

Abstract
cAMP-response element binding protein (CREB) is an oncogenic transcription factor implicated in many different types of cancer. We previously reported the discovery of 666-15 as a potent inhibitor of CREB-mediated gene transcription. In an effort to improve the aqueous solubility of 666-15, amino ester prodrugs 1 and 4 were designed and synthesized. Detailed chemical and biological studies of 1 and 4 revealed that a small portion of the prodrugs were converted into 666-15 through intermediate 3 instead of a long-range O,N-acyl transfer reaction that was initially proposed. These results provide unique insights into the activation of these ester prodrugs.
Keywords: CREB, cancer, inhibitor, prodrug, transcription
Graphical Abstract
cAMP-response element-binding protein (CREB) is a nucleus resided transcription factor involved in numerous biological processes including cellular proliferation, differentiation and memory formation.1 It belongs to a large family of basic leucine zipper (bZIP)-containing transcription factors including c-Jun, c-Fos and c-Myc. The bZIP domain in the C-terminus can homodimerize to bind the cognate DNA sequence of 5’-TGACGTCA-3’ called cAMP-response element (CRE).2 While this binding is thought to be constitutive in the cells, CREB’s transcription activity is not turned on until it is phosphorylated at Ser133 by various protein serine/threonine kinases. The first protein kinase known to phosphorylate CREB is protein kinase A (PKA).3 Since then, many other protein kinases have been shown to be able to phosphorylate CREB to turn on its transcription activity, which include protein kinase B (PKB/Akt), ribosomal S6 kinase (p90RSK) and mitogen-activated protein kinases (MAPKs).4 Phosphorylation of CREB at Ser133 is very dynamic to allow cells to respond to extracellular and intracellular signals. There are protein phosphatases that can dephosphorylate CREB. The protein phosphatases that are known to dephosphorylate CREB include protein phosphatase 1 (PP1),5 protein phosphatase 2A (PP2A),6 and phosphatase and tension homolog (PTEN).7 This dynamic and reversible phosphorylation of CREBs allows its transcription activity to be tightly regulated under normal cellular homeostasis.
In the cancer cells, however, the kinases that can phosphorylate CREB are often mutated or overexpressed to confer their oncogenic activity. As a consequence, the positive signals to drive CREB phosphorylation in the cancer cells are increased. On the other hand, the protein phosphatases known to dephosphorylate CREB are tumor suppressor proteins that are often inactivated or deleted in the cancer cells, resulting in a decrease of signals to turn off CREB-mediated gene transcription. Due to the dysregulation of both positive and negative signals to CREB in cancer cells, CREB has been shown to be consistently upregulated and activated in many different cancer tissues including breast, lung, prostate, kidney, brain, pancreas and blood.4,8–12 Because of this upregulation in many cancer tissues, CREB has been pursued as a potential cancer drug target. Consistent with this idea, genetic inhibition of CREB using both shRNA and dominant negative CREB mutants has been shown to produce profound anti-cancer effect in multiple preclinical cancer models.4,9,12 Encouraged by these promising results, we initiated development of small molecule inhibitors of CREB-mediated gene transcription.13–15 We previously developed 666-15 as a potent inhibitor of CREB-mediated gene transcription.16,17 Kikuchi group recently reported a photo-caged version of 666-15 to allow potential spatio-temporal control of CREB inhibition.18
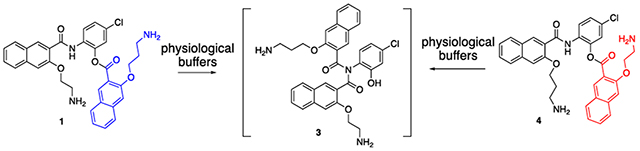
Despite 666-15’s potent CREB inhibitory activity, its aqueous solubility needs further improvement.19 In an effort to improve the aqueous solubility of 666-15, we designed amino ester compound 1 as a traceless prodrug for 666-15 based on a long-range O,N-acyl transfer reaction that we previously described (Scheme 1).20 It was anticipated that the primary amino group in 1 will nucleophilically attack the ester bond to form amide 666-15 in high yield at physiologically relevant buffers as observed before for a closely related congener compound S1 (pathway A in Scheme 1 and Scheme S1).20 Surprisingly, we found that only a small amount (<10%) of 1 was converted into 666-15 in biologically relevant buffers at pH = 7.40.19 Instead, the majority of compound 1 was converted into 653-47 and 2 (Scheme 1).19 This unexpected conversion of compound 1 into 653-47 and 2 was likely through an imide intermediate 3 (pathway B in Scheme 1). Imide 3 could then either be cleaved to generate 653-47 and 2 or rearranged to give 666-15 as a minor pathway. The unanticipated discovery of 653-47 from 1 became significant because we found that 653-47 was able to potentiate 666-15’s inhibitory activity against CREB-mediated gene transcription even though 653-47 was inactive alone.19
Scheme 1.
Pathways for converting 1 to 666-15 and 653-47
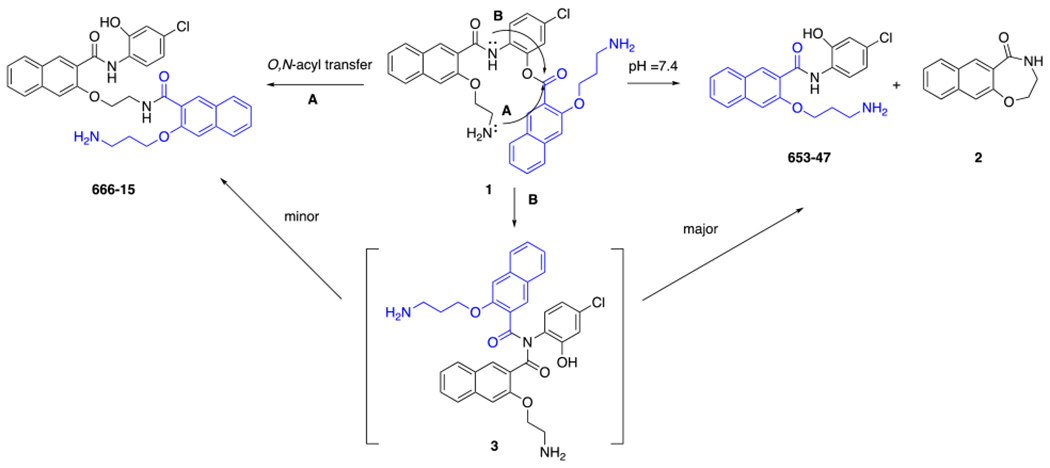
While the mechanism of activation of prodrug 1 invoking imide 3 is intriguing, further experimental evidence is still lacking. We hypothesized that the regioisomer amino ester 4 should be able to give the same intermediate imide 3 by pathway B (Scheme 2), which shall give the same products 653-47 and 2 as the major products and 666-15 as a minor species. On the other hand, if the alternative direct O,N-acyl transfer was the major mechanism for the conversion (pathway A in Scheme 2), the anticipated product would be amide 5. In this communication, we synthesized compound 4 and further studied its hydrolytical stability and biological activities to provide unique mechanistic insights into the activation of these designed ester prodrugs.
Scheme 2.
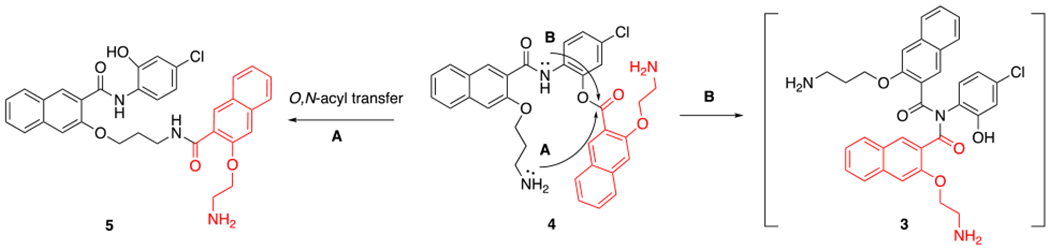
Proposed conversion of amino ester 4
The synthesis of compound 4 is shown in Scheme 3. When we employed a typical amide coupling protocol with BOP or MsCl as the activation reagent, we observed the formation of a mixture of both desired amide 8 and undesired amide 10, which were difficult to separate by column chromatography. Changing to other coupling reagents including EDCI or DCC did not improve the reaction outcome. We previously reported that lowering the reaction temperature and decreasing the reaction time could significantly inhibit the formation of undesired isomer albeit at the expense of reduced reaction conversion.19 By applying this revised protocol (BOP, 0 °C, 1 h), we were able to isolate pure compound 8 in 23% yield. Finally, deprotection of the Boc groups in 8 delivered designed compound 4 smoothly. While isomers 1 and 4 are structurally very similar, their 1H NMR spectra are quite distinct from each other in both the aromatic region and aliphatic region (Scheme 3). Similar to compound 1,19 compound 4 also exhibited dramatically improved aqueous solubility in ddI H2O (> 100 mg/mL) compared to 666-15·HCl (< 0.5 mg/mL in ddI H2O). Assessing the solubility of 1 and 4 in aqueous buffers at pH=7.4 was hampered due to its instability at this pH (see below). That the preferential formation of 8 from 6 and 7 versus preferential formation of 10 from 11 and 1219 under these conditions suggests that the proposed imide intermediate 9 was not formed directly from 6 and 7 or 11 and 12. Instead, it is more likely that 9 was formed after 8 or 10 was generated during the reaction. Decreasing the reaction time and temperature could potentially inhibit this rate-limiting step of formation of 9 from 8 or 10.
Scheme 3.
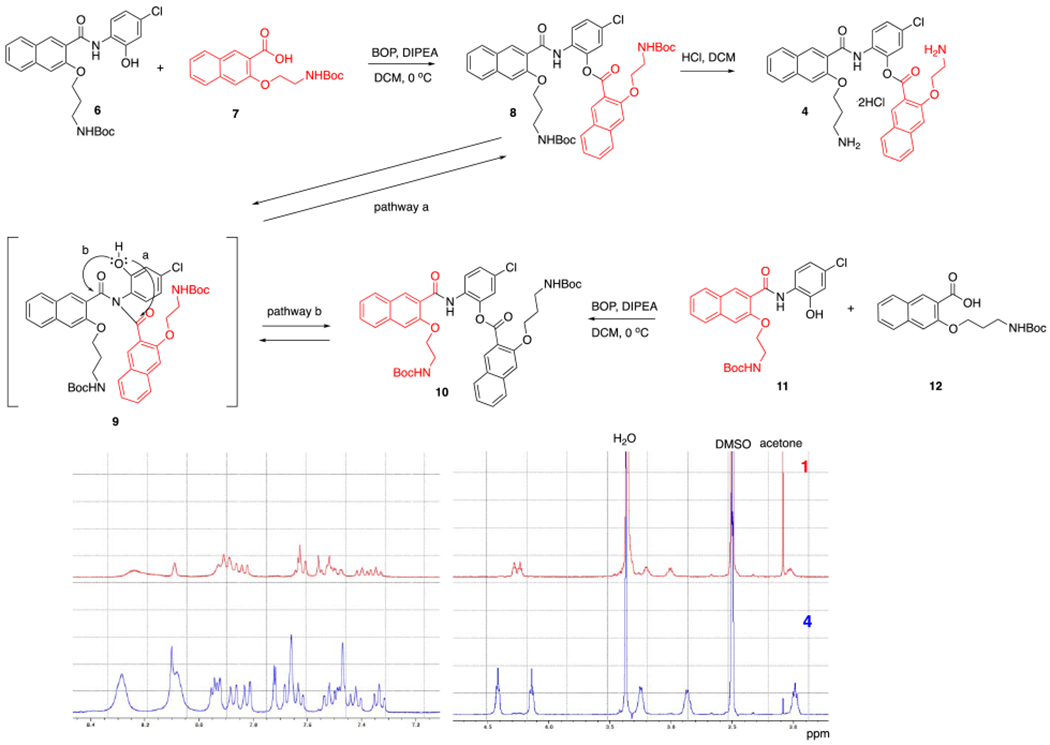
Synthesis of compound 4
With compound 4 in hand, we evaluated its stability and reaction conversion in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS). To this end, compound 4 (200 μM) was incubated in the complete tissue culture media at 37 °C for different periods of time, when an aliquot was taken for HPLC analysis. Similar to compound 1,19 compound 4 was very unstable in the complete tissue culture media and rapidly converted into multiple species (Figure 1A). The HPLC peaks generated from incubating 4 were the same as those generated from 1. After 5 minutes of incubation at 37 °C, most of 4 was converted into 653-47 and 2. Smaller amounts of 666-15 and 5 were also generated (Figure 1). The individual peaks were identified by carefully comparing with the previously synthesized authentic samples (Figure S1).16,19 Under this incubation condition, 666-15 itself was found to be very stable and unchanged even after 24 h incubation (Figure S2). To further confirm the identities of the individual species generated from 4, it was treated in phosphate buffer saline (PBS, pH 7.4) and then the reaction mixture was subjected to Boc protection to facilitate purification of compounds containing amino groups (Scheme 4). After careful chromatography separation and 1H NMR analyses, this reaction gave an inseparable mixture of 13 and 14 (23%) in a ratio of 2:1 as determined by 1H-NMR, mono-Boc protected 15 (49%), doubly Boc protected 16 (10%) and cyclic amide 2 (45%). The discovery that both 1 and 4 produced the same composition of mixtures further supports that pathway B in Scheme 2 was the pathway to contribute to the formation of 666-1519 as opposed to pathway A involving a long-range O,N-acyl transfer reaction that we previously proposed.20
Figure 1.
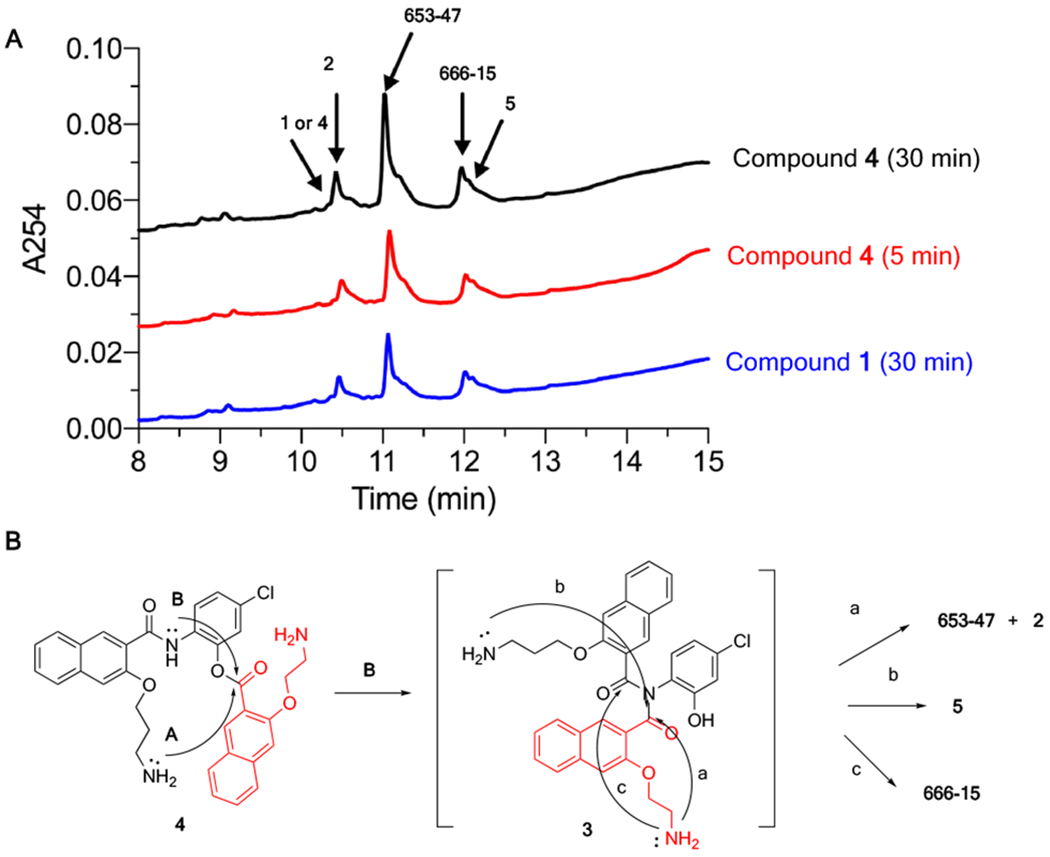
Compound 4 was converted into 666-15, 653-47, 2 and 5 in complete tissue culture media. (A) The HPLC traces of the reaction mixtures from incubating 1 or 4 in complete tissue culture media at 37 °C for different periods of time. (B) Proposed reaction pathways for generation of 666-15, 653-47, 2 and 5 from 4.
Scheme 4.
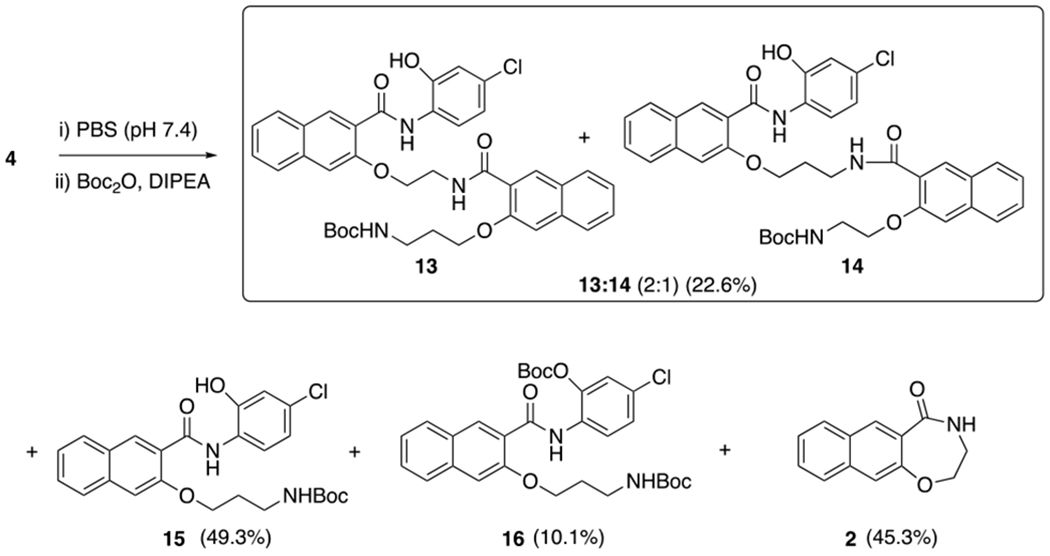
Reaction conversion of 4 in PBS
The biological activities of compounds 1 and 4 were further evaluated and compared. If both 1 and 4 were converted to the same mixture of species in the complete tissue culture media, they would be expected to have the same biological activities. To test this hypothesis, we first evaluated their activities in inhibiting CREB-mediated gene transcription using a cell-based CREB transcription reporter assay.13 In this assay, HEK293T cells were transiently transfected with a plasmid pCRE-RLuc that would express RENILLA luciferase in response to the activation of CREB. Then the cells were treated with different concentrations of the compounds followed by stimulation of the cells with forskolin (Fsk)13,21 to increase CREB phosphorylation. The results in Figure S4A and Table 1 showed that both 1 and 4 were of equal potency in inhibiting CREB-mediated gene transcription (IC50 = 0.26 ± 0.097 μM for 1 and 0.25 ± 0.16 μM for 4), which are consistent with the results that both 1 and 4 were converted to the same species under the assay conditions (Figure 1A). We previously reported that CREB inhibitor 666-15 was very potent in inhibiting triple negative breast cancer (TNBC) cell growth (Table 1).16 666-15 also inhibited the expression of endogenous CREB target gene c-Fos in the TNBC cells (Figure S3). TNBC is an aggressive subtype of breast cancer whose cells do not express estrogen receptor (ER), progesterone receptor (PR) or present amplification of human epidermal growth factor receptor 2 (HER2).22 TNBC patients do not benefit from current targeted breast cancer therapies including ER-targeting (e.g. tamoxifen) and HER2-targeting agents (e.g. trastuzumab, lapatinib). To investigate the potential anti-TNBC effect of 1 and 4, we evaluated their cell growth inhibitory activity in MDA-MB-231 and MDA-MB-468 cells, both of which are TNBC cells. The cells were treated with increasing concentrations of 1 or 4 for 72 h. Then the remaining viable cells were quantified using MTT reagent (methylthiazolyldiphenyl-tetrazolium bromide).14 The concentration needed to inhibit the cell growth by 50% was designated as GI50. As anticipated from the results in Figure 1 showing that both 1 and 4 gave the same reaction mixture upon incubation in the tissue culture media, both of the compounds displayed the same antiproliferative activity in MDA-MB-231 and MDA-MB-468 cells (Figure S4B–4C and Table 1). In MDA-MB-231 cells, the GI50s were 0.54 and 0.67 μM for 1 and 4, respectively. In MDA-MB-468 cells, both of the compounds showed higher potency with GI50 being 0.045 and 0.032 μM, which are on a par with 666-15. In both of the cell lines tested, the compounds produced net cell killing effect at higher concentrations of the drugs (> 10 μM) (Figure S4B–4C).23 Since both compounds 1 and 4 were rapidly converted into 666-15 and 653-47 in the complete tissue culture media under our biological assay conditions, we attributed the potent CREB inhibition activities and breast cancer cell growth inhibition potencies seen with 1 and 4 to the synergistic effect between 666-15 and 653-47.19 It is interesting to note that both compounds 1 and 4 were about 10-fold more potent in MDA-MB-468 cells than MDA-MB-231 cells, suggesting that the extent of synergistic effect between 666-15 and 653-47 may be cell type-dependent, the mechanism of which remains to be established.
Table 1.
The biological activities of 1, 4 and 666-15.a.
| Compound | CREB inhibition IC50 (μM)b | GI50 (μM)c |
|
|---|---|---|---|
| MDA-MB-231 | MDA-MB-468 | ||
| 1 | 0.26 ± 0.097 | 0.54 ± 0.071 | 0.045 ± 0.025 |
| 4 | 0.25 ± 0.16 | 0.67 ± 0.15 | 0.032 ± 0.028 |
| 666-15d | 0.081 ± 0.04 | 0.073 ± 0.04 | 0.046 ± 0.04 |
The stock solutions of compounds 1 and 4 were in made DMF while 666-15 was dissolved in DMSO. The use of DMF was necessary for 1 and 4 because these two compounds were found to be unstable in DMSO.
The CREB inhibition IC50 refers to the concentration needed to inhibit the CREB-mediated gene transcription reporter assay in HEK293T cells by 50%. The IC50 values are presented as mean ± SD of at least two different experiments performed in triplicates.
The GI50 refers to the concentration needed to inhibit the cancer cell growth by 50% in an MTT assay after incubating the cells with the compounds for 72 h. The GI50 values are presented as mean ± SD of at least two different experiments performed in duplicates.
These values are from reference16.
CREB has been investigated as a promising cancer drug target. We previously identified 666-15 as a potent CREB inhibitor with efficacious in vivo anticancer activity. In an effort to improve the aqueous solubility of 666-15 using prodrugs, we recently reported the design of prodrug 1,19 which revealed unexpected synergistic effect of the combination between 666-15 and 653-47. This unexpected discovery also challenged our initial proposal of the transformation from 1 to 666-15 through a long-range O,N-acyl transfer process.19,20 Instead, we proposed a modified mechanism of conversion through imide intermediate 3 (Scheme 1). In our initial proposal for the long-range O,N-acyl transfer process, the compound designed contained the same side chains that would make it hard to distinguish the two mechanisms of conversion (Scheme S1). In the current design of 1 and 4, the differences in the side chains (2-carbon and 3-carbon) allowed us to provide new insights into the mechanism of conversion. Although the formation of imide intermediate 3 could be from the mechanism of amide oxygen attacking the ester bond shown in Scheme S2, our findings that both 1 and 4 generated same mixture suggested that S4 and S5, if formed transiently, would be converted into 3. In summary, the synthesis, hydrolysis studies, biological characterization of 1 and 4 fully support that the long-range O,N-acyl transfer process is an unlikely event and transformation via imide intermediate 3 is mostly plausible. These results provide unique insights into the activation of these ester prodrugs and further suggest new strategies to design prodrugs of 666-15.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH R01GM122820. We thank the OHSU Integrated Genomics Laboratory for the STR services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interest.
Supporting Information.
The Supporting Information is available: Detailed experimental procedures, Scheme S1–S2, HPLC chromatograms, dose-response curves, NMR spectra (PDF).
REFERENCES
- (1).Shaywitz AJ; Greenberg ME Annu. Rev. Biochem 1999, 68, 821. [DOI] [PubMed] [Google Scholar]
- (2).Montminy MR; Sevarino KA; Wagner JA; Mandel G; Goodman RH Proc. Natl. Acad. Sci. U. S. A 1986, 83, 6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Montminy MR; Bilezikjian LM Nature 1987, 328, 175. [DOI] [PubMed] [Google Scholar]
- (4).Xiao X; Li BX; Mitton B; Ikeda A; Sakamoto KM Curr. Cancer Drug Targets 2010, 10, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hagiwara M; Alberts A; Brindle P; Meinkoth J; Feramisco J; Deng T; Karin M; Shenolikar S; Montminy M Cell 1992, 70, 105. [DOI] [PubMed] [Google Scholar]
- (6).Wadzinski BE; Wheat WH; Jaspers S; Peruski LF; Lickteig RL; Johnson GL; Klemm DJ Mol. Cell. Biol 1993, 13, 2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gu T; Zhang Z; Wang J; Guo J; Shen WH; Yin Y Cancer Res. 2011, 71, 2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tan X; Wang S; Zhu L; Wu C; Yin B; Zhao J; Yuan J; Qiang B; Peng X Proc. Natl. Acad. Sci. U. S. A 2012, 109, 15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rodon L; Gonzalez-Junca A; Inda MD; Sala-Hojman A; Martinez-Saez E; Seoane J Cancer Discov. 2014, 4, 1230. [DOI] [PubMed] [Google Scholar]
- (10).Zhuang H; Meng X; Li Y; Wang X; Huang S; Liu K; Hehir M; Fang R; Jiang L; Zhou JX; Wang P; Ren Y Oncotarget 2016, DOI: 10.18632/oncotarget.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Daniel P; Filiz G; Brown DV; Hollande F; Gonzales M; D’Abaco G; Papalexis N; Phillips WA; Malaterre J; Ramsay RG; Mantamadiotis T Oncogenesis 2014, 3, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Srinivasan S, Totiger T, Shi C, et al. Cancer Res. 2018, 78, 6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li BX; Xiao X Chembiochem 2009, 10, 2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li BX; Yamanaka K; Xiao X Bioorg. Med. Chem 2012, 20, 6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li BX; Xiao X Methods Enzymol. 2020, 633, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xie F; Li BX; Kassenbrock A; Xue C; Wang X; Qian DZ; Sears RC; Xiao XJ Med. Chem 2015, 58, 5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li BX; Gardner R; Xue C; Qian DZ; Xie F; Thomas G; Kazmierczak SC; Habecker BA; Xiao X Sci. Rep 2016, 6, 34513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Imoto T; Kawase A; Minoshima M; Yokoyama T; Bito H; Kikuchi K Org. Lett 2020, 22, 22. [DOI] [PubMed] [Google Scholar]
- (19).Xie F; Fan Q; Li BX; Xiao XJ Med. Chem 2019, 62, 11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li BX; Xie F; Fan Q; Barnhart KM; Moore CE; Rheingold AL; Xiao X ACS Med. Chem. Lett 2014, 5, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seamon KB; Padgett W; Daly JW Proc. Natl. Acad. Sci. U. S. A 1981, 78, 3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bianchini G; Balko JM; Mayer IA; Sanders ME; Gianni L Nat. Rev. Clin. Oncol 2016, 13, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chao B; Li BX; Xiao X Bioorg. Med Chem. Lett 2017, 27, 3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.