

Abstract
Staphylococcus aureus (S. aureus) is a leading cause of biofilm-associated prosthetic joint infection (PJI), resulting in significant disability and prolonged treatment. It is known that host leukocyte IL-10 production is required for S. aureus biofilm persistence in PJI. A S. aureus bursa aurealis Tn library consisting of 1,952 non-essential genes was screened for mutants that failed to induce IL-10 in myeloid-derived suppressor cells (MDSCs), which identified a critical role for bacterial lactic acid biosynthesis. We generated a S. aureus ddh/ldh1/ldh2 triple Tn mutant that cannot produce D- or L-lactate. Co-culture of MDSCs or macrophages with ddh/ldh1/ldh2 mutant biofilm produced substantially less IL-10 compared with wild type S. aureus, which was also observed in a mouse model of PJI and led to reduced biofilm burden. Using MDSCs recovered from the mouse PJI model and in vitro leukocyte-biofilm co-cultures we show that bacterial-derived lactate inhibits histone deacetylase 11 (HDAC11), causing unchecked HDAC6 activity and increased histone 3 acetylation at the Il-10 promoter, resulting in enhanced Il-10 transcription in MDSCs and macrophages. Finally, we show that synovial fluid of patients with PJI contains elevated amounts of D-lactate and IL-10 compared with control subjects, and bacterial lactate increases IL-10 production by human monocyte-derived macrophages.
Keywords: S. aureus, lactate, biofilm, interleukin-10, myeloid-derived suppressor cell, macrophage, histone deacetylase, prosthetic joint infection
SUMMARY PARAGRAPH
Biofilms are bacterial communities that are difficult to treat because of their tolerance to antibiotics and ability to evade immune-mediated clearance. Prosthetic joint infection (PJI), a devastating complication of arthroplasty, is characterized by biofilm formation. The current study has discovered a central role for lactic acid biosynthesis in S. aureus biofilm formation during PJI. Mechanistically, bacterial-derived lactate inhibits histone deacetylase 11 (HDAC11) activity, which causes extensive epigenetic changes at the promoters of numerous host genes, including the key anti-inflammatory cytokine Il-10. Indeed, IL-10 production by myeloid-derived suppressor cells (MDSCs) and macrophages is critical for biofilm persistence during PJI. HDAC11 inhibition by S. aureus lactate results in unchecked HDAC6 activity, a positive regulator of IL-10, thereby increasing IL-10 production by MDSCs and macrophages in vitro and in vivo. Similarly, S. aureus lactate promotes IL-10 production in human monocyte-derived macrophages following biofilm exposure. This study highlights how bacterial metabolism can influence the host immune response to promote infection persistence.
INTRODUCTION
Staphylococcus aureus (S. aureus) is a leading cause of implanted medical device infections, characterized by biofilm formation1–3. Bacterial biofilms are microbial communities that exhibit distinct metabolic properties that contribute to their chronicity and antibiotic tolerance4–6. Staphylococcal biofilms actively reprogram the host innate immune response to favor persistent infection7–13. This is mediated, in part, by the recruitment of myeloid-derived suppressor cells (MDSCs) that promote monocyte and macrophage anti-inflammatory properties14–17. In addition, S. aureus biofilm evades Toll-like receptor (TLR)-mediated recognition and inhibits macrophage phagocytosis7,10,18. Collectively, these mechanisms lead to biofilm persistence.
IL-10 is a potent anti-inflammatory cytokine19 and although it is critical for preventing excessive pro-inflammatory responses and immunopathology associated with some types of infections, dysregulated or mistimed IL-10 production can allow select pathogens to escape immune control, resulting in chronic infection16,20–22. It is well recognized that IL-10 inhibits T cell activation and Th1 polarization23–25, and MDSCs have been shown to engage in crosstalk with macrophages, in part via IL-10, to skew them toward an anti-inflammatory phenotype during tumor growth26,27. We have previously shown that MDSCs are the main source of IL-10 during early S. aureus biofilm infection, transitioning to monocytes at later stages, and IL-10 production contributes to infection persistence16.
Based on the importance of IL-10 in promoting biofilm infection, we screened the Nebraska Transposon Mutant Library (NTML)28 to identify S. aureus factors that stimulate IL-10 production by MDSCs and macrophages. Numerous genes involved in lactate biosynthesis were identified, suggesting that bacterial lactate is an important regulator of leukocyte activation. Lactate is a product of glycolysis that exists as two stereoisomers, L- and D-lactate. S. aureus encodes three lactate biosynthetic enzymes, including an inducible L-lactate dehydrogenase (Ldh1), a second L-lactate dehydrogenase (Ldh2), and a D-lactate dehydrogenase (Ddh)29,30, to produce both L- and D-lactate, respectively. In eukaryotes, L-lactate is the predominant metabolite, whereas D-lactate is present at very low concentrations31. Recently, lactate has been implicated in mechanisms of immune evasion32–34, including inhibition of monocyte and macrophage cytokine production35.
Lactate has been reported to influence chromatin function and gene expression by inhibiting histone deacetylase (HDAC) activity in immune cells36,37; however, the role of HDACs, and more specifically how they are regulated in leukocytes in the context of biofilm infection, has not been investigated. Histones are a class of highly conserved proteins (H3, H4, H2A, H2B, and H1) that are integral for regulating gene transcription through post-translational modifications of N-terminal histone tails, including methylation, phosphorylation, lactylation, and acetylation34,38. Histone acetylation is mediated by histone acetyltransferases that transfer an acetyl group to a lysine residue on the histone tail, which relaxes chromatin structure and increases promoter accessibility and gene transcription. Deacetylation is mediated by HDACs, which condense chromatin and favor gene silencing39. There are 18 HDACs in humans that are grouped into four families, including Class I (HDAC1, −2, −3, and −8), Class II (HDAC4, −5, −6, −7, −9, and −10), Class III (sirtuins; SIRT1, −2, −3, −4, −5, −6 and −7), and Class IV (HDAC11)39. HDAC6 and HDAC11 physically interact and have been identified as positive and negative regulators of IL-10 transcription, respectively40,41. In this report, we demonstrate that S. aureus biofilm-derived lactate inhibits HDAC11 to augment IL-10 transcription via unchecked HDAC6 activity, which promotes the anti-inflammatory properties of MDSCs and macrophages. Since lactate biosynthesis is a conserved pathway in bacteria42–45, this study highlights the potential broader implications of lactate as a regulator of leukocyte gene expression during infections caused by diverse bacterial species.
RESULTS
S. aureus lactate biosynthesis pathways induce IL-10 production by MDSCs and macrophages.
To determine whether S. aureus-derived factors are responsible for eliciting IL-10 production from MDSCs, we designed a screen of the NTML, a collection of 1,952 strains each possessing a single mutation of a nonessential gene in S. aureus strain USA300 JE228. This approach identified several hits in the S. aureus lactate biosynthesis pathway that were attenuated in their ability to elicit IL-10 production from MDSCs, namely Δddh, Δldh1, and Δldh2, which convert pyruvate to D- and L-lactate, respectively (Figure 1a; Extended Data 1a). Because L-lactate production in S. aureus is driven by two independent genes29, we created a Δldh1/ldh2 as well as Δddh/ldh1/ldh2 that lacks all lactate biosynthetic enzymes. Importantly, these lactate mutants showed no growth defects in liquid broth or biofilm (Extended Data 1b and c). Both Δldh1/ldh2 and Δddh/ldh1/ldh2 biofilm produced negligible L-lactate (Figure 1c) and D-lactate was undetectable in Δddh and Δddh/ldh1/ldh2 biofilm (Figure 1d). Both MDSCs and macrophages produced significantly less IL-10 when co-cultured with Δddh, Δldh1/ldh2, or Δddh/ldh1/ldh2 biofilm compared to WT (Figure 1b). The intracellular pH of leukocytes co-cultured with WT or Δddh/ldh1/ldh2 biofilm was similar, revealing that pH fluctuations were not a contributing factor for lactate effects (Extended Data 2).
Figure 1. Lactate biosynthesis pathways induce MDSC and macrophage IL-10 production.
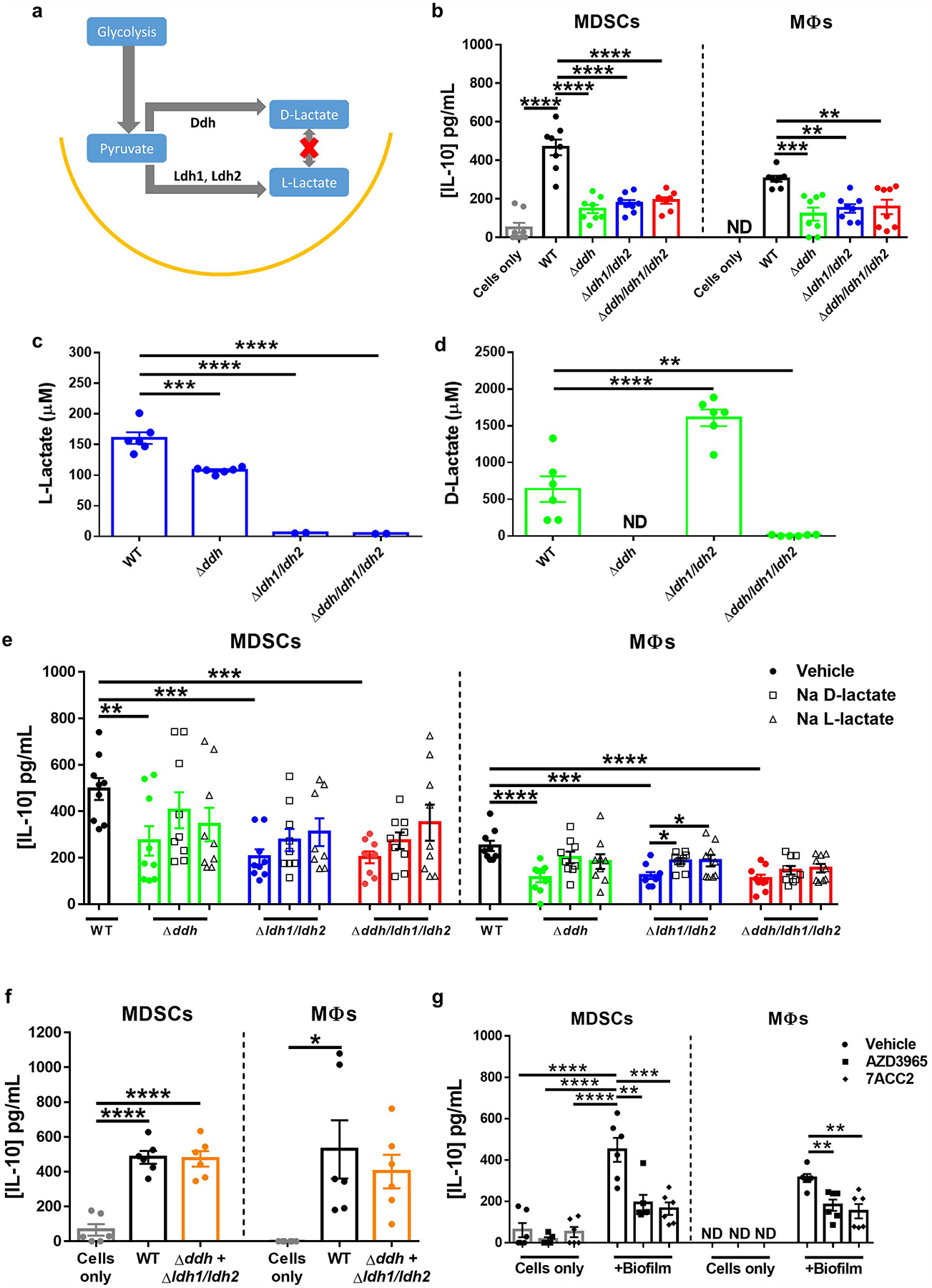
(a) Enzymes responsible for D- and L-lactate production in S. aureus. (b) MDSC and macrophage IL-10 release elicited from WT and lactate mutant biofilm. (c) L- and (d) D-lactate production by S. aureus WT and lactate mutant biofilm. (e) Sodium D- or L-lactate (2.5 μM) was added to lactate mutant biofilms during a 2 h co-culture with MDSCs or macrophages. (f) A Δddh and Δldh1/ldh2 mixed biofilm reveals the requirement for both D- and L-lactate for maximal IL-10 production. (g) MCT-1 inhibitors (AZD3965 and 7ACC2, both at 10 nM) were added during a 2 h co-culture with WT biofilm. IL-10 levels were measured for all experiments by ELISA. Results are reported as (b) the mean ± SEM from two independent experiments each with four biological replicates; (c and d) two independent experiments, each with three biological replicates; and (e) two (macrophages) and three (MDSCs) independent experiments, each with three biological replicates; and two (f, g) independent experiments, each with three biological replicates. ND, not detected. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA.
IL-10 production by both MDSCs and macrophages in response to Δddh, Δldh1/ldh2 or Δddh/ldh1/ldh2 biofilm could be partially restored with exogenous sodium D- or L-lactate (Figure 1e). However, a Δddh and Δldh1/ldh2 mixed biofilm returned MDSC and macrophage IL-10 production to levels elicited by WT biofilm (Figure 1f), a scenario where both D- and L-lactate are present. The mechanism of lactate release from S. aureus biofilm appears to be mediated by the lactate permease lctP146, since a ΔlctP1 biofilm also elicited less IL-10 production by MDSCs and macrophages (Supplementary Figure 1). The majority of lactate transport in mammalian cells occurs via the family of proton-linked monocarboxylate transporters (MCTs)47. Of this family, MCT1 is ubiquitously expressed and mediates bi-directional transport of monocarboxylates, including L- and D-lactate47–49. AZD396547 and 7ACC250 are selective MCT1 inhibitors, and both significantly decreased IL-10 production by MDSCs and macrophages following co-culture with WT S. aureus biofilm (Figure 1g).
S. aureus-derived lactate promotes bacterial persistence during PJI in an IL-10-dependent manner.
We next examined the course of biofilm infection with WT, Δddh, Δldh1/ldh2, and Δddh/ldh1/ldh2 in a mouse orthopaedic implant model. Bacterial burden in implant-associated tissue was significantly decreased for all three mutants at days 14 and 28 post-infection (Figure 2a). This coincided with fewer MDSCs and increased neutrophil and monocyte infiltrates at days 14 and 28 post-infection (Figure 2c–g); furthermore, as expected, lactate levels were reduced in animals infected with the lactate mutants at these intervals (Extended Data 3). Interestingly, only Δldh1/ldh2 and Δddh/ldh1/ldh2 were reduced in the femur at days 14 and 28, indicating an L-lactate-dependent phenotype in the bone (Figure 2b). Although IL-10 expression trended lower across the entire time course with all of the lactate mutants, IL-10 levels were not significantly decreased until day 28 in the absence of both S. aureus D- and L-lactate (Δddh/ldh1/ldh2; Figure 2h). This reduction in IL-10 was likely lactate-dependent and not influenced by decreased bacterial burden at this time point, since the expression of other inflammatory mediators was similar in WT and Δddh/ldh1/ldh2 infected tissues, as well as MDSCs and monocytes isolated from WT and Δddh/ldh1/ldh2 infected mice at day 28 post-infection (Extended Data 4 and Supplementary Table 1, respectively). During acute infection (day 3), both MDSCs and monocytes recovered from Δddh/ldh1/ldh2-infected mice expressed less IL-10 mRNA compared to WT S. aureus (Figure 2i). In addition, TNF-α expression was increased in monocytes from Δddh/ldh1/ldh2-infected mice concomitant with a reduction in the anti-inflammatory marker Arg-1 (Figure 2i), suggesting that monocytes are more pro-inflammatory in the absence of bacterial lactate. The Δddh/ldh1/ldh2 strain was utilized for all subsequent experiments to assess the biological impact of S. aureus lactate on leukocyte activation.
Figure 2. Role for Ddh, Ldh1, and Ldh2 during S. aureus orthopaedic implant infection.

Implant-associated tissues were collected at days 3, 14, and 28 post-infection, whereupon bacterial burden was quantified in (a) implant-associated soft tissue and (b) femur of mice infected with WT, Δddh, Δldh1/ldh2, and Δddh/ldh1/ldh2. (c) Representative contour plots of Ly6G and Ly6C staining demonstrate the abundance of Ly6G+Ly6C+ (inset D) and Ly6G−Ly6C+ cells (inset G) in WT or lactate mutant tissues at day 28 post-infection. (d) CD11b expression of Ly6G+Ly6C+ cells depicted in inset D, where the horizontal line demarcates CD11bhigh MDSCs from CD11blow PMNs. Quantification of (e) CD11bhighLy6G+Ly6C+ MDSCs (f) CD11blowLy6G+Ly6C+ PMNs, and (g) Ly6G−Ly6C+F4/80− monocytes in infected tissue. (h) IL-10 levels in implant-associated tissue were measured by ELISA. (i) Gene expression in CD11bhighLy6G+Ly6C+ MDSCs or Ly6G−Ly6C+F4/80− monocytes purified from implant-associated tissue of WT and Δddh/ldh1/ldh2-infected mice at day 3 by FACS, with results presented as the fold-change in Δddh/ldh1/ldh2 MDSCs or monocytes relative to leukocytes from WT-infected animals. Results represent (a-b and e-g) the mean ± SEM of three independent experiments (Day 3, n=14 for WT and Δddh and 15 for Δldh1/ldh2 and Δddh/ldh1/ldh2; Day 14, n=15/strain; Day 28, n=11 for WT, 14 for Δddh, and 15 for Δldh1/ldh2 and Δddh/ldh1/ldh2), (c and d) representative plots from three independent experiments, (h) mean ± SD of one representative experiment (Day 3, n=5 for WT and Δldh1/ldh2 and 4 for Δddh and Δddh/ldh1/ldh2; Day 14, n=5/strain; Day 28, n=4/strain) performed three times, or (i) the mean ± SEM of two independent experiments for MDSCs [n=4 (IL-10) and 5 (Arg-1 and TNF-α) biological replicates] and macrophages [n=3 (IL-10) and 4 (Arg-1 and TNF-α) biological replicates]. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA.
Co-infection with WT and Δddh/ldh1/ldh2 reversed the immune phenotypes associated with Δddh/ldh1/ldh2 monoinfection, including a return of IL-10 production (Figure 3f) and leukocyte infiltrates (Figure 3c–e) as well as D- and L-lactate (Figure 3g and h) to WT levels at days 14 or 28 post-infection, supporting the action of bacterial-derived and not host lactate. The titer of Δddh/ldh1/ldh2 during co-infection was comparable to monoinfection; revealing that WT bacteria did not outcompete Δddh/ldh1/ldh2 over the 28-day co-infection period (Figure 3a and b). To determine whether the residual IL-10 production after infection with Δddh/ldh1/ldh2 reflected the action of host lactate, mice were treated with sodium oxamate, a LDH inhibitor that has been used in cancer models51–53. Oxamate treatment of Δddh/ldh1/ldh2 infected mice had no effect on IL-10 production (Extended Data 5), demonstrating that S. aureus-derived lactate is the main driver of IL-10 expression during biofilm infection, confirming the co-infection experiments (Figure 3). Oxamate did not alter S. aureus growth or lactate production (Extended Data 6c–e). We also examined whether IL-10 was elicited by pathogen-associated molecular patterns in the biofilm, which in S. aureus primarily signal via TLRs 2 and 9 and utilize the adaptor protein MyD8854–56. IL-10 production by both MDSCs and macrophages following co-culture with WT biofilm was MyD88- and TLR2-independent (Supplementary Figure 2), emphasizing the critical role of biofilm-derived lactate in inducing IL-10 expression.
Figure 3. S. aureus lactate promotes IL-10 production and regulates leukocyte infiltrates during PJI.

Mice were infected with WT, Δddh/ldh1/ldh2, or a 1:1 ratio of WT and Δddh/ldh1/ldh2, whereupon tissues were collected at days 3, 14, and 28 post-infection to quantify bacterial burden in the (a) implant-associated soft tissue and (b) femur. Quantification of (c) CD11bhighLy6G+Ly6C+ MDSCs (d) CD11blowLy6G+Ly6C+ PMNs, (e) Ly6G−Ly6C+F4/80− monocytes, (f) IL-10, (g) D-lactate, and (h) L-lactate in implant-associated tissue. Results are combined from two independent experiments (Day 3, n=10/group; Day 14, n=9 for WT and 10 for Δddh/ldh1/ldh2 and WT+Δddh/ldh1/ldh2; Day 28, n=9 for WT and 10 for Δddh/ldh1/ldh2 and WT+Δddh/ldh1/ldh2). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA. In (a) and (b), black asterisks indicate significant differences between WT bacteria across all groups and red asterisks reflect significant differences between Δddh/ldh1/ldh2 across all groups.
To confirm that the effects of S. aureus-derived lactate in vivo were mediated by IL-10, IL-10 KO and WT mice were infected with WT S. aureus or Δddh/ldh1/ldh2. We previously reported that bacterial burden and MDSC infiltrates were significantly reduced at day 14 post-infection in IL-10 KO mice infected with WT S. aureus16, which was confirmed here (Extended Data 7a–c). In contrast, reduction in bacterial titer and changes in MDSC and monocyte infiltrates in WT mice following Δddh/ldh1/ldh2 infection were not observed in IL-10 KO animals (Extended Data 7), indicating that the main effects of S. aureus-derived lactate are IL-10-dependent.
S. aureus-derived lactate inhibits HDAC11, resulting in unchecked HDAC6-driven IL-10 production.
In eukaryotic cells, lactate can regulate gene transcription by inhibiting HDACs36,37 and the IL-10 promoter can be regulated by histone acetylation40,41,57,58. It is unknown whether S. aureus-derived lactate can affect HDACs, which would represent a metabolic virulence determinant. HDAC activity was significantly decreased in leukocytes recovered from WT compared to Δddh/ldh1/ldh2 infected mice (Figure 4a) and coincided with increased total histone 3 acetylation (H3Ac; Figure 4b), consistent with the action of S. aureus lactate as an HDACi. This was further supported by ChIP-seq, where total H3Ac within ± 10,000 bp of all genomic transcription start sites was increased by approximately two-fold in MDSCs recovered from mice infected with WT S. aureus vs. Δddh/ldh1/ldh2 (Figure 4c). scRNA-seq of CD45+ leukocytes isolated from WT and Δddh/ldh1/ldh2 infected mice identified several genes with concordant changes in H3Ac status and transcriptional expression, including IL-10, where acetylation and gene expression were both reduced in response to Δddh/ldh1/ldh2 (Figure 4d), independently confirming the findings of increased IL-10 levels in the setting of WT S. aureus infection. Other notable changes were increases in pro-inflammatory and bactericidal genes in Δddh/ldh1/ldh2-infected mice, such as cathepsin (Ctsg) and myeloperoxidase (Mpo), whereas several genes associated with MDSC recruitment and immunosuppression (IFNb1, Nfkbiz, Cxcl1, Cxcl3, Fpr1, and Ptgs2) were reduced (Figure 4d)59–64. Also of interest was reduced MCT1 (Slc16a1) expression in Δddh/ldh1/ldh2-infected mice, suggesting that lactate production by WT S. aureus augments MCT1 expression to promote lactate uptake and IL-10 production. The concordance between H3Ac levels and gene expression identified by ChIP-seq was validated by RT-qPCR (Figure 4e). ChIP-qPCR confirmed increased H3Ac in three regions near the IL-10 promoter in leukocytes from WT-infected mice that were identified by ChIP-seq, i.e. −5795 TSS, +450 TSS, and the STAT3 binding site at −507 TSS65 (Figure 4f). Treatment of Δddh/ldh1/ldh2 infected mice with the pan-HDACi trichostatin A (TSA) increased IL-10 production and bacterial burden to levels approaching that of WT S. aureus (Figure 4g–i). TSA had no effect in mice infected with WT S. aureus, which is expected if bacterial-derived lactate is already acting as an HDACi (Figure 4g–i).
Figure 4. S. aureus-derived lactate inhibits HDAC activity to regulate Histone 3 (H3) acetylation and gene expression.

Total leukocytes were recovered from WT (n=14 biological replicates) and Δddh/ldh1/ldh2-infected mice (n=12 biological replicates) at day 14 and assessed for (a) HDAC activity using a fluorescent HDAC substrate (deAc-FdL; two independent experiments; ***, p = 0.0005) and (b) relative abundance of acetylated Histone 3 (H3Ac) by ELISA (mean ± SD from six biological replicates; *, p = 0.0365). (c) Significant changes in H3Ac peaks ± 10,000 bp of the transcriptional start site (TSS) as identified by ChIP-seq in purified MDSCs collected at day 14 post-infection (two independent experiments with cells pooled from 15 mice per experiment). (d) Genes identified by single cell RNA-seq (total CD45+ leukocytes collected from 3 mice) with concordant changes in H3Ac status as determined in (c) with results expressed as the fold-change in leukocytes recovered from Δddh/ldh1/ldh2 vs. WT infected mice. Genes that appear more than once reflect those with multiple altered H3Ac ChIP peaks. (e) RT-qPCR validation of the top 3 upregulated and downregulated genes identified in (d) in purified MDSCs collected at day 14 post-infection (two independent experiments; n= 5 but some genes did not amplify in all samples). (f) ChIP-qPCR for H3Ac within three regions associated with the IL-10 promoter in purified MDSCs at day 14 after infection (n= 3 technical replicates from MDSCs that were pooled from 25 mice/group over 3 independent experiments due to limiting input ChIP DNA from low cell yields). **, p = 0.0046;*, p = 0.0261; and ***, p = 0.0006, respectively). (g-i) WT or Δddh/ldh1/ldh2-infected mice were treated with vehicle (n=10/group) or 0.5 mg/kg of the pan-HDACi trichostatin A (n=9 or 10 for WT and Δddh/ldh1/ldh2, respectively) once daily, and sacrificed at day 14 post-infection for quantification of bacterial burden in (g) implant-associated tissue and (h) femur, and (i) IL-10 in infected tissue. Results are representative of two independent experiments. Results in panels a, b, and f were analyzed using an unpaired two-tailed Student’s t-test and panels g-i by One-way ANOVA (*, p < 0.05; **, p < 0.01; ****, p < 0.0001). The results in c reflect a p-value < 0.05 as determined using a statistical test for each ChIP-enriched region as described in100 and corrected using the Benjamini and Hochberg method to control for False Discovery Rate.
HDAC6 has been shown to be a positive regulator of IL-10 production, whereas HDAC11 is a negative regulator that inhibits HDAC640,41. If S. aureus lactate inhibits HDAC11, this might alleviate the negative feedback on HDAC6 at the IL-10 promoter, effectively augmenting IL-10 production. To investigate this possibility, MDSCs and macrophages from HDAC11 KO and WT mice were co-cultured with S. aureus biofilm. HDAC11 KO leukocytes produced significantly more IL-10 in response to Δddh/ldh1/ldh2 biofilm, which likely resulted from unchecked HDAC6 activity, since IL-10 was significantly reduced by the HDAC6i tubastatin A (Figure 5b). Tubastatin A treatment of WT leukocytes co-cultured with WT biofilm also significantly reduced IL-10 production (Figure 5a), reflecting unchecked HDAC6 activity following inhibition of the negative regulator HDAC11 by S. aureus lactate. This was supported by the lack of tubastatin A effects on WT leukocytes co-cultured with Δddh/ldh1/ldh2 biofilm because the absence of lactate allowed HDAC11 to inhibit HDAC6 (Figure 5b). Similar results were obtained with a second HDAC6i (Extended Data 8). Cell-free experiments demonstrated that S. aureus-derived lactate preferentially targeted HDAC11, since the degree of enzyme inhibition by WT biofilm was greater for HDAC11 than HDAC6, whereas conditioned medium from a Δddh/ldh1/ldh2 biofilm was significantly less potent at blocking HDAC11 activity (Extended Data 9).
Figure 5. S. aureus-derived lactate inhibits the negative regulator HDAC11 to augment leukocyte IL-10 production in a HDAC6-dependent manner.
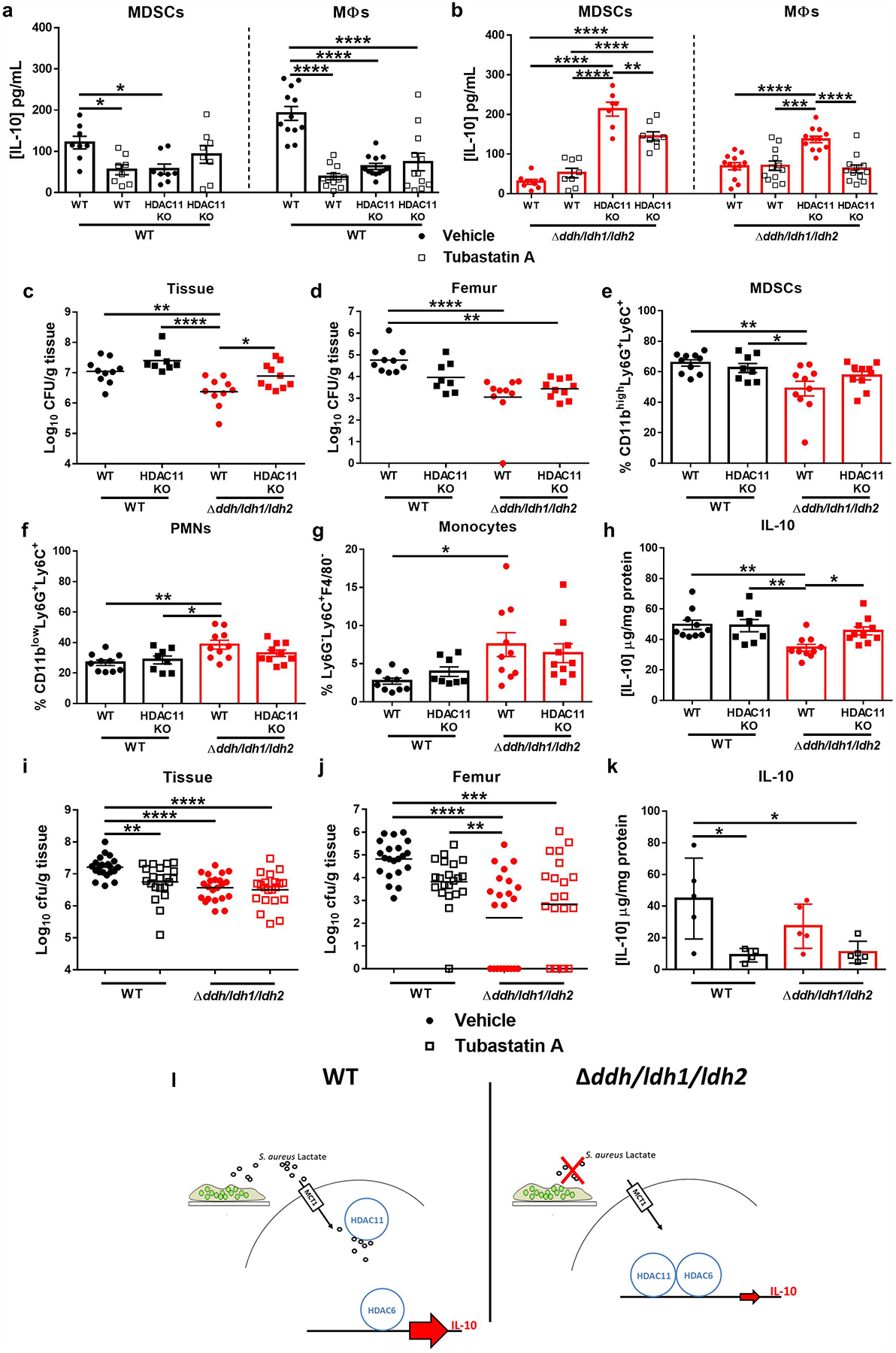
(a and b) MDSCs and macrophages from WT or HDAC11 KO mice were co-cultured with WT or Δddh/ldh1/ldh2 biofilm for 2 h in the presence or absence of the HDAC6i tubastatin A (15 nM), whereupon IL-10 production was measured by cytometric bead array. Results represent the mean ± SEM combined from two independent experiments (n=8 and 12 biological replicates for MDSCs and macrophages, respectively, except for WT MDSCs + tubastatin A in b where n=7). (c-h) WT and HDAC11 KO mice were infected with S. aureus WT (n=10 and 8 for WT and HDAC11 KO mice, respectively) or Δddh/ldh1/ldh2 (n=10/group), whereupon bacterial burden in (c) implant-associated tissue and (d) femur was quantified at day 14 post-infection. (e) CD11bhighLy6G+Ly6C+ MDSCs (f) CD11blowLy6G+Ly6C+ PMNs, (g) Ly6G−Ly6C+F4/80− monocytes, and (h) IL-10 were quantified in infected tissue at day 14 post-infection. Results represent the mean ± SEM combined from two independent experiments. (i-k) WT or Δddh/ldh1/ldh2-infected mice were treated with vehicle or 0.5 mg/kg tubastatin A once daily, and sacrificed at day 14 post-infection for quantification of bacterial burden in (i) implant-associated tissue and (j) femur as well as (k) IL-10 in implant-associated tissues by ELISA. Results are (i and j) combined from three independent experiments (vehicle, n=21 and 22 for WT and Δddh/ldh1/ldh2, respectively; tubastatin A, n=22 and 21 for WT and Δddh/ldh1/ldh2, respectively) or (k) mean ± SD of one representative experiment (vehicle, n=5/group; tubastatin A, n=4 and 5 for WT and Δddh/ldh1/ldh2, respectively) performed two times. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA. (l) During infection with wild type S. aureus (left panel), D- and L-lactate produced by the biofilm is transported into MDSCs and macrophages via the monocarboxylate transporter MCT1 where it inhibits HDAC11 activity. The lack of HDAC11 inhibition allows HDAC6-dependent IL-10 transcription to become unchecked, resulting in elevated IL-10 production. In the absence of S. aureus lactate (right panel), the inhibitory effect of HDAC11 on HDAC6 remains intact, which limits IL-10 production.
To investigate this mechanism in vivo, two complementary experiments were performed. Infection of WT and HDAC11 KO mice with WT S. aureus showed no significant differences in IL-10 production, leukocyte infiltrates, or bacterial burden (Figure 5c–h). This is expected if S. aureus-derived lactate inhibits HDAC11, making the loss of HDAC11 irrelevant, since nothing is inhibiting HDAC6 and thus IL-10 is already maximally expressed. In contrast, IL-10 production was significantly higher in HDAC11 KO mice infected with Δddh/ldh1/ldh2 compared to WT animals (Figure 5h), likely due to the lack of negative regulation of HDAC11 on HDAC6 activity that drives IL-10 production, which translated into increased bacterial burden in implant-associated tissue (Figure 5c). To demonstrate HDAC6 action in vivo, WT mice were treated with tubastatin A. Bacterial burden in the implant-associated tissue and femur as well as IL-10 production were all significantly reduced in animals infected with WT S. aureus following tubastatin A treatment, a phenotype similar to when no S. aureus lactate is produced (Δddh/ldh1/ldh2; Figure 5i–k). Moreover, tubastatin A had no effect in Δddh/ldh1/ldh2-infected WT mice (Figure 5i–k), since HDAC6 was already being inhibited by HDAC11 in the absence of S. aureus-derived lactate. Importantly, IL-10 levels were similar in HDAC11 KO mice in response to WT or Δddh/ldh1/ldh2 infection (Figure 5h) and total IL-10 was equivalent in WT and Δddh/ldh1/ldh2 infected animals following HDAC6 inhibition (Figure 5k), since once HDAC6 is inhibited the lack of HDAC11 makes no difference. Collectively, these findings support the in vitro studies, showing that bacterial-derived lactate is an HDAC11i that augments IL-10 production during S. aureus biofilm infection, by alleviating the negative feedback on HDAC6.
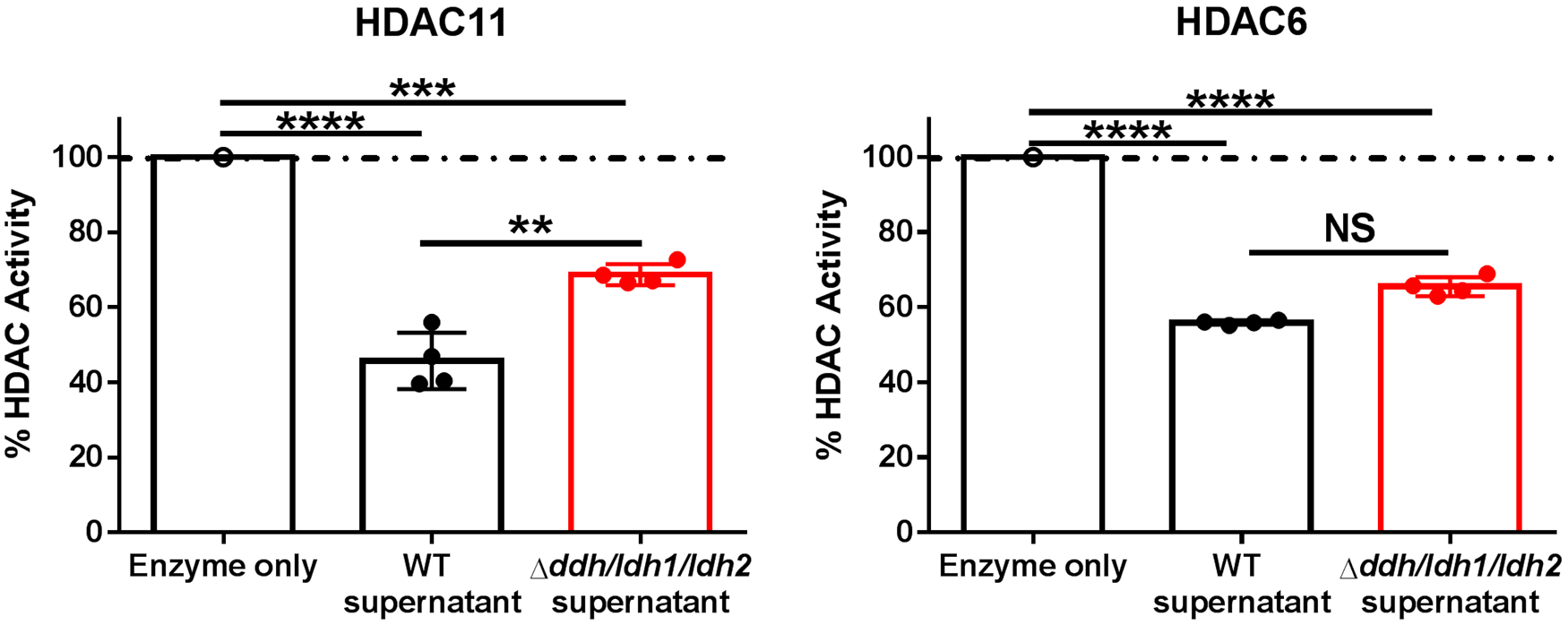
Both D-lactate and IL-10 were significantly increased in the synovial fluid of human PJIs caused by distinct bacteria, including S. aureus, S. epidermidis, and Group C Streptococcus, compared to aseptic joints; whereas, L-lactate levels were similar (Figure 6a and b). In addition, human monocyte-derived macrophages treated with clarified supernatant from Δddh/ldh1/ldh2 biofilm produced significantly less IL-10 than in the presence of lactate (Figure 6c), which was confirmed by L- and D-lactate quantification (Figure 6d and e). Similar to mouse macrophages, IL-10 production by human monocyte-derived macrophages was HDAC6-driven and lactate-dependent, since tubastatin A significantly decreased IL-10 levels following treatment with WT but not Δddh/ldh1/ldh2 clarified biofilm supernatant (Figure 6c).
Figure 6. S. aureus-derived lactate induces IL-10 production by human monocyte-derived macrophages.

Synovial fluid samples from patients with PJI vs. aseptic loosening were examined for (a) L- and D-lactate (n=12 and 9 for aseptic and PJI, respectively; ***, p = 0.0004) and (b) IL-10 (n=14 and 6 for aseptic and PJI, respectively; ***, p = 0.0003). (c) Human monocyte-derived macrophages were co-cultured with clarified supernatants from WT or Δddh/ldh1/ldh2 biofilm ± the HDAC6i tubastatin A (15 nM) for 2 h, whereupon IL-10 production was analyzed by ELISA. (d) L- and (e) D-lactate levels in clarified supernatants from WT or Δddh/ldh1/ldh2 biofilm. Results represent (c) the mean ± SD from one representative experiment (n=7 biological replicates/group) repeated with monocytes from 3 different donors and (d and e) mean ± SEM from three independent experiments. ND, not detected. Results were analyzed by (a and b) an unpaired two-tailed Student’s t-test and (c) One-way ANOVA (*, p < 0.05; ***, p < 0.001).
Collectively, these experiments demonstrate that S. aureus-derived lactate is transported into MDSCs and macrophages via MCT1. Through its action as an HDAC11i, bacterial lactate releases its negative regulation on HDAC6, a positive regulator of IL-10, resulting in increased IL-10 production during S. aureus biofilm infection (Figure 5l). Bacterial lactate also induces a global increase in H3Ac, reflecting mechanisms of epigenetic regulation extending beyond IL-10 that could also contribute to biofilm persistence.
DISCUSSION
It is well established that post-translational modifications of histones influence gene expression34,38,66. Here we demonstrate that D- and L-lactate, products of S. aureus fermentative metabolism, promote IL-10 production by biofilm-associated MDSCs and macrophages via HDAC11 inhibition. ChIP-seq and subsequent ChIP-qPCR identified the proximal region of the IL-10 promoter (−87 to −7) as a target of H3Ac by S. aureus-derived lactate, in agreement with a recent report40. This metabolic crosstalk is critical to sustain an anti-inflammatory environment that promotes bacterial persistence, demonstrating that a S. aureus metabolite can act as a virulence factor.
Recent reports have examined the role of HDAC11 in regulating MDSC expansion and function in the context of cancer67–69. MDSCs isolated from tumor-bearing mice displayed high HDAC11 expression compared to immature myeloid cells, and MDSCs from tumor-bearing HDAC11 KO mice were more suppressive with increased IL-10 levels, which was associated with enhanced tumor growth69. These observations share striking parallels to what was observed during S. aureus biofilm infection. IL-10 production was significantly higher in HDAC11 KO versus WT animals infected with Δddh/ldh1/ldh2 due to the loss of this negative regulator. This allowed us to unveil the role of HDAC6 as a driver of IL-10 expression using selective HDAC6 inhibitors. Recent evidence suggests that although HDAC6 binds to the IL-10 promoter and induces gene expression, it has minimal effects on histone deacetylation40, which could explain the preferential inhibition of S. aureus-derived lactate on HDAC11 versus HDAC6, which was supported by cell-free assays. Furthermore, we showed that S. aureus-derived lactate promotes IL-10 production in human monocyte-derived macrophages through HDAC11i and increased HDAC6 activity, reflecting the translational impact of our findings.
The mechanism whereby S. aureus lactate inhibits HDAC11 remains elusive. Pathogen targeting of HDACs has been demonstrated for other bacteria, including Mycobacterium tuberculosis, Helicobacter pylori, and Listeria monocytogenes but via distinct mechanisms70–73. Recent studies have shown that HDAC11 has other enzymatic functions in addition to deacetylase activity74–76; therefore, it is likely that the effects of S. aureus-derived lactate on HDAC11 extend beyond promoting IL-10 transcription. Furthermore, since the increase in HDAC activity during Δddh/ldh1/ldh2 infection was greater in magnitude compared to changes in H3Ac levels, it is possible that the S. aureus lactate-dependent changes in HDAC activity have effects beyond transcriptional regulation. Recently, lactate has been reported to influence the post-translational modification of histones via a process termed histone lactylation34. It is unclear what impact histone lactylation may play in the setting of S. aureus biofilm infection and represents an area for future investigation.
Although this study focused on the ability of S. aureus-derived lactate to regulate IL-10 expression, lactate also altered H3Ac at other genes, reflecting a broader mechanism of action. Notable genes that were increased in leukocytes from Δddh/ldh1/ldh2 mice included enzymes associated with neutrophil bactericidal activity (Ctsg, Mpo)77,78, which correlated with reduced bacterial burden in the absence of lactate. In addition, the expression of genes that have been linked to MDSC recruitment (Cxcl1 and Cxcl3)63,79 and suppressive activity (Ptgs2)80 were decreased in the absence of S. aureus lactate, which agrees with the reduction in MDSC infiltrates in Δddh/ldh1/ldh2-infected animals. Although several of the genes detected by ChIP-seq are typically associated with T cells (i.e. granzymes, cathepsins) they are also expressed in MDSCs81,82. Our prior work has identified T cell infiltrates in patients with PJI as well as the mouse S. aureus PJI model, although they represent a minor population in both instances (i.e. < 2% of CD45+ leukocytes)15,83.
It remains unclear whether S. aureus-derived D- and L-lactate exert distinct functions in MDSCs and macrophages, since our results have revealed actions for both stereoisomers. The MCT-1 transporter that was important for eliciting S. aureus lactate-dependent IL-10 production is more selective for L- than D-lactate84. We found that L-lactate was progressively consumed by WT S. aureus biofilm, whereas D-lactate persisted at much higher levels, since it is not utilized by S. aureus as a carbon source85,86. In addition, D-lactate was significantly increased in the synovial fluid of patients with PJI compared to aseptic cases, in agreement with a recent study87. Therefore, although MCT-1 has a higher Km for D-lactate, local concentrations in the biofilm milieu exceed that of L-lactate, making D-lactate uptake feasible. Our finding that D-lactate levels were elevated irrespective of the causative pathogen in human PJI demonstrates the broader implications of lactate on modulating host immunity during biofilm infection caused by other bacterial species.
The infection-associated epigenetic dysregulation88 by S. aureus lactate shown in this study likely culminates from a variety of interrelated factors, including chronic interactions between S. aureus and environmental signals and co-adaptation of host leukocytes with S. aureus during persistent infection. This is plausible when considering the fact that the lactate-dependent phenotypes on leukocyte activation and bacterial burden were most evident at later stages of infection (i.e. days 14 and 28), in agreement with the delayed kinetics of IL-10 action in the mouse PJI model that manifest at day 14 post-infection16. Of note, some IL-10 production was still evident in vivo in the absence of S. aureus lactate. This was not attributed to host lactate, since residual IL-10 levels were not affected after inhibiting host LDH. Therefore, lactate-independent mechanisms exist for inducing maximal IL-10 production, which is not unexpected given the large number of S. aureus virulence determinants54,89. Collectively, these findings identify S. aureus biofilm-derived lactate as a metabolic virulence factor that augments IL-10 production in MDSCs and macrophages by inhibiting HDAC11. This serves to attenuate leukocyte pro-inflammatory activity, representing one mechanism to account for biofilm persistence.
MATERIALS AND METHODS
Mice.
C57BL/6NCrl (RRID:IMSR_CRL:27), IL-10 KO (RRID:IMSR_JAX:002251), and HDAC11 KO mice (RRID:IMSR_TAC:6978)90 were used for experiments. C57BL/6NCrl and IL-10 KO animals were bred in house at the University of Nebraska Medical Center (UNMC) and mice of the same sex were randomized into standard density cages upon weaning (n= 5 animals per cage). Mice were housed in a restricted-access BSL2 room equipped with ventilated microisolator cages and maintained at 21°C under a 12 h light:12 h dark cycle with ad libitum access to water (Hydropac™; Lab Products, Seaford, DE) and Teklad rodent chow (Harlan, Indianapolis, IN) with Nestlets provided for enrichment. This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was approved by the UNMC Institutional Animal Care and Use Committee (#18-013-03).
S. aureus strains.
The Nebraska Transposon Mutant Library (NTML) was constructed in LAC JE228. Δddh, Δldh1, Δldh2, and ΔlctP1 Tn mutants were moved to the USA300 LAC 13c background by transduction with bacteriophage φ1128 (Extended Data 1a). Lactate double (Δldh1/ldh2) and triple (Δddh/ldh1/ldh) mutants were constructed from each single Tn mutant after switching antibiotic resistance cassettes associated with the Tn mutant. Transposon insertions were confirmed by PCR using chromosomal primers flanking the gene containing the Tn insertion. All S. aureus strains were transduced with a GFP plasmid (pCM13)91 and imaged using a Zeiss 710 META laser scanning microscope (Carl Zeiss) after 4 days of biofilm growth to evaluate biofilm structure.
S. aureus biofilm growth.
Single colonies of WT, Δddh, Δldh1/ldh2, and Δddh/ldh1/ldh2 were inoculated into 5mL of RPMI-1640 supplemented with 10% FBS (Cat. #SH30079.02HI; HyClone), 1% HEPES (Cat. #SH40003.01; Hyclone) and 1% L-glutamine (Cat. #25–005-CL; Corning) and grown overnight at 37°C, with shaking (250 rpm). Overnight cultures were inoculated at a 1:100 dilution in 96-well plates pre-coated with 20% human plasma (generously provided by Dr. Scott Koepsell, UNMC) as previously described92. Biofilms were grown under static conditions at 37°C in room air. After every 24 h of growth, approximately 35% of the medium was removed and replaced with fresh medium to avoid disturbing biofilm structure. Lactate levels in static biofilms ranged from 200–1500 μM, which is less than the mM concentrations reported during planktonic growth29,46. This was attributed, in part, to lactate consumption, as revealed by the progressive reduction in L-lactate over a 24 h period in WT biofilm (Extended Data 6a and b), since it can be utilized by S. aureus to form pyruvate under glucose deplete conditions46.
Generation of bone marrow-derived MDSCs and macrophages for in vitro biofilm co-culture experiments.
MDSCs and macrophages were expanded from the bone marrow of WT, HDAC11 KO, TLR2 KO (RRID:IMSR_JAX:004650), or MyD88 KO (RRID:IMSR_JAX:009088) mice as previously described16,93. Briefly, Ly6G+ MDSCs were purified using an anti-Ly6G MicroBead Kit (Cat. # 130-092-332; Miltenyi Biotec) after 4 days in culture, per the manufacturer’s instructions. The resulting Ly6G+population has been verified for Ly6C expression and is phenotypically and functionally similar to MDSCs infiltrating S. aureus implant-associated biofilm in vivo16. For macrophage cultures, FACS analysis revealed that > 95% of cells were macrophages based on CD11b and F4/80 staining after a 7 day culture period93. Bone marrow-derived MDSCs and macrophages (5×104) were added to 4 day-old S. aureus WT and lactate mutant biofilm for 2 h to assess IL-10 production. For metabolic complementation experiments, lactate mutant biofilms were treated with sodium D- or L-lactate (both at 2.5 μM; Cat. #s 71716 and 71718, respectively; Sigma-Aldrich) at the time of MDSC or macrophage addition. The MCT1 inhibitors AZD3965 and 7ACC2 (both at 10 nM; Cat. #s 19912 and 1472624-85-3, respectively; Cayman Chemical Company) and HDAC6 inhibitors tubastatin A (15nM) and HDAC6i (36nM) (Cat. #s 15785 and 1259296-46-2, respectively; Cayman Chemical Company) were included at the time of MDSC or macrophage addition to biofilm.
Gentamicin protection assay.
Bone marrow-derived macrophages were prepared as described above. Macrophages were seeded in a 96-well plate at 5×104 cells/well and incubated with WT, Δddh, Δldh1/ldh2, or Δddh/ldh1/ldh2 S. aureus strains at a MOI of 10:1 or 50:1 for 45 min at 37°C, 5% CO2. Plates were washed and incubated for another 30 min in medium containing 100 μg/mL gentamicin (Cat. #G1264, Millipore Sigma) to kill residual extracellular bacteria, whereupon cells were washed and medium containing 1 μg/mL gentamicin was added. Intracellular bacterial loads were determined at 0, 2, 4, 6 and 24 h following low dose gentamicin treatment by lysing macrophages with sterile water and plating on blood agar. This approach confirmed that the reduced survival of Δddh, Δldh1/ldh2, or Δddh/ldh1/ldh2 in vivo was not due to enhanced sensitivity to immune-mediated clearance, since macrophage bactericidal activity was similar for all mutants (Supplementary Figure 3).
D- and L-lactate and IL-10 quantification.
D- and L-lactate concentrations in supernatants from S. aureus WT and lactate mutant biofilm in vitro, mouse implant-associated tissue homogenates, and human synovial fluid were determined using D- and L-Lactate colorimetric assays according to the manufacturer’s instructions (Cat. #s K667–100 and K607–100, respectively; BioVision). Murine IL-10 levels in supernatants from MDSC- or macrophage-biofilm co-cultures and tissue homogenates were quantified using a Mouse Inflammation Cytometric Bead Array (CBA; Cat #552364, BD Biosciences) or sandwich ELISA (Mouse DuoSet; Cat. #DY417–05, R&D Systems). IL-10 in human synovial fluid samples and human monocyte-derived macrophages was measured by ELISA (Cat. #430604, BioLegend).
Mouse model of S. aureus orthopaedic biofilm infection.
To model infectious complications in patients following orthopaedic device placement, a mouse model of S. aureus PJI was used as previously described14–16,18,94–97. This model reflects biofilm growth as demonstrated by scanning electron microscopy and H&E staining14,15,18,95,96. In addition, immunophenotyping of patients with PJI, many of which were diagnosed with S. aureus, has identified similar leukocyte infiltrates as observed in the mouse model15,83, supporting its translational utility. The sample size for in vivo experiments was based on prior work from our laboratory14–16,93,94,98, which was shown to provide statistically significant findings. Both male and female mice were used for these studies between the ages of 8–10 weeks and the study was not blinded to strain or treatment status. Mice were anesthetized with ketamine/xylazine and a medial incision was made through the quadriceps. Following lateral displacement of the patellar tendon, a burr hole was created in the intercondylar notch with a 26-gauge needle, whereupon an orthopaedic-grade K-wire (0.6 mm diameter, nitinol [nickel-titanium]; Custom Wire Technologies) was inserted. A total of 1,000 CFU of S. aureus WT or lactate mutant strains were inoculated at the implant tip. For co-infection experiments, mice were infected with a 1:1 ratio of WT and Δddh/ldh1/ldh2 S. aureus (each at 1,000 CFU) and strains were differentiated based on antibiotic resistance profiles. Buprenex was administered for pain relief immediately after surgery and 24 h later, at which point mice exhibited normal ambulation and no discernable pain behaviors. For HDACi experiments, mice received daily i.p. injections of the pan-HDACi trichostatin A (0.5 mg/kg; Cat. #89730; Cayman Chemical) or the HDAC6i tubastatin A (0.5 mg/kg; Cat. #15785; Cayman Chemical Company) beginning on the day of infection. To inhibit host LDH, mice received daily i.p. injections of sodium oxamate (500 mg/kg; Cat. #19057; Cayman Chemical) dissolved in 0.5% Hydroxypropyl Methylcellulose beginning one day prior to infection.
Quantification of leukocyte infiltrates and biofilm burden.
The soft tissue surrounding the infected knee joint as well as the femur was collected after removing the skin, weighed, and homogenized, as previously described15. Serial, 10-fold dilutions of tissue and femur homogenates were plated on TSA with 5% sheep blood (Cat #R01202; Remel Products), with titers expressed as CFU per gram of tissue. Remaining homogenates were centrifuged (20,000xg, 20 min) and frozen at −80°C until analysis.
To quantify leukocyte infiltrates in the implant-associated soft tissue of mice infected with WT S. aureus or lactate mutants, homogenized tissues were filtered and red blood cells (RBCs) removed from the single cell suspension using RBC Lysis Buffer (Cat. #420301; BioLegend). After lysis, leukocytes were incubated with Fc Block (TruStain FcX, Cat. #101320; BioLegend) and stained with CD45-PacBlue (RRID:AB_493535), Ly6G-PE (RRID:AB_1186099), F4/80-PE-Cy7 (RRID:AB_893478), and CD11b-FITC (RRID:AB_312789) (all from BioLegend) or Ly6C-PerCP-Cy5.5 (RRID:AB_1727558, BD Pharmingen). Dead cells were excluded from analysis using a Live/Dead Fixable Blue Dead Cell Stain Kit (Cat. #L23105; Invitrogen) and analysis was performed using BD FACSDiva software (RRID:SCR_001456) as previously described94 using the gating strategy presented in Extended Data 10. To monitor leukocyte intracellular pH, MDSCs and macrophages were labeled with BCECF-AM (10 μM; Cat. #B1150, ThermoFisher) prior to co-culture with WT or Δddh/ldh1/ldh2 biofilm for 2 h, whereupon intracellular pH was determined by flow cytometry based on a standard curve of known pH.
Quantitative real-time reverse transcription PCR (RT-qPCR).
Ly6G−Ly6C+F4/80− monocytes and CD11bhighLy6G+Ly6C+ MDSCs were purified from mice infected with WT S. aureus or Δddh/ldh1/ldh2 by FACS, whereupon total RNA was immediately isolated using a Micro RNeasy kit (Cat. #74004; Qiagen). RT-qPCR was performed using TaqMan primer/probe mixes (ThermoScientific) for arginase-1 (Arg-1; Cat. #Mm00475988_m1), TNF-α (Cat. #Mm00443258_m1), IL-10 (Cat. #Mm00439616_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cat. #Mm99999915_g1). Gene expression levels were normalized to GAPDH and are presented as the fold-induction (2−ΔΔCt) for leukocytes from Δddh/ldh1ldh1-infected mice relative to WT.
In some experiments, gene expression was analyzed in FACS purified Ly6G−Ly6C+F4/80− monocytes and CD11bhighLy6G+Ly6C+ MDSCs by Nanostring. RNA samples (25–50 ng each) were hybridized to the nCounter® Mouse PanCancer IO 360™ Panel Reporter CodeSet and Capture CodeSets (Cat. #115000260; NanoString Technologies) overnight for 16 h at 65°C per the manufacturer’s instructions. Following hybridization, samples were loaded on the detection cartridge using the nCounter MAX/FLEX System at the High Sensitivity setting and processed on the nCounter Digital Analyzer for a counting time of 5 h. Initial sample analysis was performed using the nSolver™ 4.0 Analysis Software (RRID:SCR_003420). Quality metrics were reviewed and the results were normalized to the housekeeping genes provided by Nanostring. Following normalization, data were analyzed to determine fold-changes, where differentially expressed genes were identified based on a p-value < 0.05.
HDAC activity in cells recovered from S. aureus orthopaedic implant biofilm infection.
Implant-associated tissues from mice infected with WT or Δddh/ldh1/ldh2 S. aureus were collected at day 14 post-infection and dissociated as described above. Following RBC lysis, cells were plated at 1×105 cells/well in a flat-bottom 96-well plate. HDAC activity was measured using a FLUOR DE LYS® HDAC fluorometric activity assay kit (Cat. #BML-AK503–0001; Enzo Life Sciences). The relative amount of H3Ac in lysates of leukocytes from WT or Δddh/ldh1/ldh2-infected mice was quantified using a Pathscan ELISA kit (Cat. #7232C; Cell Signaling Technology), according to the manufacturer’s instructions.
Cell-free HDAC activity assay.
Human recombinant HDAC6 and HDAC11 were purchased in their active forms from Enzo Life Sciences (Cat #BML-SE508–0050 and BML-SE-560–0050, respectively) and incubated with conditioned medium from WT or Δddh/ldh1/ldh2 S. aureus biofilm for 30 min, whereupon enzymatic activity was quantified using a FLUOR DE LYS® HDAC fluorometric activity assay kit (Cat. #BML-AK503–0001; Enzo Life Sciences).
Chromatin immunoprecipitation sequencing (ChIP-seq), ChIP-PCR, and scRNA-seq.
ChIP-seq was performed on CD11bhighLy6G+Ly6C+ MDSCs isolated from mice infected with WT or Δddh/ldh1/ldh2 S. aureus at day 14 post-infection. For each sample, chromatin immunoprecipitation DNA was acquired using a histone pan-H3Ac antibody (RRID: AB_2793714; Active Motif) and a Zymo-Spin ChIP kit (Cat. #D5209; Zymo Research) according to the manufacturer’s instructions. The captured and purified DNA was prepared for high-throughput sequencing using the NEBNext Ultra II DNA Library Prep kit for Illumina (Cat. #E7645S; New England Biolabs). The resulting indexed libraries were sequenced by the UNMC DNA Sequencing Core Facility using an Illumina NextSeq 500 Genome Analyzer (RRID:SCR_014983). Initial raw sequence files were adaptor trimmed using Trim Galore software (RRID:SCR_016946) and the resulting fastq files were aligned to the mouse genome using the sequence aligner Bowtie2 (version 2.2.3)99. The software package Picard routine MarkDuplicates (http://broadinstitute.github.io/picard/; RRID:SCR_006525) was used to remove sequence duplications. A sequencing count of 49.7 million was acquired using SAMTOOLS (http://samtools.sourceforge.net/; RRID:SCR_002105) for each sample. For peak calling of ChIP-enriched regions, THOR peak caller100 software (RRID:SCR_001400) of each ChIP to corresponding input DNA sample was used, which utilizes a pipeline consisting of estimation of fragment size, DNA mappability, GC-content normalization, correction of signals with input DNA, and signal normalization. Density estimates from the Hidden Markov Model-based approach was employed coupled to negative binomial distribution, and an exact statistical test was run101 to assign a p-value to each ChIP-enriched region. Significant ChIP-enriched regions with a distance less than the mean of all estimated fragment sizes were merged and p-values were corrected using the Benjamini and Hochberg method of controlling for False Discovery Rate (FDR). Using this approach, ChIP-enriched binding regions were determined based on a FDR adjusted p-value (q-value) < 0.05. BigWig files were generated using the Deeptools bamCoverage routine (https://deeptools.readthedocs.io/en/develop/; RRID:SCR_016366). Alignment of significant peaks to gene-specific regions was accomplished using the BEDTools routine intersect (https://bedtools.readthedocs.io/en/latest/; RRID:SCR_006646).
For ChIP-qPCR, specific primers (Supplementary Table 2) were designed for two regions that indicated a significant change in H3Ac abundance associated with IL-10 and a region designated as an IL-10 STAT3 binding site65. Gradient annealing temperature q-PCR was performed for each primer set. The input DNA pulled from ChIP was diluted 10-fold with 10 μL Bio-Rad iTaq Universal SYBR Supermix (Cat. #1725120), 2.5 μL each forward/reverse primers (0.625 μM final concentration), and 4 μL H2O. q-PCR was performed using a Bio-Rad CFX Real-Time System with the following program: 1 min at 95°C; 45 cycles of 95°C for 15 s, 60°C for 20 s, and 72°C for 20 s. Percent Input Method was calculated as previously described102 and the delta-delta method103 was performed using two calibrator primer sets, namely 1) Negative Mouse Control Primer Set 1 (Cat #71011; Active Motif) that interrogates a gene desert region on chromosome 6; and 2) Mouse Positive Control Primer Set Actb-2 (Cat. #71017; Active Motif) that interrogates a region in the Actb promoter. ChIP-Seq data was validated in FACS-purified MDSCs collected at day 14 post-infection by RT-qPCR for the following genes; insulin-like growth factor 1 (Igf1; Cat. #Mm00439560_m1), granzyme C (Gzmc; Cat. #Mm01313651_m1), macrophage galactose N-acetyl-galactosamine specific lectin 2 (Mgl2; Cat. #Mm00460844_m1), resistin-like gamma (Retnlg, Cat. #Mm00731489_s1), interferon-beta (IFN-b1; Cat. #Mm00439546_s1), and MCT1 (Slc16a1; Cat. #Mm01306379_m1). Gene expression levels were normalized to GAPDH (Cat #Mm99999915_g1) and are presented as the fold-induction (2−ΔΔCt) for leukocytes from Δddh/ldh1ldh1-infected mice relative to WT.
For single cell RNA-sequencing (scRNA-seq), live, CD45+ leukocytes from tissues surrounding WT or Δddh/ldh1/ldh2-infected implants were isolated by FACS at day 14 post-infection and prepared for sequencing in the UNMC DNA Sequencing Core Facility. Briefly, sorted leukocytes were evaluated by light microscopy to determine concentration, percent viability, and to assess potential debris present in the suspension. Cells were loaded onto a 10X Genomics instrument and single cells were captured, lysed, RNA reverse transcribed, and RNA barcoded using a Chromium Single Cell 3’ Reagent Kit v3 (Cat. #PN-1000075; 10X Genomics), according to the manufacturer’s instructions. Illumina compatible cDNA libraries were created and quantified by qPCR using the KAPA Library Quant Kit (Illumina) (Cat. #KK4824; KAPA Biosystems) and were loaded at a concentration of 1.3 pM on an Illumina NextSeq550 instrument. Samples were sequenced following the parameters suggested by 10X Genomics to an average depth of approximately 50,000 reads per cell. Expression differences between leukocytes from WT and Δddh/ldh1/ldh2-infected mice were performed using the 10X Genomics R2 read fastq files and analyzed as single end bulk RNA-seq analysis based on the following steps. Adaptor sequences and low quality (Phred score: 20) ends were trimmed from sequences using the Trim Galore software package (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and the resulting fastq files were aligned to the mouse genome (GRCm38/mm10) using the software TopHat (v2.0.8) (http://ccb.jhu.edu/software/tophat/index/shtml/; RRID:SCR_013035). The software Cufflinks (v2.1.1) (http://cole-trapnell-lab.github.io/cufflinks/; RRID:SCR_014597) was used to estimate the expression values and Cuffdiff (V2.1.1; RRID:SCR_001647) was used to determine differential expression. Alignment of significant H3Ac peaks to gene-promoter regions was accomplished using the BEDTools routine intersect (https://bedtools.readthedocs.io/en/latest/). Excel logic formulas were used to select genes that were increased or decreased in both expression and H3Ac. The complete ChIP-Seq and scRNA-seq datasets have been deposited in the GEO repository (accession number GSE135496).
Human monocyte-derived macrophages and patient samples.
Human monocytes were obtained from healthy human donors by the UNMC Elutriation Core Facility by countercurrent centrifugal elutriation, in full compliance and approval of the Institutional Review Board (IRB). Cells were cultured at 1×106 cells/mL in RPMI-1640 supplemented with recombinant human M-CSF (100ng/mL; Cat. #574802; BioLegend), 10% human serum (Cat. #H4522; Millipore Sigma) and penicillin/streptomycin/fungizone (Cat. #30–004-CI; Corning) for 7 days. For macrophage treatment with clarified biofilm supernatants, conditioned medium was collected from mature S. aureus WT and Δddh/ldh1/ldh2 biofilm and centrifuged at 4,000xg for 10 min, followed by filtration (0.22 μM) and ultracentrifugation at 100,000xg for 2 h at 4°C. A total of 1×106 human monocyte-derived macrophages were treated with a 1:10 dilution of clarified biofilm supernatant from WT or Δddh/ldh1/ldh2 biofilm for 2 h ± the HDAC6i tubastatin A (15 nM; Cat. #15785; Cayman Chemical Company). Clarified biofilm supernatants were used for these experiments due to the rapid death of human cells during S. aureus biofilm co-culture.
Informed consent was obtained from patients undergoing primary or revision total knee or total hip arthroplasty for infectious or aseptic complications during their pre-surgical visit, following prior approval by the UNMC IRB (#177–14-FB and 657–13-EP). Subjects included both males and females between 43–81 years of age. Synovial fluid was collected intraoperatively for D- and L-lactate assays as described above and organisms associated with PJIs were identified by the Clinical Microbiology Laboratory at UNMC. Samples that were negative for bacterial growth using standard protocols were classified as aseptic. In terms of the relationship between bacterial lactate and HDACi, although a prior report demonstrated that lactate inhibits HDACs at mM concentrations in vitro36, which exceed the levels detected in human PJI, it is important to note that the synovial fluid samples in our study were collected at the time of surgery when patients were already undergoing antibiotic treatment. Therefore, these lactate measurements are likely an under-representation of levels in the infected joint prior to antibiotic treatment when the immune modulatory effects of bacterial-derived lactate would be operative.
Statistics.
Significant differences between experimental groups were determined by an unpaired two-tailed Student’s t-test or a one-way ANOVA with Tukey’s or Dunnett’s multiple comparisons post-hoc test using GraphPad Prism version 6.04 (LaJolla, CA; RRID:SCR_002798). For all analyses, p < 0.05 was considered statistically significant.
Extended Data
Extended Data Fig. 1. S. aureus lactate mutants do not display growth defects in liquid broth or biofilm in vitro.

(a) S. aureus strains used in this study. (b) The growth rate of S. aureus WT, Δddh, Δldh1/ldh2, and Δddh/ldh1/ldh2 was determined in brain-heart infusion broth over a 24 h period with constant agitation using a TECAN (7 biological replicates/strain). (c) Strains were transduced with a sarA-GFP plasmid and grown for 4 days under static growth conditions in RPMI-1640 supplemented with 10% FBS, whereupon biofilm formation was visualized by confocal microscopy. Results are representative of two independent experiments, each with 4 biological replicates. Scale bars, 100 μm.
Extended Data Fig. 2. Intracellular pH of MDSCs and macrophages is not dramatically altered by S. aureus-derived lactate during biofilm co-culture.

MDSCs or macrophages were labeled with BCECF-AM (10 μM) prior to co-culture with WT (n=4 biological replicates/group) or Δddh/ldh1/ldh2 (n=4 and 3 biological replicates for MDSCs and macrophages, respectively) biofilm for 2 h, whereupon intracellular pH was determined by flow cytometry based on a standard curve of known pH. Results shown are from one experiment.
Extended Data Fig. 3. D- and L-lactate production during S. aureus orthopaedic infection.

(a) L- and (b) D-lactate were quantified in the implant-associated tissue of mice infected with WT, Δddh, Δldh1/ldh2, or Δddh/ldh1/ldh2 at days 3, 14, and 28 post-infection (mean ± SD; n=5/group). The dashed line represents background in the assay as determined with tissues collected from animals receiving sterile implants at the same time points (n=5 at days 3 and 28 and n=4 at day 14). Results are representative of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; One-way ANOVA.
Extended Data Fig. 4. The expression of select inflammatory mediators is independent of bacterial burden during S. aureus orthopaedic infection.
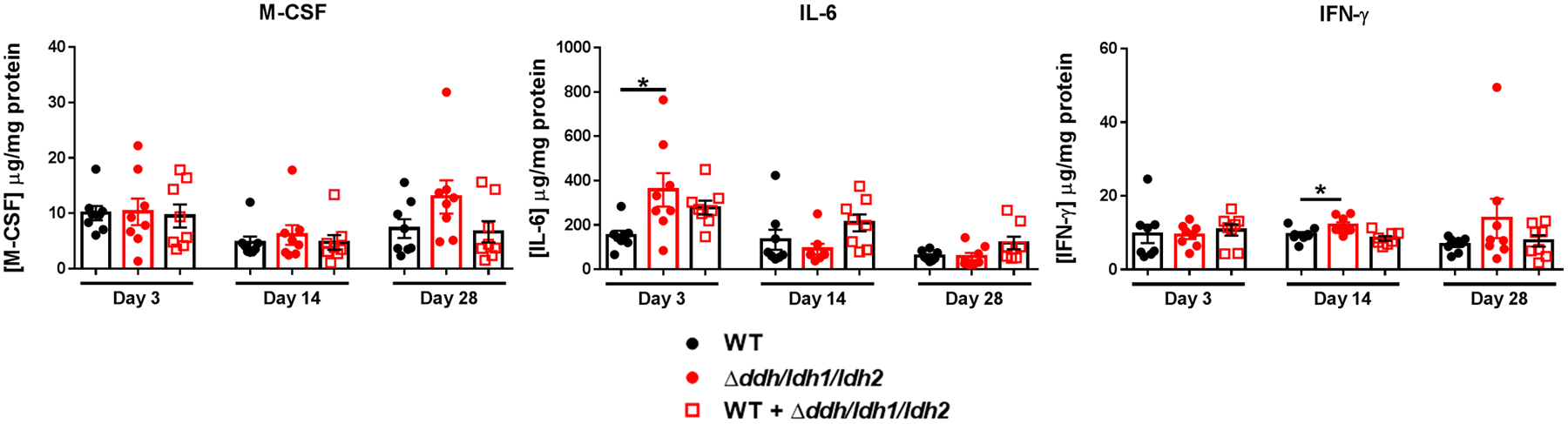
Cytokine levels were quantified in implant-associated tissue of mice infected with WT, Δddh/ldh1/ldh2, or a 1:1 ratio of WT and Δddh/ldh1/ldh2 at day 28 post-infection. Results are combined from two independent experiments (mean ± SD; n= 8/group). *, p < 0.05; One-way ANOVA.
Extended Data Fig. 5. IL-10 production during S. aureus orthopaedic infection is not influenced by host lactate.

Mice received daily i.p. injections of sodium oxamate (500 mg/kg/day) dissolved in 0.5% Hydroxypropyl Methylcellulose or vehicle (0.5% Hydroxypropyl Methylcellulose) beginning one day prior to infection with S. aureus WT (n=10/group) or Δddh/ldh1/ldh2 (n=10 or 9 for vehicle and oxamate, respectively). Mice were sacrificed at day 14 post-infection to quantify (a) D-lactate, (b) L-lactate, and (c) IL-10 in implant-associated tissue. Results are combined from two independent experiments (mean ± SD). D- and L-lactate measurements are also reported at day 14 for mice that received sterile implants (n=4/group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; One-way ANOVA. NS, not significant.
Extended Data Fig. 6. Sodium oxamate does not affect S. aureus growth or lactate production.
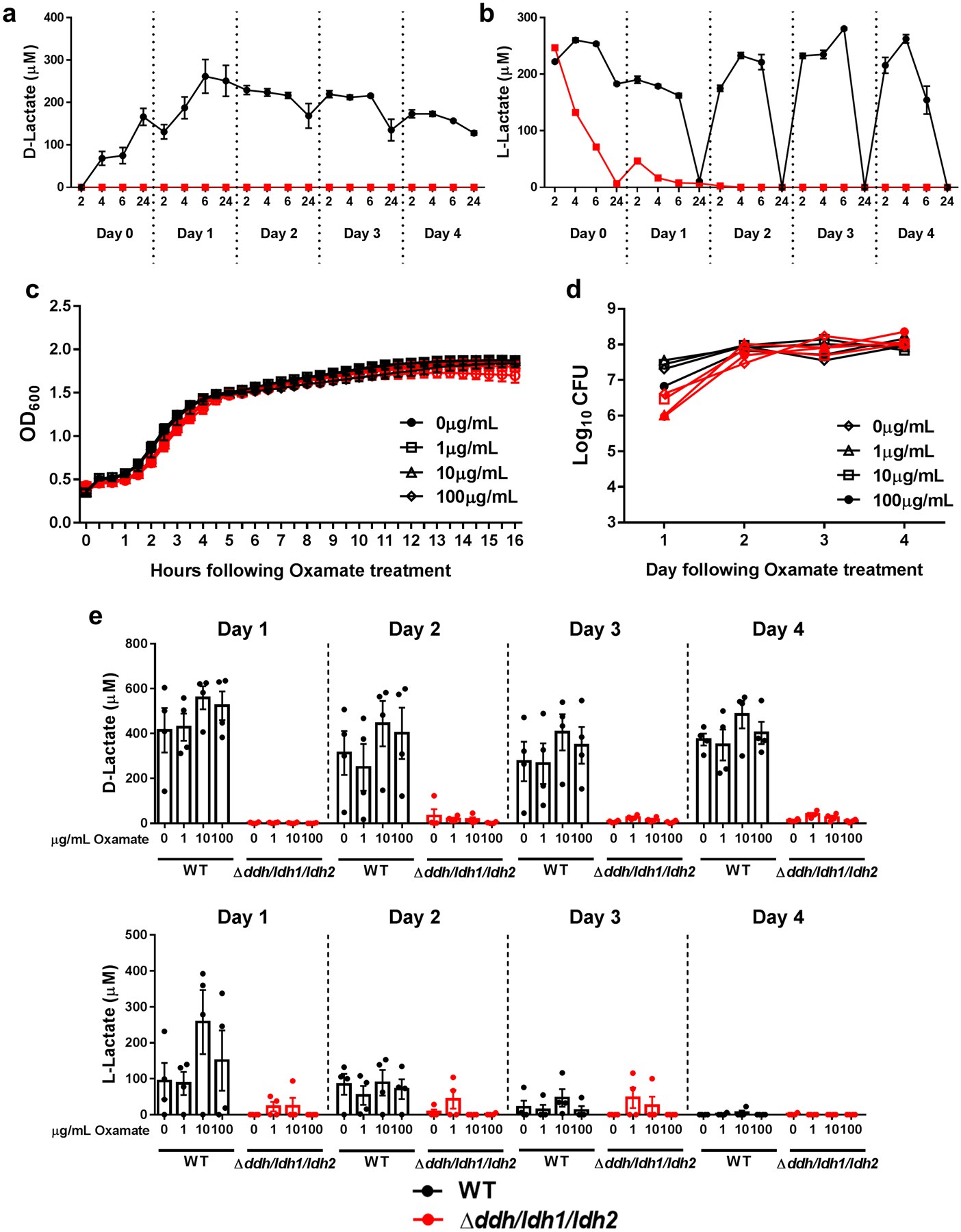
(a) D- and (b) L-lactate were quantified in S. aureus WT and Δddh/ldh1/ldh2 biofilm in 96-well plates under static growth conditions in RPMI-1640 supplemented with 10% FBS over a 4 d period. The dotted lines represent daily medium changes. Results are from one experiment with 5 biological replicates. S. aureus was exposed to various concentrations of sodium oxamate during (c) growth in liquid broth (brain-heart infusion) beginning at time 0 (n=6 biological replicates/group) or (d) throughout the 4-day biofilm maturation period (n=5 biological replicates/group). Biofilm cultures were replenished daily with fresh medium (RPMI-1640 + 10% FBS) containing sodium oxamate. Results are presented as (c) OD600 or (d) Log10 colony forming units (CFU) per well. (e) Quantification of D- and L-lactate from biofilms throughout the 4-day growth period, where the dotted lines represent daily medium changes (n=4 biological replicates/group). All results are reported as mean ± SD.
Extended Data Fig. 7. Effects of S. aureus lactate on orthopaedic implant biofilm infection are IL-10-dependent.

WT and IL-10 KO mice were infected with WT (n=10) or Δddh/ldh1/ldh2 (n=9) S. aureus, whereupon bacterial burden in (a) implant-associated tissue and (b) femur as well as (c) MDSC and (d) monocyte infiltrates were assessed at day 14 post-infection. Results represent the mean ± SEM of two independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; One-way ANOVA.
Extended Data Fig. 8. S. aureus-derived lactate inhibits the negative regulator HDAC11 to augment leukocyte IL-10 production in a HDAC6-dependent manner.

MDSCs and macrophages from WT or HDAC11 KO mice were co-cultured with (a) WT or (b) Δddh/ldh1/ldh2 biofilm for 2h ± HDAC6i (36 nM). IL-10 production was measured by cytometric bead array. Results represent the mean ± SEM of two independent experiments (n=8 and 12 biological replicates for MDSCs and macrophages, respectively). **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA. Values for untreated leukocytes are the same as those presented in Fig. 5a and b because both HDAC6i and tubastatin A were tested at the same time.
Extended Data Fig. 9. S. aureus-derived lactate preferentially inhibits HDAC11.
Purified active HDAC11 or HDAC6 were exposed to conditioned medium from WT or Δddh/ldh1/ldh2 biofilm for 30 min, whereupon HDAC activity was determined using a fluorescent HDAC substrate (deAc-FdL). Results are from one experiment with 4 biological replicates and are expressed as the percent change in HDAC activity compared to purified enzyme. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; One-way ANOVA. NS, not significant.
Extended Data Fig. 10. Gating strategy to quantitate leukocyte populations in S. aureus implant-associated soft tissue.
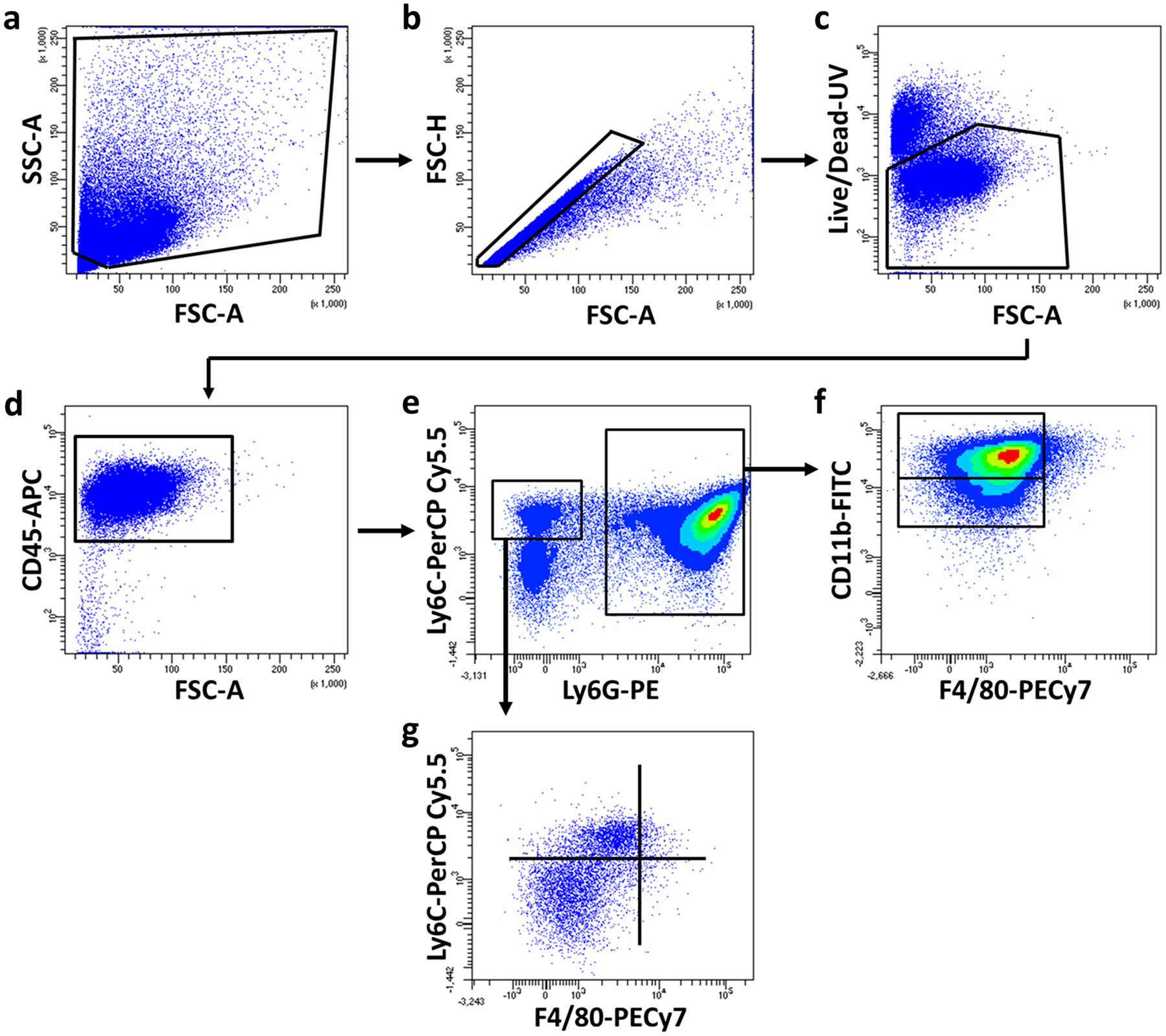
Single cells were gated from the (a) total events using (b) FSC-A vs. FSC-H, followed by (c) exclusion of dead cells. (d) Live, CD45+ leukocytes were separated into (e) Ly6G+Ly6C+ vs. Ly6G−Ly6C+. (f) MDSC and neutrophil populations were identified based on CD11b expression, while (g) monocyte and macrophage populations were identified based on Ly6C and F4/80 expression, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Disease grant P01AI083211 (Project 4 to TK) and R01AI125588 to VCT. The authors thank Rachel Fallet for managing the mouse colony and Dr. Kari Nelson for editorial review of the manuscript. The UNMC DNA Sequencing Core receives partial support from the National Institute for General Medical Science (NIGMS; INBRE - P20GM103427-14 and COBRE - 1P30GM110768-01). Both the UNMC DNA Sequencing and Flow Cytometry Research Cores receive support from The Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
DATA AVAILABILITY
The ChIP-seq and RNA-seq datasets are available in the GEO repository (accession number GSE135496) and Source Data is provided for the main figures and Extended Data in this study.
CODE AVAILABILITY
All codes utilized are published programs, with links to each provided in the Methods.
REFERENCES
- 1.Percival SL, Suleman L, Vuotto C & Donelli G Healthcare-associated infections, medical devices and biofilms: risk, tolerance and control. J Med Microbiol 64, 323–334, doi: 10.1099/jmm.0.000032 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Pulido L, Ghanem E, Joshi A, Purtill JJ & Parvizi J Periprosthetic joint infection: the incidence, timing, and predisposing factors. Clin Orthop Relat Res 466, 1710–1715, doi: 10.1007/s11999-008-0209-4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arciola CR, Campoccia D & Montanaro L Implant infections: adhesion, biofilm formation and immune evasion. Nat Rev Microbiol 16, 397–409, doi: 10.1038/s41579-018-0019-y (2018). [DOI] [PubMed] [Google Scholar]
- 4.Flemming HC et al. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14, 563–575, doi: 10.1038/nrmicro.2016.94 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Balaban NQ et al. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17, 441–448, doi: 10.1038/s41579-019-0196-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron DR, Shan Y, Zalis EA, Isabella V & Lewis K A Genetic Determinant of Persister Cell Formation in Bacterial Pathogens. J Bacteriol 200, doi: 10.1128/JB.00303-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherr TD et al. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. MBio 6, doi: 10.1128/mBio.01021-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koziel J et al. The Janus face of alpha-toxin: a potent mediator of cytoprotection in staphylococci-infected macrophages. J Innate Immun 7, 187–198, doi: 10.1159/000368048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schommer NN et al. Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A.1. Infect Immun 79, 2267–2276, doi: 10.1128/IAI.01142-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurlow LR et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186, 6585–6596, doi: 10.4049/jimmunol.1002794 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricciardi BF et al. Staphylococcus aureus Evasion of Host Immunity in the Setting of Prosthetic Joint Infection: Biofilm and Beyond. Curr Rev Musculoskelet Med 11, 389–400, doi: 10.1007/s12178-018-9501-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le KY, Park MD & Otto M Immune Evasion Mechanisms of Staphylococcus epidermidis Biofilm Infection. Front Microbiol 9, 359, doi: 10.3389/fmicb.2018.00359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He L et al. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nature microbiology 4, 1114–1119, doi: 10.1038/s41564-019-0413-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heim CE et al. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol 192, 3778–3792, doi: 10.4049/jimmunol.1303408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heim CE et al. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol 194, 3861–3872, doi: 10.4049/jimmunol.1402689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heim CE, Vidlak D & Kielian T Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol 98, 1003–1013, doi: 10.1189/jlb.4VMA0315-125RR (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tebartz C et al. A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol 194, 1100–1111, doi: 10.4049/jimmunol.1400196 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Bernthal NM et al. Protective role of IL-1beta against post-arthroplasty Staphylococcus aureus infection. J Orthop Res 29, 1621–1626, doi: 10.1002/jor.21414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ouyang W & O’Garra A IL-10 Family Cytokines IL-10 and IL-22: from Basic Science to Clinical Translation. Immunity 50, 871–891, doi: 10.1016/j.immuni.2019.03.020 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Kessler B et al. Interleukin 10 inhibits pro-inflammatory cytokine responses and killing of Burkholderia pseudomallei. Sci Rep 7, 42791, doi: 10.1038/srep42791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leech JM, Lacey KA, Mulcahy ME, Medina E & McLoughlin RM IL-10 Plays Opposing Roles during Staphylococcus aureus Systemic and Localized Infections. J Immunol 198, 2352–2365, doi: 10.4049/jimmunol.1601018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alter G et al. IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. J Clin Invest 120, 1905–1913, doi: 10.1172/JCI40913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith LK et al. Interleukin-10 Directly Inhibits CD8(+) T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 48, 299–312 e295, doi: 10.1016/j.immuni.2018.01.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akdis CA, Joss A, Akdis M, Faith A & Blaser K A molecular basis for T cell suppression by IL-10: CD28-associated IL-10 receptor inhibits CD28 tyrosine phosphorylation and phosphatidylinositol 3-kinase binding. FASEB J 14, 1666–1668, doi: 10.1096/fj.99-0874fje (2000). [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Tonkonogy SL & Sartor RB Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology 141, 653–662, 662 e651–654, doi: 10.1053/j.gastro.2011.04.053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha P, Clements VK, Bunt SK, Albelda SM & Ostrand-Rosenberg S Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179, 977–983, doi: 10.4049/jimmunol.179.2.977 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Beury DW et al. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol 96, 1109–1118, doi: 10.1189/jlb.3A0414-210R (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fey PD et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio 4, e00537–00512, doi: 10.1128/mBio.00537-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuller JR et al. Identification of a lactate-quinone oxidoreductase in Staphylococcus aureus that is essential for virulence. Front Cell Infect Microbiol 1, 19, doi: 10.3389/fcimb.2011.00019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stockland AE & San Clemente CL Multiple forms of lactate dehydrogenase in Staphylococcus aureus. J Bacteriol 100, 347–353 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondoh Y, Kawase M, Kawakami Y & Ohmori S Concentrations of D-lactate and its related metabolic intermediates in liver, blood, and muscle of diabetic and starved rats. Res Exp Med (Berl) 192, 407–414 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Puig-Kroger A et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol 73, 482–492, doi: 10.1189/jlb.0902451 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Husain Z, Huang Y, Seth P & Sukhatme VP Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol 191, 1486–1495, doi: 10.4049/jimmunol.1202702 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580, doi: 10.1038/s41586-019-1678-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratter JM et al. In vitro and in vivo Effects of Lactate on Metabolism and Cytokine Production of Human Primary PBMCs and Monocytes. Front Immunol 9, 2564, doi: 10.3389/fimmu.2018.02564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Latham T et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res 40, 4794–4803, doi: 10.1093/nar/gks066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner W, Ciszewski WM & Kania KD L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun Signal 13, 36, doi: 10.1186/s12964-015-0114-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45, doi: 10.1038/47412 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Shakespear MR, Halili MA, Irvine KM, Fairlie DP & Sweet MJ Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 32, 335–343, doi: 10.1016/j.it.2011.04.001 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Cheng F et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol 60, 44–53, doi: 10.1016/j.molimm.2014.02.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villagra A et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol 10, 92–100, doi: 10.1038/ni.1673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garvie EI Bacterial lactate dehydrogenases. Microbiol Rev 44, 106–139 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaspar P, Al-Bayati FA, Andrew PW, Neves AR & Yesilkaya H Lactate dehydrogenase is the key enzyme for pneumococcal pyruvate metabolism and pneumococcal survival in blood. Infect Immun 82, 5099–5109, doi: 10.1128/IAI.02005-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bunch PK, Mat-Jan F, Lee N & Clark DP The ldhA gene encoding the fermentative lactate dehydrogenase of Escherichia coli. Microbiology 143 (Pt 1), 187–195, doi: 10.1099/00221287-143-1-187 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Feldman-Salit A et al. Regulation of the activity of lactate dehydrogenases from four lactic acid bacteria. J Biol Chem 288, 21295–21306, doi: 10.1074/jbc.M113.458265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spahich NA, Vitko NP, Thurlow LR, Temple B & Richardson AR Staphylococcus aureus lactate- and malate-quinone oxidoreductases contribute to nitric oxide resistance and virulence. Mol Microbiol 100, 759–773, doi: 10.1111/mmi.13347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bola BM et al. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther 13, 2805–2816, doi: 10.1158/1535-7163.MCT-13-1091 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q et al. Characterization of monocarboxylate transport in human kidney HK-2 cells. Mol Pharm 3, 675–685, doi: 10.1021/mp060037b (2006). [DOI] [PubMed] [Google Scholar]
- 49.Polanski R et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res 20, 926–937, doi: 10.1158/1078-0432.CCR-13-2270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corbet C et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat Commun 9, 1208, doi: 10.1038/s41467-018-03525-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Castillo V et al. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J Cancer 8, 178–189, doi: 10.7150/jca.16387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Z, Han F, Yang S, Wu J & Zhan W Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett 358, 17–26, doi: 10.1016/j.canlet.2014.11.046 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Zhai X, Yang Y, Wan J, Zhu R & Wu Y Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol Rep 30, 2983–2991, doi: 10.3892/or.2013.2735 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Askarian F, Wagner T, Johannessen M & Nizet V Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol Rev 42, 656–671, doi: 10.1093/femsre/fuy025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanzelmann D et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat Commun 7, 12304, doi: 10.1038/ncomms12304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawasaki T & Kawai T Toll-like receptor signaling pathways. Front Immunol 5, 461, doi: 10.3389/fimmu.2014.00461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larsson L, Thorbert-Mros S, Rymo L & Berglundh T Influence of epigenetic modifications of the interleukin-10 promoter on IL10 gene expression. Eur J Oral Sci 120, 14–20, doi: 10.1111/j.1600-0722.2011.00917.x (2012). [DOI] [PubMed] [Google Scholar]
- 58.Yuan ZL, Guan YJ, Chatterjee D & Chin YE Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273, doi: 10.1126/science.1105166 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Qin G et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology 7, e1442167, doi: 10.1080/2162402X.2018.1442167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouzounova M et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 8, 14979, doi: 10.1038/ncomms14979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang CX et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ, doi: 10.1038/s41418-019-0302-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sundaram K et al. IkappaBzeta Regulates Human Monocyte Pro-Inflammatory Responses Induced by Streptococcus pneumoniae. PLoS One 11, e0161931, doi: 10.1371/journal.pone.0161931 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katoh H et al. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 24, 631–644, doi: 10.1016/j.ccr.2013.10.009 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li W, Mao Z, Fan X, Cui L & Wang X Cyclooxygenase 2 promoted the tumorigenecity of pancreatic cancer cells. Tumour Biol 35, 2271–2278, doi: 10.1007/s13277-013-1301-2 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Lucas M, Zhang X, Prasanna V & Mosser DM ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J Immunol 175, 469–477, doi: 10.4049/jimmunol.175.1.469 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Turner BM Cellular memory and the histone code. Cell 111, 285–291, doi: 10.1016/s0092-8674(02)01080-2 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Youn JI et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 14, 211–220, doi: 10.1038/ni.2526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosborough BR, Castellaneta A, Natarajan S, Thomson AW & Turnquist HR Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol 91, 701–709, doi: 10.1189/jlb.0311119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sahakian E et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol 63, 579–585, doi: 10.1016/j.molimm.2014.08.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eskandarian HA et al. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 341, 1238858, doi: 10.1126/science.1238858 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Kincaid EZ & Ernst JD Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. J Immunol 171, 2042–2049, doi: 10.4049/jimmunol.171.4.2042 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, Curry HM, Zwilling BS & Lafuse WP Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol 174, 5687–5694, doi: 10.4049/jimmunol.174.9.5687 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Pathak SK et al. TLR4-dependent NF-kappaB activation and mitogen- and stress-activated protein kinase 1-triggered phosphorylation events are central to Helicobacter pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated induction of IL-6 release from macrophages. J Immunol 177, 7950–7958, doi: 10.4049/jimmunol.177.11.7950 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Cao J et al. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci U S A 116, 5487–5492, doi: 10.1073/pnas.1815365116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moreno-Yruela C, Galleano I, Madsen AS & Olsen CA Histone Deacetylase 11 Is an epsilon-N-Myristoyllysine Hydrolase. Cell Chem Biol 25, 849–856 e848, doi: 10.1016/j.chembiol.2018.04.007 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Kutil Z et al. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem Biol 13, 685–693, doi: 10.1021/acschembio.7b00942 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Korkmaz B, Horwitz MS, Jenne DE & Gauthier F Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62, 726–759, doi: 10.1124/pr.110.002733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lau D et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A 102, 431–436, doi: 10.1073/pnas.0405193102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar V, Patel S, Tcyganov E & Gabrilovich DI The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol 37, 208–220, doi: 10.1016/j.it.2016.01.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Obermajer N & Kalinski P Key role of the positive feedback between PGE(2) and COX2 in the biology of myeloid-derived suppressor cells. Oncoimmunology 1, 762–764, doi: 10.4161/onci.19681 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dufait I et al. Perforin and Granzyme B Expressed by Murine Myeloid-Derived Suppressor Cells: A Study on Their Role in Outgrowth of Cancer Cells. Cancers 11, doi: 10.3390/cancers11060808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sawant A et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 73, 672–682, doi: 10.1158/0008-5472.CAN-12-2202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heim CE et al. Human prosthetic joint infections are associated with myeloid-derived suppressor cells (MDSCs): Implications for infection persistence. J Orthop Res 36, 1605–1613, doi: 10.1002/jor.23806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manning Fox JE, Meredith D & Halestrap AP Characterisation of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J Physiol 529 Pt 2, 285–293, doi: 10.1111/j.1469-7793.2000.00285.x (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vitko NP, Grosser MR, Khatri D, Lance TR & Richardson AR Expanded Glucose Import Capability Affords Staphylococcus aureus Optimized Glycolytic Flux during Infection. MBio 7, doi: 10.1128/mBio.00296-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vitko NP, Spahich NA & Richardson AR Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. MBio 6, doi: 10.1128/mBio.00045-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yermak K, Karbysheva S, Perka C, Trampuz A & Renz N Performance of synovial fluid D-lactate for the diagnosis of periprosthetic joint infection: A prospective observational study. J Infect, doi: 10.1016/j.jinf.2019.05.015 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Zhang Q & Cao X Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol 19, 417–432, doi: 10.1038/s41577-019-0151-6 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Turner NA et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol 17, 203–218, doi: 10.1038/s41579-018-0147-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun L et al. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci Alliance 1, e201800039, doi: 10.26508/lsa.201800039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mootz JM, Malone CL, Shaw LN & Horswill AR Staphopains modulate Staphylococcus aureus biofilm integrity. Infect Immun 81, 3227–3238, doi: 10.1128/IAI.00377-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gries CM et al. Cyclic di-AMP Released from Staphylococcus aureus Biofilm Induces a Macrophage Type I Interferon Response. Infect Immun 84, 3564–3574, doi: 10.1128/IAI.00447-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamada KJ et al. Arginase-1 Expression in Myeloid Cells Regulates Staphylococcus aureus Planktonic but Not Biofilm Infection. Infect Immun 86, doi: 10.1128/IAI.00206-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heim CE, West SC, Ali H & Kielian T Heterogeneity of Ly6G(+) Ly6C(+) Myeloid-Derived Suppressor Cell Infiltrates during Staphylococcus aureus Biofilm Infection. Infect Immun 86, doi: 10.1128/IAI.00684-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Niska JA et al. Vancomycin-rifampin combination therapy has enhanced efficacy against an experimental Staphylococcus aureus prosthetic joint infection. Antimicrob Agents Chemother 57, 5080–5086, doi: 10.1128/AAC.00702-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pribaz JR et al. Mouse model of chronic post-arthroplasty infection: noninvasive in vivo bioluminescence imaging to monitor bacterial burden for long-term study. J Orthop Res 30, 335–340, doi: 10.1002/jor.21519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Niska JA et al. Monitoring bacterial burden, inflammation and bone damage longitudinally using optical and muCT imaging in an orthopaedic implant infection in mice. PLoS One 7, e47397, doi: 10.1371/journal.pone.0047397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamada KJ et al. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PLoS Pathog 16, e1008354, doi: 10.1371/journal.ppat.1008354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Allhoff M, Sere K, J FP, Zenke M & I GC Differential peak calling of ChIP-seq signals with replicates with THOR. Nucleic Acids Res 44, e153, doi: 10.1093/nar/gkw680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Allhoff M et al. Detecting differential peaks in ChIP-seq signals with ODIN. Bioinformatics 30, 3467–3475, doi: 10.1093/bioinformatics/btu722 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Lin X, Tirichine L & Bowler C Protocol: Chromatin immunoprecipitation (ChIP) methodology to investigate histone modifications in two model diatom species. Plant Methods 8, 48, doi: 10.1186/1746-4811-8-48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pfaffl MW A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45, doi: 10.1093/nar/29.9.e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.