

Abstract
The Forkhead Box C1 (FOXC1) gene encodes a forkhead/winged helix transcription factor involved in embryonic development. Mutations in this gene cause dysgenesis of the anterior segment of the eye, most commonly Axenfeld-Rieger syndrome (ARS), often with other systemic features. The developmental mechanisms and pathways regulated by FOXC1 remain largely unknown. There are two conserved orthologs of FOXC1 in zebrafish, foxc1a and foxc1b. To further examine the role of FOXC1 in vertebrates, we generated foxc1a and foxc1b single knockout zebrafish lines and bred them to obtain various allelic combinations. Three genotypes demonstrated visible phenotypes: foxc1a−/− single homozygous and foxc1−/− double knockout homozygous embryos presented with similar characteristics comprised of severe global vascular defects and early lethality, as well as microphthalmia, periocular edema and absence of the anterior chamber of the eye; additionally, fish with heterozygous loss of foxc1a combined with homozygosity for foxc1b (foxc1a+/−;foxc1b−/−) demonstrated craniofacial defects, heart anomalies and scoliosis. All other single and combined genotypes appeared normal. Analysis of foxc1 expression detected a significant increase in foxc1a levels in homozygous and heterozygous mutant eyes, suggesting a mechanism for foxc1a upregulation when its function is compromised; interestingly, the expression of another ARS-associated gene, pitx2, was responsive to the estimated level of wild-type Foxc1a, indicating a possible role for this protein in the regulation of pitx2 expression. Altogether, our results support a conserved role for foxc1 in the formation of many organs, consistent with the features observed in human patients, and highlight the importance of correct FOXC1/foxc1 dosage for vertebrate development.
Introduction
The Forkhead Box C1 gene (FOXC1 MIM #601090) is located at human chromosome 6p25.3 and encodes a transcription factor of the forkhead/winged helix family. Mutations in FOXC1 cause several ocular developmental disorders such as Axenfeld-Rieger anomaly and syndrome type III (ARS, MIM #602482), congenital glaucoma, aniridia and Peters anomaly (1–4), characterized by variable anterior segment dysgenesis of the eye including posterior embryotoxon, corectopia or polycoria, iris hypoplasia, irido-corneal adhesions, maldevelopment of the aqueous humor drainage structures and increased intraocular pressure (1–3,5–7). Affected patients often have additional syndromic anomalies including heart defects, craniofacial dysmorphism, hearing loss and brain (white matter lesions, hydrocephalus) or skeletal (hip dysplasia or scoliosis) defects (2,8–14).
There are two genes orthologous to human FOXC1 in zebrafish, foxc1a on chromosome 2 and foxc1b on chromosome 20 (15). Both genes show significant conservation and encode proteins that are 66% (Foxc1a) and 55% (Foxc1b) identical to human FOXC1, with the highest homology in the forkhead domain and the N- and C-terminal activation domains of the protein (15). With respect to the eye, foxc1a is expressed in the periocular mesenchyme and hyaloid vasculature starting at 24-hpf in embryos and additionally in the corneal epithelium/stroma and in the retina in adults (16,17), while foxc1b has a weaker and later onset expression in the periocular mesenchyme (48-hpf). Both genes are expressed in the branchial arches and endothelial cells of the trunk vasculature in the developing embryo (17), and foxc1b is also expressed in the fin buds. Zebrafish foxc1a morpholino-mediated knockdown showed altered somitogenesis, heart edema, hemorrhages, absence of blood flow, hydrocephalus and microphthalmia, while foxc1b knockdown did not cause any visible phenotype (17,18). Consistent with this, genetic lines with zebrafish foxc1a knock-out showed altered somitogenesis, developmental defects in heart, facial cartilage and head vascular smooth muscle, impaired appendage-specific neural circuit development and embryonic lethality in homozygous embryos, while embryos with homozygous foxc1b mutations appeared normal (19–23). However, while the ocular phenotype is one of the main features seen in human patients carrying FOXC1 mutations, eye development has not been studied in any of these genetic zebrafish lines.
Here, we characterize the effects of foxc1 deficiency in zebrafish, with a focus on ocular structures, using zebrafish that we generated carrying various combinations of foxc1a and foxc1b knockout alleles. We show that foxc1a plays a major role in eye and vascular development with single foxc1a and double foxc1a/foxc1b (foxc1) homozygous mutants showing similar phenotypes; all other allelic combinations produce embryos and adults with no visible phenotype except for foxc1a+/−;foxc1b−/− adults that show skeletal, craniofacial and cardiac defects. Interestingly, expression of foxc1a was found to be significantly elevated in foxc1a+/− heterozygous and foxc1−/− homozygous embryonic eyes, suggesting a mechanism for compensatory upregulation when foxc1 function is compromised; expression of another ARS factor, pitx2, was also found to be affected.
Results
Knockout of foxc1 results in developmental eye defects
In order to evaluate the role of the foxc1 genes in eye development, we generated single foxc1a and foxc1b knockout zebrafish lines using the CRISPR-Cas9 genome editing system. For foxc1a, we isolated a mosaic founder fish (F0) carrying the germline non-sense mutation c.451-457del p.(Asp71*). Mosaic founder fish with two different germline frameshift mutations were isolated for foxc1b: c.256_257insA p.(Ala86Aspfs*30) and c.258delC p.(Ile87Serfs*11). All three mutations are predicted to result in early truncation of the Foxc1a or Foxc1b amino acid sequence producing proteins lacking functional DNA-binding (forkhead) and C-terminal domains (Fig. 1A). Similar mutations in human FOXC1 have been reported and shown to result in a complete loss-of-function (8,24–26). Thus, the foxc1a/b zebrafish alleles presented above are referred to as foxc1a− (for c.451-457del p.(Asp71*)), foxc1b− (for c.256_257insA p.(Ale86Aspfs*30) or c.258delC p.(Ile87Serfs*11)) and foxc1− (for double deficiency).
Figure 1.
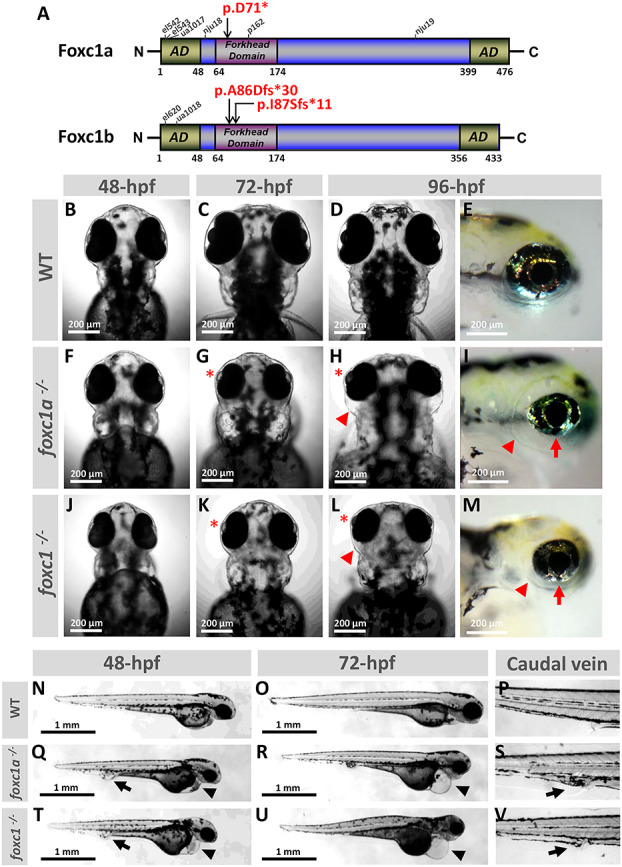
Developmental defects in foxc1a−/− and foxc1−/− knockout embryos. (A) Schematic drawing of zebrafish Foxc1a and Foxc1b proteins illustrating the positions of the variants generated in this study (in red font) as well as the positions of truncating variants in previously described lines. (B–M) Images of the head at 48-, 72- and 96-hpf of WT, foxc1a−/− and foxc1−/− zebrafish embryos. Mutant embryos showed microphthalmia with shallow/absent anterior segment (red asterisk), periocular edema (red arrowhead) and coloboma (red arrow). (N–V) Lateral views at 48- and 72-hpf of WT, foxc1a−/− and foxc1−/− embryos showing pericardial edema (black arrowhead) and caudal vein edema (black arrow). ad, activation domain.
The first visible defect in foxc1a−/− or foxc1−/− homozygous embryos was the apparent absence of blood circulation noticeable at ~34-hpf (Supplementary Material, Videos S1 and S2). This was followed by the development of heart edema and caudal vein edema/enlarged caudal vein with 100% penetrance (Fig. 1) by 48-hpf; mutant embryos also displayed reduced body length (Supplementary Material, Fig. S1). At later stages (72-hpf and 96-hpf), foxc1a−/− and foxc1−/− embryos demonstrated microphthalmia, absence of the anterior chamber, periocular edema and variable coloboma (Fig. 1G–I and K–M). By 6-dpf, both groups failed to develop swim bladders and showed progressive global swelling followed by death by 7–8-dpf. Overall, both single gene (foxc1a−/−) and double gene (foxc1−/−) knockouts presented with similar phenotypes and, particularly, both mutants showed consistent eye abnormalities. However, the foxc1−/− double homozygous embryos frequently had a smaller head and overall appeared more severely affected than the single homozygous foxc1a−/− embryos (Fig. 1T and U). The foxc1a+/−;foxc1b−/− zebrafish demonstrated a milder reduced survival and abnormal phenotype (see below); all other genotypes, including foxc1b− heterozygous or homozygous, foxc1a+/− heterozygous or foxc1+/− double heterozygous (foxc1a+/−;foxc1b+/−) larvae did not show any visible phenotype, survived to adulthood, were fertile and bred normally.
To further evaluate the developing eye, hematoxylin-and-eosin (H&E)-stained histological head sections of 48-, 72-, 96-hpf and 6-dpf foxc1−/− mutants were examined. At 48-hpf, embryos did not show any structural ocular defects (Fig. 2A and E). Consistent with the gross morphological observations, microphthalmia became noticeable at 72-hpf (Fig. 2B and F). At 96-hpf, foxc1−/− eyes continue to be small and lack visible anterior chambers; significant periocular edema is also evident (Fig. 2C and G). By 6-dpf, the edema becomes worse, the anterior chamber continues to be underdeveloped and lens opacities develop in some embryos (Fig. 2H). However, all retinal layers appear to be present in the microphthalmic eyes of foxc1−/− mutants, consistent with the absence of a primary retinal phenotype in human patients (Fig. 2D and H). We also observed brain and cartilage defects in foxc1−/− embryos such as hydrocephalus and absence of the palatoquadrate, trabecular and basihyal cartilages as well as hypoplasia of the ceratohyal cartilages (Fig. 2I and J).
Figure 2.
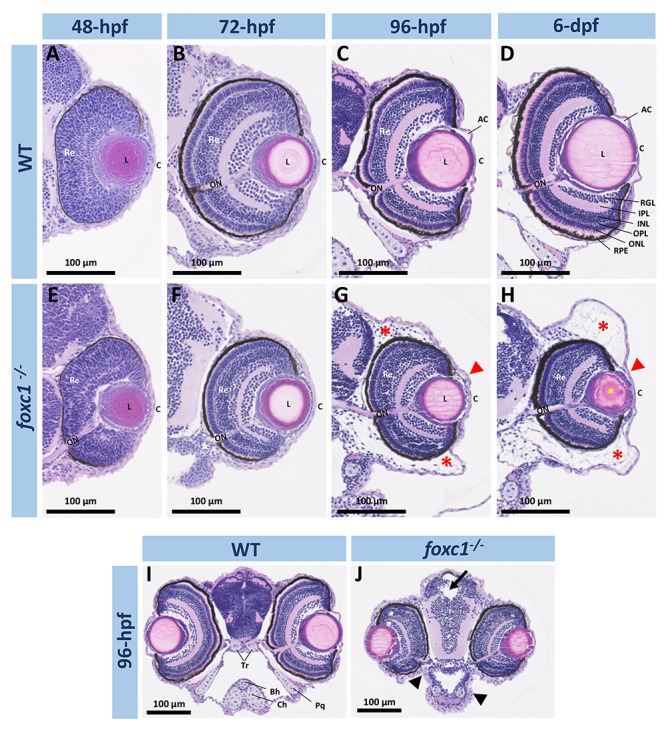
Histological analysis of ocular and craniofacial anomalies in the foxc1−/−embryos. (A–H) 20× H&E stained histology sections of the eye at 48-, 72-, 96-hpf and 6-dpf of WT and foxc1−/− double knockout embryos. Mutant embryos showed microphthalmia, absence of the anterior chamber (red arrowheads), periocular edema (red asterisks) and lens opacity (yellow asterisk). (I and J) 10× H&E sections of the head at 96-hpf showing smaller head, hydrocephalus (black arrow) and facial cartilage defects (black arrowhead). AC, anterior chamber; C, cornea; L, lens; Re, retina; ON, optic nerve; RGL, retinal ganglion layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; RPE, retinal pigmented epithelium; Bh, basihyal; Ch, ceratohyal; Tr, trabecular; Pq, palatoquadrate.
foxc1a is essential for correct development of the eye vasculature
The role of foxc1a in the formation of the eye vasculature was assessed by breeding the foxc1a− line with the transgenic Tg(fli1a:EGFP) line. The latter expresses eGFP under the control of the fli1a promoter, an early endothelial marker that allows monitoring of blood vessel formation (27). We obtained homozygous foxc1a−/− embryos that carried the fli1a:EGFP transgene. Normal development of the eye superficial choroidal vasculature involves three radial vessels, nasal (NRV), dorsal (DRV) and ventral (VRV), that project from the periphery of the optic cup towards the lens and by 44-hpf are connected by a ring-shaped vessel named the superficial annular vessel (SAV) (Fig. 3A–E) (28). Simultaneously, the intraocular hyaloid vessels, forming from the hyaloid artery (HA), branch around the lens in the hyaloid basket (HB), which will give rise to the hyaloid system (HS) (Fig. 3B–E) (29,30). At 24-hpf, foxc1a−/− homozygous embryos showed the growing hyaloid artery but absence of the dorsal radial vessel sprout (Fig. 3F) which was not visible until 44-hpf (Fig. 3H). At 34-hpf, mutant embryos showed immature organization of the vessels and absence of the hyaloid basket (Fig. 3G). Between 44- and 54-hpf, when normal embryos have already developed most of the superficial choroidal vasculature and hyaloid system, mutant embryos presented a growing DRV extending around the lens, absence of the NRV and SAV, and an amorphous hyaloid basket (Fig. 3H and I). By 80-hpf, foxc1a−/− embryos had completely abnormal eye vasculature (Fig. 3J). We also observed abnormalities in the development of the heart and brain vasculature. Unlike the control embryos, mutant embryos did not show any fli1a:EGFP fluorescent signal in the heart at 34-hpf (Fig. 3K and L) and show delayed/weaker expression at later stages. In the brain, foxc1a−/− embryos did not develop the central arteries that connect the Primordial Hindbrain Channel (PHBC) to the Basilar Artery (BA) at 44-hpf and the morphology of the Posterior Communicating Segments (PCS) was anomalous (Fig. 3M and N).
Figure 3.
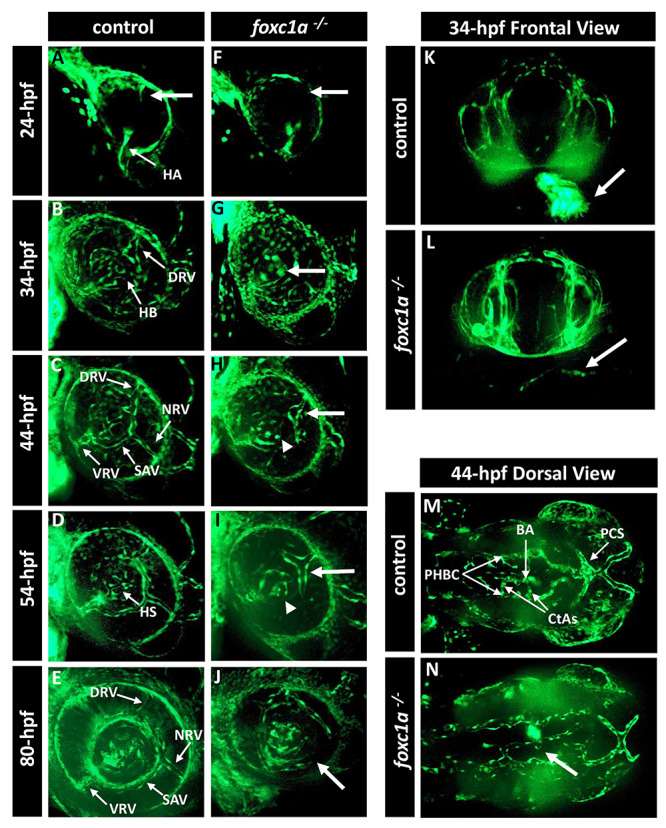
foxc1a −/− knockout embryos present with defects in the development of the eye vasculature. (A–J) Three-dimensional maximum projection images of the ocular vasculature in live control (A–E) or foxc1a−/− knockout (F–J) embryos carrying the fli1a:EGFP transgene at 24-, 34-, 44-, 54- and 80-hpf. Mutant embryos showed abnormal development of the superficial choroidal vessel (white arrow) and amorphous hyaloid system (white arrowhead). (K and L) Three-dimensional maximum projection images of the head vasculature and heart from the frontal view of 34-hpf control and mutant embryos. Mutant embryos do not show any fli1a:EGFP fluorescence in the heart region (white arrow in L). (M and N) Three-dimensional maximum projection images of the head vasculature from the dorsal view of 44-hpf control and mutant embryos. Mutant embryos show defects in the brain vasculature development such as absence of the central arteries (white arrow in N). HA, hyaloid artery; HB, hyaloid basket; DRV, dorsal radial vessel; NRV, nasal radial vessel; SAV, superficial annular vessel; VRV, ventral radial vessel; HS, hyaloid system; BA, basilar artery; CtAs, central arteries; PCS, posterior communicating segments; PHBC, primordial hindbrain channel.
Upregulation of foxc1a transcript in foxc1−/− embryonic eyes
To further explore the effects of foxc1 knockdown, we assessed expression levels of the foxc1a and foxc1b transcripts in wild-type (WT) and mutant eyes. We dissected eyes from 48- and 72-hpf WT, foxc1a+/− and foxc1−/− embryos. After RNA extraction and cDNA synthesis, we performed quantitative Real-Time PCR for both foxc1a and foxc1b transcripts. Interestingly, we found that the foxc1a transcript was significatively upregulated in heterozygous foxc1a+/− embryonic eyes with a fold change of 1.35 at 48-hpf and 1.9 at 72-hpf (Fig. 4A and B) as well as in homozygous foxc1−/− double knockout eyes with a fold change of 2.48 at 48-hpf and 2.94 at 72-hpf (Fig. 4A and B). This result indicates a possible mechanism for upregulation of foxc1a expression when its normal function (expression of downstream targets) is compromised; in heterozygous embryos (with one normal copy of foxc1a), this could lead to timely restoration of the level of functional Foxc1a protein to allow normal eye development, while in foxc1−/− double mutants this upregulation results in overexpression of non-functional protein only. Interestingly, we did not observe any significant variation in the transcript level of foxc1b in either the foxc1a+/− or the foxc1−/− 48- and 72-hpf embryonic eyes (Fig. 4C and D).
Figure 4.
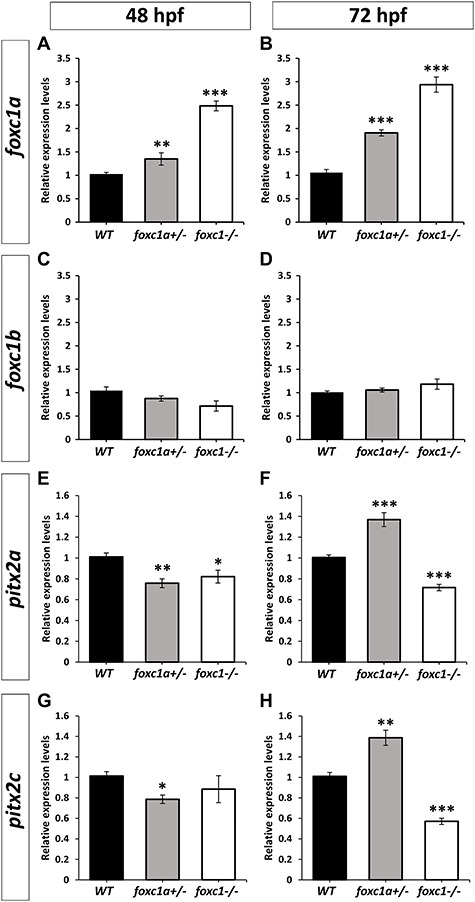
Analysis of foxc1a and pitx2 expression in foxc1 mutants. (A–D) RT-qPCR relative expression of foxc1a (A, B) and foxc1b (C, D) transcripts in 48-hpf (A, C) and 72-hpf (B, D) WT, heterozygous foxc1a+/− and double knockout homozygous foxc1−/− embryonic eyes. (E–H) RT-qPCR relative expression of pitx2a (E and F) and pitx2c (G and H) transcript in 48-hpf (E and G) and 72-hpf (F and H) WT, heterozygous foxc1a+/− and double knockout homozygous foxc1−/− embryonic eyes. β-actin (actb1) was used as the reference transcript. Three independent biological replicates were analyzed using three replicates per experiment.*: P < 0.05; **: P < 0.01; ***P < 0.001.
We next examined the expression of pitx2, an ortholog of another human gene associated with Axenfeld-Rieger syndrome, in the WT and mutant eyes. The pitx2 has two alternatively spliced isoforms, pitx2a and pitx2c; expression of each isoform was independently measured with isoform-specific primers. We observed that both the pitx2a and pitx2c transcripts were significantly downregulated at 48-hpf in heterozygous foxc1a+/− embryonic eyes (fold change of 0.76 and 0.79, respectively) (Fig. 4E and G); in contrast, both isoforms were significantly upregulated at 72-hpf (fold change of 1.37 and 1.39, respectively) (Fig. 4F and H). In homozygous foxc1−/− double knockout eyes, we similarly found a significant decrease in pitx2a expression at 48-hpf (fold change of 0.82) and somewhat decreased (but not significant) expression of pitx2c (Fig. 4E and G). However, at 72-hpf both isoforms were significantly downregulated in homozygous foxc1−/− double knockout eyes (fold change of 0.72 and 0.57, respectively) (Fig. 4F and H). The downregulation of pitx2 expression in heterozygous and homozygous eyes at 48-hpf suggests that Foxc1a may be involved in the regulation of pitx2; since approximately one half (in heterozygous samples) and all (in homozygous samples) of the total foxc1a transcript produces non-functional protein, both samples are expected to have reduced functional Foxc1a levels (~0.675 (1.35/2) and 0, respectively, in comparison with WT; see above), correlating with the lower levels of pitx2 expression. While it is possible that structural defects in the tissues normally expressing pitx2 transcripts (anterior segment) in the mutant eye could contribute to lower expression levels, this seems less likely as no visible abnormalities were detected in heterozygous embryos at any stage of development, or in 48-hpf homozygous eyes. At 72-hpf, the increase in expression of both pitx2 isoforms in the heterozygous (foxc1a+/−) eyes correlates with the increase of foxc1a expression (to ~1.9-fold and, therefore, ~0.95 for the WT allele, assuming equal expression of both alleles); however, the observed upregulation of pitx2 in comparison with WT levels cannot be fully explained by the increase in foxc1a expression alone and is likely additionally stimulated by other factors activated in the mutant eyes. The reduced expression of pitx2a and c at 72-hpf in double homozygous foxc1−/− eyes correlates with the lack of functional Foxc1a protein (despite a significant upregulation of mutant foxc1a transcript in this fish). Overall, these data suggest that zebrafish Foxc1a makes a small but measurable contribution to the regulation of pitx2 expression during eye development; additionally, upregulation of pitx2 expression at 72-hpf suggests a role for this factor in compensation for Foxc1a deficiency in heterozygotes.
Zebrafish with heterozygous loss of foxc1a combined with homozygous loss of foxc1b (foxc1a+/−;foxc1b−/−) show abnormal phenotypes
Examination of progeny produced by foxc1a/foxc1b double heterozygous (foxc1a+/−;foxc1b+/−) and foxc1b homozygous (foxc1a+/+;foxc1b−/−) parents identified reduced survival to adulthood for the foxc1a+/−;foxc1b−/− genotype, while all other genotypes were unaffected (~10% of adults had the foxc1a+/−;foxc1b−/− genotype instead of the expected 25%) (Fig. 5A). Further analysis of fish with the foxc1a+/−;foxc1b−/− genotype identified normal survival and morphology at embryonic stages 0- to 5-dpf. At 6-dpf, the foxc1a+/−;foxc1b−/− embryos remain generally indistinct from their clutchmates; however, a subset (33%; n = 9) show a slight pericardial edema that was not observed in their siblings (0%; n = 5) (Supplementary Material, Fig. S2B). By 2-mpf, foxc1a+/−;foxc1b−/− fish demonstrate highly penetrant visible anomalies such as spinal curvature defects reminiscent of scoliosis and craniofacial abnormalities including misshapen head, mandibular retrognathia and dorsally positioned eyes (Fig. 5B and C).
Figure 5.
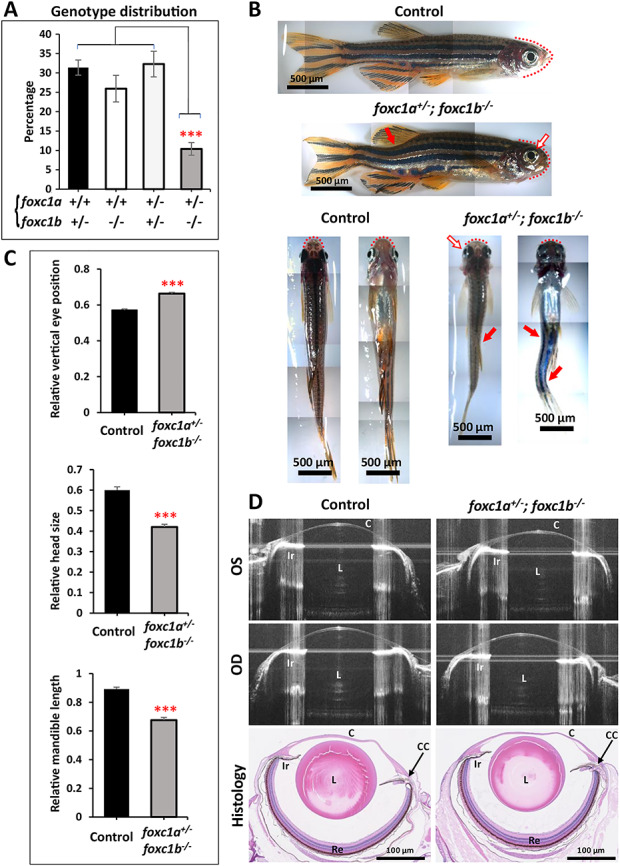
foxc1a+/−;foxc1b−/− fish present with reduced survival and developmental defects. (A) Genotype distribution (%) among adult progeny of foxc1a+/−;foxc1b+/− and foxc1a+/+;foxc1b−/− parents showing a significant reduction in survival of the foxc1a+/−;foxc1b−/− genotype (statistical significance was calculated with a one-way ANOVA analysis from 6 different spawns) (B) Representative images of 8-mpf foxc1a+/−; foxc1b−/− and foxc1a+/+; foxc1b+/− (control) sibling fish. foxc1a+/−;foxc1b−/− adults show scoliosis (red arrow) and abnormally shaped head (red dotted line) with mandibular retrognathia and mispositioning of the eyes (blank arrow with red border). (C) Relative measurements of the position of the eye in the head (vertical axis) (top), head size (horizontal axis) (middle) and mandible length (bottom) of control (n = 7) and foxc1a+/−;foxc1b−/− fish (n = 9) (measurements were performed from both sides and normalized to head height). (D) Representative spectral domain-optical coherence tomography (SD-OCT) ocular images and H&E stained histology sections of 10-mpf control and foxc1a+/−;foxc1b−/− fish. C, Cornea; CC, Ciliary Canal; Ir, Iris; L, Lens; OS, Left eye; OD, Right eye; Re, Retina. ***: P < 0.001.
To investigate for possible ocular defects, we imaged the eyes of eight foxc1a+/−;foxc1b−/− 10-mpf fish and eight foxc1a+/+; foxc1b+/− siblings using optical coherence tomography (OCT) (Fig. 5D). Using OCT images, we measured corneal thickness, pupil width, anterior chamber area, and the dorsal and ventral angles but did not find any difference in these measurements between the foxc1a+/−;foxc1b−/− fish and their control siblings (Supplementary Material, Table S2). To further investigate, we obtained histological sections of the eyes of foxc1a+/−; foxc1b−/− mutants and their foxc1a+/+;foxc1b+/− siblings but did not observe any structural differences between them (Fig. 5D). However, consistent with the gross observations presented above, we noticed facial cartilage malformations, such as rotation of the ceratohyal and basihyal bones in some of the foxc1a+/−;foxc1b−/− mutants (Supplementary Material, Fig. S2E and F).
Since heart edema was present in all foxc1a−/− and foxc1−/− homozygous embryos (Fig. 1Q–U) as well as some foxc1a+/−;foxc1b−/− larvae (Supplementary Material, Fig. S2B) and foxc1a mutants were previously shown to have various cardiac defects (20), we further examined foxc1a+/−;foxc1b−/− animals for a cardiac phenotype possibly affecting their survival. Heart tissue sections of 10-mpf foxc1a+/−;foxc1b−/− fish and their foxc1a+/+;foxc1b+/− siblings were obtained and stained with monoclonal antibody MF20 (Fig. 6A and B), which binds to sarcomeric myosin heavy chain to detect myocardial tissue (31). We quantified the compact zone and trabecular zone thickness (Fig. 6E and F), as well as the area and cells/area of the outflow tract (OFT) and atrioventricular (AV) valves (Fig. 6C–D and G–J). We observed that foxc1a+/−;foxc1b−/− fish showed a significantly larger compact zone in comparison with the foxc1a+/+;foxc1b+/− siblings (P < 0.05). The OFT valve area also appeared larger in foxc1a+/−;foxc1b−/− fish in comparison to the siblings, but this difference was not statistically significant (P = 0.0539). We did not find any difference between the two genotypes in trabecular zone thickness or other AV valve measurements (Fig. 6F and H–J). To investigate potential changes in embryonic heart development, sections of 6-dpf embryos were obtained and stained with MF20. At 6-dpf, the compact zone, noted to be affected in adults, has not formed yet. We measured the trabecular zone, which is present at this timepoint, and did not observe a significant difference between the foxc1a+/−;foxc1b−/− embryos and their foxc1a+/+;foxc1b+/− siblings, similar to adults (Supplementary Material, Fig. S2G–I).
Figure 6.
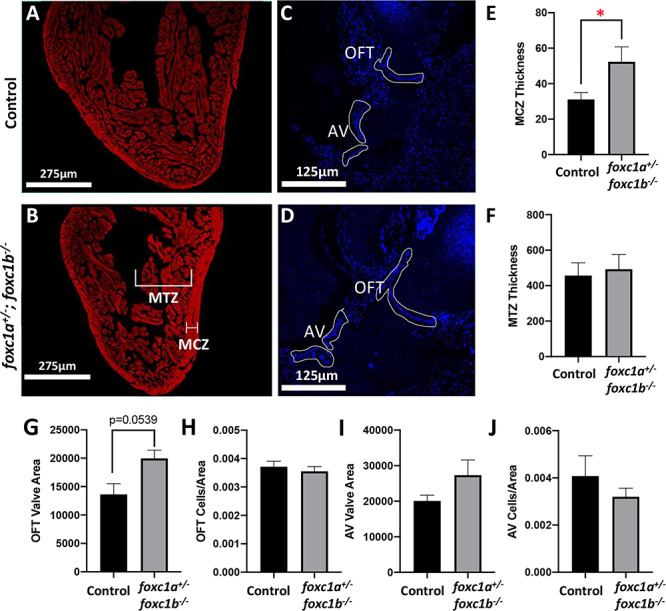
Adult foxc1a+/−; foxc1b−/− fish display myocardial defects. (A and B) MF20 staining of heart histological sections of 10-mpf foxc1a+/+;foxc1b+/− sibling (control) and foxc1a+/−;foxc1b−/− fish. (C and D) DAPI staining of heart histological sections of 10-mpf control and foxc1a+/−;foxc1b−/− fish. (E and F) Quantification (in pixels) of compact zone (MCZ) thickness and trabecular zone (MTZ) thickness in 10-mpf adult fish (n = 7). foxc1a+/−;foxc1b−/− fish have a significantly larger compact zone thickness (E) but unchanged trabecular zone thickness (F); *P = 0.047. (G–J) Quantification (in pixels) of OFT valve area, OFT valve cells/area, AV valve area and AV valve cells/area in 10-mpf control and foxc1a+/−;foxc1b−/− adult fish (n = 3–5). foxc1a+/−;foxc1b−/− fish did not show any statistically significant differences from controls; however, the OFT valve area was larger and this difference approached significance (P = 0.0539) (G). MCZ: myocardial compact zone. MTZ: myocardial trabecular zone. OFT: outflow tract. AV: atrioventricular.
Discussion
In this report, we present new foxc1a and foxc1b knockout lines, c.451-457del p.(Asp71*) for foxc1a− and c.256_257insA p.(Ala86Aspfs*30) or c.258delC p.(Ile87Serfs*11) for foxc1b−, that we generated and used to investigate the effects of foxc1 deficiency in zebrafish, with a focus on eye development. Both single foxc1a homozygous (foxc1a−/−) and foxc1a/foxc1b double-knockout homozygous (foxc1−/−) embryos were found to be severely affected with systemic and ocular defects. Fish carrying heterozygous foxc1a variants combined with homozygous foxc1b alleles, foxc1a+/−;foxc1b−/−, also demonstrated an abnormal phenotype. All other genotypes were found to be normal as embryos and adults.
The foxc1a−/− and foxc1−/− (foxc1a−/−;foxc1b−/−) embryos presented with an absence of blood flow, heart and caudal vein edema, hydrocephalus, craniofacial cartilage abnormalities and embryonic lethality. Absence of blood flow is consistent with previously published results for morpholino-mediated foxc1a knockdown (17) and foxc1ap162 homozygotes carrying the p.(Trp118*) non-sense allele (22). Other published foxc1a knockout lines such as foxc1anju19 (p.(Tyr316Aspfs*61)) and foxc1aua1017 (p.(Pro10Argfs*30)) were reported to have some blood flow but demonstrated disrupted erythrogenesis and circulatory insufficiency, respectively (20,23). Yue and coauthors also identified specific cardiac defects in the foxc1anju19 homozygous embryos such as thinner myocardium, shorter cardiac outflow tract, defective primitive valve leaflets, smaller ventricle and larger atrium and heart edema (20). The defects in brain vasculature observed in our foxc1a−/− mutants are consistent with the impaired angiogenesis in the head that was reported in the zebrafish foxc1anju19 line (20). In addition to vascular anomalies, craniofacial cartilage defects were observed, as previously reported in foxc1ael542 or foxc1ael543 homozygous embryos (carrying p.(Ser12Trpfs*57) and p.(Pro10Cysfs*55) frameshift mutations, respectively) and foxc1a/foxc1b double homozygous embryos obtained by crossing those foxc1a lines with foxc1bel620 (carrying p.(Val7Aspfs*38)) (21). While the phenotypes of the previously reported single foxc1a and double foxc1a/foxc1b lines were generally similar, some features, including those affecting the facial cartilage and ventral aorta, were noted to be more severe in the double mutant embryos (21,23). In our mutants, we similarly noted that the double mutant foxc1−/− embryos had a smaller head size compared to the single foxc1a−/− embryos. Mutations in the previously reported lines disrupt N-terminal (foxc1aua1017;foxc1ael542;foxc1ael543), forkhead (foxc1ap162), and/or C-terminal (foxc1anju19) Foxc1a domains (19,21,23); the functional effects of foxc1aua1017; foxc1ap162; foxc1ael542 and foxc1ael543 are likely to be similar to the p.(Asp71*) foxc1a allele presented here (loss-of-function), while the foxc1anju19 protein may have some residual function as it retains the DNA-binding forkhead domain. The two previously reported foxc1b mutations disrupt the N-terminal regions of the protein and likely result in loss-of-function alleles, similar to our foxc1b mutations affecting the DNA-binding forkhead domain. The craniofacial, heart and vascular anomalies observed in mutant zebrafish are consistent with the craniofacial dysmorphism, heart defects, cerebral small vessel disease and hydrocephalus found in patients with FOXC1 mutations (1,9,10).
The ocular phenotype was not characterized in the previously reported foxc1 knockout lines. In this study, the eye phenotype in single gene foxc1a−/− or double knockout foxc1−/− homozygous embryos consisted of periocular edema, microphthalmia, absence of the anterior chamber, coloboma, cataracts and maldevelopment of the ocular vasculature. We did not detect any difference in the severity of the ocular defects in the single gene foxc1a−/− compared to double foxc1−/− embryos, suggesting that foxc1a plays the major role in eye development in zebrafish. The absence of the anterior chamber seen in our mutants correlates with the anterior segment dysgenesis observed in FOXC1 patients. Similarly, knockout mice lacking Foxc1 showed absence of the anterior chamber and microphthalmia (32). In regard to the defects in ocular vasculature development in our mutants, patients with ARS caused by FOXC1 mutations show abnormal corneal angiogenesis and neovascularization; in mouse, Foxc1 has been shown to preserve corneal transparency by regulating vascular growth (33). Interestingly, the formation of the superficial vascular system in the eye has been reported to be independent of blood flow in zebrafish (28). Thus, the eye vasculature defects in foxc1a−/− embryos may be due to the specific impairment of foxc1a function in the eye. Microphthalmia, coloboma or cataracts are not characteristic phenotypes in patients with FOXC1 mutations but have been reported in some cases, including one family with a recessive FOXC1 allele (5,34,35); in zebrafish mutants, these defects may be additionally provoked by the early-onset, progressive and severe vascular phenotype (absent blood flow, edema) that likely affects the development of many organs. Ocular coloboma, in particular, may be linked to the anomalous development of the eye vasculature since abnormal blood vessels passing through the optic fissure can interfere with its closure (36).
Fish with heterozygous foxc1a alleles in a foxc1b deficient background (foxc1a+/−;foxc1b−/−) showed reduced survival and presented with craniofacial and cardiac defects as well as scoliosis, but no eye phenotype. In terms of the heart phenotype, the foxc1a+/−;foxc1b−/− adult fish showed a thicker compact zone in the myocardium in comparison with the foxc1a+/+;foxc1b+/− control siblings but had a normal trabecular zone. A thicker compact zone in the myocardium could affect the heart function and may contribute to the observed lower survival of this genotype. Interestingly, ventricular compaction defects have been described in Foxc1 homozygous mice (37). Skeletal defects have not been previously reported in zebrafish foxc1 lines, perhaps due to the embryonic lethality in previously investigated mutants, foxc1a−/− and foxc1−/−. The systemic features observed in foxc1a+/−;foxc1b−/− animals are relevant to ARS as maxillary hypoplasia, heart anomalies and scoliosis were reported in patients with dominant FOXC1 mutations (1,8,10,38).
In humans and rodents, the inheritance pattern for the FOXC1/Foxc1- associated phenotypes is primarily dominant with Foxc1 heterozygous knockout mice showing defects similar to those found in ARS patients (39). At the same time, a complete absence of Foxc1 in mice resulted in embryonic lethality with hydrocephalus, hemorrhages, and severe cardiovascular and eye defects (40). In zebrafish, foxc1a single- or foxc1 double- homozygous animals present with a severe phenotype similar to the homozygous knockout mice; foxc1a heterozygotes in a foxc1b-deficient background also show an abnormal phenotype. This indicates that foxc1b provides some compensation for foxc1a deficiency during later stages of development in zebrafish (especially craniofacial, skeletal and heart development) so that when foxc1b is completely abrogated, one functional copy of foxc1a is no longer sufficient for normal survival/phenotype. Thus, zebrafish development demonstrates a similar sensitivity to correct dosage of foxc1, which is contributed by two highly homologous duplicated genes, foxc1a and foxc1b. Genetic redundancy of the foxc1 genes in zebrafish has been previously described (15), with both genes showing high identity with human FOXC1 (66 and 55% for Foxc1a and Foxc1b, respectively).
Examination of foxc1a and foxc1b expression in the single heterozygous foxc1a+/− and double knockout foxc1−/− mutant eyes identified an increase in the expression level of the foxc1a transcript but no change for the foxc1b transcript. The increase in foxc1a expression implies that there is a mechanism for upregulating foxc1a expression when its normal function is compromised; in heterozygous embryos this could lead to a timely restoration of foxc1a transcript dosage (through upregulation of its one functional copy) to allow normal development, while in foxc1−/− double mutants this results in overexpression of non-functional alleles and thus no recovery ensues. This mechanism may explain the absence of a dominant ocular phenotype in the foxc1a+/− heterozygotes. To further explore this, we measured the expression of pitx2a and c, two isoforms of a gene orthologous to human PITX2, the second major factor in Axenfeld-Rieger syndrome (1); mutations in zebrafish pitx2 likewise result in anterior segment dysgenesis (41). We detected a decrease in pitx2 expression in the majority of foxc1 mutant samples with reduced levels of functional transcript; interestingly, a statistically significant upregulation of pitx2 expression was observed in heterozygous foxc1a+/− eyes at 72-hpf, correlating with the substantial increase in foxc1a expression at this stage (and subsequent recovery of WT Foxc1a protein level), but likely additionally stimulated by other factors. These data imply that 1) Foxc1a may be involved in the regulation of pitx2 expression, 2) increased expression of pitx2 may have a compensatory role in heterozygous mutants and 3) the decrease in pitx2 expression, in addition to foxc1a, may have an additional impact on eye development in foxc1 mutants, and, if a similar phenomenon is present in humans, in ARS patients with FOXC1 mutations.
In humans, heterozygous mutations in FOXC1 give rise to dominant phenotypes characterized by variable expressivity; while not all factors contributing to the variability are known, increased residual activity of mutant proteins/FOXC1 dosage was found to positively correlate with reduced severity in some cases (3,11,42). It is possible that similar processes (upregulation of FOXC1 and/or PITX2 expression in individuals with heterozygous FOXC1 alleles) are active in humans and contribute to the observed variability. If true, the identification of factors involved in these mechanisms will likely provide an insight into human disease.
In summary, this study supports the conserved role of FOXC1/foxc1 in the development of the eye and several other organs affected in ARS and highlights the significance of foxc1 gene dosage for proper development.
Materials and Methods
Animals
Zebrafish (Danio rerio) were maintained under standard conditions with a 14/10 h light/dark cycle in system water. Embryos were raised at 28.5°C in E2 medium up to 5-dpf. Developmental stages were determined by previously described morphological criteria (43). Before manipulation or fixation, fish were anesthetized or sacrificed in 0.2 mg/ml solution of tricaine. The Tg(fli1a:EGFP) line was used to monitor blood vessel formation (27). All experiments were conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
Generation of foxc1a and foxc1b knockout zebrafish lines
ZiFiT targeting software (http://zifit.partners.org/ZiFiT/) (44) was used to select target sites within the forkhead domain-encoding regions of the foxc1a and foxc1b genes and design complimentary oligonucleotides (Supplementary Material, Table S1). After annealing, the double-stranded oligonucleotides were cloned into a DR274 vector (Addgene#42250) (45), which contains the backbone sequence for guide RNA and T7 promoter for in vitro transcription. The vector was linearized by DraI restriction enzyme and sgRNA was synthesized using the MEGAshortscript™ T7 Kit (Invitrogen, Waltham, MA) and purified by RNA Clean&Concentrator™-5 kit (Zymo Research, Irvine, CA).
For mRNA synthesis of the Streptococcus pyogenes Cas9, the MLM3639 ZF optimized Cas9 plasmid with T7 promoter (Addgene #42252) was utilized (45). The plasmid was linearized and mRNA was transcribed using mMESSAGE mMACHINE T7 kit (Invitrogen, Waltham, MA). Following polyadenylation, which was performed with Poly(A) Tailing kit (Invitrogen, Waltham, MA), mRNA was extracted with phenol:chloroform and precipitated with isopropanol as recommended by the manufacturer. Prior to injection RNA integrity was assessed by agarose gel electrophoresis.
The single-cell stage zebrafish embryos were injected with 9.2 nl of solution containing 25 pg of sgRNA, 300 pg of Cas9 mRNA and 0.05% Phenol red (Sigma, St. Louis, MO) in Ringer buffer using the NanojectII Injector (Drummond Scientific, Broomall, PA). Mosaic breeders carrying genetic mutations in the germline were identified by analysis of their offspring via PCR-amplification and sequencing of the DNA region flanking the target site. The mutation screening of the foxc1a gene was performed with 5′-GCGCTATTCCGTCTCCAGT and 5′-TGACAAAGCACTCGTTCAGG primers and foxc1b- with 5′-CCTTTAGGCGTTGTGCCTTA and 5′-GAGTCCGGGTCTAAAGTCCA primers. Founder fish carrying c.451_457del in foxc1a (GRCz11, NM_131728.3) and c.258delC or c.256_257insA in foxc1b (GRCz11, NM_131729.2) were selected for further analysis and corresponding lines were established. For genotyping of the foxc1a mutant fish, KASP (Kompetitive allele specific PCR) assay was performed according to the manufacturer instructions (LGC genomics, Manchester, NH; ID: foxc1a_7bpdel; Project: 3294.002); BalI restriction digestion of the amplified fragment was used for foxc1b genotyping.
Morphologic analysis of embryos and adults
Embryos were immobilized in 1% low melting point agarose and imaged using a Zeiss SteREO Discovery V12 microscope (Carl Zeiss, Thornwood, NY) with either a 1.0× stereo objective or a 10× compound objective for the head imaging. Fluorescent three-dimensional maximum projection images of transgene fli1a:EGFP carrier embryos were obtained using an AxioImager.Z1 microscope with ApoTome attachment, an AxioCam MRc5 camera and ZEN pro software (Zeiss, Thornwood, NY). Adults were imaged on a Nikon SMZ-1500 with a 1.0× stereo objective. Measurements were taken using lateral images and ImageJ 1.52k (46) in the vertical and horizontal axis passing through the center of the pupil. The height of the head was measured in the vertical axis and was used to normalize other measurements. To record the positions of the eyes on the head, the distance from the bottom of the head to the center of the pupil on the vertical axis was measured. For head size, the distance from the center of the pupil to the lateral edge of the head on the horizontal axis was measured. For size of the lower jaw, the distance from the point where the vertical axis and the bottom edge of the head meet to the end of the lower jaw was measured. Student’s t-test was used to determine statistical significance.
Histology and heart tissue preparation
Whole embryos and adult bodies were immersed overnight in modified Davidson’s fixative (30% of a 37% solution of formaldehyde, 15% ethanol, 5% glacial acetic acid and 50% distilled H2O) (47). They were transferred to 70% ethanol and submitted to the Children’s Research Institute Histology Core at the Medical College of Wisconsin for paraffin sectioning and H&E staining per standard protocols. Slides were scanned on a NanoZoomer digital slide scanner, and images were viewed with NDP.view2 viewing software (Hamamatsu, Hamamatsu City, Japan). For heart examination, 10-mpf adult zebrafish and 6-dpf embryos were fixed in 4% paraformaldehyde/1× PBS overnight at 4°C. Hearts from adult fish were dissected out while whole embryos were used. Tissue was then embedded in paraffin wax and sectioned at 7 μm. Prior to tissue staining, paraffin was removed in xylenes, and tissue sections were re-hydrated through a graded ethanol series and rinsed in 1× PBS as previously described (48). Tissue sections containing hearts were then subjected to immunohistochemistry/immunofluorescence (see below for details).
Immunohistochemistry and immunofluorescence
10-mpf adult whole hearts and 6-dpf embryos were collected from foxc1a+/−;foxc1b−/− zebrafish and foxc1a+/+;foxc1b+/− siblings and prepared according to the above methods. For antibody detection, fixed paraffin tissue sections were subjected to antigen retrieval by boiling for 10 min in unmasking solution (Vector Laboratories, Burlingame, CA H-3300) and then subjected to blocking for 1 h at room temperature (1% BSA, 1% cold water fish skin gelatin, 0.1% Tween-20/PBS) as described (49). Tissue sections were then incubated for 1 h at room temperature with primary antibody against MF20 (Mouse, 1:200, eBioscience, Waltham, MA 14–6503-82). For immunofluorescent primary antibody detection of MF20, sections were incubated for 1 h at room temperature with Donkey anti-mouse Alexa-Fluor IgG secondary antibody (1:400) (Life Technologies, Waltham, MA), then mounted in Vectashield anti-fade medium with DAPI (Vector Laboratories, Burlingame, CA H-1800) to detect cell nuclei. Images were visualized and captured using the EVOS M7000 Imaging System and associated software. Image brightness and contrast were edited using Adobe Photoshop CC. Measurements of compact and trabecular zone thickness were taken on MF20 stained images, while the outflow tract (OFT) and atrioventricular (AV) valve area and OFT and AV cells/area were measured on DAPI-stained images. Quantifications were performed using ImageJ 1.52k (46). Statistical significance of quantifications was determined by unpaired t-test in GraphPad Prism 8.
Spectral domain-optical coherence tomography (SD-OCT)
Sixteen 10-mpf foxc1a+/−;foxc1b−/− and sixteen foxc1a+/+; foxc1b+/− sibling fish eyes were imaged using the Bioptigen Envisu R2200 SD-OCT imaging system with a 12 mm telecentric lens (Bioptigen, Morrisville, NC) as per Collery et al. (50). Images were assembled and measurements were done using InVivoVue software (Leica Microsystems, Buffalo Grove, IL), and ImageJ 1.52k (46). Measurements included corneal thickness, pupil width, anterior chamber area and both dorsal and ventral angles.
Quantitative RT-PCR transcript-level analysis of WT and mutant embryo eyes
48- and 72-hpf WT, foxc1a+/− and foxc1−/− (foxc1a−/−;foxc1b−/−) mutant embryos eyes were dissected and placed in TRI reagent (Zymo Research, Irvine, CA). RNA extraction was performed using Direct-zol RNA MiniPrep (Zymo Research, Irvine, CA). All samples were treated with DNase I. RNA quality and concentration were assessed using a NanoDrop 2000 UV–Vis Spectrophotometer (Thermo Fisher, Waltham, MA). cDNA was synthesized using SuperScript III reverse transcriptase (Thermo Fisher, Waltham, MA). Selected targets were analyzed by real-time qPCR using the CFX384 Touch Real-Time PCR Detection Systems (Bio Rad, Hercules, CA), SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA) and transcript specific primers (Supplementary Material, Table S1). β-actin was used as the reference gene for relative quantification of expression levels. All samples were run in triplicate to obtain average quantitation cycle (Cq) values. Total fold changes and standard deviations were calculated as the average of three independent biological repeats via the 2−ΔΔCt method (51). Student’s t-test was used to determine statistical significance.
Supplementary Material
Acknowledgements
The authors would like to thank Linda Reis, MS, CGC, for her careful reading of the manuscript and valuable comments.
Conflict of Interest statement. The authors declare no conflict of interest.
Contributor Information
Jesús-José Ferre-Fernández, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA.
Elena A Sorokina, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA.
Samuel Thompson, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA.
Ross F Collery, Department of Ophthalmology and Visual Sciences, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Emily Nordquist, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA.
Joy Lincoln, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA; Division of Pediatric Cardiology, Herma Heart Institute, Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA.
Elena V Semina, Department of Pediatrics, Children’s Research Institute, Medical College of Wisconsin and Children’s Hospital of Wisconsin, Milwaukee, WI 53226, USA; Department of Ophthalmology and Visual Sciences, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Funding
National Eye Institute (R01EY025718, R01EY015518 to E.V.S.); Children’s Research Institute Foundation at Children’s Hospital of Wisconsin (to E.V.S.); Advancing a Healthier Wisconsin (Project #5520519 to J.L.).
References
- 1. Reis L.M., Tyler R.C., Volkmann Kloss B.A., Schilter K.F., Levin A.V., Lowry R.B., Zwijnenburg P.J., Stroh E., Broeckel U., Murray J.C. and Semina E.V. (2012) PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur. J. Hum. Genet., 20, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reis L.M., Tyler R.C., Weh E., Hendee K.E., Schilter K.F., Phillips J.A. 3rd, Sequeira S., Schinzel A. and Semina E.V. (2016) Whole exome sequencing identifies multiple diagnoses in congenital glaucoma with systemic anomalies. Clin. Genet., 90, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Medina-Trillo C., Sanchez-Sanchez F., Aroca-Aguilar J.D., Ferre-Fernandez J.J., Morales L., Mendez-Hernandez C.D., Blanco-Kelly F., Ayuso C., Garcia-Feijoo J. and Escribano J. (2015) Hypo- and hypermorphic FOXC1 mutations in dominant glaucoma: transactivation and phenotypic variability. PLoS One, 10, e0119272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ansari M., Rainger J., Hanson I.M., Williamson K.A., Sharkey F., Harewood L., Sandilands A., Clayton-Smith J., Dollfus H., Bitoun P. et al. (2016) Genetic analysis of 'PAX6-Negative' individuals with Aniridia or Gillespie syndrome. PLoS One, 11, e0153757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Micheal S., Siddiqui S.N., Zafar S.N., Villanueva-Mendoza C., Cortes-Gonzalez V., Khan M.I. and den Hollander A.I. (2016) A novel homozygous mutation in FOXC1 causes Axenfeld Rieger syndrome with congenital glaucoma. PLoS One, 11, e0160016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khan A.O., Aldahmesh M.A. and Alkuraya F.S. (2011) Genetic and genomic analysis of classic aniridia in Saudi Arabia. Mol. Vis., 17, 708–714. [PMC free article] [PubMed] [Google Scholar]
- 7. Souzeau E., Siggs O.M., Zhou T., Galanopoulos A., Hodson T., Taranath D., Mills R.A., Landers J., Pater J., Smith J.E. et al. (2017) Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. Eur. J. Hum. Genet., 25, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. D'Haene B., Meire F., Claerhout I., Kroes H.Y., Plomp A., Arens Y.H., de Ravel T., Casteels I., De Jaegere S., Hooghe S. et al. (2011) Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest. Ophthalmol. Vis. Sci., 52, 324–333. [DOI] [PubMed] [Google Scholar]
- 9. French C.R., Seshadri S., Destefano A.L., Fornage M., Arnold C.R., Gage P.J., Skarie J.M., Dobyns W.B., Millen K.J., Liu T. et al. (2014) Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Invest., 124, 4877–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Avasarala J.R., Jones J.R. and Rogers C.R. (2018) Forkhead box C1 gene variant causing glaucoma and small vessel angiopathy can mimic multiple sclerosis. Mult. Scler. Relat. Disord., 22, 157–160. [DOI] [PubMed] [Google Scholar]
- 11. Saleem R.A., Murphy T.C., Liebmann J.M. and Walter M.A. (2003) Identification and analysis of a novel mutation in the FOXC1 forkhead domain. Invest. Ophthalmol. Vis. Sci., 44, 4608–4612. [DOI] [PubMed] [Google Scholar]
- 12. Strungaru M.H., Dinu I. and Walter M.A. (2007) Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest. Ophthalmol. Vis. Sci., 48, 228–237. [DOI] [PubMed] [Google Scholar]
- 13. Fuse N., Takahashi K., Yokokura S. and Nishida K. (2007) Novel mutations in the FOXC1 gene in Japanese patients with Axenfeld-Rieger syndrome. Mol. Vis., 13, 1005–1009. [PMC free article] [PubMed] [Google Scholar]
- 14. Gripp K.W., Hopkins E., Jenny K., Thacker D. and Salvin J. (2013) Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel FOXC1 mutation. Am. J. Med. Genet. A, 161A, 114–119. [DOI] [PubMed] [Google Scholar]
- 15. Topczewska J.M., Topczewski J., Solnica-Krezel L. and Hogan B.L. (2001) Sequence and expression of zebrafish foxc1a and foxc1b, encoding conserved forkhead/winged helix transcription factors. Mech. Dev., 100, 343–347. [DOI] [PubMed] [Google Scholar]
- 16. Takamiya M., Weger B.D., Schindler S., Beil T., Yang L., Armant O., Ferg M., Schlunck G., Reinhard T., Dickmeis T. et al. (2015) Molecular description of eye defects in the zebrafish Pax6b mutant, sunrise, reveals a Pax6b-dependent genetic network in the developing anterior chamber. PLoS One, 10, e0117645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skarie J.M. and Link B.A. (2009) FoxC1 is essential for vascular basement membrane integrity and hyaloid vessel morphogenesis. Invest. Ophthalmol. Vis. Sci., 50, 5026–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Topczewska J.M., Topczewski J., Shostak A., Kume T., Solnica-Krezel L. and Hogan B.L. (2001) The winged helix transcription factor Foxc1a is essential for somitogenesis in zebrafish. Genes Dev., 15, 2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Yue Y., Dong X., Jia W., Li K., Liang D., Dong Z., Wang X., Nan X., Zhang Q. and Zhao Q. (2015) Zebrafish foxc1a plays a crucial role in early somitogenesis by restricting the expression of aldh1a2 directly. J. Biol. Chem., 290, 10216–10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yue Y., Jiang M., He L., Zhang Z., Zhang Q., Gu C., Liu M., Li N. and Zhao Q. (2018) The transcription factor Foxc1a in zebrafish directly regulates expression of nkx2.5, encoding a transcriptional regulator of cardiac progenitor cells. J. Biol. Chem., 293, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu P., Balczerski B., Ciozda A., Louie K., Oralova V., Huysseune A. and Crump J.G. (2018) Fox proteins are modular competency factors for facial cartilage and tooth specification. Development, 145, dev165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Banerjee S., Hayer K., Hogenesch J.B. and Granato M. (2015) Zebrafish foxc1a drives appendage-specific neural circuit development. Development, 142, 753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whitesell T.R., Chrystal P.W., Ryu J.R., Munsie N., Grosse A., French C.R., Workentine M.L., Li R., Zhu L.J., Waskiewicz A. et al. (2019) foxc1 is required for embryonic head vascular smooth muscle differentiation in zebrafish. Dev. Biol., 453, 34–47. [DOI] [PubMed] [Google Scholar]
- 24. Carmona S., da Luz Freitas M., Froufe H., Simoes M.J., Sampaio M.J., Silva E.D. and Egas C. (2017) Novel de novo FOXC1 nonsense mutation in an Axenfeld-Rieger syndrome patient. Am. J. Med. Genet. A, 173, 1607–1610. [DOI] [PubMed] [Google Scholar]
- 25. Swiderski R.E., Reiter R.S., Nishimura D.Y., Alward W.L., Kalenak J.W., Searby C.S., Stone E.M., Sheffield V.C. and Lin J.J. (1999) Expression of the Mf1 gene in developing mouse hearts: implication in the development of human congenital heart defects. Dev. Dyn., 216, 16–27. [DOI] [PubMed] [Google Scholar]
- 26. Nishimura D.Y., Searby C.C., Alward W.L., Walton D., Craig J.E., Mackey D.A., Kawase K., Kanis A.B., Patil S.R., Stone E.M. and Sheffield V.C. (2001) A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am. J. Hum. Genet., 68, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawson N.D. and Weinstein B.M. (2002) In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol., 248, 307–318. [DOI] [PubMed] [Google Scholar]
- 28. Kaufman R., Weiss O., Sebbagh M., Ravid R., Gibbs-Bar L., Yaniv K. and Inbal A. (2015) Development and origins of zebrafish ocular vasculature. BMC Dev. Biol., 15, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kitambi S.S., McCulloch K.J., Peterson R.T. and Malicki J.J. (2009) Small molecule screen for compounds that affect vascular development in the zebrafish retina. Mech. Dev., 126, 464–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hartsock A., Lee C., Arnold V. and Gross J.M. (2014) In vivo analysis of hyaloid vasculature morphogenesis in zebrafish: a role for the lens in maturation and maintenance of the hyaloid. Dev. Biol., 394, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bader D., Masaki T. and Fischman D.A. (1982) Immunochemical analysis of myosin heavy chain during avian myogenesis in vivo and in vitro. J. Cell Biol., 95, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kidson S.H., Kume T., Deng K., Winfrey V. and Hogan B.L. (1999) The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev. Biol., 211, 306–322. [DOI] [PubMed] [Google Scholar]
- 33. Seo S., Singh H.P., Lacal P.M., Sasman A., Fatima A., Liu T., Schultz K.M., Losordo D.W., Lehmann O.J. and Kume T. (2012) Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc. Natl. Acad. Sci. USA., 109, 2015–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beby F., Des Portes V., Till M., Mottolese C. and Denis P. (2012) Chromosome 6p25 deletion syndrome: report of a case with optic disc coloboma and review of published ophthalmic findings. Ophthalmic Genet., 33, 240–248. [DOI] [PubMed] [Google Scholar]
- 35. Gould D.B., Jaafar M.S., Addison M.K., Munier F., Ritch R., MacDonald I.M. and Walter M.A. (2004) Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: relevance to ocular dysgenesis and hearing impairment. BMC Med. Genet., 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weiss O., Kaufman R., Michaeli N. and Inbal A. (2012) Abnormal vasculature interferes with optic fissure closure in lmo2 mutant zebrafish embryos. Dev. Biol., 369, 191–198. [DOI] [PubMed] [Google Scholar]
- 37. Lambers E., Arnone B., Fatima A., Qin G., Wasserstrom J.A. and Kume T. (2016) Foxc1 regulates early cardiomyogenesis and functional properties of embryonic stem cell derived cardiomyocytes. Stem Cells, 34, 1487–1500. [DOI] [PubMed] [Google Scholar]
- 38. de Vos I.J., Stegmann A.P., Webers C.A. and Stumpel C.T. (2017) The 6p25 deletion syndrome: an update on a rare neurocristopathy. Ophthalmic Genet., 38, 101–107. [DOI] [PubMed] [Google Scholar]
- 39. Hong H.K., Lass J.H. and Chakravarti A. (1999) Pleiotropic skeletal and ocular phenotypes of the mouse mutation congenital hydrocephalus (ch/Mf1) arise from a winged helix/forkhead transcriptionfactor gene. Hum. Mol. Genet., 8, 625–637. [DOI] [PubMed] [Google Scholar]
- 40. Kume T., Jiang H., Topczewska J.M. and Hogan B.L. (2001) The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev., 15, 2470–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hendee K.E., Sorokina E.A., Muheisen S.S., Reis L.M., Tyler R.C., Markovic V., Cuturilo G., Link B.A. and Semina E.V. (2018) PITX2 deficiency and associated human disease: insights from the zebrafish model. Hum. Mol. Genet., 27, 1675–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ito Y.A., Footz T.K., Berry F.B., Mirzayans F., Yu M., Khan A.O. and Walter M.A. (2009) Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Invest. Ophthalmol. Vis. Sci., 50, 3573–3579. [DOI] [PubMed] [Google Scholar]
- 43. Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B. and Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 44. Sander J.D., Maeder M.L., Reyon D., Voytas D.F., Joung J.K. and Dobbs D. (2010) ZiFiT (zinc finger targeter): an updated zinc finger engineering tool. Nucleic Acids Res., 38, W462–W468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R. and Joung J.K. (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol., 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider C.A., Rasband W.S. and Eliceiri K.W. (2012) NIH image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Latendresse J.R., Warbrittion A.R., Jonassen H. and Creasy D.M. (2002) Fixation of testes and eyes using a modified Davidson's fluid: comparison with Bouin's fluid and conventional Davidson's fluid. Toxicol. Pathol., 30, 524–533. [DOI] [PubMed] [Google Scholar]
- 48. Lincoln J., Alfieri C.M. and Yutzey K.E. (2004) Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn., 230, 239–250. [DOI] [PubMed] [Google Scholar]
- 49. Peacock J.D., Lu Y., Koch M., Kadler K.E. and Lincoln J. (2008) Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Dev. Dyn., 237, 3051–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Collery R.F., Veth K.N., Dubis A.M., Carroll J. and Link B.A. (2014) Rapid, accurate, and non-invasive measurement of zebrafish axial length and other eye dimensions using SD-OCT allows longitudinal analysis of myopia and emmetropization. PLoS One, 9, e110699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livak K.J. and Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.