

Abstract
DNA N4-methylcytosine (4mC) is a crucial epigenetic modification involved in various biological processes. Accurate genome-wide identification of these sites is critical for improving our understanding of their biological functions and mechanisms. As experimental methods for 4mC identification are tedious, expensive, and labor-intensive, several machine learning-based approaches have been developed for genome-wide detection of such sites in multiple species. However, the predictions projected by these tools are difficult to quantify and compare. To date, no systematic performance comparison of 4mC tools has been reported. The aim of this study was to compare and critically evaluate 12 publicly available 4mC site prediction tools according to species specificity, based on a huge independent validation dataset. The tools 4mCCNN (Escherichia coli), DNA4mC-LIP (Arabidopsis thaliana), iDNA-MS (Fragaria vesca), DNA4mC-LIP and 4mCCNN (Drosophila melanogaster), and four tools for Caenorhabditis elegans achieved excellent overall performance compared with their counterparts. However, none of the existing methods was suitable for Geoalkalibacter subterraneus, Geobacter pickeringii, and Mus musculus, thereby limiting their practical applicability. Model transferability to five species and non-transferability to three species are also discussed. The presented evaluation will assist researchers in selecting appropriate prediction tools that best suit their purpose and provide useful guidelines for the development of improved 4mC predictors in the future.
Keywords: DNA N4-methylcytosine site, machine learning, prediction model, sequence analysis, web server, performance validation, Epigenetic modification, Systematic evaluation, Bioinformatics, Sequence-based features
Graphical Abstract
We critically evaluate 12 publicly available 4mC site prediction tools according to species specificity, based on a huge independent validation dataset. Our results will assist researchers in selecting appropriate prediction tools that best suit their purpose and provide useful guidelines for the development of improved 4mC predictors in the future.
Introduction
DNA methylation is one of the most common epigenetic tools used by the cell to control gene expression. It alters chromatin structure, DNA conformation, DNA-protein interactions, and DNA stability.1 This modification plays important roles in the regulation of several developmental and pathological processes, such as aging, carcinogenesis, genomic imprinting, repression of transposable elements, and X chromosome inactivation.2, 3, 4, 5 Analysis of the differentiating capacity of foreign DNA and host DNA indicates that certain modifications, such as cytosine methylation, may protect the host DNA from enzyme-mediated degradation.6
Cytosine methylation is catalyzed by DNA methyl transferases and is considered a major post-replicative DNA modification in prokaryotes and eukaryotes.7 The most common types of enzymatic cytosine methylation are 5-methylcytosine and N4-methylcytosine (4mC).8, 9, 10 Unlike the former, which has been extensively studied,8,11,12 4mC has not been thoroughly investigated. 4mC formation, predominantly found in prokaryotes, is catalyzed by N4-cytosine-specific DNA methyl transferases, which methylate the amino group at the fourth position of cytosine.13 Similar to 5-methylcytosine, 4mC is an element of a restriction-modification system that protects self-DNA from enzyme-mediated degradation.14 It is also involved in the cell cycle, correction of DNA replication errors, and regulation of DNA replication.15,16
Information regarding genome-wide distribution of 4mC is crucial for deciphering the function of this modification in detail. To date, only a few experimental approaches, such as single-molecule real-time (SMRT) sequencing,17 4mC-Tet-assisted bisulfite sequencing (4mC-TAB-seq),18 and engineered transcription-activator-like effectors,9 have been developed for detecting 4mC sites in the genome. SMRT sequencing is a popular experimental approach that has been successfully implemented in the identification of 4mC modifications. Regardless of the presence or absence of an assembled genome, SMRT sequencing was designed to directly identify 4mC modification sites. However, this approach is expensive and lacks applicability to various species and large-scale genomes. Owing to this limitation, the next-generation sequencing technique 4mC-TAB-seq was designed to identify the genome-wide locations of 4mC motifs. Another group detected specific 4mC sites utilizing engineered transcription-activator-like effectors. Although these experimental methods effectively facilitate the identification of 4mC sites, they are still too laborious and expensive for genome-wide applicability.
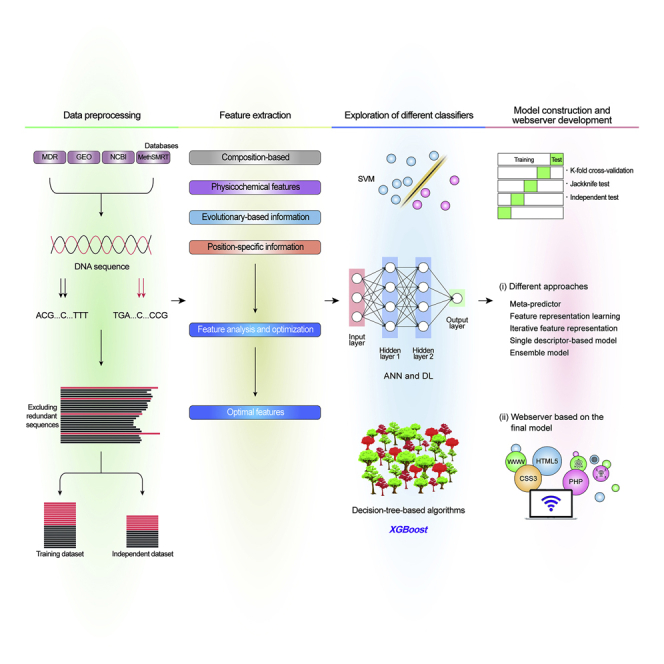
Computational approaches, particularly machine learning (ML) methods, have emerged as effective 4mC site prediction tools in multiple species. iDNA4mC,19 a pioneer ML-based method that relies on the chemical properties of nucleotides and accumulated nucleotide frequency as features to build a support vector machine (SVM)-based model, was developed in 2017. Two additional methods (4mCPred20 and 4mcPred-SVM21) were developed in 2018; five methods (Meta-4mCpred,8 4mcPred-IFL,22 4mCCNN,23 4mCpred-EL,24 and i4mCROSE25) were reported in 2019. By April 2020, five more methods (iEC4mC-SVM,26 DNA4mC-LIP,27 4mcDeep-CBI,1 iDNA-MS,2 and i4mC-Mouse28) were reported. An overview of the existing 4mC prediction methods is provided in Figure 1. Notably, most of these methods were trained on the benchmark datasets constructed by Chen et al.,19 and few were validated based on independent datasets constructed by Manavalan et al.8 Because of the recent surge in the development of 4mC prediction tools, an unbiased evaluation of these methods using a well-constructed validation dataset is necessary.
Figure 1.

Overview of the Current Computational Approaches for 4mC Site Prediction
Establishing a useful predictor for 4mC sites often involves the following steps: (1) data processing; (2) feature extraction and optimization; (3) exploration of different ML classifiers and selection of the appropriate classifier; and (4) model construction based on different approaches and web server development.
Accordingly, in this study, we considered eight species (Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Escherichia coli, Fragaria vesca, Geoalkalibacter [Geoa.] subterraneus, Geobacter [Geob.] pickeringii, and Mus musculus) having at least two or more prediction models and summarized these methods in terms of their underlying algorithms, performance evaluation strategy, feature selection, and web server utility. In total, 12 4mC prediction tools (iDNA4mC, 4mCPred_I, 4mCPred_II, 4mCpred-IFL, 4mcPred-SVM, Meta-4mCpred, 4mCCNN, DNA4mC-LIP, i4mC-ROSE, i4mc-Mouse, 4mCpred-EL, and iDNA-MS) are available as web servers. We carried out an unbiased evaluation of these existing web-based 4mC prediction tools using our own constructed validation set, which captures the overall 4mC and non-4mC pattern in the whole-genome of each individual species. While some models achieved excellent overall performance for some species, none was suitable for Geoa. subterraneus, Geob. pickeringii, and M. musculus, limiting their practical applicability. The model transferability and non-transferability were explored, and suggestions for the design and development of new prediction tools were also presented. The presented analysis will facilitate efforts to develop improved tools for the prediction of 4mC sites.
Results
In the study, an unbiased performance evaluation of existing web-based 4mC site prediction tools was performed based on validation dataset analysis. The utilized validation dataset was different from a previously reported and widely used dataset.8,24 Specifically, (1) unlike negative samples (non-4mC) analyzed before, in the new dataset, the negative samples were representative of the whole genome of each species, and (2) the new dataset was several-fold larger than the previously reported dataset. Eight species for which at least two prediction models reported are analyzed herein. Seven web servers (i.e., iDNA4mC, 4mCPred_I, 4mCPred_II, 4mcPred-SVM, Meta-4mCpred, 4mCCNN, and DNA4mC-LIP) were used to evaluate six species (A. thaliana, D. melanogaster, C. elegans, E. coli, Geoa. subterraneus, and Geob. pickeringii), two web servers (i4mC-ROSE and iDNA-MS) for F. vesca, and two web servers (4mCpred-EL and i4mC-Mouse) for M. musculus. All parameters were set to default values, as specified on their server. As an exception, the values reported for 4mCCNN were based on personal communication (J. Khanal, Chongbuk National University) because the web server was not active at the time of analysis. Computing the K-nearest neighbor (KNN) feature implemented in 4mcPred-IFL took several hours, leading to time-out errors during sequence submission. Therefore, 4mcPred-IFL was excluded from our evaluation.
Evaluation of Species-Specific Performance of the Existing 4mC Prediction Tools
Geob. pickeringii
The constructed validation set was 56-fold larger than the training set used to evaluate seven different methods (see Materials and Methods for details of the datasets and methods used). The performances of different models are shown in Figure 2A and Table S1. iDNA4mC achieved the best performance for four out of five metrics tested, i.e., Matthews correlation coefficient (MCC), balanced accuracy (BACC), specificity (Sp), and area under the curve (AUC) (0.118, 0.610, 0.687, and 0.653, respectively). These values were higher than those of the second-best method (4mCPred_I) by 0.046, 3.48%, 16.4%, and 5.5%, respectively. 4mCPred_II performance was similar to that of 4mCPred_I. The remaining four predictors (4mCCNN, 4mcPred-SVM, Meta-4mCpred, and DNA4mC-LIP) had much lower BACC values (below 50.0%), and even the random classifier performed better than these four predictors. To obtain statistically meaningful differences between the top model and other methods, AUC values for any two methods were compared, and the p value was computed based on the two-tailed t test.29 At p < 0.01, iDNA4mC significantly outperformed the other methods (Table S1).
Figure 2.

Comparison of the Prediction Performance of Six Species-Specific Web-Based 4mC Site Prediction Tools
The following validation datasets were used: (A) Geob. pickeringii; (B) E. coli; (C) C. elegans; (D) A. thaliana; (E) D. melanogaster; and (F) Geoa. subterraneus. AUC, area under the curve; BACC, balanced accuracy; MCC, Matthews correlation coefficient; Sn, sensitivity; Sp, specificity.
Most of the existing method web servers implemented a species-specific prediction model. However, the iDNA4mC web server implemented a single model based on a combined training dataset from six different species. Therefore, the improved performance of iDNA4mC may be associated with the larger training dataset. Although iDNA4mC showed superior performance, it was still far from satisfactory because, with a BACC of 61.0%, it was only slightly better than the random classifier. Recently, Tang et al.27 evaluated seven methods using a smaller validation dataset (200 4mCs and 200 non-4mCs) and demonstrated a reasonable performance for most tested methods (accuracy [ACC] range, 75.0%–85.0%). However, in our study, such performance was not replicated when using a larger validation set.
E. coli
The validation dataset used was 153-fold larger than the training dataset. As shown in Figure 2B and Table S1, 4mCCNN achieved the best performance for four out of five metrics, i.e., MCC, BACC, Sp, and AUC (0.110, 0.793, 0.805, and 0.873, respectively). The values were slightly higher than those of the second to fourth best predictors (DNA4mC-LIP, 4mCPred_II, and 4mcPred-SVM, accordingly), by 0.013–0.005, 0.5%–1.1%, 0.6%–8.3%, and 0.7%–1.5%, respectively. Of the remaining three methods, Meta-4mCpred and 4mCPred_I showed reasonable performance, with a BACC value of 77.0%, and iDNA4mC showed the worst performance. At p < 0.01, the performance of the best method (4mCCNN) was similar to that of the other methods (DNA4mC-LIP, 4mCPred_I, 4mCPred_I, Meta-4mCpred, and 4mcPred-SVM). This method significantly outperformed iDNA4mC. Although all these methods were developed using a smaller training dataset than that used in iDNA4mC (388 4mCs and 388 non-4mCs) and with different approaches, most of them performed reasonably well with respect to the genome-wide detection of 4mC sites.
C. elegans
The validation set used was 53-fold larger than the training dataset. As shown in Figure 2C and Table S1, the methods were classified based on performance into two groups, i.e., (1) four methods (4mCCNN, 4mCPred_I, 4mCPred_II, and DNA4mC-LIP), which showed similar performance in terms of MCC, BACC, and AUC values (0.453–0.486, 81.9%–82.5%, and 89.2%–90.4%, respectively), with no single best-performing method; and (3) the remaining three methods (Meta-4mCpred, 4mcPred-SVM, and iDNA4mC), which also achieved reasonable performance, with MCC and BACC values in the ranges of 0.376–0.405 and 76.3%–78.1%, respectively. However, the performance of group 2 methods was lower than that of group 1 methods. A reasonably sized training dataset was used in the model development for all methods (1,554 4mCs and 1,554 non-4mCs), and all methods were relatively robust for genome-wide analyses.
A. thaliana
The validation set used was 64-fold larger than the training dataset. As shown in Figure 2D and Table S1, DNA4mC-LIP showed the best performance for all five metrics tested, i.e., MCC, BACC, sensitivity (Sn), Sp, and AUC (0.561, 0.796, 0.778, 0.813, and 0.863, respectively). These values were higher than those of the second-best method (4mCPred_I) by 0.33, 1.6%, 1.01%, 1.5%, and 1.6%, respectively. The other four methods performed similarly well, with MCC, BACC, and AUC values in the ranges of 0.509–0.520, 77.0%–77.6%, and 84.3%–84.9%, respectively. iDNA4mC ranked at the bottom, with a BACC of 0.609, which was slightly better than that of the random predictor. At p < 0.01, the best method (DNA4mC-LIP) significantly outperformed the other six methods tested. Notably, DNA4mC-LIP considered the output of six different methods (evaluated in the current study) for the final prediction. Interestingly, this was the first time in the evaluation that a combined approach significantly outperformed the individual predictor.
D. melanogaster
The validation set was 72-fold larger than the training dataset. As shown in Figure 2E and Table S1, DNA4mC-LIP and 4mCCNN had similar performances, with MCC, BACC, Sn, Sp, and AUC values in the ranges of 0.498–0.515, 81.7%–82.2%, 88.5%–91.3%, 72.1–75.8, and 89.5%–89.6%, respectively. Accordingly, it was difficult to select the best method. Four methods (4mCPred_I, Meta-4mCpred, 4mcPred-SVM, and 4mCPred_II), ranked from third to sixth (accordingly) showed similar performance with MCC, BACC, and AUC values in the ranges of 0.449–0.490, 79.0%–80.9%, and 86.2%–88.5%, respectively. iDNA4mC ranked last, with a BACC value of 64.3%, was only slightly better than that of the random predictor. All methods (except for iDNA4mC) used a reasonably sized training dataset (1,769 4mCs and 1,769 non-4mCs), and these methods performed exceptionally well in the evaluation, especially when a larger validation set was used.
Geoa. subterraneus
The validation set used was 16-fold larger than the training dataset. The performances of the different models are shown in Figure 2F and Table S1. iDNA4mC showed the best performance for four out of five metrics, i.e., MCC, BACC, Sp, and AUC (0.150, 0.575, 0.701, and 0.611, respectively). These values were higher than those of the second-best method (4mCPred_II) by 0.053, 2.6%, 21.9%, and 3.7%, respectively. Notably, the performance of 4mCCNN was similar to that of the second-best predictor, 4mCPred_II. The remaining four predictors (4mCPred_I, 4mcPred-SVM, Meta-4mCpred, and DNAs4mC-LIP) showed similar performances, with MCC and BACC values in the ranges of 0.043–0.076 and 52.2%–53.9%, respectively. At p < 0.01, iDNA4mC significantly outperformed the other six methods. As mentioned above, the improved iDNA4mC performance may be related to the larger training dataset size.
Although iDNA4mC was superior to other methods, it is not an adequate method because the BACC value (57.5%) was only slightly higher than that of the random classifier. Recently, Tang et al.27 evaluated seven different methods (excluding 4mCCNN) using a smaller validation dataset (350 4mCs and 350 non-4mCs); most of the methods showed reasonable performance (ACC range, 80.0%–88.0%). However, such performance was not replicated when evaluating larger validation sets in the current study.
M. musculus
The constructed validation set was 155-fold larger than the training dataset, and it was used to evaluate 4mCpred-EL and i4mC-Mouse. As shown in Figure 3A and Table S1, both methods showed a similar performance, with MCC, BACC, and AUC values in the ranges of 0.018–0.020, 57.1%–57.8%, and 0.612–0.633, respectively. Furthermore, based on the Sn metric, both methods performed exceptionally well, with values exceeding 77.0%. However, the Sp value was quite low, indicating a potential drawback of the training dataset. Hence, the practical applicability of the two methods was limited because of higher false positives.
Figure 3.

Comparison of the Prediction Performance of Two Species-Specific Web-Based 4mC Site Prediction Tools
The following validation datasets were used: (A) M. musculus and (B) F. vesca.
F. vesca
The constructed validation set was 32-fold larger than the training dataset, and it was used to evaluate i4mC-Rose and iDNA-MS. Detailed performance information is shown in Figure 3B and Table S1. Notably, iDNA-MS showed the best performance for four out of five metrics, i.e., MCC, BACC, Sn, and AUC (0.417, 0.847, 0.876, and 0.930, respectively). These values were higher than those of i4mC-ROSE by 0.031, 4.5%, 11.8%, and 4.0%, respectively. At p < 0.01, iDNA-MS significantly outperformed i4mC-ROSE. Interestingly, iDNA-MS used a slightly smaller training dataset and a simpler approach (single-feature model) than i4mC-Rose, yet it showed a relatively better generalization capability.
Rationale for Model Transferability
To understand the generalization of the above methods, two-sample logos30 were used, the statistically significant position-specific composition of 4mCs and non-4mCs was determined, and comparative analysis between training and validation sets was performed. Interestingly, few nucleotide stretches or single nucleotides at specific positions shared the same location in the training and validation datasets for E. coli, C. elegans, A. thaliana, D. melanogaster, and F. vesca. For E. coli, nucleotides at positions 15–18 and 22–24 in positive samples, and those at positions 15, 16, and 22–24 in negative samples, were in the same position in the two sets (Figures 4A and 4B). In C. elegans, nucleotides at positions 15–18 and 22–33 in positive samples, and those at positions 17, 19, 20, and 22–32 in negative samples, were in the same position in the two sets (Figures 4C and 4D). In A. thaliana, nucleotides at positions 15–17, 22–25, and 27–31 in positive samples, and those at positions 17–20, 22–31, and 36–41 in negative samples, were in the same position in the two sets (Figures 4E and 4F). For D. melanogaster, nucleotides at positions 9–12, 19, 20, and 22–25 in positive samples, and those at positions 16, 17, and 22–25 in negative samples, were in the same position in the two sets (Figures 4G and 4H). For F. vesca, nucleotides at positions 22–30 and 33–41 in positive samples, and those at positions 22–29 in negative samples, were in the same position in the two sets (Figures 4I and 4J). The analysis revealed that the training dataset utilized for five species (E. coli, C. elegans, A. thaliana, D. melanogaster, and F. vesca) covered position-specific informative sequence patterns (4mCs and non-4mCs) from their respective genomes or representative samples from the entire genome. Hence, prediction models developed using these smaller training datasets showed robustness or transferability during the evaluation. It is therefore evident that the variations in method ranking depend on the features used, the choice of classifier, and the specific approach used.
Figure 4.

Statistically Significant Position-Specific Composition of 4mC and Non-4mC Sites
The compositional preferences of 4mCs and non-4mCs are denoted above and below the axis, respectively. Training and validation datasets for each species are compared. (A and B) E. coli; (C and D) C. elegans; (E and F) A. thaliana; (G and H) D. melanogaster; (I and J) F. vesca; (K and L) Geoa. subterraneus; (M and N) Geob. pickeringii; (O and P) M. musculus. (A, C, E, G, I, K, M, and O) Training datasets. (B, D, F, H, J, L, N, and P) Validation datasets.
Rationale for Model Non-transferability
To understand the failures of the existing methods on three species (Geoa. subterraneus, Geob. Pickeringii, and M. musculus), the same approach, with two-sample logos,30 was used as that for analyzing the rationale for model transferability above. Most of the nucleotides surrounding the cytosine at position 21 in both positive and negative samples differed completely in the training and validation sets for Geoa. subterraneus (Figures 4K and 4L) and Geob. pickeringii (Figures 4M and 4N). This indicated that the training dataset position-specific sequence patterns (4mCs and non-4mCs) did not cover informative sequence patterns from the entire genome or were not representative of the entire genome. Consequently, the two species-specific models suffered from generalization or had low robustness. For M. musculus, a similar pattern was observed for upstream positive samples, and a dissimilar pattern was found for negative samples (Figures 4O and 4P). Notably, both 4mCpred-EL and i4mC-Mouse were developed using a relatively small training dataset; however, covering the informative sequence patterns around non-4mCs in the entire genome was challenging. Hence, the different analytical approaches had low Sp. Overall, the three species-specific (for Geoa. subterraneus, Geob. pickeringii, and M. musculus) models performed marginally better than the random predictor, with a limited applicability.
Comparison of 4mC Site Prediction Web Servers
Because the user experience of web servers is important for experimentalists, user-friendliness of the web servers was then evaluated. Several limitations were noted. First, most of the existing web servers (except for iDNA4mC and 4mCpred-EL) could only handle 41-bp sequences with a cytosine in a central location; this limited the application, particularly for genome-wide analyses. Second, the number of FASTA sequences handled during a single request varied. In particular, Meta-4mCpred, 4mCpred-EL, iDNA-MS, i4mC-Mouse, i4mC-ROSE, and iDNA4mC handle up to 10,000 sequences; 4mCPred_I and 4mCPred_II handle up to 25,000 sequences; DNA4mC-LIP handles up to 400 sequences; and 4mcPred-SVM handles up to 5,000 sequences. Third, for batch processing, half of the servers did not support the upload of the FASTA sequence files. Only Meta-4mCpred, 4mcPred-SVM, DNA4mC-LIP, i4mC-Mouse, iDNA-MS, and i4mC-Rose offered the option of uploading FASTA sequence files. Fourth, the run times varied, ranging from 3 to 20 min. 4mCPred_I and 4mCPred_II were the only two servers that could handle large numbers of sequences in a single run and return the prediction results quickly (within 3 min). Finally, a user lacking programming knowledge would not be able to use most of the existing methods because the whole genome has to be processed into 41-bp fragments prior to analysis.
Discussion
In the current study, eight species-specific computational tools for predicting 4mC sites were surveyed and assessed herein. Some methods for E. coli, A. thaliana, F. vesca, D. melanogaster, and C. elegans showed an excellent overall performance. However, none of the existing methods was suitable for Geoa. subterraneus, Geob. pickeringii, and M. musculus, limiting their applicability. Model robustness for five species and non-transferability to three species could be explained by the position-specific compositional information between training and validation sets. During our evaluation, we observed that current methods have limitations. Seven predictors used the same training dataset to develop six species-specific prediction models, and 4mCpred-EL and i4mC-Mouse used a similar training dataset. However, the training dataset between iDNA-MS and i4mC-Rose was entirely different. Generally, construction of a high-quality dataset is the first and most important step of ML-based prediction model development.31 Surprisingly, none of the ensuing methods tested the training dataset quality of the preceding methods, and there was no attempt to enhance the training dataset quality. Based on the evaluation, the proposed training dataset for Geoa. subterraneus, Geob. pickeringii, and M. musculus did not cover the informative sequence patterns around 4mC and non-4mC sites in the whole genomes. Hence, no existing methods for these three species replicated the training performance during our evaluation, indicating that a high-quality dataset construction is important for the development of future predictors. Interestingly, some methods for the remaining five species replicated the training results during evaluation. Although the training dataset covered the informative sequence patterns around 4mC and non-4mC sites for the entire genome, it was quite small compared with the validation dataset generated in the current study. Therefore, apart from dataset quality, dataset size is another important factor that should be considered when developing future predictors.
Based on single-feature encoding and SVM, Chen et al.19 reported the optimal fragment length to 41 bp. Subsequent studies used this length regardless of the species analyzed. In the future, different feature-encoding schemes and classifiers should be explored to identify the optimal fragment length. Based on the non-transferability performance of species-specific models tested with other species, Chen et al.19 concluded the essentials of species-specific models for 4mC site prediction. Subsequent studies followed species-specific model concepts rather than attempting to develop a common 4mC site predictor using a multiple-species training dataset. Future studies in this direction are needed to understand the necessity of species-specific models rather than a common predictor. Furthermore, in terms of web server utility, most of the existing methods are not user-friendly because they can handle only a small number of sequences in a single run, require a long processing run time, lack the options for result file download and genome-wide 4mC site screening, and sometimes show time-out errors.
To improve the prediction performance, several points should be considered. First, highly homologous sequences should be excluded to reduce bias in the training dataset. Second, feature-encoding algorithms focusing on position-specific information may be useful for differentiating 4mCs from non-4mCs. Third, several feature-representation schemes have been recently proposed, such as iterative feature representation,22 adaptive feature learning,32 effective feature representation,8 and fused multi-view information.33 Application of two or more schemes to the same dataset and selection of the most appropriate one may improve model robustness. Fourth, the reliable and robust performance of deep-learning algorithm is often dependent on a huge training dataset.34, 35, 36, 37 Considering the large size of the dataset constructed in the current study, it would be interesting to apply the deep-learning algorithm to 4mC site prediction by exploring different feature representation schemes, and to ultimately compare the performance of deep-learning and conventional ML-based models. Overall, this study will assist scientists with an interest in the field of developing improved tools for the prediction of 4mC sites.
Materials and Methods
A General Framework of the Existing Computational Approaches for 4mC Site Prediction
The general framework for the existing computational approaches is shown in Figure 1 and comprises four steps. As the first step, a high-quality 4mC dataset based on validated databases38,39 and literature search is constructed. Because not many experimentally characterized non-4mC sites are available, 41-bp fragments are generated from chromosomal DNA, containing cytosine at the central position. These fragments should not overlap with 4mC sites detected experimentally and are considered negative (non-4mC) samples. To avoid overestimation of the predictions, CD-HIT is generally applied to discard redundant sequences.40 From the final dataset, 80% or 75% of samples are randomly selected and treated as the training dataset for prediction model development, and the remaining samples are treated as an independent dataset to check model robustness. The second step involves feature representation and optimization. For the former, a variety of feature descriptors are generally used to capture meaningful information that distinguishes between positive and negative samples, including composition-based features41, 42, 43, 44 (Kmer nucleotide frequency [Kmer], reverse complementary Kmer, enhanced nucleic acid composition, composition of k-spaced nucleic acid pairs, pseudo-dinucleotide composition [PseDNC], pseudo-trinucleotide composition [PseTNC], pseudo k-tuple composition, parallel correlation PseDNC, parallel correlation PseTNC, series correlation PseDNC, and series correlation PseTNC), position-specific-based features45 (mononucleotide binary encoding [MBE], dinucleotide binary encoding [DBE], numerical representation of nucleotides, position-specific trinucleotide propensity, electron-ion interaction pseudopotential [EIIP]), physicochemical property-based features46 (ring-function hydrogen chemical properties and accumulated nucleotide frequency [RFHCP], dinucleotide physicochemical properties [DPCP], trinucleotide physicochemical properties [TPCP], dinucleotide-based autocovariance [DAC], and dinucleotide-based cross-correlation [DCC]), and evolutionary-based features (KNN features). To discard irrelevant and redundant features from the original feature descriptor, feature optimization using sequential forward search (SFS) or other approaches is generally performed.47, 48, 49 In the third step, the prediction model is constructed based on the exploration of different classifiers and different approaches. Specifically, the optimal features from each descriptor (from the second step) are input to several ML classifiers (SVM, extreme gradient boosting, deep learning, and random forest) to develop a prediction model. In the fourth step, while developing the prediction model, different types of approaches are explored, including meta-predictor,33,50 feature representation learning,51,52 iterative feature representation,22,53 single descriptor-based model,54,55 ensemble model,56, 57, 58 and stacking approach.59,60 Finally, the web server is constructed based on the final prediction model, to predict whether a specific sequence represents a 4mC or non-4mC sample.
Validation Dataset Construction
To evaluate the performance of the existing 4mC prediction tools and to enable a fair comparison between them, positive and negative samples were constructed in the current study. First, positive samples were extracted for seven species (C. elegans, D. melanogaster, A. thaliana, Geoa. subterraneus, Geob. pickeringii, E. coli, and M. musculus) from the MethSMRT database,38 which includes data for 156 species (149 prokaryotes and seven eukaryotes). Epigenetic modification (4mC or N6-methyladenine [6mA]) data denoted in MethSMRT were compiled either from the Sequence Read Archive or NCBI Gene Expression Omnibus. Second, the downloaded raw data from MethSMRT were processed. Sequences lacking relevant information (i.e., with 6mA and 4mC sites lacking any modification [modQV] score) were excluded, yielding 41-bp sequences containing central cytosine (i.e., 4mC sites) with varying modQV scores. For F. vesca, sorted data (41-bp fragments with different modQV scores) were downloaded from the MDR database.39 Selecting the optimal modQV threshold was challenging, as thresholds of 2061 and 30,62 corresponding to p values of 0.01 and 0.001, respectively, are recommended in different studies. The existing methods utilized positive samples with a modQV greater than 30. However, average cutoffs greater than or equal to 25 were used for six species (C. elegans, D. melanogaster, A. thaliana, Geoa. subterraneus, Geob. pickeringii, and F. vesca), and thresholds greater than or equal to 20 were used for the remaining two species (E. coli and M. musculus). As the analysis of the latter two species at the original cutoff (≥25) resulted in a very small positive sample size, a slightly smaller modQV cutoff value was used. Finally, positive samples for each species that shared greater than 75% sequence identity with the training dataset used by the existing methods were removed using CD-HIT.40
To construct the negative samples, the same protocol as that used in previous studies was followed.8,25 Briefly, the entire single circular chromosome was considered for three species (E. coli, Geoa. subterraneus, and Geob. pickeringii), and 41-bp fragments with a central cytosine were generated. For species with multiple chromosomes, similar fragments were generated and 50% of sequences from each chromosome were randomly selected. In this manner, all chromosomes for the remaining species (C. elegans, D. melanogaster, A. thaliana, M. musculus, and F. vesca) were covered. Subsequently, sequences that shared more than 75% sequence identity with the training dataset used by the existing methods, the above generated positive samples, and positive samples with low modQV values were excluded. The validation dataset can be downloaded at http://thegleelab.org/Meta-4mCpred/EvaluationData.html.
A statistical summary of positive and negative samples for each species is shown in Table 1. On average, the numbers of positive and negative samples generated in the current study were 14-fold and 137-fold higher than the numbers of respective samples used in the training dataset.
Table 1.
Summary of the Newly Constructed Validation Dataset
| Species | Positive | Negative |
|---|---|---|
| E. colia | 670 | 118,266 |
| C. elegans | 19,289 | 144,553 |
| D. melanogaster | 45,809 | 209,690 |
| A. thaliana | 73,785 | 178,711 |
| Geoa. subterraneus | 17,573 | 11,404 |
| Geob. pickeringii | 4,445 | 60,005 |
| M. musculusa | 942 | 247,823 |
| F. vesca | 21,350 | 290,857 |
The first column represents species name. The second and third columns respectively represent positive and negative samples constructed in this study.
modQV cutoff slightly reduced for the positive samples.
Existing 4mC Prediction Methods
ML algorithms have been widely applied in various fields,63, 64, 65, 66, 67 such as post-replication DNA modification site predictions, particularly 4mC, 6mA, and 5-hydroxymethylcytosine sites.2,56,68 Table 2 summarizes the existing 4mC prediction methods that utilize a wide range of ML algorithms, feature encodings, and different approaches. The methods are described in detail below, according to the year of publication.
Table 2.
List of Currently Available Tools for 4mC Sites Prediction Assessed in This Study
| Year | Toola | Classifier | Training/Independent Dataset Size | Features | Web Server | Evaluation Strategy | File Upload |
|---|---|---|---|---|---|---|---|
| 2017 | iDNA4mCb | SVM | Chen dataset/– | RFHCP | yes | LOOCV | no |
| 2018 | 4mCPredb | SVM | Chen dataset/– | PSTNP, EIIP | yes | LOOCV | no |
| 4mcPred-SVMb | SVM | Chen dataset/– | Kmer, MBE, DBE, LPDF | yes | 10-fold CV | yes | |
| 2019 | Meta-4mCpredb | RF, ERT, GB, SVM | Chen dataset/Manavalan dataset | Kmer, MBE, DPE, LPDF, RFHCP, DPCP, TPCP | yes | 10-fold CV | yes |
| 4mcPred-IFLb | SVM | Chen dataset/– | Kmer+MBE, DBE+LPDF, PCPs, PseDNC, KNN, EIIP, MMI, RFHCP | yes | 10-fold CV | yes | |
| 4mCCNNb | CNN | Chen dataset/– | MBE | yesc | 10-fold CV | – | |
| 4mCpred-ELd | RF, GB, ERT, SVM | (800 4mCs and 800 non-4mCs)/(180 4mCs and 180 non-4mCs) | Kmer, DPE+LPDF, RFHC, EIIP, MBE, DPCP, TPCP | yes | 10-fold CV | yes | |
| i4mC-ROSEe | RF | (4854 4mCs and 4854 non-4mCs)/(1617 4mCs and 1617 non-4mCs) | KSNC, MBE, EIIP | yes | 10-fold CV | yes | |
| 2020 | iEC4mC-SVMd | SVM | (388 4mCs and 388 non-4mCs)/(134 4mCs and 134 non-4mCs) | MBE, RFHC, DAE, X-k-YCF, Kmer | – | 10-fold CV | – |
| DNA4mC-LIPd | – | –/Manavalan dataset | integration of six existing predictors | yes | independent evaluation | yes | |
| 4mcDeep-CBId | CNN, BLSTM | (1,173 4mCs and 6,635 non-4mCs)/ – | same as used in 4mcPred-IFL | – | 3-fold CV | – | |
| iDNA-MSf | RF | 7,899 samples/7,898 samples | Kmer, RFHCP, MBE | yes | 5-fold CV | yes | |
| i4mC-Moused | RF | (746 4mCs and 746 non-4mCs)/(160 4mCs and 160 non-4mCs) | Kmer, KSNC, MBE, EIIP | yes | 10-fold CV | yes |
Chen dataset contains C. elegans (4mCs, 1,554; non-4mCs, 1,554), D. melanogaster (4mCs, 1,769; non-4mCs, 1,769), A. thaliana (4mCs, 1,978; non-4mCs, 1,978), E. coli (4mCs, 388; non-4mCs, 388), Geoa. subterraneus (4mCs, 906; non-4mCs, 906), and Geob. pickeringii (4mCs, 569; non-4mCs, 569). Manavalan dataset contains C. elegans (4mCs, 750; non-4mCs, 750), D. melanogaster (4mCs, 1,000; non-4mCs, 1,000), A. thaliana (4mCs, 1,250; non-4mCs, 1,250), E. coli (4mCs, 134; non-4mCs, 134), Geoa. subterraneus (4mCs, 350; non-4mCs, 350), and Geob. pickeringii (4mCs, 200; non-4mCs, 200). SVM, support vector machine; RF, random forest; GB, gradient boosting; CNN, convolutional neural network; BLSTM, bidirectional long short-term memory network; ERT, extremely randomized tree; RFHCP, ring-function-hydrogen-chemical properties, PSTNP, position-specific trinucleotide propensity; EIIP, electron-ion interaction pseudopotential; Kmer, Kmer nucleotide frequency; MBE, mononucleotide binary encoding, DBE, dinucleotide binary encoding, LPDF, local position-specific dinucleotide frequency; DPE, dinucleotide binary profile encoding; DPCP, dinucleotide physicochemical properties; TPCP, trinucleotide physicochemical properties; PCP, physicochemical property; PseDNC, pseudo-dinucleotide composition; KNN, K-nearest neighbor; KSNC, k-space nucleotide composition; DAC, dinucleotide physicochemical properties autocorrelation; X-k-YCF, Xmer-kGap-Ymer composition frequency; ANF, accumulated nucleotide frequency; LOOCV, leave-one-out cross-validation; CV, cross-validation.
The listed tool URL addresses are as follows: iDNA4mC, http://lin-group.cn/server/iDNA4mC/; 4mCPred, http://server.malab.cn/4mCPred/; 4mcPred-SVM, http://server.malab.cn/4mcPred-SVM/; Meta-4mCpred, http://thegleelab.org/Meta-4mCpred/; 4mcPred-IFL, http://server.malab.cn/4mcPred-IFL/; 4mCCNN, https://home.jbnu.ac.kr/NSCL/4mCCNN.htm; 4mCpred-EL, http://thegleelab.org/4mCpred-EL/; i4mC-ROSE, http://kurata14.bio.kyutech.ac.jp/i4mC-ROSE/; DNA4mC-LIP, http://i.uestc.edu.cn/DNA4mC-LIP/; iDNA-MS, http://lin-group.cn/server/iDNA-MS/; i4mC-Mouse, http://kurata14.bio.kyutech.ac.jp/i4mC-Mouse/.
Tools contain six species-specific prediction models, namely A. thaliana, C. elegans, D. melanogaster, E. coli, Geoa. subterraneus, and Geob. pickeringii.
Web server is not functional.
Tool contains one prediction model to compute 4mC site from specific species.
Tool contains two prediction models for F. vesca and Rosa chinensis.
Tool contains four different species-specific models, namely F. vesca, Casuarina equisetifolia, Saccharomyces cerevisiae, and Ts. SUP5-1.
4mC Prediction Methods Developed in 2017
iDNA4mC
Chen et al.19 proposed iDNA4mC, the first method for 4mC site prediction. First, the authors constructed a nonredundant training dataset for six species, where each species was represented by an equal number of positive (4mCs) and negative (non-4mCs) samples. The positive samples were obtained from the MethSMRT database; the fragment length was 41 bp, with a cytosine base at position 21 and a modQV score greater than 30. Negative samples were constructed from the respective genomes; the fragment length and cytosine positioning were the same as in the positive samples. Most importantly, the fragments were not detected by the SMRT sequencing technology. To reduce the bias and over-fitting, the authors applied CD-HIT40 and excluded sequences that shared more than 80% sequence identity with sequences in each species. They then experimented with the different sequence lengths and evaluated the model performance. Eventually, they identified 41 bp as the optimal length for consistently obtaining the best performance, regardless of species. Surprisingly, the same optimal sequence length has been used in later studies69,70 of other species.24,25 Ultimately, a set of 3,108, 3,538, 3,956, 776, 1,812, and 1138 samples for C. elegans, D. melanogaster, A. thaliana, E. coli, Geoa. subterraneus, and Geob. pickeringii species, respectively, was generated. Herein, this dataset is referred to as the “Chen dataset.”
RFHCP encoding was then applied to convert DNA samples into 164-dimensional feature vectors. Then, SVM was utilized to develop a prediction model independently for each species using the leave-one-out cross-validation (LOOCV) procedure. iDNA4mC showed an average ACC, MCC, Sn, and Sp of 0.601, 0.800, 0.808, and 0.792, respectively. Detailed information on the performance of iDNA4mC and other methods (listed below) for each species is provided in Table 3. This prediction model is freely accessible at http://lin-group.cn/server/iDNA4mC. Furthermore, the authors performed cross-species validation (via species-specific prediction and testing other species). The analysis revealed that the individual species model was not transferrable to other species, thus indicating the need for species-specific 4mC prediction models. Notably, iDNA4mC served as a base for the development of later prediction models.
Table 3.
The Existing Method Performances Reported in the Literature Based on the Training and Independent Test
| Species | Methods | Training |
Independent test |
||||||
|---|---|---|---|---|---|---|---|---|---|
| MCC | ACC | Sn | Sp | MCC | ACC | Sn | Sp | ||
| C. elegans | iDNA4mC | 0.572 | 0.786 | 0.797 | 0.775 | – | – | – | – |
| 4mCPred_I | 0.740 | 0.870 | 0.871 | 0.869 | – | – | – | – | |
| 4mCPred_II | 0.750 | 0.877 | 0.875 | 0.879 | – | – | – | – | |
| 4mcPred-SVM | 0.631 | 0.815 | 0.824 | 0.807 | – | – | – | – | |
| Meta-4mCpred | 0.652 | 0.826 | 0.840 | 0.812 | 0.741 | 0.870 | 0.843 | 0.897 | |
| 4mcPred-IFL | 0.761 | 0.880 | 0.890 | 0.871 | – | – | – | – | |
| 4mCCNN | 0.694 | 0.842 | 0.895 | 0.825 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.786 | 0.893 | 0.885 | 0.901 | |
| 4mcDeep-CBI | 0.850 | 0.929 | 0.949 | 0.894 | – | – | – | – | |
| D. melanogaster | iDNA4mC | 0.625 | 0.812 | 0.833 | 0.791 | – | – | – | – |
| 4mCPred_I | 0.740 | 0.869 | 0.869 | 0.869 | – | – | – | – | |
| 4mCPred_II | 0.760 | 0.878 | 0.876 | 0.880 | – | – | – | – | |
| 4mcPred-SVM | 0.661 | 0.830 | 0.838 | 0.822 | – | – | – | – | |
| Meta-4mCpred | 0.685 | 0.842 | 0.831 | 0.854 | 0.812 | 0.906 | 0.913 | 0.899 | |
| 4mcPred-IFL | 0.745 | 0.873 | 0.865 | 0.88 | – | – | – | – | |
| 4mCCNN | 0.687 | 0.854 | 0.864 | 0.854 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.849 | 0.924 | 0.943 | 0.905 | |
| A. thaliana | iDNA4mC | 0.519 | 0.760 | 0.757 | 0.762 | – | – | – | – |
| 4mCPred_I | 0.650 | 0.823 | 0.813 | 0.832 | – | – | – | – | |
| 4mCPred_II | 0.670 | 0.834 | 0.830 | 0.838 | – | – | – | – | |
| 4mcPred-SVM | 0.573 | 0.787 | 0.778 | 0.796 | – | – | – | – | |
| Meta-4mCpred | 0.584 | 0.792 | 0.761 | 0.822 | 0.711 | 0.855 | 0.876 | 0.834 | |
| 4mcPred-IFL | 0.644 | 0.822 | 0.803 | 0.840 | – | – | – | – | |
| 4mCCNN | 0.622 | 0.797 | 0.804 | 0.792 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.720 | 0.859 | 0.883 | 0.836 | |
| E. coli | iDNA4mC | 0.598 | 0.799 | 0.820 | 0.778 | – | – | – | – |
| 4mCPred_I | 0.890 | 0.945 | 0.956 | 0.933 | – | – | – | – | |
| 4mCPred_II | 0.900 | 0.950 | 0.951 | 0.949 | – | – | – | – | |
| 4mcPred-SVM | 0.666 | 0.833 | 0.858 | 0.807 | – | – | – | – | |
| Meta-4mCpred | 0.697 | 0.848 | 0.869 | 0.827 | 0.650 | 0.825 | 0.806 | 0.843 | |
| 4mcPred-IFL | 0.789 | 0.894 | 0.907 | 0.881 | – | – | – | – | |
| 4mCCNN | 0.688 | 0.859 | 0.881 | 0.789 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.676 | 0.837 | 0.803 | 0.871 | |
| iEC4mC-SVM | 0.711 | 0.854 | 0.820 | 0.889 | 0.665 | 0.832 | 0.851 | 0.813 | |
| Geoa. subterraneus | iDNA4mC | 0.630 | 0.815 | 0.822 | 0.808 | – | – | – | – |
| 4mCPred_I | 0.800 | 0.900 | 0.899 | 0.900 | – | – | – | – | |
| 4mCPred_II | 0.820 | 0.910 | 0.912 | 0.909 | – | – | – | – | |
| 4mcPred-SVM | 0.674 | 0.837 | 0.840 | 0.834 | – | – | – | – | |
| Meta-4mCpred | 0.711 | 0.855 | 0.856 | 0.854 | 0.701 | 0.850 | 0.817 | 0.883 | |
| 4mcPred-IFL | 0.773 | 0.887 | 0.886 | 0.887 | – | – | – | – | |
| 4mCCNN | 0.704 | 0.860 | 0.852 | 0.843 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.674 | 0.837 | 0.814 | 0.860 | |
| Geob. pickeringii | IDNA4mC | 0.663 | 0.831 | 0.824 | 0.838 | – | – | – | – |
| 4mCPred_I | 0.810 | 0.907 | 0.900 | 0.914 | – | – | – | – | |
| 4mCPred_II | 0.820 | 0.909 | 0.903 | 0.915 | – | – | – | – | |
| 4mcPred-SVM | 0.721 | 0.860 | 0.863 | 0.858 | – | – | – | – | |
| Meta-4mCpred | 0.782 | 0.891 | 0.884 | 0.898 | 0.700 | 0.850 | 0.835 | 0.865 | |
| 4mcPred-IFL | 0.812 | 0.906 | 0.902 | 0.910 | – | – | – | – | |
| 4mCCNN | 0.750 | 0.872 | 0.858 | 0.893 | – | – | – | – | |
| DNA4mC-LIP | – | – | – | – | 0.624 | 0.811 | 0.773 | 0.849 | |
| M. musculus | 4mCpred-EL | 0.591 | 0.795 | 0.804 | 0.787 | 0.596 | 0.798 | 0.804 | 0.792 |
| i4mC-Mouse | 0.651 | 0.793 | 0.683 | 0.902 | 0.633 | 0.816 | 0.807 | 0.825 | |
| F. vesca | iDNA-MS | 0.868 | 0.934 | 0.943 | 0.925 | 0.648 | 0.824 | 0.830 | 0.818 |
| i4mC-ROSE | 0.545 | 0.767 | 0.635 | 0.899 | 0.601 | 0.797 | 0.721 | 0.873 | |
The first and second columns respectively represent the species and method names. The third and fourth columns respectively represent cross-validation performance reported in the literature based on the training dataset and independent test performance reported in the literature. MCC, Matthews correlation coefficient; ACC, accuracy; Sn, sensitivity; Sp, specificity.
4mC Prediction Methods Reported in 2018
Two methods were proposed in 2018, relying on different approaches to predict 4mC sites in different species. Both methods utilize the Chen dataset and SVM for model construction.
4mCPred
He et al.20 proposed 4mCPred that consists of two prediction models (4mCPred_I and 4mCPred_II) built using different input features. 4mCPred_I was developed using single-feature descriptors, namely, position-specific trinucleotide propensity (PSTNP) features that achieved accuracies of 87.0%, 86.94%, 82.25%, 94.46%, 89.95%, and 90.69% for C. elegans, D. melanogaster, A. thaliana, E. coli, Geoa. subterraneus, and Geob. pickeringii, respectively. 4mCPred_II was developed using hybrid features (a combination of PSTNP and EIIP), where the optimal feature set was identified by a two-step feature selection protocol, with the features ranked based on F-scores, followed by SFS using SVM. 4mCPred_II achieved accuracies of 87.71%, 87.79%, 83.37%, 94.97%, 91.04%, and 90.89% for C. elegans, D. melanogaster, A. thaliana, E. coli, Geoa. subterraneus, and Geob. pickeringii, respectively. The performance of both 4mCPred models is superior to that of iDNA4mC. They are freely accessible at http://server.malab.cn/4mCPred/index.jsp.
4mcPred-SVM
Wei et al.21 proposed another predictor, 4mcPred-SVM. The authors generated a 700D feature vector by integrating four different sequence-based features, namely Kmer (336D), MBE (164D), DBE (160D), and local position-specific dinucleotide frequency (LPDF) (40D). A two-step feature selection protocol was applied to the 700D vector, and optimal feature subsets were identified individually for six species, yielding the average ACC, MCC, Sn, and Sp values of 0.654, 0.827, 0.834, and 0.821, respectively. Furthermore, the authors showed that the performance of 4mCPred was overestimated because of over-fitting. Hence, they rebuilt the 4mCPred model. The reported that metrics for the six species, i.e., the average ACC, MCC, Sn, and Sp, were 0.637, 0.817, 0.815, and 0.818, respectively. Overall, the performance of 4mcPred-SVM was superior to that of the above two methods. 4mcPred-SVM is publicly accessible at http://server.malab.cn/4mcPred-SVM.
4mC Prediction Methods Reported in 2019
Five methods were proposed in 2019. Of these, three methods were proposed simultaneously using different approaches that predict 4mC sites in six different species. Notably, all of these methods essentially relied on the same Chen dataset for prediction model development.
Meta-4mCpred
Manavalan et al.8 reported the first meta-predictor, called Meta-4mCpred. First, 14 feature descriptors were generated by exploring seven feature descriptors (Kmer, MBE, DBE, LPDF, RHCP, DPCP, and TPCP) and seven hybrid features (different combinations of the seven feature descriptors). Consequently, 14 feature descriptors were input into four different ML classifiers (RF, SVM, gradient boosting [GB], and extremely randomized tree [ERT]) and the corresponding optimal models were developed. From these models, the predicted probabilities of 4mCs were again input to SVM, and the final predictor was developed. Unlike other existing methods, an independent dataset was constructed using the same protocol as that described for iDNA4mC, comprising 1,500, 2,000, 2,500, 268, 700, and 400 samples for C. elegans, D. melanogaster, A. thaliana, E. coli, Geoa. subterraneous, and Geob. pickeringii, respectively. Among the individual species samples, 4mCs and non-4mCs were equally distributed, and these data were used to check model transferability. For the six species, Meta-4mCpred achieved the average MCC, ACC, Sn, and Sp values of 0.685, 0.842, 0.884, and 0.898, respectively, during cross-validation. The corresponding values for independent evaluations were 0.719, 0.859, 0.848, and 0.879, respectively. Both training and independent evaluations revealed that Meta-4mCpred outperformed the other three predictors. Notably, this was the first instance where different methods were evaluated using an independent dataset for six different species. Meta-4mCpred is freely accessible at http://thegleelab.org/Meta-4mCpred.
4mcPred-IFL
Wei et al.22 used an iterative feature representation (IFR) algorithm and developed a novel predictor, 4mcPred-IFL. The authors considered eight different feature descriptors, including a combination of Kmer and MBE, a combination of DPE and LPDF, PCP, PseDNC, KNN, EIIP, multivariate mutual information, and RFHCP. Essentially, IFR involved three steps, i.e., (1) a two-step feature selection protocol and optimization of each feature descriptor, for which a corresponding optimal feature set-based SVM model was developed; (2) concatenation of the predicted probabilities of 4mCs from eight models (step 1), which was considered a new feature vector; and (3) training of the 8D vector (step ) using SVM, which generated a new model for which the predicted probabilities of 4mCs were combined with the original 8D to establish a new 9D vector. This process was repeated until the performance reached convergence. For the six species, 4mCpred-IFL achieved the average MCC, ACC, Sn, and Sp values of 0.754, 0.877, 0.876, and 0.878, respectively. The performance of 4mCpred-IFL was better than that of iDNA4mC, 4mCPred, and 4mcPred-SVM. This indicated that the features learned through the iterative process had a reasonable capacity to discriminate between positive and negative samples. 4mcPred-IFL is publicly accessible at http://server.malab.cn/4mcPred-IFL.
4mCCNN
Khanal et al.23 proposed 4mCCNN, the first deep-learning-based method, based on MBE encoding and a convolutional neural network (CNN) classifier. The authors applied 10-fold cross-validation and optimized CNN regularization parameters. For the six species, 4mCCNN achieved the average MCC, ACC, Sn, and Sp values of 0.691, 0.847, 0.858, and 0.893, respectively. The cross-validation performance of 4mCCNN was superior to that of iDNA4mC, 4mCPred, and 4mcPred-SVM. The authors also constructed a web server, accessible at https://home.jbnu.ac.kr/NSCL/4mCCNN.htm. However, the 4mCCNN server was out of service during the preparation of the current manuscript.
4mCpred-EL
Manavalan et al.24 proposed 4mCpred-EL, the first 4mC site prediction method for M. musculus. First, the training and independent datasets were constructed, comprising 1,600 and 320 samples, respectively; each dataset contained an equal number of positive and negative samples. Notably, none of the samples shared greater than 80% sequence identity with other samples in the dataset. Subsequently, seven different feature descriptors (MBE, DPCP, EIIP, Kmer, a combination of DPE and LPDF, RFHCP, and TPCP) and four different ML classifiers (RF, GB, ERT, and SVM) were used to develop a prediction model. The model was developed with the following steps: (1) development of 28 prediction models based on seven descriptors and four classifiers, combining the predicted probabilities of 4mCs, and considering them a new feature vector; and (ii) inputting the 28D vector into four classifiers again, with the majority voting from the ensemble classifiers considered as the final output. 4mCpred-EL yielded MCC, ACC, Sn, and Sp values of 0.591, 0.795, 0.804, and 0.787, respectively, during cross-validation. The corresponding metrics for independent evaluation were 0.596, 0.798, 0.804, and 0.792, respectively. This method is publicly accessible at http://thegleelab.org/Meta-4mCpred/.
i4mC-ROSE
Hasan et al.25 proposed i4mC-ROSE, the first 4mC site prediction method for F. vesca and Rosa chinensis. Because the current study focuses on F. vesca, only information for this species model is provided. The authors constructed nonredundant training and independent datasets comprising 9,708 and 3,234 samples, respectively; positive samples were obtained from the MDR database,39 and negative samples were derived as described for iDNA4mC.19 Notably, none of the samples shared greater than 65% sequence identity with other samples. Subsequently, six different feature descriptors (K-space nucleotide composition [KSNC], MBE, EIIP, Kmer, DPCP, and TPCP) and RF classifier were used for model construction, which was achieved as follows: (1) development of RF-based six descriptor models; (2) integration of the predicted probabilities of 4mCs using a linear regression approach; and (3) choosing three models, KSNC, MBE, and EIIP, which contributed 25%, 45%, and 30%, respectively, for the final prediction. i4mC-ROSE achieved MCC, ACC, Sn, and Sp values of 0.545, 0.767, 0.635, and 0.899, respectively, during cross-validation. The corresponding metrics for independent evaluations were 0.601, 0.797, 0.721, and 0.873, respectively (Table 3). i4mC-ROSE is freely accessible at http://kurata14.bio.kyutech.ac.jp/i4mC-ROSE/.
4mC Prediction Methods Developed in 2020
As of April 2020, five additional methods were published for different species and approaches. A brief description of each method is given below.
iEC4mC-SVM
Lv et al.26 developed a predictor for E. coli, termed iEC4mC-SVM. The authors used the E. coli dataset of Chen et al.19 for model building and that of Manavalan et al.8 for checking model transferability. Using the training dataset, the authors considered six different descriptors, namely, MBE, DBE, DAC, DCC, RFHCP, and Xmer-kGap-Ymer composition frequency. All of these descriptors were concatenated to generate a 10060D feature vector. Subsequently, a light gradient-boosting machine was applied to rank the features; the top 250 features were selected from the sorted features, ranked from highest to lowest. SFS was applied to these 250D features. An optimal feature set (187D) was thus obtained, with the MCC, ACC, Sn, and Sp values of 0.711, 0.854, 0.820, and 0.889, respectively, during cross-validation. The corresponding metrics for independent evaluations were 0.665, 0.832, 0.851, and 0.813, respectively. The performance of iEC4mC-SVM was similar to that of Meta-4mCpred, but the metrics were significantly worse than those of the other three methods (4mCPred, 4mcPred-IFL, and 4mcPred-SVM) during cross-validation. However, iEC4mC-SVM was slightly better than 4mCPred and Meta-4mCpred, and significantly better than 4mcPred-IFL and 4mcPred-SVM during independent assessment. Unfortunately, iEC4mC-SVM is not publicly available.
DNA4mC-LIP
Tang et al.27 proposed a novel meta-predictor, called DNA4mC-LIP. Instead of developing a prediction model based on a training dataset, the authors utilized an independent dataset (for six species) of Manavalan et al.8 and generated predicted probabilities of 4mC scores from the six existing models, namely, iDNA4mC, 4mCPred_I, 4mCPred_II, 4mcPred-SVM, 4mcPred-IFL, and Meta-4mCpred. Subsequently, the predicted probabilities of 4mCs were integrated using an optimal weight to make a final prediction. For each species, the number of existing predictor contributions varied significantly. In DNA4mC-LIP, the authors did not use any ML classifiers. Instead, they used existing ML-based prediction methods; hence, the DNA4mC-LIP method is classified as an ML-based method in the current study. For the six species, DNA4mC-LIP achieved the average MCC, ACC, Sn, and Sp values of 0.721, 0.860 0.850, and 0.870, respectively. The predictor performs slightly better than the individual predictors. DNA4mC-LIP is accessible at http://i.uestc.edu.cn/DNA4mC-LIP/.
4mcDeep-CBI
Zeng et al.1 developed 4mCDeep-CBI, a predictor for E. coli. The authors constructed a new imbalanced dataset containing 11,173 4mCs and 6635 non-4mCs. Subsequently, the authors used eight feature descriptors (as for 4mCpred-IFL) input into CNN and a bidirectional long short-term memory network for the development of the final prediction model. 4mcDeep-CBI achieved MCC, ACC, Sn, and Sp values of 0.850, 0.929, 0.949, and 0.894 during cross-validation, with a significantly better performance than 4mCpred-IFL. Although the authors provided a standalone version of 4mcDeep-CBI (https://github.com/mat310/4mcDeep), they did not provide detailed information for executing this program in the READERME.md file. Therefore, this method was excluded from the current evaluation.
iDNA-MS
Lv et al.2 proposed iDNA-MS, a method that predicts not only 4mC sites but also 5-hydroxymethylcytosine and 6mA sites. In total, the authors reported 17 prediction models for different species. Considering the F. vesca-based model, the authors generated 7,899 and 7,898 samples, and used them for training and independent evaluations, respectively. Positive samples were extracted from MDR database, and negative samples were generated as in the iDNA4mC study.19 None of the sequences in the dataset shared greater than 80% sequence identity with other sequences. The RF-based MBE descriptor achieved MCC, ACC, Sn, and Sp values of 0.868, 0.934, 0.943, and 0.925, respectively. The corresponding metrics for independent evaluations were 0.648, 0.824, 0.830, and 0.818, respectively. iDNA-MS is publicly available at http://lin-group.cn/server/iDNA-MS.
i4mC-Mouse
Hasan et al.28 proposed a second tool for 4mC site prediction for M. musculus, termed i4mC-Mouse. The authors generated a stringent dataset (1,812 samples) by applying a CD-HIT of 0.7 using a previously reported 4mCpred-EL nonredundant dataset. Of these samples, 1,492 were treated as a training dataset for model building, and the remaining 320 were considered as an independent dataset for model evaluation. Subsequently, six different feature descriptors (MBE, KSNC, EIIP, DBE, Kmer, and DPCPs) and RF classifier were used for model construction by following the subsequent steps (1) development of RF-based six-descriptor-based models; (2) integration of the predicted probabilities of 4mCs via a linear regression approach; and (3) final estimation, using only four models, where Kmer, KSNC, MBE, and EIIP contributed 10%, 45%, 25%, and 20%, respectively. i4mC-Mouse achieved MCC, ACC, Sn, and Sp values of 0.651, 0.793, 0.683, and 0.902, respectively, during cross-validation, and the corresponding metrics for independent evaluation were 0.633, 0.816, 0.807, and 0.825, respectively. This method performed marginally better than 4mCpred-EL on both cross-validation and independent evaluation. i4mC-Mouse is freely available at http://kurata14.bio.kyutech.ac.jp/i4mC-Mouse/.
The comparison of ML algorithms and their performances with those of the existing methods (Tables 2 and 3) revealed the following similarities and dissimilarities. (1) Five methods (iDNA4mC, 4mCPred, 4mcPred-SVM, 4mcPred-IFL, and iEC4mC-SVM) used the SVM classifier; three algorithms (i4mC-ROSE, iDNA-MS, and i4mC-Mouse) used RF; two methods (4mCCNN and 4mcDeep-CBI) applied deep learning; and the remaining methods (Meta-4mCpred and 4mCpred-EL) utilized multiple ML classifiers. (2) Two methods (iDNA4mC ad 4mCCNN) used single-feature encoding, whereas the remaining methods (except for DNA4mC-LIP) explored multiple-feature encodings. While developing a prediction model, most methods used feature-selection techniques and identified the optimal feature set during model development. (3) Five existing models (Meta-4mCpred, 4mCpred-EL, i4mC-Mouse, iDNA-MS, and i4mC-Rose) evaluated method transferability using an independent dataset, whereas the remaining methods focused on improving the training ACC. (4) Based on the training ACC reported in the literature, 4mcDeep-CBI achieved the best performance for C. elegans; 4mCPred_II had the best performance for five different species (D. melanogaster, A. thaliana, E. coli, Geoa. subterraneus, and Geob. pickeringii); 4mCpred-EL and i4mC-Mouse achieved similar performance for M. musculus; and iDNA-MS achieved the best performance for F. vesca.
Evaluation Metrics
To quantify and evaluate the performance of the developed models, five commonly used evaluation metrics were used:71, 72, 73, 74 Sn, Sp, ACC, BAAC, and MCC. Each metric is defined as follows:
| (1) |
where TP is the number of 4mCs correctly predicted as 4mCs, TN is the number of non-4mCs correctly predicted as non-4mCs, FP is the number of 4mCs incorrectly predicted as non-4mCs, and FN is the number of non-4mCs incorrectly predicted as 4mCs.
Author Contributions
B.M. and G.L. conceived the project and designed the experiments. M.M.H., S.B., V.G., and T.-H.S. constructed the validation dataset. B.M., V.G., and S.B. performed the experiments and analyzed the data. B.M., M.M.H., and S.B. wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (MSIT) (2018R1D1A1B07049572, 2019R1I1A1A01062260, 2020R1A4A4079722, and 2020M3E5D9080661).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.010.
Contributor Information
Balachandran Manavalan, Email: bala@ajou.ac.kr.
Gwang Lee, Email: glee@ajou.ac.kr.
Supplemental Information
References
- 1.Zeng F., Fang G., Yao L. A deep neural network for identifying DNA N4-methylcytosine sites. Front. Genet. 2020;11:209. doi: 10.3389/fgene.2020.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lv H., Dao F.Y., Zhang D., Guan Z.X., Yang H., Su W., Liu M.L., Ding H., Chen W., Lin H. iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. iScience. 2020;23:100991. doi: 10.1016/j.isci.2020.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergman Y., Cedar H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013;20:274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg M.V.C., Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019;20:590–607. doi: 10.1038/s41580-019-0159-6. [DOI] [PubMed] [Google Scholar]
- 5.Smith Z.D., Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter M.A., Li M., Rathore A., Lackey L., Law E.K., Land A.M., Leonard B., Shandilya S.M., Bohn M.F., Schiffer C.A. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 2012;287:34801–34808. doi: 10.1074/jbc.M112.385161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bart A., van Passel M.W., van Amsterdam K., van der Ende A. Direct detection of methylation in genomic DNA. Nucleic Acids Res. 2005;33:e124. doi: 10.1093/nar/gni121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol. Ther. Nucleic Acids. 2019;16:733–744. doi: 10.1016/j.omtn.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathi P., Maurer S., Summerer D. Selective recognition of N4-methylcytosine in DNA by engineered transcription-activator-like effectors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018;373:20170078. doi: 10.1098/rstb.2017.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pataillot-Meakin T., Pillay N., Beck S. 3-Methylcytosine in cancer: an underappreciated methyl lesion? Epigenomics. 2016;8:451–454. doi: 10.2217/epi.15.121. [DOI] [PubMed] [Google Scholar]
- 11.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 12.Casadesús J., Low D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006;70:830–856. doi: 10.1128/MMBR.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timinskas A., Butkus V., Janulaitis A. Sequence motifs characteristic for DNA [cytosine-N4] and DNA [adenine-N6] methyltransferases. Classification of all DNA methyltransferases. Gene. 1995;157:3–11. doi: 10.1016/0378-1119(94)00783-o. [DOI] [PubMed] [Google Scholar]
- 14.Schweizer H. Bacterial genetics: past achievements, present state of the field, and future challenges. Biotechniques. 2008;44:633–634, 636–641. doi: 10.2144/000112807. [DOI] [PubMed] [Google Scholar]
- 15.Iyer L.M., Abhiman S., Aravind L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- 16.Modrich P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 17.Flusberg B.A., Webster D.R., Lee J.H., Travers K.J., Olivares E.C., Clark T.A., Korlach J., Turner S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu M., Ji L., Neumann D.A., Chung D.H., Groom J., Westpheling J., He C., Schmitz R.J. Base-resolution detection of N4-methylcytosine in genomic DNA using 4mC-Tet-assisted-bisulfite- sequencing. Nucleic Acids Res. 2015;43:e148. doi: 10.1093/nar/gkv738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W., Yang H., Feng P., Ding H., Lin H. iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics. 2017;33:3518–3523. doi: 10.1093/bioinformatics/btx479. [DOI] [PubMed] [Google Scholar]
- 20.He W., Jia C., Zou Q. 4mCPred: machine learning methods for DNA N4-methylcytosine sites prediction. Bioinformatics. 2019;35:593–601. doi: 10.1093/bioinformatics/bty668. [DOI] [PubMed] [Google Scholar]
- 21.Wei L., Luan S., Nagai L.A.E., Su R., Zou Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics. 2019;35:1326–1333. doi: 10.1093/bioinformatics/bty824. [DOI] [PubMed] [Google Scholar]
- 22.Wei L., Su R., Luan S., Liao Z., Manavalan B., Zou Q., Shi X. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics. 2019;35:4930–4937. doi: 10.1093/bioinformatics/btz408. [DOI] [PubMed] [Google Scholar]
- 23.Khanal J., Nazari I., Tayara H., Chong K.T. 4mCCNN: identification of N4-methylcytosine sites in prokaryotes using convolutional neural network. IEEE Access. 2019;7:145455–145461. [Google Scholar]
- 24.Manavalan B., Basith S., Shin T.H., Lee D.Y., Wei L., Lee G. 4mCpred-EL: an ensemble learning framework for identification of DNA N4-methylcytosine sites in the mouse genome. Cells. 2019;8:1332. doi: 10.3390/cells8111332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasan M.M., Manavalan B., Khatun M.S., Kurata H. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 2020;157:752–758. doi: 10.1016/j.ijbiomac.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Lv Z., Wang D., Ding H., Zhong B., Xu L. Escherichia coli DNA N-4-methycytosine site prediction accuracy improved by light gradient boosting machine feature selection technology. IEEE Access. 2020;8:14851–14859. [Google Scholar]
- 27.Tang Q., Kang J., Yuan J., Tang H., Li X., Lin H., Huang J., Chen W. DNA4mC-LIP: a linear integration method to identify N4-methylcytosine site in multiple species. Bioinformatics. 2020;36:3327–3335. doi: 10.1093/bioinformatics/btaa143. [DOI] [PubMed] [Google Scholar]
- 28.Hasan M.M., Manavalan B., Shoombuatong W., Khatun M.S., Kurata H. i4mC-Mouse: improved identification of DNA N4-methylcytosine sites in the mouse genome using multiple encoding schemes. Comput. Struct. Biotechnol. J. 2020;18:906–912. doi: 10.1016/j.csbj.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanley J.A., McNeil B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 30.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sessions V., Valtorta M. Proceedings of the 11th International Conference on Information Quality. 2006. The effects of data quality on machine learning algorithms; pp. 485–498. [Google Scholar]
- 32.Wei L., Zhou C., Su R., Zou Q. PEPred-Suite: improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics. 2019;35:4272–4280. doi: 10.1093/bioinformatics/btz246. [DOI] [PubMed] [Google Scholar]
- 33.Hasan M.M., Schaduangrat N., Basith S., Lee G., Shoombuatong W., Manavalan B. HLPpred-Fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics. 2020;36:3350–3356. doi: 10.1093/bioinformatics/btaa160. [DOI] [PubMed] [Google Scholar]
- 34.Cao R., Bhattacharya D., Hou J., Cheng J. DeepQA: improving the estimation of single protein model quality with deep belief networks. BMC Bioinformatics. 2016;17:495. doi: 10.1186/s12859-016-1405-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao R., Adhikari B., Bhattacharya D., Sun M., Hou J., Cheng J. QAcon: single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics. 2017;33:586–588. doi: 10.1093/bioinformatics/btw694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conover M., Staples M., Si D., Sun M., Cao R. AngularQA: protein model quality assessment with LSTM networks. Comput. Math. Biophys. 2019;7:1–9. [Google Scholar]
- 37.Smith J., Conover M., Stephenson N., Eickholt J., Si D., Sun M., Cao R. TopQA: a topological representation for single-model protein quality assessment with machine learning. Int. J. Comput. Biol. Drug Des. 2020;13:144–153. [Google Scholar]
- 38.Ye P., Luan Y., Chen K., Liu Y., Xiao C., Xie Z. MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 2017;45(D1):D85–D89. doi: 10.1093/nar/gkw950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z.Y., Xing J.F., Chen W., Luan M.W., Xie R., Huang J., Xie S.Q., Xiao C.L. MDR: an integrative DNA N6-methyladenine and N4-methylcytosine modification database for Rosaceae. Hortic. Res. 2019;6:78. doi: 10.1038/s41438-019-0160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z.Y., Yang Y.H., Ding H., Wang D., Chen W., Lin H. Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief. Bioinform. 2020 doi: 10.1093/bib/bbz177. Published online January 28, 2020. [DOI] [PubMed] [Google Scholar]
- 42.Yang H., Yang W., Dao F.Y., Lv H., Ding H., Chen W., Lin H. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 2019 doi: 10.1093/bib/bbz123. Published online October 21, 2019. [DOI] [PubMed] [Google Scholar]
- 43.Feng C.Q., Zhang Z.Y., Zhu X.J., Lin Y., Chen W., Tang H., Lin H. iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. 2019;35:1469–1477. doi: 10.1093/bioinformatics/bty827. [DOI] [PubMed] [Google Scholar]
- 44.Dao F.Y., Lv H., Wang F., Feng C.Q., Ding H., Chen W., Lin H. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. 2019;35:2075–2083. doi: 10.1093/bioinformatics/bty943. [DOI] [PubMed] [Google Scholar]
- 45.Dao F.Y., Lv H., Zulfiqar H., Yang H., Su W., Gao H., Ding H., Lin H. A computational platform to identify origins of replication sites in eukaryotes. Brief. Bioinform. 2020 doi: 10.1093/bib/bbaa017. Published online February 17, 2020. [DOI] [PubMed] [Google Scholar]
- 46.Lai H.Y., Zhang Z.Y., Su Z.D., Su W., Ding H., Chen W., Lin H. iProEP: a computational predictor for predicting promoter. Mol. Ther. Nucleic Acids. 2019;17:337–346. doi: 10.1016/j.omtn.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. AtbPpred: a robust sequence-based prediction of anti-tubercular peptides using extremely randomized trees. Comput. Struct. Biotechnol. J. 2019;17:972–981. doi: 10.1016/j.csbj.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu M., Zhang J., Li Y., Wang C., Liu Z., Ding H., Zou Q., Ma Q. CirRNAPL: a web server for the identification of circRNA based on extreme learning machine. Comput. Struct. Biotechnol. J. 2020;18:834–842. doi: 10.1016/j.csbj.2020.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z.M., Tan J.X., Wang F., Dao F.Y., Zhang Z.Y., Lin H. Early diagnosis of hepatocellular carcinoma using machine learning method. Front. Bioeng. Biotechnol. 2020;8:254. doi: 10.3389/fbioe.2020.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics. 2019;35:2757–2765. doi: 10.1093/bioinformatics/bty1047. [DOI] [PubMed] [Google Scholar]
- 51.Wei L., Hu J., Li F., Song J., Su R., Zou Q. Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. 2020;21:106–119. doi: 10.1093/bib/bby107. [DOI] [PubMed] [Google Scholar]
- 52.Wei L., Zhou C., Chen H., Song J., Su R. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics. 2018;34:4007–4016. doi: 10.1093/bioinformatics/bty451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y., Zhao X., Liu S., Liu S., Niu Y., Zhang W., Wei L. 2019. LncPred-IEL: a long non-coding RNA prediction method using iterative ensemble learning. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) pp. 555–562. [Google Scholar]
- 54.Manavalan B., Subramaniyam S., Shin T.H., Kim M.O., Lee G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018;17:2715–2726. doi: 10.1021/acs.jproteome.8b00148. [DOI] [PubMed] [Google Scholar]
- 55.Tan J.X., Li S.H., Zhang Z.M., Chen C.X., Chen W., Tang H., Lin H. Identification of hormone binding proteins based on machine learning methods. Math. Biosci. Eng. 2019;16:2466–2480. doi: 10.3934/mbe.2019123. [DOI] [PubMed] [Google Scholar]
- 56.Basith S., Manavalan B., Shin T.H., Lee G. SDM6A: a web-based integrative machine-learning framework for predicting 6mA Sites in the rice genome. Mol. Ther. Nucleic Acids. 2019;18:131–141. doi: 10.1016/j.omtn.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J., Li J., Yang B., Xie R., Marquez-Lago T.T., Leier A., Hayashida M., Akutsu T., Zhang Y., Chou K.C. Bastion3: a two-layer ensemble predictor of type III secreted effectors. Bioinformatics. 2019;35:2017–2028. doi: 10.1093/bioinformatics/bty914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J., Yang B., Leier A., Marquez-Lago T.T., Hayashida M., Rocker A., Zhang Y., Akutsu T., Chou K.C., Strugnell R.A. Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics. 2018;34:2546–2555. doi: 10.1093/bioinformatics/bty155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu X., Cai L., Zeng X., Zou Q. StackCPPred: a stacking and pairwise energy content-based prediction of cell-penetrating peptides and their uptake efficiency. Bioinformatics. 2020;36:3028–3034. doi: 10.1093/bioinformatics/btaa131. [DOI] [PubMed] [Google Scholar]
- 60.Xiong Y., Wang Q., Yang J., Zhu X., Wei D.Q. PredT4SE-Stack: prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 2018;9:2571. doi: 10.3389/fmicb.2018.02571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang X., Hu L., Shen P., Li R., Liu D. SMRT sequencing revealed mitogenome characteristics and mitogenome-wide DNA modification pattern in Ophiocordyceps sinensis. Front. Microbiol. 2017;8:1422. doi: 10.3389/fmicb.2017.01422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu G., Jiang Y.M., Liu Y.C., Han L.L., Feng H. A novel DNA methylation motif identified in Bacillus pumilus BA06 and possible roles in the regulation of gene expression. Appl. Microbiol. Biotechnol. 2020;104:3445–3457. doi: 10.1007/s00253-020-10475-5. [DOI] [PubMed] [Google Scholar]
- 63.Cao R., Freitas C., Chan L., Sun M., Jiang H., Chen Z. ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules. 2017;22:1732. doi: 10.3390/molecules22101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Si D., Moritz S.A., Pfab J., Hou J., Cao R., Wang L., Wu T., Cheng J. Deep learning to predict protein backbone structure from high-resolution cryo-EM density maps. Sci. Rep. 2020;10:4282. doi: 10.1038/s41598-020-60598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen W., Feng P., Liu T., Jin D. Recent advances in machine learning methods for predicting heat shock proteins. Curr. Drug Metab. 2019;20:224–228. doi: 10.2174/1389200219666181031105916. [DOI] [PubMed] [Google Scholar]
- 66.Chen W., Feng P., Song X., Lv H., Lin H. iRNA-m7G: identifying N7-methylguanosine sites by fusing multiple features. Mol. Ther. Nucleic Acids. 2019;18:269–274. doi: 10.1016/j.omtn.2019.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu K., Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics. 2020;36:3336–3342. doi: 10.1093/bioinformatics/btaa155. [DOI] [PubMed] [Google Scholar]
- 68.Hasan M.M., Manavalan B., Shoombuatong W., Khatun M.S., Kurata H. i6mA-Fuse: improved and robust prediction of DNA 6 mA sites in the Rosaceae genome by fusing multiple feature representation. Plant Mol. Biol. 2020;103:225–234. doi: 10.1007/s11103-020-00988-y. [DOI] [PubMed] [Google Scholar]
- 69.Liu Q., Chen J., Wang Y., Li S., Jia C., Song J., Li F. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief. Bioinform. 2020 doi: 10.1093/bib/bbaa124. Published online July 1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu H., Jia P., Zhao Z. Deep4mC: systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Brief. Bioinform. 2020 doi: 10.1093/bib/bbaa099. Published online June 24, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Basith S., Manavalan B., Hwan Shin T., Lee G. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med. Res. Rev. 2020;40:1276–1314. doi: 10.1002/med.21658. [DOI] [PubMed] [Google Scholar]
- 72.Su R., Hu J., Zou Q., Manavalan B., Wei L. Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief. Bioinform. 2020;21:408–420. doi: 10.1093/bib/bby124. [DOI] [PubMed] [Google Scholar]
- 73.Zhu X.J., Feng C.Q., Lai H.Y., Chen W., Lin H. Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl.-Based Syst. 2019;163:787–793. [Google Scholar]
- 74.Liu M.L., Su W., Guan Z.X., Zhang D., Chen W., Liu L., Ding H. An overview on predicting protein subchloroplast localization by using machine learning methods. Curr. Protein Pept. Sci. 2020 doi: 10.2174/1389203721666200117153412. Published online January 17, 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.