

Abstract
Despite their small size, bacteria have a remarkably intricate internal organization. Bacteria deploy proteins and protein complexes to particular locations and do so in a dynamic manner in lockstep with the organized deployment of their chromosome. The dynamic subcellular localization of protein complexes is an integral feature of regulatory processes of bacterial cells.
Bacteria were once viewed as amorphous reaction vessels with chromosomes that wandered freely and randomly throughout the cell. The advent of genetically encoded fluorescent reporters harnessed to powerful cell-imaging technologies has enabled in vivo tracking of protein movement and revealed a strikingly complex inner world within bacteria. This inner environment is exquisitely organized, in a highly controlled state of flux, and responsive to changing functions demanded of the cell. For example, some proteins oscillate rapidly from one end of the cell to the other, whereas others form dynamic helices along the length of the cell or rings across its midsection, and yet others form distributed focal complexes on the cell’s surface or clusters at specific intracellular sites. Processes controlled at multiple levels construct (and remove) subsystems and surface structures at specific times and places in response to internal and external signals. This dynamic internal architecture facilitates behaviors as diverse as symmetric and asymmetric division, motility, chemotaxis, morphological differentiation, assembly into multicellular communities, and interactions with animal and plant hosts. In one bacterial species, Caulobacter crescentus, at least 10% of predicted encoded proteins exhibited specific subcellular organization (1). Here, we explore why bacteria dynamically deploy key regulatory proteins to particular sites in the cell and how this positioning and repositioning is achieved.
Why Are Proteins Localized?
Polar positioning of chemotaxis arrays.
Since the first observation that bacterial chemoreceptors, along with CheA histidine kinases and CheW adapter proteins, are located at or near one cell pole of C. crescentus (Caulobacter hereafter) and Escherichia coli (2, 3), fluorescence microscopy and cryoelectron microscopy images have revealed the exquisite architecture of these polar complexes (4, 5). The chemotaxis sensor system controls the activity of the flagellar motor so that cells move toward attractants and away from repellents. The chemoreceptor array comprises thousands of receptors arranged in a “trimer of dimers” configuration (6–8). The unit cell of this hexagonal lattice is formed by three receptor dimers (5). Why has the cell evolved this elaborate localized array? One proposal is that the close spacing of the components of the chemosensory array promotes signal amplification (9). The Caulobacter chemoreceptor array is always positioned somewhat away from the pole on the convex side of the crescent-shaped cell (Fig. 1B) (5), whereas a linear array of crescentin intermediate filaments (which confer on Caulobacter its distinctive shape) is always positioned on the concave side of the crescent (Fig. 1B) (10). Thus, the Caulobacter cell has dorsal-ventral asymmetry as well as anterior-posterior asymmetry exhibited by the differential polar placement of the flagellum and stalk, the chromosomal origin complex, and signaling kinases (Fig. 1A).
Fig. 1.
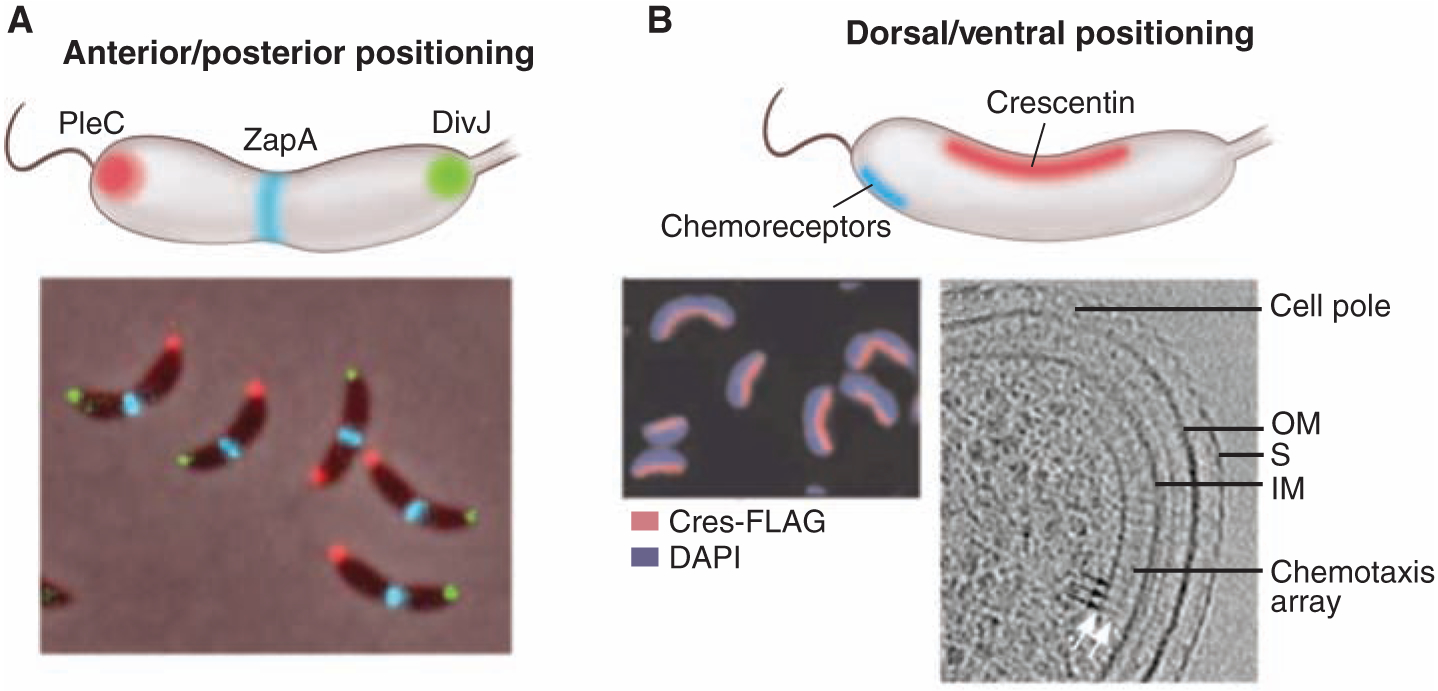
Localization patterns. (A) An anterior-posterior cellular localization axis is exhibited by the Caulobacter histidine kinases PleC (red) and DivJ (green) that dynamically and selectively localize to specific cell poles. The ZapA cell division protein (blue) localizes to the FtsZ ring. (B) In these same cells, a dorsal-ventral localization axis is exhibited by crescentin (cres) intermediate filaments that localize along the inner concave side of the crescent-shaped cell and are responsible for this distinctive cell shape (10); in contrast, the chemotaxis sensor array localizes at the convex outer side of the crescent near the cell pole (5). DAPI, 4′,6-diamidino-2-phenylindole.
Chromosome organization.
Subcellular protein localization also mediates the highly organized deployment of bacterial chromosomes. Bacterial DNA, if stretched out linearly, would be about 1000 times the length of the cell. To deal with these spatial constraints, bacteria have evolved a highly ordered deployment of the chromosome within the cell. Notably, the chromosome has a specific orientation within the cell. For many bacteria, including Caulobacter, Vibrio cholerae chromosome I, and sporulating Bacillus subtilis, the DNA sequence around the replication origin is positioned at the cell pole (11–13). In Caulobacter, upon replication of the chromosomal origin region, the origin-proximal parS centromeric sequence bound to the ParB partition protein is transported rapidly across the long axis of the cell and is captured by a polar polymeric network of the PopZ protein (Fig. 2A) (14, 15). In growing cells of B. subtilis, origins are located at the outer edge of the nucleoids. But in cells that have entered the pathway to sporulation, a hemispheric array of the DivIVA protein at the cell pole captures the RacA protein decorating the origin-proximal portion of the newly duplicated chromosome (Fig. 2B) (16, 17). In addition to specific cellular positioning of the origin and terminus sequences, the entire chromosome is organized within the cell. When positions of fluorescently tagged foci in the cell were measured in more than 100 Caulobacter strains, each with a different tagged locus (18), a linear correspondence was observed between the position of a given locus along the length of the cell and its position on the chromosome. Analysis of the B. subtilis and E. coli genome organization also suggested a linear correspondence of gene position in the cell and gene order on the chromosome (19–21).
Fig. 2.
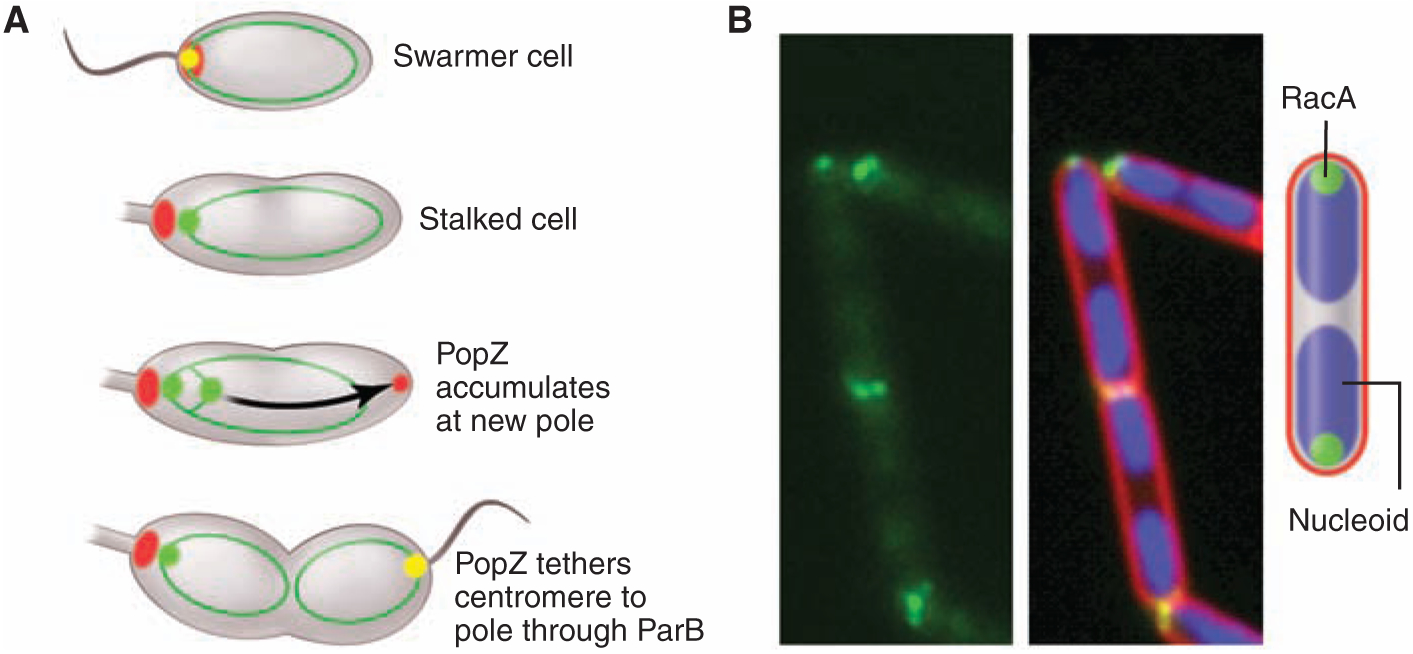
Chromosome attachment to the cell pole. (A) The Caulobacter parS centromere bound to the ParB partition protein is attached to the cell pole by interaction with the polar PopZ polymeric network (14, 15). The initiation of replication triggers the assembly of PopZ at the opposite pole, where it captures the duplicated copy of parS/ParB. The diagram shows PopZ (red), ParB (green), PopZ + ParB (yellow), and chromosomes depicted as rings (dark green). (B) Sporulating cells of B. subtilis anchor chromosomes to the cell poles via the sporulation protein RacA, which binds to sites near the replication origin and to DivIVA at the cell poles (16, 17). The images show RacA tagged with green fluorescent protein (green), the nucleoids (blue), and the cell membrane (red).
Cell division.
When cells divide, a key challenge is to ensure that cytokinesis commences at the right time and at the right place along the cell length. How is this achieved? In many rod-shaped bacteria, assembly of the cell division ring is guided to the center of the cell by the action of inhibitors of the polymerization of the tubulin-like protein FtsZ that drives cytokinesis (Fig. 2). These inhibitors are dynamically localized to the cell poles to create an inhibitor-free zone at the cell center. In the case of E. coli, this process involves rapid pole-to-pole oscillation of the MinCD inhibitor complex, so that the cell center is the site of lowest average inhibitor concentration, thus allowing midcell assembly of FtsZ (Fig. 3A) (22). The MinE protein restricts the MinCD membrane-associated complex to the cell poles. In B. subtilis, which lacks MinE, MinCD is recruited to sites of septation and diffuses away from completed septa, preventing septation from occurring near new cell poles (23). The MinJ protein tethers MinCD to DivIVA, which is positioned at septa and in a hemispheric array at the poles (24, 25). In both cases, nucleoid occlusion, in which the FtsZ ring is precluded from assembling over the chromosome, augments the action of the polar inhibitors (26–29). Caulobacter, in contrast, takes advantage of the concurrent initiation of DNA replication and polar segregation of the chromosomal origin to establish a gradient of the MipZ cytokinesis-inhibiting protein emanating from the cell poles, thus determining the midcell placement of the FtsZ ring (Fig. 2B) (30). MipZ, which interacts with ParB bound to the parS centromere anchored at the cell poles, inhibits the polymerization of FtsZ. The dynamic interaction of MipZ and ParB results in a gradient of MipZ, with the highest concentration at the cell poles. As the newly duplicated centromere is moved rapidly along the long axis of the cell to the opposite pole, FtsZ localizes to the site of lowest concentration of the MipZ inhibitor. The division plane is established at this point. The use of protein gradients emanating from cell poles is not restricted to bacterial cells. The fission yeast Schizosaccharomyces pombe establishes a gradient of polar Pom1 kinases to tie the onset of mitosis to cell length (Fig. 3C) (31, 32).
Fig. 3.
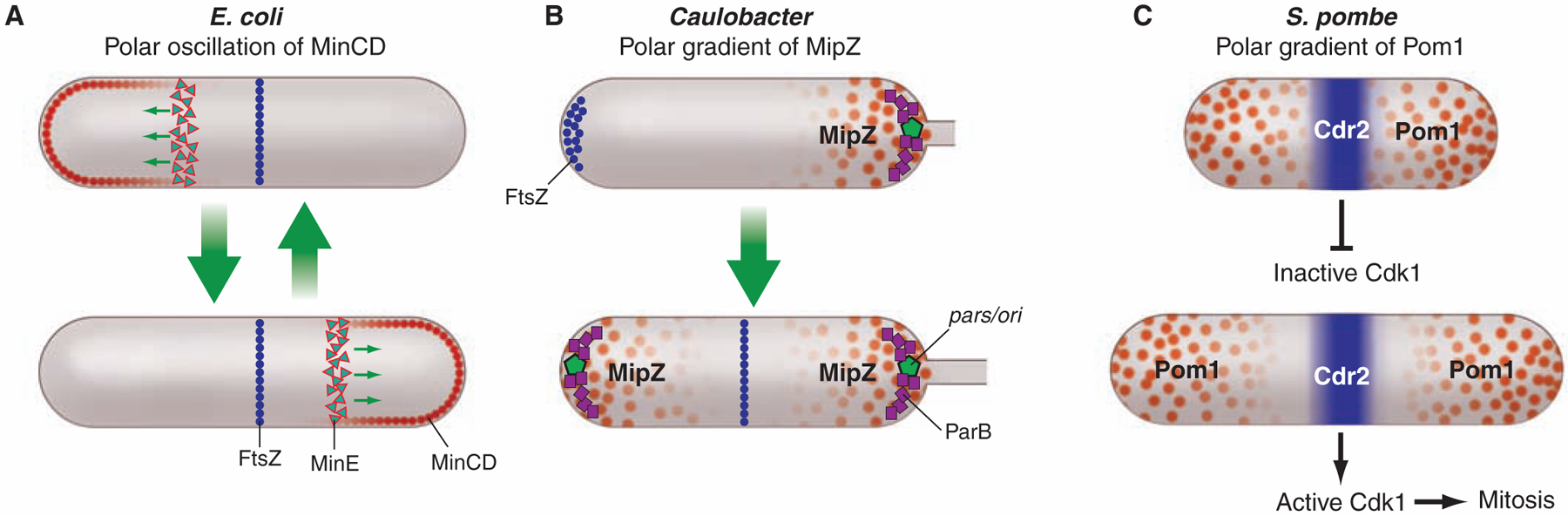
Cell topology and inhibitor gradients control place and time of cell division. Repression of FtsZ polymerization by polar localized proteins that exhibit a minimum of the repressor at midcell restricts the site of division ring assembly in both E. coli and Caulobacter. However, E. coli’s strategy depends on oscillation of the MinCD repressor from pole to pole (A), whereas Caulobacter establishes a gradient of the MipZ repressor with the highest concentration at the cell poles (B). In S. pombe, the Pom1 repressor is localized to the poles (C). In small cells, the gradient of Pom1 extending from the poles overlaps at Cdr2 located at midcell and represses its activity. Consequently, the Cdk1 pathway is blocked, preventing entry into mitosis. As the cell grows, the midcell repressor concentration diminishes until Cdr2 (and thus the Cdk1 pathway) is no longer repressed and entry into mitosis is facilitated.
These examples illustrate how different cell types use the cell’s instantaneous topology and internal organization to coordinate spatially and temporally controlled events crucial to cell cycle progression.
Using geometry to control proteolysis and drive membrane movement.
Protease complexes that clear proteins from the cell at the right time and place are key members of the cell’s regulatory tool kit. With impressive destructive power, these proteases are held in tight control. Somewhat surprisingly, in bacterial cells one of the control mechanisms uses the dynamic positioning of protease complexes to the cell poles (33, 34). This spatial control of proteolysis is seen most clearly in Caulobacter, where it is intimately linked to cell cycle progression (33).
Entry into S phase is a critical event in all cells, and no less so in bacteria. Chromosome replication in Caulobacter is restricted to once per cell cycle, and the decision to enter S phase is governed in part by the proteolysis of CtrA (33, 35), an inhibitor of replication initiation (36). Clearance of CtrA from the cell allows DnaA-mediated replication initiation. CtrA, a substrate of the essential protease ClpXP (37), is degraded only in the stalked cell. Because the ClpXP protease is present throughout the cell cycle and has many other substrates, the cell is faced with the challenge of providing both temporal and spatial specificity to ClpXP proteolysis of CtrA. Specificity is provided, in part, by localizing both the protease complex and its CtrA substrate to the stalked cell pole at specific times in the cell cycle (33). The time in the cell cycle when these protein complexes are localized to the pole is governed by a dynamically localized phospho-signaling pathway and a second messenger system (38, 39). The culmination of these events is assembly of a polar complex in which the ClpXP proteolytic machinery and its substrate are brought together at just the right time to clear CtrA from the cell so that DNA replication can be initiated. This use of polar positioning to control proteolysis is also observed in the soil bacterium Sinorhizobium meliloti, where protease polar localization is essential for coordinating cell cycle progression and for differentiation into bacteroids upon infection of plant roots (40).
How Are Proteins Localized?
Diffusion and capture.
Proteins are localized to their correct position within the bacterial cell by signals encoded in their primary amino acid sequence. But how do these signals work? Eukaryotic cells have sorting machinery based on vesicles that deliver protein cargos to their proper destination, but bacteria lack vesicle-mediated sorting. Instead, bacterial protein localization is mediated principally by diffusion and capture. In the small bacterial cell, proteins can diffuse rapidly throughout the cytoplasm in three dimensions or throughout the membrane in two dimensions, encountering other proteins in all possible configurations. When the protein encounters another protein to which it specifically adheres, that configuration will tend to persist. Thus, a localized “target” protein complex can capture and localize individual proteins or groups of proteins. Depending on the energetics of the protein-protein binding, the localization interaction may be persistent or transient; in the latter case, there can be continuous exchange between the cytoplasmic and bound-protein state. Overexpression of membrane-bound docking proteins can result in their presence throughout the membrane even though the wild-type cell exhibits specific foci, suggesting saturation of the target complex docking sites in the overexpression strain (41).
Direct evidence in support of the diffusion-and-capture mechanism has been obtained for the localization of the PleC histidine kinase to the cell pole in Caulobacter and the assembly of the BofA-SpoIVFA-SpoIVFB protease complex in sporulating B. subtilis. Observation of the movement of single fluorescently tagged PleC molecules in live cells revealed a stationary PleC focus at the cell pole while PleC molecules away from the pole defused in the membrane without a directional bias, supporting the hypothesis that PleC diffuses randomly until captured by a target at the pole (42). In sporulating B. subtilis, SpoIVFA is the anchor that captures BofA and SpoIVFB. A diffusion-and-capture mechanism was deduced from the observation that when membrane containing the target protein becomes topologically isolated from the cytoplasmic membrane after completion of the phagocytic-like engulfment process, BofA and SpoIVFB in the cytoplasmic membrane can no longer reach SpoIVFA (43). This finding demonstrated that localization required an uninterrupted membrane through which BofA and SpoIVFB can diffuse to the SpoIVFA target and be captured.
Ultimate cues.
As we have seen, proteins often localize by binding to another protein or proteins that are already sequestered at a particular location in the cell. This dependency raises the question of the prior cue that dictates the position of the target protein in the ensemble of proteins, and the one before that and so on, until the position of the first localized protein is established. Consider again the cytokinetic FtsZ ring, which in Caulobacter assembles only at a site near the midpoint between the cell poles because the MipZ inhibitor of FtsZ polymerization interacts with the chromosomal origin bound to the cell poles to form a gradient of the inhibitor. In this case, therefore, confinement of the FtsZ ring to the cell middle ultimately derives from the organization of the chromosome and the placement of origins at the poles.
Dynamic self-assembly appears to contribute to the clustering of proteins at subcellular locations. As discussed earlier, chemoreceptors form large polar clusters. In E. coli, in which large clusters are seen at both poles, receptors stochastically self-assemble into small clusters all around the cell, including along the side walls, in an exponential distribution with the largest clusters at the poles (44). According to this self-assembly model, receptor molecules are most likely to be captured by existing clusters if one is nearby, and to nucleate new clusters otherwise. As a consequence, and in steady state, larger clusters will tend to assemble as far apart from each other as possible—that is, at the poles.
What other ultimate cues dictate protein localization in bacteria? A different type of cue has emerged from studies of two peripheral membrane proteins in B. subtilis: SpoVM and DivIVA. Spore formation takes place in a sporangium that consists of a cell-within-a-cell generated by an engulfment process, as noted above. The outer cell—the mother cell—nurtures the developing spore contained within it (Fig. 4). The SpoVM protein is produced in the mother cell but localizes specifically to the outer membrane surface of the developing spore rather than the cytoplasmic membrane that surrounds the mother cell. How does SpoVM discriminate between these two membranes? Multiple lines of evidence indicate that SpoVM preferentially adheres to membrane with positive (convex) curvature: Only the surface of the spherical spore is positively curved; the cytoplasmic membrane is concave (45). So the position of this protein—and, in turn, the position of other proteins that it recruits to the outer surface of the spore—is determined by geometry, specifically the shape of the spore membrane.
Fig. 4.
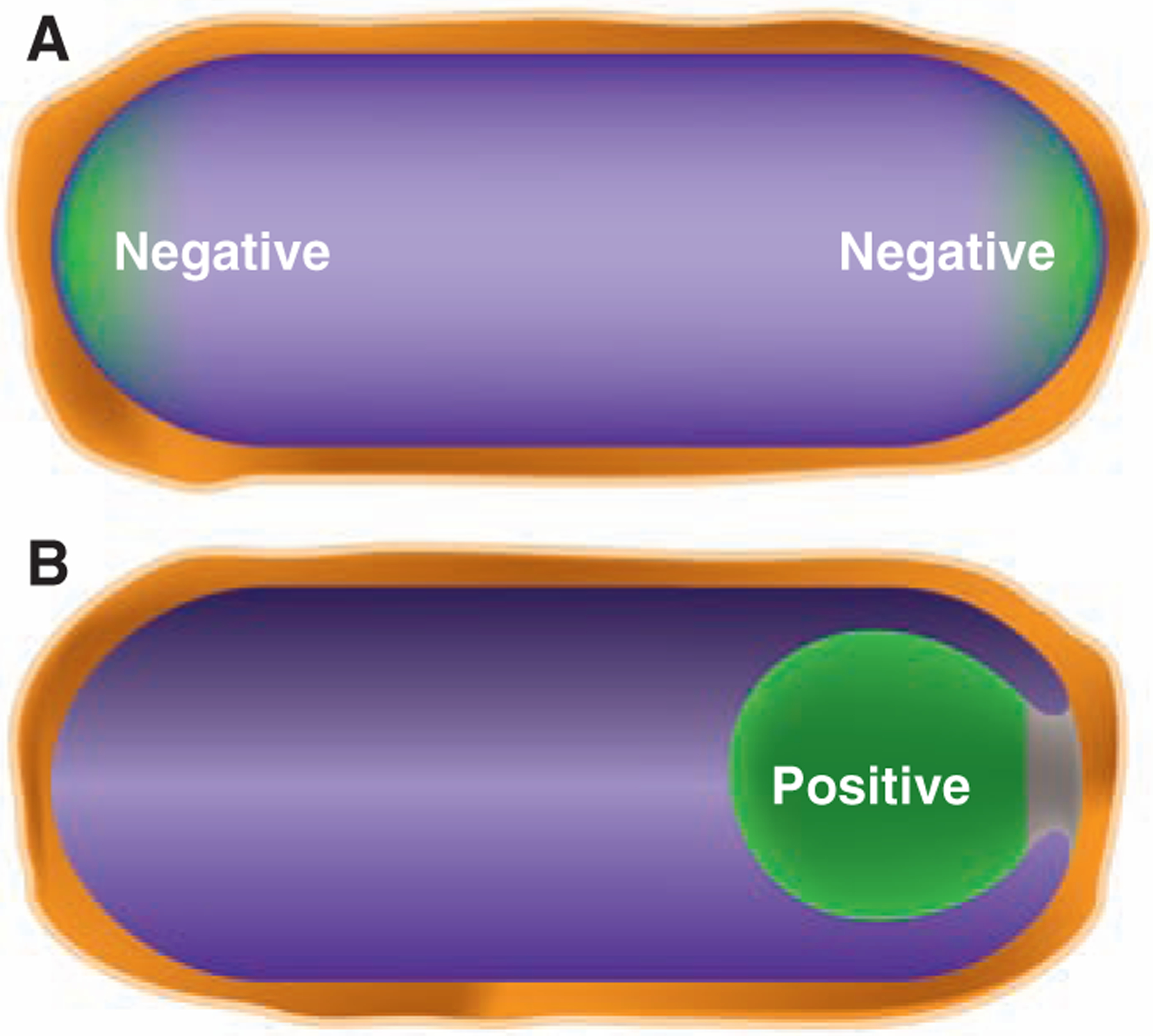
Geometric cues for protein localization. (A) Schematic depiction of the hemispherical poles of the cell. The more extreme negative curvature at the inside surface of the poles (relative to the inside surface of the lateral walls of the cell) could be a geometric cue for proteins, such as the cell division protein DivIVA, that localize to the poles. (B) The process of engulfment during spore formation in B. subtilis in which the membrane of the large mother cell (on the left) migrates around to surround and eventually pinch off the nascent spore. The positive curvature of the surface of the engulfment membrane from within the mother cell is a geometric cue for the sporulation protein SpoVM. The green color indicates the localization of DivIVA in (A) and SpoVM in (B) to regions of negative and positive curvature, respectively.
Conversely, negative membrane curvature seems to be the ultimate cue for DivIVA, a multifunctional protein that contributes to Z-ring positioning during growth (by recruiting MinCD at the B. subtilis cell poles) and localizing chromosome origin regions at the poles during sporulation (by capturing chromosome-bound RacA). DivIVA preferentially localizes as a disk around the outer edge of the developing septum and at the poles. Yet apparently it does not interact with any divisome or polar protein. What does it recognize? The poles and the junction of the septum to the lateral wall of the cell are sites of more extreme negative curvature than the sides of the cell, which are also negatively curved. Evidence suggests that DivIVA localizes in a hierarchical fashion favoring the most extreme concavity of the septum, next the hemispherical curvature of the inner surface of the poles, and finally the more gently curved inner surface of the sides of the cell (46, 47). These findings raise the possibility that geometry may play a widespread role in protein localization in bacteria. Perhaps other proteins also localize principally to the cell ends by preferring hemispherical curvature.
Some protein localization may depend on a never-ending cyclical cascade of cues from one cell generation to the next. Cell division gives rise to the new pole of the resulting daughter cells, so certain proteins that localize to the septum could be left behind at the cell poles after cytokinesis is complete and the divisome is disassembled. These proteins could provide targets for polar-localized proteins in the daughter cells. Just such a scenario has been proposed for TipN in Caulobacter, a landmark protein that provides a positional cue for the assembly of a flagellum at the pole and for controlling the size asymmetry of the two daughter cells (48, 49). Hemispherical curvature and perhaps differences in cell envelope molecular composition are also potential ultimate cues for placing proteins at the cell poles. Conceivably, some proteins, including DivIVA and TipN, rely on both cues in their localization.
Understanding dynamic changes in protein localization.
Finally, we touch on an important challenge for the future. So far we have focused on how proteins come to localize at a particular site, but not on the events that precipitate changes in protein localization. For example, what changes in the cell so that specific histidine kinases transiently localize to a cell pole rather than remaining dispersed throughout the cell membrane? What signals the replacement of that kinase with a different polar kinase? A contributing factor is changes in the phosphorylation state of one or more proteins that affects their collective binding affinity for the localization site. In other cases, the precipitating event is the de novo synthesis of a protein that then localizes by diffusion-and-capture at a target site. In each case of localized proteins involved in progression of the cell cycle or other serial developmental processes, the precipitation of localization is the consequence of an upstream event, such as a change in the cell’s topology, the prior positioning of a target binding factor, a transcriptional cascade, a series of phospho-transfer reactions, and so on. These are all causal events involving physical processes, such as diffusion and stereochemistry, that are just beginning to be understood. The challenge now is to assemble the emerging evidence into a systems-level understanding of protein localization within the overall operation of the cell.
Conclusions
Bacteria have an intricate and dynamic three-dimensional organization that is central to their capacity to grow and divide, to respond to the environment, and to develop into specialized cells. This high state of organization is not limited to proteins; the chromosome too is maintained in a strikingly organized manner. Why have bacteria evolved this intricate architecture, and how is it achieved? As we have seen, the inner life of the cell is inextricably linked to how the cell works. Proteins assemble at specific sites (such as the poles) to amplify signals from the environment, to prevent inappropriate cytokinesis, to capture chromosomal regions, to control proteolysis, and to dictate cell shape. But what are the ultimate cues that dictate position in the cell? The answers seem to lie in the interactions of certain proteins with each other through processes of dynamic self-assembly and in the very geometry of the cell itself. Clearly, a complete picture of the cell requires us to continually ask where proteins are, why they localize where they do, and how this localization is achieved.
Acknowledgments
Supported by NIH grants GM51426 and GM325062 (L.S.), NIH grant GM07311 (H.H.M. and L.S.), U.S. Department of Energy Office of Science grant DE-FG02–05ER64136 (H.H.M. and L.S.), and NIH grant GM18568 (R.L.).
References and Notes
- 1.Werner JN et al. , Proc. Natl. Acad. Sci. U.S.A 106, 7858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alley MR, Maddock JR, Shapiro L, Genes Dev. 6, 825 (1992). [DOI] [PubMed] [Google Scholar]
- 3.Maddock JR, Shapiro L, Science 259, 1717 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Zhang P, Khursigara CM, Hartnell LM, Subramaniam S, Proc. Natl. Acad. Sci. U.S.A 104, 3777 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briegel A et al. , Mol. Microbiol 69, 30 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu TS et al. , Nat. Cell Biol 2, 792 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Ames P, Studdert CA, Reiser RH, Parkinson JS, Proc. Natl. Acad. Sci. U.S.A 99, 7060 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim KK, Yokota H, Kim SH, Nature 400, 787 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Ames P, Parkinson JS, Proc. Natl. Acad. Sci. U.S.A 103, 9292 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ausmees N, Kuhn JR, Jacobs-Wagner C, Cell 115, 705 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Jensen RB, Shapiro L, Proc. Natl. Acad. Sci. U.S.A 96, 10661 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb CD et al. , Cell 88, 667 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Fogel MA, Waldor MK, Mol. Microbiol 55, 125 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Bowman GR et al. , Cell 134, 945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebersbach G, Briegel A, Jensen GJ, Jacobs-Wagner C, Cell 134, 956 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Yehuda S, Rudner DZ, Losick R, Science 299, 532 (2003); published online 19 December 2002 ( 10.1126/science.1079914). [DOI] [PubMed] [Google Scholar]
- 17.Wu LJ, Errington J, Mol. Microbiol 49, 1463 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Viollier PH et al. , Proc. Natl. Acad. Sci. U.S.A 101, 9257 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teleman AA, Graumann PL, Lin DC, Grossman AD, Losick R, Curr. Biol 8, 1102 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Wu LJ, Errington J, Science 264, 572 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Niki H, Yamaichi Y, Hiraga S, Genes Dev. 14, 212 (2000). [PMC free article] [PubMed] [Google Scholar]
- 22.Lutkenhaus J, Annu. Rev. Biochem 76, 539 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Marston AL, Thomaides HB, Edwards DH, Sharpe ME, Errington J, Genes Dev. 12, 3419 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bramkamp M et al. , Mol. Microbiol 70, 1556 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Patrick JE, Kearns DB, Mol. Microbiol 70, 1166 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Sun Q, Margolin W, J. Bacteriol 186, 3951 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernhardt TG, de Boer PA, Mol. Cell 18, 555 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu LJ et al. , EMBO J. 28, 1940 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu LJ, Errington J, Cell 117, 915 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Thanbichler M, Shapiro L, Cell 126, 147 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Moseley JB, Mayeux A, Paoletti A, Nurse P, Nature 459, 857 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Martin SG, Berthelot-Grosjean M, Nature 459, 852 (2009). [DOI] [PubMed] [Google Scholar]
- 33.McGrath PT, Iniesta AA, Ryan KR, Shapiro L, McAdams HH, Cell 124, 535 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Kain J, He GG, Losick R, J. Bacteriol 190, 6749 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domian IJ, Quon KC, Shapiro L, Cell 90, 415 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Quon KC, Yang B, Domian IJ, Shapiro L, Marczynski GT, Proc. Natl. Acad. Sci. U.S.A 95, 120 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenal U, Fuchs T, EMBO J. 17, 5658 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L, Proc. Natl. Acad. Sci. U.S.A 103, 10935 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duerig A et al. , Genes Dev. 23, 93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi H, De Nisco NJ, Chien P, Simmons LA, Walker GC, Mol. Microbiol 73, 586 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lybarger SR, Johnson TL, Gray MD, Sikora AE, Sandkvist M, J. Bacteriol 191, 3149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deich J, Judd EM, McAdams HH, Moerner WE, Proc. Natl. Acad. Sci. U.S.A 101, 15921 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudner DZ, Losick R, Proc. Natl. Acad. Sci. U.S.A 99, 8701 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenfield D et al. , PLoS Biol. 7, e1000137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramamurthi KS, Lecuyer S, Stone HA, Losick R, Science 323, 1354 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lenarcic R et al. , EMBO J. 28, 2272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramamurthi KS, Losick R, Proc. Natl. Acad. Sci. U.S.A 106, 13541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huitema E, Pritchard S, Matteson D, Radhakrishnan SK, Viollier PH, Cell 124, 1025 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Lam H, Schofield WB, Jacobs-Wagner C, Cell 124, 1011 (2006). [DOI] [PubMed] [Google Scholar]