine (Sigma #A8094–25G) for light or K8 (Silantes #211604102) and R10 isotopes (Silantes #201604102) for heavy, and 120 mg/L Proline (Sigma #P0380–100G). Media was then filtered and adjusted to 10% dialyzed FBS (HyClone #SH30079.03) and 1% penicillin-streptomycin. Heavy isotope labeling in HeLa cells was confirmed by mass spectrometry. Cells were either mock-treated or infected with Ad5 WT Ad5 or ΔE1B at an MOI of 10, each in biological triplicate. At 20 hpi, media was exchanged and heavy-labeled cells were pulsed with 500 μM 4sU, with light-labeled cells serving as non-treated controls. At 24 hpi, all samples were washed with cold PBS and crosslinked at 1.0 J/cm2 with 310 nm UV-B. Cells were then harvested, heavy/light pairs were combined 1:1, and aliquoted for further analysis.
hnRNP-C IP
Approximately 1×107 pooled heavy and light HeLa cells were used for one hnRNP-C IP. 50 μl of Protein G Dynabeads (Thermo Fisher) per sample were washed 3x in IP buffer (20 mM HEPES-KOH pH 7.4, 110 mM potassium acetate, 2 mM MgCl2, 0.1% Tween-20, 0.1% Triton, 150 mM NaCl, 1 mM DTT, 0.1 mM PMSF, 1x cOmplete Protease Inhibitor Cocktail (Roche)) and incubated with 5 μg of α-hnRNP-C antibody rotating at RT for 1 h. Cell pellets were resuspended in 500 μl IP buffer, after 10 min 1.5 μl benzonase (Sigma-Aldrich, Cat#: E1014) were added and the sample was incubated for 1 h on ice. Samples were sonicated with a Diagenode Biorupter on low setting for 30 s on and 30 s off for ten rounds at 4 °C and spun at max speed (21,130 g) for 10 min at 4 °C. 450 μl of sample were added to washed beads and incubated rotating at 4 °C for 2 h. Beads were washed 3x with IP buffer before proteins were eluted in 0.1 M glycine (pH 2.4) for 10 min at RT, and elution was quenched with an equal volume of 0.1 M Tris-HCl (pH 8.0).
Mass spectrometry sample prep
Immunoprecipitated samples were reduced with 10 mM DTT for 30 min at RT and alkylated with 20 mM iodoacetamide for 45 min at RT in the dark. Samples were adjusted to 10 mM CaCl2 and split into two aliquots. One set of aliquots was digested at RT with chymotrypsin at a ~1:25 ratio and the other with trypsin at a ~1:30 ratio. Digestions were quenched after ~16 h by addition of TFA to pH 2. Samples were then desalted over Oligo R3 reverse-phase resin (Thermo Scientific, Cat#1-1339-03). All samples were run in technical duplicate.
Data acquisition
Peptide quantification by LC-MS/MS was performed on a Thermo Fisher Ultimate 3000 Dionex™ liquid chromatography system and a Thermo Q-Exactive HF-X™ mass spectrometer. The mobile phases consisted of 0.1% formic acid aqueous (mobile phase A) and 0.1% formic acid 80% acetonitrile (mobile phase B) with a gradient of 5–45% over 48 min and a 60 min total gradient. Samples were quantified by A280 absorbance and 1 μg of each was injected. Trypsin samples were run with MS1 settings of 250–1100 m/z window, a resolution of 60,000, AGC target of 5e5, and MIT (maximum inject time) of 54 ms. MS2 scans were collected in data dependent mode with a TopN loop count of 10, resolution of 15,000, AGC target of 1e5, and MIT of 100ms. Chymotrypsin samples were run on the same LC gradient with MS1 settings of 250–1100 m/z window, a resolution of 60,000, AGC target of 1e6, and MIT of 60 ms. MS2 scans were collected in data dependent mode with a TopN loop count of 10, resolution of 15,000, AGC target of 5e5, and MIT of 120ms. Fragmentation was performed with HCD using stepped normalized collision energies (NCE) of 25, 27, 30%94.
Data processing
Data files were processed by Sequest™ within Proteome Discoverer™ (PD) 2.3 workflow nodes. Searching parameters were set to find mass offsets of 8.014 Da for heavy K(+8) lysine and 10.008 Da for heavy R(+10) arginine for the SILAC heavy pairs55. Additionally, phosphorylation (79.966Da) and methylation (14.015Da) were searched on both viral and host proteins. A human protein FASTA and adenovirus type 5 specific FASTA files downloaded directly from Uniprot were used to process the raw files95. No imputation was used across data files. A 1% FDR level cutoff was applied at the peptide level by Percolator and the protein level. The use of Minora Feature Detector™ was used to identify SILAC pairs and identify non-sequenced peptides between runs96. Post-processing of the data files was performed in R Studio and peptide abundances were normalized to their respective proteins. For peptides identified in all samples (n=12 for each condition), the heavy/light pairs p-values were determined by a Student’s t-test as previously used in both the original RBR-ID paper53 and the subsequent SILAC targeted RBR-ID paper54. Score plots and fold change plots were generated using the mapping function from the original RBR-ID paper53.
hnRNP-C interactome analysis
In the targeted RBR-ID experiment, the “light” control sample contains the global interactome data for the targeted protein, hnRNP-C, in the absence of any RNA-protein crosslinking. The “light” control data for mock, Ad5 WT, and ΔE1B infections were compared to identify the proteins that interact with hnRNP-C in each of these conditions and to quantify changes in interactions induced by WT or ΔE1B infection. Three biological replicates were generated for each condition and each biological replicate was analyzed in two technical replicates. The “light” protein abundance data for each replicate was transformed by log2 and the identified protein abundances were normalized by the abundance of hnRNP-C in the respective replicate. The protein abundance values for the two technical replicates were averaged within each biological replicate. The protein abundance values for the three biological replicates within each condition were then averaged. When computing averages, unidentified values were not included in the calculation. Z-scores were calculated by comparing the average abundance of the protein in the respective condition to the mean and standard deviation of all averaged abundances for that condition. The z-score was calculated only for proteins with abundance quantified in at least 2 replicates for the respective condition. Fold changes were calculated by comparing protein average abundance values for compared conditions. Hypothesis testing was performed by using an unpaired t-test to compare log2 normalized protein abundance values for each replicate within compared conditions. Correlations were analyzed for log2 normalized abundances using the cor function (Pearson correlation coefficient) and visualized using the corrplot package in the R software environment97.
Statistics and reproducibility
Each experiment was carried out at least in triplicate with reproducible results. The sample size was chosen to provide enough statistical power apply parametric test (unpaired, two-tailed Student’s t-test unless otherwise noted). Details regarding statistical analysis are reported in each figure legend and p-values for each analysis can be found in Source Data.
Data availability
All mass spectrometry data for this study are deposited in the CHORUS database under project ID 1648, experiment IDs 3476 (KeGG), 3477 (KeGG-matched whole cell proteome), and 3478 (RBR-ID of hnRNP-C). The seCLIP-Seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE145411. The seCLIP-Seq data have been deposited in NCBI’s Gene Expression Omnibus98 and are accessible through GEO Series accession number GSE145411 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145411). Additional supporting data are available from the corresponding author upon request.
Code availability
The proteomics data were analyzed using standard methods. The implementation of the analysis was performed using R software. The scripts are available from the corresponding author upon request or can be accessed via GitHub. https://github.com/JosephDybas/AdenovirusProteomics
Extended Data
Extended Data Fig. 1. E1B55K deletion or inhibition of Cullin-mediated ubiquitination does not decrease late RNA transcription, decay, or early RNA processing.
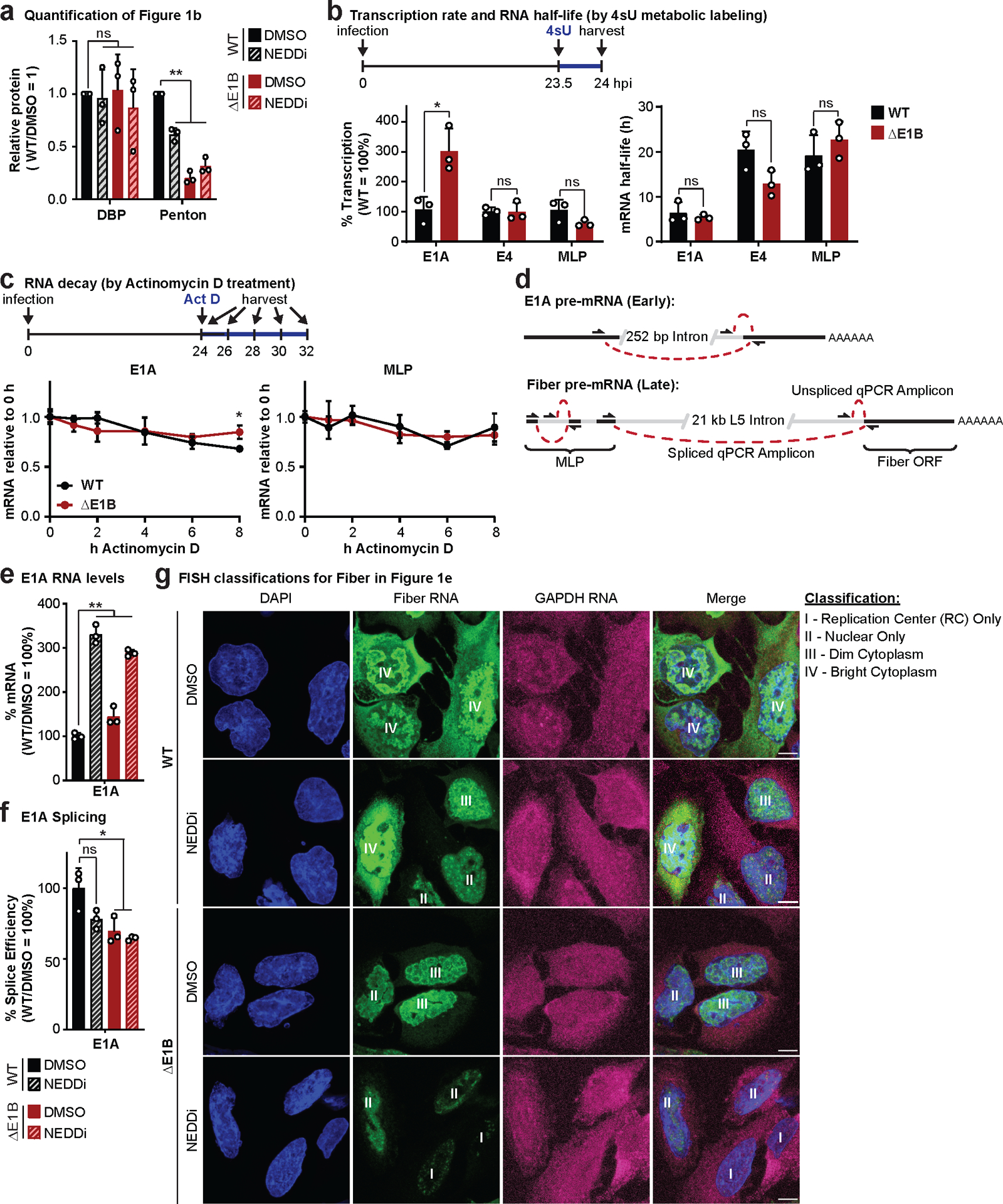
a, Quantification of results shown in Figure 1b. b, Analysis of nascent transcription and mRNA half-life by labeling RNA with 4-thiouridine (4sU) for 30 min at 23.5 hpi in HeLa cells infected with WT or ΔE1B Ad5 at MOI=10. Nascent 4sU-labeled RNA was purified for RT-qPCR to determine relative transcription rates of two early (E1A and E4) and one late (MLP) viral transcripts. mRNA half-life was approximated using the ratio of nascent and total input RNA levels normalized to GAPDH. c, Analysis of decay of viral early (E1A) and late (MLP) RNA species by Actinomycin D pulse at 24 hpi in HeLa cells infected with WT or ΔE1B Ad5 at MOI 10 by normalization to input levels. d, Schematic illustrating primer design to differentiate spliced and unspliced viral transcripts. e-g, HeLa cells infected with WT or ΔE1B Ad5 (MOI=10) in the presence of DMSO or NEDDi (neddylation inhibitor MLN4924) added at 8 hours post-infection (hpi). Cells were harvested for RNA analysis at 24 hpi. e, Bar graph representing spliced RNA levels of viral early transcripts E1A by RT-qPCR, f, Bar graph representing splicing efficiency as the ratio of spliced to unspliced transcripts of E1A relative to the WT DMSO control by RT-qPCR. g, Representative RNA FISH visualizing the localization of fiber (green) and GAPDH (magenta) transcripts in relation to nuclear DNA stained with DAPI (blue). Nuclei are labeled with the classification of each cell according to the pattern of fiber used for Figure 1d. Scale bar=10 μm. All data are representative of two (g) or three (others) biologically independent experiments. All graphs show mean+SD. Statistical significance was calculated using a paired (a) or unpaired (others), two-tailed Student’s t-test, * p <0.05, ** p <0.01.
Extended Data Fig. 2. Quantification for number of peptides and proteins identified in di-glycine remnant profiling and whole cell proteome data sets.
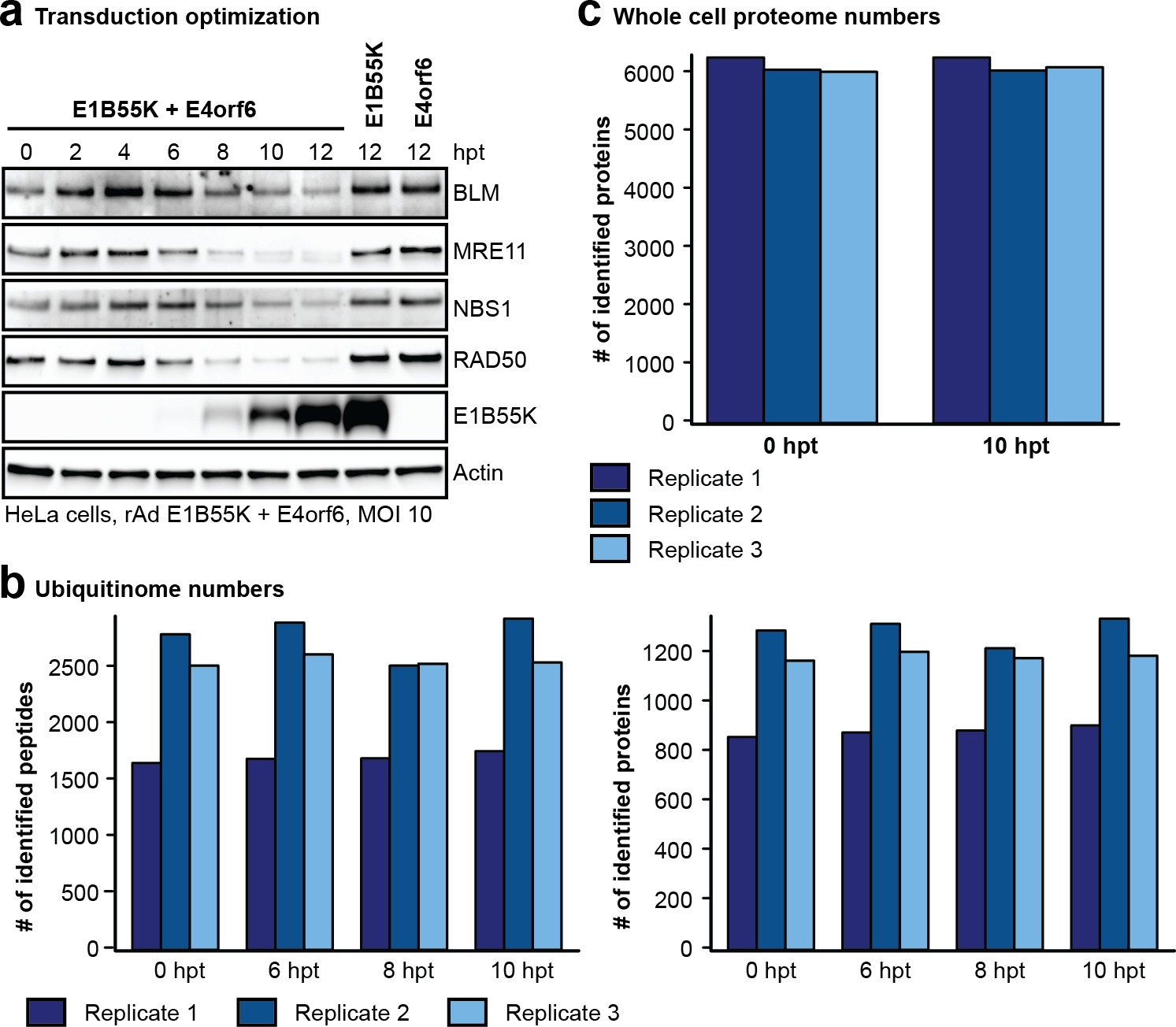
a-c, HeLa cells transduced with rAd E1B55K/E4orf6 (MOI=10). a, Immunoblot of time course of E1B55K/E4of6 expression showing degradation kinetics of known substrates. hpt=hours post-transduction. Representative of 3 biologically independent experiments. b-c, Peptide and protein counts for three biologically independent replicates (dark, medium, and light blue bars). b, Total peptides and corresponding proteins identified following K-ɛ-GG antibody enrichment combined with mass spectrometry analysis at 0, 6, 8, and 10 hours post E1B55K/E4orf6 expression. c, Total proteins identified by whole cell proteomics analysis at time 0 and 10 h of E1B55K/E4orf6 expression.
Extended Data Fig. 3. Di-glycine remnant profiling and whole cell proteome data for RNA-binding proteins enriched within predicted E1B55K/E4orf6 substrates.
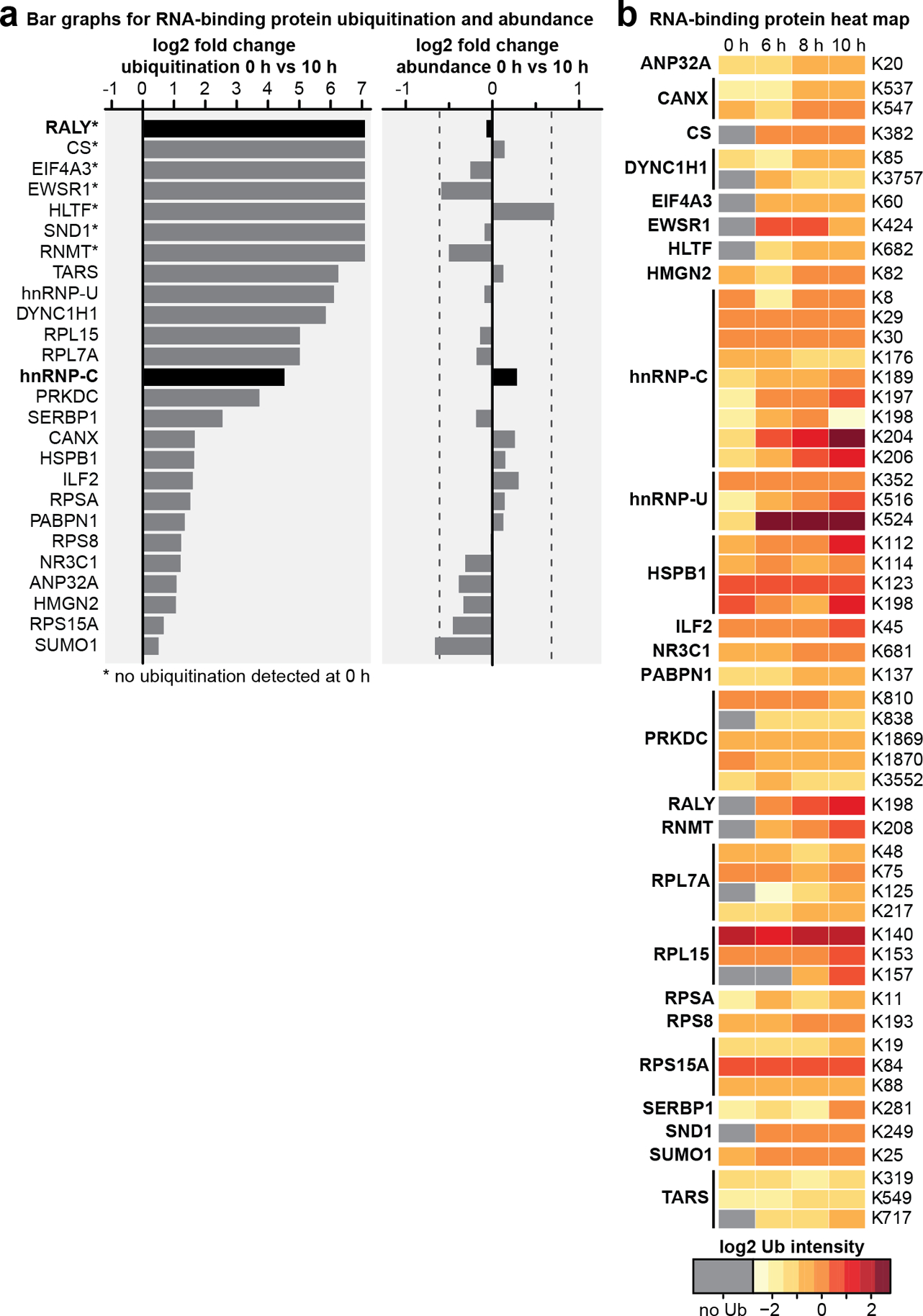
a-b, Gene ontology analysis identified RNA-binding proteins enriched in proteins that exhibited an increase in normalized protein-based ubiquitin abundance of log2 fold change >1 following 10 h expression of E1B55K/E4orf6. a, Enriched RNA-binding proteins ubiquitination log2 fold changes (left) and whole cell protein abundance log2 fold changes (right) comparing 0 and 10 h of E1B55K/E4orf6 expression. b, Heat map showing relative ubiquitination of indicated lysine residues at 0, 6, 8, and 10 h of E1B55K/E4orf6 expression within enriched RNA-binding proteins. Colors correspond to the average z-score of the ubiquitination. All data are representative of three biologically independent experiments.
Extended Data Fig. 4. Network analysis of predicted E1B55K/E4orf6 substrates identifies a “RALY/hnRNP-C module” enriched for RNA-binding proteins.
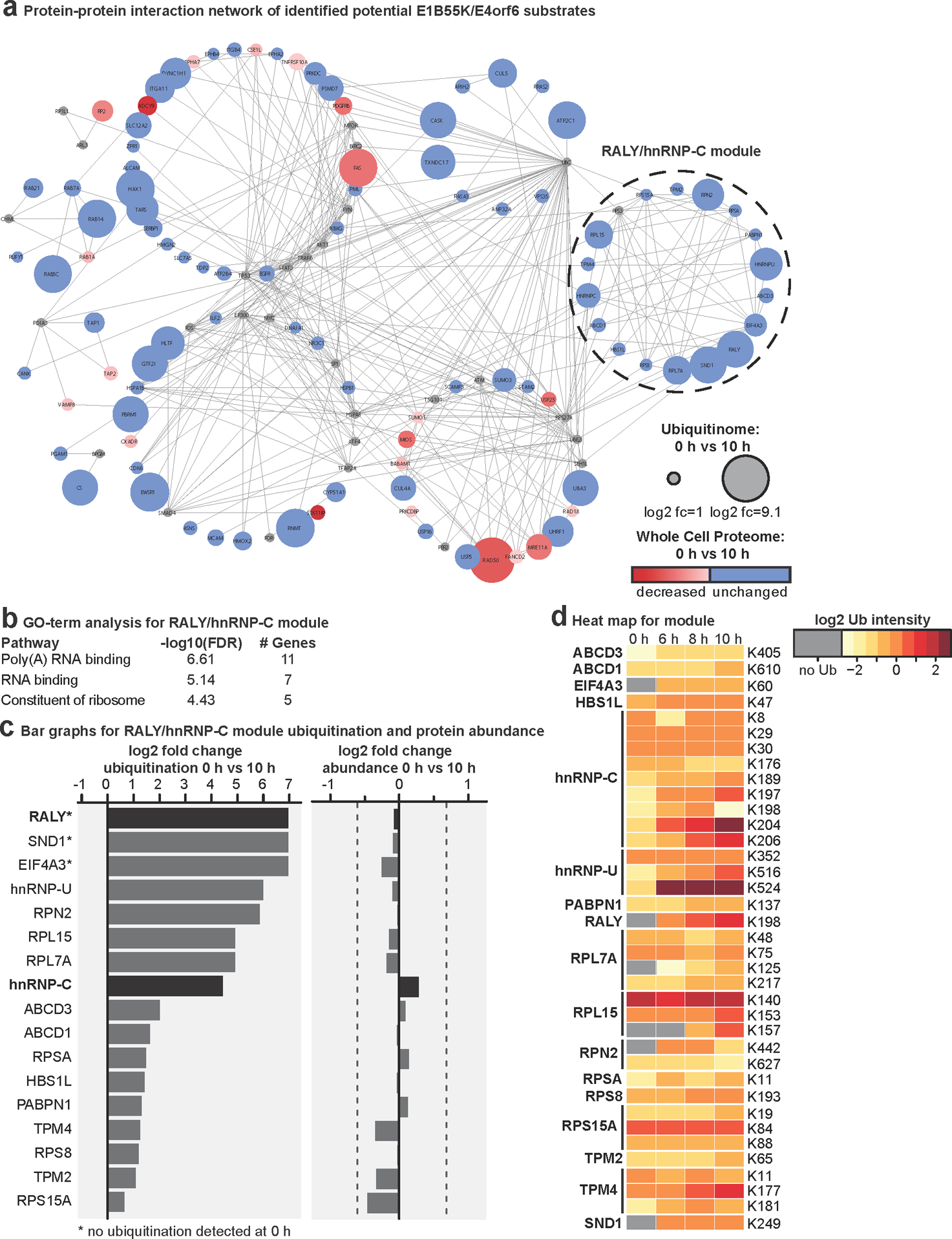
a, The Reactome-FI application in Cytoscape was utilized to generate a protein-protein interaction network for proteins increased in ubiquitination following E1B55K/E4orf6 transduction. Nodes represent proteins and edges represent Reactome-based protein-protein interactions. Node size corresponds to relative protein-based ubiquitination log2 fold change and node color corresponds to whole cell proteome log2 fold change following 10 h expression of E1B55K/E4orf6. Reactome-FI interaction module analysis was performed to identify clusters of highly interacting proteins. b, Gene ontology analysis for molecular function, as generated by ReactomeFI (FDR is Benjamini-Hochberg method), identified enrichment of RNA-binding and Poly(A) RNA-binding proteins within the RALY/hnRNP-C network module. c, RALY/hnRNP-C network module protein ubiquitin log2 fold changes (left) and whole cell protein abundance log2 fold changes (right) comparing 0 and 10 h of E1B55K/E4orf6 expression. d, Heat map showing relative ubiquitin abundance of indicated lysine residues at 0, 6, 8, and 10 h of E1B55K/E4orf6 expression from proteins within the RALY/hnRNP-C network module. Colors correspond to the average z-score of ubiquitination. All data are representative of three biologically independent experiments.
Extended Data Fig. 5. MS2 evidence for ubiquitination site localization in RALY (a) and hnRNP-C (b-f) peptides.
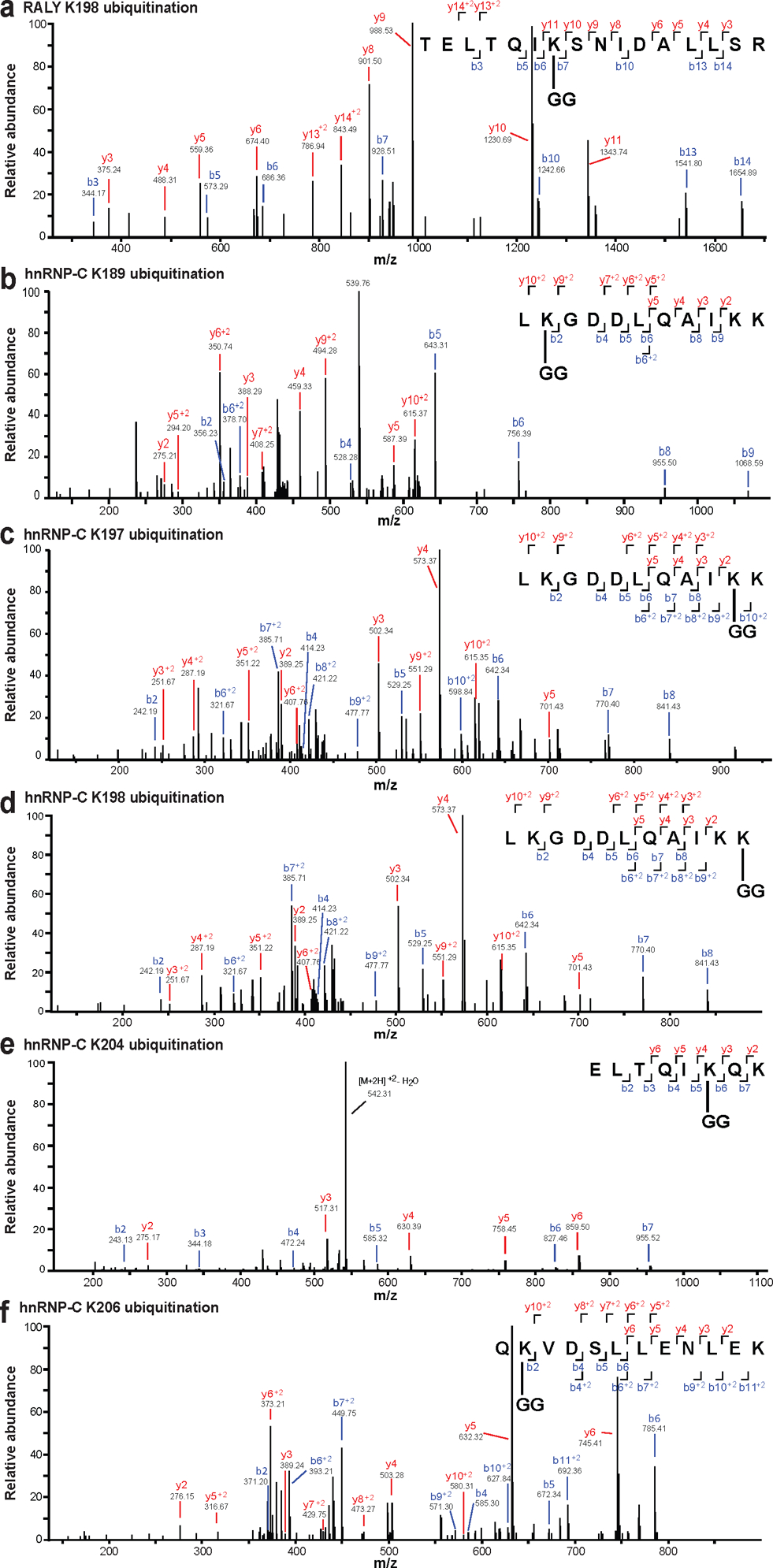
Spectra were obtained from LC-MS/MS analyses using collision-induced dissociation (CID) at 35%, and identified in MaxQuant 1.6.0.1. All modified residues can be confidently identified by confirming ions, except for hnRNP-C K198 (d), which lacks ions to distinguish between K197 and K198. Best evidence spectra were selected for annotation of b-ion (blue) and y-ion (red) series and their masses for singly- and doubly-charged fragments. All data are representative of three biologically independent experiments.
Extended Data Fig. 6. RALY and hnRNP-C are not decreased upon transduction in multiple cell lines.
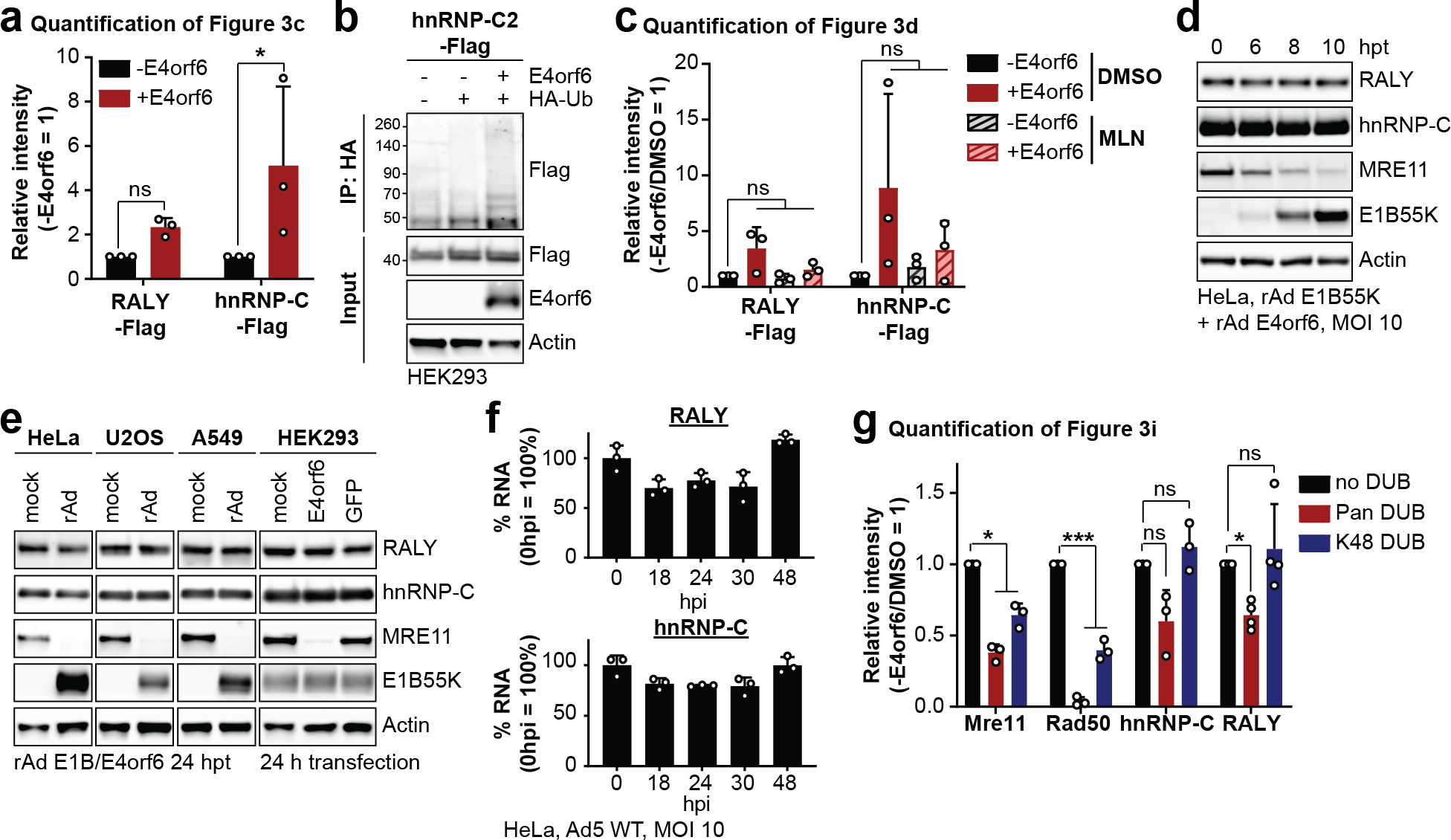
a, Quantification of results shown in Figure 3c. b, HEK293 cells transfected with the indicated constructs for 24 h, followed by denaturing IP with HA antibody, and immunoblot analysis of hnRNP-C2-Flag. c, Quantification of results shown in Figure 3d. d, Immunoblot analysis of protein levels in HeLa cells over a time course of transduction with recombinant Ad vectors expressing only E1B55K and E4orf6 (MOI=10). e, Immunoblot analysis of protein levels in HeLa, U2OS, A549 and HEK293 cells. HeLa, U2OS and A459 cells were transduced with recombinant Ad vectors expressing only E1B55K and E4orf6 for 24 h. HEK293 cells, which contain an endogenous copy of E1B55K, were mock transfected or transfected with plasmids expressing E4orf6 or GFP. f, Bar graphs of RALY and hnRNP-C RNA levels over a time course of infection with Ad5 WT (MOI=10) relative to mock as determined by RT-qPCR. g, Quantification of results shown in Figure 3i. All data are representative of three (four for RALY in g) biologically independent experiments. All graphs show mean+SD. Statistical significance was calculated using a paired, two-tailed Student’s t-test, * p <0.05, ** p <0.01, *** p <0.005.
Extended Data Fig. 7. RALY and hnRNP-C single knockdowns rescue late protein, RNA and splice efficiency during infection with Ad ΔE1B.
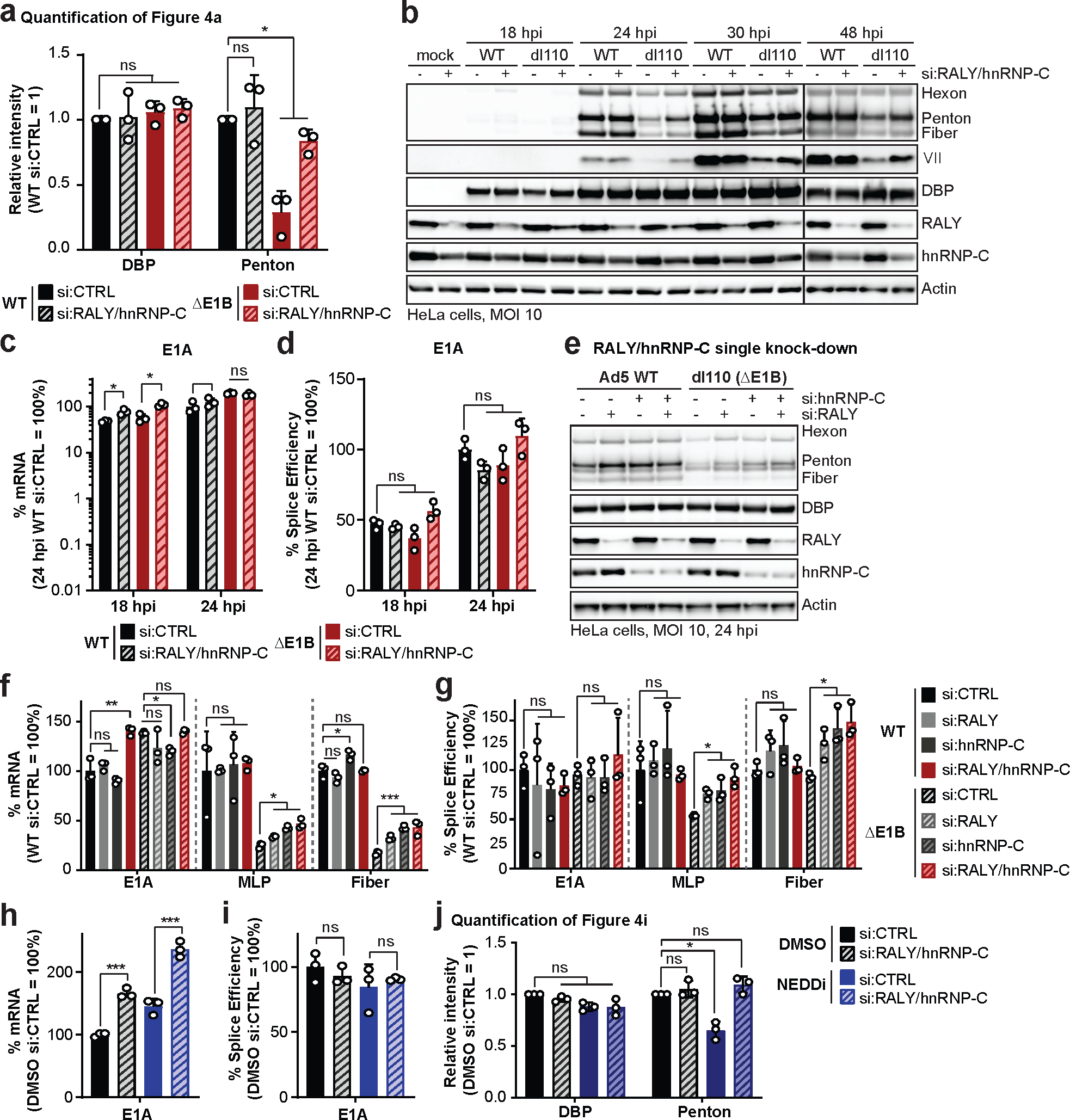
a-d, HeLa cells transfected with control (siCTRL) or RALY and hnRNP-C (siRALY/hnRNP-C) siRNA 24 h prior to infection with Ad5 WT or ΔE1B (MOI=10), harvested at respective time points. a, Quantification of results shown in Figure 4a. b, Extended immunoblot analysis of viral and cellular protein levels. c, Bar graph representing spliced RNA levels of viral early transcript E1A measured by RT-qPCR. d, Bar graph representing splicing efficiency (ratio of spliced to unspliced transcripts) of E1A measured by RT-qPCR. e-g, HeLa cells transfected with control siRNA (siCTRL), siRNA for RALY (siRALY), siRNA for hnRNP-C (sihnRNP-C) or siRNA for both RALY and hnRNP-C (siRALY/hnRNP-C) 24 h prior to infection with Ad5 WT or ΔE1B (MOI=10) and harvested at 24 hpi. e, Immunoblot analysis of viral and cellular protein levels. f, Bar graph representing spliced RNA levels of E1A, MLP and fiber measured by RT-qPCR. g, Bar graph representing splicing efficiency of E1A, MLP and fiber measured by RT-qPCR. h-j. HeLa cells transfected with control (siCTRL) or RALY and hnRNP-C (siRALY/hnRNP-C) siRNA 24 h prior to infection with Ad5 WT (MOI=10), treated with either DMSO or NEDDi at 8 hpi and processed at 24 hpi. h, Bar graph representing spliced RNA levels of E1A measured by RT-qPCR. i, Bar graph representing splicing efficiency as defined as the ratio of spliced to unspliced transcripts of E1A measured by RT-qPCR. j, Quantification of results shown in Figure 4i. All data are representative of three biologically independent experiments. All graphs show mean+SD. Statistical significance was calculated using paired (a and j) or unpaired (others), two-tailed Student’s t-test, * p <0.05, ** p <0.01, *** p <0.005.
Extended Data Fig. 8. No dramatic difference in protein localization and protein-complex formation of RALY and hnRNP-C between Ad WT and ΔE1B infection.
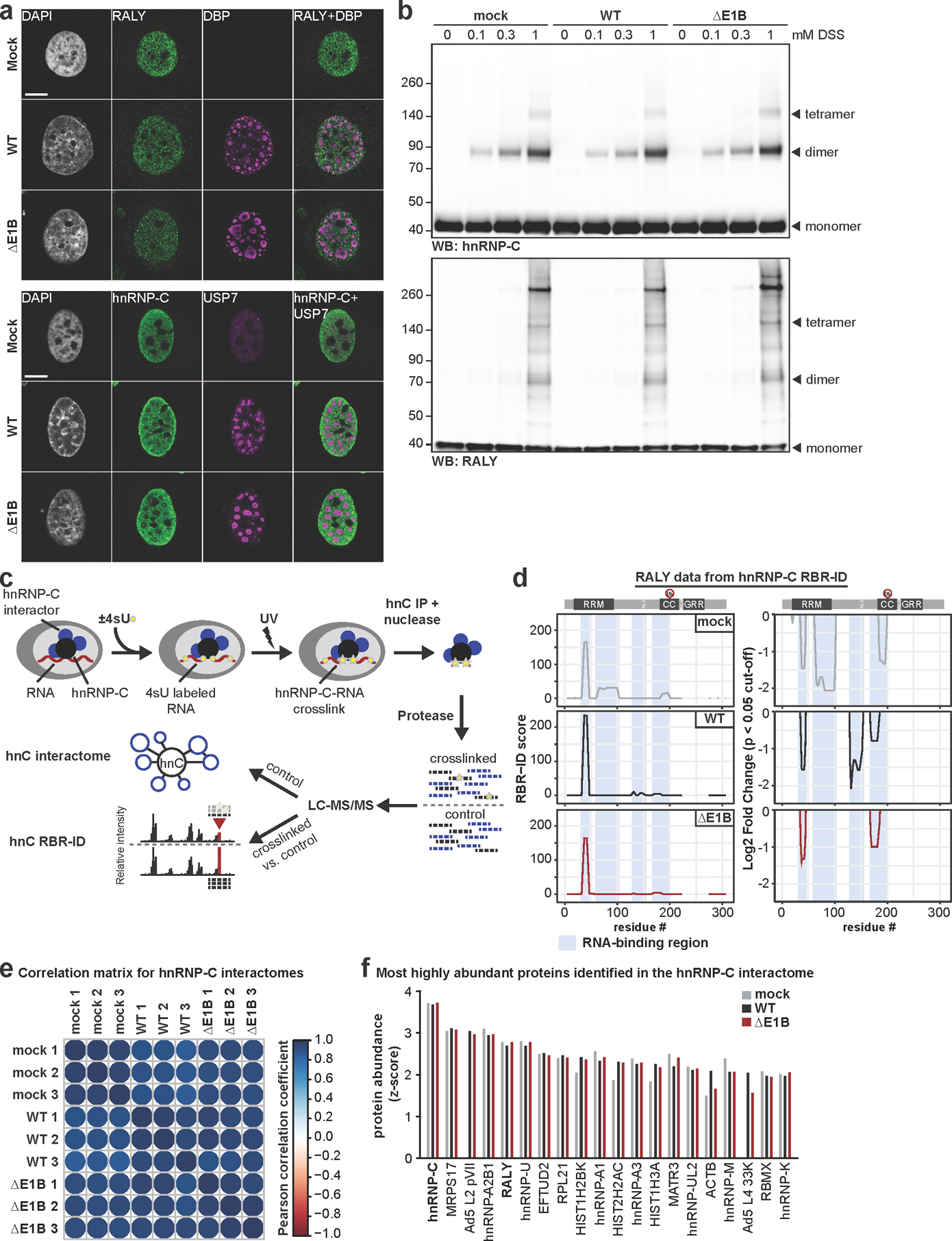
a, Representative images of immunofluorescence comparing the localization of RALY and hnRNP-C (both green) in mock, Ad WT and ΔE1B infection of HeLa cells (MOI=10, 24 hpi). Viral replication centers are stained by DBP or USP7 (both magenta) and nuclear DNA by DAPI (grey). Scale bar=10 μm. b, Immunoblot analysis of RALY and hnRNP-C protein complexes formed upon mock, Ad WT and ΔE1B infection of HeLa cells (MOI=10) and treatment with indicated concentrations of disuccinimidyl suberate (DSS) for 30 min at 24 hpi. c, Schematic for targeted hnRNP-C RNA-binding region identification (RBR-ID) and interactome. d, Data for RALY from hnRNP-C RBR-ID experiment comparing mock (grey), Ad5 WT (black), and ΔE1B (red) at 24 hpi (MOI=10). Shown are smoothed residue-level RBR-ID score plotted along the primary sequence (left) and smoothed residue-level fold-change between crosslinked and control conditions with a significance threshold of p <0.05 (right). RALY domain structure with ubiquitination site is shown above graphs. RNA-binding regions are highlighted in blue. e, Correlation matrix for hnRNP-C interactome between replicates of mock, Ad5 WT, and Ad5 ΔE1B. Color gradient is based on the Pearson correlation coefficient. f, Comparison of z-scores for top 20 proteins identified in hnRNP-C interactome during WT Ad5 infection (MOI=10, 24 hpi). Mock=grey, Ad5 WT=black, Ad5 ΔE1B=red. All data are representative of three biologically independent experiments.
Extended Data Fig. 9. In the absence of E1B55K, hnRNP-C and RALY interact more with viral late RNA.
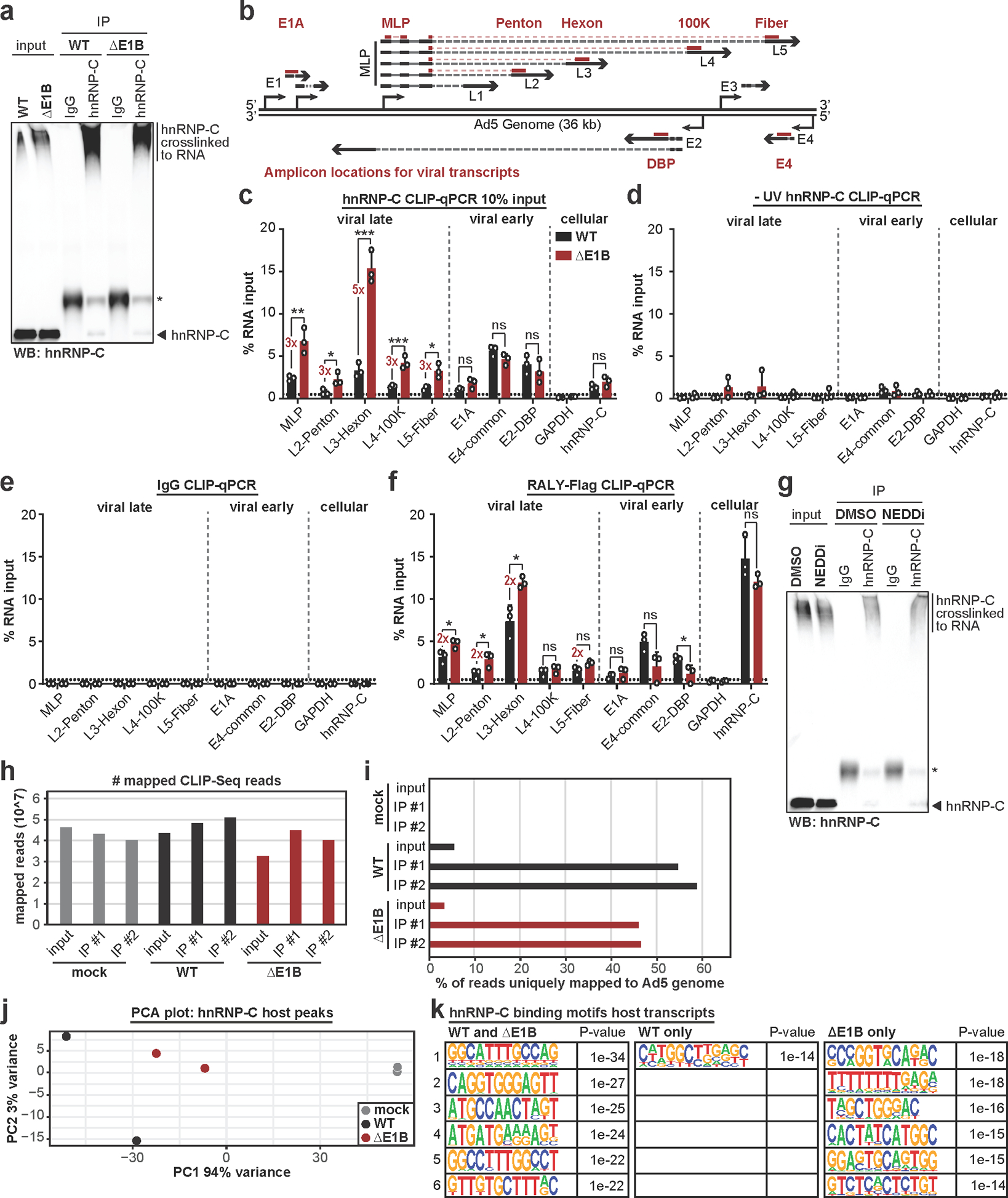
a, Control immunoblot for hnRNP-C CLIP-qPCR in Figure 5b. Higher molecular weight complexes represent hnRNP-C crosslinked to RNA. * indicates antibody heavy chain. b, Schematic of Ad5 genome and viral transcription units. Amplicons for early (E1A, DBP, E4) and late (MLP, Penton, Hexon, 100K, Fiber) transcription units indicated. c, HeLa cells infected with WT Ad5 or ΔE1B (MOI=10), UV-crosslinked and harvested at 24 hpi, subjected to hnRNP-C CLIP with 10% input as compared to Figure 5b and RT-qPCR of indicated transcripts. d, HeLa cells infected with WT Ad5 or ΔE1B (MOI=10), without UV-crosslinking, and harvested at 24 hpi, subjected to hnRNP-C CLIP and RT-qPCR of indicated transcripts. e, HeLa cells infected with WT Ad5 or ΔE1B (MOI=10), UV-crosslinked and harvested at 24 hpi, subjected to IgG CLIP and RT-qPCR of indicated transcripts. f, HeLa cells induced for RALY-Flag expression with doxycycline for 3 days total, infected with WT Ad5 or ΔE1B (MOI=10), UV-crosslinked and harvested at 24 hpi, subjected to Flag CLIP and RT-qPCR of indicated transcripts. For all CLIP-qPCR experiments: GAPDH = negative control, hnRNP-C = positive control. g, Immunoblot for hnRNP-C CLIP-qPCR in Figure 5c. Higher molecular weight complexes represent hnRNP-C crosslinked to RNA. * indicates antibody heavy chain. h, Number of mapped hnRNP-C eCLIP-Seq reads for the indicated conditions. i, Percentage of mapped reads that uniquely mapped to Ad5 genome for different hnRNP-C eCLIP-Seq conditions. j, PCA plot for hnRNP-C peaks mapped to host transcripts comparing mock (grey), Ad5 WT (black), and Ad5 ΔE1B (red). k, Top 6 hnRNP-C binding motifs in WT and ΔE1B infection, WT infection only, and ΔE1B infection only. n=1043, 162 peaks, and 267 peaks, respectively. Enriched motifs were identified with HOMER Software Package using hypergeometric enrichment calculations and adjustments for multiple comparisons. All data are representative of two (h-k) or three (a-g) independent experiments. Graphs in c-f show mean+SD. Statistical significance was calculated using unpaired, two-tailed Student’s t-test, * p <0.05, ** p <0.01, *** p <0.005.
Extended Data Fig. 10. Non-degradative ubiquitination of RNA-binding proteins promotes efficient adenoviral RNA processing.
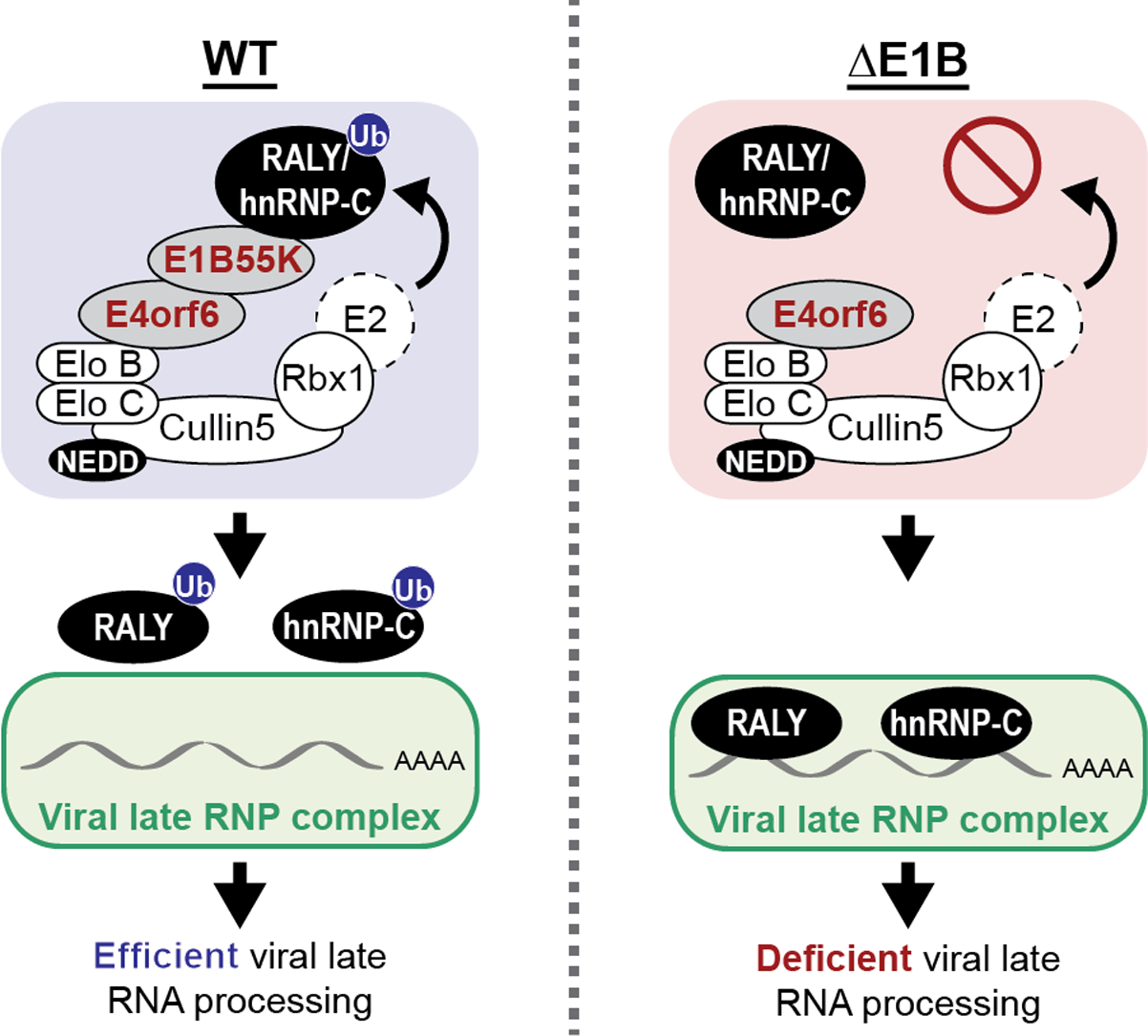
During wild-type (WT) Ad5 infection the E1B55K/E4orf6 complex induces ubiquitination of RNA-binding proteins RALY and hnRNP-C to facilitate efficient viral late RNA processing. Ubiquitination regulates interaction of these host proteins with viral RNA to facilitate viral infection. In the absence of the E1B55K/E4orf6 complex ubiquitin ligase activity, the RBPs bind more to viral late mRNAs and limit RNA processing and protein production. RNP=ribonucleoprotein.
Supplementary Material
Supplementary Table 1: Complete Di-glycine remnant and whole cell proteomics data. Normalized and log2 transformed proteomics data for K-ɛ-GG peptide based quantification (tab 1), transformation to K-e-GG protein based quantification (tab 2), and whole cell proteome quantification. Data were generated from three biologically independent experiments. Fold changes were calculated in log2 and p-values were calculated using unpaired, two-sided Student’s t-tests.
Supplementary Table 2: Di-glycine remnant and whole cell proteomics data for predicted E1B55K/E4orf6 substrates. Normalized and log2 transformed proteomics data for K-ɛ-GG protein based (tab 1) and peptide based (tab 2) quantifications for predicted E1B55K/E4orf6 substrates. Data were generated from three biologically independent experiments. Significance tests for whole cell proteome abundance changes are calculated using unpaired, two-sided Student’s t-tests. K-ɛ-GG abundance changes are normalized to whole cell proteome abundance changes and significance is calculated by one-sided Student’s t-tests with a null hypothesis of 0 fold change.
Supplementary Table 3: Reactome network protein-protein interactome analysis results. Reactome FI network analysis results of the proteins that exhibited increased protein-based ubiquitination (n=120). Reactome FI clustering algorithm was used to divide the interactome into modules (tab 1). Reactome FI was used to perform Gene Ontology and Pathway enrichment analysis. The Gene Ontology “Molecular Functions” is included for modules (tab 2) and the entire network (tab 5). The Gene Ontology “Biological Processes” is included for modules (tab3) and the entire network (tab 6). The Reactome Pathway analysis is included for modules (tab 4) and the entire network (tab 7). Data were generated from three biologically independent experiments.
Supplementary Table 4: SILAC ratios for chymotryptic and tryptic peptides from targeted hnRNP-C RBR-ID analysis. Heavy/light ratios indicate the proportion of peptides cross-linked to RNA upon UV treatment. Data represent 3 biological replicates and 2 technical replicates (n=6) per digestion condition. Chymotryptic (tab 1) and tryptic (tab 2)data were processed independently in Proteome Discoverer v2.3. Results were filtered to exclude peptides with any missing values across replicates.
Supplementary Table 5: hnRNP-C interactome derived from RBR-ID data. The non-crosslinked sample from the hnRNP-C RBR-ID is used to generate the hnRNP-C interactome. Normalized, log2 transformed data quantification for proteins identified in association with hnRNP-C. Data were generated from 3 biological replicates and 2 technical replicates (n=6) per digestion condition. Fold changes were calculated in log2 and p-values were calculated using unpaired, two-sided Student’s t-tests.
Acknowledgements
We thank members of the Weitzman Lab for insightful discussions and input. We thank P. Choi for help with seCLIP-Seq, and G. Yeo and E. Van Nostrand for their input regarding analysis of corresponding data. We are grateful to A. Berk, P. Branton, R. Greenberg, G. Ketner, A. Levine, K. Lynch, D. Ornelles, J. Wilson, and H. Wodrich for generous gifts of reagents. We thank S. Schaffer for technical advice, and D. Avgousti, L. Busino, P. Choi, K. Lynch, and J. Weitzman for careful reading of the manuscript. We thank the UPenn Cell and Developmental Biology Microscopy Core for imaging assistance. This research was supported by NIAID grants R01-AI145266 (MDW), R01-AI121321 (MDW) and R01-AI118891 (BAG), and NCI grant R01-CA97093 (MDW). Additional support came from the NCI T32 Training Grant in Tumor Virology T32-CA115299 (AMP) and Individual National Research Service Awards F32-AI147587 (JMD) and F32-AI138432 (AMP) from the National Institutes of Health.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Isaacson MK & Ploegh HL Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell host & microbe 5, 559–570 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo H Interplay between the virus and the ubiquitin-proteasome system: molecular mechanism of viral pathogenesis. Curr Opin Virol 17, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dybas JM, Herrmann C & Weitzman MD Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr Opin Virol 32, 40–47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komander D & Rape M The ubiquitin code. Annu Rev Biochem 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Oh E, Akopian D & Rape M Principles of Ubiquitin-Dependent Signaling. Annu Rev Cell Dev Biol 34, 137–162 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Kulathu Y & Komander D Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nature reviews. Molecular cell biology 13, 508–523 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Petroski MD & Deshaies RJ Function and regulation of cullin-RING ubiquitin ligases. Nature reviews. Molecular cell biology 6, 9–20 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Querido E et al. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15, 3104–3117 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada JN, Shevchenko A, Shevchenko A, Pallas DC & Berk AJ Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76, 9194–9206 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babiss LE & Ginsberg HS Adenovirus type 5 early region 1b gene product is required for efficient shutoff of host protein synthesis. J Virol 50, 202–212 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babiss LE, Ginsberg HS & Darnell JE Jr. Adenovirus E1B proteins are required for accumulation of late viral mRNA and for effects on cellular mRNA translation and transport. Mol Cell Biol 5, 2552–2558 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halbert DN, Cutt JR & Shenk T Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J Virol 56, 250–257 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pilder S, Moore M, Logan J & Shenk T The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol Cell Biol 6, 470–476 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridge E & Ketner G Redundant control of adenovirus late gene expression by early region 4. J Virol 63, 631–638 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridge E & Ketner G Interaction of adenoviral E4 and E1b products in late gene expression. Virology 174, 345–353 (1990). [DOI] [PubMed] [Google Scholar]
- 16.Sandler AB & Ketner G Adenovirus early region 4 is essential for normal stability of late nuclear RNAs. J Virol 63, 624–630 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo JL & Berk AJ Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J Virol 81, 575–587 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanchette P et al. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J Virol 82, 2642–2651 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Querido E et al. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol 71, 3788–3798 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stracker TH, Carson CT & Weitzman MD Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418, 348–352 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Baker A, Rohleder KJ, Hanakahi LA & Ketner G Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol 81, 7034–7040 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dallaire F, Blanchette P, Groitl P, Dobner T & Branton PE Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J Virol 83, 5329–5338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orazio NI, Naeger CM, Karlseder J & Weitzman MD The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J Virol 85, 1887–1892 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cathomen T & Weitzman MD A functional complex of adenovirus proteins E1B-55kDa and E4orf6 is necessary to modulate the expression level of p53 but not its transcriptional activity. J Virol 74, 11407–11412 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carson CT et al. The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo j 22, 6610–6620 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz RA et al. Distinct requirements of adenovirus E1b55K protein for degradation of cellular substrates. J Virol 82, 9043–9055 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu G, Paige JS & Jaffrey SR Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nature biotechnology 28, 868–873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Udeshi ND, Mertins P, Svinkina T & Carr SA Large-scale identification of ubiquitination sites by mass spectrometry. Nat Protoc 8, 1950–1960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soucy TA et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Hori T et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 18, 6829–6834 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Schwechheimer C NEDD8-its role in the regulation of Cullin-RING ligases. Curr Opin Plant Biol 45, 112–119 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zhou P & Howley PM Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol Cell 2, 571–580 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Tudek A, Schmid M & Jensen TH Escaping nuclear decay: the significance of mRNA export for gene expression. Curr Genet 65, 473–476 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Stewart M Polyadenylation and nuclear export of mRNAs. The Journal of biological chemistry 294, 2977–2987 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcellus RC, Teodoro JG, Charbonneau R, Shore GC & Branton PE Expression of p53 in Saos-2 osteosarcoma cells induces apoptosis which can be inhibited by Bcl-2 or the adenovirus E1B-55 kDa protein. Cell Growth Differ 7, 1643–1650 (1996). [PubMed] [Google Scholar]
- 36.Fabregat A et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. www.proteinatlas.org.
- 38.Konig J et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17, 909–915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCloskey A, Taniguchi I, Shinmyozu K & Ohno M hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 335, 1643–1646 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Zarnack K et al. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152, 453–466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cornella N et al. The hnRNP RALY regulates transcription and cell proliferation by modulating the expression of specific factors including the proliferation marker E2F1. The Journal of biological chemistry 292, 19674–19692 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi A et al. Identification and dynamic changes of RNAs isolated from RALY-containing ribonucleoprotein complexes. Nucleic Acids Res 45, 6775–6792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondy-Chorney E et al. RNA binding protein RALY promotes Protein Arginine Methyltransferase 1 alternatively spliced isoform v2 relative expression and metastatic potential in breast cancer cells. Int J Biochem Cell Biol 91, 124–135 (2017). [DOI] [PubMed] [Google Scholar]
- 44.van Eekelen C et al. Sequence dependent interaction of hnRNP proteins with late adenoviral transcripts. Nucleic Acids Res 10, 7115–7131 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tenzer S et al. Proteome-wide characterization of the RNA-binding protein RALY-interactome using the in vivo-biotinylation-pulldown-quant (iBioPQ) approach. J Proteome Res 12, 2869–2884 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Graham FL, Smiley J, Russell WC & Nairn R Characteristics of a human cell line transformed by DNA from human adenovirus type 5. The Journal of general virology 36, 59–74 (1977). [DOI] [PubMed] [Google Scholar]
- 47.Mevissen TET & Komander D Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem 86, 159–192 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Chahal JS & Flint SJ Timely synthesis of the adenovirus type 5 E1B 55-kilodalton protein is required for efficient genome replication in normal human cells. J Virol 86, 3064–3072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mashtalir N et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell 54, 392–406 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Bridge E & Pettersson U Nuclear organization of adenovirus RNA biogenesis. Exp Cell Res 229, 233–239 (1996). [DOI] [PubMed] [Google Scholar]
- 51.Pombo A, Ferreira J, Bridge E & Carmo-Fonseca M Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J 13, 5075–5085 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang M et al. The C-protein tetramer binds 230 to 240 nucleotides of pre-mRNA and nucleates the assembly of 40S heterogeneous nuclear ribonucleoprotein particles. Mol Cell Biol 14, 518–533 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He C et al. High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol Cell 64, 416–430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Q et al. RNA exploits an exposed regulatory site to inhibit the enzymatic activity of PRC2. Nat Struct Mol Biol 26, 237–247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ong SE et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Gorlach M, Burd CG & Dreyfuss G The determinants of RNA-binding specificity of the heterogeneous nuclear ribonucleoprotein C proteins. The Journal of biological chemistry 269, 23074–23078 (1994). [PubMed] [Google Scholar]
- 57.Van Nostrand EL et al. A Large-Scale Binding and Functional Map of Human RNA Binding Proteins. bioRxiv, 179648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dallaire F, Blanchette P & Branton PE A proteomic approach to identify candidate substrates of human adenovirus E4orf6-E1B55K and other viral cullin-based E3 ubiquitin ligases. J Virol 83, 12172–12184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evans VC et al. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat Methods 9, 1207–1211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu YR et al. Comparison of protein expression during wild-type, and E1B-55k-deletion, adenovirus infection using quantitative time-course proteomics. The Journal of general virology 98, 1377–1388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hung G & Flint SJ Normal human cell proteins that interact with the adenovirus type 5 E1B 55kDa protein. Virology 504, 12–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chanarat S & Mishra SK Emerging Roles of Ubiquitin-like Proteins in Pre-mRNA Splicing. Trends Biochem Sci 43, 896–907 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Vassileva MT & Matunis MJ SUMO modification of heterogeneous nuclear ribonucleoproteins. Mol Cell Biol 24, 3623–3632 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan CH et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee SW et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell 57, 1022–1033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q et al. FBXW7 Facilitates Nonhomologous End-Joining via K63-Linked Polyubiquitylation of XRCC4. Mol Cell 61, 419–433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bischoff JR et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274, 373–376 (1996). [DOI] [PubMed] [Google Scholar]
- 69.Goodrum FD & Ornelles DA p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol 72, 9479–9490 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Shea CC et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6, 611–623 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Khandelia P, Yap K & Makeyev EV Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 108, 12799–12804 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grifman M et al. Overexpression of cyclin A inhibits augmentation of recombinant adeno-associated virus transduction by the adenovirus E4orf6 protein. J Virol 73, 10010–10019 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sobhian B et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316, 1198–1202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kozarsky KF, Jooss K, Donahee M, Strauss JF 3rd & Wilson JM Effective treatment of familial hypercholesterolaemia in the mouse model using adenovirus-mediated transfer of the VLDL receptor gene. Nat Genet 13, 54–62 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Komatsu T, Dacheux D, Kreppel F, Nagata K & Wodrich H A Method for Visualization of Incoming Adenovirus Chromatin Complexes in Fixed and Living Cells. PLoS One 10, e0137102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reich NC, Sarnow P, Duprey E & Levine AJ Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128, 480–484 (1983). [DOI] [PubMed] [Google Scholar]
- 77.Sarnow P, Sullivan CA & Levine AJ A monoclonal antibody detecting the adenovirus type 5-E1b-58Kd tumor antigen: characterization of the E1b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology 120, 510–517 (1982). [DOI] [PubMed] [Google Scholar]
- 78.Marton MJ, Baim SB, Ornelles DA & Shenk T The adenovirus E4 17-kilodalton protein complexes with the cellular transcription factor E2F, altering its DNA-binding properties and stimulating E1A-independent accumulation of E2 mRNA. J Virol 64, 2345–2359 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shaffer SM et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poling BC, Price AM, Luftig MA & Cullen BR The Epstein-Barr virus miR-BHRF1 microRNAs regulate viral gene expression in cis. Virology 512, 113–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Russo J, Heck AM, Wilusz J & Wilusz CJ Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability. Methods 120, 39–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Price AM, Messinger JE & Luftig MA c-Myc Represses Transcription of Epstein-Barr Virus Latent Membrane Protein 1 Early after Primary B Cell Infection. J Virol 92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dolken L et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Van Nostrand EL et al. Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). Methods Mol Biol 1648, 177–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3 (2011). [Google Scholar]
- 87.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu TD & Nacu S Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lovci MT et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol 20, 1434–1442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dallaire F et al. The Human Adenovirus Type 5 E4orf6/E1B55K E3 Ubiquitin Ligase Complex Can Mimic E1A Effects on E2F. mSphere 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schwanhausser B et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jedrychowski MP et al. Evaluation of HCD- and CID-type Fragmentation Within Their Respective Detection Platforms For Murine Phosphoproteomics. Molecular & Cellular Proteomics 10, M111.009910 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Consortium, T. U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research 47, D506–D515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.The M, MacCoss MJ, Noble WS & Kall L Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J Am Soc Mass Spectrom 27, 1719–1727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wei T & Simko V R package “corrplot”: Visualization of a Correlation Matrix (Version 0.84) Available from https://github.com/taiyun/corrplot (2017).
- 98.Edgar R, Domrachev M & Lash AE Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Complete Di-glycine remnant and whole cell proteomics data. Normalized and log2 transformed proteomics data for K-ɛ-GG peptide based quantification (tab 1), transformation to K-e-GG protein based quantification (tab 2), and whole cell proteome quantification. Data were generated from three biologically independent experiments. Fold changes were calculated in log2 and p-values were calculated using unpaired, two-sided Student’s t-tests.
Supplementary Table 2: Di-glycine remnant and whole cell proteomics data for predicted E1B55K/E4orf6 substrates. Normalized and log2 transformed proteomics data for K-ɛ-GG protein based (tab 1) and peptide based (tab 2) quantifications for predicted E1B55K/E4orf6 substrates. Data were generated from three biologically independent experiments. Significance tests for whole cell proteome abundance changes are calculated using unpaired, two-sided Student’s t-tests. K-ɛ-GG abundance changes are normalized to whole cell proteome abundance changes and significance is calculated by one-sided Student’s t-tests with a null hypothesis of 0 fold change.
Supplementary Table 3: Reactome network protein-protein interactome analysis results. Reactome FI network analysis results of the proteins that exhibited increased protein-based ubiquitination (n=120). Reactome FI clustering algorithm was used to divide the interactome into modules (tab 1). Reactome FI was used to perform Gene Ontology and Pathway enrichment analysis. The Gene Ontology “Molecular Functions” is included for modules (tab 2) and the entire network (tab 5). The Gene Ontology “Biological Processes” is included for modules (tab3) and the entire network (tab 6). The Reactome Pathway analysis is included for modules (tab 4) and the entire network (tab 7). Data were generated from three biologically independent experiments.
Supplementary Table 4: SILAC ratios for chymotryptic and tryptic peptides from targeted hnRNP-C RBR-ID analysis. Heavy/light ratios indicate the proportion of peptides cross-linked to RNA upon UV treatment. Data represent 3 biological replicates and 2 technical replicates (n=6) per digestion condition. Chymotryptic (tab 1) and tryptic (tab 2)data were processed independently in Proteome Discoverer v2.3. Results were filtered to exclude peptides with any missing values across replicates.
Supplementary Table 5: hnRNP-C interactome derived from RBR-ID data. The non-crosslinked sample from the hnRNP-C RBR-ID is used to generate the hnRNP-C interactome. Normalized, log2 transformed data quantification for proteins identified in association with hnRNP-C. Data were generated from 3 biological replicates and 2 technical replicates (n=6) per digestion condition. Fold changes were calculated in log2 and p-values were calculated using unpaired, two-sided Student’s t-tests.
Data Availability Statement
All mass spectrometry data for this study are deposited in the CHORUS database under project ID 1648, experiment IDs 3476 (KeGG), 3477 (KeGG-matched whole cell proteome), and 3478 (RBR-ID of hnRNP-C). The seCLIP-Seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE145411. The seCLIP-Seq data have been deposited in NCBI’s Gene Expression Omnibus98 and are accessible through GEO Series accession number GSE145411 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145411). Additional supporting data are available from the corresponding author upon request.