

SUMMARY
Neuronal hyperactivity is an early primary dysfunction in Alzheimer’s disease (AD) in humans and animal models, but effective neuronal hyperactivity-directed anti-AD therapeutic agents are lacking. Here we define a previously unknown mode of ryanodine receptor 2 (RyR2) control of neuronal hyperactivity and AD progression. We show that a single RyR2 point mutation, E4872Q, which reduces RyR2 open time, prevents hyperexcitability, hyperactivity, memory impairment, neuronal cell death, and dendritic spine loss in a severe early-onset AD mouse model (5xFAD). The RyR2-E4872Q mutation upregulates hippocampal CA1-pyramidal cell A-type K+ current, a well-known neuronal excitability control that is downregulated in AD. Pharmacologically limiting RyR2 open time with the R-carvedilol enantiomer (but not racemic carvedilol) prevents and rescues neuronal hyperactivity, memory impairment, and neuron loss even in late stages of AD. These AD-related deficits are prevented even with continued β-amyloid accumulation. Thus, limiting RyR2 open time may be a hyperactivity-directed, non-β-amyloid-targeted anti-AD strategy.
Graphical Abstract
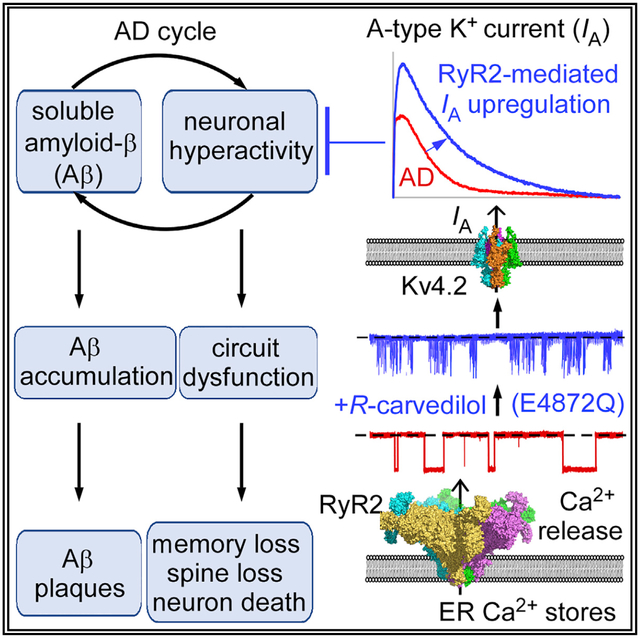
In Brief
Yao et al. show that genetically or pharmacologically limiting the open duration of ryanodine receptor 2 upregulates the A-type potassium current and prevents neuronal hyperexcitability and hyperactivity, memory impairment, neuronal cell death, and dendritic spine loss in a severe early-onset Alzheimer’s disease mouse model, even with continued accumulation of β-amyloid.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia and afflicts a rapidly growing population globally. Despite a substantial worldwide effort, there is currently no effective treatment for AD. Over the past several decades, AD research has largely targeted the amyloid cascade, which is thought to drive AD progression because of deposition of β-amyloid (Aβ) plaques in the brain (Berridge, 2010; Hardy and Selkoe, 2002; Karran et al., 2011). Hence, the dominating therapeutic anti-AD strategy has been reducing Aβ depositions (Demattos et al., 2012; Kennedy et al., 2016; Sevigny et al., 2016). Unfortunately, all major Aβ-targeted clinical trials to date have been unsuccessful (Chakroborty and Stutzmann, 2014; Honig et al., 2018; Karran et al., 2011), highlighting the urgent need to develop new non-Aβ-targeted AD therapy strategies.
Compelling evidence points to neuronal hyperactivity as an early primary neuronal dysfunction in human AD patients as well as animal models of AD (Busche et al., 2008, 2012; Busche and Konnerth, 2015; Dickerson et al., 2005; Keskin et al., 2017; Lerdkrai et al., 2018; Nuriel et al., 2017; O’Brien et al., 2010; Stargardt et al., 2015; Zott et al., 2019). Neuronal hyperactivity can be induced in vivo by acute treatment with exogenous soluble Aβ (Busche et al., 2012; Keskin et al., 2017; Zott et al., 2019). Importantly, neuronal hyperactivity itself can trigger release of endogenous soluble Aβ (Cirrito et al., 2005; Kamenetz et al., 2003; Yamamoto et al., 2015). Thus, soluble Aβ not only makes active neurons more active (hyperactive) but also triggers soluble Aβ release, which, in turn, further promotes hyperactivity. This vicious cycle is believed to drive Aβ accumulation, neuronal hyperactivity, circuit dysfunction, and AD progression (Busche and Konnerth, 2015, 2016; Stargardt et al., 2015; Zott et al., 2019). Targeting Aβ has proven to be insufficient to stop this vicious cycle in humans. An alternative, logical approach is to target neuronal hyperactivity (another component of the vicious cycle).
Ryanodine receptor 2 (RyR2) is an intracellular Ca2+ release channel. RyR2 is predominantly expressed in the heart and brain, especially in the hippocampus and cortex (Bers, 2002; Furuichi et al., 1994; Giannini et al., 1995; Murayama and Ogawa, 1996). RyR2-mediated Ca2+ release plays an important role in regulating membrane excitability of various cells (Alkon et al., 1998; Bogdanov et al., 2001; Mandikian et al., 2014; Nelson et al., 1995). Enhanced RyR2 function can cause cardiac arrhythmias and sudden death and has also been implicated in AD pathogenesis (Bruno et al., 2012; Chakroborty et al., 2009; Kelliher et al., 1999; Lacampagne et al., 2017; Oulès et al., 2012; Priori and Chen, 2011; SanMartín et al., 2017; Smith et al., 2005). Thus, targeting RyR2 may be a means to control membrane excitability and AD-related neuronal hyperactivity. However, given the multiple essential physiological roles of RyR2, dramatically blocking RyR2 function or expression would be detrimental (Bround et al., 2012; Liu et al., 2014; Takeshima et al., 1998). The challenge is how to suppress overactive RyR2 without detrimental effects.
We have shown that the E4872 residue is important for RyR2 channel gating. Mutating this residue (E4872Q) substantially shortens RyR2 open time without altering the channel’s ion conduction (Chen et al., 2014). Importantly, limiting RyR2 open time completely protects against ventricular tachyarrhythmias without detrimental effects on the heart (Chen et al., 2014). This raises the intriguing possibility that the same RyR2 suppression may also protect against AD, which is associated with enhanced RyR2 function. To test this possibility, we crossed heterozygous RyR2-E4872Q+/− mutant mice with the robust early-and rapid-onset AD mouse model 5xFAD (Oakley et al., 2006). We showed that genetically limiting RyR2 open time prevented intrinsic membrane hyperexcitability, neuronal hyperactivity, long-term potentiation (LTP) deficits, learning and memory impairments, neuronal cell death, and dendritic spine loss in 5xFAD mice. Further, we found that limiting RyR2 open time upregulates the A-type K+ current but not the commonly known RyR-regulated afterhyperpolarization current (IAHP) of hippocampal CA1 neurons. Moreover, we discovered that the RyR2 open time-limiting R-carvedilol enantiomer (Zhang et al., 2015; Zhou et al., 2011), but not a racemic carvedilol mixture, prevented and rescued neuronal hyperactivity, learning and memory impairments, and neuron loss in 5xFAD mice. Importantly, these AD-associated deficits were prevented even in late stages of AD with extensive Aβ accumulation. Therefore, limiting RyR2 open time may be a hyperactivity-directed strategy for combating AD irrespective of Aβ accumulation.
RESULTS
Limiting RyR2 Open Time Prevents AD-Associated Intrinsic Hyperexcitability of Hippocampal CA1 Pyramidal Neurons
Intrinsic hyperexcitability of hippocampal CA1 pyramidal neurons has been implicated in AD pathogenesis in animal models (Brown et al., 2011; Kerrigan et al., 2014; Scala et al., 2015; Šišková et al., 2014). Thus, it is of interest to determine whether limiting RyR2 open time affects AD-associated intrinsic hyperexcitability of CA1 cells. To this end, we crossbred heterozygous 5xFAD+/− mice (Oakley et al., 2006) with heterozygous RyR2 E4872Q+/− mutant mice (Chen et al., 2014). This breeding generated four genotypes: 5xFAD+/−, 5xFAD+/−/E4872Q+/− (5xFAD+/−/EQ+/−), E4872Q+/− (EQ+/−), and wild type (WT). Of note, RyR2 is predominantly expressed in the hippocampus and cortex as well as the soma and dendrites of CA1 pyramidal neurons, as revealed by fluorescence imaging of GFP-tagged RyR2 brain sections (Hiess et al., 2015; Figures 1A–1D).
Figure 1. The RyR2 E4872Q+/− Mutation Decreases Neuronal Excitability and Upregulates A-type K+ Current of CA1 Neurons.
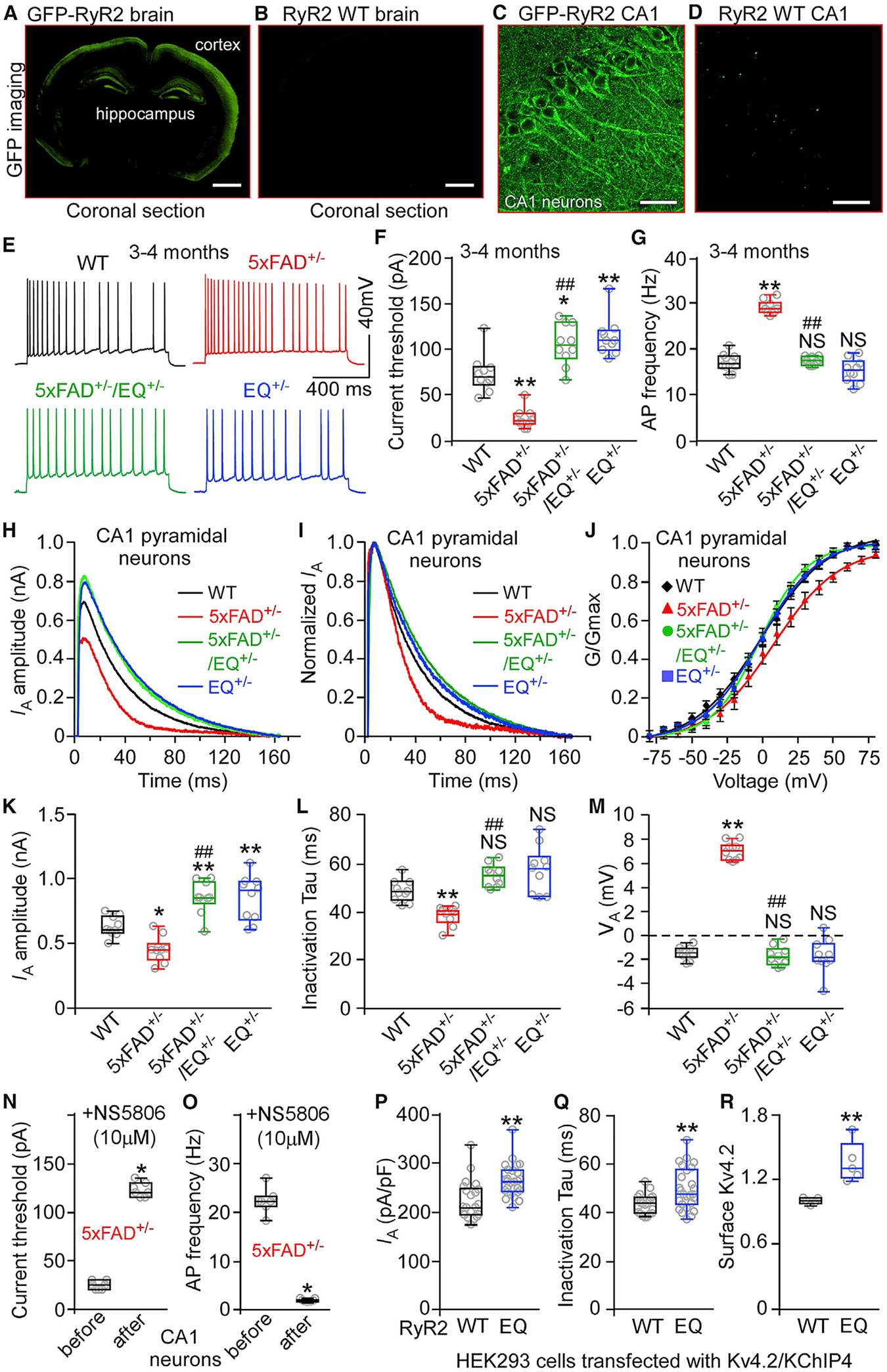
(A–D) Images of GFP-tagged RyR2 (A and C) and RyR2-WT (GFP-negative; B and D) mouse brain sections.
(E) Traces of action potential (AP) firing.
(F) Current thresholds for AP firing in WT (10 mice, 30 neurons), 5xFAD+/− (10 mice, 30 neurons), 5xFAD+/−/EQ+/− (10 mice, 30 neurons), and EQ+/− (10 mice, 30 neurons) CA1 neurons.
(G) Current injection-triggered AP firing frequency in WT (10 mice, 30 neurons), 5xFAD+/− (10 mice, 30 neurons), 5xFAD+/− /EQ+/− (10 mice, 30 neurons), and EQ+/− (10 mice, 30 neurons) CA1 neurons.
(H) Traces of A-type K+ current (IA) from 3- to 4-month-old WT, 5xFAD+/−, 5xFAD+/−/EQ+/−, and EQ+/− CA1 neurons.
(I) Normalized IA traces.
(J) Steady-state activation curves of IA.
(K) IA amplitude in 3- to 4-month-old WT (10 mice, 30 neurons), 5xFAD+/− (10 mice, 30 neurons), 5xFAD/EQ+/− (10 mice, 30 neurons), and EQ+/− (10 mice, 30 neurons) CA1 neurons.
(L) IA inactivation kinetics (Tau) from the same number of neurons as in (K).
(M) The midpoints of voltage-dependent activation of IA (VA) for WT (10 mice, 30 neurons), 5xFAD+/− (10 mice, 30 neurons), 5xFAD+/−/EQ+/− (10 mice, 30 neurons), and EQ+/− (10 mice, 30 neurons) CA1 neurons.
(N and O) Current threshold for AP firing (N) and current injection-triggered AP firing frequency (O) without or with the Kv4 channel agonist NS5806 (10 μM) in 5xFAD+/− CA1 neurons (7 mice, 14 neurons).
(P and Q) Whole-cell IA (P) and inactivation time constant Tau (Q) in Kv4.2/KChIP4-transfected HEK293 cells expressing RyR2 WT (n = 21) or the RyR2 E4872Q+/− mutation (n = 26).
(R) Biotin-labeled surface Kv4.2 in Kv4.2/KChIP4-transfected HEK293 cells expressing RyR2 WT or the RyR2 E4872Q+/− mutation.
Scale bars, 2 mm in (A) and (B) and 10 μm in (C) and (D). Data shown are the median and range (Kruskal-Wallis test with Dunn-Bonferroni post hoc test, Wilcoxon matched-pairs signed rank test, and Mann-Whitney U test; *p < 0.5, **p < 0.01 versus WT, ##p < 0.01; 5xFAD+/−/EQ+/− versus 5xFAD+/−; NS, not significant). See also Figure S1.
We performed whole-cell patch-clamp recordings of 3- to 4-month-old CA1 pyramidal neurons in brain slices to assess intrinsic excitability. In 5xFAD+/− CA1 neurons, the threshold current for inducing action potential (AP) firing was markedly reduced and the frequency of current-induced AP firing was increased compared with WT cells (Figures 1E–1G). Notably, the E4872Q+/− mutation substantially increased the threshold current for AP firing and prevented the increased frequency of AP firing in 5xFAD+/−/EQ+/− and EQ+/− CA1 pyramidal neurons (Figures 1E–1G). Thus, limiting RyR2 open time markedly constrains the intrinsic excitability of 5xFAD+/−/EQ+/− and EQ+/− CA1 pyramidal neurons.
Limiting RyR2 Open Time Upregulates the A-type K+ Current of CA1 Pyramidal Neurons
We next measured the IAHP because IAHP influences intrinsic excitability (Bodhinathan et al., 2010; van de Vrede et al., 2007). In 5xFAD+/− CA1 pyramidal neurons, the medium and slow components of IAHP were substantially reduced compared with the WT (Figures S1A–S1C; Figures 1E–1G). There was no significant difference in IAHP between 5xFAD+/− and 5xFAD+/−/EQ+/− and between WT and EQ+/− neurons (Figures S1A–S1C). Thus, the E4872Q+/− mutation did not significantly affect IAHP of 5xFAD+/− or WT CA1 pyramidal neurons even though it strongly inhibited intrinsic excitability. Thus, the action of E4872Q+/− in intrinsic excitability is unlikely to be mediated by IAHP.
Hippocampal A-type K+ current downregulation has been implicated in AD-related neuronal hyperactivity (Chen, 2005; Good et al., 1996; Hall et al., 2015; Scala et al., 2015). This led us to assess whether the E4872Q+/− mutation affects the A-type K+ current. We found that the A-type K+ current was decreased, the decay time (Tau) was shortened, and the midpoint for voltage-dependent activation (VA) was increased, but the midpoint for voltage-dependent inactivation (VH) was unchanged in 3- to 4-month-old 5xFAD+/− CA1 pyramidal neurons compared with the WT (Figures 1H–1M; Figures S1E and S1F), consistent with previous reports (Chen, 2005; Good et al., 1996; Hall et al., 2015; Scala et al., 2015). The E4872Q+/− mutation markedly upregulated the A-type K+ current of 5xFAD+/−/EQ+/− and EQ+/− hippocampal CA1 neurons compared with the WT. Specifically, the E4872Q+/− mutation increased the A-type K+ current and decay time without altering the VA or VH (Figures 1H–1M; Figures S1E and S1F). The A-type K+ current agonist NS5806 (10 μM), dramatically increased the threshold current for inducing AP firing and decreased the frequency of AP firing in 5xFAD+/− CA1 pyramidal neurons (Figures 1N and 1O; Figure S1D). Thus, limiting RyR2 open time upregulates the A-type K+ current of CA1 pyramidal neurons.
Hippocampal CA1 neurons express the Kv4.2/KChIP4 channel complex, which is thought to contribute significantly to the A-type K+ current (Lin et al., 2010; Rhodes et al., 2004; Serôdio and Rudy, 1998; Xiong et al., 2004). KChIP4 is a Ca2+ binding protein known to modulate the activity of Kv4.2 (Morohashi et al., 2002). Thus, RyR2-mediated Ca2+ release may regulate the A-type K+ current by modulating Kv4.2 via KChIP4. To test this, we assessed the action of RyR2-E4872Q+/− on the A-type K+ current in Kv4.2/KChIP4-transfected HEK293 cells. The E4872Q+/− mutation increased Kv4.2-mediated current and decay time without altering its voltage-dependent activation or inactivation (Figures 1P and 1Q; Figures S1G and S1H). This action of E4872Q+/− on Kv4.2-mediated current depends on KChIP4 because E4872Q+/− had no effect on Kv4.2-mediated current in HEK293 cells transfected with Kv4.2 alone (i.e., without KChIP4) (Figures S1I–S1L). The trafficking and surface expression of Kv4.2 are regulated by neuronal activity and Ca2+ influx (Kim et al., 2007a). RyR2-mediated Ca2+ release may also modulate Kv4.2 trafficking. Indeed, surface labeling experiments revealed that the E4872Q+/− mutation increased the surface expression of Kv4.2 in HEK293 cells (Figure 1R; Figure S1M). Caffeine (an agonist of RyR2) decreased Kv4.2-mediated current and decay time without altering its voltage-dependent activation or inactivation (Figures S1N–S1U). Thus, pharmacological manipulation of RyR2 also affects the A-type K+ current.
Intracellular Ca2+ release through RyRs has been shown to modulate presynaptic activity (Chakroborty et al., 2019, 2012b; Le Magueresse and Cherubini, 2007). Thus, we determined whether limiting RyR2 open time affects the spontaneous excitatory postsynaptic current (sEPSC) of CA1 pyramidal neurons. There were no significant differences in the amplitude or the inter-event intervals among WT, 5xFAD+/−, 5xFAD+/−/EQ+/−, and EQ+/− CA1 neurons (Figures S2A–S2C). This suggests that the action of limiting RyR2 open time in membrane excitability is unlikely to be mediated by changes to synaptic efficacy. Thus, genetically limiting RyR2 open time upregulates the A-type K+ current and constrains the intrinsic excitability of CA1 pyramidal neurons.
Limiting RyR2 Open Time Prevents AD-Associated Neuronal Hyperactivity of CA1 Pyramidal Neurons Ex Vivo and In Vivo
To determine whether limiting RyR2 open time can also prevent AD-associated neuronal hyperactivity of CA1 pyramidal neurons, we measured spontaneous neuronal activity (spontaneous AP firing) of CA1 pyramidal neurons in brain slices. Similar to previous reports (Leão et al., 2012; Šišková et al., 2014), the fraction of neurons displaying spontaneous AP firing and the frequency of spontaneous AP firing were markedly increased in 5xFAD+/− CA1 pyramidal neurons compared with the WT (Figures S2D–S2F). Notably, the E4872Q+/− mutation prevented the enhancement of spontaneous AP firing of 5xFAD+/−/EQ+/− CA1 pyramidal neurons ex vivo in brain slices (Figures S2D–S2F).
To assess whether limiting RyR2 open time can suppress AD-associated neuronal hyperactivity in vivo, we crossed double-heterozygous 5xFAD+/−/E4872Q+/− mice with heterozygous Thy-1 GCaMP6f+/− transgenic mice to introduce the GCaMP6f+/− transgene into each of the four genotypes. GCaMP6f is a fast, ultrasensitive Ca2+-sensing protein capable of detecting individual APs in neurons with high reliability (Chen et al., 2013; Dana et al., 2014; Peron et al., 2015). We performed in vivo two-photon imaging of GCaMP6f-expressing CA1 pyramidal neurons to monitor spontaneous Ca2+ transients, which are widely used to assess spontaneous neuronal activity of cell populations (Busche et al., 2008, 2012; Chen et al., 2013; Dana et al., 2014; Kerr et al., 2005; Peron et al., 2015; Sato et al., 2007; Zott et al., 2019). We found that anesthetized 5xFAD+/− mice 5–6 months old exhibited neuronal hyperactivity, as evidenced by a significant increase in the fraction of hyperactive neurons (as defined by Busche et al., 2012) and in the mean frequency of spontaneous Ca2+ transients and a significant decrease in the fraction of normal neurons compared with the WT (Figures 2A, 2B, 2E, 2F, 2I, 2J, 2N, 2P, and 2Q). There is no significant difference in the fraction of silent neurons between WT and 5xFAD+/− mice (Figure 2O). The frequency distributions of hyperactive neurons in WT and 5xFAD+/− mice are similar (Figures 2I and 2J). This is consistent with previous reports (Busche et al., 2012, 2019; Lerdkrai et al., 2018). Cumulative probability analysis also revealed that 5xFAD+/− mice have markedly increased overall spontaneous neuronal activity compared with the WT (p < 0.0001) (Figure 2M). Notably, the RyR2 E4872Q+/− mutation markedly decreased the fraction of hyperactive neurons and the mean frequency of spontaneous Ca2+ transients, increased the fraction of normal neurons, and reduced the overall spontaneous neuronal activity (as revealed by the cumulative probability analysis) in 5xFAD+/−/EQ+/− and EQ+/− mice compared with 5xFAD+/− and WT mice, respectively (Figure 2). On the other hand, consistent with a previous study (Lerdkrai et al., 2018), there is no significant difference in the amplitude of spontaneous Ca2+ transients among different genotypes (Figure S2G). Neuronal hyperactivity already occurred in 3- to 4-month-old 5xFAD+/− mice, as evidenced by a significant increase in the fraction of hyperactive neurons, the mean frequency of spontaneous Ca2+ transients, and the overall neuronal activity compared with the WT at the same age (Figure S3). Similarly, the RyR2 E4872Q+/− mutation decreased the fraction of hyperactive neurons and the mean frequency of spontaneous Ca2+ transients and reduced the overall spontaneous neuronal activity in 3- to 4-month-old 5xFAD+/−/EQ+/− mice compared with 5xFAD+/− mice (Figure S3). Overall, this shows that limiting RyR2 open time in 5xFAD+/− mice prevents AD-associated neuronal hyperactivity of CA1 pyramidal neurons ex vivo and in vivo.
Figure 2. RyR2-E4872Q+/− Prevents Neuronal Hyperactivity of 5xFAD+/− Hippocampal CA1 Neurons In Vivo.
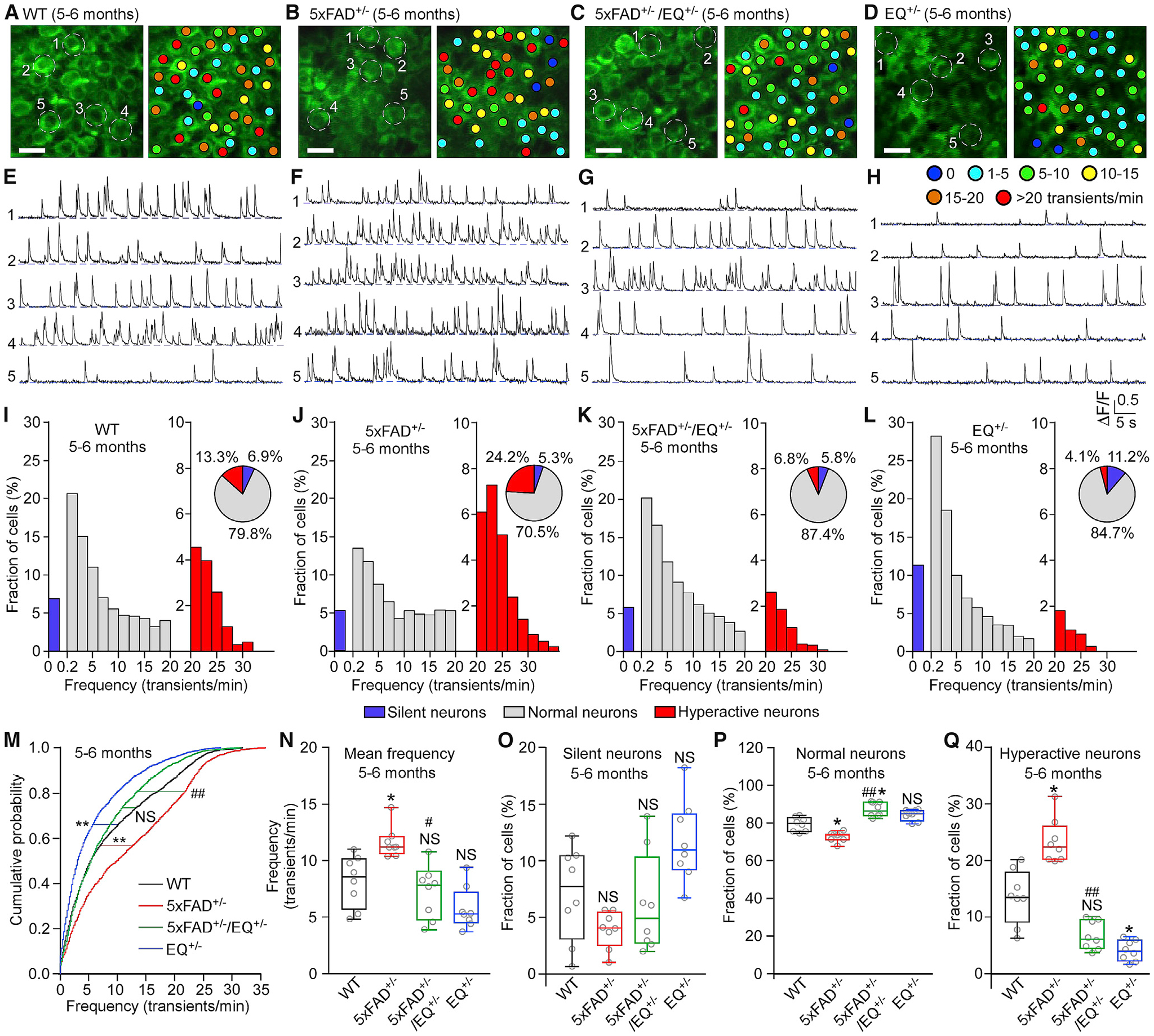
(A–D) Two-photon Ca2+ images of the hippocampal CA1 region of 5- to 6-month-old WT (A), 5xFAD+/− (B), 5xFAD+/−/EQ+/− (C), and EQ+/− (D) mice in vivo. Colored dots indicate the number of Ca2+ transients per minute.
(E–H) Ca2+ traces of the five neurons circled in (A)–(D), respectively.
(I–L) Histograms showing the frequency distribution of Ca2+ transients in (I) WT (8 mice, 2,375 cells), (J) 5xFAD+/− (8 mice, 1,708 cells), (K) 5xFAD+/−/EQ+/− (8 mice, 2,283 cells), and (L) EQ+/− (8 mice, 2,392 cells). Pie charts show the relative proportions of silent, normal, and hyperactive neurons as defined previously (Busche, 2018; Busche et al., 2008, 2012).
(M) Cumulative probability functions showing frequency distributions of spontaneous Ca2+ transients in the CA1 region of WT (black), 5xFAD+/− (red), 5xFAD+/−/EQ+/− (green), and EQ+/− (blue) mice (Kruskal-Wallis test with Dunn’s multiple comparisons test).
(N) Mean Ca2+ transient frequency in the WT, 5xFAD+/−, 5xFAD+/−/EQ+/−, and EQ+/− CA1 region.
(O–Q) Percentage of silent (O), normal (P), and hyperactive (Q) cells in the WT, 5xFAD+/−, 5xFAD+/−/EQ+/−, and EQ+/− CA1 region.
Scale bars: 10 μm. Data shown are the median and range (Kruskal-Wallis test with Dunn-Bonferroni post hoc test; *p < 0.5, **p < 0.01 versus WT, #p < 0.05, ##p < 0.01; 5xFAD+/−/EQ+/− versus 5xFAD+/−). See also Figures S2 and S3.
Limiting RyR2 Open Time Prevents AD-Associated Enhanced RyR2 Function of CA1 Pyramidal Neurons
We carried out 2-photon Ca2+ imaging of CA1 pyramidal neurons in brain slices prepared from GCaMP6f-expressing WT, 5xFAD+/−, 5xFAD+/−/EQ+/−, and EQ+/− mice. Consistent with other AD mouse models (Bruno et al., 2012; Chakroborty et al., 2009, 2012a), caffeine-induced Ca2+ release in 5xFAD+/− CA1 pyramidal neurons was markedly enhanced compared with WT cells (Figure S2H). On the other hand, caffeine-induced Ca2+ release in 5xFAD+/−/EQ+/− or EQ+/− CA1 pyramidal neurons was unchanged or reduced (respectively) compared with WT cells (Figure S2H). These data indicate that RyR2 function is markedly enhanced in 5xFAD+/− CA1 pyramidal neurons and that this aberrant RyR2 activation is prevented by the RyR2-E4872Q+/− mutation.
We also assessed the effect of presenilin 1 (PS1) WT and the PS1 mutations M146L and L286V on RyR2 function by measuring the propensity for spontaneous Ca2+ release in RyR2-expressing HEK293 cells transfected with or without PS1 WT or mutants (Chen et al., 2014; Jiang et al., 2004, 2005). Consistent with early studies (Chan et al., 2000; Rybalchenko et al., 2008; Wu et al., 2013), we found that PS1 WT and mutations markedly enhanced RyR2-mediated spontaneous Ca2+ release (Figure S2J). The E4872Q+/− mutation diminished this spontaneous Ca2+ release in all RyR2-expressing cells (Figure S2K). Thus, limiting RyR2 open time prevents AD-induced aberrant activation of RyR2-mediated Ca2+ release.
Limiting RyR2 Open Time Prevents AD-Associated Memory Loss and Hippocampal LTP Deficit
We performed Morris water maze (MWM) and novel object recognition (NOR) tests to assess whether the RyR2 E4872Q+/− mutation prevents the characteristic memory loss in 5xFAD+/− mice. 5xFAD mice (5–6 months old) have significant impairment in learning and memory (Figures 3A–3C), as evidenced by a significantly increased latency to target, decreased time spent in the target zone in the MWM, and reduced discrimination index in the NOR test compared with the WT. Remarkably, the E4872Q+/− mutation prevented these deficits in 5xFAD+/−/EQ+/− mice. There were no significant differences in MWM or NOR tests between 5- to 6-month-old 5xFAD+/−/EQ+/− and WT mice or between EQ+/− and WT mice (Figures 3A–3C). Similar MWM and NOR test outcomes were also observed in 3- to 4-month-old mice (Figures S4A–S4C). To determine whether the E4872Q+/− mutation could prevent learning and memory deficits in aged 5xFAD+/− mice, we performed Barnes maze (BM) and MWM tests on 10- to 15-month-old mice. As expected, the older 5xFAD+/− mice had severe learning and memory impairments (Figures 3D and 3E; Figures S4D–S4F). Importantly, the E4872Q+/− mutation prevented these learning and memory impairments in 5xFAD+/−/EQ+/− mice even in this late stage of AD.
Figure 3. The E4872Q+/− Mutation Prevents Memory Loss and LTP Impairment of 5xFAD+/− Mice.
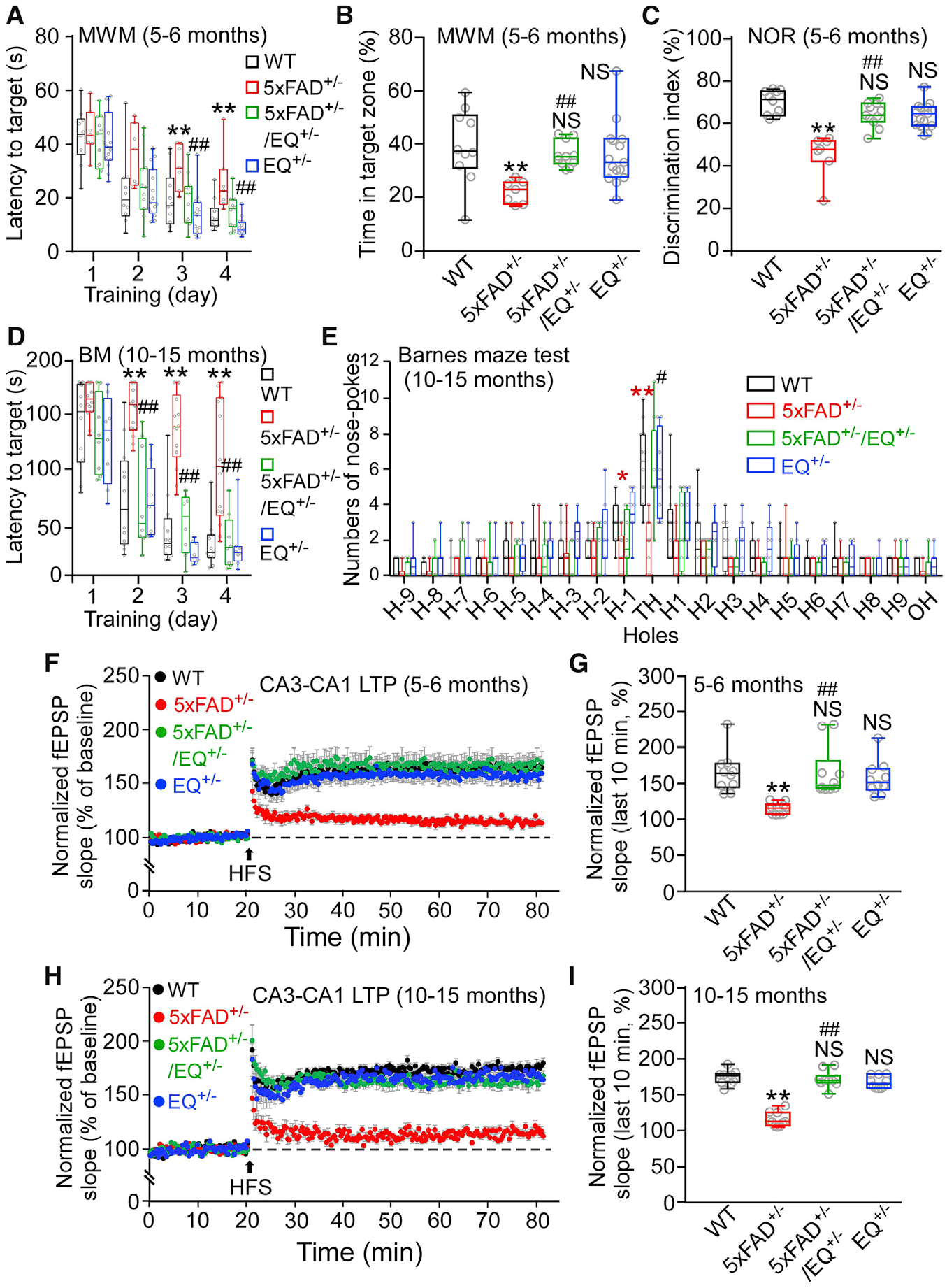
(A) The latency to reach the target platform of 5- to 6-month-old WT (n = 10), 5xFAD+/− (n = 7), 5xFAD+/−/EQ+/− (n = 11), and EQ+/− (n = 15) mice in the Morris water maze (MWM) test.
(B) The time spent in the target quadrant.
(C) The percentage of time spent on the novel object in the novel object recognition (NOR) test in 5- to 6-month-old WT (n = 10), 5xFAD+/− (n = 7), 5xFAD+/−/EQ+/− (n = 11), and EQ+/− (n = 15) mice.
(D) The latency to reach the target hole of 10- to 15-month-old WT (n = 12), 5xFAD+/− (n = 14), 5xFAD+/−/EQ+/− (n = 8), and EQ+/− (n = 8) mice in the Barnes maze (BM) test.
(E) The number of nose pokes to each hole on the BM test platform (**5xFAD versus WT).
(F) Effect of 100-Hz high-frequency stimulation (HFS) on the mean Schaffer collateral-evoked fEPSP slope in hippocampal slices from 5- to 6-month-old WT (10 mice, 20 slices), 5xFAD+/− (10 mice, 20 slices), 5xFAD+/−/EQ+/− (10 mice, 20 slices), and EQ+/− (10 mice, 20 slices) mice.
(G) The averaged normalized fEPSP slope.
(H) Effect of HFS on the fEPSP slope of 10- to 15-month-old WT (10 mice, 20 slices), 5xFAD+/− (10 mice, 20 slices), 5xFAD+/−/EQ+/− (10 mice, 20 slices), and EQ+/− (10 mice, 20 slices) mice.
(I) The averaged normalized fEPSP slope.
Data shown are the median and range (Kruskal-Wallis test with Dunn-Bonferroni post hoc test; *p < 0.5, **p < 0.01 versus WT, #p < 0.05, ##p < 0.01; 5xFAD+/−/EQ+/− versus 5xFAD+/−). See also Figures S4 and S5.
The effect of the E4872Q+/− mutation on learning and memory was also assessed at the cellular level by measuring hippocampal LTP in brain slices. Consistent with our behavioral studies, 5xFAD+/− mice at 3–4, 5–6, and 10–15 months of age showed little or no hippocampal LTP (Figures 3F–3I; Figure S4G and S4H). The E4872Q+/− mutation prevented these hippocampal LTP impairments, as evidenced by the similar levels of hippocampal LTP in WT, 5xFAD+/−/EQ+/−, and EQ+/− mice (Figures 3F–3I; Figures S4G and S4H). 5xFAD+/− mice also displayed slightly reduced hippocampal basal synaptic transmission, as revealed by the reduced field excitatory postsynaptic potential (fEPSP) slope in relation to the current input (Figures S5A–S5F), similar to a previous report (Waring et al., 2012). Thus, limiting RyR2 open time prevents hippocampal LTP impairment and memory loss in 5xFAD+/− mice.
R-carvedilol Prevents and Rescues Neuronal Hyperactivity and Memory Loss in 5xFAD+/− Mice
We showed previously that R-carvedilol shortens RyR2 open time (Zhang et al., 2015; Zhou et al., 2011). Given the effect of the E4872Q+/− mutation on hippocampal neuronal activity, this agent may provide a pharmacological means to limit RyR2 open time, neuronal hyperactivity, and AD progression. To test this, we pretreated 2- to 3-month-old 5xFAD+/− mice (i.e., before the occurrence of AD pathology) or 3- to 4-month-old 5xFAD+/− mice (i.e., after the occurrence of AD pathology) (Oakley et al., 2006) with R-carvedilol (3.2 mg/kg/day) or its vehicle control (DMSO) for 1 month. R-carvedilol pretreatment prevented and rescued neuronal hyperactivity of 5xFAD+/− hippocampal CA1 neurons in vivo, as evidenced by the observation that the fraction of hyperactive neurons, the mean frequency of spontaneous Ca2+ transients, and overall spontaneous neuronal activity were significantly lower in R-carvedilol-pretreated 5xFAD+/− mice than in vehicle-pretreated mice at both ages (Figure 4) but similar to those in the WT (Figure S3). Caffeine-induced Ca2+ release was also markedly reduced in R-carvedilol-pre-treated 5xFAD+/− CA1 pyramidal neurons compared with DMSO-treated (control) cells in brain slices (Figure S2I). Thus, R-carvedilol, like the RyR2-E4872Q+/− mutation, can prevent AD-induced aberrant activation of RyR2-mediated Ca2+ release.
Figure 4. R-carvedilol Prevents and Rescues Neuronal Hyperactivity of 5xFAD+/− Hippocampal CA1 Neurons In Vivo.
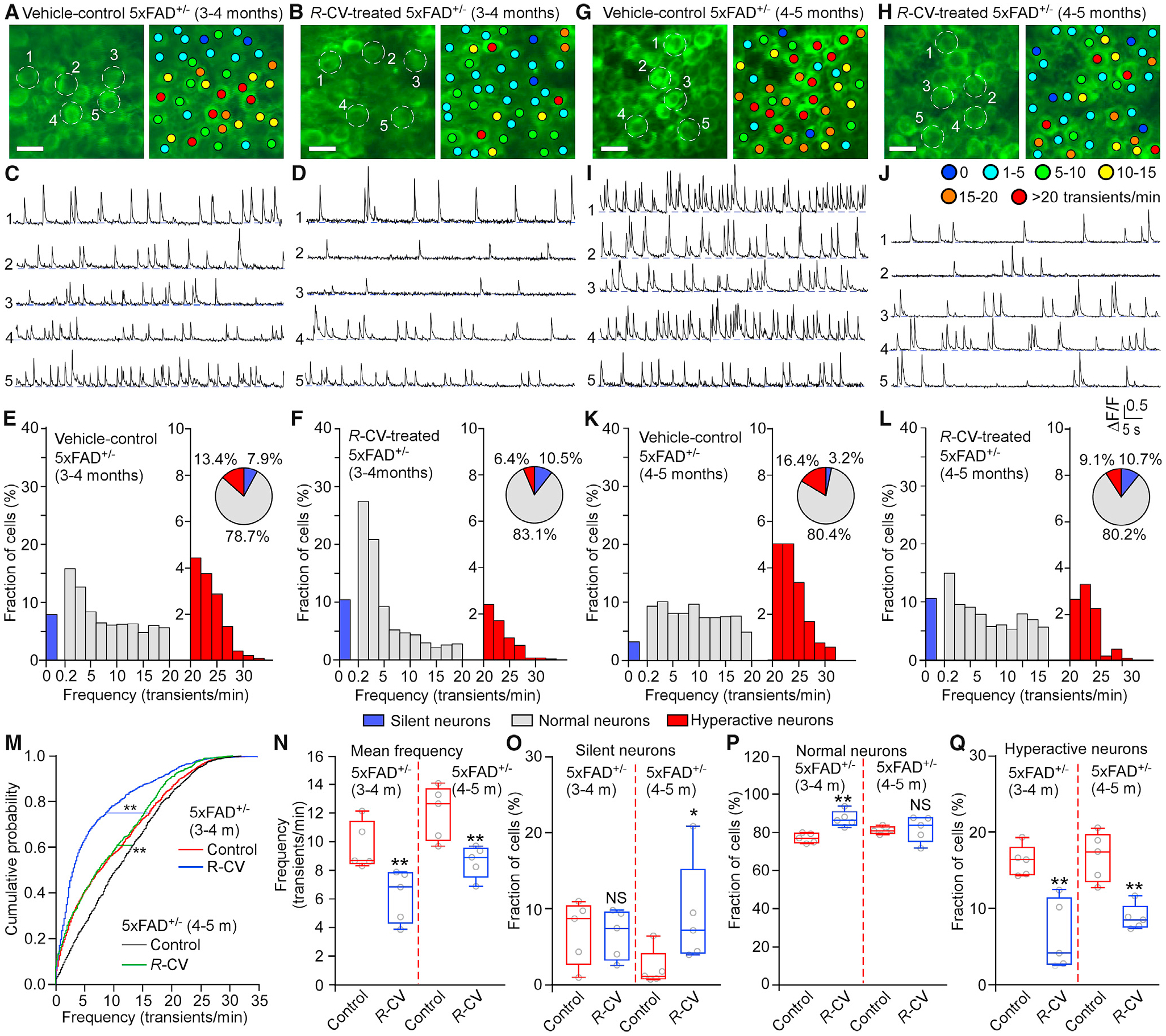
(A, B, G, and H) Two-photon in vivo Ca2+ imaging of the hippocampal CA1 region of 3- to 4-month-old (A and B) or 4- to 5-month-old (G and H) 5xFAD+/− mice treated with vehicle control (DMSO; A and G) or R-carvedilol (R-CV; 3.2 mg/kg/day; B and H). Colored dots indicate the number of Ca2+ transients per minute.
(C, D, I, and J) Ca2+ traces of the five neurons circled in (A), (B), (G), and (H), respectively.
(E, F, K, and L) Histograms showing the frequency distribution of Ca2+ transients in DMSO-treated mice (E and K, 5 mice, 1,066 cells and 5 mice, 897 cells, respectively) and R-CV-treated mice (F and L; 5 mice, 1,181 cells and 5 mice, 757 cells, respectively). Pie charts show the relative proportions of silent, normal, and hyperactive neurons.
(M) Cumulative probability functions showing frequency distributions of spontaneous Ca2+ transients in the CA1 region of 3- to 4-month-old 5xFAD+/− mice treated with DMSO (red) or R-CV (blue) and 5- to 6-month-old 5xFAD+/− mice treated with DMSO (black) or R-CV (green) (Kruskal-Wallis test with Dunn’s multiple comparisons test).
(N) Mean Ca2+ transient frequency in the CA1 region of 3- to 4-month-old and 4- to 5-month-old 5xFAD+/− mice treated with DMSO or R-CV.
(O–Q) Percentage of silent (O), normal (P), and hyperactive (Q) cells in the CA1 region of 3- to 4- and 4- to 5-month-old 5xFAD+/− mice treated with DMSO or R-CV. Scale bars, 10 μm. Data shown are the median and range (Kolmogorov-Smirnov test with Dunn-Bonferroni post hoc test and Mann-Whitney U test; **p < 0.01 versus control).
R-carvedilol pretreatment of 2- to 3-month-old 5xFAD+/− mice (before AD symptoms) also prevented memory loss and LTP impairments (Figures 5A–5E), and R-carvedilol pretreatment of 3- to 4-month-old 5xFAD+/− mice (after AD symptoms) rescued these defects (Figures 5F–5J). To further assess whether R-carvedilol pretreatment is still effective in late stages of AD, we tested the drug on 6- to 7- and 10- to 12-month-old 5xFAD+/− mice. R-carvedilol pretreatment rescued memory deficits even in 6- to 7- and 10- to 12-month-old 5xFAD+/− mice (i.e., late stages of AD) (Figures S6A–S6H). The reduced basal synaptic transmission in 5xFAD+/− hippocampal brain slices was also restored by R-carvedilol pretreatment (Figures S5G–S5L). We also tested the clinically used carvedilol racemic mixture (3.2 mg/kg/day). This carvedilol mixture did not prevent learning and memory deficits or LTP impairment in 3- to 4-month-old 5xFAD+/− mice (Figures S5M, S5N, and S6I–S6M). Thus, R-carvedilol, but not the carvedilol racemic mixture, may be a promising hyperactivity-directed anti-AD agent.
Figure 5. R-CV Prevents and Rescues Memory Loss and LTP Impairment in 5xFAD+/− Mice.
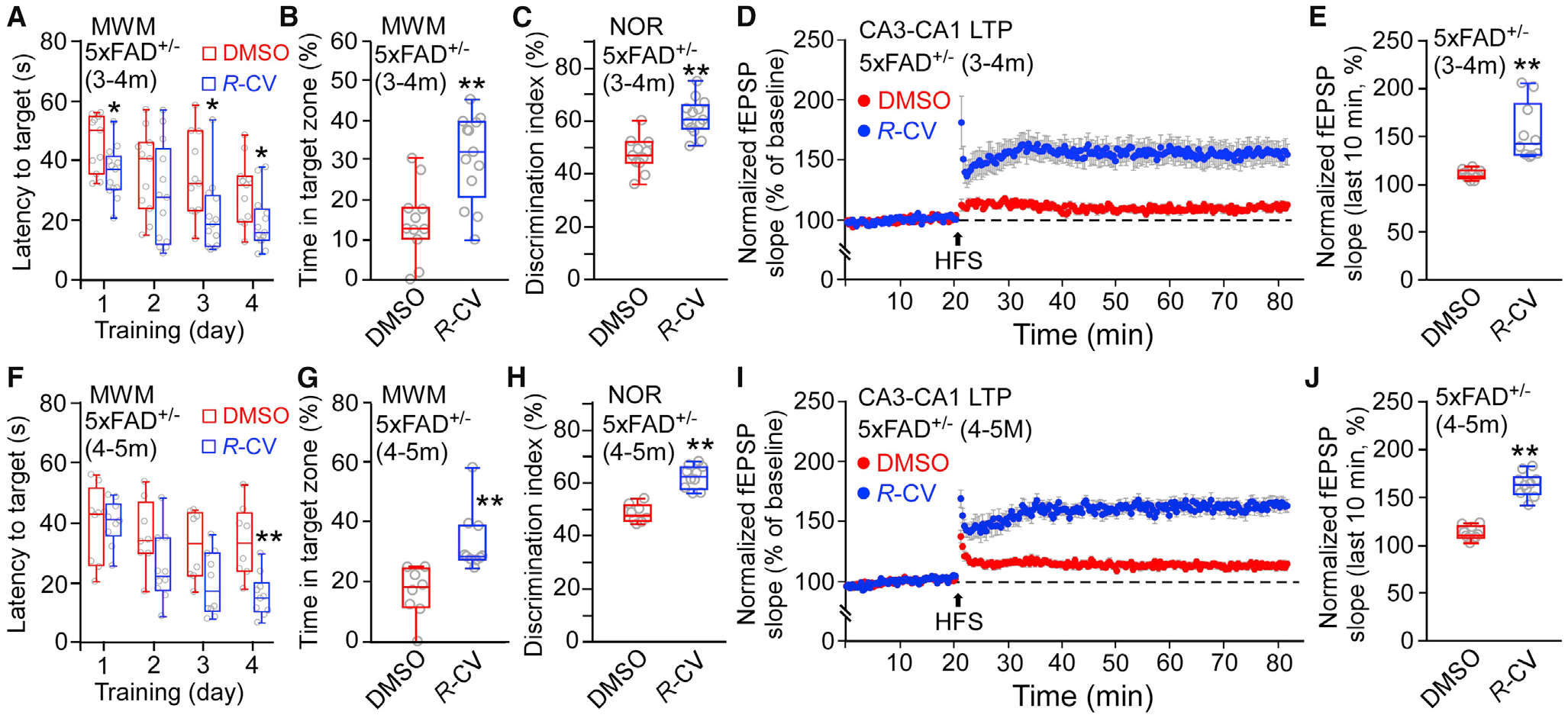
(A and F) The latency to reach the target platform of 3- to 4-month-old (A) and 4- to 5-month-old (F) 5xFAD+/− mice treated with DMSO (3–4 months old, n = 11; 4–5 months old, n = 8) or R-CV (3–4 months old, n = 13; 4–5 months old, n = 10) in the MWM test.
(B and G) The time spent in the target quadrant.
(C and H) The percentage of time spent on the novel object for 5xFAD+/− mice treated with DMSO (3–4months old, n = 11; 4–5 months old, n = 8) or R-CV (3–4 months old, n = 13; 4–5 months old, n = 10).
(D and I) Effect of HFS on mean CA3-CA1 fEPSP slope in hippocampal slices from 5xFAD+/− mice treated with DMSO (3–4 months old, 10 mice, 20 slices; 4–5 months old, 10 mice, 20 slices) or R-CV (3–4 months old, 10 mice, 20 slices; 4–5 months old, 10 mice, 20 slices).
(E and J) The averaged normalized fEPSP slope of 3- to 4-month-old (E) or 4- to 5-month-old (J) 5xFAD+/− mice treated with DMSO or R-CV.
Data shown are the median and range (Mann-Whitney U test; *p < 0.5, **p < 0.01 compared with the DMSO group). See also Figures S5 and S6.
Limiting RyR2 Open Time Does Not Affect Aβ Accumulation but Protects against Neuronal Cell Death and Dendritic Spine Loss
We performed immunohistochemical staining and immunoblotting analyses to assess the effect of the RyR2-E4872Q+/− mutation on Aβ accumulation. There was no significant difference in the number or the area of Aβ plaques detected in the hippocampus of 10- to 15-month-old 5xFAD+/− and 5xFAD+/−/EQ+/− mice (Figures 6A–6C). Immunoblot analyses also showed no significant difference in total Aβ or Aβ (1–42) levels in 10- to 15-month-old 5xFAD+/− and 5xFAD+/−/EQ+/− mouse brain tissue homogenates (Figures 6D–6F). No Aβ plaques were detected in WT or EQ+/− brain slices, and no specific Aβ signals were detected in WT or EQ+/− brain tissue homogenates. Similar results were also observed in 5- to 6-month-old mice (Figures S7A–S7F). Thus, limiting RyR2 open time does not significantly alter Aβ accumulation.
Figure 6. E4872Q+/− Has No Significant Effects on Aβ Accumulation but Protects against Neuron Loss in 5xFAD+/− Mice.
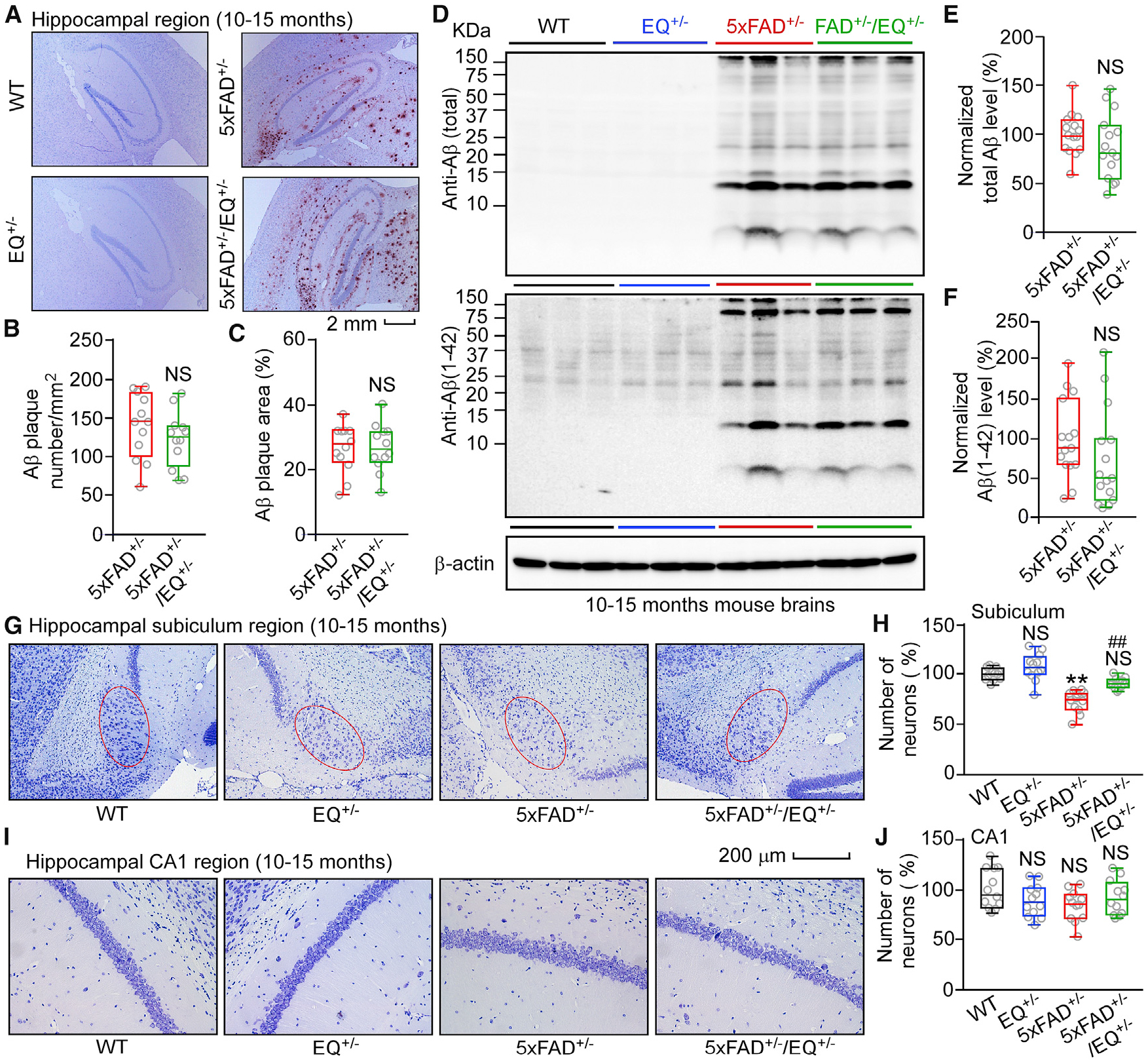
(A) Aβ deposition in the hippocampal region of 10- to 15-month-old WT, EQ+/−, 5xFAD+/−, and 5xFAD+/−/EQ+/− mice.
(B) Averaged Aβ plaque numbers in the hippocampal region of 10- to 15-month-old 5xFAD+/− (12 mice, 36 slices) and 5xFAD+/−/EQ+/− (12 mice, 36 slices) mice.
(C) Percentage of the hippocampal area showing positive Aβ staining.
(D) Immunoblot analysis of brain homogenates from 10- to 15-month-old WT, EQ+/−, 5xFAD+/−, and 5xFAD+/−/EQ+/− mice.
(E and F) Normalized total Aβ levels (E) and normalized Aβ (1–42) levels (F) in WT (n = 15), EQ+/− (n = 11), 5xFAD+/− (n = 21), and 5xFAD+/−/EQ+/− (n = 20) brains.
(G) Images of Nissl staining of 10- to 15-month-old WT, EQ+/−, 5xFAD+/− and 5xFAD+/−/EQ+/− mouse brain sections.
(H) Numbers of pyramidal neurons in the subiculum region (red oval) of WT (12 mice, 36 slices), EQ+/− (12 mice, 36 slices), 5xFAD+/− (12 mice, 36 slices), and 5xFAD+/−/EQ+/− (12 mice, 36 slices) mice.
(I) Images of Nissl staining of 10- to 15-month-old WT, EQ+/−, 5xFAD+/−, and 5xFAD+/−/EQ+/− mouse brain sections.
(J) Numbers of pyramidal neurons in the CA1 region of WT (12 mice, 36 slices), EQ+/− (12 mice, 36 slices), 5xFAD+/− (12 mice, 36 slices), and 5xFAD+/−/EQ+/− (12 mice, 36 slices) mice.
Scale bars, 2 mm in (A) and 200 μm in (G) and (I). Data shown are the median and range (Mann-Whitney U test and Kruskal-Wallis test with Dunn-Bonferroni post hoc test; **p < 0.01 versus WT, ##p < 0.01; 5xFAD+/−/EQ+/− versus 5xFAD+/−). See also Figure S7.
We also assessed whether the RyR2 E4872Q+/− mutation can influence neuronal cell death. Consistent with previous reports (Jawhar et al., 2012; Oakley et al., 2006), the number of pyramidal neurons in the subiculum (but not CA1) region was reduced significantly in 10- to 15-month-old 5xFAD+/− mouse brain slices (Figures 6G–6J). The E4872Q+/− mutation prevented this subiculum neuronal cell loss, as evidenced by the similar number of subiculum pyramidal neurons in 5xFAD+/−/EQ+/− and WT brains (Figure 6H). Like the E4872Q+/− mutation, R-carvedilol pretreatment also protected against subiculum neuronal cell loss without affecting Aβ accumulation (Figure 7; Figure S7G–S7L).
Figure 7. R-CV Treatment Has No Significant Effects on Aβ Accumulation but Protects against Neuron loss in 6- to 7-Month-Old 5xFAD+/− Mice.
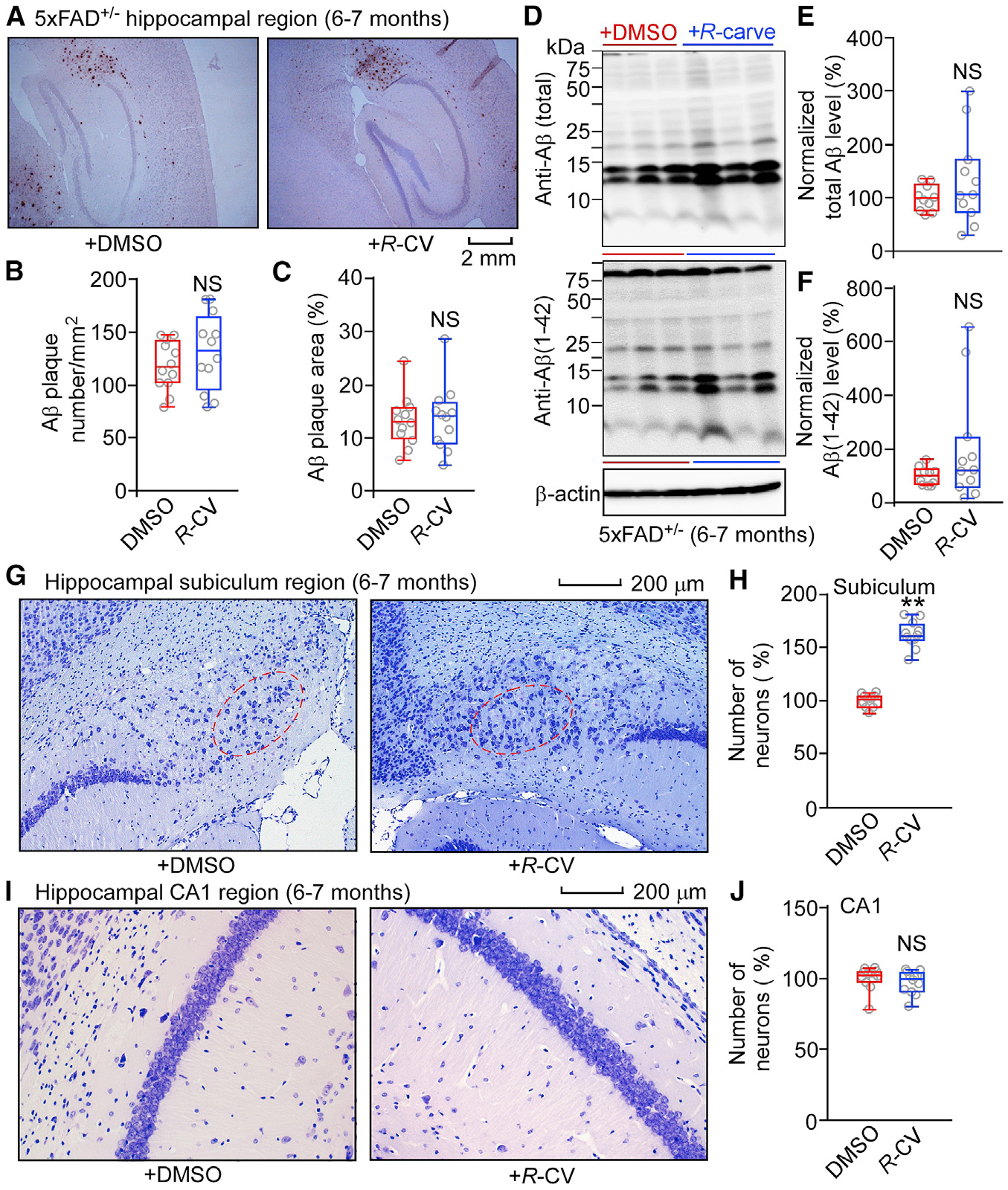
(A) Aβ deposition in 6- to 7-month-old 5xFAD+/− mice treated with DMSO or R-CV.
(B) Averaged Aβ plaque numbers in the hippocampal region of 6- to 7-month-old 5xFAD+/− mice treated with DMSO (12 mice, 36 slices) or R-CV (12 mice, 36 slices).
(C) Percentage of the hippocampal area showing positive Aβ staining.
(D) Immunoblot analysis of brain tissue homogenates from 6- to 7-month-old 5xFAD+/− mice treated with DMSO or R-CV.
(E and F) Normalized total Aβ levels (E) and normalized Aβ (1–42) levels (F) in 5xFAD+/− mice treated with DMSO (n = 10) or R-CV (n = 11).
(G) Images of Nissl staining of 6- to 7-month-old 5xFAD+/− mice treated with DMSO or R-CV.
(H) Numbers of pyramidal neurons in the subiculum region (red oval) of 6- to 7-month-old 5xFAD+/− mice treated with DMSO (12 mice, 36 slices) or R-CV (12 mice, 36 slices).
(I) Images of Nissl staining of 6- to 7-month-old 5xFAD+/− mice treated with DMSO or R-CV.
(J) Numbers of pyramidal neurons in the CA1 region of 6- to 7-month-old 5xFAD+/− mice treated with DMSO (12 mice, 36 slices) or R-CV (12 mice, 36 slices).
Scale bars, 2 mm in (A) and 200 μm in (G) and (I).
Data shown are the median and range (Mann-Whitney U test; **p < 0.01 compared with the DMSO group). See also Figure S7.
We performed Golgi staining to determine whether the RyR2 E4872Q+/− mutation affects the spine density and morphology of 5xFAD+/− CA1 pyramidal neurons. Consistent with previous studies (de Pins et al., 2019; Kim et al., 2020; Yang et al., 2018), the density of overall protrusions and specifically the density of mushroom and branched spines were reduced significantly in 5xFAD+/− CA1 pyramidal neurons compared with WT cells (Figure S7M). The E4872Q+/− mutation (limiting RyR2 open time) prevented the loss of overall protrusions and branched spines, but not the loss of mushroom spines, of 5xFAD+/−/EQ+/− CA1 pyramidal cells (Figure S7M). The E4872Q+/− mutation also decreased the density of filopodial spines and increased the density of branched spines of the EQ+/− CA1 cells compared with WT cells. This indicates that limiting RyR2 open time prevents dendritic spine loss in 5xFAD+/− mice despite extensive Aβ accumulation.
DISCUSSION
An increasing body of evidence indicates that AD progression is driven by a vicious cycle of soluble Aβ-induced neuronal hyperactivity (Busche et al., 2012; Busche and Konnerth, 2015; Stargardt et al., 2015; Zott et al., 2019). Intuitively, reducing the Aβ level would be expected to dampen Aβ-dependent neuronal hyperactivity and AD progression. However, achieving a sufficient Aβ reduction to prevent induction of neuronal hyperactivity and, consequently, AD progression may be an obstacle, which explains, in part, the failure of some recent Aβ-targeted clinical trials (Chakroborty and Stutzmann, 2014; Honig et al., 2018; Karran et al., 2011). Suppressing neuronal hyperactivity may be an alternative means to break the Aβ-hyperactivity cycle, even in the face of Aβ deposition. Here we tested this concept and demonstrate that limiting RyR2 open time (genetically or pharmacologically) prevents neuronal hyperactivity of CA1 pyramidal neurons ex vivo and in vivo in a severe early- and rapid-onset AD mouse model (5xFAD). We also show that limiting RyR2 open time prevents LTP deficit, learning and memory impairment, neuronal cell death, and dendritic spine loss in 5xFAD mice. Importantly, all of these AD-related deficits were prevented even with continued accumulation of Aβ. These findings identify RyR2 open time as a promising target for suppressing neuronal hyperactivity and AD progression irrespective of Aβ deposition.
The exact mechanism(s) underlying RyR2-mediated constraint of neuronal hyperactivity of hippocampal CA1 neurons has yet to be defined, but it is likely to be complex and multifactorial. Here we focused on the effect of limiting RyR2 open time on the intrinsic excitability of CA1 pyramidal neurons, a key determinant of neuronal activity, learning, and memory (Brager and Johnston, 2007; Kerrigan et al., 2014; Tamagnini et al., 2015; Xu et al., 2005). Intracellular RyR-mediated Ca2+ release is known to regulate neuronal intrinsic excitability by modulating the Ca2+-activated K+ channel-mediated IAHP (Bodhinathan et al., 2010; van de Vrede et al., 2007). RyR-mediated Ca2+ release also affects neuronal activity by modulating presynaptic activity (Chakroborty et al., 2012b, 2019; Le Magueresse and Cherubini, 2007). Thus, one possibility is that limiting RyR2 open time may contribute to suppression of neuronal hyperactivity by decreasing membrane excitability through modulation of IAHP and/or presynaptic activity. To test this possibility, we employed a genetic approach to assess whether limiting RyR2 open time alters IAHP and/or presynaptic activity. Surprisingly, genetically limiting RyR2 open time did not have a major effect on IAHP or spontaneous synaptic input in hippocampal CA1 neurons.
The A-type K+ current plays a major role in controlling neuronal excitability of hippocampal CA1 neurons (Hall et al., 2015; Jung and Hoffman, 2009; Kim et al., 2005; Magee et al., 1998; Varga et al., 2004). Interestingly, the A-type K+ current is downregulated by activity- and Ca2+ influx-dependent reduction in the surface expression of the A-type K+ channel subunit Kv4.2, which is modulated by a Ca2+ binding protein called KChIP4 (Kim et al., 2007a; Morohashi et al., 2002). This Ca2+ dependence of Kv4.2 trafficking raises the possibility that RyR2-mediated intracellular Ca2+ release may also regulate Kv4.2 surface expression and, thus, the A-type K+ current, considering the close relationship between Ca2+ influx and RyR2-mediated Ca2+ release (Kim et al., 2007b; Sandler and Barbara, 1999). Because RyR2 is abundantly expressed in hippocampal CA1 pyramidal neurons, it is reasonable to propose that limiting RyR2 open time and, thus, RyR2-mediated Ca2+ release in these neurons may affect Kv4.2 trafficking and consequently the A-type K+ current. Indeed, we found that limiting RyR2 open time increased the surface expression of Kv4.2, upregulated the A-type K+ current in a KChIP4-dependent manner, and decreased membrane excitability. The A-type K+ current is downregulated in AD mouse models (Chen, 2005; Good et al., 1996; Hall et al., 2015; Scala et al., 2015). This is consistent with our finding that the A-type K+ current was significantly reduced in 5xFAD+/− CA1 neurons, and this was completely restored by limiting RyR2 open time. Thus, our work establishes a previously unknown mode of RyR2 control of the A-type K+ current. Interestingly, loss-of-function variants in the Kv4.2-associated protein, dipeptidyl-peptidase 6 (DPP6), have recently been associated with early-onset AD (Cacace et al., 2019). DPP6 is known to enhance the surface expression of Kv4.2 and regulate its gating (Sun et al., 2011). Thus, loss of DPP6 may contribute to neuronal hyperactivity and AD progression by downregulating Kv4.2. Indeed, the mRNA level of hippocampal A-type K+ channels in human AD patients is downregulated (Noh et al., 2019).
Enhanced RyR2 function and expression have been implicated in AD pathogenesis (Bruno et al., 2012; Chakroborty et al., 2009; Kelliher et al., 1999; Lacampagne et al., 2017; Oulès et al., 2012; SanMartín et al., 2017; Smith et al., 2005). An effective and safe means to suppress this enhanced RyR2 function, however, remains to be identified. We have shown previously that limiting RyR2 open time prevents ventricular arrhythmias in animal models without detrimental effects on the heart (Chen et al., 2014; Zhang et al., 2015). Here we show that limiting RyR2 open time prevents cognitive dysfunction in 5xFAD+/− mice without detrimental effects on normal brain function. This indicates that limiting RyR2 open time (as opposed to blocking channel conduction) is a promising and effective means to combat neuronal disorders associated with overactive RyR2.
We reported previously that carvedilol limits RyR2 open time (Zhou et al., 2011). This carvedilol’s action is analogous to that of the RyR2 E4872Q+/− mutation, suggesting that carvedilol, like E4872Q+/−, may suppress AD progression. Unfortunately, RyR2 inhibition requires a high carvedilol dose (Zhou et al., 2011), which produces excessive β-blockade and severe adverse effects (Ko et al., 2004; Packer et al., 1996). Carvedilol is a racemic mixture of 50% S-carvedilol (a β-blocker) and 50% R-carvedilol (a non-β-blocker) (Bartsch et al., 1990; Nichols et al., 1989; Stoschitzky et al., 2001; van Zwieten, 1993). Importantly, the non-β-blocking R-carvedilol enantiomer also limits RyR2 open time (Zhang et al., 2015). Thus, application of just R-carvedilol may be a means to limit neuronal hyperactivity without severe β-blockade. Indeed, we showed that R-carvedilol pretreatment of 5xFAD+/− mice before or after appearance of AD symptoms prevented and rescued neuronal hyperactivity and memory loss. On the other hand, pretreatment with the carvedilol racemic mixture did not prevent learning and memory deficits in 5xFAD+/− mice, which may explain the negative outcome of a recent AD clinical trial of the carvedilol racemic mixture (https://www.clinicaltrials.gov/ct2/show/study/NCT01354444). The exact reason why the carvedilol racemic mixture is ineffective in suppressing AD progression is unknown. One possible explanation is that the potent β-blocking action of the racemic carvedilol mixture (especially at high doses) may adversely influence neuronal and cognitive function. Notably, R-carvedilol pretreatment rescued learning and memory impairments even in aged 5xFAD+/− mice (6–7 and 10–12 months old) with extensive Aβ accumulation. This shows that limiting RyR2 open time can restore AD-deficits even in late stages of AD. Thus, the R-carvedilol enantiomer is a non-Aβ-targeted, hyperactivity-directed, anti-AD therapeutic agent that warrants additional preclinical studies and even clinical trial.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, S. R. Wayne Chen (swchen@ucalgary.ca).
Materials Availability
All materials will be made available upon reasonable request.
Data and Code Availability
All data generated in this study will be available upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All animal studies were approved by the Institutional Animal Care and Use Committees at the University of Calgary and were performed in accordance with US National Institutes of Health guidelines. Adult genetically engineered mice, 5xFAD+/− (Oakley et al., 2006), RyR2-E4872Q+/− (EQ+/−) (Chen et al., 2014), 5xFAD+/−/RyR2-E4872Q+/− (5xFAD+/−/EQ+/−), and WT littermates of both sexes were used. All animal models were housed in the mouse facility of Cumming School of Medicine, University of Calgary. Mice were maintained in a 129E background and housed on a 12hr light/dark cycle with ad libitum access to food and water. R-carvedilol (R-CV) (3.2 mg/kg/day), carvedilol (racemic mixture) (3.2 mg/kg/day), and vehicle control (DMSO) were delivered to 5xFAD+/− mice in drinking water for one month, starting at different ages before (2–3 months old) or after (3–4 months old) the occurrence of AD pathologies (Oakley et al., 2006). To assess the effects of genetically and pharmacologically limiting RyR2 open time on the prevention and rescue of AD deficits in different stages (early, moderate, and late) of AD progression, animals at different ages (from 2–15 months) were used. As reported (Oakley et al., 2006), there is an age-dependence of AD progression in the 5xFAD+/− mice. However, we did not observe sex-dependent differences in AD progression in these mice. As shown previously, only heterozygous RyR2-E4872Q+/− mutant mice were produced as homozygous E4872Q+/+ mutation is embryonic lethal (Chen et al., 2014). Functional RyR2s are tetrameric channels formed by 4 RyR2 monomers. The heterozygous E4872Q+/− mutant mice (harboring one WT allele and one E4872Q mutant allele) will produce a mixture of homo- and hetero-tetrameric channels that contain the WT, E4872Q mutant, or both WT/E4872Q mutant monomers. Thus, the RyR2-E4872Q mutation can exert its negative impact not only on the function of the E4872Q homo-tetramers, but also on the function of the WT monomer in the WT/E4872Q hetero-tetrameric channels. For 2-photon Ca2+ imaging experiments, 5xFAD, RyR2 WT, and RyR2 mutant mice were cross-bred with the heterozygous Thy1-GCaMP6f transgenic mice (Chen et al., 2012, 2013) (GP5.17, JAX 025393) to express the GCaMP6f Ca2+ sensing probe (driven by the Thy1 promotor) in hippocampal neurons in each of the genotypes used.
Cell lines
The Flp-In T-REx HEK293 cell line was obtained from Invitrogen. HEK293 cell lines expressing RyR2 WT or the RyR2 E4872Q mutation were generated using the Flp-In T-Rex HEK293 cell line. HEK293 cell lines were cultured in Dulbecco’s Modified Eagle Medium (GIBCO) supplemented with 10% fetal bovine serum (GIBCO), 100 units/ml penicillin and 100 μg/ml streptomycin (GIBCO), 4 mM L-glutamin (GIBCO) and 0.1 mM MEM Non-Essential Amino Acids Solution (GIBCO). All cell lines were cultivated in a humidified incubator with 5% CO2 at 37°C and were tested negative for mycoplasma contamination.
METHOD DETAILS
Acute brain slice preparation
Acute brain slices were prepared according to the published procedures with some modifications (Ting et al., 2014, 2018). Briefly, mice were anesthetized with isoflurane (5%) and perfused through the heart with 20 mL ice-cold, carbogenated (95% O2, 5% CO2), N-methyl-D-glucamine (NMDG)-cutting solution, containing NMDG, 93 mM; KCl, 2.5 mM; NaH2PO4, 1.2 mM; NaHCO3, 30 mM; HEPES, 20 mM; glucose, 25 mM; thiourea, 2 mM; Na-ascorbate, 5 mM; Na-pyruvate, 3 mM; CaCl2·2H2O, 0.5 mM, and MgSO4·7H2O, 10 mM, pH to 7.3–7.4 with HCl. Brains were rapidly removed and placed in the ice-cold, carbogenated cutting solution for 30 s. Transverse hippocampal slices (260 or 300 μm thick) were prepared using a Vibratome (VT1200S, Leica) and then transferred to a homemade incubation chamber at 32°C with the carbogenated cutting solution. We then performed Na+ spike-in schedule according to the mouse age. The slices were then kept in HEPES containing aCSF (NaCl, 92 mM; KCl, 2.5 mM; NaH2PO4, 1.25 mM; NaHCO3, 30 mM; HEPES, 20 mM; glucose, 25 mM; thiourea, 2 mM; Na-ascorbate, 5 mM; Na-pyruvate, 3 mM; CaCl2·2H2O, 2 mM, and MgSO4·7H2O, 2 mM, pH to 7.3–7.4 with NaOH) at room temperature for at least 60 min before use.
Whole-cell patch-clamp recordings
For whole-cell patch-clamp recordings, slices were transferred to a submerged recording chamber perfused with carbogenated external solutions at a flow rate of 4–6 mL/min at room temperature (23°C). Action potentials (APs) were measured in hippocampal CA1 pyramidal neurons in transverse hippocampal slices (260 μm) from all genotypes of 3–4 months old mice using whole-cell patch-clamp with an Axopatch 700B amplifier (Axon Instruments). We focused on hippocampal CA1 pyramidal neurons as they play a critical role in neuronal activity, learning and memory (Brager and Johnston, 2007; Kerrigan et al., 2014; Tamagnini et al., 2015; Xu et al., 2005). We did not measure membrane potentials in 5–6 months old or older neurons as aged neurons are difficult to patch. AP firing was recorded in an external solution (NaCl, 124 mM; KCl, 2.5 mM; NaH2PO4, 1.25 mM; NaHCO3, 24 mM; HEPES, 5 mM; glucose, 12.5 mM; MgCl2, 2 mM; and CaCl2, 2 mM; pH 7.4 adjusted with NaOH) and soft-glass recording pipettes (Sutter Instruments; Novato CA) filled with an internal solution (potassium gluconate, 135 mM, KCl, 10 mM, HEPES, 10 mM, CaCl2, 1 mM, MgCl2, 1 mM, EGTA, 10 mM, ATP, 1 mM, GTP, 0.1 mM, and pH 7.3 adjusted with KOH). The pipette resistance was 4–6 MΩ after filling with internal solution. For spontaneous AP recording, cells were hold at −70 mV and recorded for 3 min. For the measurement of current-injection triggered APs, 0.05 mM 2-amino-5-phosphonovaleric acid (APV), 0.02 mM 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 0.1 mM picrotoxin were added to the external solution to block synaptic activity. APs were initiated by injecting current from 0 to 300 pA for 1 s in 10 pA steps at 10 s intervals. For testing the Kv4 channel agonist NS5806, APs were measured before and 15 min after perfusion of 10 μM NS5806 in the external solution. For recording of spontaneous excitatory post-synaptic currents (sEPSCs), same external and internal solutions for AP recording were used. Picrotoxin (0.1 mM) was added to the external solution to block inhibitory current. CA1 neuron were held at 70 mV for 2 min.
Previous studies used whole-cell patch clamp recordings at the soma of CA1 pyramidal cells to measure the whole-cell A-type K+ current (Chen, 2005; Good et al., 1996; Hall et al., 2015; Scala et al., 2015). To make comparisons to earlier studies, we employed the same approach. Briefly, whole-cell A-type K+ current (IA) was elicited by depolarizing pulses to +40 mV from a holding potential of −100 mV in the presence of 20 mM tetraethylammonium (TEA) and 100 nM tetrodotoxin (TTX). In steady-state activation experiments, membrane potential was held at −100 mV, and IA was evoked by a 200-ms depolarizing pulse from a first pulse potential of −80 mV to +80 mV in 10-mV steps at 10 s intervals. Data were analyzed using the equation GK = IK/(Vm Vrev), where GK is the membrane K+ conductance, Vm is the membrane potential, and Vrev is the reversal potential for K+. To study steady-state inactivation of IA, currents were elicited using 1 s conditioning pre-pulses from −110 mV to 0 mV before a 200-ms test pulse of +50 mV. After normalizing each current amplitude to the maximal current, amplitude obtained from the −110 mV pre-pulse was used as a function of the conditioning pre-pulse potential and fitted with the function IA / IA − max = 1/(1 + exp((Vm1/2 − Vm)/k)), from which, an inactivation curve of IA was obtained, and the VH value (the voltage at which the current amplitude was half-inactivated) was calculated. Note that in other studies and ours, the A-type K+ current was measured by performing whole-cell current recordings at the soma of CA1 pyramidal neurons to assess the role of somatic whole-cell A-type K+ current in somatic excitability and spike output. Given the well-known somatodendritic gradient of A-type K+ current density distribution (Hoffman et al., 1997), the use of somatic whole-cell recordings would limit our ability to identify changes in the A-type K+ current density distribution over the soma-dendritic axis. Further studies will be needed to define the contribution of dendritic A-type K+ current to AD-associated neuronal hyperactivity and RyR2-mediated control of neuronal excitability. Nevertheless, somatic whole-cell recordings still provide valuable information on the magnitude of somatic A-type K+ current, an important determinant of somatic excitability.
For HEK293 cell experiments, HEK293 cell lines were maintained as previously described (Jiang et al., 2004) and transiently transfected with cDNAs of Kv4.2 and KChIP4 together with cDNA encoding for GFP to identify cells successfully transfected. 12–16 h before recording, tetracycline was added to culture media to induce the expression of RyR2 WT or RyR2 E4872Q mutant. IA was recorded with the same protocols as described above. Prior to IA recording, the culture medium was replaced with a bath solution (NaCl, 125 mM; KCl, 2.5 mM; HEPES, 10 mM; MgCl2, 1 mM; glucose, 10 mM; TEA, 20 mM; pH 7.4 adjusted with NaOH).
For recording the afterhyperpolarization current (IAHP), brain slices were perfused with the carbogenated aCSF and pipettes were filled with IAHP inner solution (KMeSO3, 130 mM; EGTA, 0.1 mM; HEPES, 10 mM; NaCl, 7 mM; MgCl2, 0.3 mM; di-tris-creatine, 5 mM; Tris-ATP, 2 mM and Na-GTP, 0.5 mM, pH 7.3 with KOH). IAHP was evoked by a 100 ms depolarizing voltage step to +60 mV from a holding potential of −85 mV. Medium (ImAHP) and slow (IsAHP) amplitudes were measured at the peak of the current and 1 s after the end of the depolarizing pulse, respectively. All cells had a resting membrane potential more hyperpolarized than −60 mV, leak current smaller than 100 pA, and an input resistance of 150–350 MΩ. Input resistance was determined from a −5 mV (100 ms) hyperpolarizing pulse applied at the beginning of each sweep. Access resistance was 80% electronically compensated and stable at < 20 MΩ.
In vivo 2-photon Ca2+ imaging of CA1 neurons
To assess the impact of genetically limiting RyR2 open time on AD progression, we crossbred heterozygous 5xFAD+/− mice that overexpress 5 human familial AD mutations (Oakley et al., 2006) with heterozygous RyR2 mutant mice harboring the RyR2 mutation E4872Q+/− that markedly shortens RyR2 open time (Chen et al., 2014). This breeding generated four genotypes: 5xFAD+/−, 5xFAD+/−/E4872Q+/− (5xFAD+/−/EQ+/−), E4872Q+/− (EQ+/−), and wild-type (WT). To determine whether limiting RyR2 open time can suppress AD-associated neuronal hyperactivity in vivo as reported previously (Busche et al., 2008, 2012; Delekate et al., 2014; Eichhoff and Garaschuk, 2011; Kim et al., 2016; Takano et al., 2007; Zott et al., 2019), we then crossed the double heterozygous 5xFAD+/−/E4872Q+/− mice with the heterozygous Thy-1 GCaMP6f+/− transgenic mice (GP5.17, JAX 025393) to introduce the GCaMP6f+/− transgene into each of the four genotypes (driven by the Thy1 promotor). Note GCaMP6f is a fast, ultrasensitive Ca2+ sensing protein that is capable of detecting individual action potentials in neurons with high reliability (Chen et al., 2013; Dana et al., 2014; Peron et al., 2015). Also, in the hippocampal CA1 region of the Thy-1 GCaMP6f+/− transgenic mice, GCaMP6f is predominantly expressed in pyramidal neurons (not in GABAergic neurons or glial cells). We performed in vivo two-photon imaging of GCaMP6f-expressing CA1 pyramidal neurons in each of the four genotypes to monitor spontaneous Ca2+ transients. These spontaneous Ca2+ transients are widely used to assess the spontaneous neuronal activity of cell populations (Busche et al., 2008, 2012; Chen et al., 2013; Dana et al., 2014; Kerr et al., 2005; Peron et al., 2015; Sato et al., 2007; Zott et al., 2019).
Neuronal hyperactivity has been reported in anesthetized AD model mice in vivo using two-photon Ca2+ imaging (Busche et al., 2008, 2012, 2019; Lerdkrai et al., 2018). To be able to compare our data to this considerable body of existing literature, we employed the same approach described originally by Busche et al. and used by others in the field (Busche et al., 2008, 2012; Delekate et al., 2014; Eichhoff and Garaschuk, 2011; Kim et al., 2016; Takano et al., 2007; Zott et al., 2019). Craniotomy was performed according to the protocol reported previously (Busche, 2018; Busche et al., 2008, 2012) with some modifications. Briefly, mice at different ages were anesthetized with 1%–2% isoflurane (vol/vol in pure oxygen), and placed onto a heating plate (Homeothermic Monitor, Harvard Apparatus). The body temperature was monitored and controlled at 36.5–37.5°C during the entire surgery and imaging procedure. After the removal of the skin, the skull was rinsed with artificial cerebral spinal fluid (aCSF: NaCl, 125 mM; KCl, 4.5 mM; NaH2PO4, 1.25 mM; NaHCO3, 26 mM; glucose, 20 mM; CaCl2·2H2O, 2 mM, and MgCl2, 1 mM, pH to 7.3–7.4 with NaOH) and dried with cotton tips. A custom-made plastic recording chamber was glued to the skull with dental cement. The chamber was filled and kept perfusing with warm (37°C) aCSF. The stereotactic coordinates of the hippocampus were located according to the mouse brain atlas. A 1–2 mm wide cranial window centered 2.5 mm posterior to bregma and 2.2 mm lateral to the midline was opened in the skull with a dental drill. The dura was removed with fine forceps and the cortical tissue above the hippocampus was carefully removed by aspiration. When the dorsal surface of the hippocampus was seen, the aspiration was stopped and the imaging area was thoroughly irrigated with warm aCSF for about 30 min to make sure no bleeding and the blood was removed as much as possible. The craniotomy was filled with agarose (2%–3%) and stabilized with a cover glass. Then, the animal was moved to the recording platform, and the isoflurane was gradually reduced to 0.5%–0.8%. Besides the core body temperature, the respiratory and pulse rate were continuously monitored (MouseOx plus. STARR Life Science Corp.).
In vivo two-photon recordings were made using a custom-built two-photon microscope fed by a Ti:Sapph laser (Ultra II, ~4W average power, 670–1080 nm, Coherent), using a water dipping Nikon objective lens (16x, NA 0.8) and Hamamatsu GaAsP PMT detectors. Image data were acquired using MATLAB, running on an open source scanning microscope control software named ScanImage (version 3.8.1, Howard Hughes Medical Institute/Janelia Farms, RRID:SCR_014307) (Pologruto et al., 2003). Imaging was performed at an excitation wavelength of 920 nm for GCaMP6f and fluorescence was captured using a 560nm secondary dichroic and a 525–40nm bandpass emission filter (Chroma Technologies). Time-series images were acquired at 15.63 Hz with a pixel density of 256 × 256 and a field of view size of ~110 μm. For each view, spontaneous Ca2+ transients of hippocampal CA1 neurons were recorded for 5–10 min.
Image analyses were performed offline using ImageJ (https://imagej.nih.gov/ij) and an open-source MATLAB program NeuroSeg (Guan et al., 2018). First, images were stabilized with ImageJ to reduce the x-y vibration, then regions of interest (ROI) were drawn around individual somata and the relative fluorescence change (ΔF/F) versus time traces for each ROI was generated using NeuroSeg. Ca2+ transients were identified as changes in ΔF/F that were three times larger than the standard deviation of the noise band. We classified all recorded neurons based on their activity rates as silent (0–0.2 transients/min), normal (0.2–20 transients/min), and hyperactive (≥20 transients/min) neurons following the definitions by Busche and colleagues (Busche, 2018; Busche et al., 2008, 2012). Note that analyses of frequency distributions were performed using cells pooled from all animals, while analyses of mean frequency and fraction of silent, normal, and hyperactive cells were based on data from individual animals.
Ex vivo 2-photon Ca2+ imaging of CA1 neurons
Ex vivo two-photon Ca2+ imaging was carried out as described previously with some modifications (Chen-Engerer et al., 2019). 5xFAD, RyR2 WT, and RyR2 mutant mice were cross-bred with the heterozygous Thy1-GCaMP6f transgenic mice (Chen et al., 2012, 2013) (GP5.17, JAX 025393) to express the GCaMP6f Ca2+ sensing probe (driven by the Thy1 promotor) in hippocampal neurons in each of the genotypes used. Transverse hippocampal slices (260 μm) were prepared as described above and kept in the carbogenated HEPES containing aCSF for at least 60 min before recording. Slices were then moved to a recording chamber containing carbogenated external solution (NaCl, 124 mM; KCl, 2.5 mM; NaH2PO4, 1.25 mM; NaHCO3, 24 mM; HEPES, 5 mM; glucose, 12.5 mM; MgCl2, 2 mM; and CaCl2, 2 mM; pH 7.4 adjusted with NaOH) and put under an up-right two-photon imaging system (SP8 DIVE, Leica, Germany) with CHAMELEON HEAD/PSU: ULTRA (II): 80MHz (RoHS) laser (Coherent, UK). A 25x water-immersion objective with NA 0.95 (Leica, Germany) was used for imaging. Laser wavelength was set at 920 nm. Images were recorded with a resolution of 296 × 296 pixels at 16.77 fps. During recording, 0.5 μM tetrodoxin (TTX), 0.03 mM 2-amino-5-phosphonovaleric acid (APV), 0.02 mM 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 0.1 mM picrotoxin were added to the external solution. Local drug application was performed by using a glass pipette with a resistance of ~8 MΩ, which was connected to a modified pressurized perfusion system (ALA Scientific Instruments, Inc., USA). The pipette was filled with caffeine ringer solution (caffeine, 40 mM; CaCl2, 2 mM; HEPES, 10 mM; KCl, 2.5 mM; MgCl2, 1 mM; NaCl, 120 mM; NaH2PO4, 1.25 mM; pH 7.4 adjusted with NaOH). The pipette tip was placed at 15–20 μm from the soma of CA1 neuron. Caffeine (40 mM) was applied for 3 s to induce Ca2+ release. The fluorescence intensity in each somatic ROI was corrected by background subtraction. A ROI immediately outside of the neuron was taken as the background. Temporal fluorescence intensity changes in ROIs were expressed as relative changes in fluorescence intensity: ΔF/F = ((F − F0)/F0). F0 is defined as baseline fluorescence, which is the fluorescence intensity before a given stimulus, and F is the fluorescence recorded over time. ΔF/F values were calculated and plotted using NeuroSeg.
Long-term potentiation recording
Schaffer collateral fibers were stimulated at the CA3 subfield to record field excitatory postsynaptic potentials (fEPSPs) in the CA1 stratum radiatum of transverse hippocampal slices (300 μm) from all genotypes and drug-treated mice at different ages. After recovering at room temperature for 1 h (or 2 h for brain slices from drug-treated mice), hippocampal slices were allowed to recover in the recording chamber for additional 10 min. To evaluate basal synaptic transmission, we applied different stimulation strengths (0 μA to 200 μA in steps of 20 μA) and plotted fEPSP slopes versus the current input to compare the slope of input/output (I/O) curves of fEPSP. In the experiments that followed, stimulus current was adjusted so that fEPSP stabilized at 40%–50% of maximum. Baseline was recorded for at least 20 min until the differences among fEPSP slopes were within 10%. Long-term potentiation (LTP) was induced using a tetanic high-frequency stimulation (HFS; 4 trains of 100 pulses at 100 Hz, with 20 s intervals). Synaptic responses were recorded for at least 60 min after tetanization and quantified as the slope of the evoked fEPSP as percentage of the baseline.
Learning and memory tests
The learning and memory of mice with all genotypes were evaluated using the Morris water maze (MWM) test, the Barnes maze (BM) test, and/or the novel object recognition (NOR) test. Experiments were carried out blindly. For the MWM test, mice at different ages were trained to localize a hidden escape platform (10 × 10 cm) in a circular pool (116.84 cm in diameter, 50 cm in depth) (San Diego Instruments, CA) via distal visual cue. The platform was submerged 1–2 cm beneath the surface in water (22–24°C), which was rendered opaque by addition of milk powder. The localization of the pool in relation to visual cues was maintained constantly during the entire task. The cues were distinct in color and shape. Digital division of the tracking area (pool) into four quadrants was performed by the SMART video tracking system, Smart 3.0 (Panlab Harvard Apparatus; Barcelona Spain). The escape platform was placed in the center of the south-west quadrant for the entirety of the learning phase (4 training days) and digitally defined as target. Spatial training consisted of 4 days with 5 trials per mouse per day. Mice were released with their heads facing the pool wall at one of four entry locations (north, east, south and west) in a non-repetitive random order. Swimming was automatically video-tracked until the subject found the escape platform and remained on it (≥5sec), or until a maximum of 60 s. Mice that did not locate the hidden platform within the time limit of 60 s were guided to the escape platform until they spent ≥ 10 s on it. In between trials (inter-trial interval ≥ 10min), mice were housed in heated cages to avoid performance deficits due to exhaustion or hypothermia. The latency and swimming speed to reach the escape platform were recorded for comparison. After the learning phase, memory retention was evaluated by one probe trial 24 hours after the last training session. The escape platform was removed before mice were released from the north entry point into the pool. Their swimming was video-tracked for 60 s. The area at the location of the removed hidden platform was defined as the target and the south-west quadrant the target quadrant. The percentage of time mice spent in the target quadrant (including the target) were measured for comparison.
For the BM test, the size and characteristics of the device are as follows: a 92 cm diameter platform; the platform contains 20 holes, each 5 cm in diameter, equally distributed around the platform and separated by 7.5 cm; the device stands 105 cm above the floor. In one hole there is an escape box communicated with the platform through transparent plastic tunnels arranged in such a way that they cannot be seen from the platform. Similar to the MWM test the simultaneous use of a video-monitoring system is used to obtain automated behavioral recordings. Each trial lasts 3 min per mouse, with an inter-trial interval of 15 min, with four trials per day during the acquisition phase. The first phase (habituation), consisted of placing the mouse on the center of the platform and then, turning on the bright light as an aversive stimulus. Then the mouse was gently taken to the escape hole; once in the escape chamber, the light was turned off, and the mouse was kept inside for two additional minutes. During acquisition, mice were placed on the center of the platform and the light was turned on for 3 min, the latency to find the escape hole was recorded. If the mouse did not reach the escape hole within 3 min, the experimenter placed it at the entrance of the escape hole for 1 min, and then took it back to its home-cage. This protocol continued for 4 days. On day 5, 24 h after the last training day, the probe trial was conducted. The target hole was closed. The maze was rotated so that the target hole was closed and the maze was readjusted so that the holes were in the same position as during the training days. The mouse was then placed in the middle of the maze and allowed to explore the maze as before. The mouse was removed after a 90 s. The probe trial was done in order to determine if the animal remembered where the target hole was located. The numbers of nose pokes to each hole were measured.
For the NOR test, mice were habituated for 10 min per mouse in an equally illuminated, odor-free, white, plastic box (40 × 40 × 50 cm3) embedded with fresh aspen shavings and shreds. In between each mouse trial the box was wiped with ethanol to avoid odor-induced stress. 24 hours after habituation, two identical objects ware placed at equal distance to each other and the corners of the box. Each mouse was placed into the center, and allowed to move freely for 10 min. Mice were video recorded during this familiarizing phase. Side preferences was evaluated by dividing the time a mouse spent exploring one object by the time they spent at the other object. Twenty-four hours later, one of the objects was replaced by a novel object. The other object remained constant. The selection of a familiar object to be replaced was random. Each mouse was again placed into the center of the box and allowed to move freely for another 10 min while videotaped. General exploration was evaluated by determining the time spent exploring the objects. The discrimination ratio describes the time a mouse explored the novel object divided by the total time it spent exploring (novel and familiar objects). The above experiments were carried out blindly.
Biotinylation assays
Biotinylation assays were performed according to the protocol described previously (Lin et al., 2010) with some modifications. Briefly, after 24–48 h of transfection with the Kv4.2 and KChIP4 cDNAs, transfected HEK293 cell lines expressing RyR2 WT or RyR2 E4872Q mutation were rinsed with ice-cold PBS for three times, surface proteins were biotinylated with 1.5 mg/ml sulfo-NHS-SS-biotin reagent (Pierce, Cat# PG82077) in PBS for 30 min on ice. Unbound biotin was quenched with ice-cold 50 mM glycine in PBS. Cells were lysed with ice-cold lysis buffer: 150 mM NaCl, 20 mM Tris–HCl, 1% NP40 and protease inhibitor cocktail (Roche, Cat# 4693159001), sonicated and centrifuged at 12,000 g for 10 min. Cell lysates were incubated overnight at 4°C with immobilized-Streptavidin agarose beads (Pierce, Cat# 20349), unbound proteins were removed from the beads with 3 washes in lysis buffer. The bound proteins were eluted with 2 × SDS sample buffer. Surface expressed proteins were separated by electrophoresis in 12% Tris-glycine SDS-PAGE and transferred to PVDF membranes. Western blots were probed with the following antibodies: rabbit anti-Kv4.2 (1:1000, abcam, Cat# ab 16719), rabbit anti-Rab4 (1:1000, Cell Signaling Technology, Cat# 2167), goat anti-rabbit IgG secondary antibodies conjugated with horseradish peroxidase (1:10000, ThermoFisher, Cat#31460). The bound antibodies were detected using an enhanced chemiluminescence kit from Pierce.
Immunoblotting
Immunoblotting analysis was carried out using the method described previously (Rosen et al., 2010). Briefly, the frozen mouse brain was homogenized with ice-cold buffer, containing 50 mM Tris-HCl (pH 7.5) plus 4 μM leupeptin, 1 mM benzamidine, 0.1 mM phenyl-methylsulfonyl fluoride (PMSF), and 0.5 mM dithiothreitol (DTT). The homogenates were sonicated 3 times and spun at 3000xg for 5 min. The supernatants were mixed with sample buffer (sodium dodecyl sulfate (SDS), 12%; β-merceptoethanol, 6%; DTT, 200mM; Tris-HCl, 150mM; and glycerol, 30%; pH 7.0) and heated at 95°C for 15min. Samples were subjected to Tricine/SDS polyacrylamide gel electrophoresis (15%), and were transferred onto 0.2 μm polyvinylidene difluoride (PVDF) membranes (Bio-Rad) at 100 V for 1h at 4°C. The membrane was blocked with 5% non-fat milk powder for 30min. The blocked membrane was incubated with a primary antibody (anti-Aβ (total) antibody (Cell Signaling Technology, Cat# 8243), anti-Aβ (1–42) antibody (Biolegend, Cat# 803001), or anti-β-actin (Sigma, Cat# A5316)), and then the goat anti-rabbit IgG (ThermoFisher, Cat#31460) or the goat anti-mouse IgG (ThermoFisher, Cat# 31430) secondary antibody conjugated with horseradish peroxidase (1:20000 dilution). After washing 3 times, 10 min each, the bound antibodies were detected using an enhanced chemiluminescence kit from Pierce. The band intensities were determined from its intensity profile obtained by ImageQuant LAS 4000 (GE Healthcare Life Sciences), analyzed by using the Image-J software, and normalized to that of β-actin. Note that genetically or pharmacologically limiting RyR2 open time prevents AD-related deficits without altering Aβ accumulation. This differs from previous studies showing that dantrolene (a potent RyR1) significantly reduced Aβ load (Chakroborty et al., 2012a; Oulès et al., 2012; Peng et al., 2012; Wu et al., 2015). The reason for this difference remains to be determined. Dantrolene treatment has been associated with hepatotoxicity in humans (Chan, 1990; Utili et al., 1977) and worsening AD pathologies in mice (Zhang et al., 2010). The brain expresses all three RyR isoforms. RyR1 and RyR3 are readily inhibited by dantrolene (Zhao et al., 2001), but dantrolene inhibition of RyR2 is subtle and varies with the RyR2 oxidative and modulated states (Maxwell et al., 2012; Oda et al., 2015; Zhao et al., 2001). Hence, whether dantrolene can effectively suppress RyR2 in the brain is unclear. Dantrolene by interacting with multiple targets (Oo et al., 2015; Zhao et al., 2001, 2006) may affect Aβ metabolism differently from limiting RyR2 open time.
Immunohistochemical and Nissl staining
Mice of different ages and genotypes were anesthetized and transcardially perfused with 10% neutral buffered formalin (NBF). Whole brains were removed and post-fixed in NBF for at least 24 h. The fixed brains were then embedded in paraffin after dehydration and diaphanization. For the IHC staining, paraffin-embedded brain tissue sections (5 μm) were immersed in xylene (5 min, 3 times), rehydrated in absolute ethanol (5 min, 3 times) followed by immersion in 95%, 80% and 70% solutions of ethanol (in water) (5 min each). Antigens were reactivated by treatment with 0.01 M citrate buffer (pH 6.0) for 2 min in microwave. Slides were washed in phosphate buffered saline (PBS: NaCl, 137 mM; KCl, 2.7 mM; Na2HPO4, 10 mM; KH2PO4, 2 mM, pH 7.4) and blocked with 10% normal horse serum in PBS for 10 min, then incubated with the primary antibody, the anti-β-amyloid peptide (total) antibody (Cell Signaling Technology, Cat# 8243), for 12–16 h at 2–8°C. After washing with PBS, slides were incubated with biotinylated secondary antibody (Vector Laboratories, Cat# BA-1000) for 10 min, washed twice with PBS, and incubated with 3% H2O2 for 25 min for inactivation of endogenous peroxidase. Slides were then incubated with streptavidin–biotin–peroxidase for 30 min. Slides were covered with 3, 3′-diaminobenzidine (DAB) solution (0.06% in PBS containing 0.018% H2O2) for 1 to 5 min or until a brown precipitate could be observed. Identical conditions and reaction times were used for slides from different samples to allow comparison between immunoreactivity densities. Reaction was stopped by immersion of slides in distilled water. Counterstaining was performed with Harris hematoxilin. Coverslips were mounted with resinous mounting medium.
For the Nissl staining, paraffin-embedded brain tissue sections (5 μm) were immersed in xylene (5 min, 2 times), rehydrated in absolute ethanol (5 min, 2 times) followed by 95%, 75% and 50% solutions of ethanol in water (5 min each), then washed in distilled water for 2 times, 5 min each. Slides were stained in FD cresyl violet solution (FD Neurotechnologies, Baltimore, MD, USA) for 10 min, then, briefly rinsed in 100% ethanol and differentiated in 100% ethanol containing 0.1% glacia acetic acid for 1 min. Slides were then dehydrated in absolute ethanol (2 min, 4 times) followed by clearance in xylene (3 min, 2 times). Coverslip were mounted with resinous mounting medium.
Single-cell Ca2+ imaging of HEK293 cells
Intracellular cytosolic Ca2+ changes in stable, inducible HEK293 cells expressing RyR2 WT or RyR2 E4872Q mutant, transfected with presenilin 1 (PS1) WT, PS1-M146L, PS1-L286V or control plasmid (pcDNA3) were monitored using single-cell Ca2+ imaging and the fluorescent Ca2+ indicator dye Fura-2 AM, as described previously (Chen et al., 2014; Jiang et al., 2004, 2005). Briefly, transfected cells grown on glass coverslips for 18–22 h after induction by 1 μg/ml tetracycline were loaded with 5 μM Fura-2 AM in KRH (Krebs-Ringer-HEPES) buffer (NaCl, 125 mM; KCl, 5 mM; KH2PO4, 1.2 mM; glucose, 6 mM; MgCl2, 1.2 mM; HEPES, 25 mM; pH 7.4) plus 0.02% Pluronic F-127 and 0.1 mg/ml BSA for 20 min at room temperature (23°C). Coverslips were then mounted onto a perfusion chamber (Warner Instruments) on an inverted microscope (TE2000-S, Nikon, Japan). The cells were continuously perfused with KRH buffer containing increasing extracellular Ca2+ concentrations (0–2.0 mM). Increasing the extracellular Ca2+ concentration will lead to increased Ca2+ entry and subsequent Ca2+ loading of internal stores; when store Ca2+ content reaches a threshold level, the RyR2 opens, resulting in a spontaneous Ca2+ release event. Caffeine (10 mM) was applied at the end of each experiment to confirm the expression of active RyR2 channels. Time-lapse images (0.5 fps) were captured and analyzed with Compix Simple PCI 6 software (Compix Inc.). Fluorescence intensities were measured from regions of interest centered on individual cells. Only cells that responded to caffeine were used in analyses.
Dendritic spine density analysis
A FD Rapid GolgiStain kit (FD Neurotechnologies, Baltimore, MD, USA) was used for dendritic spine histological analysis by following the manufacturer’s instructions as previously described (Zhao et al., 2015). Briefly, mouse brains were removed, placed in an appropriate impregnation solution and stored in the dark for 14 days at room temperature. The brains were then transferred into a solution containing sucrose and incubated at 4°C until the residual water was removed from the tissues (2–7 days). Finally, a vibratome (VT-1200, Leica, Germany) was used to obtain 100μm thick, transverse hippocampal slices. Slices were then mounted onto gelatin-coated slides and allowed to air dry at room temperature for about two days. Once sufficiently dried, the slices were rinsed with distilled water and incubated for ten minutes in a solution containing silver nitrate. Slices were then rinsed with distilled water before being dehydrated in absolute alcohol, cleared with xylene and covered with non-acidic synthetic balsam and coverslips. Three independent slices per mouse were imaged. Z stacks of Golgi-stained CA1 neuron apical dendrites (up to 80 μm total on Z axis; optical section thickness = 0.5 μm) were taken at 100x magnification on a Zeiss AxioImager (Zeiss, Germany).
The secondary dendrites that were 100–250 μm away from the soma were used for analysis. Dendritic segments were identified and measured with ImageJ and RECONSTRUCT as described previously (Risher et al., 2014). Dendritic spines were identified as “Filopodia,” “Long Thin,” “Thin,” “Stubby,” “Mushroom” and “Branched.” The densities of total protrusion and each type of spines were analyzed. Spine density was calculated by quantifying the number of spines per dendritic segment and normalized to 10 μm of dendritic length.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed blindly to genotype, age and treatment. All data shown are medians and range (min and max), unless indicated otherwise. For small datasets (n number less than 15) or non-Gaussian distributed data, non-parametric methods were used. For large datasets and normally distributed data, parametric tests were performed. With respect to non-parametric analyses, for experiments with two groups, Mann-Whitney U test was used for unpaired samples. Wilcoxon matched-pairs signed rank test was used for paired samples. For experiments with 3 or more groups, Kruskal-Wallis test with Dunn-Bonferroni post hoc test and Friedman test with Dunn-Bonferroni post hoc test were used for independent samples or repeat measurements, respectively. With respect to parametric analyses, for experiments with two groups, Student’s t test was used for unpaired samples. Paired t test was used for paired samples. For experiments with 3 or more groups, one-way ANOVA or two-way ANOVA test followed by Bonferroni post hoc test and repeated-measure ANOVA test with Bonferroni post hoc test were used for independent samples or repeat measurements, respectively. Sample sizes and p values can be found in figure legends. P values smaller than 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Kv4.2 | Abcam | Cat# ab16719; RRID: AB_443443 |
| Rabbit polyclonal anti-Rab4 | Cell Signaling Technology | Cat# 2167; RRID: AB_2253579 |
| Rabbit monoclonal anti-β-Amyloid | Cell Signaling Technology | Cat# 8243; RRID: AB_2797642 |
| Mouse monoclonal anti-β-Amyloid | Biolegend | Cat# 803001; RRID: AB_2564653 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A5316; RRID: AB_476743 |
| HRP Goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# 31460; RRID: AB_228341 |
| HRP Goat anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# 31430; RRID: AB_228307 |
| Biotinylated Goat anti-Rabbit IgG | Vector Laboratories | Cat# BA-1000; RRID: AB_2313606 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (R)-(+)-carvedilol | Zhang et al., 2015 | N/A |
| D(−)-2-Amino-5-phosphonopentanoic acid | Sigma-Aldrich | Cat# A8054; CAS# 79055-68-8 |
| 6,7-dinitroquinoxaline-2,3-dione | Sigma-Aldrich | Cat# D0540; CAS# 2379-57-9 |
| Picrotoxin | TOCRIS | Cat# 1128; CAS# 124-87-8 |
| Tetrodotoxin citrate | Abcam | Cat# ab 120055; CAS# 18660-81-6 |
| Tetraethylammonium chloride | Sigma-Aldrich | Cat# T2265; CAS# 56-34-8 |
| NS5806 | Sigma-Aldrich | Cat# N2540; CAS# 426834-69-7 |
| Critical Commercial Assays | ||
| Premium Grade Sulfo NHS-SS-Biotin | Pierce | Cat# PG82077 |
| Streptavidin Agarose | Pierce | Cat# 20349 |
| FD Cresyl Violet Solution | FD NeuroTechnologies | Cat# PS102–01 |
| FD Rapid GolgiStain Kit | FD NeuroTechnologies | Cat# PK401 |
| Experimental Models: Cell Lines | ||
| Flp-In T-Rex HEK293 cell | Invitrogen | Cat# R78007; RRID: CVCL_U427 |
| RyR2 WT HEK293 cell line | Jiang et al., 2004 | N/A |
| RyR2 E4872Q HEK293 cell line | Chen et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: 5xFAD+/− | The Jackson Laboratory | MMRRC Stock No: 34848-JAX |
| Mouse: RyR2 E4872Q+/− | Chen et al., 2014 | N/A |
| Mouse: Thy1-GCaMP6f+/− | The Jackson Laboratory | Stock No: 025393 |
| Recombinant DNA | ||
| Plasmid: Kv4.2 | This paper | N/A |
| Plasmid: EGFP | This paper | N/A |
| Plasmid: KChIP4 | This paper | N/A |
| Plasmid: PS1 | This paper | N/A |
| Plasmid: PS1 M146L | This paper | N/A |
| Plasmid: PS1 L286V | This paper | N/A |
| Software and Algorithms | ||
| pCLAMP 10.6 | Molecular Devices | https://www.moleculardevices.com/ |
| OriginPro 9.0 | OriginLab | https://www.originlab.com/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| ScanImage 3.8.1 | Pologruto et al., 2003 | RRID: SCR_014307 |
| ImageJ | https://imagej.nih.gov/ij | |
| NeuroSeg | Guan et al., 2018 | N/A |
| RECONSTRUCT | Risher et al., 2014 | N/A |
| Graphpad Prism 7 | Graphpad Software | https://www.graphpad.com/ |
Highlights.
Limiting RyR2 open time prevents neuronal hyperactivity as well as major AD deficits
Limiting RyR2 open time enhances A-type K+ current to constrain neuronal excitability
R-carvedilol rescues AD-related neuronal hyperactivity, memory loss, and cell death
AD deficits can be prevented in the face of continued β-amyloid accumulation
ACKNOWLEDGMENTS
This work was supported by research grants from the Canadian Institutes of Health Research to R.J.T. (PJT-169174; PJT-168968), R.W.T. (PJT-156153), G.R.G. (FDN-148471), and S.R.W.C. (PJT-152914), the National Institutes of Health to M.F. (R01 HL057832; R01 GM111397), and the Heart and Stroke Foundation Chair in Cardiovascular Research to S.R.W.C. We gratefully acknowledge generous donations from the Alvin and Mona Libin Foundation, Canadian Pacific Railway Company (CP), the Mozell Family, and the Libin Cardiovascular Institute. We thank Dr. Xiaowei Chen (Army Medical University, China) for invaluable advice and discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108169.
DECLARATION OF INTERESTS
We are planning to submit a Provisional Application for Patent, entitled “METHODS OF TREATING ALZHEIMER’S DISEASE OR DELAYING PROGRESSION THEREOF BY LIMITING RYANODINE RECEPTOR OPEN TIME.”
REFERENCES
- Alkon DL, Nelson TJ, Zhao W, and Cavallaro S (1998). Time domains of neuronal Ca2+ signaling and associative memory: steps through a calexcitin, ryanodine receptor, K+ channel cascade. Trends Neurosci. 21, 529–537. [DOI] [PubMed] [Google Scholar]
- Bartsch W, Sponer G, Strein K, Müller-Beckmann B, Kling L, Böhm E, Martin U, and Borbe HO (1990). Pharmacological characteristics of the stereoisomers of carvedilol. Eur. J. Clin. Pharmacol 38 (Suppl 2), S104–S107. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (2010). Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 459, 441–449. [DOI] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, and Foster TC (2010). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J. Neurophysiol 104, 2586–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov KY, Vinogradova TM, and Lakatta EG (2001). Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ. Res 88, 1254–1258. [DOI] [PubMed] [Google Scholar]
- Brager DH, and Johnston D (2007). Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in I(h) in hippocampal CA1 pyramidal neurons. J. Neurosci 27, 13926–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bround MJ, Asghari P, Wambolt RB, Bohunek L, Smits C, Philit M, Kieffer TJ, Lakatta EG, Boheler KR, Moore ED, et al. (2012). Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc. Res 96, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Chin J, Leiser SC, Pangalos MN, and Randall AD (2011). Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer’s disease. Neurobiol. Aging 32, 2109.e1–2109.e14. [DOI] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, and Stutzmann GE (2012). Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 33, 1001.e1–1001.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA (2018). In Vivo Two-Photon Calcium Imaging of Hippocampal Neurons in Alzheimer Mouse Models. Methods Mol. Biol 1750, 341–351. [DOI] [PubMed] [Google Scholar]
- Busche MA, and Konnerth A (2015). Neuronal hyperactivity–A key defect in Alzheimer’s disease? BioEssays 37, 624–632. [DOI] [PubMed] [Google Scholar]
- Busche MA, and Konnerth A (2016). Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, and Garaschuk O (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, and Konnerth A (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 109, 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, and Hyman BT (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci 22, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace R, Heeman B, Van Mossevelde S, De Roeck A, Hoogmartens J, De Rijk P, Gossye H, De Vos K, De Coster W, Strazisar M, et al. ; BELNEU Consortium (2019). Loss of DPP6 in neurodegenerative dementia: a genetic player in the dysfunction of neuronal excitability. Acta Neuropathol. 137, 901–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, and Stutzmann GE (2014). Calcium channelopathies and Alzheimer’s disease: insight into therapeutic success and failures. Eur. J. Pharmacol 739, 83–95. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, and Stutzmann GE (2009). Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci 29, 9458–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, and Stutzmann GE (2012a). Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PLoS ONE 7, e52056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, and Stutzmann GE (2012b). Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J. Neurosci 32, 8341–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Hill ES, Christian DT, Helfrich R, Riley S, Schneider C, Kapecki N, Mustaly-Kalimi S, Seiler FA, Peterson DA, et al. (2019). Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol. Neurodegener 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH (1990). Dantrolene sodium and hepatic injury. Neurology 40, 1427–1432. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mayne M, Holden CP, Geiger JD, and Mattson MP (2000). Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem 275, 18195–18200. [DOI] [PubMed] [Google Scholar]
- Chen C (2005). beta-Amyloid increases dendritic Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1 pyramidal neurons. Biochem. Biophys. Res. Commun 338, 1913–1919. [DOI] [PubMed] [Google Scholar]
- Chen Q, Cichon J, Wang W, Qiu L, Lee SJ, Campbell NR, Destefino N, Goard MJ, Fu Z, Yasuda R, et al. (2012). Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron 76, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, et al. (2014). The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med 20, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Engerer HJ, Hartmann J, Karl RM, Yang J, Feske S, and Konnerth A (2019). Two types of functionally distinct Ca2+ stores in hippocampal neurons. Nat. Commun 10, 3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, and Holtzman DM (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Dana H, Chen TW, Hu A, Shields BC, Guo C, Looger LL, Kim DS, and Svoboda K (2014). Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS ONE 9, e108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pins B, Cifuentes-Díaz C, Farah AT, López-Molina L, Montalban E, Sancho-Balsells A, López A, Ginés S, Delgado-García JM, Alberch J, et al. (2019). Conditional BDNF Delivery from Astrocytes Rescues Memory Deficits, Spine Density, and Synaptic Properties in the 5xFAD Mouse Model of Alzheimer Disease. J. Neurosci 39, 2441–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delekate A, Füchtemeier M, Schumacher T, Ulbrich C, Foddis M, and Petzold GC (2014). Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun 5, 5422. [DOI] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, et al. (2012). A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 76, 908–920. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, et al. (2005). Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhoff G, and Garaschuk O (2011). Two-photon imaging of neural networks in a mouse model of Alzheimer’s disease. Cold Spring Harb. Protoc 2011, 1206–1216. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Furutama D, Hakamata Y, Nakai J, Takeshima H, and Mikoshiba K (1994). Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J. Neurosci 14, 4794–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G, Conti A, Mammarella S, Scrobogna M, and Sorrentino V (1995). The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol 128, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good TA, Smith DO, and Murphy RM (1996). Beta-amyloid peptide blocks the fast-inactivating K+ current in rat hippocampal neurons. Biophys. J 70, 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Li J, Liang S, Li R, Li X, Shi X, Huang C, Zhang J, Pan J, Jia H, et al. (2018). NeuroSeg: automated cell detection and segmentation for in vivo two-photon Ca2+ imaging data. Brain Struct. Funct 223, 519–533. [DOI] [PubMed] [Google Scholar]
- Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, Hoffman DA, and Roberson ED (2015). Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J. Neurosci 35, 6221–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hiess F, Vallmitjana A, Wang R, Cheng H, ter Keurs HE, Chen J, Hove-Madsen L, Benitez R, and Chen SR (2015). Distribution and Function of Cardiac Ryanodine Receptor Clusters in Live Ventricular Myocytes. J. Biol. Chem 290, 20477–20487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, and Johnston D (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. [DOI] [PubMed] [Google Scholar]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, et al. (2018). Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med 378, 321–330. [DOI] [PubMed] [Google Scholar]
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA, and Wirths O (2012). Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 196.e29–196.e40. [DOI] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, and Chen SRW (2004). RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. USA 101, 13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, and Chen SRW (2005). Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res 97, 1173–1181. [DOI] [PubMed] [Google Scholar]
- Jung SC, and Hoffman DA (2009). Biphasic somatic A-type K channel downregulation mediates intrinsic plasticity in hippocampal CA1 pyramidal neurons. PLoS ONE 4, e6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, and De Strooper B (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, and O’Neill C (1999). Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience 92, 499–513. [DOI] [PubMed] [Google Scholar]
- Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med 8, 363ra150. [DOI] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, and Helmchen F (2005). Imaging input and output of neocortical networks in vivo. Proc. Natl. Acad. Sci. USA 102, 14063–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrigan TL, Brown JT, and Randall AD (2014). Characterization of altered intrinsic excitability in hippocampal CA1 pyramidal cells of the Aβ-overproducing PDAPP mouse. Neuropharmacology 79, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin AD, Kekuš M, Adelsberger H, Neumann U, Shimshek DR, Song B, Zott B, Peng T, Förstl H, Staufenbiel M, et al. (2017). BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 114, 8631–8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, and Hoffman DA (2005). Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J. Physiol 569, 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, and Hoffman DA (2007a). Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yun HM, Baik JH, Chung KC, Nah SY, and Rhim H (2007b). Functional interaction of neuronal Cav1.3 L-type calcium channel with ryanodine receptor type 2 in the rat hippocampus. J. Biol. Chem 282, 32877–32889. [DOI] [PubMed] [Google Scholar]
- Kim D, Baik SH, Kang S, Cho SW, Bae J, Cha MY, Sailor MJ, Mook-Jung I, and Ahn KH (2016). Close Correlation of Monoamine Oxidase Activity with Progress of Alzheimer’s Disease in Mice, Observed by in Vivo Two-Photon Imaging. ACS Cent. Sci 2, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim B, Kim HS, and Cho JY (2020). Nicotinamide attenuates the decrease in dendritic spine density in hippocampal primary neurons from 5xFAD mice, an Alzheimer’s disease animal model. Mol. Brain 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DT, Hebert PR, Coffey CS, Curtis JP, Foody JM, Sedrakyan A, and Krumholz HM (2004). Adverse effects of beta-blocker therapy for patients with heart failure: a quantitative overview of randomized trials. Arch. Intern. Med 164, 1389–1394. [DOI] [PubMed] [Google Scholar]
- Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, Teich AF, Zalk R, Saint N, Arancio O, et al. (2017). Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 134, 749–767. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, and Cherubini E (2007). Presynaptic calcium stores contribute to nicotine-elicited potentiation of evoked synaptic transmission at CA3-CA1 connections in the neonatal rat hippocampus. Hippocampus 17, 316–325. [DOI] [PubMed] [Google Scholar]
- Leão RN, Colom LV, Borgius L, Kiehn O, and Fisahn A (2012). Medial septal dysfunction by Aβ-induced KCNQ channel-block in glutamatergic neurons. Neurobiol. Aging 33, 2046–2061. [DOI] [PubMed] [Google Scholar]
- Lerdkrai C, Asavapanumas N, Brawek B, Kovalchuk Y, Mojtahedi N, Olmedillas Del Moral M, and Garaschuk O (2018). Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 115, E1279–E1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Wikenheiser AM, Kung F, and Hoffman DA (2010). KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol. Cell. Neurosci 43, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Supnet C, Sun S, Zhang H, Good L, Popugaeva E, and Bezprozvanny I (2014). The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels (Austin) 8, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J, Hoffman D, Colbert C, and Johnston D (1998). Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu. Rev. Physiol 60, 327–346. [DOI] [PubMed] [Google Scholar]
- Mandikian D, Bocksteins E, Parajuli LK, Bishop HI, Cerda O, Shigemoto R, and Trimmer JS (2014). Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol 522, 3555–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JT, Domeier TL, and Blatter LA (2012). Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am. J. Physiol. Heart Circ. Physiol 302, H953–H963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morohashi Y, Hatano N, Ohya S, Takikawa R, Watabiki T, Takasugi N, Imaizumi Y, Tomita T, and Iwatsubo T (2002). Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem 277, 14965–14975. [DOI] [PubMed] [Google Scholar]
- Murayama T, and Ogawa Y (1996). Properties of Ryr3 ryanodine receptor isoform in mammalian brain. J. Biol. Chem 271, 5079–5084. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, and Lederer WJ (1995). Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637. [DOI] [PubMed] [Google Scholar]
- Nichols AJ, Sulpizio AC, Ashton DJ, Hieble JP, and Ruffolo RR Jr. (1989). The interaction of the enantiomers of carvedilol with alpha 1- and beta 1-adrenoceptors. Chirality 1, 265–270. [DOI] [PubMed] [Google Scholar]
- Noh W, Pak S, Choi G, Yang S, and Yang S (2019). Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Front. Cell. Neurosci 13, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, Emrani S, Liu L, Herman M, Barrett G, et al. (2017). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun 8, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, and Sperling RA (2010). Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 74, 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Yang Y, Uchinoumi H, Thomas DD, Chen-Izu Y, Kato T, Yamamoto T, Yano M, Cornea RL, and Bers DM (2015). Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol 85, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo YW, Gomez-Hurtado N, Walweel K, van Helden DF, Imtiaz MS, Knollmann BC, and Laver DR (2015). Essential Role of Calmodulin in RyR Inhibition by Dantrolene. Mol. Pharmacol 88, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, et al. (2012). Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci 32, 11820–11834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, and Shusterman NH; U.S. Carvedilol Heart Failure Study Group (1996). The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N. Engl. J. Med 334, 1349–1355. [DOI] [PubMed] [Google Scholar]
- Peng J, Liang G, Inan S, Wu Z, Joseph DJ, Meng Q, Peng Y, Eckenhoff MF, and Wei H (2012). Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett 516, 274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peron S, Chen TW, and Svoboda K (2015). Comprehensive imaging of cortical networks. Curr. Opin. Neurobiol 32, 115–123. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, and Svoboda K (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, and Chen SR (2011). Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res 108, 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, et al. (2004). KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J. Neurosci 24, 7903–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Ustunkaya T, Singh Alvarado J, and Eroglu C (2014). Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE 9, e107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen RF, Tomidokoro Y, Ghiso JA, and Walker LC (2010). SDS-PAGE/immunoblot detection of Abeta multimers in human cortical tissue homogenates using antigen-epitope retrieval. J. Vis. Exp (38), 1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Hwang SY, Rybalchenko N, and Koulen P (2008). The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol 40, 84–97. [DOI] [PubMed] [Google Scholar]
- Sandler VM, and Barbara JG (1999). Calcium-induced calcium release contributes to action potential-evoked calcium transients in hippocampal CA1 pyramidal neurons. J. Neurosci 19, 4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMartín CD, Veloso P, Adasme T, Lobos P, Bruna B, Galaz J, García A, Hartel S, Hidalgo C, and Paula-Lima AC (2017). RyR2-Mediated Ca2+ Release and Mitochondrial ROS Generation Partake in the Synaptic Dysfunction Caused by Amyloid β Peptide Oligomers. Front. Mol. Neurosci 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TR, Gray NW, Mainen ZF, and Svoboda K (2007). The functional microarchitecture of the mouse barrel cortex. PLoS Biol. 5, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scala F, Fusco S, Ripoli C, Piacentini R, Li Puma DD, Spinelli M, Laezza F, Grassi C, and D’Ascenzo M (2015). Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K(+) current inhibition mediated by activation of caspases and GSK-3. Neurobiol. Aging 36, 886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serôdio P, and Rudy B (1998). Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J. Neurophysiol 79, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dun-stan R, Salloway S, Chen T, Ling Y, et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, and Remy S (2014). Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Smith IF, Hitt B, Green KN, Oddo S, and LaFerla FM (2005). Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neurochem 94, 1711–1718. [DOI] [PubMed] [Google Scholar]
- Stargardt A, Swaab DF, and Bossers K (2015). Storm before the quiet: neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer’s disease. Neurobiol. Aging 36, 1–11. [DOI] [PubMed] [Google Scholar]
- Stoschitzky K, Koshucharova G, Lercher P, Maier R, Sakotnik A, Klein W, Liebmann PM, and Lindner W (2001). Stereoselective effects of (R)- and (S)-carvedilol in humans. Chirality 13, 342–346. [DOI] [PubMed] [Google Scholar]
- Sun W, Maffie JK, Lin L, Petralia RS, Rudy B, and Hoffman DA (2011). DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 71, 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Han X, Deane R, Zlokovic B, and Nedergaard M (2007). Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann. N Y Acad. Sci 1097, 40–50. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Hirose K, Nishi M, Noda T, and Iino M (1998). Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 17, 3309–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnini F, Novelia J, Kerrigan TL, Brown JT, Tsaneva-Atanasova K, and Randall AD (2015). Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell. Neurosci 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, and Lein E (2018). Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J. Vis. Exp (132), 53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utili R, Boitnott JK, and Zimmerman HJ (1977). Dantrolene-associated hepatic injury. Incidence and character. Gastroenterology 72, 610–616. [PubMed] [Google Scholar]
- van de Vrede Y, Fossier P, Baux G, Joels M, and Chameau P (2007). Control of IsAHP in mouse hippocampus CA1 pyramidal neurons by RyR3-mediated calcium-induced calcium release. Pflugers Arch. 455, 297–308. [DOI] [PubMed] [Google Scholar]
- van Zwieten PA (1993). Pharmacodynamic profile of carvedilol. Cardiology 82 (Suppl 3), 19–23. [DOI] [PubMed] [Google Scholar]
- Varga AW, Yuan LL, Anderson AE, Schrader LA, Wu GY, Gatchel JR, Johnston D, and Sweatt JD (2004). Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J. Neurosci 24, 3643–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring JF, Anderson DJ, Kroeger PE, Li J, Chen SP, Hooker BA, Gopalakrishnan M, and Briggs CA (2012). Gene Expression Changes Related to Synaptic Deficits in the Tg2576 Mouse Model of Alzheimer’s Disease. J. Drug Metab. Toxicol 3, 1000115. [Google Scholar]
- Wu B, Yamaguchi H, Lai FA, and Shen J (2013). Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Natl. Acad. Sci. USA 110, 15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yang B, Liu C, Liang G, Eckenhoff MF, Liu W, Pickup S, Meng Q, Tian Y, Li S, and Wei H (2015). Long-term dantrolene treatment reduced intraneuronal amyloid in aged Alzheimer triple transgenic mice. Alzheimer Dis. Assoc. Disord 29, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Kovacs I, and Zhang Z (2004). Differential distribution of KChIPs mRNAs in adult mouse brain. Brain Res. Mol. Brain Res 128, 103–111. [DOI] [PubMed] [Google Scholar]
- Xu J, Kang N, Jiang L, Nedergaard M, and Kang J (2005). Activity-dependent long-term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J. Neurosci 25, 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Tanei ZI, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, et al. (2015). Chronic optogenetic activation augments aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 11, 859–865. [DOI] [PubMed] [Google Scholar]
- Yang EJ, Mahmood U, Kim H, Choi M, Choi Y, Lee JP, Cho JY, Hyun JW, Kim YS, Chang MJ, and Kim HS (2018). Phloroglucinol ameliorates cognitive impairments by reducing the amyloid β peptide burden and pro-inflammatory cytokines in the hippocampus of 5XFAD mice. Free Radic. Biol. Med 126, 221–234. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B, and Bezprozvanny I (2010). Role of presenilins in neuronal calcium homeostasis. J. Neurosci 30, 8566–8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou Q, Smith CD, Chen H, Tan Z, Chen B, Nani A, Wu G, Song LS, Fill M, et al. (2015). Non-β-blocking R-carvedilol enantiomer suppresses Ca2+ waves and stress-induced ventricular tachyarrhythmia without lowering heart rate or blood pressure. Biochem. J 470, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Li P, Chen SR, Louis CF, and Fruen BR (2001). Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J. Biol. Chem 276, 13810–13816. [DOI] [PubMed] [Google Scholar]
- Zhao X, Weisleder N, Han X, Pan Z, Parness J, Brotto M, and Ma J (2006). Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J. Biol. Chem 281, 33477–33486. [DOI] [PubMed] [Google Scholar]
- Zhao QR, Lu JM, Yao JJ, Zhang ZY, Ling C, and Mei YA (2015). Neuritin reverses deficits in murine novel object associative recognition memory caused by exposure to extremely low-frequency (50 Hz) electromagnetic fields. Sci. Rep 5, 11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, et al. (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med 17, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, and Konnerth A (2019). A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 365, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study will be available upon reasonable request.