

Abstract
Teeth are comprised of three unique mineralized tissues, enamel, dentin, and cementum, that are susceptible to developmental defects similar to those affecting bone. X-linked hypophosphatemia (XLH), caused by PHEX mutations, leads to increased fibroblast growth factor 23 (FGF23)-driven hypophosphatemia and local extracellular matrix disturbances. Hypophosphatasia (HPP), caused by ALPL mutations, results in increased levels of inorganic pyrophosphate (PPi), a mineralization inhibitor. Generalized arterial calcification in infancy (GACI), caused by ENPP1 mutations, results in vascular calcification due to decreased PPi, later compounded by FGF23-driven hypophosphatemia. In this perspective, we compare and contrast dental defects in primary teeth associated with XLH, HPP, and GACI, briefly reviewing genetic and biochemical features of these disorders and findings of clinical and preclinical studies to date, including some of our own recent observations. The distinct dental defects associated with the three heritable mineralization disorders reflect unique processes of the respective dental hard tissues, revealing insights into their development and clues about pathological mechanisms underlying such disorders.
Keywords: Mineralized tissues, enamel, dentin, cementum, hypophosphatemia
1. Introduction
In vertebrates, biomineralized tissues featuring hydroxyapatite are recognized as bones and teeth, which make up the skeleton and dentition. The associated process of biomineralization is complex, widespread, and varied. Under controlled conditions, organisms direct deposition of inorganic minerals and formation of organic-inorganic composite materials employing molecules such as silicates, oxalates, calcium carbonates, phosphates, and other minerals (Boskey 2003). Biomineralization is complex and energetically expensive for organisms, as they must synthesize extracellular matrices (ECM) to direct mineral deposition and regulate ion transport. Nevertheless, the evolutionary benefits of biomineralization are apparent in the functions provided by bones and teeth, which include protection, locomotion, mastication, and participation in endocrine and mineral metabolism.
Proper formation of the skeleton and dentition requires integration of numerous complex processes beginning in early embryonic development and extending throughout the lifespan. These include head, limb, and dental arch patterning, cell migration and proliferation, differentiation from stem/progenitor cells into specialized mineralizing cells, ECM secretion and modification, controlled biomineralization of bones and teeth, and remodeling of bone (Foster et al. 2014). With these multiple levels of regulation and developmental complexity in biomineralization, there are many possibilities for the process to go awry. In simplistic terms, mineralization defects can be discussed as those that cause insufficient mineral deposition or hypomineralization, while the inverse problem includes disorganized, excessive, or inappropriately located mineralization, i.e. ectopic or hypermineralization. A variety of hereditary conditions affecting all aspects of mineralization have been discovered, which highlight the complexity and provide considerable opportunities to better understand the biomineralization processes in bones and teeth (Foster et al. 2014; Foster, Nociti, and Somerman 2014; Luder 2015; Opsahl Vital et al. 2012; Hu and Simmer 2007; Mitsiadis and Luder 2011).
The oral cavity is home to four unique mineralized tissues, the only such location in the body (Foster, Nociti, and Somerman 2014). Enamel is the most highly mineralized tissue in the body, covering the clinically visible surfaces of the tooth crown (Fig. 1). In secretory stage, ameloblasts secrete a partially mineralized organic enamel matrix composed of a semi-specific suite of proteins (e.g., amelogenin, ameloblastin, and enamelin) that are largely degraded and removed during maturation stage, leaving mature enamel at >95% hydroxyapatite by weight (Bartlett 2013; Hu et al. 2007). Dentin, which is less mineralized than enamel, provides a tough, fibrous tissues that forms the bulk of the tooth. The initial dentin organic matrix secreted by odontoblasts is unmineralized predentin composed of type I collagen and other ECM proteins and proteoglycans, roughly equivalent to the osteoid layer in osteogenesis. Predentin mineralizes to dentin with the help of selectively expressed factors including dentin sialoprotein (DSP) and dentin phosphoprotein (DPP) (Goldberg et al. 2011). Dentin is not uniform, being composed of an initial mantle dentin region that transitions to the circumpulpal dentin that comprises the majority of the tissue. Cementum is a relatively thin tissue that envelopes the tooth root and provides anchorage of the tooth to the surrounding jawbone by inclusion of mineralized Sharpey’s fibers that are continuous with collagen fibers in the periodontal ligament (PDL). Cementum is present in two main varieties, the cervically located acellular cementum (most critical for tooth attachment), and apically located cellular cementum that includes cementocytes. Acellular cementum will be the primary type discussed in this report as it is more clinically relevant and more often present and amenable to analysis on exfoliated teeth. Cementoblasts secrete an ECM very similar to that in bone, including bone sialoprotein (BSP) and osteopontin (OPN) (Foster et al. 2007; Bosshardt 2005). The alveolar bone of the tooth socket distributes masticatory forces and responds to mechanical stimuli by continuous and rapid remodeling (Sodek and McKee 2000).
Fig. 1.
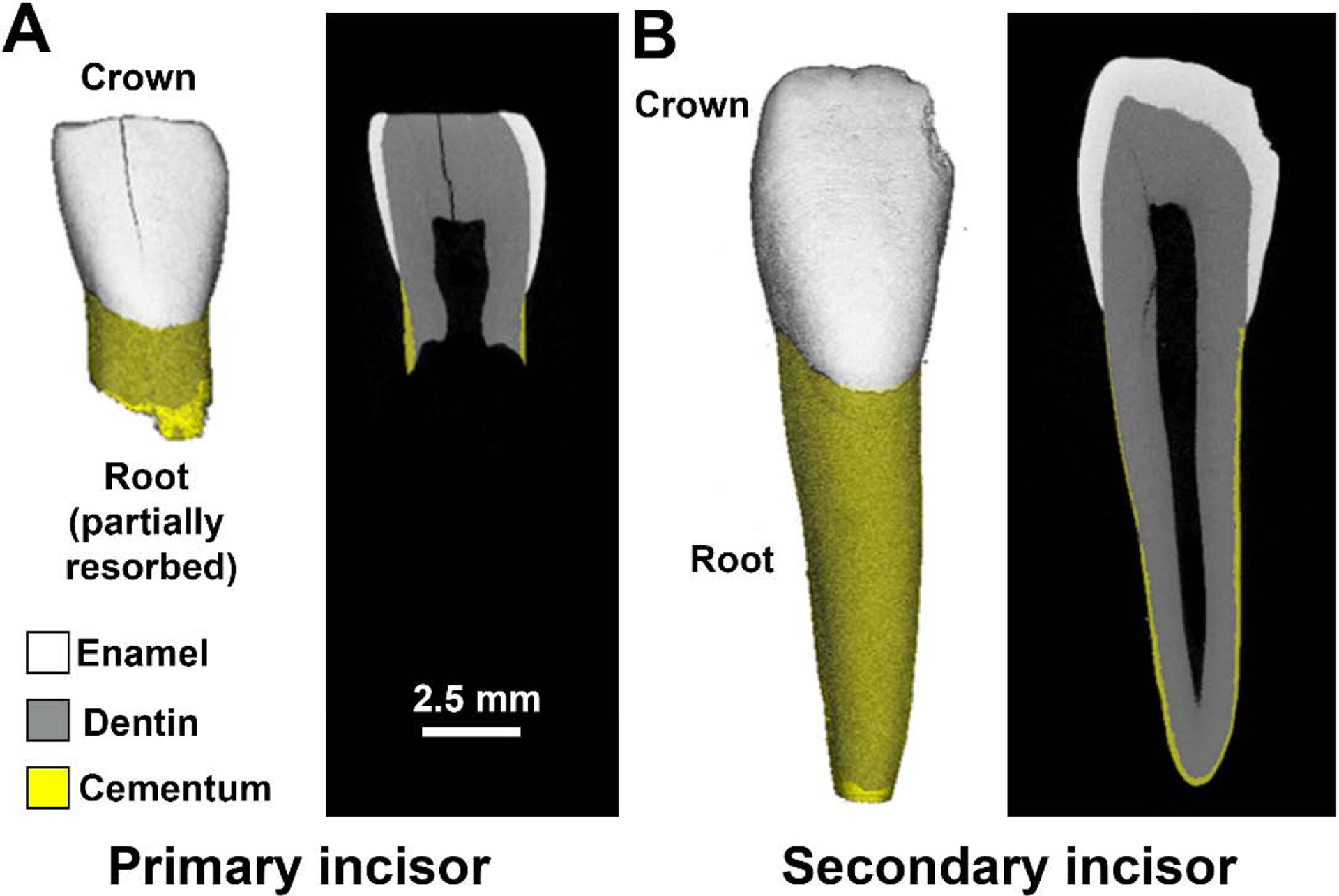
Dental Mineralized Tissues in Healthy Teeth. A) Human primary incisor shown by micro-CT in 3D and 2D exhibiting enamel (white), dentin (gray), and cementum (yellow). Note the root has undergone partial resorption as part of the physiological process of exfoliation. B) Human secondary incisor shown by micro-CT in 3D and 2D exhibiting the crown and a full-length root.
Although odontogenesis differs from osteogenesis in several aspects, tooth mineralization occurs through processes parallel to skeletal mineralization and is susceptible to failures similar to those appearing in bone caused by developmental disturbances of mineral metabolism. While the processes are parallel, different mechanisms of cell differentiation, physiology, and function underpin odontogenesis. The unique developmental biology of enamel-forming ameloblasts, dentin-forming odontoblasts, and cementum-forming cementoblasts results in distinct effects of mineralization disorders in each of these tissues. Understanding their pathologies provides unique insights into their biology and may provide essential clues to prevent and ameliorate disease and promote repair and regeneration of tissues lost to trauma or insult.
Rickets, an old term referring to a group of disorders that weaken and deform the bones, strikes during childhood skeletal development when bone is rapidly modeling and remodeling. Disturbances in mineralization cause accumulation of mechanically unsound hypomineralized bone osteoid and a host of mineralization problems that include tibial and femoral bowing (genu valgum and genu varum), metaphyseal cartilage dysplasia, costochondral swelling, growth disturbance, enlarged cranial sutures and fontanelles, and deformations of the cranium (Foster, Nociti, and Somerman 2014; Imel 2020; Rowe 2004). While nutritional rickets have been described for centuries, research in recent decades has clarified multiple forms of hereditary rickets. In this perspective, we focus on describing and interpreting the dental effects of three different forms of hereditary rickets that fall under the broader mantle of mineralization defects in more updated disease nosology of genetic skeletal disorders (Mortier et al. 2019): X-linked hypophosphatemia (XLH), hypophosphatasia (HPP), and generalized arterial calcification in infancy (GACI) (Table 1). This perspective summarizes findings of several studies and some our own recent observations to make a comparison of how these disorders specifically and disparately affect enamel, dentin, and cementum.
Table 1.
Inherited disorders presented in this perspective and their genetic and biochemical characteristics and dental mineralization defects.
| X-linked hypophosphatemia (XLH) | Hypophosphatasia (HPP) | Generalized arterial calcification in infancy (GACI) | |
|---|---|---|---|
| Genetic component | |||
| Gene | PHEX | ALPL | ENPP1 |
| Protein | PHEX | TNAP, TNSALP | ENPP1 |
| Biochemistry | |||
| Phosphate | Low | Normal | Low |
| Calcium | Normal | Normal | Normal |
| Pyrophosphate | Normal (or high) | High | Low |
| 1,25-dihydroxyvitamin D | Low or normal | Normal | Low or normal |
| Fibroblast growth factor 23 (FGF23) | High | Normal | High |
| Dental mineralization defects | |||
| Gene/protein expression | Odontoblasts, cementocytes | Ameloblasts, odontoblasts, cementoblasts | Cementoblasts |
| Primary dental mineralization effects | Primarily dentin, with lesser changes in enamel and acellular and cellular cementum | Primarily acellular cementum, with variable effects on enamel and dentin | Acellular cementum |
| Preclinical models | Hyp mutant mouse | Alpl−/− mouse, Alpl+/A116T mouse, Alpl conditional knockout mouse, Alpl+/A116T sheep | Enppl−/− mouse |
| Key references | Opsahl Vital et al., 2012; Biosse-Duplan et al., 2017; Coyac et al., 2017; Coyac et al., 2018; Zhang et al., 2020 | Reibel et al., 2009; Foster et al., 2012; Foster et al., 2015; Bloch-Zupan, 2016; Foster et al., 2017 | Thumbigere-Math et al., 2018 |
2. X-linked Hypophosphatemia (XLH)
X-linked hypophosphatemia (XLH; OMIM #307800), caused by inactivating mutations in PHEX (Phosphate regulating endopeptidase homolog, X-linked), affects approximately 1:20,000 births (Eicher et al. 1976; Beck-Nielsen et al. 2009). XLH features increased levels of fibroblast growth factor 23 (FGF23), hypophosphatemia, low 1,25-dihydroxyvitamin D, and elevated parathyroid hormone (PTH), with combined disturbances in local and systemic mineralization that manifest as osteomalacia and growth plate disturbances (Foster, Nociti, and Somerman 2014). PHEX is highly expressed by odontoblasts and osteocytes, with low level expression by cementocytes (Zhang et al. 2020). Although the total range and identities of the physiologic substrates for PHEX remain uncertain, the enzyme exhibits the ability to cleave and inactivate acidic serine- and aspartate-rich motif (ASARM) peptides derived from ECM proteins including OPN and matrix extracellular phosphoprotein (MEPE) that can act as mineral inhibitors (Salmon et al. 2014; Addison et al. 2010; Barros et al. 2013). Therefore, XLH may encompass systemic disturbance in mineral metabolism as well as local changes in the ECM mineralization milieu.
Reports on dental pathology in XLH highlight most consistently abnormal dentin mineralization marked by unmerged calcospherites (mineralization foci) and responsible for most of the clinical manifestations described (e.g. dental abscesses and necrosis), however descriptions may also be found for enamel defects, thin dentin, enlarged pulp chambers, altered root size and shape, alveolar bone hypomineralization, and increased prevalence of periodontal disorders that have not been well defined to date (Foster, Nociti, and Somerman 2014; Foster et al. 2014; Biosse Duplan et al. 2017; Chesher et al. 2018; Skrinar et al. 2019; Boukpessi et al. 2017; Opsahl Vital et al. 2012; Coyac et al. 2018). XLH broadly impacts all the dental mineralized tissues in primary teeth. Primary teeth from individuals more severely affected by XLH are more frequently extracted due to abscesses and pulpal necrosis (Fig. 2A, B). Dentin and enamel densities are both substantially reduced in many affected individuals, indicating severe defects in these tissues (Fig. 2E), though it should be noted that highly acidic phosphate supplements taken by individuals with XLH can also negatively affect enamel. So-called “interglobular” patterns in dentin observed by histology, micro-CT, or electron microscopy reveal inability of discreet calcospherites to fuse into a unified mineralization front and are characteristic of XLH. Negative effects of XLH on dentin density extend into both mantle dentin and circumpulpal dentin regions (Fig. 2E). This subdivision is of interest because of developmental and mechanistic implications. Mantle dentin is reported to depend on matrix vesicle initiated mineralization, while mineralization of circumpulpal dentin is guided by phosphoproteins such as DSP and DPP (Takano et al. 2000; Takano et al. 1998; Stratmann et al. 1997; Goldberg et al. 2011). In the case of XLH, both dentin regions appear to be adversely affected to a similar degree, indicating commonalities in the mechanisms of mineralization for mantle and circumpulpal dentin that responded similarly in this disorder. Very few reports on XLH have analyzed cementum. Acellular cementum was shown to be reduced in thickness in a study on the permanent dentition in XLH (Biosse Duplan et al. 2017) and cellular cementum mineralization was defective (Coyac et al. 2017). Reduced acellular cementum thickness also appears to accompany enamel and dentin defects in primary teeth from individuals with XLH (Fig. 2B).
Fig. 2.

Effects on primary human teeth of three different forms of rickets. Quantitative micro-CT and qualitative histology approaches were combined to analyze dental mineralized tissues. A) Control primary incisor shown by 3D and 2D micro-CT images and H&E stain of a histology section showing acellular cementum (AC). B) A cohort of 6 children (4 females and 2 males) with XLH provided 14 primary incisors and 1 primary canine for analysis. Shown here is a primary incisor from a child with XLH with apparent dentin mineralization defects. Cementum was undetectable by micro-CT and reduced thickness by H&E stain. C) One 2.5-year-old female with a history of mild skeletal manifestations and premature tooth loss featured two ALPL variants (c.346G>A/p. Ala116Thr and c.1077C>G/p.ISO359Met) and reduced circulating alkaline phosphatase activity (ALP) of 29 U/L (Normal range: 150–440 U/L). Shown here is a primary incisor this HPP subject with apparent radiolucency in the outer dentin region. Cementum was undetectable by micro-CT and absent by H&E stain (* indicates plaque accumulation on root surface). D) A cohort of 3 children (2 females and 1 male) with GACI provided 10 primary incisors for analysis. Cementum thickness in GACI teeth was dramatically increased by micro-CT and H&E stain. E) Quantitative analyses of dental mineralized tissues by micro-CT, showing mineral densities of enamel, dentin, mantle dentin (Mt.D; the outer 100 μm), and circumpulpal dentin (Cp.D; the remaining dentin minus the inner 100 μm of recently deposited dentin matrix), and cementum thickness and density. Each bar features individual data points and mean ± standard deviation and the 95% confidence interval (CI) generated from control teeth is shown as a gray shaded region. For XLH, mean enamel density was below the 95% CI and mean dentin density was near the lower limit of the 95% CI, with many individual teeth falling below the 95% CI. Dentin defects in XLH were present in both Mt.D and Cp.D regions, with nearly all measurements below the 95% CI. For HPP, enamel and overall dentin densities were unaffected, however Mt.D density was substantially reduced below the 95% CI. For GACI, enamel and dentin were wholly unaffected, while cementum thickness and density were dramatically increased well above the 95% CI for each parameter. Images in A and D adapted and reproduced with permission from Thumbigere-Math et al., 2018.
The Hyp mutant mouse model of XLH harbors Phex mutations and phenocopies biochemical and skeletal features of XLH (Eicher et al. 1976). Dental defects in Hyp mice are well described and replicate many aspects of human dental pathology in XLH, including enamel hypoplasia, defects in dentin volume and mineralization, and accumulation of interglobular dentin (Zhang et al. 2020; Coyac et al. 2018). While premature loss of teeth is generally not described for XLH (though extractions for abscessed teeth are common), Hyp mice exhibit thin acellular cementum. regions of PDL detachment, and significantly altered periodontal mechanical properties (Zhang et al. 2020). These types of accumulated periodontal defects may contribute to increased periodontal disease later in life in individuals with XLH (Biosse Duplan et al. 2017).
Though there is variation in severity of effects across individuals, it is notable that XLH impacts all dental tissues despite PHEX being highly expressed in only odontoblasts and osteocytes among dental cells (Zhang et al. 2020). It has been hypothesized that this profound impact stems from a combination of hypophosphatemia, altered vitamin D metabolism, and local ECM disturbances. Dissection of contributing factors has been attempted in Hyp mice, but with limited success to date. Substantial persistent mineralization defects in teeth of Hyp mice wherein Fgf23 was ablated and hypophosphatemia was corrected suggest these are not predominant factors in the dental pathology of XLH (Foster, Nociti, and Somerman 2014). Contribution of local disturbances has been supported by studies in Hyp mice (and limited experiments with human dental tissues) indicating abnormal accumulation in dentin of OPN, a regulator of hydroxyapatite crystal growth and potential source of ASARM peptides (Boukpessi et al. 2017; Salmon et al. 2014; Zhang et al. 2010; Zhang et al. 2020). Attempts to ameliorate dental defects in Hyp mice by genetically over-expressing or ablating additional factors are ongoing (Guirado et al. 2020), but have not yet clarified the pathological mechanisms.
3. Hypophosphatasia (HPP)
HPP is an inborn error-of-metabolism with a broad range of severity (OMIM #241500, #241510, and #146300) (Bowden and Foster 2019; Whyte 2016). ALPL encodes tissue non-specific alkaline phosphatase (TNAP), an enzyme expressed in bones, teeth, liver, and kidney (Millan and Whyte 2016). TNAP hydrolyzes the mineralization inhibitor inorganic pyrophosphate (PPi) and loss-of-function mutations in ALPL leads to deficiency in TNAP activity and increased PPi concentrations, leading to mineralization defects. HPP is inherited in autosomal dominant or recessive fashion and has a very broad range of clinical presentations from severe life-threatening forms (infantile and perinatal) to milder forms (prenatal benign, childhood, adult, and odontohypophosphatasia). Skeletal defects include growth plate disturbances and osteomalacia, resulting in fractures and bone pain. More than 400 mutations have been reported to date (http://www.sesep.uvsq.fr/03_hypo_mutations.php), where severe forms of HPP affect about 1:300,000 live births and milder forms affect about 1:6,370 in Europe (Mornet 2017).
Dental disorders have been described across the spectrum of mild and severe clinical forms of HPP (Reibel et al. 2009; Foster et al. 2014), however reports vary in their descriptions of which tissues are affected and the severity of the defects. Dental defects reported for HPP include acellular cementum hypoplasia or aplasia associated with premature loss of fully rooted primary teeth and/or loss of secondary teeth, delayed eruption, periodontal disease, enamel alterations, thin and hypomineralized dentin, widened pulp chambers, and short and/or malformed roots (Foster et al. 2014; Reibel et al. 2009; Bloch-Zupan 2016; Luder 2015). Ameloblasts, odontoblasts, cementoblasts, osteoblasts, and periodontal ligament (PDL) cells express TNAP (Bowden and Foster 2019; Zweifler et al. 2015), indicating the enzyme may function in all aspects of dental and periodontal mineralization.
Of the clinical signs listed above, premature exfoliation of fully rooted primary teeth is the most common manifestation and is highly characteristic of HPP. Primary teeth prematurely exfoliated from children with HPP generally exhibit substantial remaining root structure compared to healthy control teeth, reflecting exfoliation prior to physiological tooth root resorption (Fig. 2C). HPP teeth are reported to lack a recognizable cementum layer, reflecting the most severe effects of HPP on any of the dental tissues. Roots sometimes show accumulation of dental plaque, an unusual observation that confirms lack of periodontal attachment allowing bacterial invasion deep into the periodontal tissues (Fig. 2C) (Luder 2015).
While many descriptive reports on HPP dental pathology have been published, there is a paucity of quantitative data that contributes to lack of understanding how HPP differentially affects enamel, dentin, and cementum. In mild cases of HPP, enamel and dentin properties may not be substantially altered in comparison to control teeth (Fig. 2E), even when cementum formation is potently inhibited. Recent observations in human teeth suggest that mineralization defects in HPP localize preferentially to the mantle dentin, while circumpulpal dentin remains unaffected (Fig. 2E), pointing to a specific importance of TNAP function in mantle dentin mineralization.
Several HPP animal models have been described. The most extensively reported model has been the global knockout Alpl−/− mouse that phenocopies a severe and lethal infantile form of HPP (Narisawa, Frohlander, and Millan 1997; Bowden and Foster 2019). A series of studies reported cementum hypoplasia, disturbed enamel mineralization, and dentin hypomineralization (Yadav et al. 2012; Foster et al. 2013; Beertsen, VandenBos, and Everts 1999; Foster et al. 2012; Zweifler et al. 2015). TNAP enzyme has been localized to matrix vesicles (Bottini et al. 2018), and failure of hydroxyapatite crystals to grow and merge into a unified mineralization front was described in mantle dentin regions of an HPP mouse model, similar to observations in long bones (Foster et al. 2013; Anderson et al. 2004). The severity of the disease and early lethality in Alpl−/− mice have been limitations for understanding the broader spectrum of HPP dental defects, therefore additional animal models have been developed. Knock-in of a human mutation into mice (Alpl+/A116T) created a very mild HPP manifestation (Foster et al. 2015), while conditional ablation of the Alpl gene under either the Col1a1 promoter (to delete Alpl in osteoblasts and dental cells) or Prx1 promoter (to delete Alpl in limb buds, chondrocytes, osteoblasts, and craniofacial mesenchyme) recapitulated key aspects of HPP-associated dental defects, including cementum hypoplasia and hypomineralized dentin, and these conditional Alpl knockouts were the first to demonstrate periodontal breakdown and alveolar bone loss, likely due to their longer lifespans (Foster et al. 2017). Inherent limitations in mice for the study of bones and teeth led to the knock-in of an HPP-associated ALPL mutation into sheep. Alpl+/A116T heterozygous sheep exhibited primary incisors with thin and short roots and reduced alveolar bone levels, with additional dental analyses yet to be published (Williams et al. 2018). This model may provide additional insights into dental pathology and therapies because unlike in mice, sheep develop both primary and secondary dentitions.
While HPP can potentially affect enamel and dentin, cementum and mantle dentin can be affected even in mild cases, pointing to the sensitivity of these tissues to HPP, presumably due to elevated PPi levels (as discussed under part 4 below for cementum). Studies of mantle dentin in HPP are in their infancy, so it remains unclear how this type of localized mineralization disturbance might affect overall tooth structure and function. In the human teeth quantitatively analyzed so far, mantle dentin defects did not appear to translate into deleterious effects on the circumpulpal dentin, however additional quantitative studies in a larger cohort of subjects are warranted. It is notable that mantle dentin and cementum directly interface one another in the tooth root, suggesting potential for interactions in the mineralization of these adjacent hard tissues. While the dentin-cementum junction has been described using various modalities (Ho et al. 2004; Bosshardt and Selvig 1997), many developmental and functional aspects of this region remain unstudied and unknown.
4. Generalized Arterial Calcification in Infancy (GACI)
Ectonucleotide pyrophosphatase phosphodiesterase 1 (ENPP1) is a membrane-bound enzyme expressed by mineralizing cells that contributes to regulation of skeletal mineralization (Stefan, Jansen, and Bollen 2005; Millan 2013). ENPP1 generates intracellular and extracellular PPi via hydrolysis of adenosine triphosphate (ATP). Inactivating ENPP1 mutations cause generalized arterial calcification of infancy (GACI; OMIM #208000), a rare autosomal recessive disorder characterized by prenatal-onset vascular calcification (Ferreira, Ziegler, and Gahl 2014; Rutsch et al. 2003; Boyce, Gafni, and Ferreira 2020). ENPP1-derived PPi is critical for preventing soft tissue calcification, and decreased PPi levels account for GACI-associated pathologic calcifications. In this sense, GACI is the inverse of HPP with low vs. high levels of PPi and excess mineral deposition as opposed to hypomineralization. Individuals with GACI who survive infancy may develop autosomal recessive hypophosphatemic rickets type 2 (ARHR2; OMIM #613312) in childhood. ARHR2 is marked by elevated FGF23 and hypophosphatemia mirroring some aspects of XLH and other forms of inherited mineralization disorders (Lorenz- Depiereux et al. 2010; Levy-Litan et al. 2010; Rutsch et al. 2008; Ferreira et al. 2016). The etiology of ARHR2 in GACI remains unclear.
Primary teeth from individuals with GACI exhibit a massive amount of cementum, with no apparent effects on enamel or dentin (Fig. 2D, E) (Thumbigere-Math et al. 2018). Investigation of ENPP1 expression by immunostaining indicates strong, selective expression by cementoblasts in human and mouse tissues (Thumbigere-Math et al. 2018). These results indicate that cementogenesis is very responsive to modulation by PPi levels. Interestingly, several individuals affected by GACI reported delayed tooth exfoliation, primary tooth-bone ankylosis, and/or slow orthodontic tooth movement. Histological evidence suggested that physiologic resorption of primary tooth roots may have been “reversed” by reparative cementum production in GACI, an insight that may in part explain irregularities in tooth eruption and exfoliation in this cohort. These observations suggest that increased cementum formation has functional implications on the periodontal apparatus that deserve further investigation. While enamel and dentin are susceptible to defects under elevated PPi in HPP, they are apparently insensitive to the reduced PPi levels in GACI.
Whereas high PPi in HPP inhibits acellular cementum, low PPi in GACI promotes abundant production of cementum. These effects are recapitulated in the Enpp1−/− mouse model of GACI, indicating it is an ancient evolutionarily conserved mechanism (Thumbigere-Math et al. 2018; Zweifler et al. 2015; Foster et al. 2012). Further exploration of the role of PPi in directing cementogenesis has been accomplished in additional mouse models. Mice ablated for the progressive ankylosis protein (ANK) feature reduced PPi and hypercementosis identical to Enpp1−/− mice (Ao et al. 2017; Foster et al. 2012), whereas relative correction of PPi levels in mice ablated for both Alpl and Ank normalized acellular cementum formation (Chu et al. 2020).
In contrast to XLH and HPP, GACI teeth exhibited no perturbations in enamel or dentin density (Fig. 2E). Surprisingly, there was no sign of mineralization defects in enamel or dentin of affected individuals, though GACI was accompanied by elevated FGF23, hypophosphatemia, and disturbed vitamin D metabolism in all subjects investigated. Onset of ARHR2 can be as early as one year of age, when primary and secondary teeth during this period are sensitive to mineral disturbances as evidenced by other forms of nutritional and inherited rickets, e.g. XLH and vitamin D-dependent rickets (VDDR1A; OMIM#264700) (Davit-Beal et al. 2014; Opsahl Vital et al. 2012). These metabolic disturbances cause severe enamel and dentin defects, making teeth susceptible to caries and abscesses. One explanation for lack of deleterious effects in enamel and dentin in GACI may be that primary tooth mineralization was already established or sufficiently advanced when ARHR2 set in. In that case, the unerupted secondary dentition forming during childhood would be expected to be more susceptible to mineralization defects. Though the secondary dentition has not been quantitatively analyzed to date, no apparent mineralization defects have been noted by oral exam or radiographs in any individuals with GACI, including the original 5 subjects (Thumbigere-Math et al. 2018) and additional subjects examined since then (personal observations). This raises the interesting proposition that high FGF23 and resultant low phosphate and vitamin D are not sufficient to drive the mineralization disturbances, i.e. lack of defects in ARHR2/GACI may represent additional evidence that local dysfunction of PHEX contributes directly to dental pathology in XLH (McKee et al. 2013). Alternatively, perhaps reduced PPi in GACI somehow offsets the reduction in phosphate and vitamin D in mineralization of dental tissues. These hypotheses require further investigation.
5. Conclusions
Disparate effects on dental mineralized tissues are observed when comparing teeth affected by the inherited conditions, XLH, HPP, and GACI (Table 1). While XLH severely affects dentin, and to a lesser extent enamel and cementum, HPP appears to selectively impact acellular cementum and mantle dentin, while GACI negatively affects neither enamel nor dentin, rather promoting dramatically increased cementum production. These selective effects likely reflect a combination of differential expression of affected genes as well as unique physiologies of enamel, dentin, and cementum that render them sensitive to different types of metabolic disturbances. These disparate effects are also being explored in mouse models that phenocopy the conditions, where controlled experiments and interventions have provided additional mechanistic insights (Thumbigere-Math et al. 2018; Zhang et al. 2020; Bowden and Foster 2019; Foster et al. 2017; Zweifler et al. 2015; Foster et al. 2015; Foster, Nociti, and Somerman 2014; Foster et al. 2013; Foster et al. 2012; Yadav et al. 2012; Guirado et al. 2020; Coyac et al. 2018; Wolf et al. 2018; Chu et al. 2020).
Limitations of this perspective include small sample sizes reported for the recent quantitative analyses of inherited mineralization disorders. This results from a combination of rarity of the conditions and novelty of the quantitative approach, and observations will benefit from accumulation of studies. Case reports over several decades have been compiled and summarized in this perspective. More recent additions of quantitative studies are essential for better defining dental defects in light of new treatments. A recombinant mineral-targeted TNAP (Asfotase alfa; Strensiq®) was approved for use in 2015 to treat HPP (Bowden and Foster 2019; Whyte et al. 2012). An FGF23-targeting monoclonal antibody (Burosumab; Crysvita®) was approved in 2018 for treatment of adult and pediatric XLH patients (Carpenter et al. 2018; Imel et al. 2019; Insogna et al. 2018). Preclinical studies of a recombinant ENPP1-Fc fusion protein in a mouse model of GACI showed promising results (Albright et al. 2015; Boyce, Gafni, and Ferreira 2020). In therapeutic approaches for all these conditions, careful assessment of treatment benefits on dental tissues have not been included in clinical study designs, representing missed opportunities to understand ramifications of interventions to prevent, ameliorate, or reverse dental pathology and improve oral health and quality of life. Furthermore, the study and insights gained from investigating rare genetic mineralization disorders has a much wider impact, providing us with increased knowledge and understanding of the mechanisms that drive and control mineralization that may spur medical innovation into reparative and regenerative strategies for all mineralized tissues.
Highlights.
Teeth are composed of three unique mineralized tissues: Enamel, dentin, and cementum, which exhibit disparate responses to mineralization disorders
X-linked hypophosphatemia (XLH) broadly affects dentin density and to a lesser degree enamel density, and reduces cementum thickness
Hypophosphatasia (HPP) has variable effects on dental tissues, with targeted effects on cementum and mantle dentin
Generalized arterial calcification in infancy (GACI) does not affect enamel or dentin, but causes hypercementosis
Acknowledgements
We thank the families for participation in this research. We thank Dr. Sasigarn Bowden (Nationwide Children’s Hospital, Columbus, OH), Dr. Dana Gaddy and Dr. Larry Suva (Texas A&M University, TX), Dr. Carlos Ferreira (National Human Genome Research Institute, National Institutes of Health, Bethesda, MD), and Dr. Martha J. Somerman (National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD), and Ms. Anh Tran, Ms. Delaney Clayton, Ms. Michelle H. Tan, and Ms. Tamara Kolli (The Ohio State University College of Dentistry, Columbus, OH). We would also like to acknowledge Dr. Cathrine Hoyo, Ms. Rachel Maguire, and Dr. Sarah Park for providing control tooth samples (North Carolina State University; Newborn Epigenetics Study funded by R24ES028531 to CH). This work was funded by research grants from Soft Bones, Inc. (to BLF), a research grant from Ultragenyx Pharmaceutical (to BLF), R03 grants DE028632 and DE028411 (to BLF), K99/R00 grant AR073926 and NIAMS intramural research program funding (to EYC), K99/R00 grant DE028439 and start-up funds from the University of Maryland School of Dentistry (to VTM), and T32 grant DE014320-19 (support for MBC). We thank Dr. Stephen Weiner, Dr. Marc McKee, Dr. Dobrawa Napierala, and the three anonymous reviewers for their invitation to submit this perspective, and their essential feedback the contents of the paper.
Abbreviations:
- ALP
circulating alkaline phosphatase activity
- ALPL
alkaline phosphatase tissue-nonspecific isozyme gene
- CI
confidence interval
- CT
computed tomography
- ECM
extracellular matrix
- ENPP1
ectonucleotide pyrophosphatase phosphodiesterase 1
- FGF23
fibroblast growth factor 23
- GACI
generalized arterial calcification in infancy
- HPP
hypophosphatasia
- PDL
periodontal ligament
- PHEX
phosphate regulating endopeptidase homolog X-linked
- PPi
inorganic pyrophosphate
- PTH
parathyroid hormone
- TNAP
tissue-nonspecific alkaline phosphatase
- XLH
X-linked hypophosphatemia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Addison WN, Masica DL, Gray JJ, and McKee MD. 2010. ‘Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage’, J Bone Miner Res, 25: 695–705. [DOI] [PubMed] [Google Scholar]
- Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, Covo MS, Tehan M, Yang G, Cheng Z, Bouchard K, Yu ZX, Thorn S, Wang X, Folta-Stogniew EJ, Negrete A, Sinusas AJ, Shiloach J, Zubal G, Madri JA, De La Cruz EM, and Braddock DT. 2015. ‘ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy’, Nat Commun, 6: 10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HC, Sipe JB, Hessle L, Dhanyamraju R, Atti E, Camacho NP, Millan JL, and Dhamyamraju R. 2004. ‘Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice’, Am J Pathol, 164: 841–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao M, Chavez MB, Chu EY, Hemstreet KC, Yin Y, Yadav MC, Millan JL, Fisher LW, Goldberg HA, Somerman MJ, and Foster BL. 2017. ‘Overlapping functions of bone sialoprotein and pyrophosphate regulators in directing cementogenesis’, Bone, 105: 134–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, and McKee MD. 2013. ‘Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia’, J Bone Miner Res, 28: 688–99. [DOI] [PubMed] [Google Scholar]
- Bartlett JD 2013. ‘Dental enamel development: proteinases and their enamel matrix substrates’, ISRN Dent, 2013: 684607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, and Jensen TK. 2009. ‘Incidence and prevalence of nutritional and hereditary rickets in southern Denmark’, Eur J Endocrinol, 160: 491–7. [DOI] [PubMed] [Google Scholar]
- Beertsen W, VandenBos T, and Everts V. 1999. ‘Root development in mice lacking functional tissue non-specific alkaline phosphatase gene: inhibition of acellular cementum formation’, J Dent Res, 78: 1221–9. [DOI] [PubMed] [Google Scholar]
- Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, Briot K, Linglart A, and Chaussain C. 2017. ‘Phosphate and Vitamin D Prevent Periodontitis in X-Linked Hypophosphatemia’, J Dent Res, 96: 388–95. [DOI] [PubMed] [Google Scholar]
- Bloch-Zupan A 2016. ‘Hypophosphatasia: diagnosis and clinical signs - a dental surgeon perspective’, Int J Paediatr Dent, 26: 426–38. [DOI] [PubMed] [Google Scholar]
- Boskey AL 2003. ‘Biomineralization: an overview’, Connect Tissue Res, 44 Suppl 1: 5–9. [PubMed] [Google Scholar]
- Bosshardt DD 2005. ‘Are cementoblasts a subpopulation of osteoblasts or a unique phenotype?’, J Dent Res, 84: 390–406. [DOI] [PubMed] [Google Scholar]
- Bosshardt DD, and Selvig KA. 1997. ‘Dental cementum: the dynamic tissue covering of the root’, Periodontol 2000, 13: 41–75. [DOI] [PubMed] [Google Scholar]
- Bottini M, Mebarek S, Anderson KL, Strzelecka-Kiliszek A, Bozycki L, Simao AMS, Bolean M, Ciancaglini P, Pikula JB, Pikula S, Magne D, Volkmann N, Hanein D, Millan JL, and Buchet R. 2018. ‘Matrix vesicles from chondrocytes and osteoblasts: Their biogenesis, properties, functions and biomimetic models’, Biochim Biophys Acta Gen Subj, 1862: 532–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukpessi T, Hoac B, Coyac BR, Leger T, Garcia C, Wicart P, Whyte MP, Glorieux FH, Linglart A, Chaussain C, and McKee MD. 2017. ‘Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia’, Bone, 95: 151–61. [DOI] [PubMed] [Google Scholar]
- Bowden SA, and Foster BL. 2019. ‘Alkaline Phosphatase Replacement Therapy for Hypophosphatasia in Development and Practice’, Adv Exp Med Biol, 1148: 279–322. [DOI] [PubMed] [Google Scholar]
- Boyce AM, Gafni RI, and Ferreira CR. 2020. ‘Generalized Arterial Calcification of Infancy: New Insights, Controversies, and Approach to Management’, Curr Osteoporos Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, Padidela R, Van’t Hoff W, Mao M, Chen CY, Skrinar A, Kakkis E, San Martin J, and Portale AA. 2018. ‘Burosumab Therapy in Children with X-Linked Hypophosphatemia’, N Engl J Med, 378: 1987–98. [DOI] [PubMed] [Google Scholar]
- Chesher D, Oddy M, Darbar U, Sayal P, Casey A, Ryan A, Sechi A, Simister C, Waters A, Wedatilake Y, Lachmann RH, and Murphy E. 2018. ‘Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations’, J Inherit Metab Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu EY, Vo TD, Chavez MB, Nagasaki A, Mertz EL, Nociti FH, Aitken SF, Kavanagh D, Zimmerman K, Li X, Stabach PR, Braddock DT, Millan JL, Foster BL, and Somerman MJ. 2020. ‘Genetic and pharmacologic modulation of cementogenesis via pyrophosphate regulators’, Bone, 136: 115329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyac BR, Falgayrac G, Baroukh B, Slimani L, Sadoine J, Penel G, Biosse-Duplan M, Schinke T, Linglart A, McKee MD, Chaussain C, and Bardet C. 2017. ‘Tissue-specific mineralization defects in the periodontium of the Hyp mouse model of X-linked hypophosphatemia’, Bone, 103: 334–46. [DOI] [PubMed] [Google Scholar]
- Coyac BR, Falgayrac G, Penel G, Schmitt A, Schinke T, Linglart A, McKee MD, Chaussain C, and Bardet C. 2018. ‘Impaired mineral quality in dentin in X-linked hypophosphatemia’, Connect Tissue Res, 59: 91–96. [DOI] [PubMed] [Google Scholar]
- Davit-Beal T, Gabay J, Antoniolli P, Masle-Farquhar J, and Wolikow M. 2014. ‘Dental complications of rickets in early childhood: case report on 2 young girls’, Pediatrics, 133: e1077–81. [DOI] [PubMed] [Google Scholar]
- Eicher EM, Southard JL, Scriver CR, and Glorieux FH. 1976. ‘Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets.’, Proc Natl Acad Sci U S A, 73: 4667–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira CR, Ziegler SG, Gupta A, Groden C, Hsu KS, and Gahl WA. 2016. ‘Treatment of hypophosphatemic rickets in generalized arterial calcification of infancy (GACI) without worsening of vascular calcification’, Am J Med Genet A, 170A: 1308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira C, Ziegler S, and Gahl W. 2014. ‘Generalized Arterial Calcification of Infancy’ in Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH and Stephens K (eds.), GeneReviews(R) (Seattle (WA)). [PubMed] [Google Scholar]
- Foster BL, Kuss P, Yadav MC, Kolli TN, Narisawa S, Lukashova L, Cory E, Sah RL, Somerman MJ, and Millan JL. 2017. ‘Conditional Alpl Ablation Phenocopies Dental Defects of Hypophosphatasia’, J Dent Res, 96: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Nagatomo KJ, Nociti FH Jr., Fong H, Dunn D, Tran AB, Wang W, Narisawa S, Millan JL, and Somerman MJ. 2012. ‘Central role of pyrophosphate in acellular cementum formation’, PLoS One, 7: e38393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Nagatomo KJ, Tso HW, Tran AB, Nociti FH Jr., Narisawa S, Yadav MC, McKee MD, Millan JI, and Somerman MJ. 2013. ‘Tooth root dentin mineralization defects in a mouse model of hypophosphatasia’, J Bone Miner Res, 28: 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Nociti FH Jr., and Somerman MJ. 2014. ‘The rachitic tooth’, Endocr Rev, 35: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Popowics TE, Fong HK, and Somerman MJ. 2007. ‘Advances in defining regulators of cementum development and periodontal regeneration’, Curr Top Dev Biol, 78: 47–126. [DOI] [PubMed] [Google Scholar]
- Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, Wright JT, Akintoye SO, Somerman MJ, and Collins MT. 2014. ‘Rare bone diseases and their dental, oral, and craniofacial manifestations’, J Dent Res, 93: 7S–19S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Sheen CR, Hatch NE, Liu J, Cory E, Narisawa S, Kiffer-Moreira T, Sah RL, Whyte MP, Somerman MJ, and Millan JL. 2015. ‘Periodontal Defects in the A116T Knock-in Murine Model of Odontohypophosphatasia’, J Dent Res, 94: 706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Kulkarni AB, Young M, and Boskey A. 2011. ‘Dentin: structure, composition and mineralization’, Front Biosci (Elite Ed), 3: 711–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirado E, Chen Y, Ross RD, Zhang Y, Chaussain C, and George A. 2020. ‘Disrupted Protein Expression and Altered Proteolytic Events in Hypophosphatemic Dentin Can Be Rescued by Dentin Matrix Protein 1’, Front Physiol, 11: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SP, Balooch M, Marshall SJ, and Marshall GW. 2004. ‘Local properties of a functionally graded interphase between cementum and dentin’, J Biomed Mater Res A, 70: 480–9. [DOI] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, and Simmer JP. 2007. ‘Enamel formation and amelogenesis imperfecta’, Cells Tissues Organs, 186: 78–85. [DOI] [PubMed] [Google Scholar]
- Hu JC, and Simmer JP. 2007. ‘Developmental biology and genetics of dental malformations’, Orthod Craniofac Res, 10: 45–52. [DOI] [PubMed] [Google Scholar]
- Imel EA 2020. ‘Congenital Conditions of Hypophosphatemia in Children’, Calcif Tissue Int. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Hogler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M, Chen CY, Skrinar A, San Martin J, and Portale AA. 2019. ‘Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial’, Lancet, 393: 2416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, Imanishi Y, Ito N, Lachmann RH, Tanaka H, Perwad F, Zhang L, Chen CY, Theodore-Oklota C, Mealiffe M, San Martin J, Carpenter TO, and Axles Investigators. 2018. ‘A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis’, J Bone Miner Res, 33: 1383–93. [DOI] [PubMed] [Google Scholar]
- Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, and Parvari R. 2010. ‘Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene.’, Am J Hum Genet, 86: 273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, and Strom TM. 2010. ‘Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets.’, Am J Hum Genet, 86: 267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder HU 2015. ‘Malformations of the tooth root in humans’, Front Physiol, 6: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee MD, Hoac B, Addison WN, Barros NM, Millan JL, and Chaussain C. 2013. ‘Extracellular matrix mineralization in periodontal tissues: Noncollagenous matrix proteins, enzymes, and relationship to hypophosphatasia and X-linked hypophosphatemia’, Periodontol 2000, 63: 102–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan JL 2013. ‘The role of phosphatases in the initiation of skeletal mineralization’, Calcif Tissue Int, 93: 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan JL, and Whyte MP. 2016. ‘Alkaline Phosphatase and Hypophosphatasia’, Calcif Tissue Int, 98: 398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiadis TA, and Luder HU. 2011. ‘Genetic basis for tooth malformations: from mice to men and back again’, Clin Genet, 80: 319–29. [DOI] [PubMed] [Google Scholar]
- Mornet E 2017. ‘Genetics of hypophosphatasia’, Arch Pediatr, 24: 5S51–5S56. [DOI] [PubMed] [Google Scholar]
- Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, Sillence D, Superti-Furga A, Unger S, and Warman ML. 2019. ‘Nosology and classification of genetic skeletal disorders: 2019 revision’, Am J Med Genet A, 179: 2393–419. [DOI] [PubMed] [Google Scholar]
- Narisawa S, Frohlander N, and Millan JL. 1997. ‘Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia’, Dev Dyn, 208: 432–46. [DOI] [PubMed] [Google Scholar]
- Opsahl Vital S, Gaucher C, Bardet C, Rowe PS, George A, Linglart A, and Chaussain C. 2012. ‘Tooth dentin defects reflect genetic disorders affecting bone mineralization’, Bone, 50: 989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reibel A, Maniere MC, Clauss F, Droz D, Alembik Y, Mornet E, and Bloch-Zupan A. 2009. ‘Orodental phenotype and genotype findings in all subtypes of hypophosphatasia’, Orphanet J Rare Dis, 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe PS 2004. ‘The wrickkened pathways of FGF23, MEPE and PHEX’, Crit Rev Oral Biol Med, 15: 264–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Böyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW, Davies JH, Loirat C, Strom TM, Schnabel D, Nürnberg P, Terkeltaub R, and GACI Study Group. 2008. ‘Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy.’, Circ Cardiovasc Genet, 1: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, and Nürnberg P. 2003. ‘Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification.’, Nat Genet, 34: 379–81. [DOI] [PubMed] [Google Scholar]
- Salmon B, Bardet C, Coyac BR, Baroukh B, Naji J, Rowe PS, Opsahl Vital S, Linglart A, McKee MD, and Chaussain C. 2014. ‘Abnormal osteopontin and matrix extracellular phosphoglycoprotein localization, and odontoblast differentiation, in X-linked hypophosphatemic teeth’, Connect Tissue Res, 55 Suppl 1: 79–82. [DOI] [PubMed] [Google Scholar]
- Skrinar A, Dvorak-Ewell M, Evins A, Macica C, Linglart A, Imel EA, Theodore-Oklota C, and San Martin J. 2019. ‘The Lifelong Impact of X-Linked Hypophosphatemia: Results From a Burden of Disease Survey’, J Endocr Soc, 3: 1321–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodek J, and McKee MD. 2000. ‘Molecular and cellular biology of alveolar bone’, Periodontol 2000, 24: 99–126. [DOI] [PubMed] [Google Scholar]
- Stefan C, Jansen S, and Bollen M. 2005. ‘NPP-type ectophosphodiesterases: unity in diversity.’, Trends Biochem Sci, 30: 542–50. [DOI] [PubMed] [Google Scholar]
- Stratmann U, Schaarschmidt K, Wiesmann HP, Plate U, Hohling HJ, and Szuwart T. 1997. ‘The mineralization of mantle dentine and of circumpulpal dentine in the rat: an ultrastructural and element-analytical study’, Anat Embryol (Berl), 195: 289–97. [DOI] [PubMed] [Google Scholar]
- Takano Y, Sakai H, Baba O, Sakamoto Y, Terashima T, Ohya K, and Kurosaki N. 1998. ‘Demonstration of putative Ca-binding domains in dentin matrix of rat incisors after daily injections of 1-hydroxyethylidene-1,1-bisphosphonate (HEBP)’, Eur J Oral Sci, 106 Suppl 1: 274–81. [DOI] [PubMed] [Google Scholar]
- Takano Y, Sakai H, Baba O, and Terashima T. 2000. ‘Differential involvement of matrix vesicles during the initial and appositional mineralization processes in bone, dentin, and cementum’, Bone, 26: 333–9. [DOI] [PubMed] [Google Scholar]
- Thumbigere-Math V, Alqadi A, Chalmers NI, Chavez MB, Chu EY, Collins MT, Ferreira CR, FitzGerald K, Gafni RI, Gahl WA, Hsu KS, Ramnitz MS, Somerman MJ, Ziegler SG, and Foster BL. 2018. ‘Hypercementosis Associated with ENPP1 Mutations and GACI’, J Dent Res, 97: 432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MP 2016. ‘Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment’, Nat Rev Endocrinol, 12: 233–46. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar AM, Crine P, and Landy H. 2012. ‘Enzyme-replacement therapy in life-threatening hypophosphatasia’, N Engl J Med, 366: 904–13. [DOI] [PubMed] [Google Scholar]
- Williams DK, Pinzon C, Huggins S, Pryor JH, Falck A, Herman F, Oldeschulte J, Chavez MB, Foster BL, White SH, Westhusin ME, Suva LJ, Long CR, and Gaddy D. 2018. ‘Genetic engineering a large animal model of human hypophosphatasia in sheep’, Sci Rep, 8: 16945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M, Ao M, Chavez MB, Kolli TN, Thumbigere-Math V, Becker K, Chu EY, Jager A, Somerman MJ, and Foster BL. 2018. ‘Reduced Orthodontic Tooth Movement in Enpp1 Mutant Mice with Hypercementosis’, J Dent Res, 97: 937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav MC, de Oliveira RC, Foster BL, Fong H, Cory E, Narisawa S, Sah RL, Somerman M, Whyte MP, and Millan JL. 2012. ‘Enzyme replacement prevents enamel defects in hypophosphatasia mice’, J Bone Miner Res, 27: 1722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Sun Y, Chen L, Guan C, Guo L, and Qin C. 2010. ‘Expression and distribution of SIBLING proteins in the predentin/dentin and mandible of hyp mice’, Oral Dis, 16: 453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chavez MB, Kolli TN, Tan MH, Fong H, Chu EY, Li Y, Ren X, Watanabe K, Kim DG, and Foster BL. 2020. ‘Dentoalveolar Defects in the Hyp Mouse Model of X-linked Hypophosphatemia’, J Dent Res: 22034520901719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifler LE, Patel MK, Nociti FH Jr., Wimer HF, Millan JL, Somerman MJ, and Foster BL. 2015. ‘Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis’, Int J Oral Sci, 7: 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]