

Abstract
Tropane alkaloids (TAs) from nightshade plants are neurotransmitter inhibitors used for treating neuromuscular disorders and are classified as essential medicines by the World Health Organization1,2. Global supply challenges have resulted in frequent drug shortages3,4. Further supply-chain vulnerabilities are revealed by events like the Australian wildfires5 and the COVID-19 pandemic6. Rapidly deployable production strategies that are robust to environmental and socioeconomic upheaval7,8 are needed. Here, we engineered baker’s yeast to produce the medicinal TAs hyoscyamine and scopolamine starting from simple sugars and amino acids. We combined functional genomics to identify a missing pathway enzyme, protein engineering to enable functional acyltransferase expression via trafficking to the vacuole, heterologous transporters to facilitate intracellular routing, and strain optimization to improve titers. Our integrated system positions >20 proteins adapted from yeast, bacteria, plants, and animals across six sub-cellular locations to recapitulate the spatial organization of TA biosynthesis in plants. Microbial biosynthesis platforms can facilitate discovery of TA derivatives as novel therapeutics for neurological disease and, once scaled, enable robust and agile supply of these essential medicines.
Tropane alkaloids (TAs) such as cocaine and atropine are present in plants from the nightshade (Solanaceae), coca (Erythroxylaceae), and bindweed (Convolvulaceae) families. Some TAs, including hyoscyamine and scopolamine, are used to treat neuromuscular disorders ranging from nerve agent poisoning to Parkinson’s disease1,2. Direct chemical syntheses of TAs are not economically viable due to challenging stereochemistries9. Thus, intensive cultivation of Duboisia shrubs from the nightshade family in Australia, India, Brazil, and Saudi Arabia undergirds the global supply for medicinal TAs2,10,11. This agriculture-based supply chain poses three risks to public health. First, overall increasing demand for TA-based medicines already results in recurring supply shortages3,4. Second, regional events, such as the 2019–20 Australian wildfires, can threaten global supply5. Third, global crises, such as the ongoing COVID-19 pandemic, can threaten local availability due to demand spikes and supply-chain disruption6,12. The urgency of having options for quickly scaling production of essential medicines to match regional and local demand, free of geopolitical dependencies and robust to environmental and socioeconomic upheaval, is widely recognized7,8.
Phytochemical production using engineered yeast can address many of the vulnerabilities associated with crop cultivation. The rapid generation times and high cell densities achieved in microbial fermentations enable production of target compounds with reduced time, space, and resource requirements relative to plant extraction. Cultivation in closed bioreactors can also reduce supply chain susceptibility to environmental and geopolitical disruption, while providing improved batch-to-batch consistency and active ingredient purity.
However, TA biosynthesis in Solanaceae exhibits extensive intra- and inter-cellular compartmentalization, with enzymes active across specific sub-cellular compartments (cytosol, mitochondrion, chloroplast, peroxisome, ER membrane, vacuole), cell types (root pericycle, endodermis, cortex), and tissues (secondary roots)11. Reconstitution of such pathways in yeast is thus made challenging by incompatibilities of enzymes adapted for specific spatial or regulatory contexts, and metabolite transport strategies that are not readily realized in microbial hosts.
Hyoscyamine and scopolamine comprise an arginine-derived 8-azabicyclo[3.2.1]octane (‘tropine’) acyl acceptor esterified with a phenylalanine-derived phenyllactic acid (PLA) acyl donor (Fig. 1a). The identification of a type III polyketide synthase (PYKS) and cytochrome P450 (CYP82M3) catalyzing the cyclization of N-methylpyrrolinium to tropinone in Atropa belladonna13 enabled us and others to engineer yeast strains for de novo production of tropine14,15. The recent report of a UDP-glucosyltransferase (UGT84A27) and serine carboxypeptidase-like (SCPL) acyltransferase (littorine synthase, LS) catalyzing the condensation of tropine and phenyllactate to littorine16 resolved a debate about the nature of the acyl transfer reaction9. However, functional expression of plant SCPL acyltransferases (SCPL-ATs) in non-plant hosts has not been reported. Also, while the cytochrome P450 (CYP80F1) catalyzing rearrangement of littorine to hyoscyamine aldehyde17,18 and the 2-oxoglutarate-dependent hydroxylase/dioxygenase (H6H) catalyzing epoxidation of hyoscyamine to scopolamine are established19,20, no enzymatic activity for reduction of hyoscyamine aldehyde to hyoscyamine is known, necessitating discovery of such an enzyme (Fig. 1a).
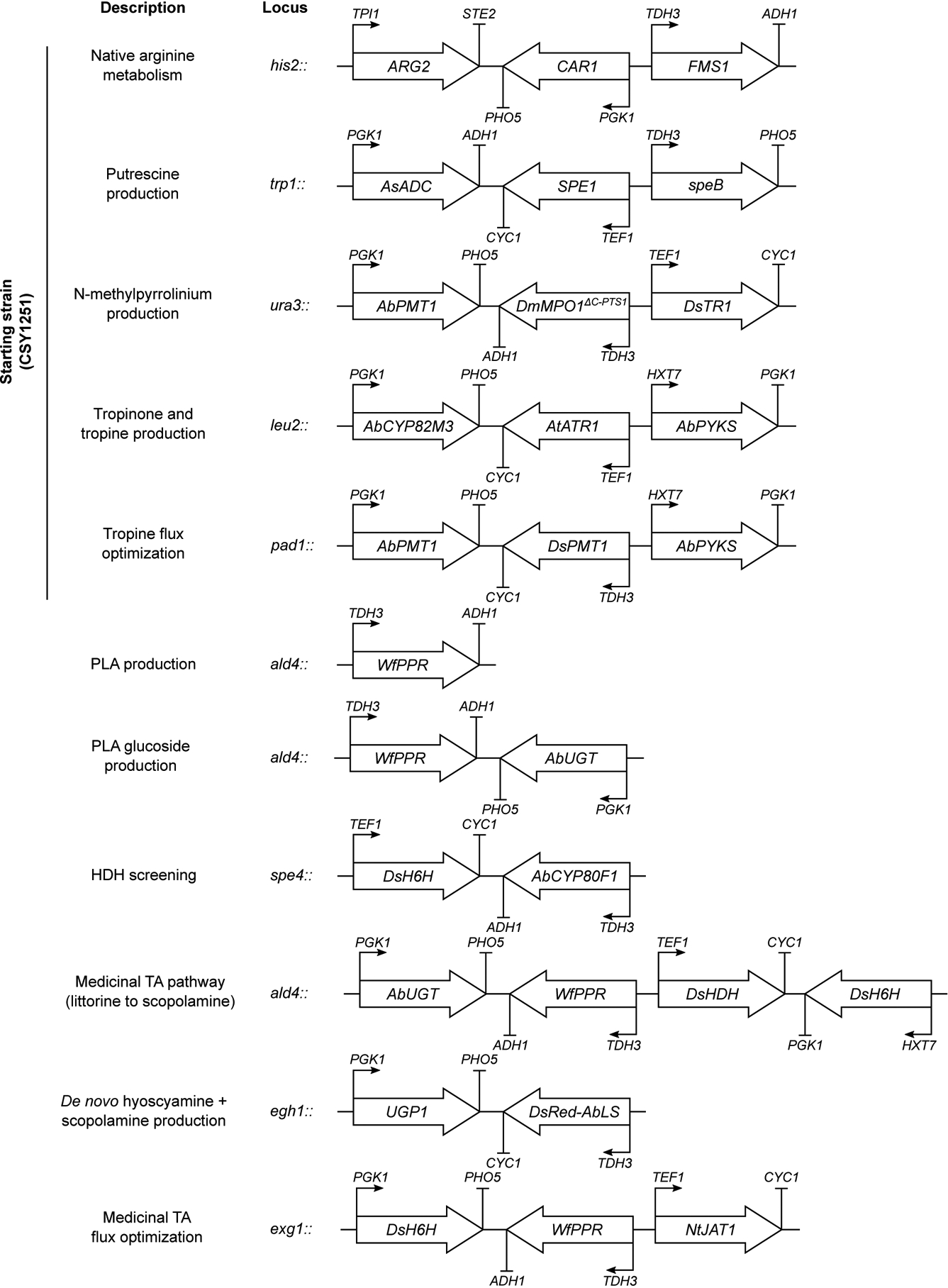
Figure 1. Engineered biosynthetic pathway for de novo production of scopolamine in yeast and optimization of PLA-glucoside biosynthesis.
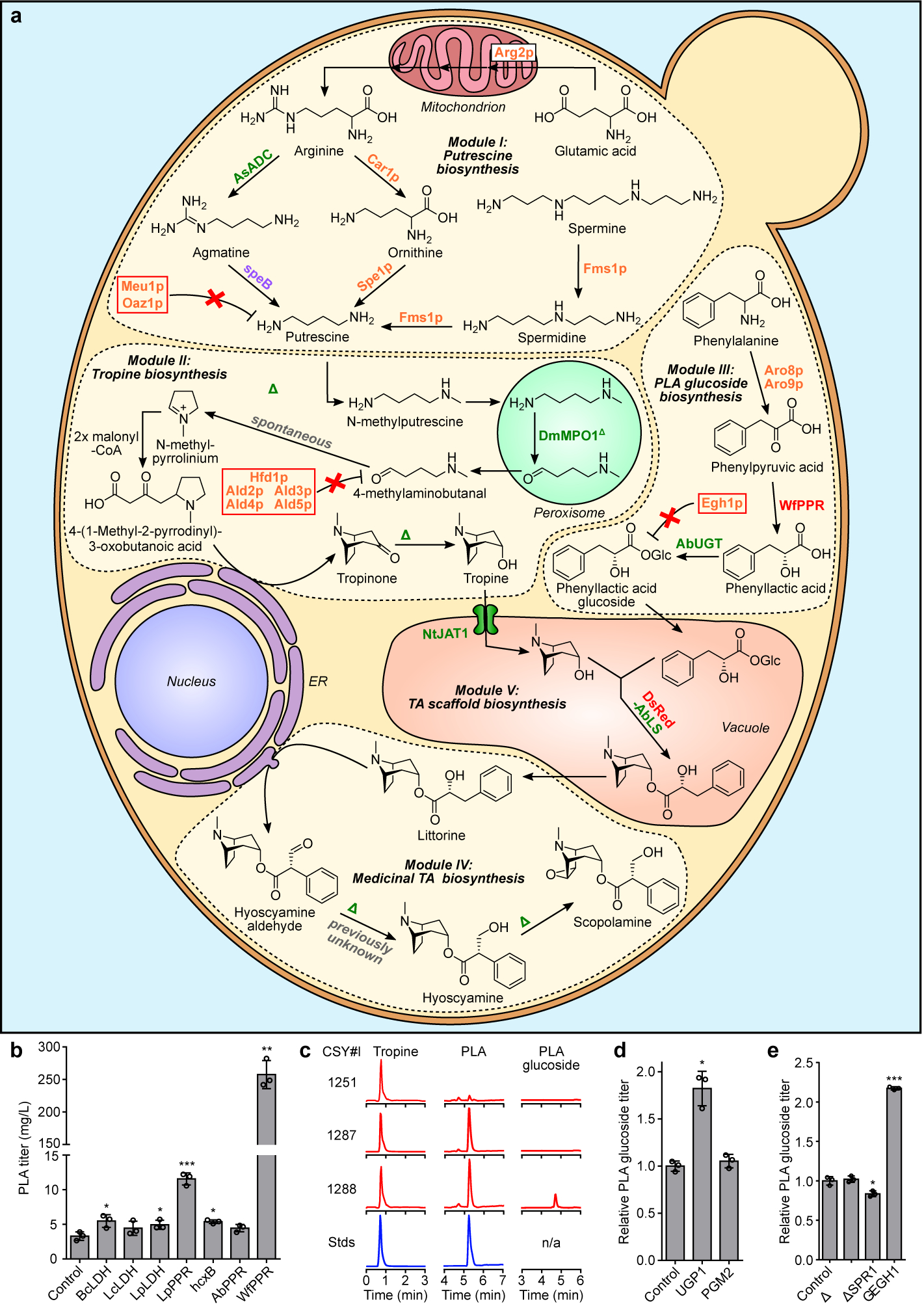
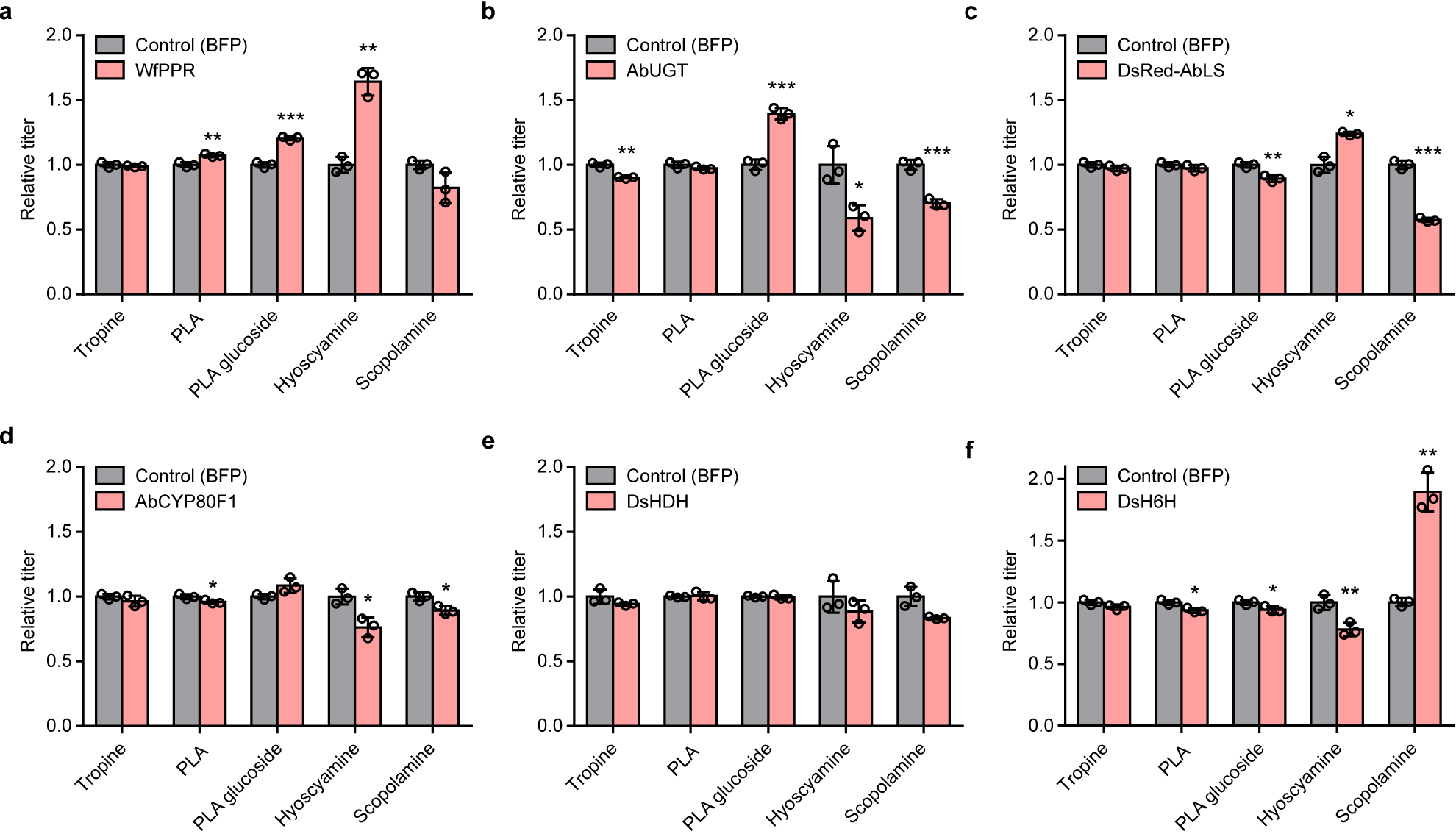
(a) Modular pathway construction for scopolamine biosynthesis in yeast. Enzyme/protein color scheme: orange, yeast (overexpressed); green, plant; purple, bacteria; red, other eukaryote; grey, spontaneous/non-enzymatic. Red boxes indicate disrupted yeast proteins; dotted lines/solid line of vacuole membrane delineate functional biosynthetic modules. Enzymes: Arg2p, glutamate N-acetyltransferase; Car1p, arginase; AsADC, Avena sativa arginine decarboxylase; speB, agmatine ureohydrolase; Spe1p, ornithine decarboxylase; Fms1p, polyamine oxidase; Meu1p, methylthioadenosine phosphorylase; Oaz1p, ornithine decarboxylase antizyme-1; AbPMT1/DsPMT1, Atropa belladonna/Datura stramonium putrescine N-methyltransferase 1; DmMPO1ΔC-PTS1, Datura metel N-methylputrescine oxidase 1 with peroxisome targeting sequence 1 and truncated C-terminus; AbPYKS, A. belladonna pyrrolidine ketide synthase; AbCYP82M3, A. belladonna tropinone synthase; AtATR1, Arabidopsis thaliana NADP+-cytochrome P450 reductase; DsTR1, D. stramonium tropinone reductase I; Hfd1p/Ald2p-Ald5p, aldehyde dehydrogenases; Aro8p/Aro9p, aromatic amino acid aminotransferases; WfPPR, Wickerhamia fluorescens 3-phenylpyruvate reductase; AbUGT, A. belladonna UDP-glucosyltransferase 84A27; Egh1p, steryl-β-glucosidase; DsRed-AbLS, Discosoma sp. red fluorescent protein fused to the N-terminus of A. belladonna littorine synthase; NtJAT1, Nicotiana tabacum jasmonate-inducible alkaloid transporter 1; AbCYP80F1, A. belladonna littorine mutase; DsHDH, D. stramonium hyoscyamine dehydrogenase; DsH6H, D. stramonium hyoscyamine 6β-hydroxylase/dioxygenase. (b) PLA production in yeast engineered for expression of phenylpyruvate reductases (PPRs) or lactate dehydrogenases (LDHs). Heterologous enzymes or negative control (BFP) were expressed from low-copy plasmids in strain CSY1251. (c) Multiple reaction monitoring (MRM) and extracted ion chromatogram (EIC) traces from culture media of yeast engineered for step-wise reconstitution of PLA glucoside biosynthesis via Module III. Chromatogram traces are representative of three biological replicates. (d) Relative PLA glucoside titers in yeast engineered for overexpression of UDP-glucose biosynthetic enzymes. Enzymes or negative control (BFP) were expressed from low-copy plasmids in strain CSY1288. (e) Relative PLA glucoside titers in CSY1288 with disruptions to endogenous glucosidases. For d and e, PLA glucoside accumulation was compared using relative titers due to lack of an authentic chemical standard. Strains were cultured for 72 h before LC-MS/MS analysis of metabolites in culture supernatant. Data in b, d, and e represent the mean of n = 3 biologically independent samples (open circles), error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Statistical significance is shown relative to controls. Exact P-values are in Supplementary Table 5.
TA acyl acceptor and donor biosynthesis
We designed a biosynthetic pathway comprising five functional modules for hyoscyamine and scopolamine production from simple precursors in yeast (Fig. 1a). Modules I/II and III enable de novo biosynthesis of the acyl acceptor and donor moieties; module IV enables TA scaffold modifications to produce hyoscyamine and scopolamine; module V comprises the central acyltransferase reaction linking upstream acyl acceptor/donor biosynthesis to downstream scaffold modifications. As a starting point, we used a yeast platform strain (CSY1251) that was previously engineered for de novo production of the acyl acceptor tropine via Modules I and II14. A putrescine biosynthesis module (I) designed to increase putrescine accumulation incorporated (i) overexpression of glutamate N-acetyltransferase (Arg2p), arginase (Car1p), ornithine decarboxylase (Spe1p), and polyamine oxidase (Fms1p); (ii) a parallel plant/bacterial pathway encoded by Avena sativa arginine decarboxylase (AsADC) and Escherichia coli agmatine ureohydrolase (speB); and (iii) disruptions to polyamine regulatory mechanisms encoded by methylthioadenosine phosphorylase (Meu1p) and ornithine decarboxylase antizyme-1 (Oaz1p). A tropine biosynthesis module (II) incorporated (i) seven enzymes: A. belladonna and Datura stramonium putrescine N-methyltransferases (AbPMT1, DsPMT1), Datura metel N-methylputrescine oxidase engineered for improved peroxisomal activity (DmMPO1ΔC-PTS1), A. belladonna pyrrolidine ketide synthase (AbPYKS) and tropinone synthase (AbCYP82M3), Arabidopsis thaliana cytochrome P450 reductase (AtATR1), and D. stramonium tropinone reductase 1 (DsTR1); and (ii) disruptions to five aldehyde dehydrogenases (Hfd1p, Ald2p, Ald3p, Ald4p, Ald5p) to reduce loss of pathway intermediates.
We designed a third module (III) for production of the acyl donor 1-O-β-phenyllactoylglucose (PLA glucoside) from phenylalanine via aromatic aminotransferases Aro8p/Aro9p, phenylpyruvate reductase (PPR), and PLA UDP-glucosyltransferase (UGT84A27)16. Yeast produce 3-phenylpyruvate from phenylalanine via Aro8p and Aro9p21, and wild-type yeast and CSY1251 produce trace levels of PLA, potentially via nonspecific activity of a lactate dehydrogenase (LDH) acting on 3-phenylpyruvate22. We screened PPRs from E. coli23, Lactobacillus (UniProt A0A2U9AUW1), A. belladonna24, and Wickerhamia fluorescens25 and LDHs from Bacillus and Lactobacillus with reported activity on 3-phenylpyruvate22,26,27 via expression from a plasmid in CSY1251. All screened enzymes yielded modest (1.3- to 3.5-fold) improvements in PLA production relative to control, except for W. fluorescens PPR, which resulted in a nearly 80-fold increase to ~250 mg/L (Fig. 1b) and was integrated into CSY1251 to make strain CSY1287.
In A. belladonna, PLA is activated for acyl transfer to tropine via glucosylation by UGT84A27 (AbUGT)16. Plant UGTs participate in the biosynthesis of diverse phenylpropanoids and often exhibit broad substrate scope28. We expressed AbUGT from a plasmid in CSY1251 and measured conversion of three phenylpropanoid acyl donors (PLA, cinnamic acid, ferulic acid) to their respective glucosides (Extended Data Fig. 2a,c). While AbUGT glucosylated ~60% and 90% of cinnamic acid and ferulic acid, respectively, <3% of PLA was glucosylated (Extended Data Fig. 2b). AbUGT orthologs identified from transcriptomes of other TA-producing Solanaceae (Supplementary Note 1) and structure-guided active site mutants (Supplementary Note 2) exhibited poor activity on PLA (Extended Data Fig. 2b,d–f), suggesting PLA glucosylation may constitute a key limitation in TA production. We constructed strain CSY1288 by integrating codon-optimized WfPPR and AbUGT into the genome of CSY1251, and verified PLA production (66 mg/L) and minimal PLA glucoside accumulation (Fig. 1c).
We increased PLA glucoside levels by incorporating genetic modifications promoting UDP-glucose accumulation and decreasing glycoside degradation. We overexpressed PGM2 and UGP1, whose gene products catalyze the isomerization of glucose-6-phosphate to glucose-1-phosphate and conversion of glucose-1-phosphate to UDP-glucose, respectively, from plasmids in CSY1288. While PGM2 overexpression yielded no improvement relative to control, overexpression of UGP1 resulted in ~1.8-fold increase in PLA glucoside production (Fig. 1d). We disrupted three native glucosidase genes—EXG1, SPR1, and EGH1—in CSY1288, as glucosidases have been shown to hydrolyze heterologous glucosides in yeast29. The disruption of EGH1 more than doubled PLA glucoside production (Fig. 1e), indicating that hydrolysis by Egh1p constitutes a substantial loss of TA precursor from the pathway. We thus incorporated both UGP1 overexpression and EGH1 disruption into a complete TA production strain.
HDH discovery and scopolamine biosynthesis
We used a functional genomics approach to discover the enzyme, hyoscyamine dehydrogenase (HDH), which catalyzes reduction of hyoscyamine aldehyde to hyoscyamine. We searched for genes that co-express with TA biosynthetic genes in secondary root tissues by mining a publicly available A. belladonna transcriptome dataset30. Starting from >40,000 identified transcripts, we removed transcripts without putative dehydrogenase or reductase-like domains, and further filtered by clustering tissue-specific expression profiles with those of bait genes AbCYP80F1 and AbH6H (Extended Data Fig. 4a). Nearly all candidates exhibited the secondary root-specific expression pattern observed for TA biosynthetic genes. Due to missing sequence regions, we repeated the de novo transcriptome assembly from raw RNAseq reads30 using the Trinity software package31 and reconstituted missing fragments for twelve HDH candidates via alignment of incomplete regions against the newly assembled transcriptome (Supplementary Table 1).
We identified the missing HDH activity by screening candidates generated via transcriptome mining in yeast. Lack of an authentic commercial standard for hyoscyamine aldehyde and insufficient yield from chemical syntheses, as well as similar chromatographic and mass spectrometric properties of littorine and hyoscyamine, necessitated screening of HDH candidates by detection of scopolamine (m/z+ 304) from fed littorine (m/z+ 290) via a three-step biosynthetic pathway (Fig. 1a). We constructed an HDH screening strain (CSY1292) by integrating codon-optimized AbCYP80F1 and an optimal H6H ortholog from D. stramonium (DsH6H; Extended Data Fig. 3) into the genome of CSY1251, and expressed codon-optimized HDH candidates from a plasmid. One of the candidates, HDH2 (i.e., AbHDH), exhibited a 35% decrease in hyoscyamine aldehyde levels and accumulation of scopolamine (7.2 μg/L), indicating the missing HDH activity (Fig. 2a).
Figure 2. Identification and characterization of hyoscyamine dehydrogenase in A. belladonna.
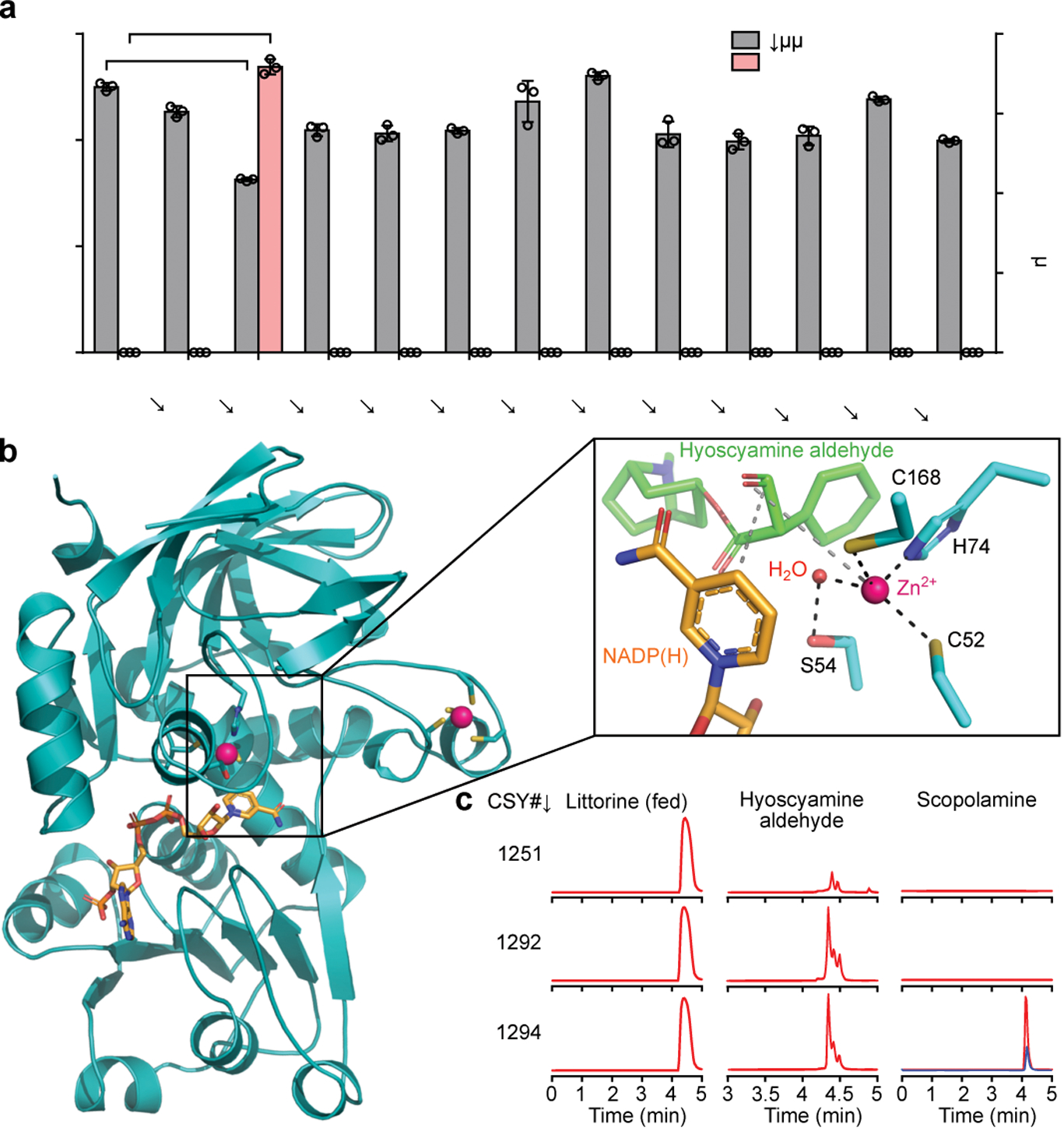
(a) Hyoscyamine aldehyde and scopolamine production in yeast engineered for expression of A. belladonna HDH candidates. Candidates or a negative control (BFP) were expressed from low-copy plasmids in CSY1292. Hyoscyamine aldehyde accumulation was compared using relative titers due to lack of an authentic chemical standard. Amino acid sequences are in Supplementary Table 1. Data represent the mean of n = 3 biologically independent samples (open circles), error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Statistical significance is shown relative to control. Exact P-values are in Supplementary Table 5. (b) Homology model of AbHDH. NADPH and Zn2+ are shown in orange and pink, respectively. Box: zoomed view of AbHDH active site with NADPH and docked hyoscyamine aldehyde. Dashed lines indicate interactions important for catalysis. (c) Multiple reaction monitoring (MRM) traces from culture media of yeast engineered for step-wise reconstitution of Module IV for conversion of littorine to scopolamine. Blue trace represents 125 nM (38 μg/L) scopolamine standard. Chromatogram traces are representative of three biological replicates. For a and c, strains were cultured for 72 h with 1 mM littorine prior to LC-MS/MS analysis of metabolites in culture supernatant.
Structural and phylogenetic analyses provided insight into the catalytic mechanism and evolutionary history of HDH (Supplementary Notes 3–4). Homology modeling indicated that AbHDH is a zinc-dependent alcohol dehydrogenase of the medium-chain dehydrogenase/reductase (MDR) superfamily and likely uses NADPH as the hydride donor for hyoscyamine aldehyde reduction (Fig. 2b; Supplementary Note 3). Ligand docking simulations and active site mutants suggested a mechanism wherein the oxyanion intermediate formed upon hydride attack of hyoscyamine aldehyde is stabilized by a catalytic Zn2+, which is bound by Cys52, His74, Cys168, and a displaceable water molecule positioned by polar interactions with Ser54 (Fig. 2b; Extended Data Fig. 4b; Supplementary Note 3). We identified orthologs of AbHDH from transcriptomes of Datura innoxia (DiHDH) and Datura stramonium (DsHDH)32, and verified their activity via co-expression with an additional copy of DsH6H from plasmids in CSY1292. DsHDH showed the highest substrate depletion and product accumulation of the variants tested (Extended Data Fig. 4c,d).
We reconstituted the medicinal TA biosynthetic branch (Module IV) comprising optimal enzyme variants and overexpression of a limiting enzyme into our platform strain. Strain CSY1294 was constructed by integrating codon-optimized WfPPR and AbUGT (Module III), DsHDH, and an additional copy of DsH6H, which limits scopolamine accumulation (Extended Data Fig. 4d), into CSY1292. Scopolamine production from fed littorine was verified in CSY1294 (Fig. 2c). Strain CSY1294 incorporates the enzymes for producing the acyl acceptor (tropine; Modules I/II) and acyl donor (PLA glucoside; Module III) for littorine biosynthesis, and the enzymes for modification of the TA scaffold to scopolamine (Module IV), leaving the central acyltransferase reaction catalyzed by littorine synthase (Module V) as the final enzymatic step to implement.
Engineering vacuolar littorine biosynthesis
Recently, littorine biosynthesis in A. belladonna was demonstrated to occur via esterification of glucosylated PLA with tropine by an acyltransferase of the SCPL family (littorine synthase, AbLS)16. Few plant SCPL-ATs have been studied and no reports of in vivo activity in non-plant hosts have emerged, due to difficulties of extensive post-translational processing and trafficking in microbial hosts33. SCPL-ATs are expressed via the secretory pathway and localize to the plant tonoplast33 (Extended Data Fig. 6a). An N-terminal signal peptide (SP) directs the nascent polypeptide to the ER, wherein it undergoes processing steps for folding—SP cleavage, disulfide bond formation, and, in some cases, proteolytic removal of propeptide sequences. The partially folded SCPL-AT protein is transported through the Golgi, wherein it acquires N-glycosylation on asparagine residues within N-X-S/T motifs (where X is not proline). Recognition of cryptic signal sequences by vacuole-associated transport factors directs SCPL-AT to the vacuole lumen34. Although the yeast secretory pathway possesses much the same compartments and processing steps as in plants, it is unlikely that yeast transport factors recognize the same signal sequences and yeast protein glycosylation patterns differ from those of plants35. Our initial attempts to express wild-type AbLS in CSY1294 resulted in a severe growth defect and no detectable TA biosynthesis.
We then showed that terminal and internal peptide sequences impact processing and localization of SCPL-ATs in yeast. A putative N-terminal signal peptide in AbLS suggested that it follows the expected SCPL-AT ER-to-vacuole trafficking pathway in planta. Fluorescence microscopy of N- and C-terminal GFP fusions of AbLS expressed from plasmids in CSY1294 revealed that the N-terminal fusion (GFP-AbLS) co-localized with a vacuolar membrane stain (Fig. 3a, Extended Data Fig. 6b), whereas no fluorescence was detected for the C-terminal fusion (AbLS-GFP), consistent with reports that a native C-terminus is crucial for SCPL-AT folding36. To identify possible failure points in AbLS expression, maturation, and trafficking in yeast, we screened AbLS variants engineered for localization to subcellular compartments (Supplementary Note 5) and compared AbLS N-glycosylation patterns in tobacco and yeast (Extended Data Fig. 6c–h; Supplementary Note 6), which did not implicate mis-targeting or mis-glycosylation as primary factors impeding activity in yeast. Characterization of AbLS endoproteolytic processing based on identification of a putative internal propeptide sequence suggested that the enzyme may become stalled in the yeast secretion pathway upstream of the trans-Golgi network (TGN) (Extended Data Figs. 6g,h, 7; Supplementary Note 7). This potential disruption of TGN sorting may account for the lack of activity and growth defect observed in CSY1294 expressing wild-type AbLS.
Figure 3. Engineering littorine synthase for activity in yeast.
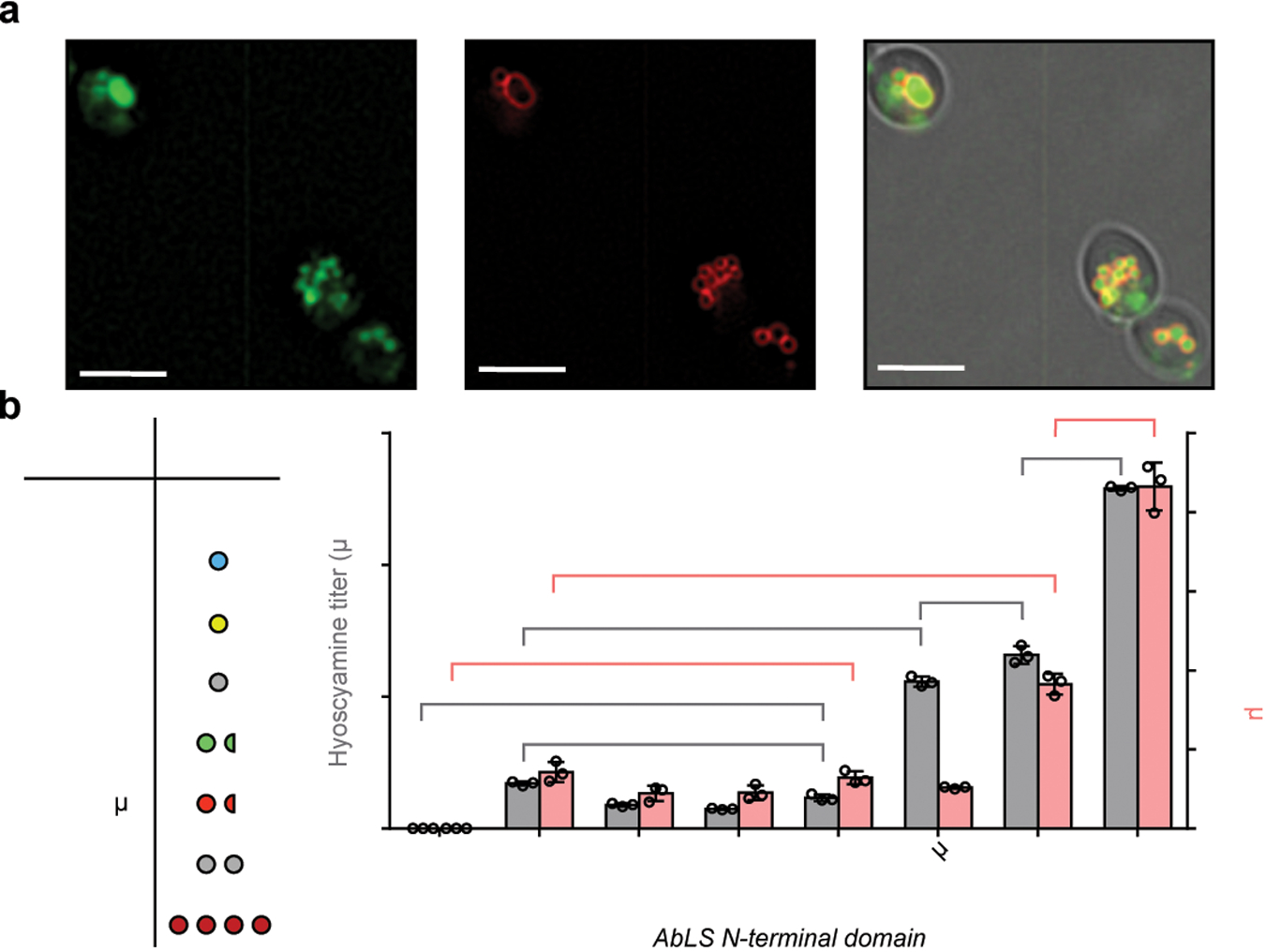
(a) Yeast epifluorescence microscopy showing N-terminal GFP-tagged AbLS (GFP-AbLS), vacuolar membrane stain FM4–64, and brightfield merged images. Microscopy was performed on CSY1294 expressing GFP-AbLS from a low-copy plasmid. 2D deconvolution was performed as described in Online Methods. Scale bar, 5 μm. Images are representative of two independent experiments. (b) De novo hyoscyamine and scopolamine production in engineered yeast expressing AbLS N-terminal fusions. Table shows expected oligomerization state of each N-terminal domain. Wild-type (control) or AbLS fusions were expressed from low-copy plasmids in CSY1294. Transformed strains were cultured for 96 h prior to LC-MS/MS analysis of metabolites in culture supernatant. No littorine was detected, indicating complete conversion to downstream TAs. Data represent the mean of n = 3 biologically independent samples (open circles), error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Exact P-values are in Supplementary Table 5.
Functional expression of AbLS in yeast was achieved by engineering N-terminal fusions that may alter sorting from the TGN. Transport of soluble proteins from the TGN to the vacuole requires recognition of a typically N-terminal signal sequence by vacuole protein sorting (Vps) cargo transport proteins, whereas integral membrane proteins that reach the yeast TGN are sorted to the vacuole by default37,38. We hypothesized that conversion of AbLS into a transmembrane protein by masking the SP with an N-terminally fused soluble domain might resolve the putative obstruction in TGN sorting (Fig. 3a; Supplementary Note 8). We constructed AbLS variants with N-terminally fused soluble domains, including fluorescent proteins from Aequoria (EGFP, tagBFP, mVenus) and Discosoma (mCherry, DsRed.T3); small ubiquitin-related modifier (Smt3p) with a mutated protease cleavage site (SUMO*)39; and AbUGT. We expressed these variants and wild-type AbLS from plasmids in CSY1294. Enhancement of AbLS activity appeared to be correlated with the N-terminal domain oligomerization state, with scopolamine production increasing from monomeric or weakly dimeric (GFP, BFP, mVenus, mCherry, SUMO*) to homodimeric (AbUGT) to homotetrameric (DsRed) domains; reaching de novo hyoscyamine and scopolamine titers up to 10.3 μg/L and 0.87 μg/L, respectively (Fig. 3b). To generate a strain harboring all five metabolic modules for complete TA biosynthesis (Modules I-V; Fig. 1a), we integrated a codon-optimized DsRed-AbLS and an additional copy of UGP1 into the genome of CSY1294 at the disrupted EGH1 site to generate CSY1296. CSY1296 exhibited de novo hyoscyamine and scopolamine titers of 10.2 μg/L and 1.0 μg/L, respectively.
Inter-compartment transport limitations were addressed by incorporation of plant transporters. Vacuolar compartmentalization of DsRed-AbLS in CSY1296 (Extended Data Fig. 8) necessitates import of cytosolic tropine and PLA glucoside to the vacuole lumen and export of vacuolar littorine to the cytosol. Several multidrug and toxin extrusion (MATE) transporters responsible for vacuolar alkaloid and glycoside sequestration have been identified in Solanaceae, including three with observed or predicted activity on TAs40,41. We expressed N. tabacum jasmonate-inducible alkaloid transporter 1 (NtJAT1) and two MATEs (NtMATE1, NtMATE2) from plasmids in CSY1296. Expression of NtJAT1 and NtMATE2 improved TA production; the former resulting in 74% and 18% increases in hyoscyamine and scopolamine titers, respectively (Fig. 4a). Fluorescence microscopy of CSY1296 expressing C-terminal GFP fusions of NtJAT1 or NtMATE2 from plasmids supports that NtJAT1 localizes to the vacuolar membrane (co-localizing with DsRed-AbLS), whereas NtMATE2 is partitioned between vacuolar and plasma membranes (Extended Data Fig. 8), suggesting both transporters might function to alleviate vacuolar substrate transport limitations while the latter might also improve cellular TA export (Fig. 4b).
Figure 4. Optimization of substrate transport limitations and medicinal TA production.
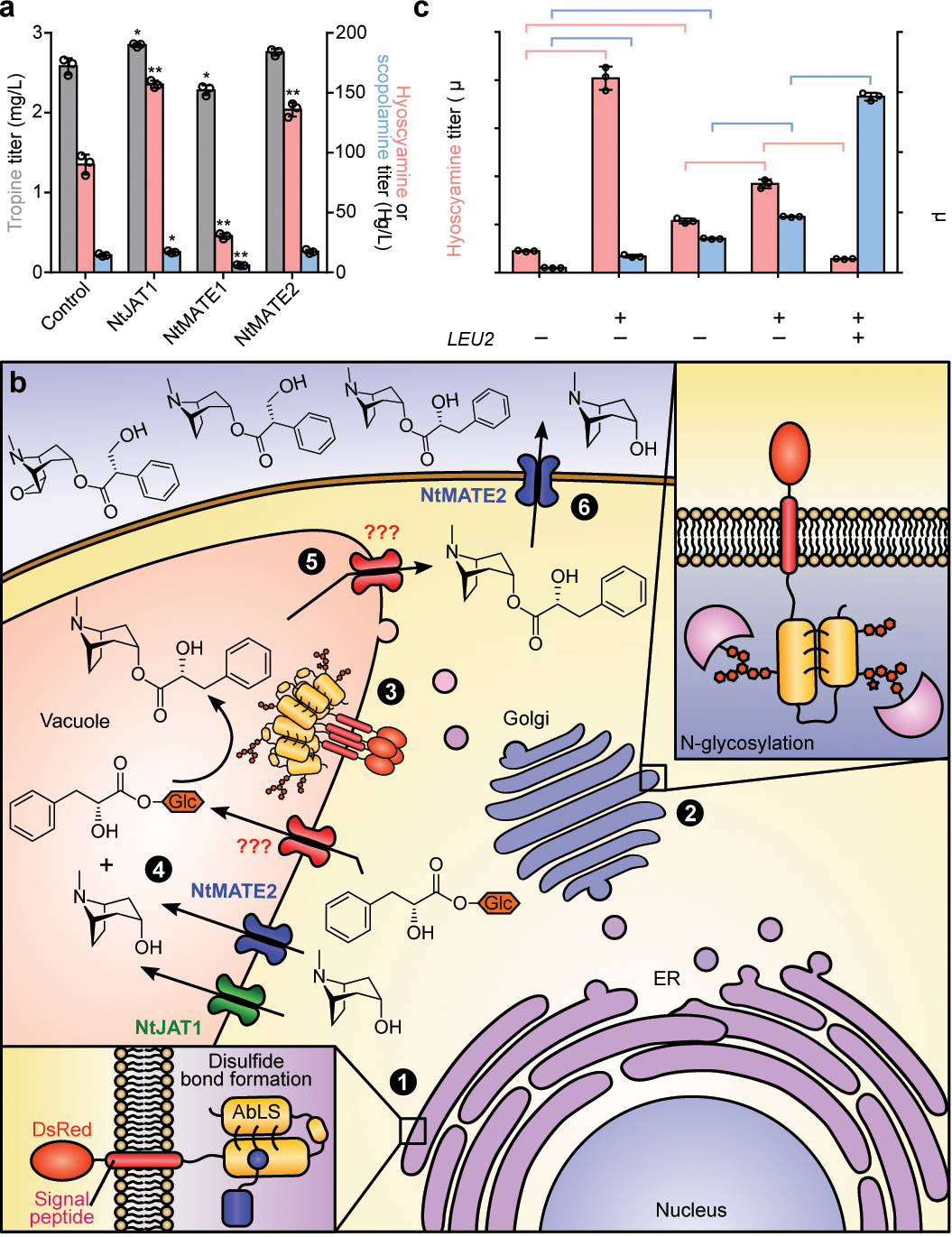
(a) Production of tropine, hyoscyamine, and scopolamine in CSY1296 engineered for expression of heterologous alkaloid transporters. Nicotiana tabacum jasmonate-inducible alkaloid transporter 1 (NtJAT1), multidrug and toxin extrusion (MATE) transporters 1/2, or a negative control (BFP) were expressed from low-copy plasmids in CSY1296 and transformed strains were cultured for 96 h. (b) Illustration of proposed DsRed-AbLS trafficking and alleviation of substrate transport limitations via heterologous transporter expression in engineered yeast. Putative transport activities based on microscopy studies are indicated; ‘???’ indicates unknown transport mechanism. Circled numbers indicate major proposed steps in DsRed-AbLS expression and activity, including maturation in (1) ER lumen and (2) Golgi, (3) trafficking to vacuole membrane, vacuolar (4) substrate import and (5) product export, and (6) cellular TA export. (c) Summary of strain and media optimization for de novo scopolamine production in engineered yeast. Strains were cultured in non-selective (CSY1296, CSY1297) or selective (CSY1298: leucine dropout) media with or without cofactors (50 mM 2-oxoglutarate, 15 mg/L Fe2+) at 25 °C for 96 h. Strain CSY1298 is prototrophic for leucine and harbors a blank plasmid with LEU2 gene (pCS4213). For a and c, metabolite titers in culture supernatant were quantified by LC-MS/MS. Data indicate the mean of n = 3 biologically independent samples, error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Exact P-values are in Supplementary Table 5.
Improvements in TA production were achieved via overexpression of limiting enzymes and media optimization. Additional copies of WfPPR and DsH6H expressed from plasmids in CSY1296 resulted in 64% and 89% increases in hyoscyamine and scopolamine titers, respectively (Extended Data Fig. 9). Supplementation with iron and 2-OG, required for H6H activity19,42, resulted in 9.0- and 3.4-fold increases in hyoscyamine and scopolamine titers from CSY1296 (Fig. 4c). We constructed an optimized strain (CSY1297) by integrating NtJAT1 and additional copies of WfPPR and DsH6H into CSY1296, which showed 2.4- and 7.1-fold respective increases in hyoscyamine and scopolamine accumulation (Fig. 4c). Removing leucine auxotrophy by expressing 3-isopropylmalate dehydrogenase (Leu2p) from a plasmid in CSY1297 (denoted CSY1298) increased conversion of hyoscyamine (85% decrease) to scopolamine (>3-fold increase) (Fig. 4c), potentially by improving access to Fe2+ via increased NADH regeneration43. Pseudo-fed-batch, high-density, shake-flask cultures grown in optimized media showed no littorine accumulation, hyoscyamine and scopolamine titers of ~30 μg/L in CSY1297 and CSY1298, respectively, and tropine and PLA accumulation up to ~3 mg/L and 160 mg/L (Extended Data Fig. 10; Supplementary Note 9), suggesting incorporation of PLA into littorine is a major limitation and target for future improvement.
Discussion
Our final strain comprises 34 chromosomal modifications (26 genes, 8 gene disruptions), resulting in an integrated whole-cell system that expresses enzymes and transporters in diverse sub-cellular locations (cytosol, mitochondria, peroxisome, vacuole, ER and vacuolar membranes) (Supplementary Note 10). Combining functional genomics with our synthesis platform, we identified an oxidoreductase that catalyzes the remaining uncharacterized step in the biosynthesis of hyoscyamine and scopolamine. We developed an N-terminal fusion strategy to achieve functional expression of the key TA scaffold-generating enzyme AbLS. Our strategy may improve folding and trafficking of the engineered transmembrane AbLS through the secretory pathway to the vacuole (Supplementary Note 8), potentially enabling heterologous expression of plant SCPL-ATs and expanding the diversity of natural product biosyntheses in yeast33. We used a plant vacuolar alkaloid importer to address import restrictions in that compartment. Although plasma membrane transporters have been used to improve cellular export and import of metabolites44,45, our work demonstrates that incorporation of plant transporters can facilitate intracellular transport and help reconstruct sub-cellular compartmentalization inherent to many plant biosynthetic pathways.
Our demonstration of total biosynthesis of hyoscyamine and scopolamine via engineered yeast suggests that centralized, plantation-based supply of medicinal TAs can be complemented or replaced by industrial fermentation. Process improvements to increase productivities from titers reported here (~30–80 μg/L; Fig. 4c), which are typical of first implementations of complex plant natural product pathways46–48, to commercial production (~5 g/L) are becoming routine49, and we anticipate would take ~1–2 years of focused effort by a professional team (Supplementary Note 11). From a land-use perspective, we estimate that a fermentation-based process sourcing sugar from sugarcane would require at least ~10-fold less land compared to the existing Duboisia farming-based approach (Supplementary Note 12). Transitioning from agriculture- to fermentation-based production could have many indirect impacts ranging from land-use and natural biodiversity, to labor markets and livelihoods, to supply-chain decouplings and geopolitical interdependencies50. Practically, because a fermentation-based approach can be implemented where needed and operated with a process time of days, our results support development of flexible manufacturing platforms enabling robust and agile supply of essential medicines.
Methods
Chemical compounds and standards
Tropine, (S)-hyoscyamine hydrobromide, and (S)-scopolamine hydrobromide were purchased from Santa Cruz Biotechnology (Dallas, TX). (R)-littorine hydrochloride was purchased from Toronto Research Chemicals (Toronto, ON). All other chemicals were purchased from Sigma (St. Louis, MO).
Plasmid construction
DNA oligonucleotides used in this study were synthesized by the Stanford Protein and Nucleic Acid Facility (Stanford, CA) and are listed in Supplementary Data 1. Genes encoding biosynthetic enzymes used in this study are listed by source and accession number in Supplementary Table 2; for HDH genes newly identified in this work, full amino acid sequences are given in Supplementary Table 1. Endogenous yeast genes were amplified from Saccharomyces cerevisiae CEN.PK2–1D51 genomic DNA via colony PCR52. Gene sequences encoding heterologous enzymes were codon-optimized for expression in S. cerevisiae using GeneArt GeneOptimizer software (Thermo Fisher Scientific) and synthesized as double-stranded gene fragments (Twist Bioscience). Plasmids used in this study are listed in Supplementary Data 2. Three types of plasmids were used in this work: yeast expression plasmids, yeast integration plasmids, and Agrobacterium tumefasciens binary vectors.
Yeast expression plasmids harbored a gene of interest flanked by a constitutive promoter and terminator, an auxotrophic selection marker, and either a low-copy CEN6/ARS4 or a high-copy 2μ yeast origin of replication. These plasmids were constructed by addition of 5’ and 3’ restriction sites to genes of interest using PCR, restriction digestion of PCR amplicons or synthesized gene fragments, and ligation of digested inserts into similarly digested vectors pAG414GPD-ccdB, pAG415GPD-ccdB, pAG416GPD-ccdB, pAG424GPD-ccdB, pAG425GPD-ccdB, or pAG426GPD-ccdB53 using T4 DNA ligase (New England Biolabs, NEB). Yeast expression plasmids expressing fusions of multiple proteins or enzymes were prepared by PCR-amplification of each gene of interest with 15–25 bp of overlap to adjacent fragments, assembly of fragments into single inserts with 5’ and 3’ restriction sites using overlap-extension PCR, and ligation cloning into digested vectors as described.
Yeast integration plasmids comprised a gene of interest flanked by a constitutive promoter and terminator, but lacked a selection marker and origin of replication for yeast expression. These plasmids were constructed by PCR linearization of the empty holding vectors pCS2656, pCS2657, pCS2658, pCS2661, or pCS2663 using primers complementary to the 3’ and 5’ ends of the promoter and terminator, respectively. Genes intended for yeast genomic integration were PCR-amplified to append 5’ and 3’ overhangs with 35–40 bp of homology to the termini of the linearized holding vectors and then assembled using Gibson assembly.
For transient expression of littorine synthase variants in Nicotiana benthamiana, A. tumefasciens binary vectors harbored a transfer-DNA (T-DNA) region comprising a gene of interest flanked by the constitutive Cauliflower Mosaic Virus (CaMV) 35S promoter/Cowpea Mosaic Virus (CPMV) 5’UTR and a nopaline synthase terminator, as well as an analogous expression cassette for the p19 RNAi-suppressor protein. These plasmids were constructed via addition of 5’ AgeI and 3’ XhoI restriction sites to a gene of interest via PCR, followed by digestion and ligation into the pEAQ-HT binary vector pCS335254.
All PCR amplification was performed using Q5 DNA polymerase (NEB) and linear DNA was purified using the DNA Clean and Concentrator-5 kit (Zymo Research). Assembled plasmids were transformed into chemically competent Escherichia coli (TOP10, Thermo Fisher Scientific) via heat-shock and propagated with selection in Luria-Bertani (LB) broth or on LB-agar plates with either carbenicillin (100 μg/mL) or kanamycin (50 μg/mL) selection. E. coli plasmid DNA was isolated by alkaline lysis from overnight cultures grown at 37 °C and 250 rpm in selective LB media using Econospin columns (Epoch Life Science) according to the manufacturer’s protocol. Plasmid sequences were verified by Sanger sequencing (Quintara Biosciences).
Yeast strain construction
Yeast strains used in this study (Supplementary Table 3) were derived from our previously reported tropine-producing strain CSY125114, which is in turn derived from the parental strain CEN.PK2–1D51. Strains were grown non-selectively in yeast-peptone media supplemented with 2% w/v dextrose (YPD media), yeast nitrogen base (YNB) defined media (Becton, Dickinson and Company (BD)) supplemented with synthetic complete amino acid mixture (YNB-SC; Clontech) and 2% w/v dextrose, or on agar plates of the aforementioned media. Strains transformed with plasmids bearing auxotrophic selection markers (URA3, TRP1, and/or LEU2) were grown selectively in YNB media supplemented with 2% w/v dextrose and the appropriate dropout solution (YNB-DO; Clontech) or on YNB-DO agar plates.
Yeast genomic modifications were performed using the CRISPRm method55. CRISPRm plasmids expressing Streptococcus pyogenes Cas9 and a single guide RNA (sgRNA) targeting a genomic locus were constructed by assembly PCR and Gibson assembly of DNA fragments encoding SpCas9 (pCS3410), tRNA promoter and HDV ribozyme (pCS3411), a 20-nt guide RNA sequence oligonucleotide, and tracrRNA and terminator (pCS3414) (Supplementary Data 2). For gene insertions, integration fragments comprising one or more genes of interest flanked by unique promoters and terminators were PCR-amplified from yeast integration plasmids using Q5 DNA polymerase (NEB) with flanking 40 bp microhomology regions to adjacent fragments and/or to the yeast genome at the integration site (Supplementary Data 1, Extended Data Fig. 1). For gene disruptions, integration fragments comprised 6–8 stop codons in all three reading frames flanked by 40 bp of microhomology to the disruption site, which was located within the first half of the open reading frame. Approximately 0.5–1 μg of each integration fragment was co-transformed with 500 ng of CRISPRm plasmid targeting the desired genomic site. Positive integrants were identified by yeast colony PCR52, Sanger sequencing, and/or functional screening by liquid chromatography and tandem mass spectrometry (LC-MS/MS).
Yeast transformations
Yeast strains were chemically transformed using the Frozen-EZ Yeast Transformation II Kit (Zymo Research) as per the manufacturer’s instructions, with the following modifications. For competent cell preparation, individual colonies were inoculated into YPD media and grown overnight at 30 °C and 460 rpm. Saturated cultures (~14–18 h) were back-diluted between 1:10 and 1:50 in fresh YPD media and grown to exponential phase (~5–7 h). Cultures were pelleted by centrifugation at 500 × g for 4 min, washed twice with 50mM Tris-HCl buffer (pH 8.5), and then resuspended in 20–50 μL of EZ2 solution per transformation. For transformation, competent cells were mixed with 250–1000 ng of total DNA and 200–500 μL of EZ3 solution. Cell suspensions were incubated at 30 °C with slow rotation for 1–1.5 h. For plasmid transformations, the transformed yeast were directly plated onto YNB-DO agar plates. For CRISPRm genomic modifications, yeast suspensions were instead mixed with 1mL YPD media, pelleted by centrifugation at 500 × g for 4 min, and then resuspended in 300–500 μL of fresh YPD media. Suspensions were incubated at 30 °C with gentle rotation for 2–3 h to allow expression of geneticin resistance and then spread on YPD plates supplemented with 200–400 mg/L G418 (geneticin) sulfate. Plates were incubated at 30 °C for 72 h to allow sufficient colony formation before downstream applications.
Growth conditions for metabolite assays
Small-scale metabolite production assays were performed in YNB-SC or YNB-DO media supplemented with 2% dextrose and 5% glycerol (YNB-G) for optimal tropine production14 in at least three replicates. Our previous work showed that tropine biosynthesis is significantly enhanced by higher starting cell densities14. Therefore, yeast colonies were initially inoculated in triplicate into 1 mL YPD or YNB-DO and grown to saturation (~18–22 h) at 30 °C and 460 rpm, pelleted by centrifugation at 500 × g for 4 min and 3,000 × g for 1 min, resuspended in 1 mL of fresh selective or non-selective YNB-G media (for some experiments, additionally supplemented with 15 mg/L Fe2+ from iron (II) sulfate and 50 mM 2-oxoglutarate56), and then 300 μL transferred into 2mL deep-well 96-well plates sealed with AeraSeal gas-permeable film (Excel Scientific). Cultures were grown for 72–96 h at 25 °C, 460 rpm, and 80% relative humidity in a Lab-Therm LX-T shaker (Adolf Kuhner).
Growth conditions for time courses
To simulate high-density batch culture conditions, strains were inoculated in triplicate into 10 mL of YPD media or selective YNB-G media and grown overnight to saturation at 30 °C and 250 rpm. Saturated cultures were pelleted by centrifugation at 500 × g for 4 min and 3000 × g for 1 min and then resuspended in 10 mL of fresh selective or non-selective YNB-G media supplemented with 50 mM 2-oxoglutarate and 15 mg/L Fe2+, and grown in 50-mL shake flasks with 10 mL starting volume in triplicates at 25 °C and 300 rpm for 120 h. Where indicated, fed-batch conditions were approximated by supplementing cultures after 72 h of growth with appropriate carbon sources and amino acids at 2% and 1X final concentrations, respectively. At appropriate time points, 250 μL samples were removed from cultures for analysis; 100 μL of culture was diluted 10× and used for optical density measurement at 600 nm on a Nanodrop 2000c spectrophotometer, and 150 μL of culture was used for metabolite quantification.
Analysis of metabolite production
Yeast cultures were pelleted by centrifugation at 3500 × g for 5 min at 12 °C and 150 μL aliquots of supernatant were removed for analysis. Metabolites were analyzed by LC-MS/MS using an Agilent 1260 Infinity Binary HPLC and an Agilent 6420 Triple Quadrupole mass spectrometer. Chromatography was performed using a Zorbax EclipsePlus C18 column (2.1 × 50 mm, 1.8 μm; Agilent Technologies) with 0.1% v/v formic acid in water as mobile phase solvent A and 0.1% v/v formic acid in acetonitrile as solvent B. The column was operated with a constant flow rate of 0.4 mL/min at 40 °C and a sample injection volume of 10 μL. Chromatographic separation was performed using the following gradient14: 0.00–0.75 min, 1% B; 0.75–1.33 min, 1–25% B; 1.33–2.70 min, 25–40% B; 2.70–3.70 min, 40–60% B; 3.70–3.71 min, 60–95% B; 3.71–4.33 min, 95% B; 4.33–4.34 min, 95–1% B; 4.34–5.00 min, equilibration with 1% B. For separation and detection of phenylpropanoid acyl donors (PLA, cinnamate, ferulate) and corresponding glucosides, the final equilibration step at 1% B was extended to 4.34–7.50 min. The LC eluent was directed to the MS from 0.01–5.00 min operating with electrospray ionization (ESI) in positive mode, source gas temperature 350 °C, gas flow rate 11 L/min, and nebulizer pressure 40 psi. Data collection was performed using MassHunter Workstation LC/MS Data Acquisition software (Agilent). Metabolites were identified and quantified by integrated peak area in MassHunter Workstation Qualitative Analysis Navigator software (Agilent) using the mass fragment/transition parameters in Supplementary Table 4 and standard curves. Primary MRM transitions were identified by analysis of 0.1–1 mM aqueous standards using MassHunter Workstation Optimizer software (Agilent) and corroborated against published mass transitions if available, and/or against predicted transitions determined using the CFM-ID fragment prediction utility57 and the METLIN database58. As both phenyllactic acid (PLA) and its glucoside formed strong ammonium adducts, these metabolites were detected and quantified in positive mode using the corresponding [M + H + 17]+ ions, m/z 184 (PLA) and 346 (PLA glucoside) (Supplementary Table 4).
Fluorescence microscopy
Individual colonies of yeast strains transformed with plasmids encoding biosynthetic enzymes fused to fluorescent protein reporters were inoculated into 1 mL selective or non-selective YNB-G media and grown overnight (~14–18 h) at 30 °C and 460 rpm. Overnight cultures were back-diluted between 1:2 and 1:4 into fresh YNB-G media and grown to exponential phase at 30 °C and 460 rpm for an additional 6–8 h to allow slow-maturing fluorescent proteins to fold before imaging.
Yeast vacuoles were co-imaged with fluorescent reporter-fused biosynthetic enzymes using the FM4–64 stain (Thermo Fisher) and pulse-chase fluorescence microscopy. FM4–64 is a red-fluorescent lipophilic styryl dye that intercalates into the yeast plasma membrane and is endocytosed during growth on rich media, accumulating in vacuolar membranes59. Transformed yeast colonies were inoculated into 1 mL selective or non-selective YNB-G and grown overnight (~14–18 h) at 30 °C and 460 rpm, then back-diluted between 1:10 and 1:3 into 1 mL of fresh YNB-G and grown for an additional 2–4 h until OD600 ~ 0.5–0.8. Cultures were pelleted by centrifugation at 5,000 × g for 5 min, resuspended in 500 uL fresh YPD with 8 μM (5 ng/μL) FM4–64, and incubated at 30 °C for 30 min with gentle rotation. Stained cells were pelleted by centrifugation at 3,000 × g for 5 min (pellets were visibly red), washed twice with 1 mL YPD, resuspended in 5 mL YPD, and then incubated at 30 °C and 460 rpm for 90–120 min to allow endocytosis and vacuolar accumulation of the dye. Cultures were pelleted by centrifugation at 500 × g for 4 min followed by 3,000 × g for 1 min, then resuspended in 250 μL of 40 mM MES buffer (pH 6.5) and imaged immediately.
For imaging, approximately 5–10 μL of cell suspension was spotted onto a glass microscope slide and covered with a glass coverslip (Thermo Fisher) and then imaged using an upright Zeiss AxioImager Epifluorescence/Widefield microscope with a ×64 oil immersion objective. Fluorescence excitation was performed using an EXFO X-Cite 120 illumination source and the following Semrock Brightline filter settings: GFP, 472/30 excitation and 520/35 emission; mCherry/DsRed/Cy3/TexasRed, 562/40 excitation and 624/40 emission. Emitted light was captured with a Zeiss Axiocam 503 mono camera and Zen Pro software, and subsequent image analysis was performed in ImageJ/Fiji (NIH). Images were converted to pseudocolor using the ‘Merge Channels’ and ‘Split Channels’ functions (Image→ Color→Merge/Split Channels). For each sample, linear histogram stretching was applied across all images for a given channel to improve contrast.
To reduce the interference of light from other focal planes when imaging sub-cellular organelles, we performed 2D digital deconvolution analysis, a common computational technique used for removing out-of-focus light distortion from 2D images of 3D structures60. First, a theoretical point-spread function (PSF), which mathematically describes the diffraction of light from a point source in a specific imaging setup, was computed using the ‘Diffraction PSF 3D’ plugin for ImageJ (available from http://fiji.sc/Diffraction_PSF_3D) for the green and red channels using the following parameters: index of refraction of the media, 1.518 (lens oil); numerical aperture, 1.40; wavelength (nm), 520 (green) or 624 (red); longitudinal spherical aberration at max. aperture (nm), 0.00 (default); image pixel spacing (nm), 72; slice spacing (nm), 0; width (pixels), 240; height (pixels), 242; depth (slices), 1; normalization, sum of pixel values = 1. Next, green and red channel images were separately deconvolved against the corresponding PSFs using the ‘Parallel Spectral Deconvolution 2D’ plugin for ImageJ (available from http://fiji.sc/Parallel_Spectral_Deconvolution) with default settings and auto regularization.
Identification of HDH candidates
Tissue-specific abundances (fragments per kilobase of contig per million mapped reads, FPKM) and putative protein structural and functional annotations for each of 43,861 unique transcripts identified from the A. belladonna transcriptome were obtained from the MSU Medicinal Plant Genomics Resource30. Transcripts encoding hyoscyamine dehydrogenase candidates were identified based on clustering of tissue-specific expression profiles with those of the bait genes CYP80F1 (littorine mutase) and H6H (hyoscyamine 6β-hydroxylase/dioxygenase), which respectively precede and follow the dehydrogenase step in the TA biosynthetic pathway, using a custom R script which is described below.
First, the complete list of 43,861 transcripts was filtered for those annotated with any of the following oxidoreductase protein family (PFAM) IDs: PF00106, PF13561, PF08659, PF08240, PF00107, PF00248, PF00465, PF13685, PF13823, PF13602, PF16884, PF00248; or any of the following functional annotation keywords: alcohol dehydrogenase, aldehyde reductase, short chain, aldo/keto. Additionally, any transcripts with functional annotations containing the keywords putrescine, tropinone, and tropine were included in the filter as positive control TA-associated genes to validate clustering with bait genes. Next, mean tissue-specific expression profiles were generated for the CYP80F1 and H6H bait genes. For each of the two bait genes, linear regression models were constructed to express the bait gene expression profile (in FPKM) as a linear function of each candidate gene profile and correlation p-values were computed for each candidate. The candidates identified using each of the two bait genes were pooled and duplicates were removed. Combined p-values for each candidate were computed as the sum of the log10 p-values of the correlations with each of the two bait genes. Transcripts matching known dehydrogenases in the TA biosynthetic pathway (i.e., tropinone reductases I and II) were removed, and the remaining candidates were ranked by combined p-value and by distance from bait genes via hierarchical clustering of tissue-specific expression profiles.
De novo transcriptome assembly
All data pre-processing, analysis, and de novo transcriptome assembly was performed on the Sherlock2.0 high-performance computing cluster (HPCC) at Stanford University (Stanford, CA). Paired-end raw RNA-seq reads corresponding to A. belladonna secondary roots (Accession #SRX060267, Run #SRR192881) and sterile seedling tissue (Accession #SRX060269, Run #SRR192882) were retrieved from the NCBI Sequence Read Archive (SRA) using the SRA Toolkit (NIH). The paired-end raw reads were analyzed via FastQC (Babraham Bioinformatics) and then trimmed using the BBDuk.sh utility (Joint Genome Institute, Department of Energy) using the following parameters: k-mer trimming, right end only (“ktrim=r”); k-mer length, 23 (“k=23”); minimum k-mer for end-trimming, 11 (“mink=11”), Hamming distance for k-mer matching, 1 (“hdist=1”); trim paired reads to same length (“tpe”), trim adapters using pair overlap detection (“tbo”); quality trimming, both right and left ends (“qtrim=rl”); quality cut-off, 5 (“trimq=5”); minimum permissible read length after trimming, 25 (“minlen=25”). Two independent de novo transcriptome assemblies were generated from the processed paired-end reads from secondary root (SRR192881) and seedling (SRR192882), respectively, using the Trinity software suite with default settings31,61.
Transcript functional annotation for each of the two assemblies (secondary root and seedling) was performed using the Trinotate package62. Following coding region prediction using the TransDecoder.LongOrfs and TransDecoder.Predict commands, annotations were generated using a BLASTp search against the UniProt/SwissProt databases and a protein homology search using HMMER. Complete ORF sequences for each of the candidate transcripts identified from co-expression analysis were generated by performing tBLASTn and tBLASTx searches against the Trinity transcriptome assemblies; hits with protein percent identity of at least 98%, accounting for sequencing errors, were assumed to be identical.
Identification of orthologs from transcriptome databases
Orthologs of A. belladonna UGT84A27 (UGT) were identified using tBLASTn searches of the transcriptomes of Brugmansia sanguinea and Datura metel in the 1000Plants (1KP) database63. This search yielded two unique, full-length amino acid sequences (i.e., within 5% of the length of the query sequence) and with expectation value 0.0: scaffold-AIOU-2012986-Brugmansia_sanguinea (B. sanguinea, BsUGT) and scaffold-JNVS-2051323-Datura_metel (D. metel, DmUGT).
Orthologs of hyoscyamine dehydrogenase (HDH) were identified using tBLASTn searches of the transcriptomes of several Datura species in the Medicinal Plant RNAseq database32. This search yielded two unique, full-length amino acid sequences (i.e., within 5% of the length of the query sequence) and with expectation value 0.0: medp_datin_20101112|6354 (D. innoxia, DiHDH) and medp_datst_20101112|10433 (D. stramonium, DsHDH).
Coding sequences for all putative orthologs were optimized for yeast expression and then cloned into expression vectors as described in the section ‘Plasmid construction’.
Protein structural analysis and substrate docking
Homology models of AbUGT, AbHDH, and AbLS were constructed using RaptorX with default modeling parameters64. For docking simulations, the binding of cosubstrates (UDP-glucose for AbUGT) or cofactors (NADPH for AbHDH) was first predicted based on structural alignment with the crystal structures of Arabidopsis thaliana salicylate UDP-glucosyltransferase UGT74F2 with bound UDP (PDB: 5V2K) and Populus tremuloides sinapyl alcohol dehydrogenase with bound NADPH (PDB: 1YQD) respectively, as the binding pockets for these cosubstrates are tightly conserved. Geometry optimizations of substrate structures (PLA or hyoscyamine aldehyde) prior to docking simulations were conducted using the Gaussian 16 software package on the Stanford Sherlock2.0 HPCC (run parameters: DFT, B3LYP, LANL2DZ). The energy-minimized ligand structures were then docked into the corresponding cosubstrate/cofactor-bound homology models using the Maestro and Glide XP software packages (Schrodinger) with default parameters; for the docking of hyoscyamine aldehyde into AbHDH, a spatial constraint of maximum 6 Å separation between the aldehyde carbon and the catalytic Zn2+ was additionally imposed65. Enzyme structures and docking results were visualized using PyMOL software (Schrodinger).
Phylogenetic analysis of HDH orthologs
Phylogenetic tree construction was based on a BLASTp search using AbHDH as a query against the UniProt/SwissProt database (annotated sequences only). Sequences chosen for tree construction included the top 50 BLASTp hits based on E-value, as well as 10 additional hits selected from among the next 100 ranks. Phylogenetic relationships were derived via bootstrap neighbor-joining with n = 1000 trials in ClustalX2 and the resulting tree was visualized with FigTree software.
Expression of littorine synthase HA-tagged variants in tobacco
Binary vector (pEAQ-HT-based) plasmids were transformed into A. tumefasciens (GV3101) using the freeze-thaw method66. Transformants were grown on LB-agar plates supplemented with 50 μg/mL kanamycin and 30 μg/mL gentamicin at 30 °C for 48 h. Colonies were inoculated into 5 mL liquid cultures of LB with 50 μg/mL kanamycin and 30 μg/mL gentamicin and grown for 18–24 hours at 30 °C and 250 rpm in a shaking incubator. Saturated cultures were pelleted by centrifugation at 5000 × g for 5 min. Pellets were resuspended in the same volume (~5 mL) of freshly prepared infiltration buffer (10 mM MES buffer, pH 5.6, 10 mM MgCl2, 150 μM acetosyringone), incubated at room temperature for 2–3 hours with gentle rocking to prevent settling, and then diluted in infiltration buffer to OD600 ~ 0.8–1.0. N. benthamiana plants were grown for 4 weeks under a 16 h light/8 h dark cycle prior to infiltration. Approximately 1–2 mL of Agrobacterium cell suspension was infiltrated into the underside of each of the three largest leaves of each plant using a needleless 1 mL syringe. Leaves were harvested four days post-infiltration, flash-frozen in liquid nitrogen, and stored at -80 °C for downstream processing. All infiltrations were performed in triplicate, where one biological replicate comprised three infiltrated leaves from a single plant.
Deglycosylation of yeast- and tobacco-expressed littorine synthase
Removal of N- and O-linked glycosylation from littorine synthase in yeast and N. benthamiana crude cell lysate was performed using PNGase F and O-glycosidase (New England Biolabs), respectively, following the manufacturer’s protocols. Briefly, ~30 μg of total protein containing littorine synthase in crude cell lysate was denatured in 1X glycoprotein denaturing buffer at 100 °C for 10 min, followed by immediate chilling on ice. Denatured lysates were deglycosylated using PNGase F or O-glycosidase as per manufacturer instructions at 37 °C for 1 h, then stored at -20 °C until analysis.
Analysis of protein expression by Western blot
For immunoblot analysis of yeast-expressed proteins, strain CSY1294 was transformed with HA-tagged AbLS expression vectors as described in the Yeast transformations section. Three days post-transformation, transformed colonies were inoculated into 2 mL YNB-DO media and grown overnight (~16–20 h) to stationary phase at 30 °C and 460 rpm. Cells were pelleted by centrifugation at 3,000 × g for 5 min, resuspended in 200 μL H2O, mixed with 200 μL of 0.2 M NaOH, and incubated at room temperature for 5 min to allow hydrolysis of cell wall glycoproteins67. Cells were re-pelleted at 3,000 × g for 5 min, resuspended in 75 μL H2O, mixed with 25 μL of 4X NuPAGE LDS sample buffer (Thermo Fisher), and then boiled at 95 °C for 3 min to lyse cells. Suspensions were pelleted by centrifugation at 16,000 × g for 5 min to remove insoluble debris and supernatants were transferred to pre-chilled tubes. Samples were stored at -20 °C until further analysis.
For analysis of tobacco-expressed proteins, all three infiltrated leaves from a single plant were ground together to a fine powder under liquid nitrogen and resuspended in 4–5 mL of 25 mM potassium phosphate buffer (pH 8.0) with HALT protease inhibitor cocktail (Thermo Fisher). Leaf homogenate slurries (final volume 7–8 mL) were incubated at 4 °C with gentle rotation for 45–60 min and then clarified by centrifugation at 9,000 × g for 10 min. Supernatant fractions were transferred to new tubes and re-clarified. Lysate protein concentrations were estimated using the Bio-RAD Protein Assay kit. Samples were stored at -80 °C until further analysis.
For analysis under reducing conditions, protein lysates were mixed with β-mercaptoethanol (final concentration 10%) and incubated at 70 °C for 10 min. Approximately 20–40 μg of total protein was loaded onto NuPAGE Bis-Tris 4–12% acrylamide gels (Thermo Fisher) with Precision Plus Dual Color protein molecular weight marker (BioRad). Electrophoresis was conducted in 1X NuPAGE MOPS SDS running buffer at 150 V for 90 min. Transfer of protein to a nitrocellulose membranes was performed at 15 V for 15 min using a Trans-Blot Semi-Dry apparatus (BioRad) and NuPAGE transfer buffer (Thermo Fisher) per manufacturer instructions. For reducing conditions, NuPAGE antioxidant (Thermo Fisher) was added to a final concentration of 1X to both the running buffer and transfer buffer. Membranes with transferred protein were washed for 5 min in Tris-buffered saline with Tween (TBS-T; 137 mM NaCl, 2.7 mM KCl, 19 mM Tris base, 0.1% Tween20, pH 7.4) and then blocked with 5% skim milk in TBS-T for 1 h at room temperature. Membranes were incubated overnight at 4 °C with a 1:1500 dilution of chimeric rabbit IgGκ anti-HA HRP-conjugated antibody (Absolute Antibody, 16.43/Ab00828–23.0) in TBS-T with 5% milk, washed three times for 5 min each with TBS-T, and then visualized using Western Pico PLUS HRP substrate (Thermo Fisher) and a G:BOX gel imager (Syngene).
Statistics
Where indicated, the statistical significance of any differences in metabolite titer between conditions was verified using Student’s two-tailed t-test in Microsoft Excel Professional 2013. For yeast experiments, biological replicates are defined as independent cultures inoculated from separate yeast colonies or streaks and cultivated in separate containers. For tobacco experiments, one biological replicate is defined as all infiltrated leaves from a single plant.
Additional software
All figures were prepared using Graphpad Prism 7, ImageJ, PyMOL, and Inkscape.
Extended Data
Extended Data Figure 1. Design of genomic integrations for pathway construction in yeast.
Block arrows represent gene expression cassettes with unique promoter and terminator for each locus. Genus and species sources for heterologous genes are indicated by two letters preceding the gene symbol. Superscript annotations on gene symbols indicate N- or C-terminal modifications; dash (-) indicates fusion protein. See Supplementary Tables 1–2 for gene sources and Supplementary Table 3 for yeast strain genotypes.
Extended Data Figure 2. Substrate specificity and structure-guided active site engineering of UDP-glucosyltransferase 84A27 (UGT84A27) in engineered yeast.
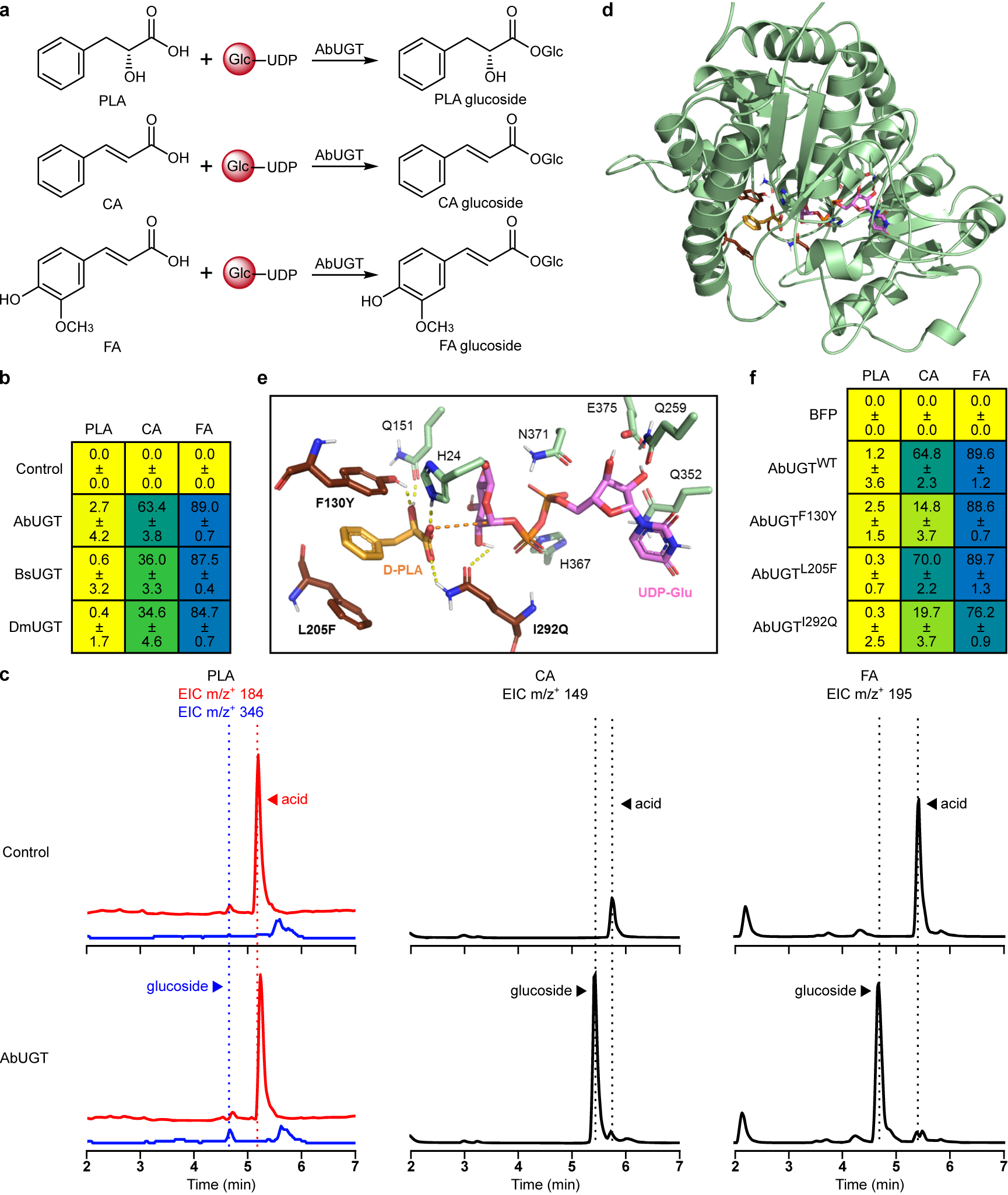
(a) Phenylpropanoids tested as glucose (Glc) acceptors for UGT84A27 in engineered yeast. Top, (D)-3-phenyllactic acid (PLA); middle, trans-cinnamic acid (CA); bottom, trans-ferulic acid (FA). (b) Heatmap of percent conversion of fed phenylpropanoids to glucosides by yeast engineered for UGT84A27 expression. UGT84A27 orthologs or a BFP negative control were expressed from low-copy plasmids in CSY1251. Transformed cells were cultured in selective media supplemented with 500 μM PLA, CA, or FA for 72 h prior to LC-MS/MS analysis of culture supernatant. Data represent the mean of n = 3 biologically independent samples ± standard deviation. (c) Representative LC-MS/MS traces showing conversion of PLA, CA, and FA to cognate glucosides by AbUGT in CSY1251 cultured as in b for 120 h to enable more complete glucosylation. For PLA, acid and glucoside were distinguished by different NH4+ adduct parent masses as well as different retention times. For CA and FA, rapid fragmentation necessitated detection of the glucosides based on the lower-retention peaks produced by their phenylpropanoid fragments. (d) Homology model of AbUGT84A27 constructed based on the crystal structure of Arabidopsis thaliana salicylate UDP-glucosyltransferase UGT74F2 with bound UDP (PDB: 5V2K). PLA (orange) is shown in the preferred binding pose with UDP-glucose (pink) based on docking simulations. (e) Zoomed view of AbUGT active site with docked D-PLA and UDP-glucose. Potential mutations identified to improve PLA selectivity (F130Y, L205F, I292Q) are shown; dashed lines indicate putative polar/hydrogen bond interactions. (f) Heatmap of percent conversion of fed phenylpropanoids to glucosides by yeast engineered for expression of AbUGT mutants. AbUGT wild-type, active site mutants, or a BFP negative control were expressed from low-copy plasmids in CSY1251. Transformed cells were cultured in selective media supplemented with 500 μM PLA, CA, or FA for 72 h prior to LC-MS/MS analysis of culture supernatant. Data represent the mean of n = 3 biologically independent samples ± standard deviation.
Extended Data Figure 3. Screening H6H orthologs from TA-producing Solanaceae in yeast.
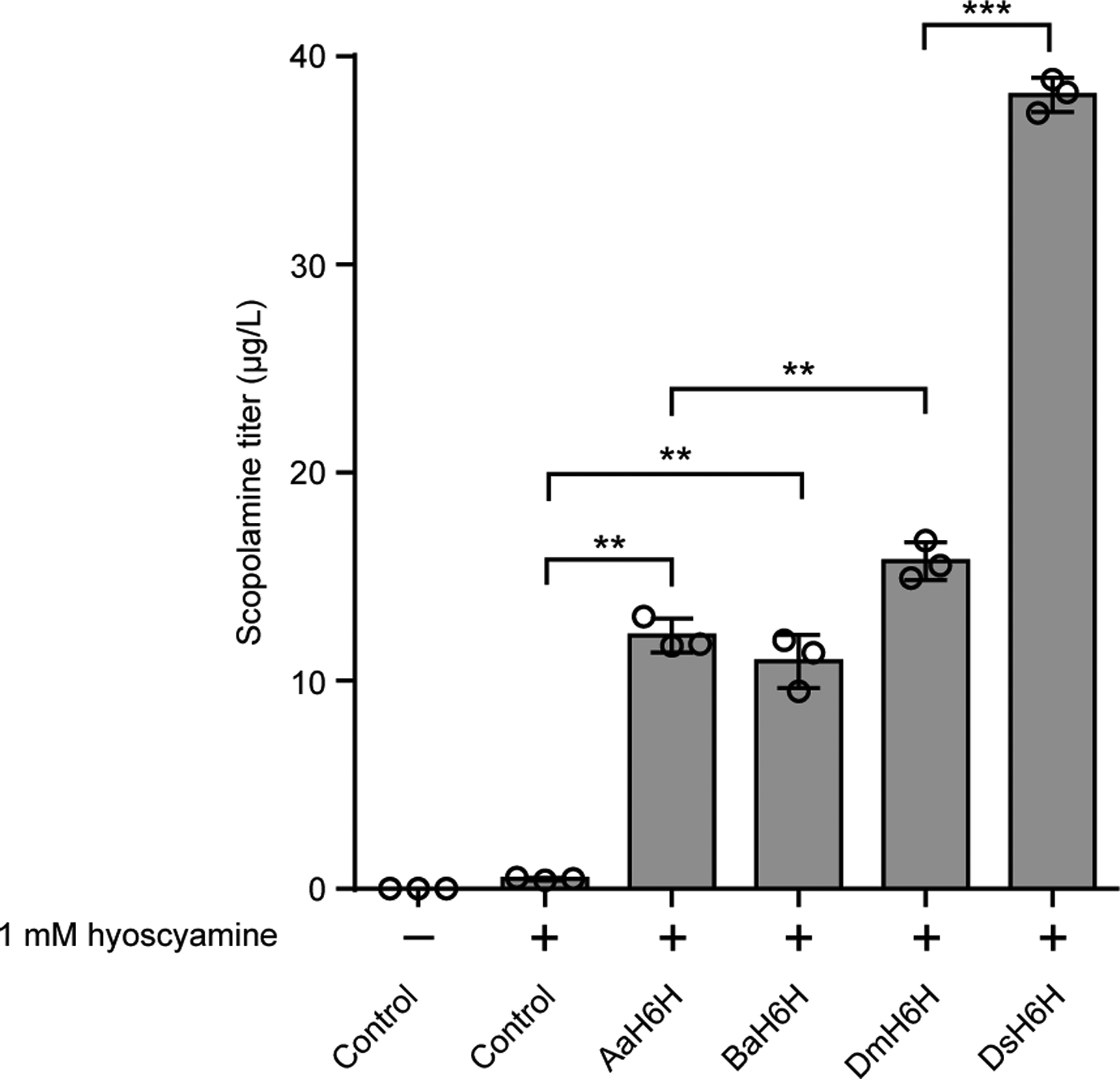
H6H orthologs from Anisodus acutangulus (AaH6H), Brugmansia arborea (BaH6H), Datura metel (DmH6H), Datura stramonium (DsH6H), or a negative control (BFP) were expressed from low-copy plasmids in CSY1251. Transformed cells were cultured in selective media supplemented with 1 mM hyoscyamine for 72 h prior to LC-MS/MS analysis of culture supernatant. Data represent the mean of n = 3 biologically independent samples (open circles) and error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Exact P-values: AaH6H vs. control, 0.00146; BaH6H vs. control, 0.00486; DmH6H vs. AaH6H, 0.00739; DsH6H vs. DmH6H, 6.67 × 10−6.
Extended Data Figure 4. Coexpression analysis, active site mutagenesis, and ortholog identification for AbHDH.
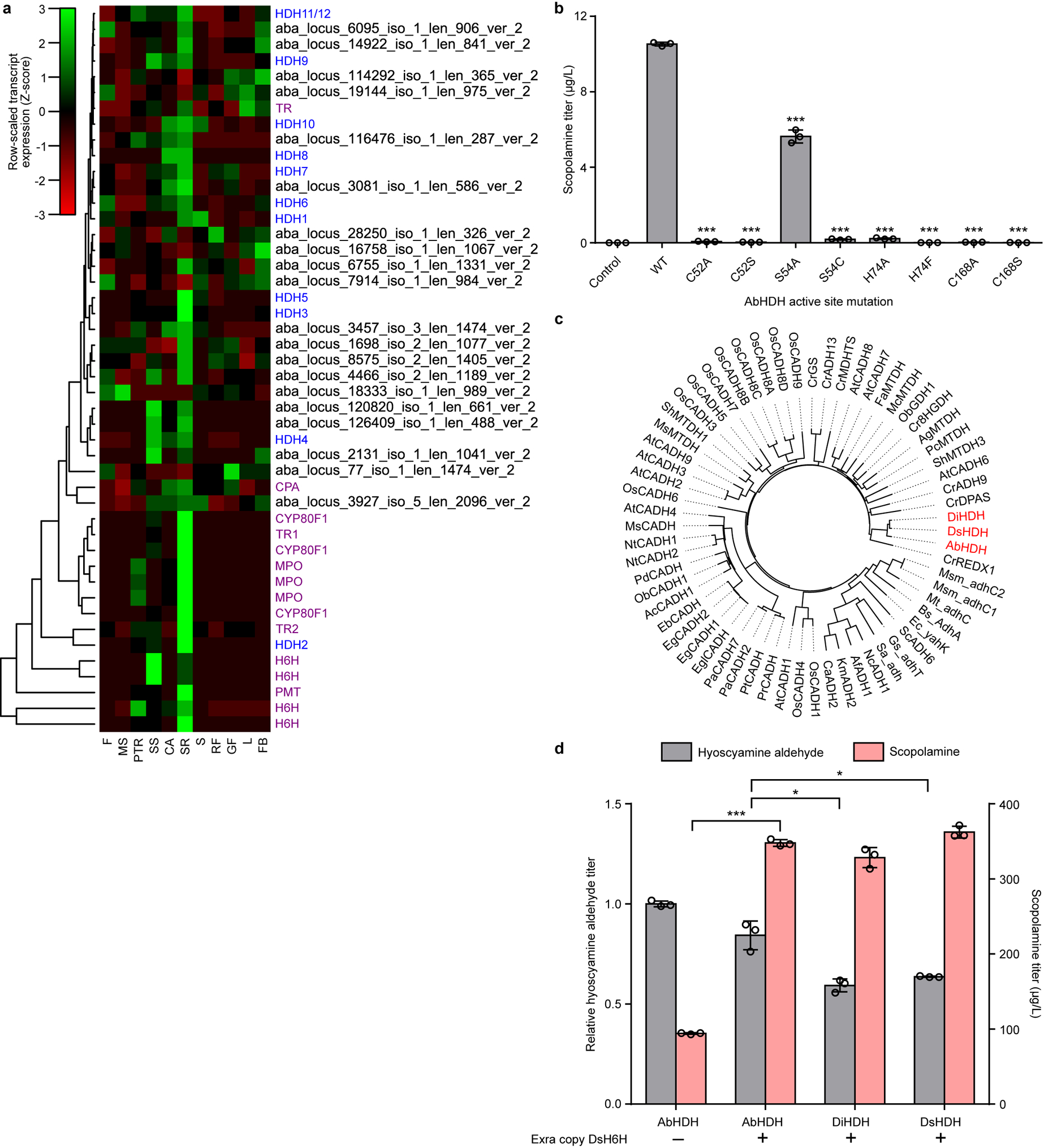
(a) Heatmap of tissue-specific expression profiles for hyoscyamine dehydrogenase (HDH) gene candidates identified from the A. belladonna transcriptome. Transcript expression is scaled by row using a normal distribution. Dendrogram indicates hierarchical clustering of candidates by tissue-specific expression profile. Color scheme for gene IDs: purple, known TA pathway genes; blue, putative HDH candidates with complete open reading frame sequences; black, putative HDH candidates with incomplete open reading frame sequences. Gene abbreviations (vertical axis): CPA, N-carbamoylputrescine amidase. Tissue abbreviations (horizontal axis): F, flower; MS, mature seed; PTR, primary tap root; SS, sterile seedling; CA, callus; SR, secondary root; S, stem; RF, ripe fruit; GF, green fruit; L, leaf; FB, flower bud. (b) Wild-type (WT) AbHDH, active site mutants, or a negative control (BFP) were expressed from low-copy plasmids in CSY1292. Transformed strains were cultured in selective media with 1 mM littorine at 25 °C for 72 h prior to quantification of scopolamine production by LC-MS/MS analysis of culture supernatant. Data indicate the mean of n = 3 biologically independent samples (open circles) and error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001; statistical significance is shown relative to wild-type. Exact P-values: C52A, 1.68 × 10−5; C52S, 2.06 × 10−5; S54A, 8.19 × 10−4; S54C, 5.27 × 10−6; H74A, 3.40 × 10−6; H74F, 2.78 × 10−5; C168A, 1.75 × 10−5; C168S, 2.39 × 10−5. (c) Phylogenetic tree of the three identified HDH orthologs (AbHDH, DiHDH, DsHDH) together with closest protein hits in the UniProt/SwissProt database. Phylogenetic relationships were derived via bootstrap neighbor-joining with n = 1000 trials in ClustalX2 and the resulting tree was visualized with FigTree software. Abbreviations: ADH, alcohol dehydrogenase; CADH, cinnamyl alcohol dehydrogenase; MTDH, mannitol dehydrogenase; DPAS, dehydroprecondylocarpine acetate synthase; 8HGDH, 8-hydroxygeraniol dehydrogenase; GDH, geraniol dehydrogenase; GS, geissoschizine synthase; REDX, unspecified redox protein. (d) HDH orthologs (AbHDH, DiHDH, DsHDH) were co-expressed with either a BFP negative control (‘–’) or an additional copy of DsH6H (‘+’) from low-copy plasmids in CSY1292. Transformed cells were cultured in selective media supplemented with 1 mM littorine for 72 h prior to LC-MS/MS analysis of culture supernatant. Data represent the mean of n = 3 biologically independent samples (open circles) and error bars show standard deviation. Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Exact P-values: AbHDH + DsH6H vs. AbHDH only, scopolamine, 4.68 × 10−5; DiHDH vs. AbHDH, hyoscyamine aldehyde, 0.0141; DsHDH vs. AbHDH, hyoscyamine aldehyde, 0.0372.
Extended Data Figure 5. Phylogenetic analysis of hyoscyamine dehydrogenase (HDH).
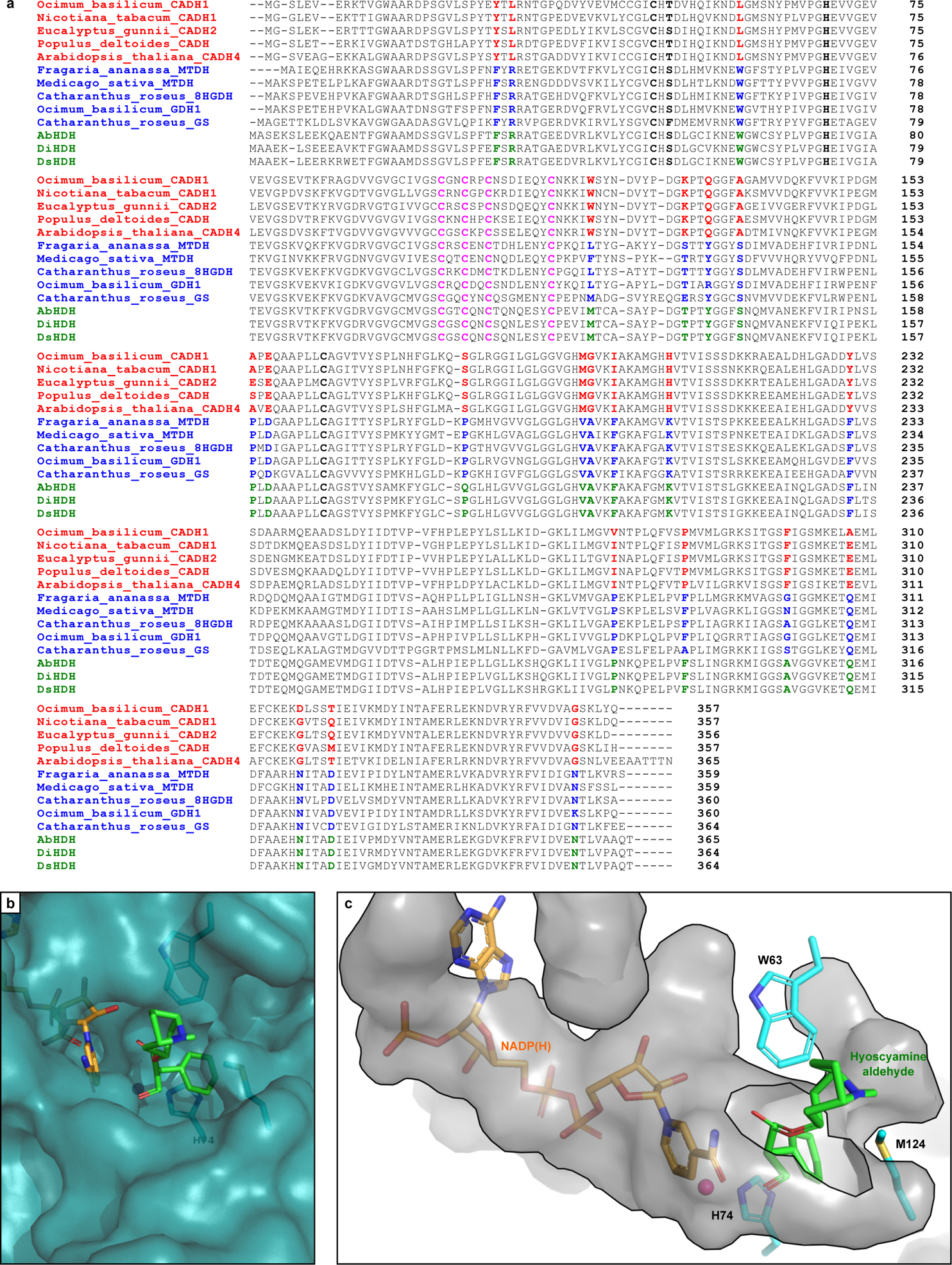
(a) Sequence alignment of AbHDH, DiHDH, and DsHDH (green) with selected BLASTp hits from the cinnamyl alcohol dehydrogenase (CAD)-like (red) and sinapyl alcohol dehydrogenase (SAD)-like (blue) families. Catalytic Zn2+ residues are highlighted in bold (black); structural Zn2+-binding residues are highlighted in pink; and key conserved residues which differentiate CAD- and SAD-like ADHs are highlighted in red/blue/green. (b) Zoomed surface view of AbHDH substrate binding pocket with bound NADPH (orange) and hyoscyamine aldehyde (green) based on docking simulations. (c) Zoomed inverse surface (cavity) view of AbHDH active site pocket with bound NADPH (orange) and hyoscyamine aldehyde (green) based on docking simulations as in b. Residues W63, H74, and M124 form a tight binding pocket for the aryl moiety of hyoscyamine aldehyde, leaving the tropine moiety to extend out of the active site.
Extended Data Figure 6. Analysis of AbLS localization, N-glycosylation, and proteolytic processing patterns in yeast and tobacco.
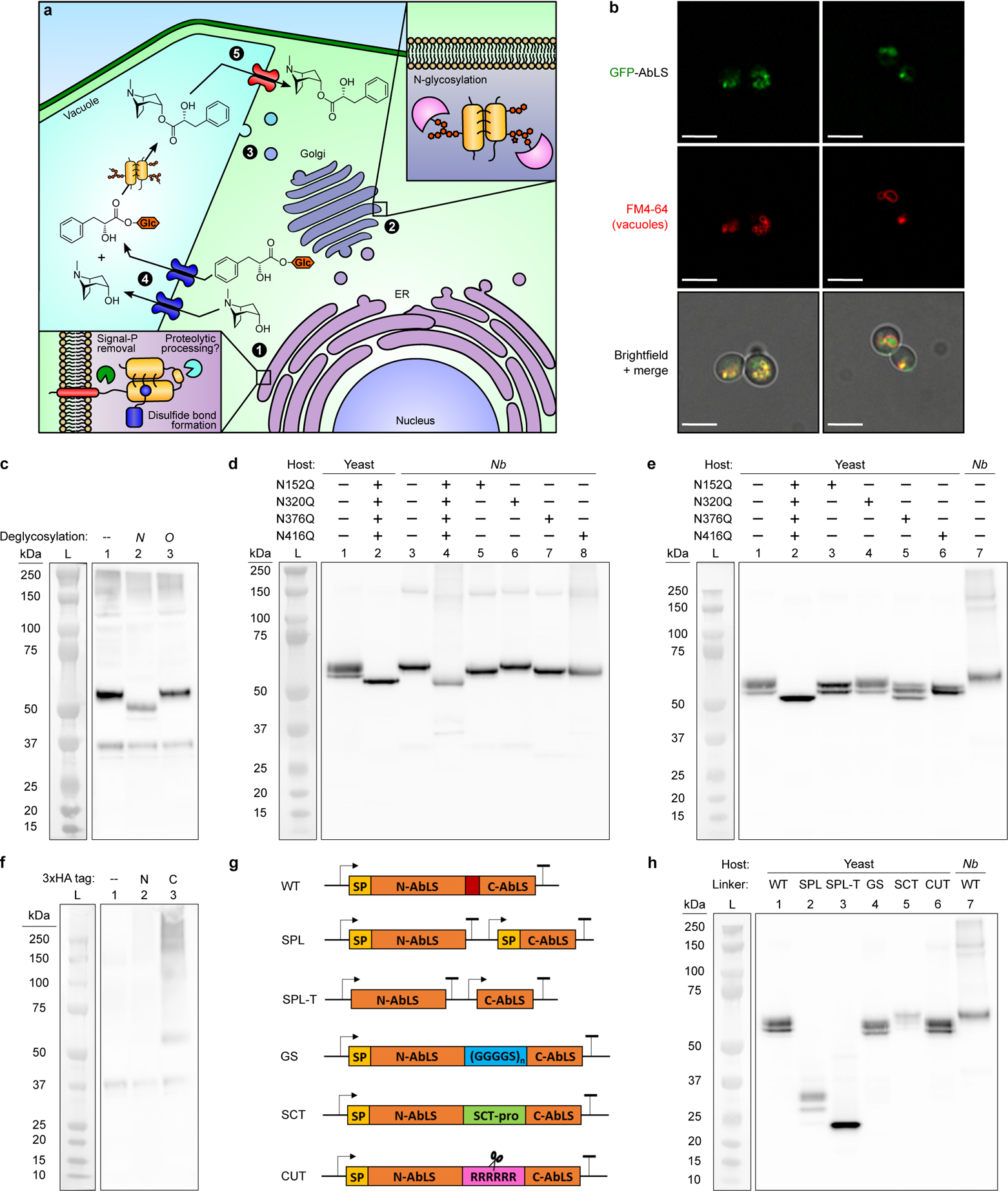
(a) Illustration of the canonical plant ER-to-vacuole trafficking and maturation pathway for SCPL acyltransferases (SCPL-ATs), with A. belladonna littorine synthase (AbLS) shown as example. Circled numbers indicate major steps in SCPL-AT expression and activity, including maturation in the (1) ER lumen and (2) Golgi, (3) trafficking to the vacuole, and vacuolar (4) substrate import and (5) product export. (b) Additional fields of view (see Fig. 3a) of yeast epifluorescence microscopy showing N-terminal GFP-tagged AbLS (GFP-AbLS), the vacuolar membrane stain FM4–64, and brightfield merged images. Microscopy was performed on CSY1294 expressing GFP-AbLS from a low-copy plasmid. 2D deconvolution analysis was performed as noted in Online Methods. Scale bar, 5 μm. Images are representative of two independent experiments. (c) Western blot of wild-type AbLS expressed in tobacco and treated with deglycosylases. C-terminal HA-tagged AbLS was transiently expressed in N. benthamiana leaves via agroinfiltration. Crude leaf extracts were either untreated (lane 1: ‘--’), or treated with peptide N-glycosidase F (PNGase F; lane 2: ‘N’) or O-glycosidase (lane 3: ‘O’) to remove N- or O-linked glycosylation, respectively. Lane ‘L’, Bio-Rad Precision Plus Dual Color protein ladder. (d,e) Western blot of AbLS glycosylation site mutants expressed in yeast and tobacco. C-terminal HA-tagged wild-type AbLS, single glycosylation site point mutants (N → Q), or a quadruple mutant were expressed transiently via agroinfiltration in N. benthamiana (‘Nb’) (d) or from low-copy plasmids in CSY1294 (‘Yeast’) (e). For d and e, corresponding yeast- and tobacco-expressed controls are included for comparison. Lane ‘L’, Bio-Rad Precision Plus Dual Color protein ladder. (f) Western blot of untagged and HA-tagged wild-type AbLS expressed in tobacco. Untagged (lane 1: ‘--’), N-terminal HA-tagged (lane 2: ‘N’), or C-terminal HA-tagged (lane 3: ‘C’) AbLS was transiently expressed in N. benthamiana leaves via agroinfiltration. Lane ‘L’, Bio-Rad Precision Plus Dual Color protein ladder. (g,h) Analysis of proteolytic cleavage patterns for AbLS split controls and putative propeptide-swapped variants (g) in yeast via Western blot (h). C-terminal HA-tagged AbLS variants were expressed from low-copy plasmids in CSY1294 (lanes 1–6); HA-tagged wild-type AbLS expressed in Nicotiana benthamiana (Nb) is shown as an additional control (lane 7). Lane symbols: L, protein molecular weight ladder; WT, wild-type AbLS; SPL, AbLS split at putative propeptide with signal peptides on both fragments; SPL-T, AbLS split at putative propeptide without signal peptides on either fragment; GS, AbLS variant with wild-type propeptide swapped for flexible Gly-Ser linker; SCT, AbLS variant with wild-type propeptide swapped for AtSCT propeptide sequence; CUT, AbLS variant with wild-type propeptide swapped for synthetic poly-arginine site recognized and cleaved by Kex2p protease. For blots in c-h, sample preparation, electrophoresis, and protein transfer steps were performed under denaturing and disulfide-reducing (c, d, e, h) or non-reducing (f) conditions; AbLS detection was performed using a chimeric rabbit IgGκ anti-HA HRP-conjugated antibody (see Online Methods). Blots are representative of three (c) or two (d, e, f, h) independent experiments. For gel source data, see Supplementary Figure 1.
Extended Data Figure 7. Analysis of putative endoproteolytic propeptide removal in AbLS.
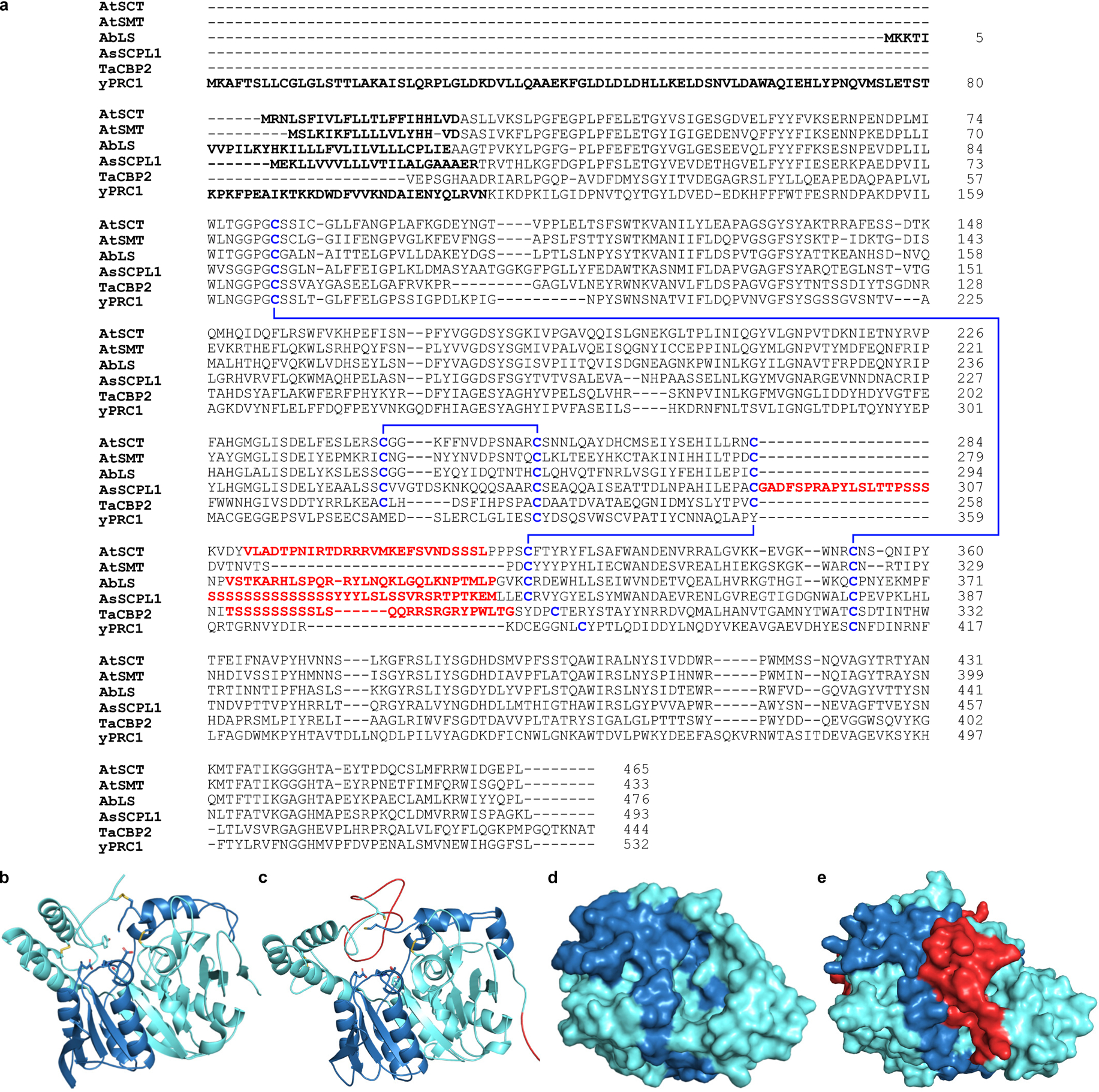
(a) Sequence alignment of AbLS with characterized serine carboxypeptidases and SCPL acyltransferases known to possess (AtSCT, AsSCPL1, TaCBP2) or lack (AtSMT, yPRC1) proteolytically-removed internal propeptide linkers (red). Putative N-terminal signal peptides are indicated in bold; disulfide bonds are indicated in blue. AtSCT, Arabidopsis thaliana sinapoylglucose: choline sinapoyltransferase; AtSMT, A. thaliana sinapoylglucose:malate sinapoyltransferase; AbLS, Atropa belladonna littorine synthase; AsSCPL1, Avena strigosa avenacin synthase; TaCBP2, Triticum aestivum carboxypeptidase 2; yPRC1, yeast carboxypeptidase Y. (b,d) Crystal structure of TaCBP2 (PDB: 1WHT) in (b) cartoon and (d) surface representation showing disulfide bonds (yellow) and internal propeptide removal sites. (c,e) Homology model of AbLS based on the crystal structure of TaCBP2 in (c) cartoon and (e) surface representation showing N-terminal signal peptide (red, bottom right in c), disulfide bonds (yellow), and putative internal propeptide (red, top/middle) which appears to block active site access. Note that surface views in d and e are rotated 90° toward the viewer.
Extended Data Figure 8. Fluorescence microscopy of tobacco alkaloid transporters expressed in CSY1296 for alleviation of vacuolar TA transport limitations.
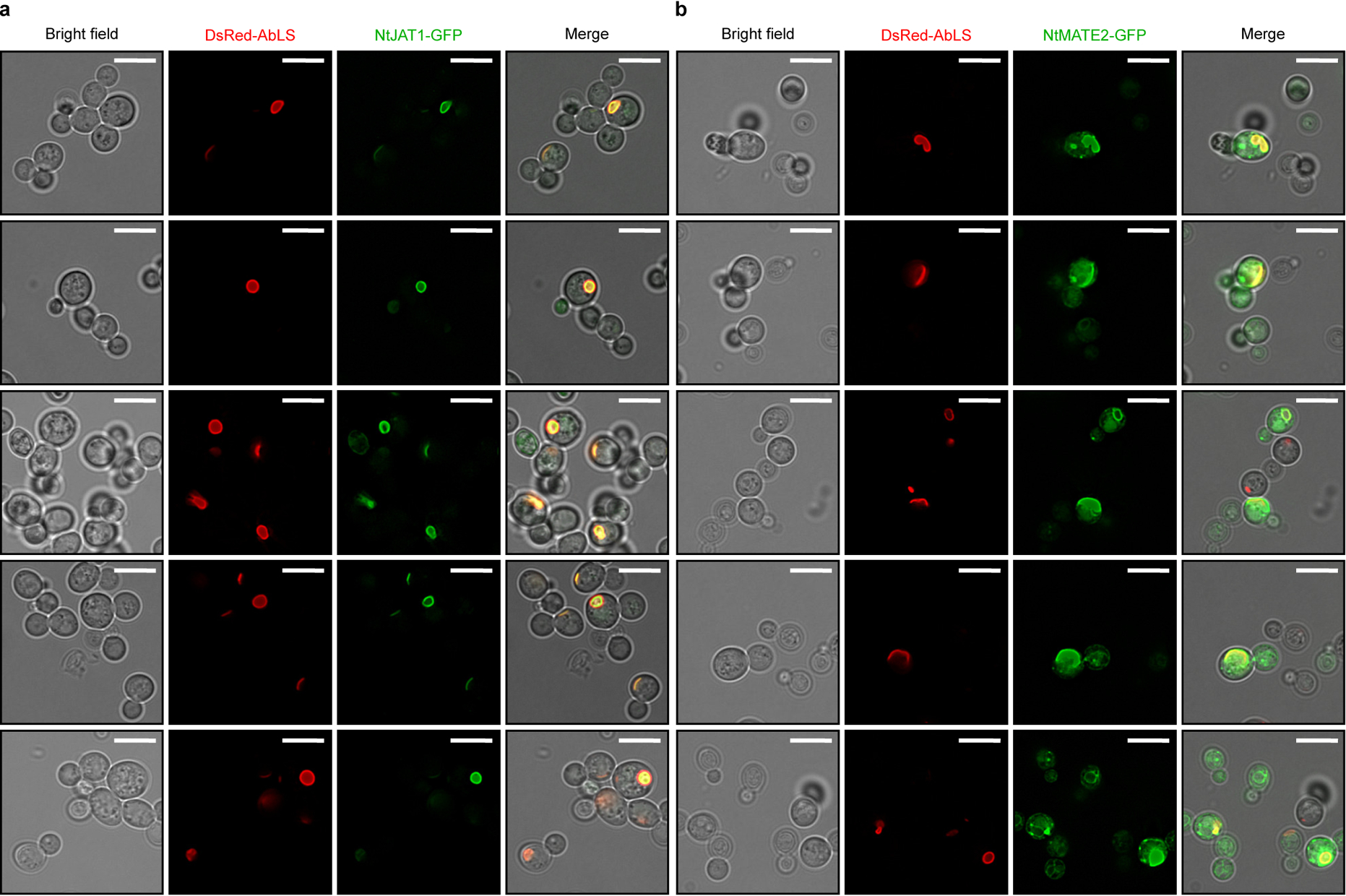
C-terminal GFP fusions of (a) NtJAT1 and (b) NtMATE2 were expressed from low-copy plasmids in CSY1296. Strains were cultured, imaged, and analyzed via widefield epifluorescence microscopy and 2D deconvolution analysis as described in Online Methods. Images are representative of two independent experiments. Scale bar, 5 μm.
Extended Data Figure 9. Effect of extra gene copies on accumulation of TA pathway intermediates and products in scopolamine-producing strain CSY1296.
An additional copy of each biosynthetic enzyme between tropine and scopolamine was expressed from the following low-copy plasmids in strain CSY1296: (a) WfPPR, pCS4436; (b) AbUGT, pCS4440; (c) DsRed-AbLS, pCS4526; (d) AbCYP80F1, pCS4438; (e) DsHDH, pCS4478; (f) DsH6H, pCS4439; or a BFP control (pCS4208, pCS4212, or pCS4213). Transformed strains were cultured in appropriate selective media at 25 °C for 96 h prior to quantification of metabolites in the growth medium by LC-MS/MS analysis of culture supernatant. Note that no littorine accumulation was observed for any samples. Data indicate the mean of n = 3 biologically independent samples (open circles) and error bars show standard deviation. Metabolite titers are shown relative to the BFP control with the same auxotrophic marker as the biosynthetic gene: WfPPR (a), DsRed-AbLS (c), AbCYP80F1 (d), and DsH6H (f) relative to the pCS4213 control (LEU2); AbUGT (b) relative to the pCS4208 control (URA3); and DsHDH (e) relative to the pCS4212 control (TRP1). Student’s two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001. Statistical significance is shown relative to corresponding controls. Exact P-values: (a) PLA, 0.00950; PLA glucoside, 3.36 × 10−4; hyoscyamine, 0.00221; (b) tropine, 0.00500; PLA glucoside, 3.59 × 10−4; hyoscyamine, 0.0192; scopolamine, 6.90 × 10−4; (c) PLA glucoside, 0.00544; hyoscyamine, 0.0165; scopolamine, 3.43 × 10−4; (d) PLA, 0.0487; hyoscyamine, 0.0154; scopolamine, 0.0159; (f) PLA, 0.0153; PLA glucoside, 0.0453; hyoscyamine, 0.00958; scopolamine, 0.00816.
Extended Data Figure 10. Time courses of de novo TA and precursor production in pseudo-fed-batch cultures of CSY1297 and CSY1298.
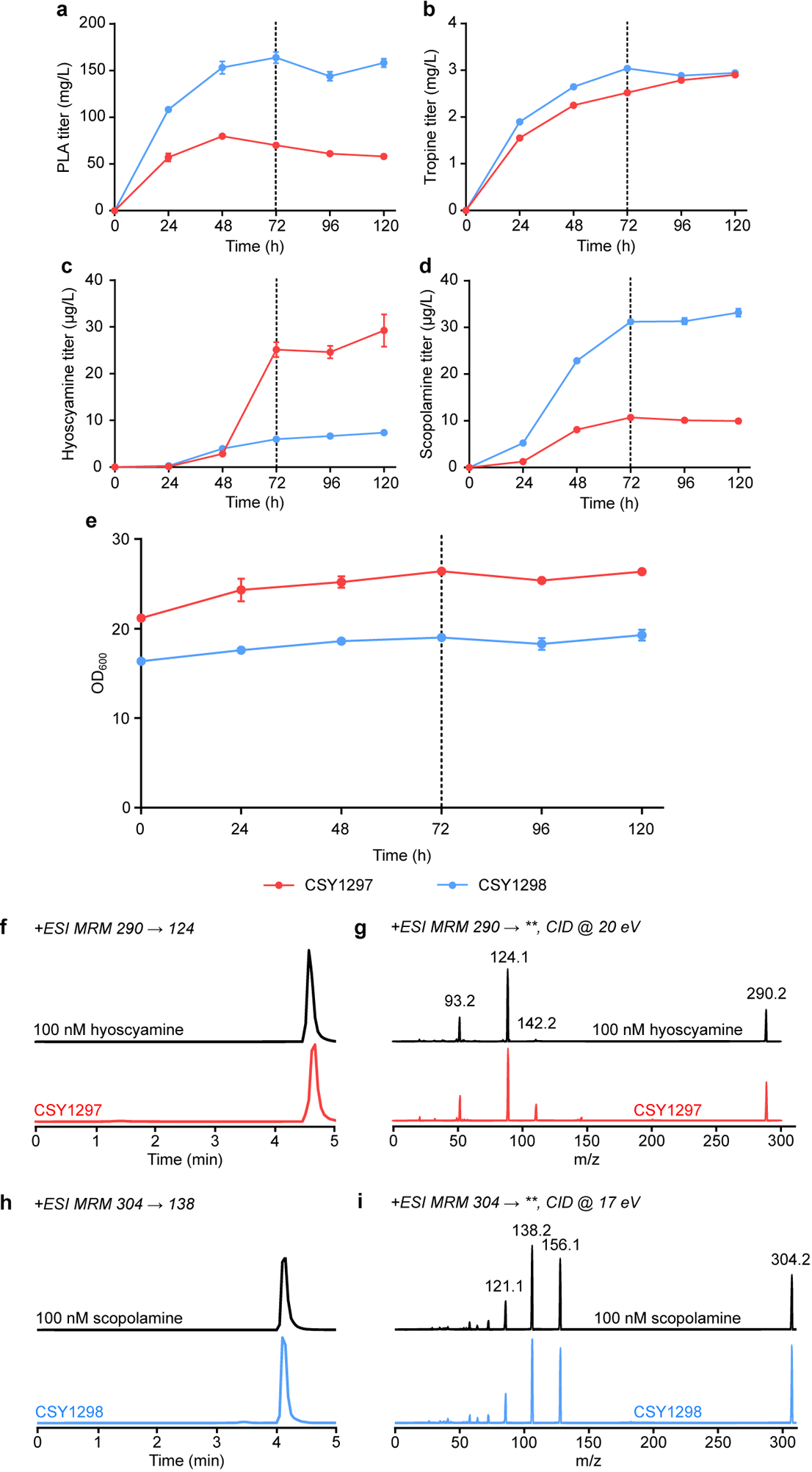
(a-e) TA-producing strains CSY1297 and CSY1298 (with pCS4213) were respectively cultured in non-selective or selective (leucine dropout) media with 50 mM 2-OG and 15 mg/L Fe2+ under pseudo-fed-batch conditions at 25 °C for 120 h. Culture supernatants were sampled for (a) PLA, (b) tropine, (c) hyoscyamine, and (d) scopolamine titers by LC-MS/MS analysis and (e) optical density at 600 nm (OD600) every 24 h. Cultures were supplemented with additional dextrose, glycerol, and amino acids to final concentrations of 2%, 2%, and 1× at 72 h (vertical dotted line). Data indicate the mean of n = 3 biologically independent samples and error bars show standard deviation. Note that no littorine accumulation was observed in any samples. (f-i) LC-MS/MS verification of de novo medicinal TA production in CSY1297 and CSY1298. Panels show LC-MS/MS chromatograms of (f,g) hyoscyamine and (h,i) scopolamine detected in CSY1297 and CSY1298 cultures grown in non-selective media or selective media, respectively (samples from experiment in c and d, 120 h), and of authentic 100 nM chemical standards. Primary MRM transitions used for detection and quantification are shown in f and h; additional characteristic transitions are shown in g and i (‘**’ indicates any detected fragment mass in product ion scan). Traces are representative of n = 3 biologically independent samples.
Supplementary Material
Acknowledgements
We thank A. Cravens for the yeast CRISPRm Cas9/sgRNA plasmids (pCS3410, 3411, 3414, and 3700-3703); T. Lozanoski (Jarosz laboratory) for a plasmid harboring yeast codon-optimized mVenus; J. Payne for assistance with the generation of DFT energy-optimized ligand structures for docking studies; P. Dykstra for assistance with fluorescence microscopy and the Stanford Cell Sciences Imaging Facility for access to microscopy equipment and training; R. Nett for suggestions regarding de novo transcriptome assembly using Trinity; W. Cody and the Sattely laboratory for providing N. benthamiana plants, Agrobacterium strains, a pEAQ-HT binary vector backbone plasmid, and suggestions for tobacco agroinfiltration experiments; D. Endy and J. Payne for discussions and feedback in the preparation of this manuscript. This work was supported by the National Institutes of Health and the Natural Sciences and Engineering Research Council of Canada (doctoral postgraduate scholarship to P.S.).
Footnotes
Competing interests
The authors declare the following competing interests: P.S. and C.D.S. are inventors on a pending patent application; C.D.S. is a founder and CEO of Antheia, Inc.
Additional information
This article includes the following supplementary materials:
Data and code availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request. This article includes raw source data files associated with Figs. 1–4 and Extended Data Figs. 3, 4, 6 (Supplementary Figure 1), 9, and 10. Novel genetic sequences identified and characterized in this study are available from the following public databases. 1000Plants (1KP) database63: scaffold-AIOU-2012986-Brugmansia_sanguinea (BsUGT); scaffold-JNVS-2051323-Datura_metel (DmUGT). Medicinal Plant RNAseq database32: medp_datin_20101112|6354 (DiHDH); medp_datst_20101112|10433 (DsHDH). MSU Medicinal Plant Genomics Resource30: full amino acid sequences and database accession numbers (IDs) for all tested HDH candidates are provided in Supplementary Table 1. Accession numbers for previously reported gene and protein sequences in the GenBank/UniProt databases are provided in Supplementary Table 2. Protein crystal structures used for homology modeling are available from the RCSB protein data bank: Arabidopsis thaliana salicylate UDP-glucosyltransferase UGT74F2 with bound UDP, PDB: 5V2K; Populus tremuloides sinapyl alcohol dehydrogenase with bound NADPH, PDB: 1YQD.
The custom R script used for identification of HDH candidates via coexpression analysis of A. belladonna RNA sequencing data is available from the Smolke Laboratory GitHub: github.com/smolkelab/Oxidoreductase_coexpression_analysis.
References
- 1.World Health Organization. WHO Model List of Essential Medicines - 19th List (April 2015). (2015). doi: 10.1016/S1473-3099(14)70780-7 [DOI] [Google Scholar]
- 2.Grynkiewicz G & Gadzikowska M Tropane alkaloids as medicinally useful natural products and their synthetic derivatives as new drugs. Pharmacol. Reports 60, 439–463 (2008). [PubMed] [Google Scholar]
- 3.United States Food and Drug Administration. FDA Drug Shortages: Atropine sulfate injection. FDA Drug Shortages Database (2020). at <https://www.accessdata.fda.gov/scripts/drugshortages/dsp_ActiveIngredientDetails.cfm?AI=AtropineSulfateInjection&st=c>
- 4.United States Food and Drug Administration. FDA Drug Shortages: Scopolamine transdermal system. FDA Drug Shortages Database (2020). at <https://www.accessdata.fda.gov/scripts/drugshortages/dsp_ActiveIngredientDetails.cfm?AI=ScopolamineTransdermalSystem&st=r>
- 5.The Climate Council of Australia. ‘This is Not Normal’: Climate change and escalating bushfire risk. (2019).
- 6.Hahn SM FDA Statement: Coronavirus (COVID-19) Supply Chain Update. (2020). at <https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-supply-chain-update>
- 7.United States Food and Drug Administration. Coronavirus (COVID-19) Update: FDA takes further steps to help mitigate supply interruptions of food and medical products. US FDA Press Annoucements; (2020). at <https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-takes-further-steps-help-mitigate-supply-interruptions-food-and> [Google Scholar]
- 8.Agrawal G, Ahlawat H & Dewhurst M Winning against COVID-19: The implications for biopharma. (2020). at <https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/winning-against-covid-19-the-implications-for-biopharma?from=groupmessage&isappinstalled=0#>
- 9.Kohnen KL, Sezgin S, Spiteller M, Hagels H & Kayser O Localization and Organization of Scopolamine Biosynthesis in Duboisia myoporoides R. Br. Plant Cell Physiol. 59, 107–118 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ullrich SF, Hagels H & Kayser O Scopolamine: a journey from the field to clinics. Phytochemistry Reviews 16, 333–353 (2017). [Google Scholar]
- 11.Kohnen-Johannsen KL & Kayser O Tropane alkaloids: Chemistry, pharmacology, biosynthesis and production. Molecules 24, 1–23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.American Society of Anesthesiologists (ASA). ASA Urges Federal Government to Take Action on Drug Shortages. (2020). at <https://www.asahq.org/advocacy-and-asapac/fda-and-washington-alerts/washington-alerts/2020/04/asa-urges-federal-government-to-take-action-on-drug-shortages>
- 13.Bedewitz MA, Jones AD, D’Auria JC & Barry CS Tropinone synthesis via an atypical polyketide synthase and P450-mediated cyclization. Nat. Commun 9, 5281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srinivasan P & Smolke CD Engineering a microbial biosynthesis platform for de novo production of tropane alkaloids. Nat. Commun 10, 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ping Y et al. De Novo Production of the Plant-Derived Tropine and Pseudotropine in Yeast. ACS Synth. Biol 8, 1257–1262 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Qiu F et al. Functional genomics analysis reveals two novel genes required for littorine biosynthesis. New Phytol. nph.16317 (2019). doi: 10.1111/nph.16317 [DOI] [PubMed] [Google Scholar]
- 17.Li R et al. Functional Genomic Analysis of Alkaloid Biosynthesis in Hyoscyamus niger Reveals a Cytochrome P450 Involved in Littorine Rearrangement. Chem. Biol 13, 513–520 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Nasomjai P et al. Mechanistic insights into the cytochrome P450-mediated oxidation and rearrangement of littorine in tropane alkaloid biosynthesis. ChemBioChem 10, 2382–2393 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Matsuda J, Okabe S, Hashimoto T & Yamada Y Molecular cloning of hyoscyamine 6β-hydroxylase, a 2-oxoglutarate-dependent dioxygenase, from cultured roots of Hyoscyamus niger. J. Biol. Chem 266, 9460–9464 (1991). [PubMed] [Google Scholar]
- 20.Hashimoto T, Matsuda J & Yamada Y Two-step epoxidation of hyoscyamine to scopolamine is catalyzed by bifunctional hyoscyamine 6β-hydroxylase. FEBS Lett. 329, 35–39 (1993). [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Jiang M, Guo X, Liu Z & He X Reconstruction of metabolic module with improved promoter strength increases the productivity of 2-phenylethanol in Saccharomyces cerevisiae. Microb. Cell Fact 17, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Zhang S, Shi Y, Shen F & Wang H A new high phenyl lactic acid-yielding Lactobacillus plantarum IMAU10124 and a comparative analysis of lactate dehydrogenase gene. FEMS Microbiol. Lett 356, 89–96 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Sévin DC, Fuhrer T, Zamboni N & Sauer U Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods 14, 187–194 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Qiu F et al. A Phenylpyruvic Acid Reductase Is Required for Biosynthesis of Tropane Alkaloids. Org. Lett acs.orglett.8b03236 (2018). doi: 10.1021/acs.orglett.8b03236 [DOI] [PubMed] [Google Scholar]
- 25.Fujii T et al. Novel fungal phenylpyruvate reductase belongs to d-isomer-specific 2-hydroxyacid dehydrogenase family. Biochim. Biophys. Acta - Proteins Proteomics 1814, 1669–1676 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Zheng Z et al. Efficient conversion of phenylpyruvic acid to phenyllactic acid by using whole cells of bacillus coagulans SDM. PLoS One 6, 23–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li JF et al. Directed modification of L-LcLDH1, an L-lactate dehydrogenase from Lactobacillus casei, to improve its specific activity and catalytic efficiency towards phenylpyruvic acid. J. Biotechnol 281, 193–198 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Ross J, Li Y, Lim E-K & Bowles DJ Higher plant glycosyltransferases. Genome Biol. 2, 3004.1–3004.6 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H et al. Engineering Saccharomyces cerevisiae with the deletion of endogenous glucosidases for the production of flavonoid glucosides. Microb. Cell Fact 15, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michigan State University. Medicinal Plant Genomics Resource. at <http://medicinalplantgenomics.msu.edu>
- 31.Grabherr MG et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 29, 644–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.NIH. Medicinal Plant RNA Seq Database. at <https://medplantrnaseq.org>
- 33.Bontpart T, Cheynier V, Ageorges A & Terrier N BAHD or SCPL acyltransferase? What a dilemma for acylation in the world of plant phenolic compounds. New Phytol. 208, 695–707 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Carqueijeiro I et al. in Plant Vacuolar Trafficking: Methods and Protocols 1789, 33–54 (2018). [Google Scholar]
- 35.Strasser R Plant protein glycosylation. Glycobiology 26, 926–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stehle F, Stubbs MT, Strack D & Milkowski C Heterologous expression of a serine carboxypeptidase-like acyltransferase and characterization of the kinetic mechanism. FEBS J. 275, 775–787 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Stack JH Receptor-Mediated Protein Sorting to the Vacuole in Yeast: Roles for Protein Kinase, Lipid Kinase and GTP-Binding Proteins. Annu. Rev. Cell Dev. Biol 11, 1–33 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Roberts CJ, Nothwehr SF & Stevens TH Membrane protein sorting in the yeast secretory pathway: Evidence that the vacuole may be the default compartment. J. Cell Biol 119, 69–83 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu L, Spurrier J, Butt TR & Strickler JE Enhanced protein expression in the baculovirus/insect cell system using engineered SUMO fusions. Protein Expr. Purif 62, 21–28 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morita M et al. Vacuolar transport of nicotine is mediated by a multidrug and toxic compound extrusion (MATE) transporter in Nicotiana tabacum. Proc. Natl. Acad. Sci. U. S. A 106, 2447–2452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shoji T et al. Multidrug and toxic compound extrusion-type transporters implicated in vacuolar sequestration of nicotine in tobacco roots. Plant Physiol. 149, 708–718 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cardillo AB, Perassolo M, Sartuqui M, Rodríguez Talou J & Giulietti AM Production of tropane alkaloids by biotransformation using recombinant Escherichia coli whole cells. Biochem. Eng. J 125, 180–189 (2017). [Google Scholar]
- 43.Lesuisse E, Crichton RR & Labbe P Iron-reductases in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol 1038, 253–259 (1990). [DOI] [PubMed] [Google Scholar]
- 44.Hu Y, Zhu Z, Nielsen J & Siewers V Heterologous transporter expression for improved fatty alcohol secretion in yeast. Metab. Eng 45, 51–58 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Dastmalchi M et al. Purine Permease-Type Benzylisoquinoline Alkaloid Transporters in Opium Poppy. Plant Physiol. 181, 916–933 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ro DK et al. Induction of multiple pleiotropic drug resistance genes in yeast engineered to produce an increased level of anti-malarial drug precursor, artemisinic acid. BMC Biotechnol. 8, 1–14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown S, Clastre M, Courdavault V & O’Connor SE De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci 112, 3205–3210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galanie S et al. Complete biosynthesis of opioids in yeast. Science 349, 1095–100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cravens A, Payne J & Smolke CD Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat. Commun 10, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Redford KH, Brooks TM, Macfarlane NBW & Adams JS Genetic frontiers for conservation: an assessment of synthetic biology and biodiversity conservation. (2019). at https://portals.iucn.org/library/node/48408 [Google Scholar]
- 51.Entian KD & Kötter P 25 Yeast Genetic Strain and Plasmid Collections. Methods Microbiol. 36, 629–666 (2007). [Google Scholar]
- 52.Kwiatkowski TJ, Zoghbi HY, Ledbetter SA, Ellison KA & Chinault AC Rapid identification of yeast artificial chromosome clones by matrix pooling and crude iysate PCR. Nucleic Acids Res. 18, 7191 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alberti S, Gitler AD & Lindquist S A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24, 913–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sainsbury F, Thuenemann EC & Lomonossoff GP pEAQ: Versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J 7, 682–693 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Ryan OW et al. Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thodey K, Galanie S & Smolke CD A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol 10, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen F, Pon A, Wilson M, Greiner R & Wishart D CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 42, 94–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guijas C et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem 90, 3156–3164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amberg DC, Burke DJ & Strathern JN Yeast Vital Stains: Visualizing Vacuoles and Endocytic Compartments with FM4–64. Cold Spring Harb. Protoc 2006, pdb.prot4165 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Wallace W, Schaefer LH & Swedlow JR A workingperson’s guide to deconvolution in light microscopy. Biotechniques 31, 1076–1097 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Haas BJ et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc 8, 1494–512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bryant DM et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 18, 762–776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matasci N et al. Data access for the 1,000 Plants (1KP) project. Gigascience 3, 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Källberg M et al. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc 7, 1511–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bomati EK & Noel JP Structural and kinetic basis for substrate selectivity in Populus tremuloides sinapyl alcohol dehydrogenase. Plant Cell 17, 1598–1611 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weigel D & Glazebrook J Transformation of Agrobacterium Using the Freeze-Thaw Method. Cold Spring Harb. Protoc 2006, pdb.prot4666–pdb.prot4666 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Kushnirov VV Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.