

Abstract
Many neurodegenerative diseases such as Alzheimer’s disease (AD), multiple sclerosis, and traumatic brain injury (TBI) are associated with systemic inflammation. Inflammation itself results in increased blood content of fibrinogen (Fg), called hyperfibrinogenemia (HFg). Fg is not only considered an acute phase protein and a marker of inflammation, but has been shown that it can cause inflammatory responses. Fibrin deposits have been associated with memory reduction in neuroinflammatory diseases such as AD and TBI. Reduction in short-term memory has been seen during the most common form of TBI, mild-to-moderate TBI. Fibrin deposits have been found in brains of patients with mild-to-moderate TBI. The vast majority of the literature emphasizes the role of fibrin-activated microglia as the mediator in the neuroinflammation pathway. However, the recent discovery that astrocytes, which constitute approximately 30% of the cells in the mammalian central nervous system (CNS), warrants further investigations in the causative role of HFg in astrocyte-mediated neuroinflammation. Our previous study showed that Fg deposited in the vasculo-astrocyte interface activated astrocytes. However, little is known of how Fg directly affects astrocytes and neurons. In this review, we summarize studies that show the effect of Fg on different types of cells in the vasculo-neuronal unit. We will also discuss the possible mechanism of HFg-induced neuroinflammation during TBI.
Keywords: Astrocytes, cortical contusion injury, Fg-PrPC complex, neurodegeneration, short-term memory
Introduction
Fg is a soluble glycoprotein that is comprised of two sets of disulfide-bridged Aα-, Bβ-, and γ-chains. It is widely known for its central role of providing scaffolding in the coagulation cascade in hemostasis [1]. Fg is known to interact with a variety of cell receptor molecules on an array of cell types with different cellular membrane protein composition and gene expression profiles. This could be due to the nature of its molecular structure with multiple binding sites for cell receptors with different biological functions [2]. On the platelet, Fg interactions with its integrin receptors lead to platelet aggregation during coagulation events. One of its integrin receptors, GP IIb/IIIa (αIIbβ3), on the activated platelet will undergo a modification that renders it receptive to binding with ligands such as Fg or von Willebrand factor [3]. This is followed by Fg’s conversion to insoluble fibrin via the action of thrombin and subsequently the generation of platelet thrombus, colloquially called a clot formation [4]. The cryptic binding epitope C-terminal γ377–395 sequence of the Fg molecule that enables it to recognize and bind to the CD11b (αM) integrin receptor is exposed via the polymerization of Fg to fibrin or Fg’s immobilization on a substrate [5]. In addition to its effects on platelets, fibrin’s interaction with αIIbβ3 receptor on mast cells is involved in systemic blood pressure regulation [6,7]. Fg’s ability to interact with different cell types is mostly accomplished by binding to integrins, like the αIIbβ3 receptor mentioned above. Integrins are a family of heterodimeric receptors that integrate the extracellular matrix with the intracellular cytoskeleton to mediate cell migration and adhesion. Other integrins that serve as Fg receptors are α5β1, αMβ2 (CD11b/CD18, Mac-1, CR3, Mo-1), αvβ3, and αxβ2 (CD11c/CD18) [8]. Fg’s ability to bind to a variety of integrin receptors may elicit activation of different pathways, which in turn, regulate downstream signaling that mediate cell migration, proliferation, cytokine production, and cell survival or apoptosis. While the activation of vascular smooth muscle αvβ3 integrin with arginylglycylaspartic acid (RGD) peptide causes vasodilation, activation of endothelial α5β1 integrin results in vasoconstriction [9]. Since Fg has two RDG sequences on its Aα chain [10], we tested its intraluminal effect on microvascular reactivity that resulted in vasoconstriction [11]. This Fg-induced vasoconstriction can be explained by the fact that, among its integrin receptors, only α5β1 but not β3 and therefore not the αvβ3 integrin, was found on the apical side of the endothelial cell (EC) and at the EC-to-cell contact border [12]. In addition, localization of α5β1 integrin at cell-cell borders makes it one of the important determinants of EC monolayer integrity and thus, one of the major players in the vascular permeability changes [12].
In addition to binding to integrins mentioned above, Fg binds to intracellular adhesion molecule-1 (ICAM-1) [13,14]. Fg binds to ICAM-1 via a sequence in the γ chain (γ residues 117–133) [15]. We found that at high level Fg caused vasoconstriction [11], exocytosis of Weibel-Palade bodies releasing endothelin-1 [16], and an increase in EC layer permeability [17]. All these effects could have been a result of Fg binding to ICAM-1 and α5β1 on the apical side of ECs. Fg bound to CD11b/CD18, CD11c/CD18, and the non-integrin toll-like receptor-4 (TLR-4) on macrophages and microglia leads to macrophage activation and the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƘB) pathway that mediate cytokine release [6,2].
Normal concentration of Fg in blood is close to 2 g/L with a half-life of approximately 4 days [18]. In various pathological conditions where there is injury and inflammation present, Fg concentration is elevated. Therefore, it has been used a reliable inflammatory biomarker [19]. As a large (340-kD) macromolecular blood protein, Fg usually stays in the circulation and is kept from the brain parenchyma by the blood-brain barrier (BBB) [20]. However, in a pathological condition where the BBB is compromised, Fg gains access to the CNS [8,17] and accelerates vasculo-neuronal damage-causing neuroinflammation [21].
Fg and fibrin depositions have been found in the brains of patients with inflammatory diseases as TBI [22,23], AD [24], and multiple sclerosis (MS) [25]. In the animal models of these diseases, it has been shown that the depletion of Fg ameliorates the development and severity of the diseases [20,26,27]. Therefore, it became evident that Fg is not only a biomarker but may play a more active role in the development and/or exacerbation of neurodegenerative complications of the neuroinflammatory diseases mentioned above [27,6,23].
Plaque formations that contain Fg/fibrin are typical for inflammatory neurodegenerative diseases associated with memory reduction such as AD [28], MS [29], and TBI [30]. Like the amyloid-β (Aβ) plaques found in AD patients, plaques are found in TBI patients [30]. However, the plaques are formed more rapidly during TBI relative to slower developing ones during AD [30]. A strong contribution of Fg in the development of AD has been shown [28,31]. Deposits of fibrin were found postmortem in humans after some years of head injury [32,23]. Deposition of Fg exacerbates neurovascular damage and accelerates neuroinflammation [32,20]. Reduction in short-term memory persists longer in patients with mild to moderate TBI that is characterized with non-ruptured vessels [33]. We found that TBI-induced inflammation resulted in increased BBB permeability of non-ruptured vessels, causing extravasation of Fg and its deposition in extravascular space [34,35]. These effects were associated with neurodegeneration and short-term memory reduction in mice with head trauma [34,35]. Enhanced extravasation and accumulation of Fg in the interstitium could potentially lead to the formation of protein complexes containing Fg, such as Fg and Aβ (Fg-Aβ) complex [36,30] or Fg and a cellular prion protein (PrPC) complex (Fg-PrPC) [34], and maybe others. This causes subsequent pathology in the brain. Formation of both Fg-Aβ and Fg-PrPC complexes are associated with reduced cognitive function and memory [30,34].
Thus, it is evident that there is a strong connection between the inflammation-induced HFg-mediated memory impairment during neurodegenerative diseases (e.g. TBI, AD, and MS). Therefore, the primary focus of this review will be on the effects of HFg on neurons and other cell types of the nervous system during TBI, following the movement of Fg from blood, through the vascular endothelium, toward the brain parenchyma, and ultimately affecting the neurons.
Fg effects on vascular and EC layer permeability
Integrins α5β1 and αvβ3 [9,37] and ICAM-1 [14] act as Fg receptors on EC surface. At elevated levels, Fg has been shown to increase cardiac microvascular EC permeability, which seems to be mediated by Fg binding to its EC apical surface receptors ICAM-1 and α5β1 [17]. This was manifested as the translocation of large proteins such as albumin and Fg through the EC monolayer, which could occur by paracellular [17] or transcellular transport involving caveolae formation [38,39]. Acute HFg was shown to compromise microvascular integrity through activation of matrix metalloproteinases 9, downregulate an adherence junction protein, vascular endothelial cadherin, and upregulate a plasmalemmal vesicle-associated protein-1, an integral membrane-associated protein found in caveolae [40]. These results suggested that acute elevation of blood level of Fg may alter cerebrovascular and EC layer permeability via both paracellular and transcellular transport pathways. Regardless of its possible dual effect, chronic increase in blood level of Fg that occurs during mild-to-moderate TBI, induces enhanced cerebrovascular permeability resulting in extravasation of large molecular weight proteins, particularly Fg, mainly via caveolar transcytosis [36]. We found that Fg crossed the vascular wall and deposited in the vasculo-astrocyte endfeet interface where it was immobilized [35] and most likely converted to fibrin.
Effects of Fg and fibrin
Fg in its soluble form has been shown to disrupt cultured brain EC layer integrity [41] and astrocyte activation [42]. Infusion of nondegraded Fg into the systemic circulation of mice increased pial venular permeability [40]. Chronic elevation of blood Fg level resulted in increased cerebrovascular permeability to proteins, increased deposition of Fg in extravascular space, and the formation of Fg-Aβ and/or Fg-PrPC complexes that were associated with cognitive impairment manifested by reduction in short-term memory [36].
It was shown that while soluble Fg at a concentration of 50 μg/ml (lower than physiological concentration) did not have an effect, immobilized Fg altered microglia morphology in vitro [43]. A later study, by the same group, has shown that intravenous administration of Fg resulted in activation of microglia, neuronal dendritic loss, and dendritic spine elimination; all being associated with cognitive decline [44]. All these results confirm our findings that to have an effect on microglia and/or neurons, first, Fg has to cross vascular wall and deposit in extravascular space. It is noteworthy that lyophilized Fg was used in the studies mentioned above. An administration of Fg to animals could have increased blood content of Fg by no more than 0.5 mg/ml [44], while the blood content of Fg during inflammatory conditions is equal to or more than double its normal level [45,46]. While immobilized, both Fg and fibrin can bind to their receptor, αMβ2 with high-affinity/avidity, while soluble Fg is a relatively poor ligand [47,48]. However, our study suggest that soluble Fg specifically binds an unidentified, integrin-type receptor on the surface of rat erythrocytes [49]. Therefore, in all our in vitro studies we used hirudin (inhibits thrombin from converting Fg to fibrin) to test the effects of Fg before it has been converted to fibrin [16,17,50,51,39,42]. Others have shown that the use of hirudin, and thus, inhibiting fibrin formation after the onset of intracerebral hemorrhage reduces neuroinflammation and improves long-term outcome [26]. In many reports Fg and fibrin are mentioned interchangeably. For example, it has been reported that Fg deposits in brains of patients with TBI [22,23]. However, we would like to emphasize that if extravascular Fg deposits are found, they are most likely fibrin deposits that formed after conversion of extravasated Fg.
Fg and astrocytes
Astrocytes, specialized glial cells named after its star-like shaped, are 5 times more abundant in brain than neurons [52,53]. They act as the connection between neurons and the CNS vasculature. In response to neurologic insult, astrocytes become reactive, resulting in astrocytosis that is marked by astrocyte overexpression of the glial fibrillary acidic protein (GFAP), an intermediate filament cytoskeletal protein expressed primarily by astroglia [54].
Previously, our group showed that HFg-induced activation of astrocytes in vitro caused increased expression of ICAM-1 and tyrosine receptor kinase B (TrkB) [42]. TrkB receptors are expressed in the central and peripheral nervous system. Their signaling is indispensable for the survival and maintenance of neurons and their synaptic plasticity [55]. TrkB stimulation in astrocytes induces nitric oxide (NO) production and nitrotyrosine deposition, which potentially promote neurodegeneration [56]. It has been shown that NO modulates and could increase the level of reactive oxygen species (ROS) in cells [57], which is known to cause neurodegeneration [58].
ICAM-1 is a transmembrane glycoprotein that is expressed on many types of cells, including leukocytes and ECs, and has been aberrantly expressed in diseased glial cells [59]. Our study showed that blocking of ICAM-1 function resulted in reduction of Fg and astrocyte interactions and the resultant overexpression of TrkB in vitro, suggesting that ICAM-1 can be a receptor for Fg on the surface of astrocytes [42]. ICAM-1 ligation was shown to induce the expression of pro-inflammatory cytokines on rat astrocytes, including interleukin-1α (IL-1α), IL-1β, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) [60]. Another study has shown that Fg causes astrogliosis, where it increases GFAP and neurocan immunoreactivity [61]. Neurocan is a member of the chondroitin sulfate proteoglycans that is often found upregulated in CNS injuries and it inhibits axonal and neurite growth [62]. Therefore, Fg-induced increase in expressions of TrkB, ICAM-1, and neurocan support the notion that Fg contributes to neuroinflammation development.
As astrocytes are a glial cell type, they may possess phagocytic properties. It has been shown that astrocytes remove Fg that has been coated on the surface of a Petri dish [63]. However, although it was not specifically discussed in the paper, the uptake of Fg by astrocytes resulted in their activation and death [63], suggesting toxic effect of Fg/fibrin.
There is a growing body of evidence which indicates that inflammation is linked to both post-traumatic neurodegeneration and functional deficits in animal [64–66] and clinical studies [67,68]. It has been shown that reactive, toxic A1 astrocytes, formed after inflammation, deposit an inhibitory extracellular matrix consisting primarily of chondroitin sulphate proteoglycan, which is involved in the formation of glial scarring [69]. Our data showed that astrocytes were activated during mild-to-moderate TBI [35]. These data were correlated with neurodegeneration and reduction in short-term memory in mice after CCI [35]. Others have confirmed these findings by showing expression of complement component 3, the marker of toxic astrocytes [70], which is a highly upregulated gene in neurotoxic, A1 astrocytes that exacerbate disease pathogenesis and not in A2 astrocytes that promote neuronal survival [71].
Fg and microglia
Microglia serve as the macrophage of the CNS parenchyma [72]. Effect of Fg on microglia has been tested in vivo, in the experimental autoimmune encephalomyelitis (EAE) model for MS [73]. A local injection of Fg in the healthy mouse cortex induced significantly higher microglial responses than those caused by an injection of albumin or artificial cerebrospinal fluid [73]. Fg-activated microglia formed perivascular clusters in areas of fibrin deposition, which mediated axonal damage and release of ROS [73]. The use of transgenic mice Fibγ3940–396A with a mutation on Fg lacking binding site for CD11b/CD18 receptor confirmed that the axonal damage and microglial activation requires fibrin signaling via the receptor.
Fg binding to CD11b/CD18 integrin receptor on leukocytes causes activation of innate immune responses [74,47] including activation of NF-ƘB and mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K), v-Akt Murine Thymoma Viral Oncogene (Akt), and Rho signaling. These, in turn, leads to adhesion, migration, chemotaxis, and phagocytosis of leukocytes, macrophages, and monocytes [2]. It has also been shown that fibrin interaction with the CD11b/CD18 receptor activates CNS immune cells responsible for innate immunity, namely microglia and perivascular macrophages, inducing M-1 type activation with increased expression of major histocompatibility complex class II, CD86, and IL-12, which suggest activation of antigen presenting cells in response to infection or inflammation [75]. This might lead to activation of TLRs and the overexpression of chemokines, such as C-X-C motif chemokine 10 (CXCL10) and C-C motif chemokine 2 (CCL2), suggesting that extravascular deposition of fibrin mediates the recruitment of peripheral macrophages into the CNS. This exacerbates neuroinflammation and associated damage, such as demyelination [21].
The role of fibrin deposition has been studied in the transgenic TgCRND8 mice, a mouse model for AD that mimics human pathological brain aging, showing Aβ-associated pathology as a result of the neuroinflammation [20]. Depletion of Fg by genetic or therapeutic treatment reduces vascular pathology as well as the activation of microglia, whereas increasing fibrin deposition exacerbates these effects [20].
Astrocyte and microglia activation
Neuroinflammation causes microglia and astrocytes to become “reactive”. Similar to the well-characterized M1 and M2 polarization in macrophages, their CNS counterparts, microglia, could exhibit an “M1-like” pro-inflammatory phenotype with upregulation of TNF and interleukin-1 beta (IL-1β) or an “M2-like” immunosuppressive phenotype with upregulation of chitinase-like-3 (Chil3), frizzled class receptor-1 (Fzd1), and arginase-1 (Arg1) [76,77]. Since colony stimulating factor 1 receptor knockout mice that lack microglia were unable to generate astrocyte activation after lipopolysaccharide (LPS) injection, it has been suggested that microglia activation is necessary to induce astrocyte reactivity [71]. However, our in vitro study suggested that astrocytes alone still can be activated [42] suggesting a more complex functionality of astrocytes.
Activated microglia produce molecular agonists including IL-1β, TNF and complement component 1q, that consequently activate astrocytes [71]. Activated astrocytes, characterized by increased expression of the GFAP, loose their function and ability to support neurons. Such astrocytes, identified as “A1”, act as a potent neurotoxin [77]. Whereas ischemia-induced reactive astrocytes, defined as “A2”, are thought to have a reparative function via upregulation of neurotrophic factors [77].
How the phenotypic polarization of the activated astrocytes occurs remains unknown. In the classical inflammatory activation of astrocytes, there has been a suggestion that lipocalin-2 plays a critical role as an autocrine modulator of the functional polarization [78]. Interestingly, astrocytes that are classically activated using LPS and interferon-gamma have shown opposite reactions to astrocytes that are activated with interleukin-4 or interleukin-10. The former two are neurotoxic as opposed to the latter ones, which protect neurons against excitotoxic and oxidative injuries [78].
Fg and neuron
Neurons are the fundamental units of the brain and nervous system [79]. Although there are some reports that neurons may innervate brain blood vessels [80], in general, it is accepted that neurons do not directly innervate brain microvessels. Their action upon microvessels occurs mainly via astrocytes. It has been shown that conditioned medium from astrocytes treated with 2.5 mg/ml of Fg inhibit neurite outgrowth in the cultured cortical neurons [61]. However, when conditioned media from astrocytes that has been pre-treated with transforming growth factor beta inhibitor was used on the neurons, the neurite outgrowth inhibitory effect was reversed, which suggest the receptor participation in the activation of astrocytes and subsequent neurite inhibitory effect of the neurons [61].
Fg is shown to be neurotoxic [81]. Fg treatment showed dose-dependent cell death measured by the 3-(4,5-dimethylthiazol-2-yl) 2–5,-diphenyltetrazolium bromide reduction method with about 10% cell death after incubation with 5 μg/μl Fg for 24 hour compared to almost 50% cell death with double the concentration at 10 μg/μl of Fg [81]. Although the study was not performed on cultured primary neurons, the SH-SY5Y neuroblastoma cells that were used were originally derived from human metastatic bone tumor biopsy and had neuron-like phenotype [82]. In the same study, Fg treatment caused increased expression of caspase-3 after just 45 min of treatment, suggesting that Fg induces neuronal apoptosis [83]. As to our knowledge, there are no studies that test the direct effect of Fg on neurons. However, this interaction may still need to be investigated since, in nature, besides conditions when Fg deposited in extravascular space may result in astrocyte activation and death causing neuroinflammatory pathologies, a massive accumulation of Fg/fibrin may create a condition when Fg/fibrin would come into a direct contact with neurons.
Fg causing oxidative damage
It is known that fibrin(ogen) induces microglial activation and ROS generation in vitro [7] and in vivo mouse models of AD [44] and MS [7]. Fibrin causes activation of microglia by interaction with CD11b, which subsequently generates ROS and causes oxidative damage [7,44]. Increased production of ROS results in neuronal injury and axonal damage believed to be caused by impairment of mitochondria and the resulting energy impairment in neurons [84]. Reactive astrocyte are known to enhance the expression of inducible nitric oxide synthase and production of NO [85]. During inflammatory disease, such as TBI, an increase in ROS production usually comes concomitant with an increase in NO and, when combined, they produce a potent reactive nitrogen species [86]. It has been shown that axon exposure to NO causes neuronal degeneration [87]. Thus, Fg/fibrin-activated astrocytes may result neurodegeneration.
Increased expression of TrkB on astrocytes is associated with NO production and neurodegeneration [56]. The increased expression of the truncated isoform of the neurotropin receptor TrkB-T1 was observed in MS lesions from human patients, which is confirmed by immunohistochemistry, mRNA expression, and flow cytometry methods [56]. Brain-derived neurotrophic factor (BDNF) is a neurotrophin and a well-known agonist for TrkB [88]. BDNF functions as a major regulator of activity-dependent plasticity at excitatory synapses in the CNS and plays an important role in neuronal synaptic regulation [89]. However, it has been shown that BDNF dose-dependently stimulates activation of astrocytes, and when the conditioned media was used on neurons, it caused neuronal degeneration [56]. The neurodegeneration was initiated by NO produced by the activated astrocytes, that were treated with 25 ng/ml of BDNF, showing significantly higher NO than 5 ng/ml, the control group [56].
Another source of ROS formation can be astrocyte PrPC. It is well accepted that PrPC exhibits a dual effect in brain: when it is unaltered, it provides neuroprotective effects, while when ligated, it results in neurotoxicity [90]. It has been identified that PrPC is involved in cell-redox homeostasis through ROS production via Nicotinamide adenine dinucleotide phosphate oxidase and extracellular regulated kinases 1/2 signaling stress [91]. This PrPC-induced ROS formation results in neuronal and other brain cell toxicity and oxidative stress [91]. It has been shown that Fg-containing protein complexes, like Fg-Aβ, that are highly resistant to degradation [92,93], contribute to increased inflammation and are associated with reduction in short-term memory [92,36]. We have recently found that Fg associates with PrPC (unpublished data) on astrocyte surface, which resulted in Fg-PrPC complex formation in brain extravascular space during HFg [36] and TBI [35]. This formation of Fg-PrPC complex that may result in PrPC property changes, may be a mechanism for neurodegeneration and the resultant short-term memory reduction found in our studies [36,35]. Combined, these results suggest that increased expression of astrocyte PrPC and its association with extravasated Fg can result in formation of Fg-PrPC complex, which can have a role in memory reduction during inflammatory cerebrovascular diseases, e.g. TBI (Figure 1).
Figure 1. Mechanism of traumatic brain injury (TBI)-induced hyperfibrinogenemia (HFg)-mediated production of reactive oxygen species (ROS).
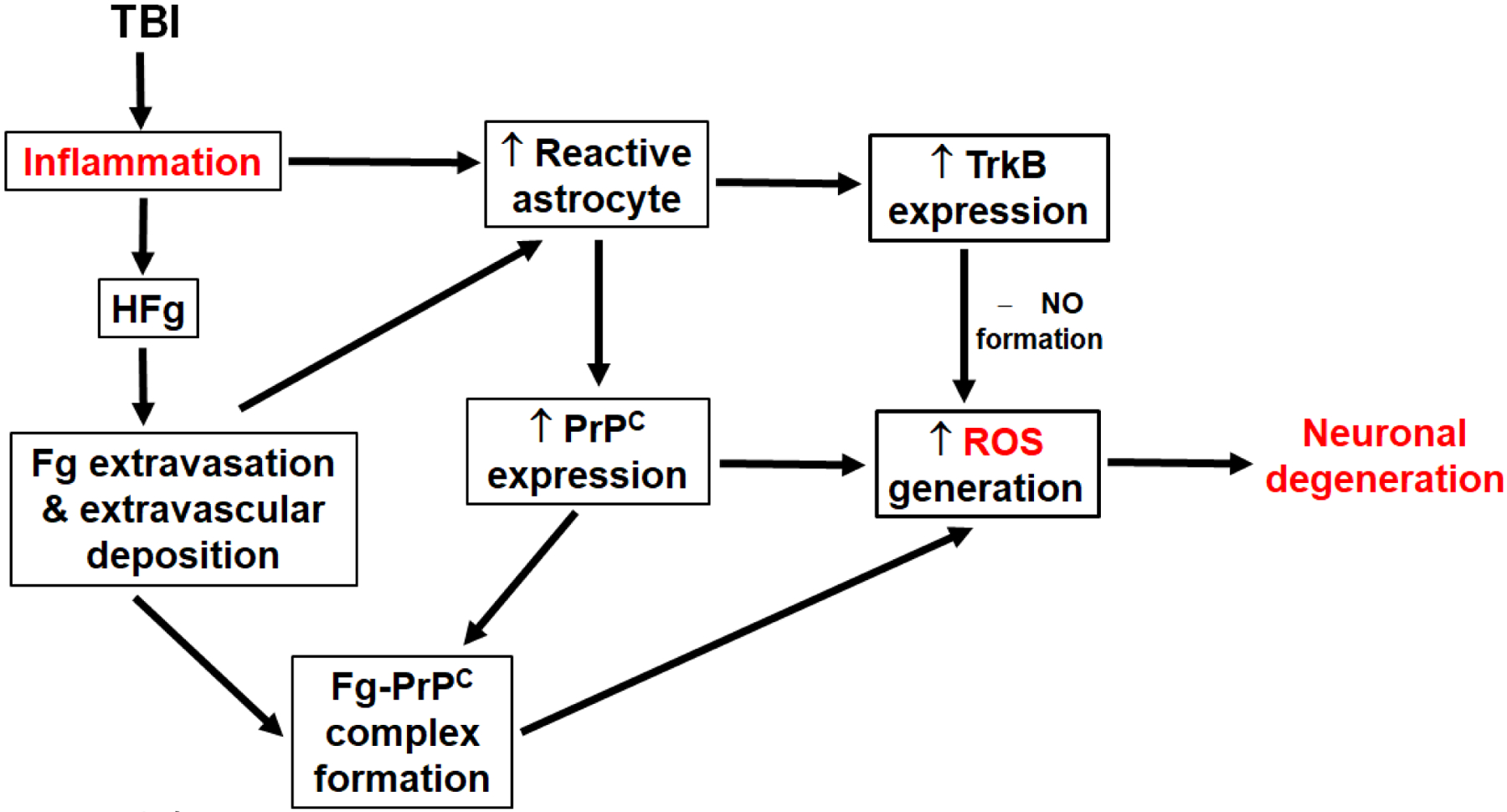
TBI results in inflammation causing astrocyte activation and HFg. The latter leads to Fg extravasation and its deposition in extravascular space. Fg deposited in vasculo-astrocyte interface also activates astrocytes. Reactive, toxic astrocytes overexpress cellular prion protein (PrPC) and tropomyosin receptor kinase B (TrkB). Close proximity of Fg and PrPC predisposes formation of Fg-PrPC complex. Overexpressed PrPC and Fg-PrPC complex result in production of ROS, while TrkB, via formation of nitric oxide (NO), also results in ROS generation. Increased ROS in neurovascular unit contributes to neurodegeneration.
Generation of ROS was also associated with increased brain endothelial layer permeability by activation of the protein kinase B signaling pathway via the Ras homolog family member A [94]. Generation of ROS in ECs by itself can promote Fg extravasation, as mentioned above.
Fg in traumatic brain injury
During TBI, immediate and delayed dysfunction of the BBB is observed. Disruption of the tight junction complexes and the integrity of the basement membrane results in increased paracellular permeability of the BBB [95,96,34]. The resultant inflammation induces HFg [97] that leads to translocation of Fg to the extravascular space [34]. The level of Fg increases in human patients during TBI to a level above 4 g/L two days after injury and remains high for as long as 14 days [98,97]. This agrees to our finding that after cortical contusion injury (CCI) in mice, an animal model of TBI, there is higher Fg deposition that could be found in the vasculo-astrocyte endfeet interface even after 14 days [96,35]. Furthermore, there was increased neuronal degeneration in the brain of mice from the CCI group when compared to sham, which was shown by reduced expression of neuronal marker NeuN, while there was an increase Fluoro-Jade C staining in the CCI injury group. Although it is tempting to connect these two findings and point out that they are due to the increase of Fg disposition in extravascular space, the effects of other contributing factors could not be ruled out.
TBI and inflammation
TBI causes the induction of a series of the inflammatory cascade, which could exacerbate the already altered homeostasis of the injured brain with the detrimental, potentially progressive effects [99–101]. Studies have shown that the effect of inflammation and microglial activation could persist for as long as 17–18 years after the insult [102]. Although the traumatic event that causes the brain injury itself is a factor that is hard to control, this finding suggests that the window of opportunity for therapeutic intervention following brain injury might be longer than was expected. Therefore, by investigating further mechanisms contributing to the progression of neuroinflammation and associated neurodegeneration, there could be room for intervention to prevent further damages.
To our knowledge there are no other studies than ours that specifically addressed TBI-associated inflammation cascade that involves Fg-mediated inflammation mechanism. Increased blood level of Fg is considered as a marker of inflammation [19]. Previously we have showed that HFg results in vasoconstrictions of brain vessels, which is also is a marker of inflammation. In our studies, we have shown that in mouse model of mild-to-moderate TBI, animals with CCI exhibited an increase in blood content of Fg and increase in cerebrovascular permeability. These effects are by themselves are indications of inflammation. As a result, we found an increased deposition of Fg in the vasculo-astrocyte interface, which was associated with increased neuronal degeneration [35].
It is known that within a few minutes after trauma to the brain, immune mediators are released which orchestrate a further sequence of inflammatory events. It was shown that 6 hours after CCI, the levels of IL-1β, IL-6, and TNF-α were significantly increased [103]. IL-6 was found to be increased by 40 to 100-fold in the CSF compared to that in serum, with detectable concentration as early as 2 hours post-injury [97,104]. The peak concentration was found between 4–6 hours and decreased by 16–24 hours in the animal model [104]. Whereas in human patients, it reached the maximal concentration within 1 to 2 days after trauma, before decreasing to a still detectable level for the whole length of the 14-day study [104]. At all times in the study, the CSF concentration of IL-6 was higher than that in the plasma, suggesting that IL-6 is produced in the CNS and that it plays a role in moderating the acute-phase response [97]. The maximal increase of IL-6 was found to be correlated with the increase levels of acute-phase proteins including α1-antitrypsin and Fg [104]. On the other hand, it is possible that Fg, as an inflammatory agent, is involved in the elevation of IL-6 levels and, since IL-6 is needed for Fg enhanced synthesis [105], that it triggers a positive feedback loop exacerbating the inflammatory responses.
TNF-α is released very rapidly immediately (1–6 hours) after injury and later decreases to an almost undetectable level within a few hours [100,103]. TNF-α is involved in the acute phase reaction during TBI, apoptosis, cell death, an increase in BBB permeability, and vasogenic brain edema. However, it’s effect are pleiotropic, as shown by a transgenic mouse model lacking the gene for TNF, which suggests that TNF-α action in the acute phase might be more neurotoxic, but that later it becomes neuroprotective [106].
Insignificant IL-1 was detected in both CSF and serum in a clinical study from human patients with TBI [104]. On the contrary, in a CCI animal model of TBI, IL-1β was significantly increased with the peak concentration at 6 hours after head injury, but this quickly reduced in 12 hours [103].
Primary rat astrocytes express a low basal level of surface ICAM-1 protein. However, in the presence of the cytokines TNF-α or IL-1β, astrocyte expression of ICAM-1 increases and reaches maximal level in 12–16 hours after treatment for both cytokines [107]. In another study, ICAM-1 ligation by monoclonal antibody to rat ICAM-1 induced mRNA expression of IL-1α and IL-1β through activation of extracellular signal-regulated kinases 1/2 (ERK 1/2), and expression of IL-6 through activation of both ERK 1/2 and p38 MAPK. TNF-α was also induced but to a lower level. These findings suggest that ICAM-1 activation could be a result of or a contributor to the ongoing inflammatory processes [60]. We have previously shown that HFg induced an increased expression of ICAM-1 in the cultured astrocytes [42] and in the mouse brain microvessels in vivo [40].
Signaling molecules and pathways during neuroinflammation
In general, little is known of signal transduction pathways activated by HFg in extravascular cells of the neurovascular unit. Activated astrocytes produce cytokines, chemokines and bioactive complexes that trigger signaling pathways and transcription factors. Activation of astrocytes leads to translocation of the proinflammatory transcription factor NF-ƘB [108] and this could result in a heterogeneous response towards protective or detrimental pathways [109,77,110].
When the CNS is injured, the expression of NF-ƘB-dependent genes is activated [110]. The detrimental role of NF-ƘB in neuroinflammatory pathology has been shown [111]. Blocking astroglial NF-ƘB significantly reduces disease severity and improves functional recovery during experimental autoimmune encephalomyelitis in a mouse model for MS [110]. On the other hand, another study that used a mouse model with neuronal deletion of NF-ƘB inhibition showed a worse outcome after induced TBI with increased mortality, neurological deficit, and neuronal cell death. The differential activation of NF-ƘB dimers [111] and difference in target cell type (astrocyte vs. neuron) might play a part in the opposing responses described above. It has been shown that physiologic NF-kB activation in neurons results in neuroprotection after closed head injury and neuronal NF-kB inhibition is detrimental for TBI outcome [112]. However, activation of NF-kB in astrocytes and oligodendrocytes leads to worsening of brain damage after CCI [113]. Thus, these results suggest a complex role of NF-kB signaling depending on its brain cell type origin and experimental injury model.
Inhibition of astroglial NF-ƘB also resulted in significantly reduced expression of adhesion molecules such as ICAM-1, vascular adhesion molecule 1 (VCAM-1), and integrins β5 and β7 when compared to the control group [110]. We have previously shown that Fg could upregulate expression of and binding to ICAM-1 protein on astrocytes, causing their activation [42,35]. It is possible that Fg binding to astrocyte ICAM-1 can trigger NF-ƘB activation and associated signaling mechanisms in the cell resulting in further effects on neurons leading to their degeneration found in our study [35].
There is more evidence of astrocyte proinflammatory activation that involves NF-ƘB signaling. Both, the chemokine CCL2 and CXCL10 mentioned above have been shown to be regulated by NF-ƘB [110,108]. It has been shown that activated astrocytes upregulate CXCL10 [78] and CCL2 [114]. NF-ƘB is a central mediator of inflammation that induces and controls the expression of various inflammatory genes, including cytokines and chemokine. It is known that CCL2 is a proinflammatory chemokine [115]. In response to injury, cells in the CNS, like astrocytes and microglia, produce CCL2, a proinflammatory chemokine, that orchestrates recruitment of leukocytes to the injury site. A study in transgenic mice with astrocyte-specific inhibition of NF-ƘB activation has been shown to reduce CCL2 expression and leukocyte infiltration suggesting the critical role of NF-ƘB for injury-induced CCL2 transcription in astrocytes [108].
CXCL10 (also called interferon gamma-inducible protein 10) is also a chemoattractant for T-lymphocytes, which is upregulated in the cerebrospinal fluid and brain lesion area during MS [116]. In a transgenic mouse model of MS with the deletion of CXCL10, the disease deficits were milder and acute spinal cord demyelination was reduced [117]. Based on information presented above, we hypothesize NF-ƘB-mediated signaling can play a central role in Fg-induced astrocyte activation that can affect neurons.
Besides effects of NF-kB described here, Fg/fibrin are known to be involved in activation of other inflammatory pathway such as Rho guanosine triphosphatase seen on Chinese hamster ovary cells transfected to investigate Fg effect on ICAM-1 [118]. Fg is seen to directly stimulate profibrogenic and proinflammatory function on pancreatic stellate cells through MAPKs and Akt pathways during pancreatic cancer [119]. In astrocytes, treatment with Fg resulted in an increased expression of TrKB that was accompanied by changes in morphology of astrocytes suggesting their activation [42].
Fg depletion as a potential therapeutic approach
In some studies related to inflammatory pathologies, blood levels of Fg was decreased with ancrod, a defibrinogenating agent derived from the venom of the Malayan pit viper [20,120,61]. However, it is known that ancrod decreases levels of other high molecular weight proteins including fibronectin, von Willebrand factor, and globulins. Therefore, ancrod cannot be considered as an agent specifically targeting level of Fg in blood. Fenofibrate (brand name Tricor) used in rodents suppressed Fg gene expression in liver and decreased the Fg content in blood [121]. However, fenofibrate is mainly used to reduce low density lipoproteins and triglycerides [122]. Therefore, it cannot be considered as a specific inhibitor of Fg synthesis. In most of studies related to Fg effects, Fg gene knock out (Fg−/−) mice were used [123,124]. The results show that other proteins, e.g. fibronectin, can “assume” role of Fg in coagulation and/or thrombogenesis [123]. Thus, all the approaches mentioned above mask Fg effects in circulation. On the other hand, Fg antisense oligonucleotide (Fg-ASO) specifically and effectively decreases synthesis of Fg, and thus, blood level of Fg in mice [125]. In fact, we found that mice treated with Fg-ASO post-head injury exhibited lesser cerebrovascular permeability and lesser reduction in short-term memory (unpublished data). Therefore, as of today, use of Fg-ASO would be the best potential therapeutic approach for Fg depletion in animal models and possibly in patients with TBI, AD, and MS.
Conclusion
The studies reviewed here support our overall hypothesis (Figure 2) that during inflammatory diseases, such as TBI, increased blood content of Fg, which promotes extravasation of Fg and its deposition in the vasculo-astrocyte endfeet interface in the brain, results in neurodegeneration. This occurs through the binding of Fg to ICAM-1 and PrPC on astrocytes causing astrocyte activation. Fg-induced astrocyte activation causes inflammatory signaling, in part, through NF-ƘB pathway. The activated astrocytes increase production of inflammatory agents such as IL-6 and CCL10. Overexpression of IL-6 by itself can contribute to further increase synthesis of Fg and worsen deleterious effects of HFg. Fg-activated astrocytes also generate ROS that could itself contribute to neuroinflammation. The formation of a Fg-PrPC complex that can be highly resistant to degradation could lead to plaque formation associated with loss of short-term memory. Further studies are necessary to clarify the inflammatory effect of Fg in the crosstalk between coagulation and inflammation and between different types of cells and functional units ultimately causing neurodegeneration and cognitive decline, particularly reduction in short-term memory.
Figure 2. Mechanism of fibrinogen (Fg)-induced neurodegeneration during traumatic brain injury (TBI).
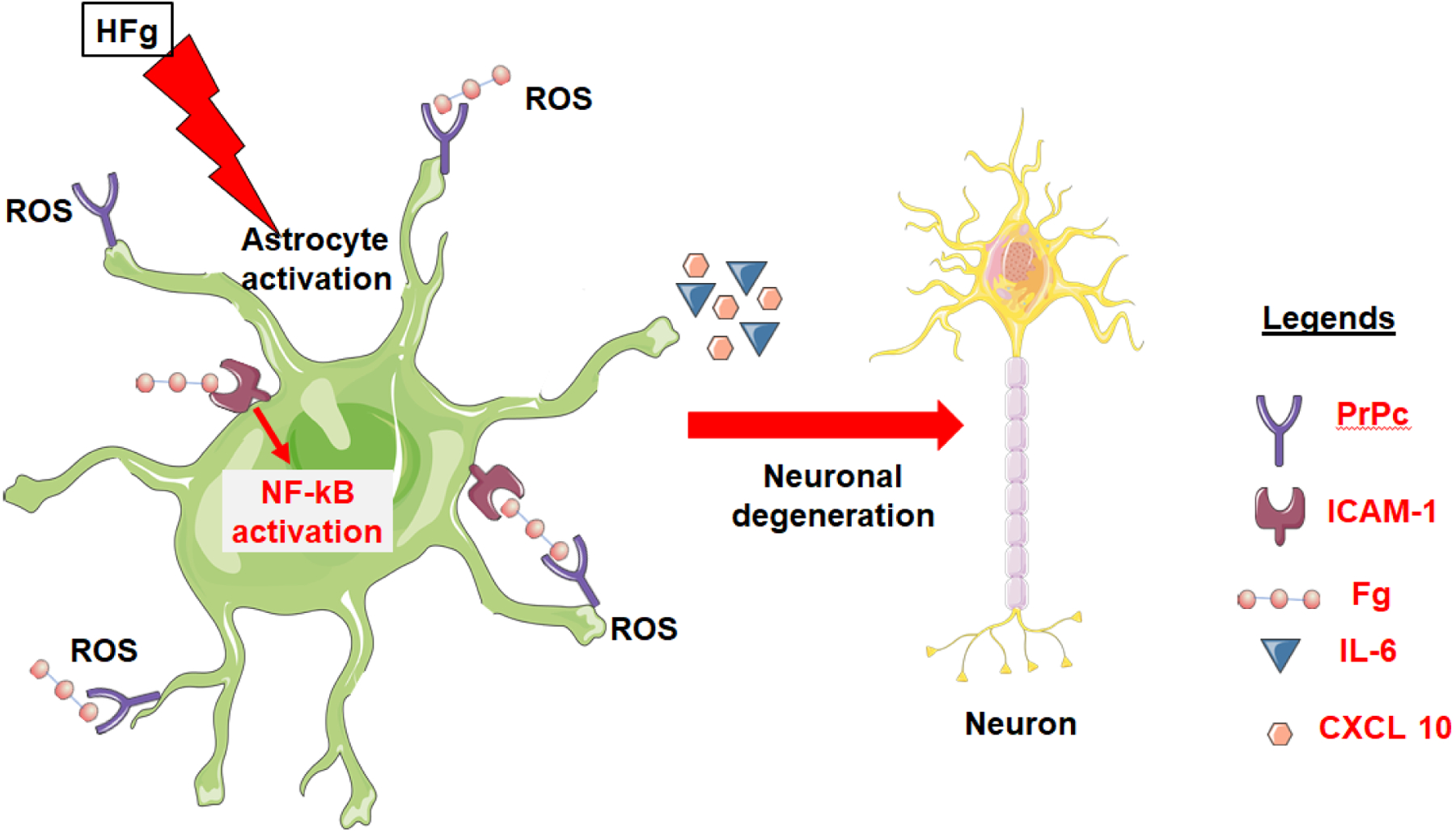
TBI-induced hyperfibrinogenemia (HFg) promotes extravasation of Fg and its deposition in the vasculo-astrocyte endfeet interface. Deposited Fg activates astrocytes in part, through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƘB) pathway. Activated astrocytes overexpress cellular prion protein (PrPC) and intracellular adhesion molecule-1 (ICAM-1), release interleukin-6 (IL-6) and C-X-C motif chemokine 10 (CXCL10) promoting inflammatory cascade and generate reactive oxygen species (ROS). Fg binds to astrocyte ICAM-1 and cellular prion protein (PrPC) further activating astrocytes and forming Fg-PrPC complex. All these effects lead to neurodegeneration. In parallel, Fg-PrPC complex that can be forming undegradable protein clamp could lead to plaque formation and further contribute to neurodegeneration and thus, short-term memory reduction.
Acknowledgement
This work was supported by the NIH grant # HL146832.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Mosesson MW (2005) Fibrinogen and fibrin structure and functions. Journal of Thrombosis and Haemostasis 3 (8):1894–1904 [DOI] [PubMed] [Google Scholar]
- 2.Akassoglou K, Strickland S (2002) Nervous system pathology: the fibrin perspective. Biol Chem 383 (1):37–45. doi: 10.1515/bc.2002.004 [DOI] [PubMed] [Google Scholar]
- 3.Leclerc JR (2002) Platelet glycoprotein IIb/IIIa antagonists: Lessons learned from clinical trials and future directions. Critical Care Medicine 30 (5):S332–S340 [DOI] [PubMed] [Google Scholar]
- 4.Ferguson JJ, Waly HM, Wilson JM (1998) Fundamentals of coagulation and glycoprotein IIb/IIIa receptor inhibition. American Heart Journal 135 (4):s35–s42. doi: 10.1016/S0002-8703(98)70296-0 [DOI] [PubMed] [Google Scholar]
- 5.Ugarova TP, Solovjov DA, Zhang L, Loukinov DI, Yee VC, Medved LV, Plow EF (1998) Identification of a Novel Recognition Sequence for Integrin αMβ2 within the γ-chain of Fibrinogen. Journal of Biological Chemistry 273 (35):22519–22527. doi: 10.1074/jbc.273.35.22519 [DOI] [PubMed] [Google Scholar]
- 6.Adams R, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K (2007) Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Current Medicinal Chemistry 14 (27):2925–2936 [DOI] [PubMed] [Google Scholar]
- 7.Davalos D, Akassoglou K (2012) Fibrinogen as a key regulator of inflammation in disease. Seminars In Immunopathology 34 (1):43–62 [DOI] [PubMed] [Google Scholar]
- 8.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. The Journal of Biological Chemistry 275 (29):21785–21788 [DOI] [PubMed] [Google Scholar]
- 9.Mogford JE, Davis GE, Meininger GA (1997) RGDN peptide interaction with endothelial alpha 5 beta 1 integrin causes sustained endothelial-dependent vasoconstrictionto rat skeletal muscle arterioles . . Journal of Clinical Investigation 100 (6):1647–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suehiro K, Gailit J, Plow EF (1997) Fibrinogen is a ligand for integrin alpha beta 5 beta1 on endothelial cells. The Journal of Biological Chemistry 272 (8):5360–5366 [DOI] [PubMed] [Google Scholar]
- 11.Lominadze D, Tsakadze N, Sen U, Falcone JC, D’Souza SE (2005) Fibrinogen- and fragment D-induced vascular constriction. American Journal of Physiology 288 (3):H1257–H1264 [DOI] [PubMed] [Google Scholar]
- 12.Lampugnani MG, Resnati M, Dejana E, Marchisio PC (1991) The role of integrins in the maintenance of endothelial monolayer integrity. The Journal of Cell Biology 112 (3):479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harley SL, Sturge J, Powell JT (2000) Regulation by fibrinogen and its products of intercellular adhesion molecule-1 expression in human saphenous vein endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology 20 (3):652–658 [DOI] [PubMed] [Google Scholar]
- 14.Languino LR, Plescia J, Duperrray A, Brian AA, Plow EF, Geltosky JE, Alteri DC (1993) Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell 73 (7):1423–1434 [DOI] [PubMed] [Google Scholar]
- 15.Altieri DC, Duperray A, Plescia J, Thornton GB, Languino LR (1995) Structural recognition of a novel fibrinogen gamma chain sequence (117–133) by intercellular adhesion molecule-1 mediates leukocyte-endothelium interaction. J Biol Chem 270 (2):696–699 [DOI] [PubMed] [Google Scholar]
- 16.Sen U, Tyagi N, Patibandla PK, Dean WL, Tyagi SC, Roberts AM, Lominadze D (2009) Fibrinogen-induced endothelin-1 production from endothelial cells. Am J Physiol Cell Physiol 296 (4):C840–C847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D (2008) Fibrinogen induces endothelial cell permeability. Molecular & Cellular Biochemistry 307 (1–2):13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tennent GA, Brennan SO, Stangou AJ, O’Grady J, Hawkins PN, Pepys MB (2007) Human plasma fibrinogen is synthesized in the liver. Blood 109 (5):1971–1974. doi: 10.1182/blood-2006-08-040956 [DOI] [PubMed] [Google Scholar]
- 19.Castell J, Gómez-Lechón M, David M, Fabra R, Trullenque R, Heinrich P (1990) Acute-phase response of human hepatocytes: regulation of acute-phase protein synthesis by interleukin-6. Hepatology 12 (5):1179–1186. doi: 10.1002/hep.1840120517 [DOI] [PubMed] [Google Scholar]
- 20.Paul J, Strickland S, Melchor JP (2007) Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. The Journal of Experimental Medicine 204 (8):1999–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu JK, Davalos D, Akassoglou K (2009) Fibrinogen signal transduction in the nervous system. Journal of Thrombosis and Haemostasis 7:151–154. doi: 10.1111/j.1538-7836.2009.03438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Wang J, Wu X, Xi C, Gai Y, Liu H, Yuan Q, Wang E, Gao L, Hu J, Zhou L (2011) Validating the incidence of coagulopathy and disseminated intravascular coagulation in patients with traumatic brain injury – analysis of 242 cases. British Journal of Neurosurgery 25 (3):363–368. doi:doi: 10.3109/02688697.2011.552650 [DOI] [PubMed] [Google Scholar]
- 23.Jenkins DR, Craner MJ, Esiri MM, DeLuca GC (2018) The contribution of fibrinogen to inflammation and neuronal density in human traumatic brain injury. Journal of Neurotrauma 35 (19):2259–2271. doi: 10.1089/neu.2017.5291 [DOI] [PubMed] [Google Scholar]
- 24.Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV (2002) Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood–brain barrier. European Journal of Clinical Investigation 32 (5):360–371. doi: 10.1046/j.1365-2362.2002.00994.x [DOI] [PubMed] [Google Scholar]
- 25.Yates RL, Esiri MM, Palace J, Jacobs B, Perera R, DeLuca GC (2017) Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Annals of Neurology 82 (2):259–270. doi: 10.1002/ana.24997 [DOI] [PubMed] [Google Scholar]
- 26.Li X, Zhu Z, Gao S, Zhang L, Cheng X, Li S, Li M (2019) Inhibition of fibrin formation reduces neuroinflammation and improves long-term outcome after intracerebral hemorrhage. International Immunopharmacology 72:473–478. doi: 10.1016/j.intimp.2019.04.029 [DOI] [PubMed] [Google Scholar]
- 27.Adams R, Passino M, Sachs B, Nuriel T, Akassoglou K (2004) Fibrin mechanisms and functions in nervous system pathology. Molecular Interventions 4 (3):163–176. doi: 10.1124/mi.4.3.6 [DOI] [PubMed] [Google Scholar]
- 28.Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S (2010) Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA 107 (50):21812–21817. doi: 10.1073/pnas.1010373107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vos CMP, Geurts JJG, Montagne L, van Haastert ES, Bö L, van der Valk P, Barkhof F, de Vries HE (2005) Blood–brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiology of Disease 20 (3):953–960. doi: 10.1016/j.nbd.2005.06.012 [DOI] [PubMed] [Google Scholar]
- 30.Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-[beta] pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 11 (5):361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cortes-Canteli M, Strickland S (2009) Fibrinogen, a possible key player in Alzheimer’s disease. Journal of Thrombosis and Haemostasis 7:146–150. doi: 10.1111/j.1538-7836.2009.03376.x [DOI] [PubMed] [Google Scholar]
- 32.Hay J,R, Johnson VE, Young AM, Smith DH, Stewart W (2015) Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. Journal of Neuropathology and Experimental Neurology 74 (12):1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuah YML, Maybery MT, Fox AM (2004) The long-term effects of mild head injury on short-term memory for visual form, spatial location, and their conjunction in well-functioning university students. Brain and Cognition 56 (3):304–312. doi: 10.1016/j.bandc.2004.08.002 [DOI] [PubMed] [Google Scholar]
- 34.Muradashvili N, Benton RL, Saatman KE, Tyagi SC, Lominadze D (2015) Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab Brain Dis 30 (2):411–426. doi: 10.1007/s11011-014-9550-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muradashvili N, Tyagi SC, Lominadze D (2017) Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sciences 7 (7):77. doi: 10.3390/brainsci7070077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muradashvili N, Tyagi R, Tyagi N, Tyagi SC, Lominadze D (2016) Cerebrovascular disorders caused by hyperfibrinogenemia. The Journal of Physiology 594 (20):5941–5957. doi: 10.1113/JP272558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mogford JE, Davis GE, Platts SH, Meininger GA (1996) Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res 79 (4):821–826 [DOI] [PubMed] [Google Scholar]
- 38.Muradashvili N, Benton R, Tyagi R, Tyagi S, Lominadz D (2014) Elevated level of fibrinogen increases caveolae formation; Role of matrix metalloproteinase-9. Cell Biochem Biophys 69 (2):283–294. doi: 10.1007/s12013-013-9797-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muradashvili N, Khundmiri SJ, Tyagi R, Gartung A, Dean WL, Lee M-J, Lominadze D (2014) Sphingolipids affect fibrinogen-induced caveolar transcytosis and cerebrovascular permeability. Am J Physiol Cell Physiol 307 (2):C169–C179. doi: 10.1152/ajpcell.00305.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muradashvili N, Qipshidze N, Munjal C, Givvimani S, Benton RL, Roberts AM, Tyagi SC, Lominadze D (2012) Fibrinogen-induced increased pial venular permeability in mice. J Cereb Blood Flow Metab 32 (1):150–163. doi: 10.1038/jcbfm.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muradashvili N, Tyagi N, Tyagi R, Munjal C, Lominadze D (2011) Fibrinogen alters mouse brain endothelial cell layer integrity affecting vascular endothelial cadherin. Biochemical and Biophysical Research Communications 413 (4):509–514. doi: 10.1016/j.bbrc.2011.07.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark VD, Layson A, Charkviani M, Muradashvili N, Lominadze D (2018) Hyperfibrinogenemia-mediated astrocyte activation. Brain Research 1699:158–165. doi: 10.1016/j.brainres.2018.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K (2007) The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. The Journal of Experimental Medicine 204 (3):571–582. doi: 10.1084/jem.20061931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, Seeley WW, Mucke L, Nelson RB, Akassoglou K (2019) Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 101 (6):1099–1108.e1096. doi: 10.1016/j.neuron.2019.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letcher RL, Chien S, Pickering TG, Sealey JE, Laragh JH (1981) Direct relationship between blood pressure and blood viscosity in normal and hypertensive subjects. Role of fibrinogen and concentration. The American Journal of Medicine 70:1195–1202 [DOI] [PubMed] [Google Scholar]
- 46.Lominadze D, Schuschke DA, Joshua IG, Dean WL (2002) Increased ability of erythrocytes to aggregate in spontaneously hypertensive rats. Clin Exp Hypertens 24 ((5)):397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL (2004) Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113 (11):1596–1606. doi: 10.1172/JCI20741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP (2002) Regulated unmasking of the cryptic binding site foriIntegrin αMβ2 in the γC-domain of fibrinogen. Biochemistry 41 (43):12942–12951. doi: 10.1021/bi026324c [DOI] [PubMed] [Google Scholar]
- 49.Lominadze D, Dean WL (2002) Involvement of fibrinogen specific binding in erythrocyte aggregation. FEBS Letters 517 (1–3):41–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patibandla PK, Tyagi N, Dean WL, Tyagi SC, Roberts AM, Lominadze D (2009) Fibrinogen induces alterations of endothelial cell tight junction proteins. Journal of Cellular Physiology 221 (1):195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muradashvili N, Tyagi R, Lominadze D (2012) A dual-tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Frontiers in Physiology 3:166–172. doi: 10.3389/fphys.2012.00166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimelberg HK, Norenberg MD (1989) Astrocytes. Scientific American 260 (4):66–72, 74, 76. doi: 10.1038/scientificamerican0489-66 [DOI] [PubMed] [Google Scholar]
- 53.Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathologica 119 (1):7–35. doi: 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes - Implications for their role in neurologic disease. Neuroscience 54 (1):15–36. doi: 10.1016/0306-4522(93)90380-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta V, You Y, Gupta V, Klistorner A, Graham S (2013) TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. International Journal of Molecular Sciences 14 (5):10122–10142. doi: 10.3390/ijms140510122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada L, Medico E, Hohlfeld R, Meinl E, Farina C (2012) Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. Journal of Experimental Medicine 209 (3):521–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Genius J, Fandrey J (2000) Nitric oxide affects the production of reactive oxygen species in hepatoma cells: implications for the process of oxygen sensing. Free Radical Biology and Medicine 29 (6):515–521. doi:Doi: 10.1016/s0891-5849(00)00343-9 [DOI] [PubMed] [Google Scholar]
- 58.Ishii T, Takanashi Y, Sugita K, Miyazawa M, Yanagihara R, Yasuda K, Onouchi H, Kawabe N, Nakata M, Yamamoto Y, Hartman PS, Ishii N (2017) Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: a new model for aging brain. Aging Cell 16 (1):39–51. doi: 10.1111/acel.12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brosnan C, Cannella B, Battistini L, Raine C (1995) Cytokine localization in multiple sclerosis lesions: correlation with adhesion molecule expression and reactive nitrogen species. Neurology 45 (6 Suppl 6):S16–S21 [DOI] [PubMed] [Google Scholar]
- 60.Lee SJ, Drabik K, Van Wagoner NJ, Lee S, Choi C, Dong Y, Benveniste EN (2000) ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. The Journal of Immunology 165 (8):4658–4666. doi: 10.4049/jimmunol.165.8.4658 [DOI] [PubMed] [Google Scholar]
- 61.Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. The Journal of Neuroscience 30 (17):5843–5854. doi: 10.1523/jneurosci.0137-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW (2000) Neurocan Is Upregulated in Injured Brain and in Cytokine-Treated Astrocytes. The Journal of Neuroscience 20 (7):2427. doi: 10.1523/JNEUROSCI.20-07-02427.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsiao TW, Swarup VP, Kuberan B, Tresco PA, Hlady V (2013) Astrocytes specifically remove surface-adsorbed fibrinogen and locally express chondroitin sulfate proteoglycans. Acta Biomaterialia 9 (7):7200–7208. doi: 10.1016/j.actbio.2013.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Webster KM, Wright DK, Sun M, Semple BD, Ozturk E, Stein DG, O’Brien TJ, Shultz SR (2015) Progesterone treatment reduces neuroinflammation, oxidative stress and brain damage and improves long-term outcomes in a rat model of repeated mild traumatic brain injury. J Neuroinflammation 12:238. doi: 10.1186/s12974-015-0457-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shultz SR, Sun M, Wright DK, Brady RD, Liu S, Beynon S, Schmidt SF, Kaye AH, Hamilton JA, O’Brien TJ, Grills BL, McDonald SJ (2015) Tibial Fracture Exacerbates Traumatic Brain Injury Outcomes and Neuroinflammation in a Novel Mouse Model of Multitrauma. Journal of Cerebral Blood Flow & Metabolism 35 (8):1339–1347. doi: 10.1038/jcbfm.2015.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Webster KM, Sun M, Crack P, O’Brien TJ, Shultz SR, Semple BD (2017) Inflammation in epileptogenesis after traumatic brain injury. Journal of Neuroinflammation 14 (1):10–10. doi: 10.1186/s12974-016-0786-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faden AI, Wu J, Stoica BA, Loane DJ (2016) Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol 173 (4):681–691. doi: 10.1111/bph.13179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Corps KN, Roth TL, McGavern DB (2015) Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72 (3):355–362. doi: 10.1001/jamaneurol.2014.3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cafferty WBJ, Yang S-H, Duffy PJ, Li S, Strittmatter SM (2007) Functional Axonal Regeneration through Astrocytic Scar Genetically Modified to Digest Chondroitin Sulfate Proteoglycans. The Journal of Neuroscience 27 (9):2176. doi: 10.1523/JNEUROSCI.5176-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clark DPQ, Perreau VM, Shultz SR, Brady RD, Lei E, Dixit S, Taylor JM, Beart PM, Boon WC (2019) Inflammation in Traumatic Brain Injury: Roles for Toxic A1 Astrocytes and Microglial-Astrocytic Crosstalk. Neurochem Res 44 (6):1410–1424. doi: 10.1007/s11064-019-02721-8 [DOI] [PubMed] [Google Scholar]
- 71.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541 (7638):481–487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ransohoff RM, Perry VH (2009) Microglial Physiology: Unique Stimuli, Specialized Responses. Annual Review of Immunology 27 (1):119–145. doi: 10.1146/annurev.immunol.021908.132528 [DOI] [PubMed] [Google Scholar]
- 73.Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227. doi:http://www.nature.com/ncomms/journal/v3/n11/suppinfo/ncomms2230_S1.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fan ST, Edgington TS (1993) Integrin regulation of leukocyte inflammatory functions. CD11b/CD18 enhancement of the tumor necrosis factor-alpha responses of monocytes. The Journal of Immunology 150 (7):2972. [PubMed] [Google Scholar]
- 75.Sela U, Sharabi A, Dayan M, Hershkoviz R, Mozes E (2009) The role of dendritic cells in the mechanism of action of a peptide that ameliorates lupus in murine models. Immunology 128 (1pt2):e395–e405. doi: 10.1111/j.1365-2567.2008.02988.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boche D, Perry VH, Nicoll JAR (2013) Review: Activation patterns of microglia and their identification in the human brain. Neuropathology and Applied Neurobiology 39 (1):3–18. doi: 10.1111/nan.12011 [DOI] [PubMed] [Google Scholar]
- 77.Liddelow SA, Barres BA (2017) Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46 (6):957–967. doi: 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- 78.Jang E, Kim J-H, Lee S, Kim J-H, Seo J-W, Jin M, Lee M-G, Jang I-S, Lee W-H, Suk K (2013) Phenotypic Polarization of Activated Astrocytes: The Critical Role of Lipocalin-2 in the Classical Inflammatory Activation of Astrocytes. The Journal of Immunology 191 (10):5204. doi: 10.4049/jimmunol.1301637 [DOI] [PubMed] [Google Scholar]
- 79.Sardi S, Vardi R, Sheinin A, Goldental A, Kanter I (2017) New Types of Experiments Reveal that a Neuron Functions as Multiple Independent Threshold Units. Scientific Reports 7 (1):18036. doi: 10.1038/s41598-017-18363-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jójárt I, Joó F, Siklós L, László FA (1984) Immunoelectronhistochemical evidence for innervation of brain microvessels by vasopressin-immunoreactive neurons in the rat. Neuroscience Letters 51 (2):259–264. doi: 10.1016/0304-3940(84)90561-5 [DOI] [PubMed] [Google Scholar]
- 81.Ill-Raga G, Palomer E, Ramos-Fernández E, Guix FX, Bosch-Morató M, Guivernau B, Tajes M, Valls-Comamala V, Jiménez-Conde J, Ois A, Pérez-Asensio F, Reyes-Navarro M, Caballo C, Gil-Gómez G, Lopez-Vilchez I, Galan AM, Alameda F, Escolar G, Opazo C, Planas AM, Roquer J, Valverde MA, Muñoz FJ (2015) Fibrinogen nitrotyrosination after ischemic stroke impairs thrombolysis and promotes neuronal death. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1852 (3):421–428. doi: 10.1016/j.bbadis.2014.12.007 [DOI] [PubMed] [Google Scholar]
- 82.Kovalevich J, Langford D (2013) Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology In: Methods in Molecular Biology, vol 1078. Neuronal Cell Culture Humana Press, Totowa, N.J., pp 9–21. doi: 10.1007/978-1-62703-640-5_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nuñez G, Benedict MA, Hu Y, Inohara N (1998) Caspases: the proteases of the apoptotic pathway. Oncogene 17 (25):3237–3245. doi: 10.1038/sj.onc.1202581 [DOI] [PubMed] [Google Scholar]
- 84.Lassmann H (2010) Axonal and neuronal pathology in multiple sclerosis: what have we learnt from animal models. Exp Neurol 225 (1):2–8. doi: 10.1016/j.expneurol.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 85.Pandey HS, Seth P (2019) Friends Turn Foe-Astrocytes Contribute to Neuronal Damage in NeuroAIDS. J Mol Neurosci 69 (2):286–297. doi: 10.1007/s12031-019-01357-1 [DOI] [PubMed] [Google Scholar]
- 86.Hall ED, Vaishnav RA, Mustafa AG (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7 (1):51–61. doi: 10.1016/j.nurt.2009.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith KJ, Kapoor R, Hall SM, Davies M (2001) Electrically active axons degenerate when exposed to nitric oxide. Annals of Neurology 49 (4):470–476. doi: 10.1002/ana.96 [DOI] [PubMed] [Google Scholar]
- 88.Haniu M, Montestruque S, Bures EJ, Talvenheimo J, Toso R, Lewis-Sandy S, Welcher AA, Rohde MF (1997) Interactions between brain-derived neurotrophic factor and the TrkB receptor: Identification of Two Ligand Binding Domains in Soluble TRKB by Affinity Separation and Chemical Cross-Linking. Journal of Biological Chemistry 272 (40):25296–25303. doi: 10.1074/jbc.272.40.25296 [DOI] [PubMed] [Google Scholar]
- 89.Leal G, Afonso PM, Salazar IL, Duarte CB (2015) Regulation of hippocampal synaptic plasticity by BDNF. Brain Research 1621:82–101. doi: 10.1016/j.brainres.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 90.Onodera T (2017) Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc Jpn Acad Ser B Phys Biol Sci 93 (4):155–173. doi: 10.2183/pjab.93.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schneider B, Mutel V, Mathéa Pietri M, Ermonval M, Mouillet-Richard S, Kellermann O (2003) NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells Proc Natl Acad Sci USA 100 (23):13326–13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S (2010) Fibrinogen and β-Amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s Disease. Neuron 66 (5):695–709. doi: 10.1016/j.neuron.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zamolodchikov D, Strickland S (2012) Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 119 (14):3342–3351. doi: 10.1182/blood-2011-11-389668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud P-O, Piontek J, Blasig IE, Dijkstra CD, Ronken E, de Vries HE (2007) Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J 21 (13):3666–3676. doi: 10.1096/fj.07-8329com [DOI] [PubMed] [Google Scholar]
- 95.Chodobski A, Zink BJ, Szmydynger-Chodobska J (2011) Blood-brain barrier pathophysiology in traumatic brain injury. Translational stroke research 2 (4):492–516. doi: 10.1007/s12975-011-0125-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muradashvili N, Lominadze D (2013) Role of fibrinogen in cerebrovascular dysfunction after traumatic brain injury. Brain Injury 27 (13–14):1508–1515. doi: 10.3109/02699052.2013.823562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kossmann T, Hans VH, Imhof HG, Stocker R, Grob P, Trentz O, Morganti-Kossmann C (1995) Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 4 (5):311–317. doi: 10.1097/00024382-199511000-00001 [DOI] [PubMed] [Google Scholar]
- 98.Gabay C, Kushner I (1999) Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 340 (6):448–454. doi:doi: 10.1056/NEJM199902113400607 [DOI] [PubMed] [Google Scholar]
- 99.Ghirnikar RS, Lee YL, Eng LF (1998) Inflammation in Traumatic Brain Injury: Role of Cytokines and Chemokines. Neurochem Res 23 (3):329–340. doi: 10.1023/A:1022453332560 [DOI] [PubMed] [Google Scholar]
- 100.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T (2002) Inflammatory response in acute traumatic brain injury: a double-edged sword. Current Opinion in Critical Care 8 (2):101–105 [DOI] [PubMed] [Google Scholar]
- 101.Cederberg D, Siesjö P (2010) What has inflammation to do with traumatic brain injury? Child’s Nervous System 26 (2):221–226. doi: 10.1007/s00381-009-1029-x [DOI] [PubMed] [Google Scholar]
- 102.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ (2011) Inflammation after trauma: Microglial activation and traumatic brain injury. Annals of Neurology 70 (3):374–383. doi: 10.1002/ana.22455 [DOI] [PubMed] [Google Scholar]
- 103.Harting MT, Jimenez F, Adams SD, Mercer DW, Cox CS Jr. (2008) Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery 144 (5):803–813. doi: 10.1016/j.surg.2008.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Morganti-Kossman MC, Lenzlinger PM, Hans V, Stahel P, Csuka E, Ammann E, Stocker R, Trentz O, Kossmann T (1997) Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Molecular Psychiatry 2 (2):133–136. doi: 10.1038/sj.mp.4000227 [DOI] [PubMed] [Google Scholar]
- 105.Vasse M, Paysant J, Soria J, Collet JP, Vannier JP, Soria C (1996) Regulation of fibrinogen biosynthesis by cytokines, consequences on the vascular risk. Haemostasis 26 ((Suppl 4)):331–339 [DOI] [PubMed] [Google Scholar]
- 106.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK (1999) Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A 96 (15):8721–8726. doi: 10.1073/pnas.96.15.8721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shrikant P, Il Yup C, Ballestas ME, Benveniste EN (1994) Regulation of intercellular adhesion molecule-1 gene expression by tumor necrosis factor-α, interleukin-1β, and interferon-γ in astrocytes. Journal of Neuroimmunology 51 (2):209–220. doi: 10.1016/0165-5728(94)90083-3 [DOI] [PubMed] [Google Scholar]
- 108.Khorooshi R, Babcock AA, Owens T (2008) NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. Journal of Immunology 181 (10):7284–7291. doi: 10.4049/jimmunol.181.10.7284 [DOI] [PubMed] [Google Scholar]
- 109.Colombo E, Farina C (2016) Astrocytes: Key Regulators of Neuroinflammation. Trends in Immunology 37 (9):608–620. doi: 10.1016/j.it.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 110.Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR (2009) Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol 182 (5):2628–2640. doi: 10.4049/jimmunol.0802954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shih R-H, Wang C-Y, Yang C-M (2015) NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Frontiers in Molecular Neuroscience 8:77–77. doi: 10.3389/fnmol.2015.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mettang M, Reichel SN, Lattke M, Palmer A, Abaei A, Rasche V, Huber-Lang M, Baumann B, Wirth T (2018) IKK2/NF-κB signaling protects neurons after traumatic brain injury. FASEB J 32 (4):1916–1932. doi: 10.1096/fj.201700826R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lian H, Shim DJ, Gaddam SSK, Rodriguez-Rivera J, Bitner BR, Pautler RG, Robertson CS, Zheng H (2012) IκBα deficiency in brain leads to elevated basal neuroinflammation and attenuated response following traumatic brain injury: implications for functional recovery. Molecular Neurodegeneration 7 (1):47. doi: 10.1186/1750-1326-7-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hennessy E, Griffin EW, Cunningham C (2015) Astrocytes Are Primed by Chronic Neurodegeneration to Produce Exaggerated Chemokine and Cell Infiltration Responses to Acute Stimulation with the Cytokines IL-1beta and TNF-alpha. J Neurosci 35 (22):8411–8422. doi: 10.1523/jneurosci.2745-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bettcher BM, Neuhaus J, Wynn MJ, Elahi FM, Casaletto KB, Saloner R, Fitch R, Karydas A, Kramer JH (2019) Increases in a Pro-inflammatory Chemokine, MCP-1, Are Related to Decreases in Memory Over Time. Frontiers in Aging Neuroscience 11 (Article 25):1–9. doi: 10.3389/fnagi.2019.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM (1999) Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 103 (6):807–815. doi: 10.1172/jci5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mills Ko E, Ma JH, Guo F, Miers L, Lee E, Bannerman P, Burns T, Ko D, Sohn J, Soulika AM, Pleasure D (2014) Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. Journal of Neuroinflammation 11 (1):105. doi: 10.1186/1742-2094-11-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sans E, Delachanal E, Duperray A (2001) Analysis of the Roles of ICAM-1 in Neutrophil Transmigration Using a Reconstituted Mammalian Cell Expression Model: Implication of ICAM-1 Cytoplasmic Domain and Rho-Dependent Signaling Pathway. The Journal of Immunology 166 (1):544. doi: 10.4049/jimmunol.166.1.544 [DOI] [PubMed] [Google Scholar]
- 119.Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Hamada S, Shimosegawa T (2009) Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut 58 (4):550. doi: 10.1136/gut.2008.154401 [DOI] [PubMed] [Google Scholar]
- 120.del Zoppo GJ, Levy DE, Wasiewski WW, Pancioli AM, Demchuk AM, Trammel J, Demaerschalk BM, Kaste M, Albers GW, Ringelstein EB (2009) Hyperfibrinogenemia and functional outcome from acute ischemic stroke. Stroke 40 (5):1687–1691. doi: 10.1161/strokeaha.108.527804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kockx M, Gervois PP, Poulain P, Derudas B, Peters JM, Gonzalez FJ, Princen HMG, Kooistra T, Staels B (1999) Fibrates suppress fibrinogen gene expression in rodents via activation of the peroxisome proliferator-activated receptor-α. Blood 93 (9):2991–2998 [PubMed] [Google Scholar]
- 122.Genest JJ, Nguyen N-H, Theroux P, Davignon J, Cohn JS (2000) Effect of Micronized Fenofibrate on Plasma Lipoprotein Levels and Hemostatic Parameters of Hypertriglyceridemic Patients with Low Levels of High-Density Lipoprotein Cholesterol in the Fed and Fasted State. Journal of Cardiovascular Pharmacology 35 (1):164–172 [DOI] [PubMed] [Google Scholar]
- 123.Ni H, Denis C, Subbarao S, Degen J, Sato T, Hynes R, Wagner D (2000) Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. Journal of Clinical Investigation 106 (3):385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wilberding JA, Ploplis VA, McLennan L, Liang Z, Cornelissen IVO, Feldman M, Deford ME, Rosen ED, Castellino FJ (2001) Development of pulmonary fibrosis in fibrinogen-deficient mice. Annals of the New York Academy of Sciences 936 (1):542–548. doi: 10.1111/j.1749-6632.2001.tb03542.x [DOI] [PubMed] [Google Scholar]
- 125.Yuasa M, Mignemi NA, Nyman JS, Duvall CL, Schwartz HS, Okawa A, Yoshii T, Bhattacharjee G, Zhao C, Bible JE, Obremskey WT, Flick MJ, Degen JL, Barnett JV, Cates JMM, Schoenecker JG (2015) Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. The Journal of Clinical Investigation 125 (8):3117–3131. doi: 10.1172/JCI80313 [DOI] [PMC free article] [PubMed] [Google Scholar]