

Abstract
Objectives
Aging is associated with altered immune function and chronic low‐grade inflammation, referred to as immunosenescence. As breast cancer is an age‐related disease, the impact of aging on tumor immune responses may have important consequences. However, effects of immunosenescence on breast tumor immune infiltration remain largely unknown.
Methods
This exploratory study investigated a broad panel of immune/senescence markers in peripheral blood and in the tumor microenvironment of young, middle‐aged and old patients diagnosed with early invasive luminal (hormone‐sensitive, HER2‐negative) breast cancer. In the old group, G8‐scores were computed as a correlate for clinical frailty.
Results
Significant age‐related changes in plasma levels of several inflammatory mediators (IL‐1α, IP‐10, IL‐8, MCP‐1, CRP), immune checkpoint markers (Gal‐9, sCD25, TIM‐3, PD‐L1), IGF‐1 and circulating miRs (miR‐18a, miR‐19b, miR‐20, miR‐155, miR‐195 and miR‐326) were observed. Shifts were observed in distinct peripheral blood mononuclear cell populations, particularly naive CD8+ T‐cells. At the tumor level, aging was associated with lower total lymphocytic infiltration, together with decreased abundance of several immune cell markers, especially CD8. The relative fractions of cell subsets in the immune infiltrate were also altered. Clinical frailty was associated with higher frequencies of exhausted/senescent (CD27−CD28− and/or CD57+) terminally differentiated CD8+ cells in the blood and with increased tumor infiltration by FOXP3+ cells.
Conclusion
Aging and frailty are associated with profound changes of the blood and tumor immune profile in luminal breast cancer, pointing to a different interplay between tumor cells, immune cells and inflammatory mediators at higher age.
Keywords: ageing, biomarkers, breast cancer, clinical frailty, tumor immune infiltrate
In this study, apart from age‐related changes in the immune profile of patients with luminal breast cancer, remarkable frailty‐related changes within the subgroup of older patients were observed. Our data support age‐dependent remodelling of both systemic immunity features and anti‐tumor immune responses.

Introduction
Breast cancer (BC), like all epithelial cancers, shows increased incidence with age. As the general population is ageing, the number of older patients with BC cancer is dramatically rising. 1 Additionally, ageing might also impact BC biology: compared to younger patients, older women often develop less aggressive tumors that are mostly oestrogen receptor (ER)‐positive, lack over‐expression of human epidermal growth factor receptor‐2 (HER2) and have lower proliferation rates (luminal A‐like tumors). Contrarily, older patients often present with larger and more advanced stage tumors at diagnosis.
Lifelong exposure to endogenous and exogenous factors can induce progressive oxidative stress and DNA damage, eventually resulting in cell transformation and tumor initiation. Furthermore, ageing is associated with accumulation of metabolically active senescent cells, exhibiting a senescence‐associated secretory phenotype (SASP). SASP includes various inflammatory mediators that may promote tumor growth. Lastly, ageing induces a progressive decay of immune functioning, which may result in insufficient immune responses against a developing tumor. 2 , 3
Decreased adaptive immunity and increased low‐grade chronic inflammation in older people have been defined as ‘immunosenescence’ and ‘inflammaging’. 3 Ageing strongly affects the adaptive immune system, reflected by shifts in abundance and functioning of several adaptive immune cell subsets. Decreased numbers of naive peripheral blood T cells and B cells and increases in memory cells and regulatory T cells (Tregs) have been reported. 3 , 4 , 5 , 6 T‐cell function is impaired since T cells become anergic and T‐cell receptor diversity declines. Compared to the CD4 compartment, the CD8 compartment appears to be more affected by age. 3 , 5 , 7 Also, T‐cell p16INK4a expression has been described as a hallmark of T‐cell senescence. 8 The innate immune system is reshaped as well. Chemotaxis and phagocytosis are reduced in neutrophils and macrophages. The latter produce more inflammatory cytokines, natural killer (NK) cells produce less cytokines, and their cytolytic potential decreases. 3 , 7 , 9 In plasma, a gradual increase in pro‐inflammatory cytokines and chemokines has been observed, concomitant with a decrease in anti‐inflammatory mediators. 3 , 10 Additionally, several microRNAs (miRs) may be interesting immunosenescence markers. Over the past years, numerous miRs were reported to be involved in various immunological processes such as T‐ and B‐cell proliferation, differentiation and activation. 11 , 12 , 13 , 14
Breast cancer has long been considered as non‐immunogenic, yet many recent studies have demonstrated that the tumor immune infiltrate actually is of considerable clinical importance with respect to prognosis and outcome, most particularly for triple‐negative and HER2‐positive disease. 15 Recent publications clearly show tumor‐infiltrating lymphocytes (TILs) in luminal BC 16 ; however, the immune system’s role in hormone‐sensitive (luminal) BC is less established and impact of age has hardly been studied. Therefore, we performed an in‐depth analysis of the immunological profile, both in tumor and blood, in luminal BC patients from different age and frailty categories.
Results
Patient and tumor characteristics
Between March 2014 and November 2015, 65 patients who fulfilled the inclusion criteria were included in this study: 15 patients aged 35–45 years (young group); 19 patients aged 55–65 years (middle group) and 31 patients older than 70 years (old group). A sufficient amount of tumor tissue could be collected from 62 out of the 65 patients. In the old group, 19 patients had a ‘normal’ geriatric 8 (G8) score higher than 14 and 10 patients had a decreased G8 score that was equal to or lower than 14 (indicating increased risk for significant deficits when a full geriatric assessment is performed). Table 1 summarises the main characteristics of patients and tumors.
Table 1.
Patient characteristics (age, G8 score for older patients) and tumor properties (histological subtype, grade, size and lymph node involvement)
| Variable | Statistic | All | 35–45 years | 55–65 years | ≥ 70 years |
|---|---|---|---|---|---|
| Age | |||||
| N (%) | 65 | 15 | 19 | 31 | |
| Mean | 63.4 | 40.1 | 60.4 | 76.3 | |
| (Range) | (35.0; 89.0) | (35.0; 46.0) | (55.0; 65.0) | (70.0; 89.0) | |
| G8 score | |||||
| N | 29 | 29 | |||
| Mean | 15.2 | 15.2 | |||
| (Range) | (12.0; 17.0) | (12.0; 17.0) | |||
| Tumor histological Subtype | |||||
| Ductal (IDA) | n/N (%) | 54/65 (83.1) | 15/15 (100.0) | 13/19 (68.4) | 26/31 (83.9) |
| Lobular (ILA) | n/N (%) | 5/65 (7.7) | 0/15 (0.0) | 3/19 (15.8) | 2/31 (6.5) |
| Mixed ILA‐IDA | n/N (%) | 2/65 (3.1) | 0/15 (0.0) | 1/19 (5.3) | 1/31 (3.2) |
| Invasive solid papillary | n/N (%) | 2/65 (3.1) | 0/15 (0.0) | 1/19 (5.3) | 1/31 (3.2) |
| Micro‐papillary | n/N (%) | 1/65 (1.5) | 0/15 (0.0) | 0/19 (0.0) | 1/31 (3.2) |
| Mixed micro‐papillary and mucinous | n/N (%) | 1/65 (1.5) | 0/15 (0.0) | 1/19 (5.3) | 0/31 (0.0) |
| Tumor Grade | |||||
| Grade I | n/N (%) | 1/65 (0.02) | 0/15 (0.0) | 0/19 (0.0) | 1/31 (0.03) |
| Grade II | n/N (%) | 40/65 (61.5) | 9/15 (60.0) | 10/19 (52.6) | 21/31 (67.7) |
| Grade III | n/N (%) | 24/65 (36.9) | 6/15 (40.0) | 9/19 (47.4) | 9/31 (29.0) |
| Tumor size (mm) | |||||
| N | 65 | 15 | 19 | 31 | |
| Mean | 31.8 | 27.7 | 31.4 | 34.0 | |
| (Range) | (10.0; 115.0) | (10.0; 60.0) | (15.0; 60.0) | (12.0; 115.0) | |
| Node status | |||||
| pN0 | n/N (%) | 32/65 (49.2) | 6/15 (40.0) | 9/19 (47.4) | 17/31 (54.8) |
| pN1 | n/N (%) | 29/65 (44.6) | 8/15 (53.3) | 9/19 (47.4) | 12/31 (38.7) |
| pN2 | n/N (%) | 3/65 (4.6) | 1/15 (6.7) | 1/19 (5.3) | 1/31 (3.2) |
| pN3 | n/N (%) | 1/65 (1.5) | 0/15 (0.0) | 0/19 (0.0) | 1/31 (3.2) |
The inclusion criteria were based on clinical estimate of the tumor size and on the grading based on the core needle biopsy. Enough tumor material could be collected for 62 out of the 65 patients. The table reports pathological tumor size and tumor grade measured on the resection specimen after surgery, explaining a few discrepancies between selection criteria and results on the surgical specimen reported here. For two patients in the old group, the G8 scores were not available.
This exploratory study generated enormous amounts of data. Only the most striking observations are presented in figures throughout this paper, while full data are available in Supplementary tables [Link], [Link], [Link], [Link], [Link], [Link], [Link].
Analysis of immune/senescence markers in blood and association with age
In the blood of the patients, multiple significant differences were observed between the three age groups, as summarised in Figure 1 and Supplementary table 1. Regarding PBMC subset profiling, no major differences between the age groups were noted among the principal cell populations (i.e. T cells, B cells, NK cells, dendritic cells, stem cells), except for a significant increase in the proportion of CD14highCD16+ intermediate monocytes (P = 0.019) with increasing age (Figure 1, Supplementary table 1). When looking deeper into T‐cell subsets, CD4+ subpopulations did not show any significant age‐related changes (Figure 1). Conversely, within the CD8+ pool, a significant decrease in cells expressing the costimulatory receptor CD27 (P = 0.036) was observed in the older age group. Moreover, the naive CD8+ compartment (i.e. CD8+ cells expressing both CD45RA and CD197/CCR7 and mostly CD28/CD27) showed to be highly affected by age: significant decreases were found not only for the total population of naive CD8+ cells (P = 0.035), but also for subsets of naive CD8+ cells expressing either CD27 (P = 0.018), CD28 (P = 0.031) or both (P = 0.021) (Supplementary table 1, Figure 1). With increasing age, significantly elevated plasma levels of several inflammatory cytokines [interleukin (IL)‐1α, P < 0.001) and chemokines (IP‐10, P < 0.001; IL‐8, P = 0.011 and monocyte chemoattractant protein 1 (MCP‐1), P = 0.001] were observed. Noteworthy, in our cohort, plasma levels of IL‐6 increased with ageing; however, this increase did not reach statistical significance. Moreover, no significant changes were observed in tumor necrosis factor alpha (TNFα) plasma levels (Supplementary table 1). Higher plasma levels at more advanced ages were also noted for different soluble mediators, the immune checkpoint markers galectin‐9 (Gal‐9) (P < 0.001), soluble CD25 (sCD25) (P = 0.008) and T‐cell immunoglobulin and mucin domain 3 (TIM‐3) (P = 0.017), as well as for the aspecific inflammation parameter C‐reactive protein (CRP) (P = 0.030). Contrary, significant age‐related decreases were seen for the inflammatory cytokine IL‐17A (P = 0.012), immune checkpoint marker 4‐1BB (P = 0.025) and programmed death‐ligand 1 (PD‐L1) (P = 0.008) and, most prominently, for the insulin‐like growth factor‐1 (IGF‐1) (P = 0.002). We noticed that, however, age‐related alterations did not always follow similar dynamics in function of time: whereas some markers progressively increased/decreased over the age groups (e.g. interferon gamma‐induced protein 10 (IP‐10), Gal‐9, CRP, IGF‐1), others reached a maximum/minimum in the middle age group and then remained more or less stable (e.g. IL‐8, MCP‐1, 4‐1BB) or even slightly dropped/raised again (e.g. sCD25, TIM‐3, PD‐L1). A significant increase of T‐cell p16INK4a expression with age (P = 0.014), especially in the oldest group, was also observed in our cohort. However, this marker could only be measured in 66% of patients because of the low RNA yield from the purified T cells. Finally, we also studied the evolution of the plasma miR profile in function of age. Six miRs showed significantly different plasma expression levels between the age groups: miR‐18a (P < 0.001) was decreased in the oldest age group, while miR‐19b (P < 0.001), miR‐20a (P = 0.002) and miR‐195 (P = 0.034) first peaked in the 55–65 years group but then dropped again in the ≥70 years group. A marked progressive increase with age was seen for miR‐155 (P < 0.001). Noteworthy, miR‐326 could only be measured in the majority (77%) of patients from the ≥ 70 years group, while being totally undetectable in young and middle‐aged patients (Figure 1).
Figure 1.

(a) PBMC subsets with significant age‐related variations are shown, as well as the important CD4 and CD8 T‐cell subpopulations (Naive, CM, EM, TEMRA) together with CD27/CD28 and CD57 expression on CD4+ and CD8+ T cells. (b) Blood immune/senescence plasma markers that showed significant difference between the three different age categories. Plasma markers measured via multiplex cytometric bead array technology (plasma cytokines, chemokines and immune checkpoint markers) or ELISA (IGF‐1) were run in duplicate. T‐cell p16INK4a and miRs expression were measured via RT‐PCR in triplicate. The 35–45 years category (N = 15) is represented with Y, the 55–65 years category (N = 19) is represented with M, and the ≥ 70 years (N = 31) category is represented with O. The median and IQR are shown in grey. The level of significance is represented with an *, with *: P ≤ 0.05; **: P ≤ 0.01 and ***: P ≤ 0.001. The P‐values were calculated via the Kruskal–Wallis test.
Characterisation of the tumor immune infiltrate in the different age groups
For each immune cell marker, full data, including proportion and density of positively stained cells in different tumor zones (invasive front, tumor centre and whole tumor), are summarised per age category in Supplementary table 2 and in Figure 2 with representative microphotographs of each marker. Stromal tumor‐infiltrating lymphocytes (sTILs) % ranged between 0% and 82%. Yet, only 5 tumors showed ≥ 40% sTILs. The predominant TIL subset consisted of CD3+ T cells in all patients, but CD20+ B cells also represented a considerable fraction. The proportion of CD3+ T cells seemed to expand, while the fraction of B cells decreased with increasing tumor size. We also examined the spatial distribution of the immune subtypes within the tumor and noted that, compared to the tumor centre, TILs in the invasive front were significantly enriched in CD20+, cells which was also seen by König et al., 17 while FOXP3+ cells tended to invade much deeper into the tumor core. For the markers CD3, CD4, CD5 and CD8, spatial differences were less pronounced. With increasing age, various significant changes were observed regarding the tumor immune infiltrate. sTILs % (P = 0.025) decreased with increasing age; tumors from patients of the middle‐aged group surprisingly exhibited the lowest degree of lymphocytic infiltration (Figure 2). The pie charts (Figure 2) show the % of tumors with a low, intermediate, or high CD3 and CD8 infiltration in the different age groups, revealing a striking decrease of highly infiltrated tumors (both for CD3 and CD8) with increasing age. Comprehensive results for the particular cell subsets (i.e. proportion and density of positive cells in distinct tumor regions for each age group) are displayed in the bubble chart (Figure 2). The density of cells staining positive for the T‐cell markers CD3 and CD5 decreased in all tumor regions with increasing age [CD3: tumor centre (P = 0.007), invasive front (P = 0.019) and whole tumor (P = 0.005); CD5: tumor centre (P = 0.022), invasive front (P = 0.006) and whole tumor (P = 0.006)] (Supplementary table 2, Figure 2). Likewise, an age‐related decrease in the density of the cytotoxic T‐cell marker CD8 was seen in all tumor regions (tumor centre: P = 0.002, invasive front and whole tumor: P < 0.001). The B‐cell marker CD20 was less abundant in the invasive front (P = 0.042), and whole tumor (P = 0.031) in the older age groups, although the age effect appeared to be less pronounced than for the T‐cell markers CD3, CD5 and CD8. Furthermore, the proportion of the immune infiltrate also changed with ageing: proportions of lymphocytes staining positive for CD8+ were lower in all tumor regions (all P < 0.001) at older ages. Differently, no significant age‐related changes were observed for the markers CD4, FOXP3 and CD68 (Supplementary table 2, Figure 2). Besides the immune infiltrate’s quantity and composition in the tumor regions, spatial distribution of the immune markers within the tumor area was also investigated. We compared within each age group the mean proportion of positive cells between the different tumor regions. No significant interactions were observed between the age groups and tumor regions (invasive front and tumor centre), indicating that distribution patterns of the studied immune cells within the tumor did not significantly differ between the age groups (Supplementary table 3). Irrespective of age group, several immune subtypes were unequally distributed across the tumor zones: CD20+ cells predominantly resided in the invasive front, and FOXP3+ cells represented a higher proportion of immune cells in the tumor centre (Supplementary table 3).
Figure 2.
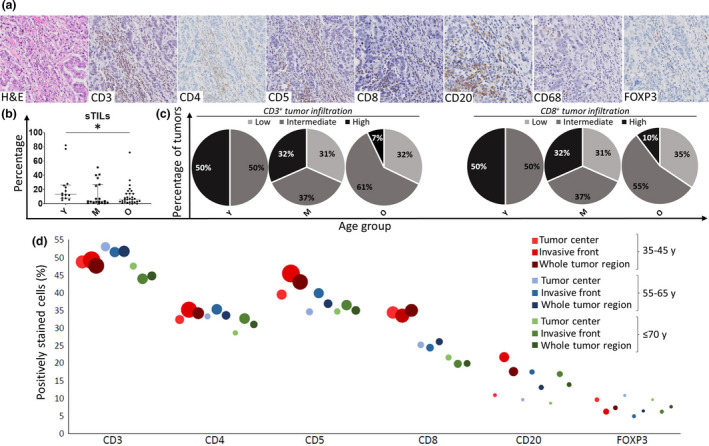
(a) Representative microphotographs of the 7 immunostainings: H&E, CD3, CD4, CD5, CD8, CD20, CD68 and FOXP3. The microphotographs were collected from the same tumor sample, which had 27.0% sTILs. (b) Stromal tumor‐infiltrating lymphocytes (sTILs) % shows a significant difference (P = 0.025) between the three different age categories: the 35–45 years ‘young’ (Y) category (N = 14), the 55–65 years ‘middle’ (M) category (N = 19) and the ≥ 70 years ‘old’ (O) category (N = 29). The median and IQR are shown. (c) The percentage of tumors with a low, intermediate or high CD3 and CD8 infiltration in the different age groups. The cut‐offs were based on the IQR of the density of CD3+ and CD8+ cells in the whole cohort. Low infiltration: minimum – lower quartile (lowest 25%), intermediate infiltration: lower quartile – upper quartile and high infiltration: upper quartile – maximum (highest 25%). (d) Tumor immune infiltration in the different tumor regions for the different age categories. The different immune cell markers (CD3, CD4, CD5, CD8, CD20 and FOXP3) are denoted on the x‐axis. Positively stained immune cells (%) can be found on the y‐axis. The area of the bubbles represents the density (absolute number per mm2) of the marker in that specific region. The 35–45 years age category is represented in red, the 55–65 years category in blue and the ≥ 70 years category in green. The tumor centre, invasive front and the whole tumor region are represented by increasing colour intensities. P‐values were calculated via the Kruskal–Wallis test.
Association of blood immune/senescence markers with G8 score
Next, we compared the blood immune/senescence profile between the G8 subgroups (i.e. G8‐normal and G8‐decreased) of the ≥ 70 years age category, as summarised in Supplementary table 4, mean age of the groups was comparable. While none of the plasma protein biomarkers (including IL‐6, TNFα and CRP) nor miRs differed significantly between both G8 subgroups, the PBMC subset profiles revealed multiple significant G8‐dependent differences (Figure 3). Firstly, fractions of total CD8+ cells expressing CD27 or CD28 were significantly lower in more frail patients (P = 0.044 and P = 0.016, respectively) as compared to fit patients. The same also applied to double‐positive CD27+CD28+ cells (P = 0.029), whereas double‐negative CD27−CD28− cells (mostly terminally differentiated cells) were concurrently increased within the CD8+ compartment of frailer patients (P = 0.013). Similar observations were made when looking specifically into the terminally differentiated effector memory re‐expressing CD45RA (TEMRA) CD8+ subpopulation: the fraction of cells co‐expressing CD27 and CD28 was again lower in the G8‐decreased than in the G8‐normal subgroup (P = 0.044), while, accordingly, the proportion of cells lacking both costimulatory receptors was higher in the frailer group (P = 0.013). Moreover, the proportion of cells expressing the senescence marker CD57 was significantly higher in frailer patients, both in the total CD8+ pool (P = 0.007) and in the TEMRA CD8+ subpopulation (P = 0.005). Additionally, the frequency of a peculiar subpopulation of CD3+CD16+ cells (NK‐like T cells) was significantly higher (P < 0.001) in the subgroup of frailer patients (G8 ≤ 14). Finally, higher T‐cell p16INK4a expression (P = 0.043) was also associated with lower G8 score and thus with frailer patients.
Figure 3.
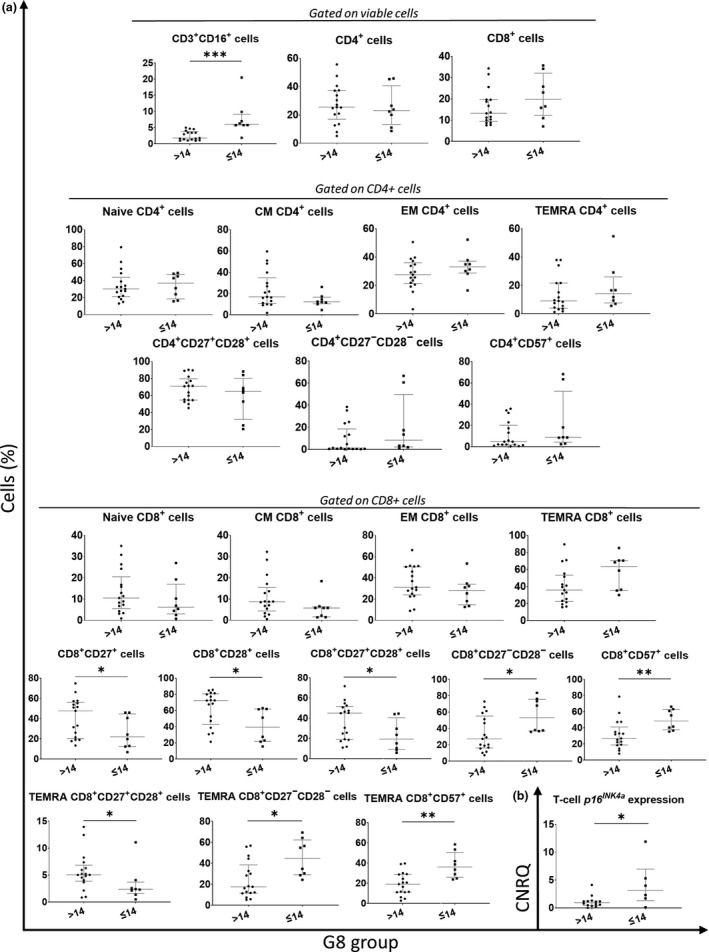
(a) PBMC subsets with significant variations between the G8 groups [G8 > 14: ‘fit’ (N = 19) and G8 ≤ 14: ‘frail’ (N = 10)] are shown, and biologically relevant CD4 and CD8 T‐cell subpopulations (Naive, CM, EM, TEMRA) together with CD27/CD28 and CD57 expression on CD4+ and CD8+ T‐cells. (b) T‐cell p16INK4a expression, showing significant difference between the two G8 groups. Plasma markers measured via multiplex cytometric bead array technology (plasma cytokines, chemokines and immune checkpoint markers) or ELISA (IGF‐1) were run in duplicate. T‐cell p16INK4a and miRNA expression were measured via RT‐PCR in triplicate. The median and IQR are shown in grey. Levels of significance: *: P ≤ 0.05; **: P ≤ 0.01 and ***: P ≤ 0.001. The P‐values were calculated via the Mann–Whitney U test.
G8‐dependent changes in the tumor immune infiltrate
Within the oldest (≥ 70 years) age category, we also examined the different tumor immune markers according to G8 score (Supplementary table 5, Figure 4). Noteworthy, tumor characteristics were comparable between the two groups. Significant changes were observed between G8‐normal and G8‐decreased subgroups with regard to FOXP3+ cell infiltration. In frailer patients (G8 ≤ 14), the proportion of FOXP3+ cells was higher in the invasive front (P = 0.015) and whole tumor (P = 0.007), as compared to fit patients (G8 > 14), and the density of FOXP3+ cells was also increased in the tumor centre (P = 0.037). Thus, breast tumors in frailer patients appeared to be more heavily infiltrated by immunosuppressive FOXP3+ cells. sTILs %, CD68 grade and infiltration of the other markers were not associated with the G8 score. Again, no significant interactions were observed between G8 groups and tumor regions (invasive front and tumor centre), indicating that spatial distribution of the studied immune cells within the tumor did not differ between the G8 groups (Supplementary table 3).
Figure 4.
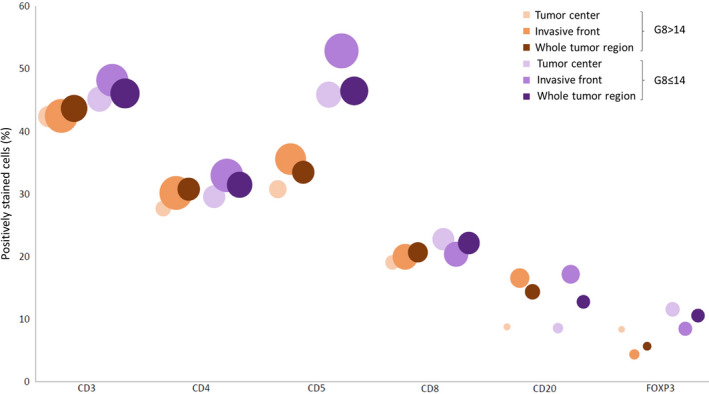
Bubble chart: tumor immune infiltration in the different tumor regions in the different G8 groups [G8 > 14: ‘fit’ (N = 19) and G8 ≤ 14: ‘frail’ (N = 10)]. The different immune cell markers (CD3, CD4, CD5, CD8, CD20 and FOXP3) are represented on the x‐axis. Positively stained immune cells (%) can be found on the y‐axis. The area of the bubbles represent the density of each marker in a specific region. The tumor centre is represented by the lightest shade, invasive front by the second lightest shade and the whole tumor region by the darkest colour. The G8 > 14 is represented in orange, the G8 ≤ 14 in purple.
Correlations between blood immune/senescence markers and tumor immune infiltrate
We also performed a detailed investigation of the relationships among and between blood and tumor immune biomarkers (Supplementary table 6). Firstly, sTILs % inversely correlated with the pro‐inflammatory plasma markers IL‐1α, IL‐6 and MCP‐1, the plasma immune checkpoint markers TIM‐3 and Gal‐9, with CRP and also with circulating Tregs. The strong negative correlation of MCP‐1 with global sTILs % was further substantiated by its inverse relationship with whole tumor density of CD3+, CD5+, CD8+ and FOXP3+ cells. The immune checkpoint marker TIM‐3, in addition to its negative association with sTILs %, also inversely correlated with CD68 staining grade and with whole tumor density of CD8+ and CD20+ cells, while IL‐1α only showed a significant inverse association with CD8. Secondly, several correlations were noted between specific tumor immune markers and PBMC subpopulations. Remarkably, circulating class‐switched memory B cells (CD27+IgD−) showed a strong inverse correlation with CD68 staining grade, while intermediate monocytes (CD14++CD16+) negatively correlated with CD3+ and CD5+ cell density in the tumor. Also, the density of CD8+ cells in the whole tumor was inversely correlated with the % Tregs in blood. CD8+ TEMRA cells expressing CD27 and/or CD28 showed multiple positive correlations with tumor infiltration by CD3+, CD4+, CD5+, CD8+ and CD20+ cells, whereas senescent CD8+ TEMRA (lacking both CD27 and CD28 and/or expressing CD57) clearly exhibited a positive association with infiltration of FOXP3+ cells in the tumor. Similar observations, albeit somewhat less pronounced, were made for the corresponding CD4+ TEMRA subsets. Lastly, several immune‐related circulating miRs also showed diverse correlations with different tumor immune infiltration markers. miR‐17 and especially miR‐19b appeared to strongly correlate with the proportion of CD3+ cells in the tumor infiltrate, while miR‐195 consistently showed positive associations with both proportion and density of CD3+, CD4+, CD5+, CD8+ and FOXP3+ cells, but not CD20+ cells. Other correlations between the blood immune/senescence markers and tumor immune infiltrate markers are not further discussed here because statistical significance was only moderate and biological relevance uncertain (Supplementary table 6). Additionally, solid correlations were found between breast tumor biology parameters (size, grade and nodal status) and the immune profile, mainly at tumor level (Supplementary results and Supplementary table 7).
Age and G8 group prediction using a panel of blood and tumor biomarkers
Additionally, we examined the ability of the blood immune/senescence and tumor immune infiltrate markers to predict a patient’s age and G8 group. For both age and G8, panels of 10 and 5 biomarkers were selected based on their individual statistical performance and biological relevance. The 50 statistically highest ranked blood immune/senescence and tumor immune infiltrate markers can be found in Table 2. The 10‐biomarker panel for age groups consisted of the blood immune/senescence markers: miR‐326, miR‐155, p16INK4a, IL‐17A, Gal‐9, IP‐10, IGF‐1 and naive CD8+CD27+CD28+ cells in combination with the tumor immune infiltrate markers: whole tumor density of CD8+ cells and tumor centre density of CD3+ cells. Using this 10‐biomarker panel, the old group could be separated quite well from the two younger groups by either dimensionality reduction tool (uMap, PCA or t‐SNE) (Figure 5). Moreover, the uMap shows a distinct cluster of older patients while a smaller group of older patients mix with the younger patients. Interestingly, all ‘frailer’ patients (G8 ≤ 14) except for one were contained within the distinct cluster of older patients and this observation was statistically significant (P = 0.018). For the G8 groups, the 10‐biomarker panel included the blood immune/senescence markers: miR‐9, p16INK4a, Gal‐9, CD3+CD16+, CD8+CD57+, CD8+CD27−CD28−, CD4+CD27−CD28− cells and CD4+CD57+ cells together with the tumor immune infiltrate markers: whole tumor proportion and tumor centre density of FOXP3+ cells. The ‘frailer’ older patients (G8 ≤ 14) could be separated relatively well from the ‘fit’ older patients (G8 > 14) using the 10‐biomarker panel (Figure 5). The uMap clearly shows that most of the ‘fit’ patients are separated from the ‘frailer’ patients, while some of the ‘fit’ patients mix with the ‘frailer’ patients. Furthermore, we checked if the same or better separation could be achieved using 5‐biomarker panels. These consisted of miR‐326, miR‐155, p16INK4a, Gal‐9 and IP‐10 for age and p16INK4a, CD3+CD16+, CD8+CD57+, CD28+CD27−CD28− and whole tumor proportion of FOXP3+ cells for the G8 groups. For both age and G8, the 5‐biomarker panel displayed an even better performance compared to the 10‐biomaker panel in the uMap, while performing comparably well in t‐SNE and PCA (Figure 5). Moreover, for age, a distinct cluster with most of the frailer patients could be observed with the 5‐biomarker panel.
Table 2.
The 50 statistically highest ranked blood immune/senescence and tumor immune infiltrate markers to classify the patients in the age groups and the G8 groups
| Position | Blood immune/senescence and tumor immune infiltrate markers | N | AUC | P‐value | log FC | AUC score | P‐value score | log FC score | Final score |
|---|---|---|---|---|---|---|---|---|---|
| Age | |||||||||
| 1 | miR‐326 | 65 | 0.110 | < 0.001 | 10.00 | 4 | 1 | 1 | 3 |
| 2 | p16INK4a | 43 | 0.240 | 0.004 | 1.87 | 44 | 44 | 51 | 46 |
| 3 | miR‐155 | 65 | 0.240 | < 0.001 | 1.01 | 43 | 15 | 193 | 74 |
| 4 | CD8+ cells – Whole tumor – Density | 62 | 0.710 | 0.004 | −1.45 | 129 | 45 | 109 | 103 |
| 5 | miR‐18a | 65 | 0.780 | < 0.001 | −0.63 | 27 | 7 | 394 | 114 |
| 6 | miR‐19b | 65 | 0.780 | < 0.001 | −0.62 | 28 | 9 | 407 | 118 |
| 7 | CD3+ cells – Tumor centre – Density | 61 | 0.700 | 0.007 | −1.49 | 161 | 60 | 99 | 120 |
| 8 | IL‐17A | 65 | 0.690 | 0.010 | −2.19 | 192 | 85 | 37 | 127 |
| 9 | Gal‐9 | 65 | 0.240 | < 0.001 | 0.61 | 42 | 14 | 419 | 129 |
| 10 | CD8+ cells – Tumor centre – Density | 62 | 0.690 | 0.010 | −1.59 | 196 | 86 | 74 | 138 |
| 11 | PBMC – Naive CD8+CD27+ | 57 | 0.710 | 0.007 | −0.85 | 127 | 66 | 253 | 143 |
| 12 | CD3+ cells – Whole tumor – Density | 61 | 0.690 | 0.011 | −1.29 | 195 | 101 | 142 | 158 |
| 13 | PBMC – Naive CD8+CD27+CD28+ | 57 | 0.700 | 0.008 | −0.84 | 160 | 72 | 256 | 162 |
| 14 | CD8+ cells – Invasive front – Proportion | 62 | 0.760 | < 0.001 | −0.48 | 45 | 19 | 577 | 172 |
| 15 | CD8+ cells – Invasive front – Density | 62 | 0.680 | 0.013 | −1.21 | 216 | 112 | 152 | 174 |
| 16 | CD8+ cells – Whole tumor – Proportion | 62 | 0.760 | 0.001 | −0.46 | 46 | 23 | 612 | 182 |
| 17 | IP‐10 | 65 | 0.240 | < 0.001 | 0.43 | 41 | 20 | 659 | 190 |
| 18 | PBMC – Naive CD8+CD28+ | 57 | 0.690 | 0.013 | −0.78 | 190 | 110 | 289 | 195 |
| 19 | PBMC – Naive CD8+ | 57 | 0.690 | 0.013 | −0.74 | 191 | 106 | 320 | 202 |
| 20 | CD5+ cells – Whole tumor – Density | 62 | 0.670 | 0.023 | −1.18 | 264 | 149 | 156 | 208 |
| 21 | CD5+ cells – Tumor centre – Density | 62 | 0.660 | 0.033 | −1.23 | 309 | 177 | 149 | 236 |
| 22 | CD8+ cells – Tumor centre – Proportion | 62 | 0.720 | 0.003 | −0.40 | 101 | 38 | 734 | 244 |
| 23 | CD5+ cells – Invasive front – Density | 62 | 0.660 | 0.036 | −1.00 | 308 | 197 | 198 | 253 |
| 24 | 4‐1BB | 65 | 0.640 | 0.019 | −2.42 | 427 | 134 | 31 | 255 |
| 25 | PBMC – CD8+CD27+ | 57 | 0.700 | 0.010 | −0.46 | 159 | 89 | 613 | 255 |
| 26 | miR‐195 | 65 | 0.670 | 0.021 | −0.64 | 262 | 138 | 384 | 262 |
| 27 | IGF‐1 | 65 | 0.710 | 0.003 | −0.37 | 128 | 37 | 805 | 275 |
| 28 | miR‐9 | 65 | 0.640 | 0.022 | −1.36 | 430 | 141 | 126 | 282 |
| 29 | CD3+ cell – Invasive front – Density | 61 | 0.650 | 0.043 | −1.01 | 359 | 216 | 194 | 282 |
| 30 | PBMC – CD8+CD27+CD28+ | 57 | 0.680 | 0.020 | −0.44 | 214 | 135 | 649 | 303 |
| 31 | CD20+ cells – Tumor centre – Density | 62 | 0.640 | 0.064 | −1.51 | 431 | 277 | 92 | 308 |
| 32 | CD20+ cells – Whole tumor – Density | 62 | 0.640 | 0.060 | −1.39 | 432 | 269 | 120 | 313 |
| 33 | PBMC – CD56brightCD16− NK cells | 57 | 0.660 | 0.040 | −0.60 | 306 | 208 | 438 | 315 |
| 34 | CD4+ cells – Whole tumor – Density | 61 | 0.640 | 0.055 | −1.07 | 434 | 249 | 179 | 324 |
| 35 | FOXP3+ cells – Tumor centre – Density | 61 | 0.640 | 0.066 | −0.97 | 435 | 287 | 205 | 341 |
| 36 | CD4+ cells – Tumor centre – Density | 61 | 0.630 | 0.087 | −1.19 | 523 | 358 | 155 | 390 |
| 37 | CD3+ cells – Invasive front – Proportion | 61 | 0.690 | 0.010 | −0.21 | 193 | 93 | 1371 | 463 |
| 38 | miR‐19a | 65 | 0.640 | 0.049 | −0.39 | 429 | 235 | 759 | 463 |
| 39 | miR‐20a | 65 | 0.660 | 0.029 | −0.28 | 307 | 163 | 1081 | 465 |
| 40 | CD20+ cells – Invasive front – Density | 62 | 0.620 | 0.121 | −0.94 | 624 | 481 | 212 | 485 |
| 41 | miR‐125b | 65 | 0.640 | 0.050 | −0.35 | 428 | 237 | 864 | 489 |
| 42 | CD3+ cells – Whole tumor – Proportion | 61 | 0.680 | 0.018 | −0.20 | 215 | 131 | 1425 | 497 |
| 43 | FOXP3+ cells – Whole tumor – Density | 61 | 0.620 | 0.114 | −0.73 | 626 | 458 | 326 | 509 |
| 44 | PBMC – EM CD8+CD27+ | 57 | 0.650 | 0.058 | −0.28 | 356 | 257 | 1082 | 513 |
| 45 | PBMC – Intermediate monocytes | 57 | 0.350 | 0.059 | 0.28 | 358 | 262 | 1083 | 515 |
| 46 | Total pDC | 57 | 0.640 | 0.079 | −0.25 | 425 | 333 | 1191 | 594 |
| 47 | let‐7e | 65 | 0.370 | 0.077 | 0.30 | 520 | 331 | 1016 | 597 |
| 48 | PBMC – TEMRA CD8+CD27−CD28− | 57 | 0.380 | 0.124 | 0.43 | 622 | 499 | 666 | 602 |
| 49 | PBMC – CD8+CD27−CD28− | 57 | 0.380 | 0.119 | 0.39 | 621 | 472 | 762 | 619 |
| 50 | miR‐21 | 65 | 0.620 | 0.095 | −0.33 | 623 | 389 | 917 | 638 |
| G8 | |||||||||
| 1 | PBMC – CD3+CD16+ | 25 | 0.940 | < 0.001 | −1.62 | 1 | 10 | 67 | 20 |
| 2 | PBMC – TEMRA CD8+CD57+ | 25 | 0.860 | 0.003 | −0.98 | 5 | 40 | 202 | 63 |
| 3 | p16INK4a | 20 | 0.800 | 0.041 | −1.94 | 21 | 211 | 47 | 75 |
| 4 | FOXP3+ cells – Whole tumor – Proportion | 27 | 0.830 | 0.005 | −0.80 | 11 | 50 | 274 | 87 |
| 5 | PBMC – TEMRA CD8+CD27−CD28− | 25 | 0.820 | 0.011 | −0.90 | 16 | 98 | 225 | 89 |
| 6 | PBMC – CD8+CD57+ | 25 | 0.850 | 0.005 | −0.67 | 7 | 49 | 356 | 105 |
| 7 | FOXP3+ cells – Invasive front – Proportion | 27 | 0.800 | 0.012 | −0.78 | 22 | 105 | 285 | 109 |
| 8 | PBMC – CD8+CD27−CD28− | 25 | 0.820 | 0.011 | −0.75 | 15 | 99 | 310 | 110 |
| 9 | FOXP3+ cells – Tumor centre – Density | 27 | 0.750 | 0.035 | −1.24 | 53 | 191 | 148 | 111 |
| 10 | PBMC – CD8+CD28+ | 25 | 0.190 | 0.013 | 0.63 | 20 | 109 | 393 | 136 |
| 11 | PBMC – CD8+CD27+CD28+ | 25 | 0.220 | 0.027 | 0.71 | 29 | 153 | 331 | 136 |
| 12 | PBMC – EM CD4+CD57+ | 25 | 0.740 | 0.057 | −1.07 | 70 | 255 | 176 | 143 |
| 13 | PBMC – TEMRA CD8+CD27+CD28+ | 25 | 0.240 | 0.044 | 0.77 | 48 | 221 | 295 | 153 |
| 14 | PBMC – TEMRA CD4+CD57+ | 25 | 0.730 | 0.076 | −1.20 | 82 | 321 | 154 | 160 |
| 15 | PBMC – CD8+CD27+ | 25 | 0.240 | 0.044 | 0.65 | 47 | 222 | 373 | 172 |
| 16 | PBMC – CD4+CD27−CD28− | 25 | 0.720 | 0.086 | −1.17 | 102 | 350 | 157 | 178 |
| 17 | PBMC – CM CD8+ | 25 | 0.280 | 0.086 | 0.90 | 103 | 351 | 228 | 196 |
| 18 | FOXP3+ cells – Tumor centre – Proportion | 27 | 0.730 | 0.060 | −0.66 | 84 | 263 | 363 | 199 |
| 19 | Gal‐9 | 29 | 0.720 | 0.062 | −0.66 | 104 | 275 | 365 | 212 |
| 20 | PBMC – CM CD8+CD27+ | 25 | 0.290 | 0.098 | 0.91 | 118 | 394 | 220 | 213 |
| 21 | PBMC – CD4+CD57+ | 25 | 0.710 | 0.110 | −1.08 | 130 | 438 | 174 | 218 |
| 22 | PBMC – CM CD8+CD28+ | 25 | 0.290 | 0.109 | 0.89 | 119 | 433 | 232 | 226 |
| 23 | miR‐9 | 29 | 0.340 | 0.056 | 10.00 | 332 | 252 | 4 | 230 |
| 24 | PBMC – TEMRA CD8+CD27+ | 25 | 0.260 | 0.057 | 0.51 | 71 | 256 | 535 | 233 |
| 25 | CD5+ cells – Tumor centre – Density | 27 | 0.720 | 0.076 | −0.62 | 106 | 322 | 408 | 236 |
| 26 | PBMC – Total Treg cells | 25 | 0.290 | 0.098 | 0.73 | 117 | 395 | 323 | 238 |
| 27 | PBMC – Naive CD8+CD27−CD28− | 25 | 0.710 | 0.097 | −0.76 | 131 | 393 | 303 | 240 |
| 28 | PBMC – CM CD8+CD27+CD28+ | 25 | 0.300 | 0.124 | 0.88 | 149 | 497 | 240 | 259 |
| 29 | PBMC – CM CD4+CD27+ | 25 | 0.300 | 0.124 | 0.76 | 148 | 498 | 304 | 275 |
| 30 | FOXP3+ cells – Whole tumor – Density | 27 | 0.690 | 0.131 | −0.96 | 199 | 525 | 209 | 283 |
| 31 | PBMC – CD56brightCD16− NK cells | 25 | 0.300 | 0.123 | 0.69 | 150 | 493 | 345 | 285 |
| 32 | PBMC – EM CD4+CD27−CD28− | 25 | 0.680 | 0.153 | −1.25 | 217 | 578 | 147 | 290 |
| 33 | sCD25 | 29 | 0.710 | 0.069 | −0.45 | 133 | 300 | 628 | 299 |
| 34 | PBMC – TEMRA CD4+CD27−CD28− | 25 | 0.680 | 0.157 | −1.09 | 218 | 590 | 173 | 300 |
| 35 | PBMC – TEMRA CD8+ | 25 | 0.710 | 0.098 | −0.50 | 132 | 396 | 545 | 301 |
| 36 | PBMC – CM CD4+CD27+CD28+ | 25 | 0.310 | 0.140 | 0.75 | 176 | 548 | 312 | 303 |
| 37 | miR‐195 | 29 | 0.710 | 0.069 | −0.43 | 134 | 301 | 660 | 307 |
| 38 | CD5+ cells – Tumor centre – Proportion | 27 | 0.730 | 0.060 | −0.37 | 83 | 264 | 803 | 308 |
| 39 | miR‐20a | 29 | 0.720 | 0.056 | −0.36 | 105 | 253 | 830 | 323 |
| 40 | FOXP3+ cells – Invasive front – Density | 27 | 0.670 | 0.176 | −0.94 | 267 | 645 | 211 | 348 |
| 41 | miR‐17 | 29 | 0.680 | 0.130 | −0.49 | 220 | 521 | 560 | 380 |
| 42 | PBMC – Naive CD8+CD28+ | 25 | 0.320 | 0.175 | 0.63 | 246 | 637 | 400 | 382 |
| 43 | PBMC – Naive CD8+CD27+CD28+ | 25 | 0.320 | 0.175 | 0.61 | 244 | 638 | 421 | 387 |
| 44 | PBMC – CM CD4+CD28+ | 25 | 0.330 | 0.194 | 0.81 | 289 | 701 | 269 | 387 |
| 45 | PBMC – Naive CD8+CD27+ | 25 | 0.320 | 0.175 | 0.59 | 245 | 639 | 448 | 394 |
| 46 | PBMC – TEMRA CD8+CD28+ | 25 | 0.320 | 0.175 | 0.54 | 247 | 640 | 495 | 407 |
| 47 | PBMC – CM CD4+ | 25 | 0.340 | 0.215 | 0.80 | 330 | 752 | 277 | 422 |
| 48 | CD4+ cells – Tumor centre – Density | 27 | 0.680 | 0.145 | −0.42 | 222 | 560 | 693 | 424 |
| 49 | CD5+ cells – Invasive front – Proportion | 27 | 0.690 | 0.118 | −0.32 | 197 | 467 | 940 | 450 |
| 50 | miR‐223 | 29 | 0.660 | 0.176 | −0.51 | 312 | 646 | 539 | 452 |
The number of patients (N) for whom a specific marker could be assessed, the area under the curve (AUC), P‐value and log fold change (FC) are reported. Based on the statistical performance, the different markers were assigned a score for AUC, P‐value and log FC. In the final score, AUC weighted double compared to P‐value and log FC. For both age and G8, a 10‐biomarker panel (combination of the markers marked in light and dark grey) and a 5‐biomarker panel (marked in dark grey) were selected based on the statistical performance of a marker together with its biological relevance.
Figure 5.
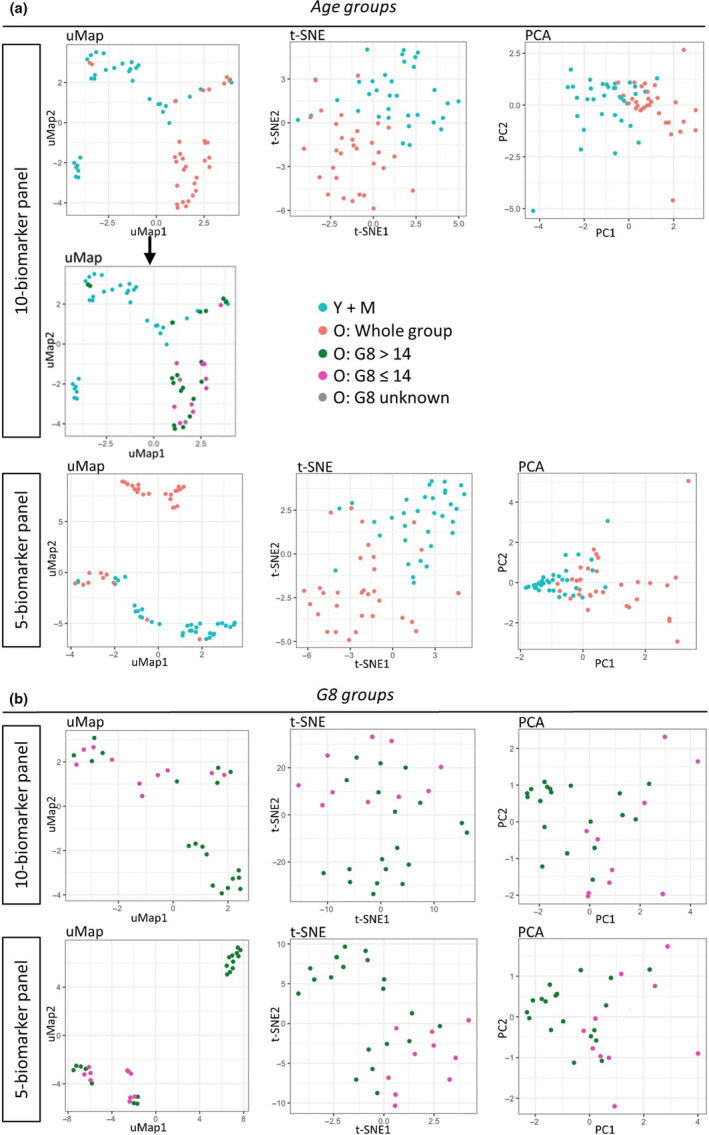
Dimensionality reduction plots. (a) A panel of 10 and 5 biomarkers was assembled to classify the patients in the old group (N = 31) or the younger age groups (N = 34). The 10 and 5 biomarkers were selected based on their statistical performance and biological relevance. uMap, t‐SNE and PCA projections are shown, with the younger age groups in blue (Y + M), the whole old group in red. The uMap projection of the 10‐biomarker age panel shows a distinct cluster of older patients. This cluster was studied in more detail in the plot below, showing ‘frail’ older patients (G8 ≤ 14) are shown in pink, ‘fit’ older patients (G8 > 14) in green and older patients with an unknown G8‐score in grey. (b) Another panel of 10 or 5 biomarkers was assembled to classify the patients in the ‘fit’ G8 > 14 group (N = 19) or the ‘frail’ G8 ≤ 14 group (N = 10). Again, the 10 or 5 biomarkers were selected based on their statistical performance and biological relevance. On the uMap, t‐SNE and PCA projections the ‘fit’ patients (G8 > 14) are shown in green and the ‘frail’ patients (G8 ≤ 14) in pink.
Discussion
In this study, we have for the first time performed a comprehensive analysis of the immune/senescence profile in blood, together with a detailed characterisation of the tumor immune infiltrate in hormone‐sensitive BC in relation to the patient’s age and clinical frailty. Our main findings regarding the observed biomarker changes in function of ageing and frailty are basically summarised in Figure 6.
Figure 6.
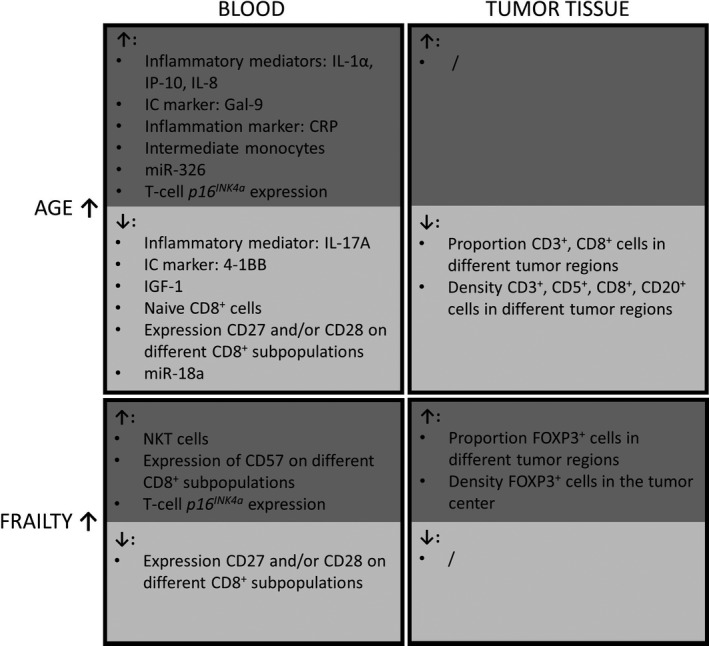
An overview of increases (↑) and decreases (↓) of the blood immune/senescence markers and the tumor immune infiltrate markers according to age and increasing frailty level. Notably, for some markers (not shown in this figure), the highest levels were present in the middle age category (e.g. blood levels of MCP‐1, sCD25, TIM‐3, miR‐19b, miR‐20a, miR‐195), while for other markers, lowest levels were present in the middle age category (PD‐L1 in blood, and sTILs % in tumor). IC, Immune checkpoint.
Overall, we observed huge variations in the degree of tumor immune infiltration in our cohort of patients with luminal breast cancer; yet, only a minority of tumors (8%) showed strong infiltration (≥ 40% sTILs). This indicates that luminal tumors are indeed less immunogenic than HER2‐positive or TNBC. 16 , 18 Interestingly, tumors of higher grade appeared to be more heavily infiltrated by immune cells, which has previously been reported by Smid et al., 19 and increasing tumor size was associated with shifts in TILs composition. Notably, nodal status (i.e. positive versus negative) was not correlated with any of the peripheral nor tumor immune biomarkers, suggesting that the impact of immune status/immunosenescence on lymph node metastasis may be limited for luminal BC.
When comparing the different age groups, our data revealed a decreased degree of lymphocytic infiltration in older patients and an altered immune infiltrate composition, with a markedly lower contribution of CD8+ cells. Within the oldest age group, we also investigated whether patient’s clinical fitness/frailty status, assessed by G8 score, had an impact on the tumor immune response. We observed a relatively more immunosuppressive FOXP3+ immune milieu, which could possibly suggest less effective immune response, in frail patients.
We also evaluated a comprehensive panel of immune/senescence markers in blood, in order (i) to investigate the impact of ageing on systemic immunity features (immunosenescence) in patients with luminal BC and (ii) to explore connections between tumor and blood immune profiles. Significant age‐related increases were noted for several plasma inflammatory cytokines (IL‐1α) and chemokines (IP‐10, IL‐8 and MCP‐1), which all play an eminent role in the migration, recruitment and activation of specific immune subsets. 20 , 21 , 22 , 23 , 24 These observations are consistent with earlier studies reporting on a gradually increasing chronic, low‐grade inflammation in the old aged. 3 , 21 , 22 , 25 , 26 , 27 Despite their well‐established role in lymphocyte and macrophage migration, elevated plasma levels of the 3 chemokines in the older patients of our cohort were not associated with increased immune infiltration in the tumor; actually, the opposite was true: MCP‐1 showed a striking inverse correlation with sTILs %, more specifically with T‐cell subsets expressing CD3, CD5, CD8 and FOXP3. It would be interesting to study intratumoral levels of MCP‐1 and other chemokines in function of age, which might be more relevant than circulating chemokines. In the context of ageing and frailty, IL‐6 is one of the most described pro‐inflammatory cytokines, involved in acute as well as chronic inflammation. 23 , 25 , 26 , 27 , 28 IL‐6 indeed showed a trend towards an age‐related increase in our cohort, although this association did not reach statistical significance. The clinical inflammation biomarker CRP, on the other hand, did significantly rise with ageing in our study. Both IL‐6 and CRP have consistently been reported to be strongly correlated with various age‐related diseases and are considered valuable predictors of physical and cognitive decline. 28 , 29 Furthermore, a significant age‐related decrease was seen for plasma IGF‐1, in accordance with the notion that IGF‐1 signalling diminishes with age and may play a role in biological ageing. 30 Several immune checkpoint markers displayed altered plasma concentrations in function of ageing: Gal‐9, TIM‐3 and sCD25 levels were elevated at more advanced age, whereas age‐related decreases were seen for 4‐1BB and PD‐L1. To date, limited research has been carried out on soluble immune checkpoint markers plasma. TIM‐3 has been recognised as a T‐cell exhaustion marker, which is shed into the extracellular milieu. 31 Gal‐9 is the most important ligand of TIM‐3 32 , 33 and sCD25 is the soluble form of IL‐2Ra, a cytokine involved in activation of Tregs. 34 As T‐cell exhaustion and elevated Treg frequency/functional potential have both been associated with old age, 35 plasma accumulation of TIM‐3, Gal‐9 and sCD25 at higher age is not unexpected. Although we could not demonstrate significantly increased frequencies of circulating Tregs nor exhausted/senescent (CD27−CD28− or CD57+) T cells in the oldest patients of our cohort, we did find significant expansion of these specific cell subsets in blood of frailer patients within the oldest age category, concomitant with higher representation of FOXP3+ cells within their tumors. These findings possibly suggest that the previously reported increases in Tregs and exhausted/senescent T cells in older patients might rather be associated with progressive frailty than barely higher age.
Previous studies in the general (non‐cancer) population have shown that the cytotoxic CD8+ T‐cell population appears to be highly affected by age. 5 Our current data further support these findings. Particularly, percentages of circulating naive CD8+ T cells significantly decreased with age, probably by immunological reserves depletion because of lifelong exposure to immune challenges. Moreover, smaller fractions of naive CD8+ T cells expressing CD27, CD28 or both were present in the higher age groups. These co‐receptors are important for T‐cell survival and activation by (tumor) antigens and are thus essential for effective T‐cell responses. Loss of CD27/CD28, which has been described as a hallmark of ageing, 5 , 9 could be partially responsible for decreased tumor infiltration by CD8+ cells observed in the older patients of our cohort. Apparently, circulating immune cells of older patients may be less capable to recognise tumor antigens, become activated and infiltrate into the tumor tissue. Interestingly, activated T cells, mainly CD8+ cells, also express the costimulatory receptor 4‐1BB. Its age‐related decrease in plasma, as observed in our study, could thus also be linked to impaired CD8+ cell activation. 36
T‐cell expression of the cell cycle inhibitor p16INK4a has been proposed as a promising biomarker of ageing. 8 Our data indeed demonstrated a significant increase of T‐cell p16INK4a expression with advancing age. However, because of its low or even unmeasurable expression in a considerable number of patients, accurate evaluation of p16INK4a was technically challenging, compromising this biomarker’s utility. Finally, six immune‐related miRs showed significantly different plasma expression levels across the age groups: miR‐18a decreased with age; miR‐155 and miR‐326 increased; and miR‐19b, miR‐20a and miR‐195 peaked in the middle group. miRs play critical roles in numerous and diverse developmental events and physiological pathways (including pathological and immune‐related processes) making it hard to determine their specific role at a given moment. 11 , 37 Especially for miR‐18a, miR‐19b and miR‐20a, the results should be considered with caution. These miRs all belong to the miR‐17‐92 cluster, which is highly involved in innate and adaptive immune responses and is important for T‐cell proliferation and survival, 12 , 14 , 37 but, simultaneously, has also been implicated in cancer through its oncogenic effects. 38 , 39 Expression levels of miRs in this cluster were previously reported to be downregulated with increasing age. 40 However, since we are considering a cancer population here, observed changes of these miRs in the age groups are difficult to interpret and opposed effects of ageing (downregulation) and cancer (upregulation) may explain the observed ambiguous dynamics of their plasma expression. The pro‐inflammatory miR‐155 showed a marked increase with age, which is in line with its reported active involvement in immune function and chronic inflammation. 14 , 41 Furthermore, plasma miR‐195 showed consistent positive correlations with the expression of immune markers within the tumor and was decreased in the oldest age group. Since this miR is known to enhance T‐cell activation in the tumor microenvironment by blocking the PD‐L1/programmed cell death protein 1 (PD‐1) immune checkpoint, 42 , 43 it may also play a role in the observed age‐related decrease in tumor immune infiltration.
Apart from age‐related changes, differences in (tumor) immunity between fit and frail patients could be equally important. Based on the G8 scores, the oldest patients were subdivided into a fitter group (G8 > 14) and a frailer group (G8 ≤ 14). In the frailer group, higher T‐cell p16INK4a expression was observed, in accordance with previous observations by Liu et al., 8 who even reported a stronger correlation of T‐cell p16INK4a expression with frailty than with chronical age. Furthermore, the G8‐score was also correlated with several PBMC subsets. In the frailer subgroup, a significantly increased percentage of a distinct CD3+CD16+ cell population (NK‐like T cells) was found, which had, however, remained relatively stable along the age groups. These cells have been described as cytotoxic effector cells involved in the anti‐cancer immune response. 44 Yet, the link between accumulating CD3+CD16+ cells and clinical frailty remains unclear. Increased frailty was also associated with depletion of fully active CD8+ cells expressing CD27 and/or CD28 and, concurrently, accumulation of senescent CD8+CD57+ cells with decreased proliferative capacity. Surprisingly, in our cohort, no significant differences could be demonstrated between frailer and fitter patients with regard to the best documented frailty markers IL‐6, TNFα and CRP. Moreover, while Ipson et al. 45 have identified circulating miR‐326 as a candidate frailty biomarker, our data only showed increased miR‐326 expression in the oldest age category, without any significant difference between G8 subgroups within the old aged. These discrepancies could possibly be explained by the relatively small numbers of fit versus frailer patients and a different frailty assessment used in our study.
A number of associations between blood biomarkers and the tumor infiltrate are interesting to be highlighted. Firstly, levels of several pro‐inflammatory mediators (e.g. MCP‐1, IL‐1α, intermediate monocytes) inversely correlated with different tumor immune infiltrate markers. Hence, low‐grade chronic inflammation associated with ageing may induce a less effective tumor immune response. Secondly, higher plasma levels of the inhibitory immune checkpoint marker TIM‐3 46 correlated with lower sTILs % and lower infiltration of different immune cell subsets. Furthermore, higher frequencies of fully functional CD27+CD28+/− TEMRA cells in blood positively correlated with all T‐ and B‐cell markers in the tumor, whereas expansion of the senescent CD27−CD28− and CD57+ TEMRA cell blood populations was associated with increased infiltration of FOXP3+ cells in the tumor. Not surprisingly, blood Tregs % also inversely correlated with sTILs % and with CD8 density in the tumor. Finally, higher plasma levels of several miRs appeared to be associated with increased abundancy of T‐cell markers in the tumor immune infiltrate.
As a next step, we compiled panels consisting of the 5 or 10 highest significant and biologically most relevant age and frailty biomarkers. These panels proved able to quite accurately separate the oldest age group from the younger age groups, as well as the more ‘fit’ older patients from the ‘frailer’ older patients. Interestingly, uMap plotting with the biomarker panels for age revealed a distinct cluster of older patients exhibiting a clearly different ageing profile compared to the remaining older patients who mingled among the younger patients. Of note, virtually all ‘frail’ older patients clustered in the distinct group. This may suggest that the patients in this separate cluster are mainly ‘frail’ older patients or patients at risk of becoming frail, whereas the older patients mixed among the younger patients are the truly ‘fit’ older patients. Within the old group, uMap plotting with the frailty biomarker panels again showed a separation of the older patients in two clusters: a purely ‘fit’ group and a mixed group comprising all ‘frailer’ patients admixed with ‘fit’ individuals. We speculate that the latter show a biomarker profile that could point to beginning frailty development, which is not yet clinically apparent.
This study has some limitations. Primarily, it is a highly exploratory study without a hard primary endpoint. Further exhaustive research and validation of the findings are required. The study cohort was relatively small, and the old group was composed of relatively fit patients; only 10 patients had a G8 ≤ 14, and none of those had a G8 lower than 12. The difference in ‘frailty’ between the G8‐normal and G8‐decreased patients was thus rather moderate, but apparently still large enough to reveal some noticeable biomarker differences. Also, our study cohort did not allow conclusions on whether the changes in blood biomarkers are driven by ageing alone or are affected by the developing tumor as well. This could be further explored by comparing the blood immune/senescence profile of the patients in our cohort to age‐matched healthy control groups, which were unfortunately not available for this study. Furthermore, no long‐term outcome data are available to date, and also in the future, the limited sample size will not allow survival analysis. Finally, only a selection of potential immune/senescence biomarkers was evaluated; for instance, data on PD‐L1 expression and other immune checkpoint and activation markers on the tumor are not yet available. An extensive multiplex immunohistochemistry analysis, using the newly developed ‘Multiple Iterative Labeling by Antibody Neodeposition (MILAN)’ technique 47 and including a multitude of additional immune‐related tumor markers is currently ongoing. These in depth analyses will allow us to characterise in more detail the phenotype and functional status of immune subsets of the tumor infiltrate, co‐expression of immune markers and the local interactions between different cell types in the tumor microenvironment. Additional multidimensional bioinformatics techniques will also be applied to the data, aiming at compiling a blood biomarker signature of immunosenescence that could accurately reflect the tumor immune response in older BC patients.
Nevertheless, our study also has major strengths. Most importantly, blood and tumor tissue has been prospectively collected from a relatively homogenous group of patients with a similar luminal breast tumor (hormone‐sensitive, HER2‐negative). In this tumor type, the tumor immune infiltrate has been poorly studied despite it is the most frequent BC subtype. Our investigations thus contributed to further disclosure of the largely unexplored immunological landscape of luminal breast tumors. To the best of our knowledge, this is also the first BC study where the tumor immune infiltrate has been characterised in concert with the peripheral blood immune profile and in function of ageing and clinical frailty. This allowed us to establish interesting correlations between immunosenescence markers in the blood, tumor immune biomarkers and ageing/clinical frailty.
In conclusion, this extensive immune biomarker study revealed several interesting age‐related modifications in the blood immunological portrait and local tumor immune response of patients with luminal BC, as well as differences between fit and frailer old patients. The observed changes can be linked to various processes occurring during the ageing process, such as inflammation, cellular senescence and immunosenescence. 2 Our data support age‐dependent remodelling of both systemic immunity features and anti‐tumor immune responses. Given this knowledge that interactions between tumor cells, immune cells and inflammatory mediators differ with age, older patients may react differently to BC treatments like chemotherapy or (in the future), immunotherapy approaches. Ageing/immunosenescence should therefore be taken into account when outlining the optimal treatment strategy for each individual patient.
Methods
Patient selection
Patients were eligible for this prospective biomarker study, named IMAGE (IMmunity‐AGE), if they met the following inclusion criteria and upon written informed consent: early BC diagnosis, planned upfront surgery; grade II or III invasive carcinoma on core needle biopsy (CNB); ER‐positive, HER2‐negative; and clinical tumor size 1.5 cm or bigger on imaging with sufficient formalin‐fixed and paraffin‐embedded (FFPE) tumor tissue. We aimed to recruit patients within 3 distinct age categories: 35–45 years (premenopausal young population), 55‐65 years (postmenopausal but not ‘old’) and patients ≥ 70 years (postmenopausal older population). To evaluate blood immune parameters, an extra blood sample was drawn and processed at inclusion (before surgery). In the ≥ 70 years group, the G8 screening tool was used at inclusion (before surgery) as a surrogate for clinical frailty.
G8 assessment
The G8 questionnaire is an easy, widely used geriatric screening tool 48 that can be considered as surrogate for frailty. 49 The G8 includes eight items: seven items selected from the mini nutritional assessment (MNA) questionnaire (i.e. nutritional status, weight loss, body mass index, motor skills, psychological status, number of medications and self‐perception of health) and one item indicating the patient’s age (<80; 80–85; >85). The G8 score ranges from 17 (not at all impaired) to 0 (heavily impaired). Patients with a score 14 or lower are considered vulnerable or frail, and patients with a score higher than 14 are considered fit. 48 , 50
Blood collection and biomarker analysis
A detailed description of the materials and methods listed below can be found in Supplementary methods.
Blood was collected in two 10‐mL EDTA tubes (BD Vacutainer®; Becton, Dickinson and company, NJ), 4 mL of the total volume was used for EDTA plasma collection, 6 mL for T‐cell isolation, and the remaining blood was used for PBMC isolation, as described in detail in Supplementary methods. Briefly, PBMCs were isolated via density gradient centrifugation with Histopaque‐1077® (Sigma‐Aldrich; Saint Louis), using SepMate‐50 tubes (StemCell Technologies, Vancouver) for easy separation of fractions. For T‐cell isolation, RosetteSep Human T‐Cell Enrichment Cocktail (StemCell Technologies, Vancouver) was additionally added.
A broad panel of plasma cytokines [IL‐1α; IL‐1β; IL‐6; IL‐10; IL‐12p70; IL‐17A; IL‐17F; IL‐27; interferon gamma (INFγ); TNFα and free active transforming growth factor‐beta 1 (TGF‐β1)], plasma chemokines (i.e. IP‐10; IL‐8; MCP‐1), immune checkpoint proteins [sCD25; 4‐1BB; CD86; cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA‐4); PD‐L1; PD‐1; TIM‐3; lymphocyte‐activation gene 3 (LAG‐3); Gal‐9; sCD27; programmed cell death‐ligand 2 (PD‐L2)] and C‐reactive protein (CRP) were measured by cytometric bead array assays (AimPlex Human Inflammation 11‐plex; ImTec Diagnostics, Antwerp and YSL AimPlex; BioLegend, San Diego). Insulin‐like growth factor (IGF‐1) levels in plasma were measured by ELISA (Human IGF‐I Quantikine ELISA kit; R&D Systems, Minneapolis). T‐cell p16INK4a expression was measured via a probe‐based RT‐qPCR. Plasma levels of 20 immune‐related miRs (i.e. let‐7e, let‐7i, miR‐9, miR‐17, miR‐18a, miR‐19a, miR‐19b, miR‐20a, miR‐21, miR‐92a, miR‐125b, miR‐126, miR‐146a, miR‐150, miR‐155, miR‐181a, miR‐195, miR‐223, miR‐326 and miR‐424) were measured via SYBR Green RT‐qPCR. By using fluorescent antibody panels (Supplementary figure 1), PBMC immune subset profiles were analysed by 8‐colour flow cytometry, using a 3‐laser FACSVerse (BD Biosciences; Becton, Dickinson and company, NJ). All biomarker assay procedures are described in detail in Supplementary methods.
Tumor immune marker analysis
For the final analysis, we used tumor grade and pathological TMN staging measured on the resection specimen, which explained some discrepancies with grading on CNB and tumor size on imaging. All stainings were performed on surgical resection specimen. FFPE tumor tissue sections were cut at a thickness of 5 µm. sTILs were microscopically assessed on representative haematoxylin and eosin (H&E) stained tumor sections, according to published guidelines. 51 Further tumor immune infiltrate characterisation was performed by evaluating 7 immune cell markers (CD3, CD4, CD5, CD8, CD20, CD68 and FOXP3) via immunohistochemistry on whole section. CD68 staining grade was determined using a reported method. 52 The remaining immune cell markers (CD3, CD4, CD5, CD8, CD20 and FOXP3) were scored using a scoring protocol in the QuPath software to compare density and proportion of immune cells in different tumor regions. 53
Statistics
This is a prospective exploratory biomarker study; thus, no upfront sample size calculation was performed. Our aim was to include at least 15 patients for the young and middle age categories and 30 for the ≥ 70 years category, allowing to subdivide older patients in fitter group (G8 > 14) and frailer group (G8 ≤ 14). Because of the exploratory character of the study, statistical tests were performed without correction for multiple testing. The Mann–Whitney U‐test was used for associations between continuous and binary variables with more than two groups. The Kruskal–Wallis test was used for associations between continuous and categorical variables. Interactions between tumor region and age or G8 groups on the proportion and density of positively stained cells were assessed using linear mixed models, accounting for clustering of regions within patients. Spearman correlations were used for associations between different biomarkers. The analyses were performed using SAS software (SAS, Cary). Additionally, R software (R project, Vienna) was used to calculate biomarker performance and generate the dimensionally reduced data. Biomarkers were ranked using a combination of quantitative [fold change (FC) and P‐value (ANOVA)] and qualitative (area under the ROC curve (AUC)) parameters. The final score was calculated using a weighted average of the individual scores. The weights were set to 2:1:1 for AUC:FC:P‐value; that is, AUC was weighted double compared to P‐value and FC. Panels of 10 and 5 biomarkers were compiled based on their individual statistical performance and biological relevance for both age groups and G8 groups. These panels were dimensionally reduced using three different methods: uniform manifold approximation and projection (uMAP), principal component analysis (PCA) and T‐distributed stochastic neighbour embedding (t‐SNE). Biomarkers were normalised using Z‐scores before dimensionality reduction. Patient clustering was performed visually on the dimensionally reduced plots. Visually defined clusters were evaluated for enrichment with specific patient groups using hypergeometric tests. All reported P‐values are two‐sided, and significance threshold was set below 5% for all tests.
Conflict of interest
The authors declare no conflict of interest
Author contributions
Lieze Berben: Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Giuseppe Floris: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Supervision; Writing‐original draft; Writing‐review & editing. Cindy Kenis: Data curation; Writing‐review & editing. Bruna Dalmasso: Data curation; Writing‐review & editing. Ann Smeets: Data curation; Writing‐review & editing. Hanne Vos: Data curation; Writing‐review & editing. Patrick Neven: Data curation; Writing‐review & editing. Asier Antoranz Martinez: Formal analysis; Software; Visualization; Writing‐review & editing. Annouschka Laenen: Formal analysis; Software; Visualization; Writing‐review & editing. Hans Wildiers: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Writing‐original draft; Writing‐review & editing. Sigrid Hatse: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Supervision; Writing‐original draft; Writing‐review & editing.
Supporting information
Acknowledgements
The research project received funding from Fond Wetenschappelijk Onderzoek (FWO), application number G.0670.14N and G.0E68.16N. LB was supported by Kom op tegen kanker via an Emmanuel van der Schueren start‐up grant. HW is a FWO postdoctoral fellow. GF is a KOOR‐UH‐Leuven postdoctoral fellow.
References
- 1. Organization WH .Ageing and health 2018. Available from: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health.
- 2. Aunan JR, Cho WC, Søreide K. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis 2017; 8: 628–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fulop T, Larbi A, Dupuis G et al Immunosenescence and inflamm‐aging as two sides of the same coin: friends or foes? Front Immunol 2018; 8: 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yanes RE, Gustafson CE, Weyand CM, Goronzy JJ. Lymphocyte generation and population homeostasis throughout life. Semin Hematol 2017; 54: 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun Ageing 2008; 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raynor J, Lages CS, Shehata H, Hildeman DA, Chougnet CA. Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol 2012; 24: 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller L, Di Benedetto S, Pawelec G. The immune system and its dysregulation with aging. Subcell Biochem 2019; 91: 21–43. [DOI] [PubMed] [Google Scholar]
- 8. Liu Y, Sanoff HK, Cho H et al Expression of p16(INK4a) in peripheral blood T‐cells is a biomarker of human aging. Aging Cell 2009; 8: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol 2012; 24: 331–341. [DOI] [PubMed] [Google Scholar]
- 10. Brouwers B, Dalmasso B, Hatse S et al Biological ageing and frailty markers in breast cancer patients. Aging 2015; 7: 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008; 27: 5959–5974. [DOI] [PubMed] [Google Scholar]
- 12. Mogilyansky E, Rigoutsos I. The miR‐17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 2013; 20: 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatse S, Brouwers B, Dalmasso B et al Circulating MicroRNAs as easy‐to‐measure aging biomarkers in older breast cancer patients: correlation with chronological age but not with fitness/frailty status. PLoS One 2014; 9: e110644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kroesen B‐J, Teteloshvili N, Smigielska‐Czepiel K et al Immuno‐miRs: critical regulators of T‐cell development, function and ageing. Immunology 2015; 144: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salgado R, Denkert C, Demaria S et al The evaluation of tumor‐infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 2015; 26: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denkert C, von Minckwitz G, Darb‐Esfahani S et al Tumour‐infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018; 19: 40–50. [DOI] [PubMed] [Google Scholar]
- 17. Konig L, Mairinger FD, Hoffmann O et al Dissimilar patterns of tumor‐infiltrating immune cells at the invasive tumor front and tumor center are associated with response to neoadjuvant chemotherapy in primary breast cancer. BMC Cancer 2019; 19: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loi S, Drubay D, Adams S et al Tumor‐infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early‐stage triple‐negative breast cancers. J Clin Oncol 2019; 37: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smid M, Rodriguez‐Gonzalez FG, Sieuwerts AM et al Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration. Nat Commun 2016; 7: 12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014; 1843: 2563–2582. [DOI] [PubMed] [Google Scholar]
- 21. Antonelli A, Rotondi M, Fallahi P et al Age‐dependent changes in CXC chemokine ligand 10 serum levels in euthyroid subjects. J Interferon Cytokine Res 2005; 25: 547–552. [DOI] [PubMed] [Google Scholar]
- 22. Sarkar D, Fisher PB. Molecular mechanisms of aging‐associated inflammation. Cancer Lett 2006; 236: 13–23. [DOI] [PubMed] [Google Scholar]
- 23. Lee JS, Lee WW, Kim SH et al Age‐associated alteration in naive and memory Th17 cell response in humans. Clin Immunol 2011; 140: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res 2009; 29: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol 2001; 8: 131–136. [DOI] [PubMed] [Google Scholar]
- 26. Minciullo PL, Catalano A, Mandraffino G et al Inflammaging and anti‐inflammaging: the role of cytokines in extreme longevity. Arch Immunol Ther Exp 2016; 64: 111–126. [DOI] [PubMed] [Google Scholar]
- 27. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age‐related diseases: role of inflammation triggers and cytokines. Front Immunol 2018; 9: 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Velissaris D, Pantzaris N, Koniari I et al C‐reactive protein and frailty in the elderly: a literature review. J Clin Med Res 2017; 9: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang Y, Fung E, Xu A, Lan HY. C‐reactive protein and ageing. Clin Exp Pharmacol Physiol 2017; 44(Suppl 1): 9–14. [DOI] [PubMed] [Google Scholar]
- 30. Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF‐1 axis in ageing and longevity. Nat Rev Endocrinol 2013; 9: 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moller‐Hackbarth K, Dewitz C, Schweigert O et al A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim‐3). J Biol Chem 2013; 288: 34529–34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreira A, Gross S, Kirchberger MC, Erdmann M, Schuler G, Heinzerling L. Senescence markers: Predictive for response to checkpoint inhibitors. Int J Cancer 2019; 144: 1147–1150. [DOI] [PubMed] [Google Scholar]
- 33. Banerjee H, Kane LP. Immune regulation by Tim‐3. F1000Res 2018; 7: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellul P, Mariotti‐Ferrandiz E, Leboyer M, Klatzmann D. Regulatory T cells as supporters of psychoimmune resilience: toward immunotherapy of major depressive disorder. Front Neurol 2018; 9: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elyahu Y, Hekselman I, Eizenberg‐Magar I et al Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes. Sci Adv 2019; 5: eaaw8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vinay DS, Kwon BS. 4–1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep 2014; 47: 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuo G, Wu C‐Y, Yang H‐Y. MiR‐17‐92 cluster and immunity. J Formos Med Assoc 2019; 118: 2–6. [DOI] [PubMed] [Google Scholar]
- 38. Fuziwara CS, Kimura ET. Insights into regulation of the miR‐17‐92 cluster of miRNAs in cancer. Front Med 2015; 2: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olive V, Jiang I, He L. mir‐17‐92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol 2010; 42: 1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grillari J, Hackl M, Grillari‐Voglauer R. miR‐17‐92 cluster: ups and downs in cancer and aging. Biogerontology 2010; 11: 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alivernini S, Gremese E, McSharry C et al MicroRNA‐155‐at the critical interface of innate and adaptive immunity in arthritis. Front Immunol 2018; 8: 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang L, Cai Y, Zhang D et al miR‐195/miR‐497 regulate CD274 expression of immune regulatory ligands in triple‐negative breast cancer. J Breast Cancer 2018; 21: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tao Z, Xu S, Ruan H et al MiR‐195/‐16 family enhances radiotherapy via T cell activation in the tumor microenvironment by blocking the PD‐L1 immune checkpoint. Cell Physiol Biochem 2018; 48: 801–814. [DOI] [PubMed] [Google Scholar]
- 44. Krijgsman D, de Vries NL, Skovbo A et al Characterization of circulating T‐, NK‐, and NKT cell subsets in patients with colorectal cancer: the peripheral blood immune cell profile. Cancer Immunol Immunother 2019; 68: 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ipson BR, Fletcher MB, Espinoza SE, Fisher AL. Identifying exosome‐derived microRNAs as candidate biomarkers of frailty. J Frailty Aging 2018; 7: 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cattoretti G, Bosisio FM, Marcelis L, Bolognesi MM. Multiple iterative labeling by antibody neodeposition (MILAN) [Protocol]. Protocol exchange 2019 [Protocol (Version 5)]. Available from: 10.21203/rs.2.1646/v5 [DOI]
- 48. Kenis C, Decoster L, Van Puyvelde K et al Performance of two geriatric screening tools in older patients with cancer. J Clin Oncol 2014; 32: 19–26. [DOI] [PubMed] [Google Scholar]
- 49. Decoster L, Van Puyvelde K, Mohile S et al Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations dagger. Ann Oncol 2015; 26: 288–300. [DOI] [PubMed] [Google Scholar]
- 50. Bellera CA, Rainfray M, Mathoulin‐Pelissier S et al Screening older cancer patients: first evaluation of the G‐8 geriatric screening tool. Ann Oncol 2012; 23: 2166–2172. [DOI] [PubMed] [Google Scholar]
- 51. Hendry S, Salgado R, Gevaert T et al Assessing tumor‐infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the International Immuno‐Oncology Biomarkers Working Group: Part 2: TILs in melanoma, gastrointestinal tract carcinomas, non‐small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv Anat Pathol 2017; 24: 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ch'ng ES, Tuan Sharif SE, Jaafar H. In human invasive breast ductal carcinoma, tumor stromal macrophages and tumor nest macrophages have distinct relationships with clinicopathological parameters and tumor angiogenesis. Virchows Arch 2013; 462: 257–267. [DOI] [PubMed] [Google Scholar]
- 53. Berben L, Wildiers H, Marcelis L et al Computerised scoring protocol for identification and quantification of different immune cell populations in breast tumour regions by the use of QuPath software. Histopathology 2020; 77: 79–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials