

Abstract
Oral delivery of protein drugs is considered a life-changing solution for patients who require regular needle injections. However, clinical translation of oral protein formulations has been hampered by inefficient penetration of drugs through the intestinal mucus and epithelial cell layer, leading to low absorption and bioavailability, and safety concerns owing to tight junction openings. Here we report a zwitterionic micelle platform featuring a virus-mimetic zwitterionic surface, a betaine side chain and an ultra-low critical micelle concentration, enabling drug penetration through the mucus and efficient transporter-mediated epithelial absorption without the need for tight junction opening. This micelle platform was used to fabricate a prototype oral insulin formulation by encapsulating a freeze-dried powder of zwitterionic micelle-insulin into an enteric-coated capsule. The biocompatible oral insulin formulation shows a high oral bioavailability of >40%, offers the possibility to fine-tune insulin acting profiles and provides long-term safety, enabling the oral delivery of protein drugs.
Oral delivery of proteins or peptides can potentially improve the quality of patients’ lives who routinely receive needle based injections of those therapeutic macromolecules.1, 2 Nevertheless, there is barely any protein drug formulation approved for oral administration, mostly due to the low bioavailability.3 For an oral protein drug to work, it must overcome three major physiological barriers. The first one is in stomach, where a protein drug needs to survive from the ultra-acidic pH (pH=1–3) and proteolytic enzymes that destabilize the formulation and denature/degrade the proteins. The second one is in intestine, where the protein drug needs to penetrate the mucus layers that protect the epithelial surfaces, and the third is to go across the epithelial cell layer of intestine to enter the blood. The former barrier has been effectively tackled by enzyme inhibitors and drug encapsulation (such as enteric coatings).3–5 The latter two barriers, however, pose a big challenge resulting in low bioavailability and poor therapeutic function of current oral protein drugs.3, 6, 7
The mucus layer and epithelial cell layer are key players of intestinal absorption and known for their protective functions of preventing foreign particles from entering.8–10 The continuously secreted mucus efficiently traps pathogens and foreign particles, and rapidly clears them from penetrating the epithelia.11–14 To address the mucus barrier, there is a significant interest in developing mucus-penetrating particle carriers to prevent the adherence to the mucus barrier and to avoid the rapid mucus clearance mechanisms.15, 16 State-of-the-art technology shows that coating particles with poly (ethylene glycol) (PEG) achieved a hydrophilic, neutral surface charge, reduced adsorption of mucins (major proteins in mucus) and improved the transport of particles within mucus by up to three orders of magnitude.15, 17–19 Despite these improvements, current oral delivery of protein drugs remains low in absorption and bioavailability.3, 6, 7, 20
To address the intestinal epithelial cell barrier, many absorption enhancers have been developed to promote the transport across the epithelial layer including surfactant type molecules21–23,24 and ionic liquid25. Nevertheless, majority of them relied on the mechanism of opening tight junctions to facilitate the paracellular transport, or target transcellular transport but inevitably damaged the tight junction21, 22. It should be noted that tight junctions account for a low amount of the intestine endothelial surface, but their opening associates with the risk of autoimmune disease, bacterial infection and inflammatory bowel diseases26, 27. There are other strategies involving cell receptors/transporters to facilitate transcellular delivery of the payload such as Fc receptor28 and bile acid transporter29, 30, but potential impact on the tight junctions has rarely been studied.
Here we report the major finding that the zwitterionic carrier platform was able to simultaneously address both mucus and epithelial barriers, and drastically enhance the transport of protein payload (insulin) without opening tight junctions or inducing leaky gut.
Although the mucus binds extremely efficiently and remove most invading particles, capsid viruses (e.g., Norwalk virus and human papillomavirus) are known for their unique, unhindered diffusion through mucus, which enables them to readily infect mucosal epithelia14, 15, 19. The external surface of capsid viruses is coated with equal densities of positive and negative charges (net-neutral) without any hydrophobic patches14. Such surface design effectively prevents the virus from interacting with mucins and facilitates its penetration through mucus14. We used zwitterionic betaine polymers to closely mimics the surface characteristics of capsid viruses (Figure 1a), and found that zwitterionic particles diffused in mucus nearly one order of magnitude faster than PEG particles at a comparable size.
Figure 1.
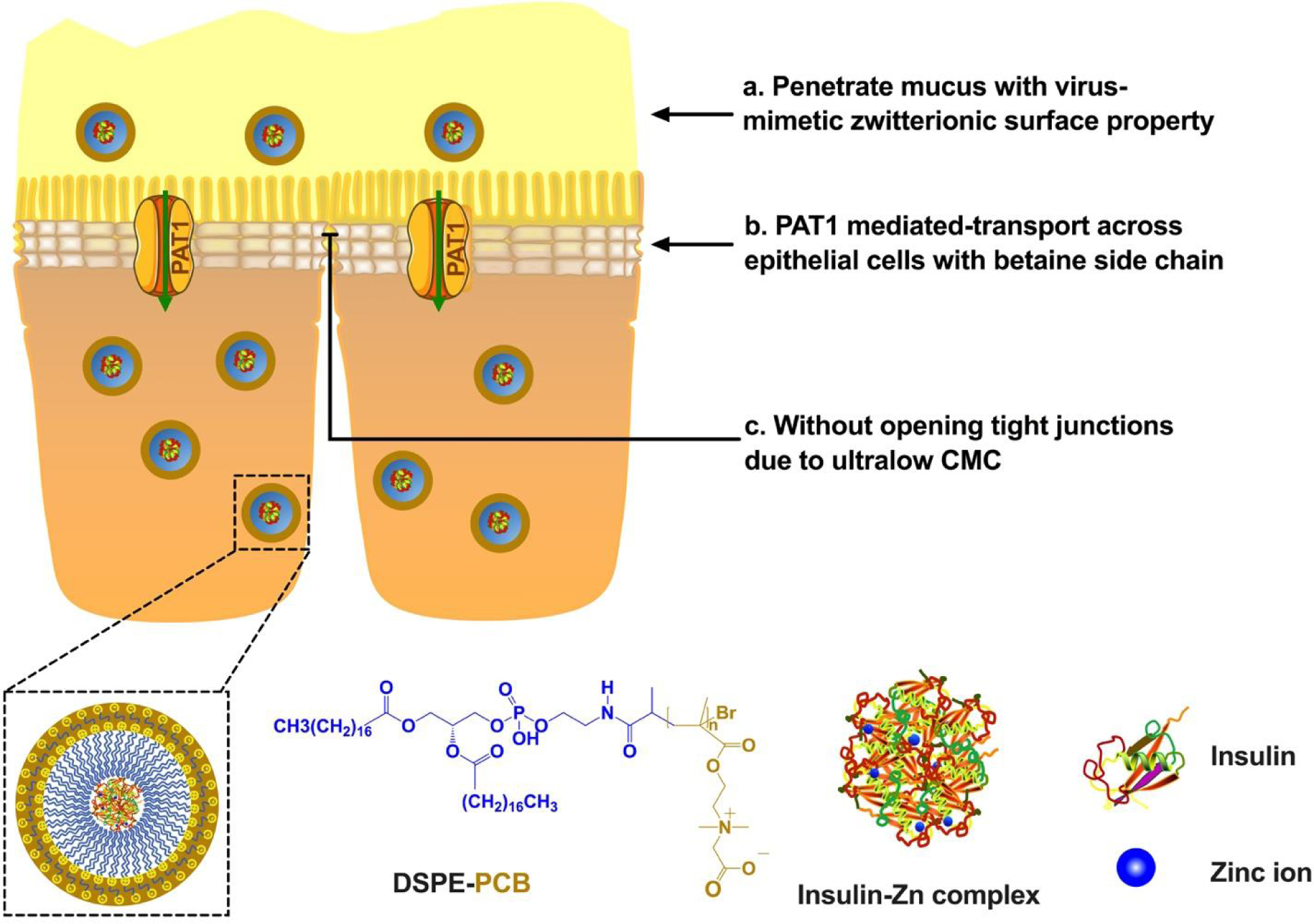
DSPE-PCB micelle platform addresses both the mucus (a) and the epithelial cell layer (b) barriers without opening tight junctions (c), contributing to an overall enhanced intestinal absorption.
We further encapsulated a model protein, insulin, with a zwitterionic betaine polymer micelle (DSPE-PCB: zwitterionic betaine polymer (PCB) of 5,000Da MW conjugated to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) lipid31, 32) for potential oral insulin delivery (Figure 1). This zwitterionic micelle formulation was found to penetrate across the epithelial cell layer through a pathway mediated by proton-assisted amino acid transporter 1 (PAT1) (Figure 1b). PAT1 is known for facilitating the penetration of betaine and betaine-derivatives through epithelial cells layer33, and expected to contribute to the enhanced transport of the betaine-containing DSPE-PCB/insulin formulation through the intestinal epithelium. This mechanism was supported by the in vivo observation that DSPE-PCB micelles were transported transcellularly rather than through tight junctions, and the in vitro assays showing drastically higher-level uptake of zwitterionic micelles by PAT1 overexpressing cells (Caco-2) and significant inhibitory uptake in presence of PAT1 substrates (betaine, L-tryptophan).
One key benefit of zwitterionic micelle DSPE-PCB is its ultra-low critical micelle concentration (CMC) below 10−6mM32. This implies low likelihood for DSPE-PCB to dissociate and play a detergent effect to the tight junction proteins and cell membranes, compared with common surfactants having orders of magnitude higher CMC values. Epithelial tissues treated with DSPE-PCB did not show any tight junction opening as indicated in a lactulose-mannitol intestinal permeability test and TEM characterization on intestinal epithelial tissues, similar to the result of healthy intestinal epithelium without micelle treatment (Figure 1c). Control surfactants polysorbate 80 (CMC=0.015mM) and sodium decanoate (CMC=50mM) significantly opened the intestinal tight junctions in this study. The sodium decanoate, a known tight junction opener34, 35, having the relatively higher CMC value, resulted in the largest level of the tight junction opening. Zwitterionic DSPE-PCB micelles potentially show a unique and superior safety profile for maintaining gut health for future oral formulations.
Lastly we fabricated a prototype of oral insulin in which a dry powder form of DSPE-PCB/insulin was packaged in an enteric-coated capsule and administered by oral gavage to diabetic rats. The freeze-drying process of DSPE-PCB/insulin formulation did not deteriorate the in vivo absorption performance even without the presence of cryoprotectants. The preparation of DSPE-PCB/insulin formulation is simple, even into a dried form, and can easily integrate with low-cost manufacturing processes. The oral insulin prototype achieved a bioavailability of 42.6% and the insulin adsorption profile (insulin action time period) can be fine-tuned by varying formulation parameters.
Zwitterionic particle increases transport through mucus.
We constructed nanogel particles made with zwitterionic monomers and other neutral, positive and negative charged monomers crosslinked with a fluorescent crosslinker (Figure 2a). This nanogel chemistry allows convenient preparation of particles that had comparable hydrodynamic size (~40 nm) but different zeta-potentials as determined by Dynamic Light Scattering (DLS) (Figure 2b). These particles were incubated with reconstituted porcine stomach mucus, and their diffusion trajectory was tracked under a spinning disc confocal fluorescent microscope. We found that the ensemble-averaged geometric mean square displacement (MSD) of the zwitterionic PCB particle was about 6.7 times of the PEG particle and 100 times or more of anionic and cationic particles (Figure 2c). These results indicate that zwitterionic particles diffuse much faster than the state-of-the-art mucus-penetrating PEG particles in the mucus.
Figure 2. Nanoparticles with virus-mimetic zwitterionic surface show the highest diffusive property in mucus compared with others including the PEG-based particles.
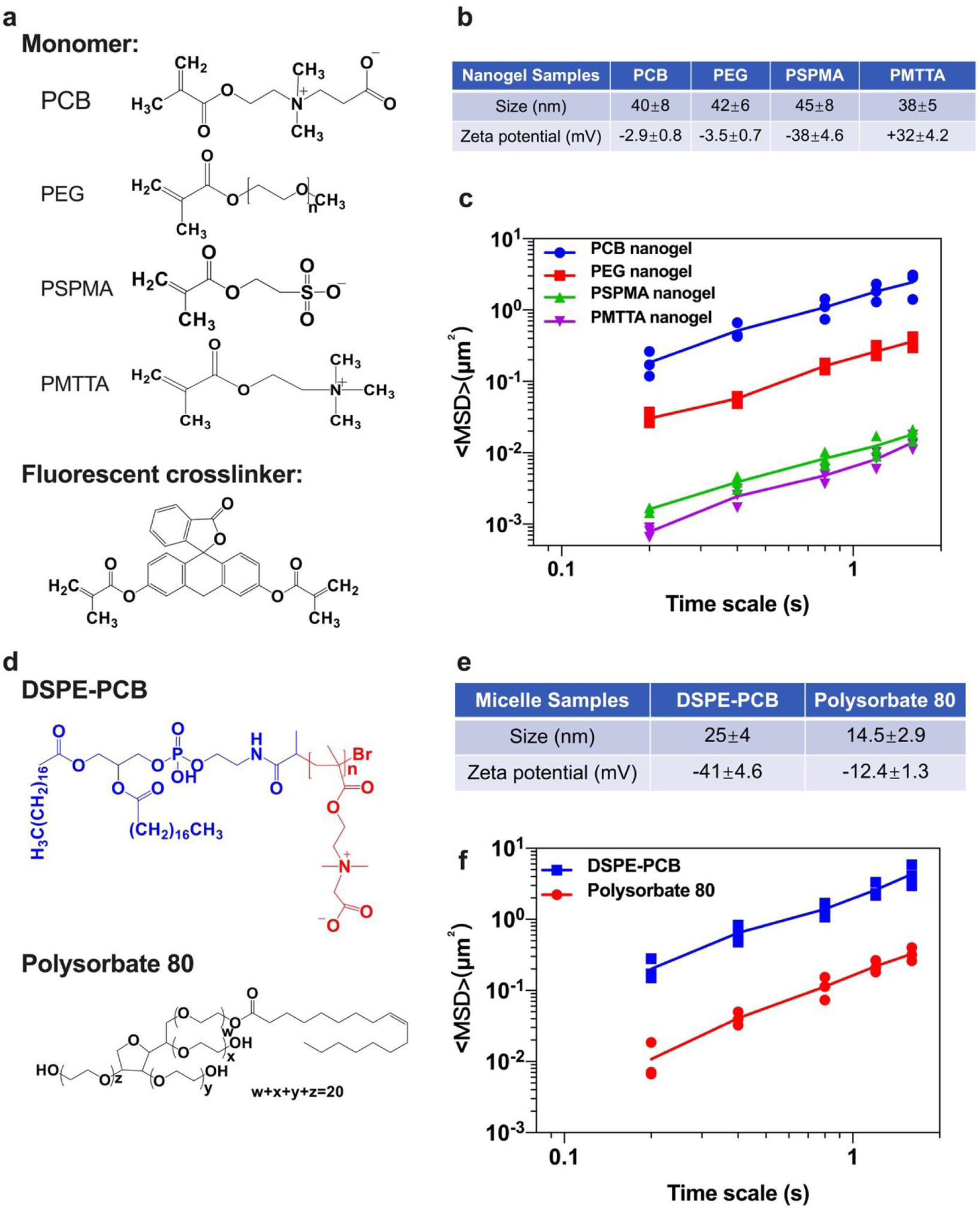
(a) Monomer and crosslinker structures for different nanogel particles. The nanogel particles were made from monomers for zwitterionic PCB (polycarboxybetaine), neutral nonionic PEG (polyethylene glycol), anionic PSPMA (poly 3-sulforpropyl methacrylate potassium salt), or cationic PMTTA (poly((2-(methacryloyloxy) ethyl) trimethylammonium chloride)), and crosslinked by a fluorescent dye (fluorescein-o’odemethacrylate). (b) Hydrodynamic size and zeta-potential for particle samples measured by DLS (N=3 independent samples, mean ± SD). (c) Transport of nanogels in reconstituted porcine stomach mucus. The graph shows the MSD as a function of time for different types of particles. Data represent three independent experiments with 50 particles tracked for each experiment (N=3 independent samples, means connected). (d) Structures for DSPE-PCB and Polysorbate 80 micelles. (e) Hydrodynamic size and zeta-potential for micelle samples with fluorescent payload encapsulated measured by DLS (N=3 independent samples, mean ± SD). (f) Transport of micelle samples in reconstituted porcine stomach mucus. The graph shows the MSD as a function of time for different types of particles. Data represent three independent experiments with 50 particles tracked for each experiment (N=3 independent samples, means connected).
We further constructed zwitterionic DSPE-PCB micelles with a fluorescent payload (polystyrene nanoparticle crosslinked by fluorescent dye) encapsulated using our previous reported method32, and compared with Polysorbate 80 micelles (containing PEG chains in the hydrophilic group) for their diffusive behaviors in the reconstituted porcine stomach mucus under the same test condition (Figure 2d,e). This is to examine the mucus-diffusive potential of DSPE-PCB which could be further utilized for drug delivery. Results showed that MSD of zwitterionic DSPE-PCB micelles was ~12 times larger than Polysorbate 80 micelles (Figure 2f). These results indicate that particles (both nanogel and micelles) with virus-mimetic zwitterionic surface property diffuse much faster than the state-of-the-art mucus-penetrating PEG based particles in mucus.
Zwitterionic micelle increases absorption via transporters.
The zwitterionic DSPE-PCB was used to encapsulate human recombinant insulin via a simple process where Zn2+ was slowly added with both DSPE-PCB and insulin in presence, and induced the insulin precipitation and the encapsulation by micelles. Both DSPE-PCB/insulin and Polysorbate 80/insulin were obtained with their hydrodynamic size below 30 nm as characterized by TEM and DLS (Figure 3a,b). The obtained micelle/insulin formulation was injected directly into the lumen of the ileum of streptozotocin (STZ)-induced C57BL/6 diabetic mice. Ileal administration was used to evaluate intestinal mucosal absorption in order to exclude the significant pH and enzyme degradation factors found in the stomach and duodenum36, which can be overcome by strategies such as enteric coatings, discussed next. We found that mouse blood glucose level was efficiently lowered by insulin delivered by DSPE-PCB—more efficiently than insulin encapsulated by PEG micelles (i.e., Polysorbate 80) (Figure 3c). Native insulin not encapsulated by any carrier hardly achieved any glucose-lowering effect, which was quantified by comparison with baseline blood glucose level of mice that received saline (Figure 3c). Insulin encapsulated by zwitterionic polymer micelles DSPE-PCB had a pharmacological activity of 41.2%, which is four times that of insulin loaded in Polysorbate 80, which was 10.6%.
Figure 3. Zwitterionic micelle/insulin formulation: characterization, high intestinal absorption efficacy, and transporter mediated absorptive mechanism.
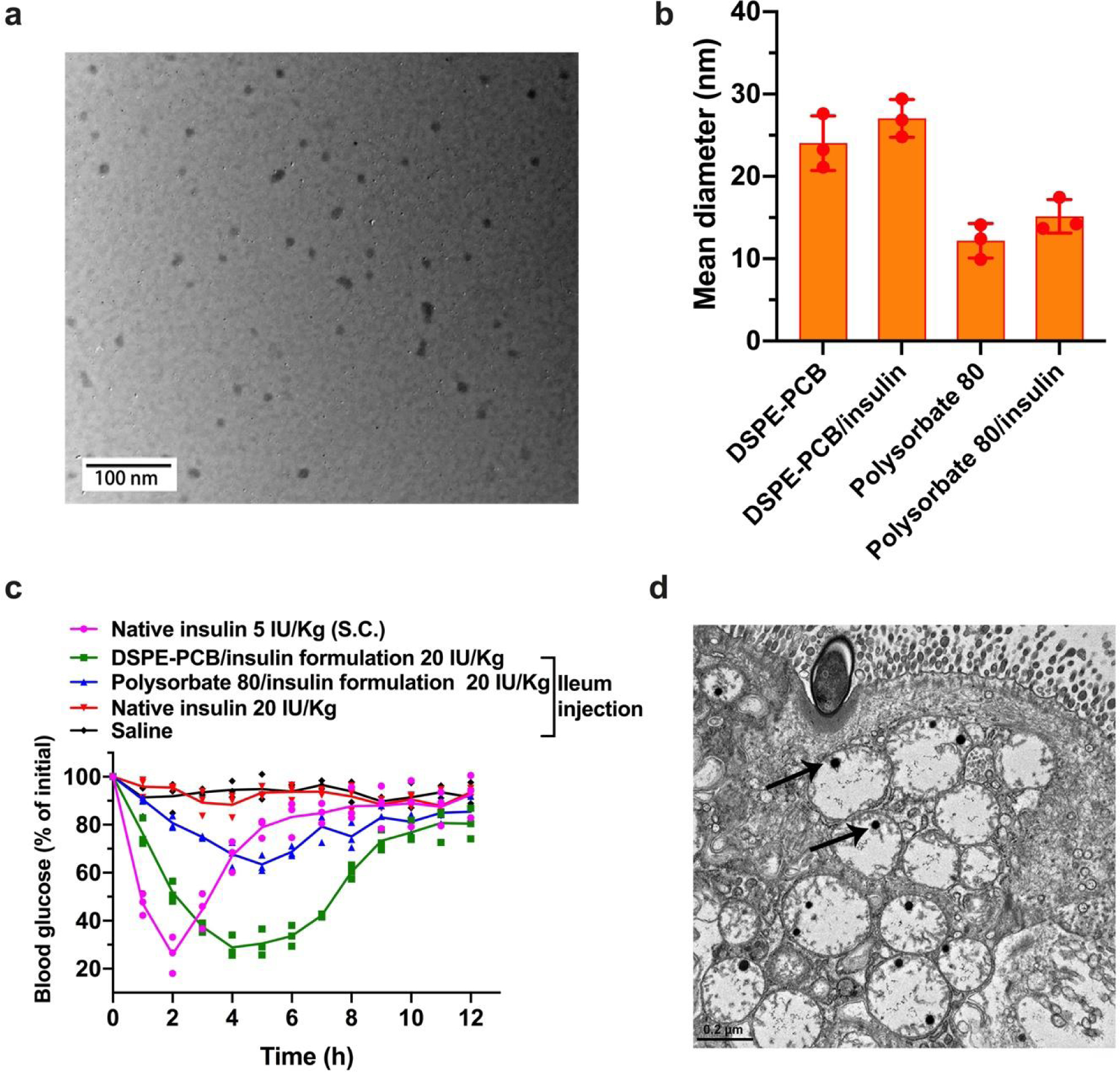
(a) Representative TEM image of DSPE-PCB/insulin formulation. This experiment was repeated independently three times with similar results. (b) Hydrodynamic size of different micelle, and micelle/insulin formulations (N=3 independent experiments, mean ± SD) measured by DLS. (c) Blood glucose lowering (pharmacological) performance for DSPE-PCB/insulin formulation on diabetic mice through ileum injection, compared with polysorbate 80/insulin formulation and non-formulated native insulin at the same dose of 20 IU/Kg (N=3 biologically independent animals, means connected). Insulin/ZnCl2 feeding ratio is 2.5/1 by weight. S.c. injected native insulin at 5 IU/Kg was used as a control (1 IU/ml). (d) Representative TEM image of epithelial tissues collected 1 hour after the ileum injection of DSPE-PCB/gold nanoparticles. Scale bar shown is 0.2 μm. This experiment was repeated independently three times with similar results.
The efficient intestinal absorption was hypothesized to be mediated by PAT1 transporter given that the surface of DSPE-PCB micelle contains a polymerized version of betaine, which is a known substrate for PAT133. To study the potential pathway for DSPE-PCB micelle to cross the epithelial layer, we constructed DSPE-PCB with gold nanoparticles encapsulated as previously reported32, and similarly administered the DSPE-PCB/gold through ileum injection. At one hour post injection, mice were sacrificed with epithelial tissue surrounding the injection site collected and processed for TEM imaging (Figure 3d). Gold nanoparticles located at the inside of the epithelial cells support potential transporter mediated pathway. We further conducted in vitro cellular uptake study of DSPE-PCB/fluorescently labeled polystyrene nanoparticles (previously used for mucus diffusion assay in Figure 2e,f) using a confocal laser scanning microscopy and flow cytometry. Significantly higher level cellular uptake of the DSPE-PCB micelle particle was observed on Caco-2 cells (human colon epithelial cells known for PAT1 overexpressing37) compared with regular 3T3 cells (Figure 4a,b,c). Such high level cellular uptake by Caco-2 was significantly inhibited with the presence of betaine or L-tryptophan, which are known as PAT1 substrates33 (Figure 4a,b,c). Polysorbate 80 was used as non-targeting control and no observable polysorbate 80 uptake by Caco-2 cells was found. Three-dimensional visualization of cells confirmed that the micelle particles (green) entered the cells (nucleus in blue) rather than binding to the cell membrane/surface (red) (Figure 4d).
Figure 4. In vitro cellular uptake of zwitterionic micelle particles showed a transporter-mediated mechanism.
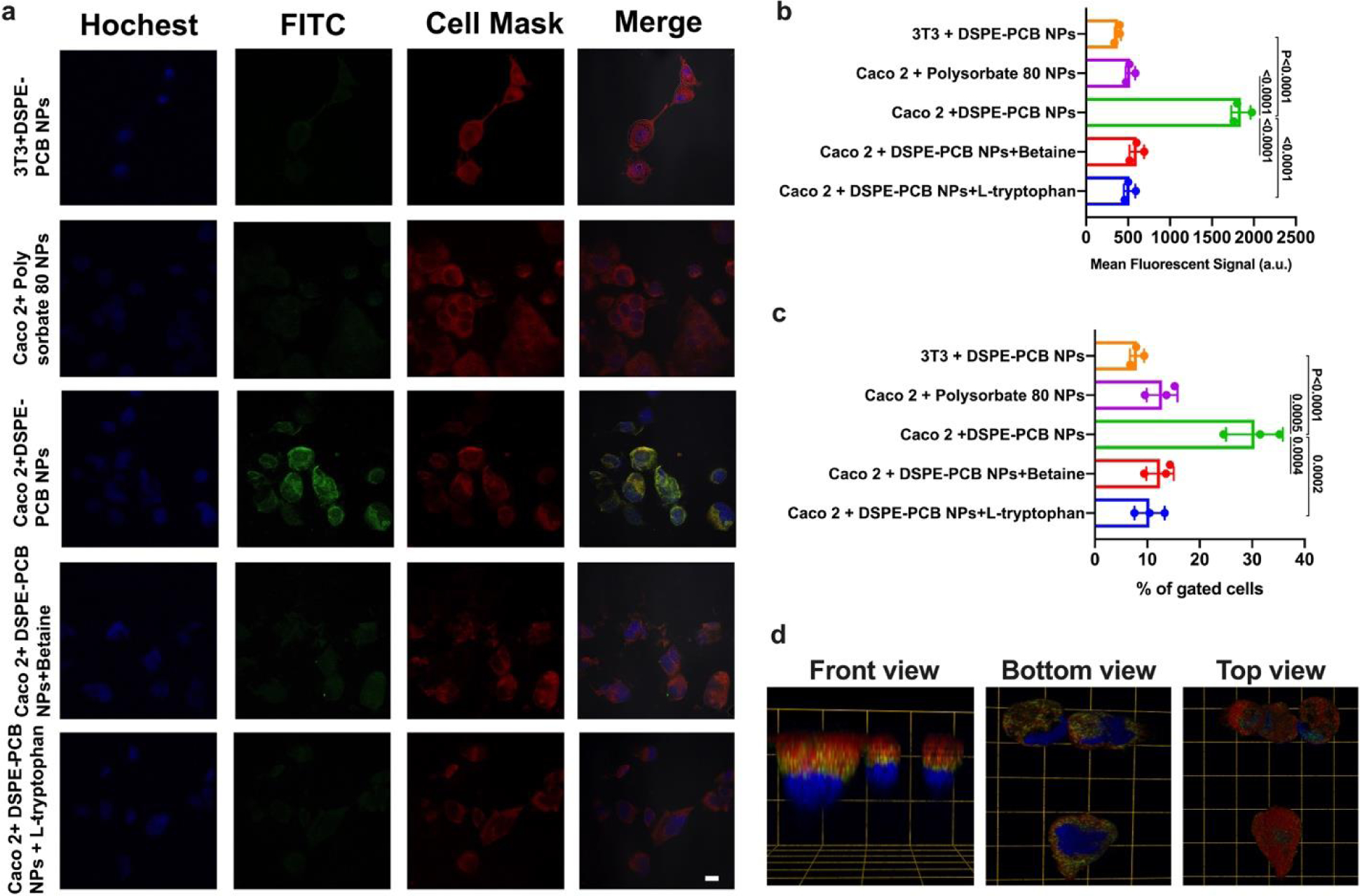
(a) Confocal laser scanning microscopy images (scale bar=20μm), (b) confocal and (c) flow cytometry quantitative data of zwitterionic DSPE-PCB and polysorbate 80 fluorescently labeled polystyrene nanoparticles incubated with 3T3 cells, Caco-2 cells, or Caco-2 cells treated with betaine or L-tryptophan (PAT1 substrates) (N=3 biologically independent samples, mean ± SD). A one-way analysis of variance with Tukey multi-comparison was used for statistical analysis. (d) 3D visualization of the Caco-2 cells incubated with DSPE-PCB/fluorescently labeled polystyrene nanoparticles (Grid unit length=20μm; Red: cell membrane, Green: the polystyrene nanoparticles, Blue: nucleus). (a) and (d) experiment was repeated independently three times with similar results.
Penetrates epithelium without opening tight junctions.
To examine potential impact on tight junctions by DSPE-PCB micelle, we co-administered lactulose, mannitol, and free micelles to both healthy and diabetic mice via ileum injection. One hour after the administration, the animals were sacrificed, and urine samples were collected in which lactulose and mannitol contents were quantified using HPLC-MS-MS (Figure 5a, b). The excretion ratio of lactulose to mannitol (L/M) in urine has been routinely studied in clinical practice as a measure for intestinal permeability and the integrity of mucosal barrier38, 39. A tight junction opening allows more lactulose (larger in size than mannitol) to pass through and overall increases the excreted L/M ratio. We did not observe any significant change in L/M ratio for DSPE-PCB micelle treated group, compared with the control group where animals only received the lactulose and mannitol but without any micelles (Figure 5a, b). By contrast, both polysorbate 80 and sodium decanoate treated groups showed drastically higher L/M ratio values, and it appeared that sodium decanoate, a known tight junction opener34, 35, with higher CMC values (50 mM, v.s. 0.015 mM for polysorbate 80) showed even higher L/M value (Figure 5a, b).
Figure 5. Zwitterionic micelle/insulin treatment did not open intestinal tight junctions.
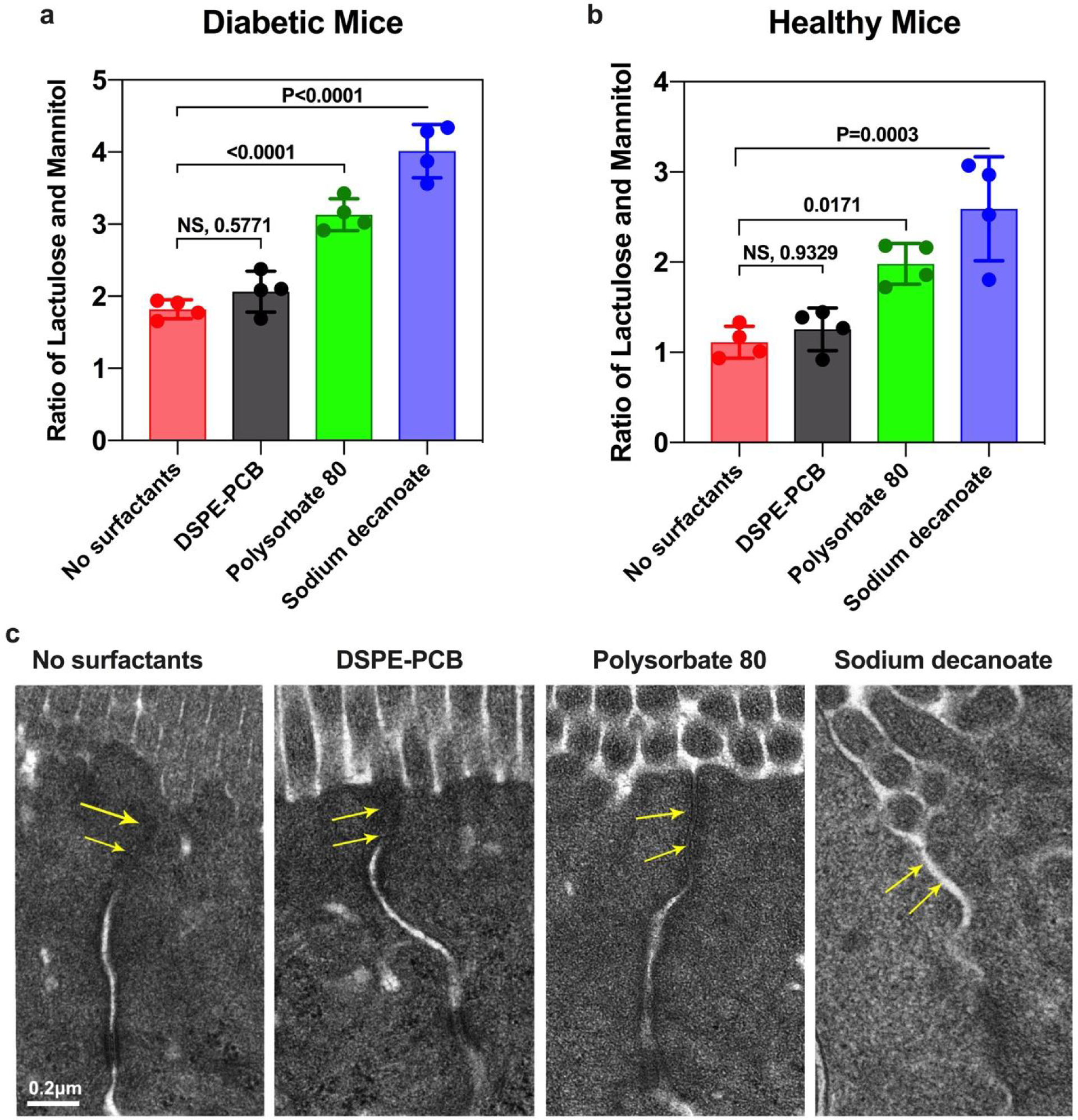
(a, b) Intestinal permeability tests on diabetic mice and healthy mice, respectively. One hour after co-administering lactulose, mannitol, and different types of surfactants through ileum injection, urine was collected to measure the ratio of lactulose and mannitol contents (N=4 biologically independent animals, mean ± SD). A one-way analysis of variance with Tukey multi-comparison was used for statistical analysis (NS: not significant). (c) Representative TEM images of epithelial tissues at 1hour post-ileum injection of different types of surfactants. Tight junctions were indicated by arrows. Scale bar shown is 0.2 μm.
The intestine epithelial tissues surrounding the injection sites were further fixed and processed for TEM imaging. DSPE-PCB micelle treated tissue showed intact tight junctions morphologically similar to the control tissue not treated by any micelle (Figure 5c). Tissues treated with polysorbate 80 and sodium decanoate showed more and even more severe tight junction opening, respectively (Figure 5c). As a separate way to confirm the integrity of tight junction after treated by DSPE-PCB micelles, we incubated DSPE-PCB and control micelles with a monolayer of Caco-2 cells for 2 h, followed by ZO-1 staining (tight junction protein) in green (Extended data Figure 2). Similar results were observed that DSPE-PCB treatment did not compromise the integrity of tight junctions while control micelles (tight junction openers) significantly disrupted the tight junction protein ZO-1. A transepithelial electrical resistance (TEER) test indicated that incubation with DSPE-PCB for 4 h did not increase the permeability of Caco-2 cell layers, while both polysorbate 80 and sodium decanoate treated groups showed drastically decreased TEER values (Extended data Figure 3).
Prototype oral insulin development.
The solution form of DSPE-PCB/insulin was able to address both intestinal mucus and epithelial barriers and achieved a pharmacological activity as high as 41.2% through ileum injections as previously mentioned. Nevertheless, direct oral gavage of the solution form to diabetic mice can hardly achieve a comparably high pharmacological activity as the ileum injected group at the same dose of 20 IU/kg, even by further increasing the oral dose to 30 IU/kg (Figure 6a). This indicates the possibility of destabilization or degradation of DSPE-PCB/insulin at the harsh stomach environment (the first barrier) and necessitates the drugability to form an oral capsule to overcome this barrier. For comparison, solution form of polysorbate 80/insulin was similarly studied and achieved a far less capability in lowering blood glucose through either ileum injection or oral gavage at comparable doses (Figure 6b).
Figure 6. Zwitterionic micelle/insulin aqueous formulation loses pharmacological activity through direct oral administration but shows stability to form dry powders for potentially oral capsule formulation.
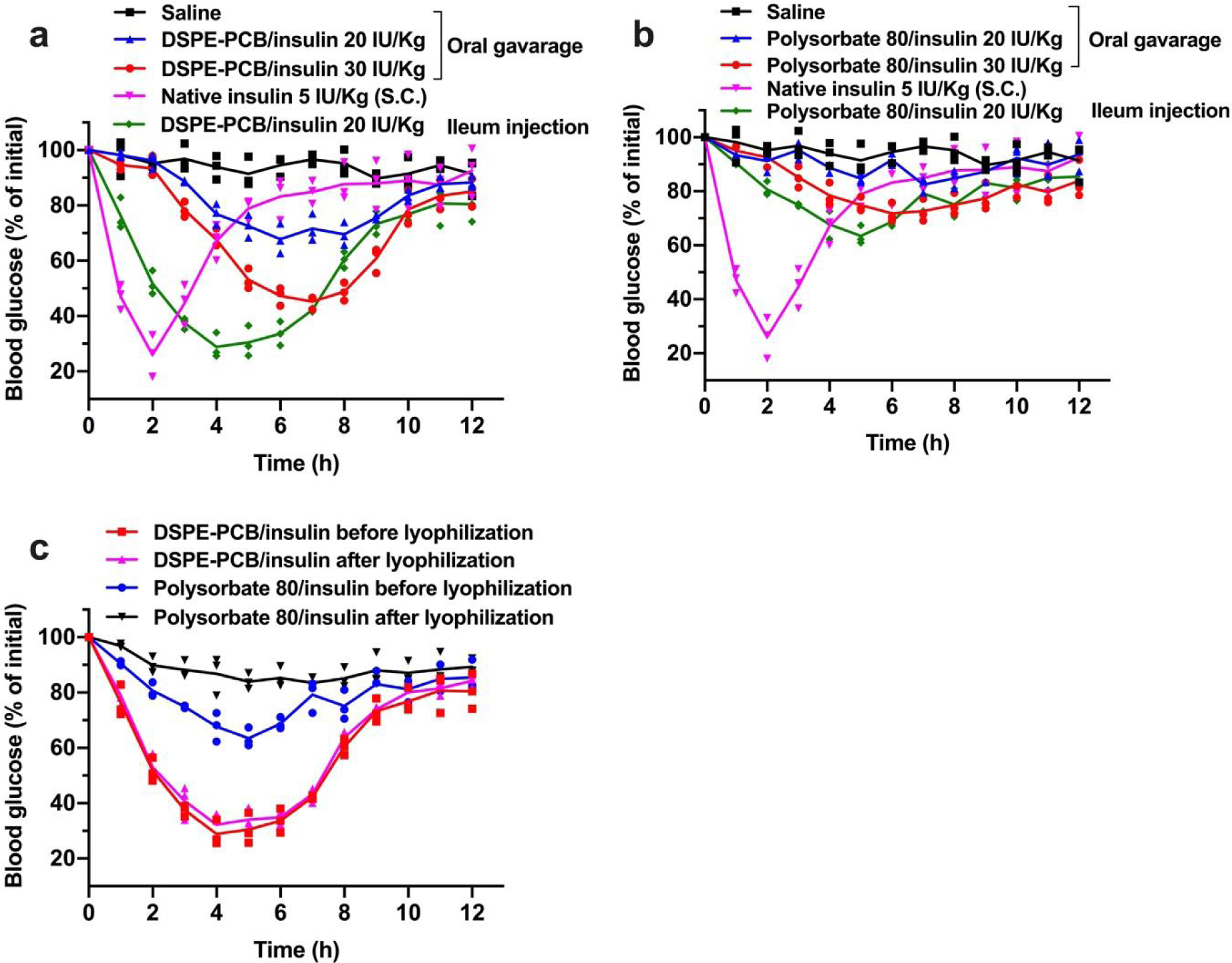
Blood glucose lowering (pharmacological) performance for (a) DSPE-PCB/insulin and (b) polysorbate 80/insulin aqueous formulation on diabetic mice through oral gavage at 20 and 30 IU/Kg compared with ileum injection at 20 IU/kg (N=3 biologically independent animals, means connected). Insulin/ZnCl2 feeding ratio is 2.5/1 by weight. S.c. injected native insulin at 5 IU/Kg was used as a control (1 IU/ml). (c) Blood glucose lowering (pharmacological) performance for DSPE-PCB/insulin and polysorbate 80/insulin aqueous formulation on diabetic mice through ileum injection at 20 IU/Kg before and after a lyophilization procedure (N=3 biologically independent animals, means connected).
An ideal oral capsule formulation should contain dry powders of DSPE-PCB/insulin for the purpose of ease of manufacturing, preventing the drug performance loss and long-term storage. We directly freeze-dried the solution form of DSPE-PCB/insulin into powders (without any additive cryoprotectant), reconstituted the powders into the solution form, and compared the pharmacological activity before and after the lyophilization through ileum injections to diabetic mice (Figure 6c). No apparent change of pharmacological activity for DSPE-PCB/insulin was observed after lyophilization. In comparison, there was an obvious loss of the blood glucose lowering activity of polysorbate 80/insulin after experiencing the lyophilization procedure (Figure 6c). The performance loss is expected to be due to insulin leakage during lyophilization: DSPE-PCB/insulin retained 98.2% while polysorbate 80/insulin leaked 25% as determined by resolubilizing the formulation in water, followed by ultracentrifugation separation (10K MWCO) and BCA quantification for insulin. The results indicate high feasibility to directly package DSPE-PCB/insulin powers to form an orally delivered capsule.
We fabricated a prototype of oral insulin by packaging the direct lyophilized powder of DSPE-PCB/insulin into a size M porcine gelatin capsule, followed by an Eudragit L100–55 enteric-coating procedure. The obtained insulin capsule (containing DSPE-PCB/insulin, or polysorbate 80/insulin) was administered to diabetic rats through oral gavage at the dose of 20 IU/kg followed by periodic measurement of blood glucose and serum insulin concentration using a glucometer and human insulin ELISA assay, respectively (Figure 7a, b). Rats receiving subcutaneous (s.c.) injection of 5 IU/kg free insulin solution were used as controls. Based on the glucose lowering profile (Figure 7a), the pharmacological activity of DSPE-PCB/insulin capsule was calculated to be 43.4%, much higher than Polysorbate 80/insulin capsule which was 8.56%. Based on the serum insulin profile (Figure 7b), the bioavailability of DSPE-PCB/insulin capsule was calculated to be 42.6%, while polysorbate 80/insulin was merely 8.35%. It should be noted that when the free insulin capsule was orally administered, almost no insulin presence in serum was observed, indicating the inability for free insulin to get absorbed through intestine.
Figure 7. Pharmacological activity and bioavailability of an oral insulin capsule containing zwitterionic micelle/insulin, and the capability in adjusting the drug acting profile.
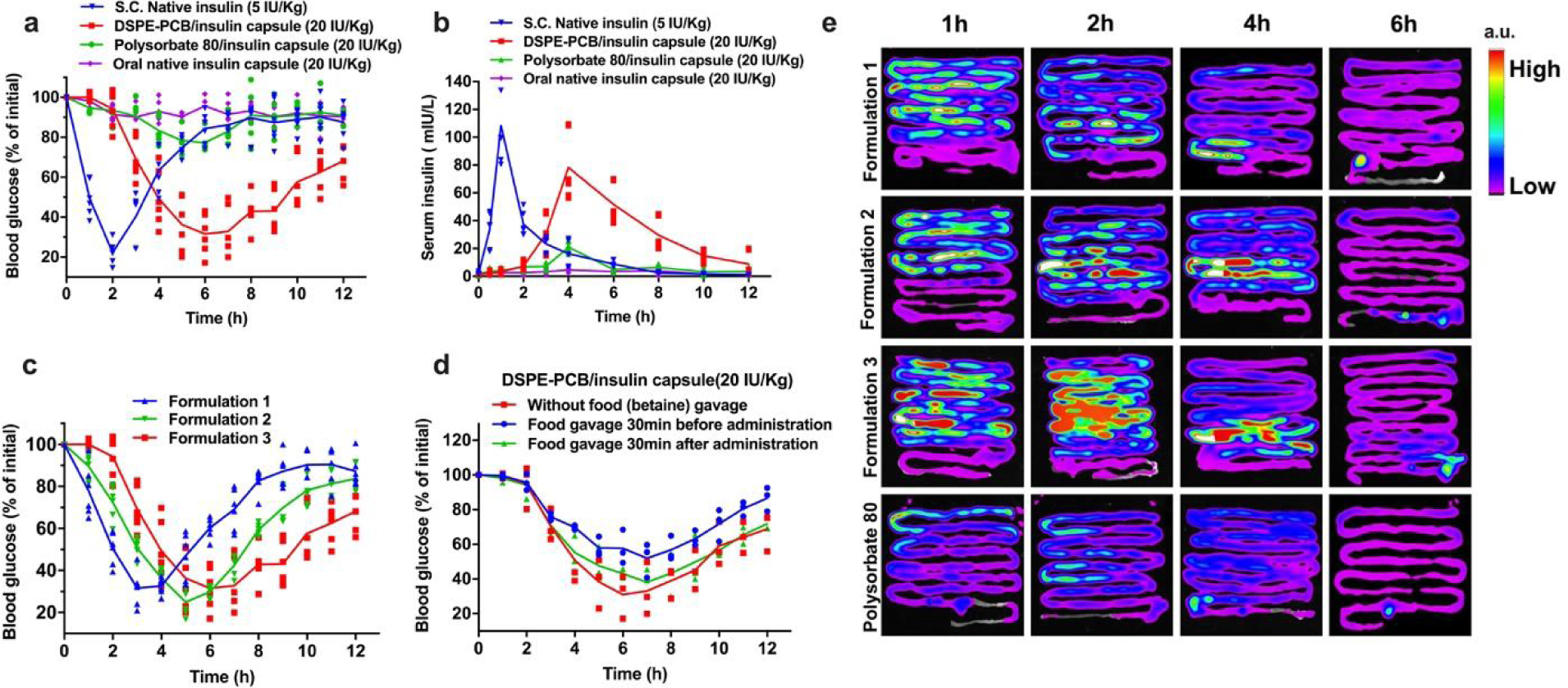
(a) Blood glucose lowering (pharmacological) performance for DSPE-PCB/insulin capsule on diabetic rats through oral gavage, compared with polysorbate 80/insulin capsule at the same dose of 20 IU/Kg (N=6 biologically independent animals, means connected). Insulin/ZnCl2 feeding ratio is 2.5/1 by weight. S.c. injected native insulin at 5 IU/Kg was used as a control (10 IU/ml). (b) Serum insulin concentration (bioavailability) for DSPE-PCB/insulin capsule on diabetic rats through oral gavage, compared with polysorbate 80/insulin capsule and native insulin capsule at the same dose of 20 IU/Kg (N=6 biologically independent animals, means connected). S.c. injected native insulin at 5 IU/Kg was used as a control. (c) Blood glucose lowering (pharmacological) performance for various formulations of DSPE-PCB/insulin capsules on diabetic rats through oral gavage (N=6 biologically independent animals, means connected). Formulation 1, 2, and 3 had an insulin/ZnCl2 feeding ratio of 50/1, 20/1, and 2.5/1 by weight during the encapsulation process, respectively. Their drug loading is 6.24%, 6.23% and 6.10%, while the corresponding particle hydrodynamic sizes are 28.52, 26.36 and 25.96 nm respectively. (d) Food effect on the glucose-lowing efficacy of DSPE-PCB/insulin capsule. Diabetic rats received oral gavage of 80 mg/kg betaine water (model food) 30 minutes before or after the oral gavage of DSPE-PCB/insulin capsule, followed by blood glucose measurement at predetermined intervals (N=3 biologically independent animals, means connected). (e) Absorption sites and kinetics of orally delivered insulin. Healthy rats were administered with DSPE-PCB/Cy7-insulin enteric capsules (Formulation 1–3) or Polysorbate 80/Cy7-insulin enteric capsule (20 IU/kg) by oral gavage after fasting for 12 h. At different post-administration time points, the entire intestine was collected and visualized with the Bruker In-Vivo Xtreme imaging system. a.u., arbitrary units. Experiments were repeated independently twice with similar results.
Insulin therapy for potential type 1 and type 2 diabetes typically requires an adjustable drug acting time profile, such as short acting and long acting, for various clinical needs. We further studied the feasibility of fine-turning the blood glucose-lowering profile by simply changing the composition of Zn2+ used in the prototype formulation. We observed that DSPE-PCB/insulin capsule after oral gavage to diabetic rats showed an accelerated action time in lowering blood glucose as less Zn2+ was used during the formulation procedure (Zn2+ composition: Formulation 1 < Formulation 2 < Formulation 3) (Figure 7c). By further decreasing Zn2+ composition, the peak action time for the oral insulin can reach 1–2 h (such as Formulation 0 in Extended data Figure 4). Our prior work indicates a dissolution dependent process for payloads to release from the ultralow CMC DSPE-PCB micelles32. The observation here could be explained that by increasing the Zn2+ composition during the insulin precipitation and encapsulation process, more condensed insulin aggregation is expected to form in the hydrophobic core of the micelle, which results in slower release rate of insulin (Extended data Figure 1).
We have further studied potential food effect on DSPE-PCB/insulin capsule (Figure 7d). Since betaine was found to decrease the uptake of DSPE-PCB micelle by blocking/inhibiting PAT1 transporter (Figure 4), we chose betaine water as a model food in this study. Betaine water was gavaged to the diabetic rats 30 mins before or after the oral insulin administration at the dose of 80 mg betaine/kg (This dose roughly equals to the average daily betaine intake for rats). Results indicate that potential food interference could be minimized by administering oral insulin capsule 30 min before the meal (betaine water) consumption.
We further examined the absorption sites of our oral insulin at different post-gavage time points in an ex vivo fluorescent imaging study (insulin labeled with Cy7) (Figure 7e). DSPE-PCB/Cy7-insulin (formulation 1–3) and polysorbate 80/Cy7-insulin control were placed into the enteric capsules and administered to the rats through oral gavage. Results showed that DSPE-PCB/insulin had improved retention and absorption in the small intestine (jejunum and ileum) compared with polysorbate 80/insulin, consistent with the finding that PAT1 is highly expressed at the small intestine40, 41. The retention/absorption sites did not differ significantly among the three DSPE-PCB/insulin formulations. Formulation 3 had the slowest release rate of encapsulated insulin (highest Zn2+ composition) and showed the highest fluorescent intensity retained.
Biocompatibility and long-term safety.
We further examined the in vitro cell viability and live/dead imaging, and in vivo long-term safety of the zwitterionic micelles. For cell viability, both DSPE-PCB and polysorbate 80 exhibited no significant cytotoxicity on the Caco-2 cells between 0.01 and 1mg/ml concentration range, which was also confirmed through live/dead staining (Figure 8a, b). To examine potential cell-membrane disruption, both DSPE-PCB and Polysorbate 80 were incubated with Caco-2 cells and no significant lactate dehydrogenase (LDH) release was observed (Figure 8c). Since single dosing with DSPE-PCB micelle did not open the tight junction (Figure 5), we further examine potential tight junction opening and safety concern under a long-term dosing regimen (mice received oral gavage of DSPE-PCB micelle twice-daily for 14 consecutive days). At day 14, the urine lactulose/mannitol test showed that there was no significant tight junction opening even after the long-term repeated dose challenge (Figure 8d). H&E staining images on day 14 showed that no significant structural damage occurred to the small intestine tissue (Figure 8e). The fingerlike villi were found intact in all the tissues examined and no significant inflammation was observed (Figure 8e).
Figure 8. DSPE-PCB micelles show no significant cytotoxicity or leaky gut-related endotoxin leakage and inflammation of repeated consecutive dosing.
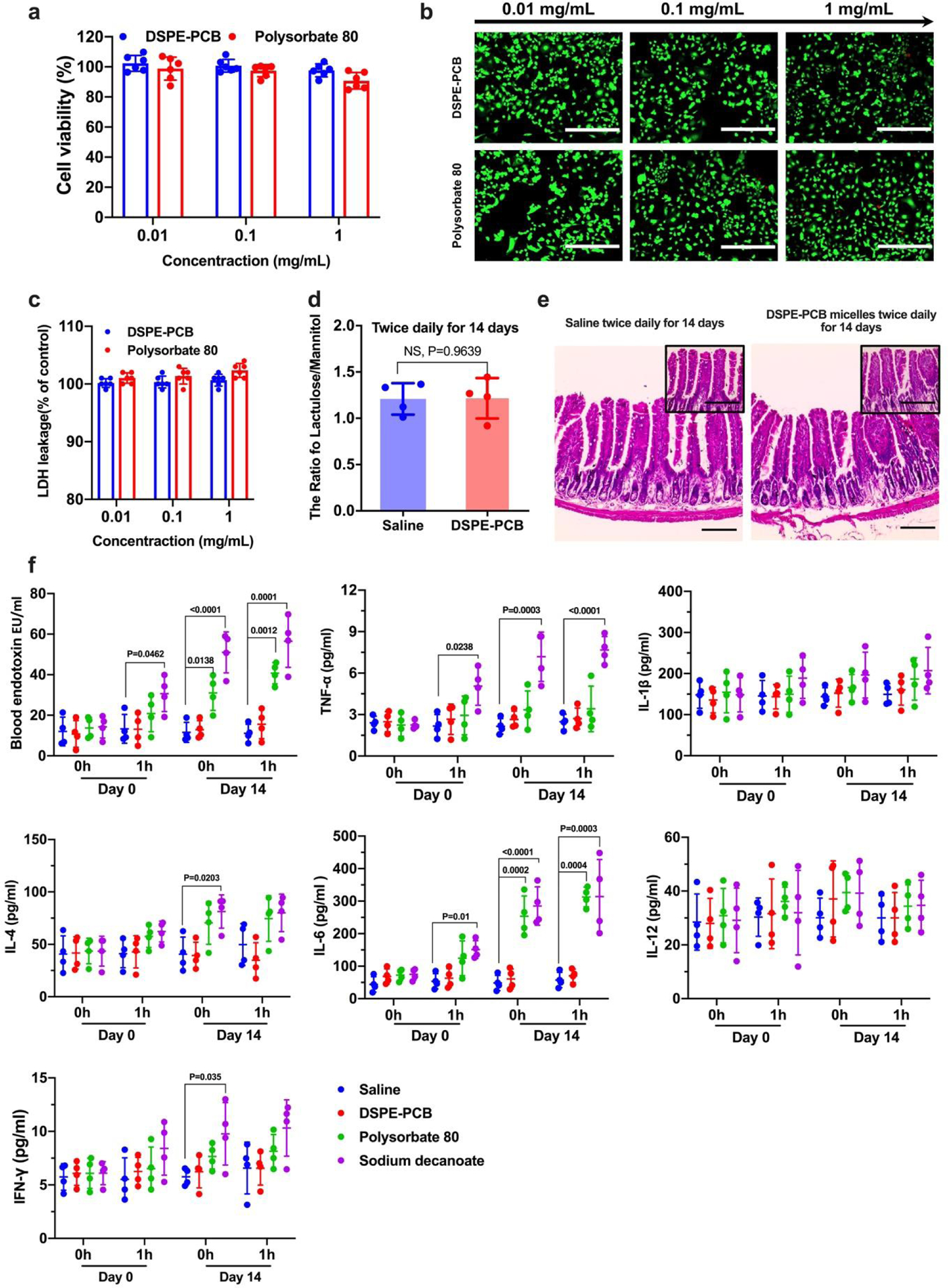
(a) Caco-2 cells were incubated with DSPE-PCB and Polysorbate 80 micelles with concentration range of 0.01–1 mg/mL for 24 h. Cell viability was measured in an MTT assay (Mean ± SD, N=6 biologically independent samples). (b) The treated Caco-2 cells were further visualized using live/dead staining (Calcein AM (green) and EthD-1 (red), respectively). Scale bar: 400 μm. Experiments were repeated three times independently with similar results. (c) Cell membrane integrity test by measuring lactate dehydrogenase (LDH) leakage. Caco-2 cells were co-cultured with DSEP-PCB, Polysorbate 80 for 24 hours, and the LDH levels were measured using LDH assay kit (Sigma) and plotted as % of untreated cell negative control (N=6 biologically independent samples, mean ± SD). As a positive control, 1% Triton X-100 and sonication were used to completely lyse the caco-2 cells and its LDH leakage level was found to be 240%. (d) Healthy mice were administered with DSPE-PCB micelle or saline through oral gavage twice daily for 14 consecutive days, followed by the intestinal permeability test. One hour after co-administering lactulose, mannitol, and DSPE-PCB micelles through ileum injection, urine was collected to measure the ratio of lactulose and mannitol contents (N=4 biologically independent samples, mean ± SD). A two-tailed t-test analysis was used for statistical analysis (NS: not significant). (e) Small intestine tissue sections were stained with Hematoxylin and Eosin (Scale bar: 200 μm). Sections at higher magnification (Scale bar: 50 μm.). Experiments were repeated four times independently with similar results. (f). Healthy mice were administered with DSPE-PCB micelle, saline, Polysorbate 80, or sodium decanoate through oral gavage twice daily for 14 consecutive days. On day 0 and 14, before and 1 h after the oral administration, blood serum was collected and the endotoxin and pro-inflammatory cytokines were measured with the mouse magnetic Luminex® assays (N=4 biologically independent samples, mean ± SD). A one-way analysis of variance with Tukey multi-comparison was used for statistical analysis; compare with saline within the same group.
We further characterized potential leaky gut by examining the endotoxin and proinflammatory cytokine levels in blood serum from the mice receiving oral gavage of DSPE-PCB micelle and control micelles twice-daily for 14 consecutive days (at the same dose that was used to formulate oral insulin) (Figure 8f). Results showed that the endotoxin and certain inflammatory cytokines such as TNF-α, IFN-γ and IL-6 had an observable increase even 1h after the mice received the oral dose of polysorbate 80 and sodium decanoate. Such high level of endotoxin and cytokines was further increased over the 14 days of consecutive administration, and did not appeared to be temporary, e.g., as characterized by the high serum level before administrating micelles on day 14. Between the control micelles, sodium decanoate induced even higher endotoxin leakage and proinflammatory response compared with polysorbate 80, which is consistent to its higher tight junction opening capability (Figure 5). By contrast, DSPE-PCB did not induce any observable increase in terms of endotoxin and pro-inflammatory cytokines, supporting its long-term safe use without opening tight junctions and inducing leaky gut.
Conclusions
In summary, we showed that zwitterionic micelle platform (DSPE-PCB) was able to simultaneously address both mucus and epithelial barriers, and drastically enhance the intestinal transport of protein payload (insulin). Our prototype oral insulin can be easily manufactured by packaging dry powder of DSPE-PCB/insulin into an enteric-coated capsule. Different from other preclinical oral insulins, our formulations uniquely utilized PAT1 as a mechanism for epithelium penetration, achieved the rarely reported high bioavailability and the controlled versatility in fine-tuning drug acting profiles, as well as not induced tight junction opening. A direct clinical impact of our findings could potentially be an oral insulin capsule for prandial and/or basal applications. With biocompatibility and long-term safety, this platform technology has the potential as a practical solution for oral delivery of other protein/peptide payloads.
Methods
Materials.
DSPE-PCB micelle was synthesized following the previously reported method31. Human recombinant insulin (Cat#: NC1063131) was purchased from Life Technologies Corporation. Polysorbate 80, porcine stomach mucin type II, streptozotocin (STZ), ZnCl2, HAuCl4, NaBH4, styrene, divinylbenzene, fluorescein-o’odemethacrylate, Poly(ethylene glycol) methyl ether methacylate (average Mn 500), SPMA (3-sulfopropyl methacrylate potassium salt) and MTTA ((2-(methacryloyloxy) ethyl) trimethylammonium chloride, betaine, lactulose, mannitol, Corning® Transwell polyester membrane cell culture inserts, Lactate dehydrogenase activity assay kit (Cat# MAK066) were purchased from Sigma-Aldrich. Eudragit L 100–55 was obtained from Evonik Industries. ZO-1 Monoclonal Antibody Alexa Fluor 488 was purchased from Invitrogen (Cata# MA3–39100-A488, Lot# UE288707), Mouse Magnetic Luminex® assays was purchased from R&D systems (Cata# LXSAMSM- 6, Lot# L131929).
Synthesis and characterization of zwitterionic nanogels and zwitterionic micelle labeled with polystyrene nanoparticles.
In a typical preparation to synthesize nanogels, 100mg monomer (PCB monomer), 0.3 mg fluorescein crosslinker (fluorescein O, O’- dimethacrylate), 3.5 mg surfactant (sodium dodecyl sulfate, SDS), 2 mg initiator (ammonium persulfate, APS) and 0.25 mg catalysts (N, N, N’, N’-tetramethylethylenediamine, TEMED) was dissolved in 3 ml water which was previously purged with nitrogen. The reaction took two hours under vigorous stirring. The resulting mixture was then purified by 0.22 μm filter membrane, centrifuged several times to remove large particles, and dialyzed against deionized water to remove unreacted monomer and surfactant. The obtained nanogel solution was condensed to an appropriate concentration with ultrafiltration. The obtained nanogels were characterized using dynamic light scattering (DLS, Zetasizer Nano ZS; Malvern Instruments) to measure the hydrodynamic size and zeta-potential. In a typical preparation to synthesize micelles containing polystyrene nanoparticle payloads, DSPE-PCB or polysorbate 80 micelles, ammonium persulfate, styrene and divinyl benzene and fluorescein crosslinker (molar ratio is typically 1: 10: 1.5: 1: 1) were sonicated in deionized water to form nano-emulsion, followed by 20 min heating in water bath at 40 °C. Micelles with polystyrene nanoparticle loaded were further purified through 10K MWCO ultrafiltration to remove rough materials, and 0.22 μm filter to remove unexpected large particles. The size and zeta-potential of the polystyrene nanoparticles were determined by DLS.
Tracking nanoparticle diffusion in mucus.
Nanoparticle diffusion behavior in mucus was studied by modifying methods as previously reported17, 42. Mucus was reconstituted by dissolving porcine stomach mucin in phosphate buffered saline (PBS) at 30 mg/ml. Nanogels were added into 100 μl mucus to reach a final concentration of 3% vol/vol (final particle concentration, 8.25×10−7 wt/vol) and incubated for 1 h on a Nunc™ glass bottom dish (0.17 mm thickness for the inside bottom glass) before observation. Movies were captured with 64X oil-immersion objective in a spinning disc confocal microscope (Yokogawa CSU-X1M 5000 microlens, Zeiss LSM 800). For each nanogel sample, trajectories of n= 50 particles were analyzed, and three samples were examined for each type of nanogel. MSD was calculated with the equation of MSD=<| r(t)−r(0) |2> at a temporal resolution of 200 ms for 20 s.
Preparation and characterization of DSPE-PCB/insulin formulations and oral insulin capsules.
In a typical preparation of DSPE-PCB/insulin formulation, 15 mg DSPE-PCB was added into 1 mL insulin solution (1 mg/mL, dissolved in 0.1 M sodium bicarbonate) under stirring at 800 r/min. Then 200 μl of ZnCl2 solution (2 mg/mL) were added dropwise to insulin-containing solution and the resulting mixture was purified by an ultrafiltration membrane (10 K Da cutoff). The encapsulation efficiency for insulin was found to be over 98% by quantifying insulin content retained by the micelle using BCA Protein Assay Kit. Polysorbate 80/insulin formulation was prepared following the same procedure. Obtained DSPE-PCB/insulin formulations were measured for hydrodynamic size with DLS, and stained with phosphotungstic acid (2%, w/v) and visualized using JEOL 2010 transmission electron spectroscopy. To prepare oral insulin capsules, dry powder of DSPE-PCB/insulin or polysorbate 80/insulin was placed into a porcine gelatin capsule (size M, Torpac Inc., USA) followed by immersing the capsule in the Eudragit L 100–55 methanol solution (15% w/w) and drying at room temperature for three times to obtain the enteric coating. The amount of DSPE-PCB/insulin or polysorbate 80/insulin loaded per capsule was varied with targeted animal weight (each animal received one capsule containing 20 IU/kg insulin).
Cell uptake of polystyrene nanoparticles for Caco-2 and 3T3 cells.
Caco-2 human colon epithelial cells and NIH 3T3 mouse embryonic fibroblast cells were used as received from the American Type Culture Collection without further authentication, and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) and Eagle’s Minimum Essential Medium (EMEM) at 37 °C in a 5% CO2 environment respectively. Human Caco-2 was tested negative for Mycoplasm contamination per supplier. It is unclear whether NIH/3T3 has been tested for mycoplasma contamination by the supplier. Cells were seeded in a 12-well plate at a density of 1×105 cells per well overnight, washed twice with pre-wared PBS, and incubated with DSPE-PCB/polystyrene nanoparticles in culture medium at 37 °C for 2 h. For inhibitory groups, cells were pretreated with proton-assisted amino acid transporter inhibitors (betaine and L-tryptophan respectively, concentration: 5 mg/ml) in culture medium at 37 °C for 1 h before incubating with DSPE-PCB/polystyrene nanoparticles. The cells were further stained with 20 μL Hochest 33342(1 mg/ml) and 700 μL of CellMask (Invitrogen, USA), each for 10 min at 37°C followed by three times wash with PBS, and fixed using 4 % glutaraldehyde for 15 min. Cellular uptake of DSPE-PCB/polystyrene nanoparticles in different groups was examined by an Zeiss LSM 780 confocal laser scanning microscope. The Z-stack method was used for three-dimensional visualization, where the section thickness was 2.5 μm, the step interval was 2 μm, and the total z-stack length was 44 μm (23 slices). Fluorescent signal was quantified by using the image J software. Cellular uptake was also quantitively measured with flow cytometry (BD FACSAria II flow cytometer) and analyzed with FlowJo X software.
In vitro cytotoxicity and live/dead assay.
In vitro cytotoxicity was evaluated using MTT assay with Caco-2 cells. Caco-2 cells were seeded on 96-well plates at a density of 2 × 104 cells per well in Eagle’s Minimum Essential Medium (EMEM) with 20% FBS and cultured in 5% CO2 atmosphere at 37 °C for 24 h. Then the cells were washed twice with HBSS and incubated with 100 μl of DSPE-PCB micelle or Polysorbate 80 solutions in EMEM with concentrations of 0.01, 0.1, and 1.0 mg/ml, respectively. After 24 h of incubation at 37 °C, the DSPE-PCB micelle and Polysorbate 80 solution were replaced with 100 μL of MTT solution (0.5 mg/ml in HBSS), and the cells were incubated for an additional 4 h at 37 °C. Subsequently, the supernatant was carefully removed and 150 μL of dimethyl sulfoxide (DMSO) was added to each well. The absorbance of the resultant solutions was measured at 570 nm using a microplate reader (Bio-Rad, USA). Untreated cells were used as negative control for 100% cell viability. Cell viability was expressed as percentage of absorbance relative to the control. Live/dead assay was conducted by staining the cells with 100 μL of PBS buffer containing 2 μM Calcein and 4 μM ethidiumhomodimer-1 (Live/Dead viability/cytotoxicity kit, Invitrogen, CA, USA). The cells were incubated at room temperature for 30 minutes and observed by the EVOS FL fluorescence microscope (AMG).
The LDH leakage assay.
Caco-2 cells were seeded in a 96-well plate at a concentration of 104 cells per well. After incubating the plate overnight at 37 °C, the medium was aspirated and replaced with DSPE-PCB or polysorbate solutions (100 μl per well at various concentrations). After 2 h of exposure, 50 μl of media from each well was transferred to a new, clear plate and was measured with the sigma lactate dehydrogenase activity assay kit. LDH level were quantified by comparing the absorbance of treated cells with that of untreated (negative control). Cells completely lysed were used as positive control.
The tight junction ZO-1 protein staining.
Caco-2 cells were seeded on 96-well plates at a density of 2 × 104 cells per well in Eagle’s Minimum Essential Medium (EMEM) with 20% FBS and cultured in 5% CO2 atmosphere at 37 °C. Then cells were washed twice with HBSS and incubated with different micelles respectively. After 2 h of incubation at 37 °C, the cells were washed with PBS solution, and the cells were further stained with 20 μL Hochest 33342(1 mg/ml) and ZO-1 Monoclonal Antibody Alexa Fluor 488 for 10 min at 37°C followed by three times wash with PBS, and fixed using 4 % glutaraldehyde for 15 min. The tight junction ZO-1 protein was examined by a Zeiss LSM 780 confocal laser scanning microscope.
Transepithelial electrical resistance (TEER) study.
Caco-2 cells were suspended in EMEM supplemented with 20% FBS, and seeded at a density of 2 × 105 cells per well on Corning® Transwell polyester membrane cell culture inserts and incubated for 96 h. The TEER was monitored to confirm proper barrier formation and only monolayers with initial TEER values of at least 300 Ω × cm2 were used for TEER or molecular permeability experiments. Prior to the study, the medium in the apical and basolateral chambers was replaced with prewarmed EMEM. After equilibration at 37 °C for 30 min, the apical solution was replaced with Sodium decanoate, polysorbate 80 or DSPE-PCB (0.2% w/v) in EMEM. After incubation for 4 h, the TEER value of each insert was measured with EVOM2 Epithelial voltohmmeter to explore the integrity of the tight junction.
Animals and diabetes induction.
All animal experiments were performed according to the National Institute of Health (NIH) guidelines for animal research under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Wayne State University. To establish streptozotocin (STZ)-induced diabetic mice model, healthy mice (C57BL/6J, male, 6–8 weeks of age, Jackson Lab) received a daily intraperitoneal injection of 5 mg/ml STZ at 50 mg/kg body weight for 5 consecutive days. 17 days after the first injection, body weight and blood glucose were measured to confirm the diabetic status. Diabetic mice with blood glucose above 300 mg/dL were selected for further in vivo test. To establish STZ-rat model, healthy rats (Sprague Dawley rat, male, ~180g, Charles River) received a one-time intraperitoneal injection of 65 mg/kg STZ. 10 days after injection, diabetic rats with blood glucose above 300 mg/dL were selected for further in vivo test.
Ileum injection surgery.
Mice received buprenorphine SR subcutaneously and surgical anesthesia was managed by isoflurane inhalation. The surgical site (abdomen) was shaved and prepped with three alternating scrubs of betadine and alcohol. On prepped animal, a single incision (1.5–2 cm) was made through abdomen skin. About 0.5–1cm ileum of intestine was pulled-out from the enterocoelia. The enteric cavity of ileum was injected with different formulations and abdominal wall was closed with 3–0 nylon suture, and the skin wound was closed with wound clips ¼” apart. The duration of procedure was 20 minutes or less. During the whole surgical procedure mice was placed on a warm pad. Mice remained fasted during this experiment (less than 12 h) and were euthanized thereafter. To evaluate glucose lowering performance of ileum injected formulations, blood samples were collected from the tails at predetermined time intervals for glucose testing.
The lactulose-mannitol intestinal permeability test.
Healthy and diabetic mice remained fasted for 8h and were restricted from water for 3 h before the experiment. Then mice received 500 μL water through oral gavage. 1 h later, the lactulose, mannitol, and free micelles were co-administered via ileum injection at the dose of 40/8/5.2 mg/ml (50 μL) and the wound is closed with wound clips. 1 h post ileum injection, the urine was collected, and the ileum tissue surrounding the injection site was collected for TEM imaging. The concentrations of lactulose and mannitol were measured using LC-MS-MS, with a mobile phase of 10 mM ammonium formate in water and acetonitrile (90:10), and an Agilent advanced bio glucan mapping column (2.1 × 100 mm × 2.7 μm) (precursor and product: mannitol 180.7→89; lactulose 341.2→160.8).
TEM imaging of tight junctions affected by the ileum injected formulations.
1 h post ileum injection of the formulations, mice were euthanized and small parts of ileum (5×5 mm) near to the injection site were collected and washed with PBS. The samples were continuously fixed in 2.5 % glutaraldehyde + 2.5% paraformaldehyde in 0.1M PBS buffer and 4% osmium tetroxide solution and transferred into Beem® embed capsule with micro-embed resin added to embed samples. The obtained blocks were further sectioned with ultra-microtomy to obtain sections with thickness of ~70 nm. These sections were retrieved via copper grid and stained with 1% lead citrate and 4% uranyl acetate separately, and then visualized under a JEM-100CX II electron microscope operated at 100 kV.
Oral administration of insulin capsules.
Diabetic rats were fasted overnight, received one capsule containing the pre-determined insulin dose through oral gavage, and remained fasted during the experiment (water ad libitum). At predetermined time intervals, blood samples were collected for glucose testing. Serum insulin level was determined using a Mercodia human insulin ELISA assay.
Absorption sites and kinetics of orally delivered insulin.
Healthy rats were fasted for 12 h and then administered with DSPE-PCB/Cy7-insulin enteric capsules (Formulation 1–3) or Polysorbate 80/Cy7-insulin enteric capsules (20 IU/kg) by oral gavage. At various post-administration time points, the entire intestine was collected and visualized with the Bruker In-Vivo Xtreme imaging system.
Long-term safety of repeated consecutive dosing.
Healthy mice were administered with saline or free DSPE-PCB micelles through oral gavage (0.258 mg DSPE-PCB per mice, the same dose in the intestinal permeability test) twice daily for 14 consecutive days. At day 14, the lactulose/mannitol experiment was conducted to examine potential tight junction opening through the lactulose-mannitol intestinal permeability test. In addition, the ileum tissue was collected and fixed in 10% zinc fixative (BD pharmingen™), dehydrated in ethanol, and embedded in paraffin. Five-μm cross-sections of intestine tissue were deparaffinized, rehydrated, and stained with Hematoxylin and Eosin. Histological morphology was examined using an EVOS XL Core Cell Imaging microscope. In addition, healthy mice received the same oral administered with DSPE-PCB micelle, saline, Polysorbate 80, or sodium decanoate twice daily for 14 consecutive days (the same dose as above). On day 0 and 14, before and 1 h after the oral administration, blood serum was collected and the endotoxin and pro-inflammatory cytokines (IFN-γ, IL-1β, IL-4, IL-6, IL-12, TNF-α) were measured with Pierce LAL Chromogenic Endotoxin Quantitation Kit and the mouse magnetic Luminex® assays (Lot No: L131929).
Statistical analysis.
Animal cohorts were randomly selected, but investigators were not blind to the experiments. The one-way ANOVA with Tukey multiple comparisons test was used to determine significant differences among multiple groups with unpaired biological replicates. A two-tailed t-test analysis was used to determine significant difference between two groups with unpaired biological replicates. P values were reported in figure legends. A value of P < 0.05 was considered statistically significant.
Extended Data
Extended Fig. 1. DSPE-PCB/insulin formulations with increasing zinc content showed increased retention of insulin release.
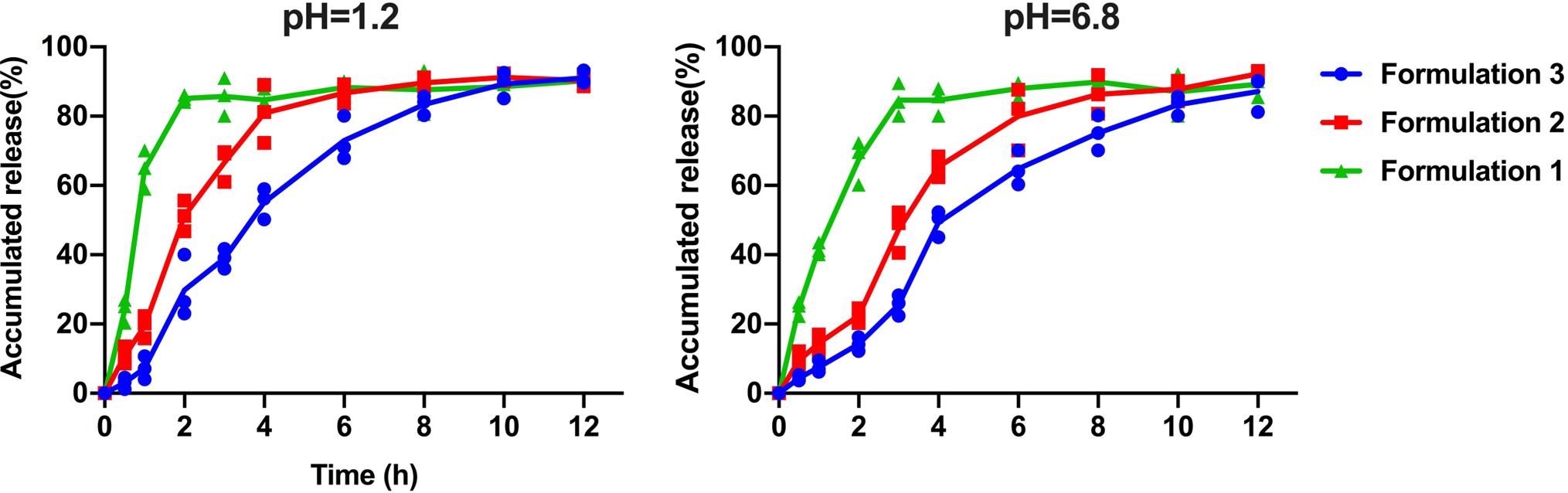
DSPE-PCB/insulin formulations were dialyzed (10 kDa MWCO) against pH 1.2 and 6.8 buffer with 5 mM bile salt at 37 °C. Formulation 1, 2, and 3 had an insulin/ZnCl2 feeding ratio of 50/1, 20/1, and 2.5/1 by weight during the encapsulation process, respectively. Their drug loading is 6.24%, 6.23% and 6.10%, while the corresponding particle sizes are 28.52, 26.36 and 25.96nm respectively. The cumulative released insulin was measured using the BCA assay (N=3 independent experiments, means connected).
Extended Fig. 2. Representative images of the tight junction protein ZO-1 of monolayer of Caco-2 cells after treated with different micelles.
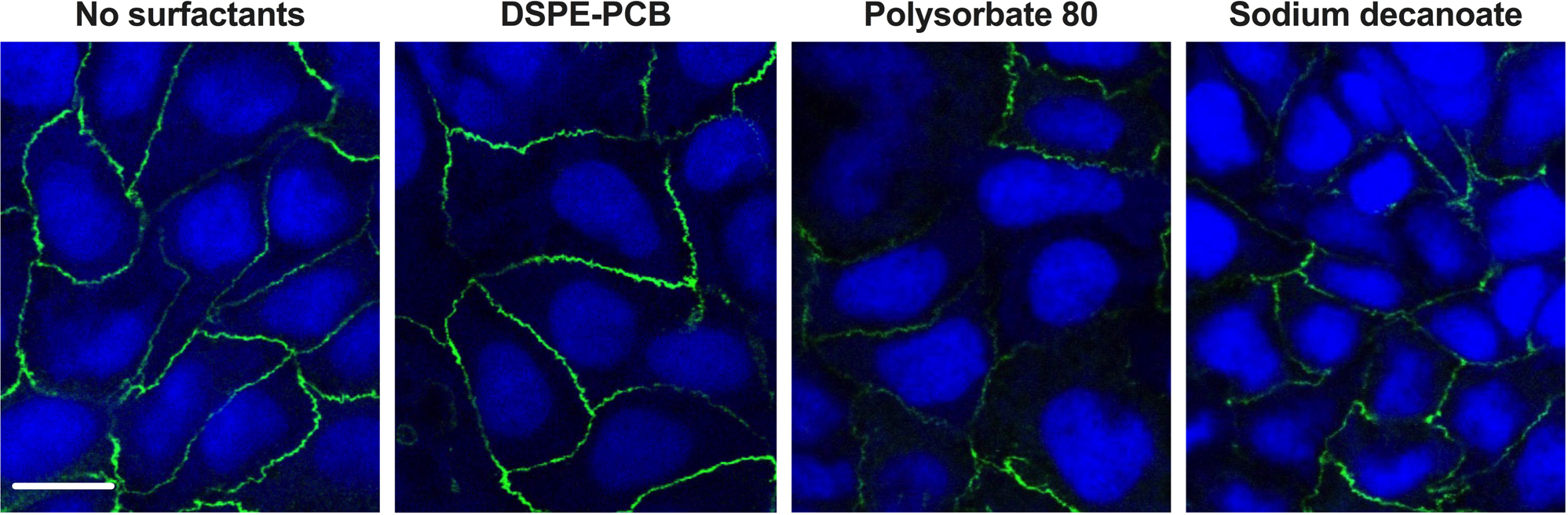
The tight junction protein ZO-1 was stained with ZO-1 Monoclonal Antibody Alexa Fluor 488 (green) while the nucleus was stained with Hochest 33342 (blue). Scale bar =20μm. Experiments were repeated three times independently with similar results.
Extended Fig. 3. DSPE-PCB micelles did not increase intestinal monolayer permeability in vitro.
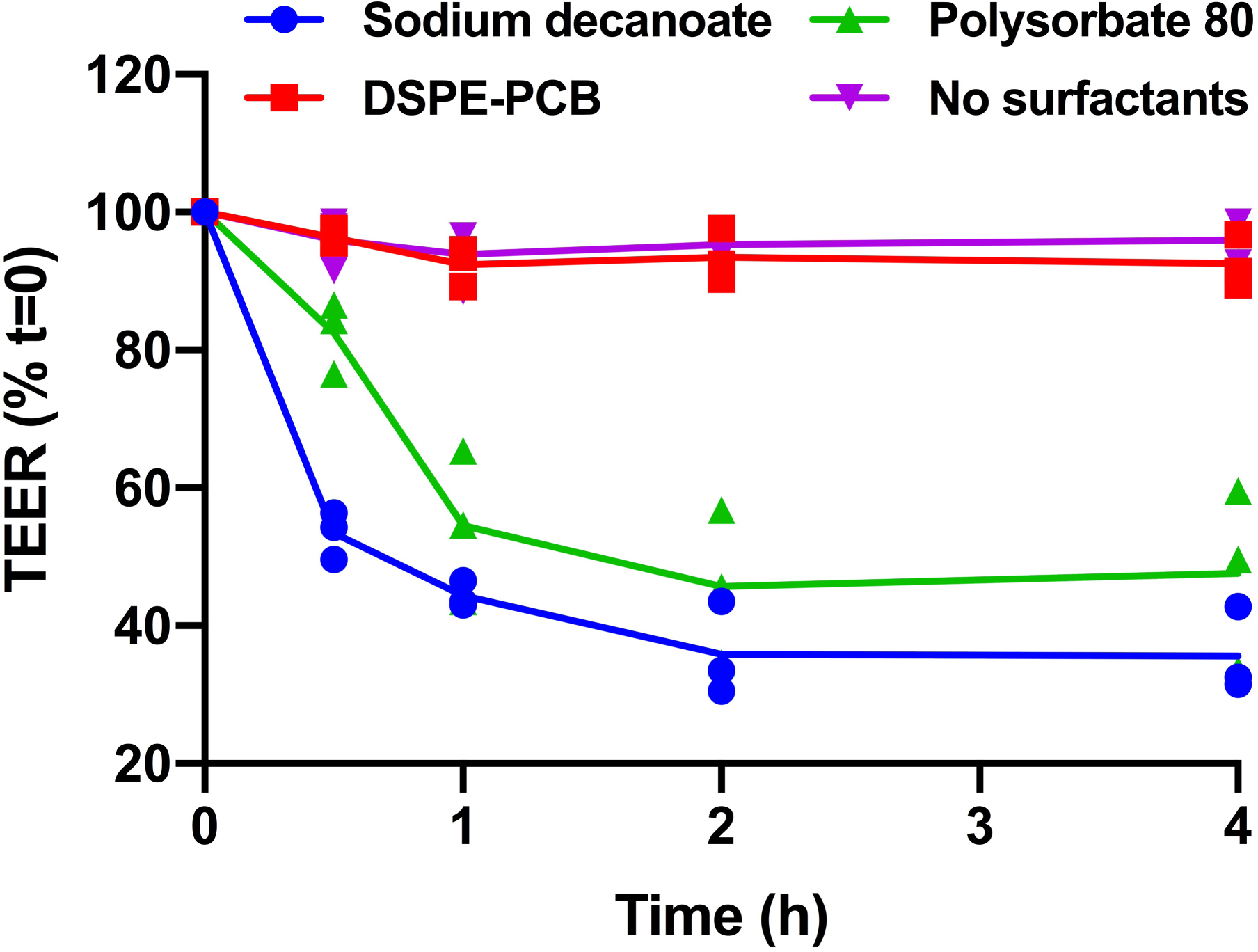
Sodium decanoate caused greater reductions in the TEER of Caco-2 monolayers than polysorbate 80. (N=3 biologically independent samples, means connected). Caco-2 cells were cultured for 96 hours to form monolayers and then treated with different micelles for 4 hours. The electrical resistance was measured at different time points.
Extended Fig. 4. Blood glucose profiles for various formulations of DSPE-PCB/insulin capsules on diabetic rats through oral gavage (N=6 biologically independent animals, means connected).
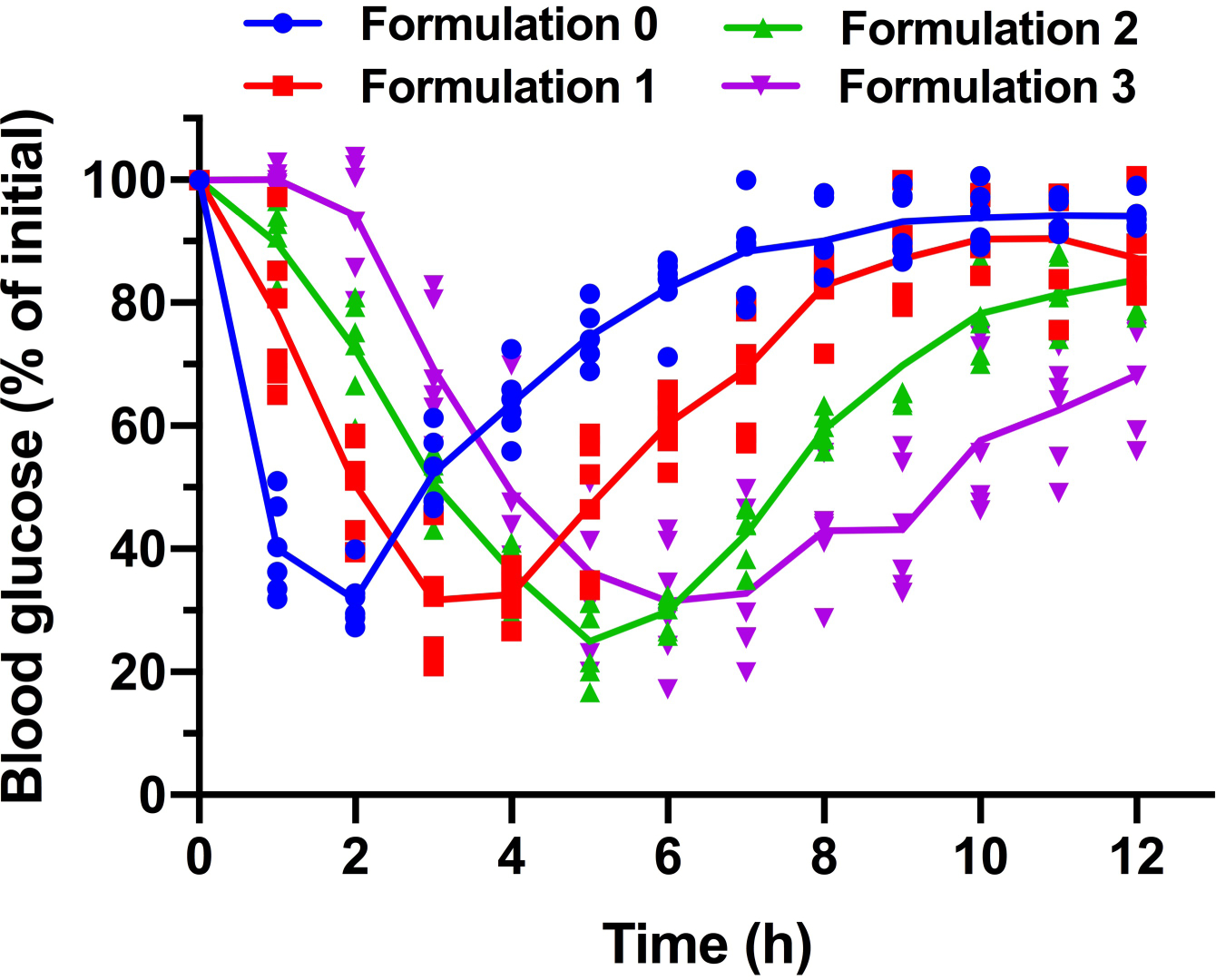
Formulation 0, 1, 2 and 3 had an insulin/ZnCl2 feeding ratio of 75/1, 50/1, 20/1, and 2.5/1 by weight during the encapsulation process, respectively. Their drug loading is 6.25%, 6.24%, 6.23% and 6.10%, while the corresponding particle hydrodynamic sizes are 28.56, 28.52, 26.36 and 25.96 nm respectively.
Acknowledgements
This work was supported by National Science Foundation (1809229) and National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (DP2DK111910 and R01DK123293). This work made use of the JEOL 2010 transmission electron microscope supported by National Science Foundation Award 0216084. We thank Alicia Withrow at Michigan State University for supporting tissue sample processing and TEM study. We thank Nicholas Peraino of the Lumigen Instrument Center Mass Spectrometry facilities to access Shimadzu 8040 (LC-MS-MS). The Microscopy and imaging are supported, in part, by NIH Center grant P30 CA22453 to the Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the Nation Institutes of Child Health and Development.
Footnotes
Data Availability
The authors declare that all data supporting the findings of this study are available within the paper and its Extended Data.
Competing Interests
The authors have declared that no competing interest exists.
Extended data figures are available in the online version of the paper.
Reference:
- 1.Sinha V et al. Oral colon-specific drug delivery of protein and peptide drugs. Crit Rev Ther Drug Carrier Syst 24, 63–92 (2007). [DOI] [PubMed] [Google Scholar]
- 2.des Rieux A, Fievez V, Garinot M, Schneider Y-J & Preat V Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release 116, 1–27 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Muheem A et al. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharmaceutical Journal 24, 413–428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serra L, Domenech J & Peppas NA Drug transport mechanisms and release kinetics from molecularly designed poly(acrylic acid-g-ethylene glycol) hydrogels. Biomaterials 27, 5440–5451 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Yu F et al. Enteric-coated capsules filled with mono-disperse micro-particles containing PLGA-lipid-PEG nanoparticles for oral delivery of insulin. Int J Pharm 484, 181–191 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Mustata G & Dinh SM Approaches to oral drug delivery for challenging molecules. Crit Rev Ther Drug Carrier Syst 23, 111–135 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Goldberg M & Gomez-Orellana I Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov 2, 289–295 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Ensign LM, Cone R & Hanes J Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv Drug Deliver Rev 64, 557–570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cu Y & Saltzman WM Mathematical modeling of molecular diffusion through mucus. Adv Drug Deliver Rev 61, 101–114 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saltzman WM, Radomsky ML, Whaley KJ & Cone RA Antibody Diffusion in Human Cervical-Mucus. Biophys J 66, 508–515 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chilvers MA & O’Callaghan C Local mucociliary defence mechanisms. Paediatric Respiratory Reviews 1, 27–34 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Knowles MR & Boucher RC Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 109, 571–577 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McAuley JL et al. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J Clin Invest 117, 2313–2324 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cone R Barrier properties of mucus. Adv Drug Deliver Rev 61, 75–85 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Lai SK, Wang Y & Hanes J Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliver Rev 61, 158–171 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ensign LM, Schneider C, Suk JS, Cone R & Hanes J Mucus Penetrating Nanoparticles: Biophysical Tool and Method of Drug and Gene Delivery Advanced Materials 24, 3887–3894 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai SK et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. P Natl Acad Sci USA 104, 1482–1487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maisel K, Ensign LM, Reddy M, Cone R & Hanes J Effect of surface chemistry on nanoparticle interaction with gastrointestinal mucus and distribution in the gastrointestinal tract following oral and rectal administration in the mouse. Journal of Controlled Release 197, 48–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cu Y & Saltzman WM Controlled Surface Modification with Poly(ethylene)glycol Enhances Diffusion of PLGA Nanoparticles in Human Cervical Mucus. Mol Pharmaceut 6, 173–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis SA, Berg JR & Kleine TJ Modulation of epithelial permeability by extracellular macromolecules. Physiol Rev 75, 561–589 (1995). [DOI] [PubMed] [Google Scholar]
- 21.McCartney F, Gleeson JP & Brayden DJ Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 4, e117682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maher S, Mrsny RJ & Brayden DJ Intestinal permeation enhancers for oral peptide delivery. Adv Drug Deliver Rev 106, 277–319 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Whitehead K, Karr N & Mitragotri S Safe and effective permeation enhancers for oral drug delivery. Pharmaceut Res 25, 1782–1788 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Kapitza C et al. Oral Insulin: A Comparison With Subcutaneous Regular Human Insulin in Patients With Type 2 Diabetes. Diabetes Care 33, 1288–1290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee A et al. Ionic liquids for oral insulin delivery. P Natl Acad Sci USA 115, 7296–7301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyer H, Khedkar A & Verma M Oral insulin - a review of current status. Diabetes Obes Metab 12, 179–185 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Lerner A & Matthias T Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun Rev 14, 479–489 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Pridgen EM et al. Transepithelial Transport of Fc-Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery. Sci Transl Med 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KS, Suzuki K, Cho H, Youn YS & Bae YH Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport. ACS Nano 12, 8893–8900 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KS, Kwag DS, Hwang HS, Lee ES & Bae YH Immense Insulin Intestinal Uptake and Lymphatic Transport Using Bile Acid Conjugated Partially Uncapped Liposome. Mol Pharmaceut 15, 4756–4763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao Z, Zhang L & Jiang S Superhydrophilic zwitterionic polymers stabilize liposomes. Langmuir 28, 11625–11632 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Lu Y et al. Micelles with ultralow critical micelle concentration as carriers for drug delivery. Nat Biomed Eng 2, 318–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thwaites DT & Anderson CMH The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. Brit J Pharmacol 164, 1802–1816 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salim SY & Soderholm JD Importance of Disrupted Intestinal Barrier in Inflammatory Bowel Diseases. Inflamm Bowel Dis 17, 362–381 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Soderholm JD et al. Reversible increase in tight junction permeability to macromolecules in rat ileal mucosa in vitro by sodium caprate, a constituent of milk fat. Digest Dis Sci 43, 1547–1552 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Brake R, Vogl AW & Mitchell AWM Gray’s anatomy for students. (Elsevier/Churchill Livingstone, Philadelphia; 2005). [Google Scholar]
- 37.Frolund S, Holm R, Brodin B & Nielsen CU The proton-coupled amino acid transporter, SLC36A1 (hPAT1), transports Gly-Gly, Gly-Sar and other Gly-Gly mimetics. Br J Pharmacol 161, 589–600 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willems D, Cadranel S & Jacobs W Measurement of Urinary Sugars by Hplc in the Estimation of Intestinal Permeability - Evaluation in Pediatric Clinical-Practice. Clin Chem 39, 888–890 (1993). [PubMed] [Google Scholar]
- 39.Jiang XH, Li N & Li JS Intestinal permeability in patients after surgical trauma and effect of enteral nutrition versus parenteral nutrition. World J Gastroentero 9, 1878–1880 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson CMH et al. H+/amino acid transporter 1 (PAT1) is the imino acid carrier: An intestinal nutrient/drug transporter in human and rat. Gastroenterology 127, 1410–1422 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Broberg ML et al. Function and expression of the proton-coupled amino acid transporter PAT1 along the rat gastrointestinal tract: implications for intestinal absorption of gaboxadol. Brit J Pharmacol 167, 654–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olmsted SS et al. Diffusion of Macromolecules and Virus-Like Particles in Human Cervical Mucus. Biophys J 81, 1930–1937 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]