

Summary
Diffuse large B-cell lymphomas (DLBCLs) are genetically heterogeneous and highly proliferative neoplasms derived from germinal center (GC) B-cells. Herein, we show that DLBCLs are dependent on mitochondrial lysine deacetylase SIRT3 for proliferation, survival, self-renewal, and tumor growth in vivo regardless of disease subtype and genetics. SIRT3 knockout attenuated B-cell lymphomagenesis in VavP-Bcl2 mice without affecting normal GC formation. Mechanistically, SIRT3 depletion impaired glutamine flux to the TCA cycle via glutamate dehydrogenase and reduction in acetyl-CoA pools, which in turn induce autophagy and cell death. We developed a mitochondrial-targeted Class I sirtuin inhibitor, YC8-02, that phenocopied the effects of SIRT3 depletion and killed DLBCL cells. SIRT3 is thus a metabolic non-oncogene addiction and therapeutic target for DLBCLs.
Keywords: SIRT3, Cancer metabolism, Glutaminolysis, TCA cycle, Autophagy, YC8-02 inhibitor, DLBCL, GDH
Introduction
Diffuse large B-cell lymphomas (DLBCLs) are an aggressive and heterogeneous group of diseases from the genetic and molecular standpoints. Though R-CHOP chemoimmunotherapy has improved DLBCL clinical outcomes, approximately 40% of patients relapse or are refractory to their treatments (Basso and Dalla-Favera, 2015; Intlekofer and Younes, 2014). DLBCLs manifest more than 150 recurrent somatic mutations and are highly genetically complex (Reddy et al., 2017). Despite this mutational diversity, there are general molecular mechanisms that drive malignant phenotype across broader cross-sections of DLBCL patients and serve as a source of non-oncogene addiction (dependencies on non-mutated genes). Targeting specific genes contributing to non-oncogene addiction may provide an attractive strategy for therapeutic vulnerability in lymphomas independent of their genetics (Luo et al., 2009).
One form of non-oncogene addiction in cancer is the dependency of tumors on particular metabolic pathways for their survival and proliferation (Luo et al., 2009; Pavlova and Thompson, 2016). It is notable that DLBCLs are highly proliferative and metabolically active tumors that must fulfill significant requirements for energy and metabolic precursors. DLBCLs rely on aerobic glycolysis to generate energy and metabolic precursors, which limits the production of metabolic precursors through the tricarboxylic acid (TCA) cycle via glucose (Caro et al., 2012; Le et al., 2012). Instead, lymphoma cells engage anaplerotic programs to supply the TCA cycle with metabolites for synthesis of fatty acids, amino acids and nucleotides (Caro et al., 2012; Le et al., 2012). However, the mechanisms that control the activity and role of these different metabolic pathways in DLBCL are poorly understood.
Sirtuin family proteins have been implicated in mediating cancer adaptive pathways relevant to metabolism and gene expression, some having oncogenic and others tumor suppressor functions (Chalkiadaki and Guarente, 2015; Houtkooper et al., 2012). Sirtuins are evolutionally conserved nicotinamide adenine dinucleotide (NAD+)-dependent lysine deacetylases. In mammals, seven sirtuin members (SIRT1-SIRT7) possess various enzymatic activities including lysine deacetylation, desuccinylation and defattyacylation. Depletion of NAD+ induces cell death in hematologic cancer cells, which implicates NAD+ dependent sirtuins as important for survival (Nahimana et al., 2009). Prior reports suggest that SIRT1 may play both oncogenic and tumor suppressive functions in lymphoma. Sirt1+/− Trp53+/− mice tend to develop lymphomas (Oberdoerffer et al., 2008), while inhibition of SIRT1 mediated deacetylation of BCL6 and p53 may promote lymphoma cell death (Bereshchenko et al., 2002; Heltweg et al., 2006). Functions of other sirtuins in lymphomas largely remain unknown, which may lead to better understanding of DLBCLs and therapeutic options.
Results
DLBCL Cells are Biologically Dependent on SIRT3 Regardless of Disease Subtype
We performed an shRNA survey of the importance of the seven sirtuin family members in two DLBCL cell lines. We observed that SIRT3 was the most robustly required, although SIRT5 and SIRT7 depletion was also deleterious (Figures S1A–S1B). SIRT3 is generally considered a tumor suppressor in solid tumors (Finley et al., 2011; Haigis et al., 2012) but its function in DLBCL has not been systematically explored. To confirm that DLBCLs are dependent on SIRT3, we assembled a panel of eight DLBCL cell lines representing the cell of origin (COO) classification of DLBCL: germinal center B-cell (GCB) and activated B-cell (ABC) DLBCLs (Alizadeh et al., 2000), as well as DLBCL cells classified as having oxidative phosphorylation (OxPhos) gene expression signatures (Monti et al., 2005). We also included two control solid tumor cell lines, given that SIRT3 may function as a tumor suppressor in these cells (Bell et al., 2011; Haigis et al., 2012). We generated lentiviruses expressing three individual SIRT3 shRNAs or control shRNA with yellow fluorescent protein (YFP) (Figure S1C). All DLBCL cell lines manifested significant depletion of YFP+ cells expressing SIRT3 shRNA as compared to control (Figure 1A). In contrast, depletion of SIRT3 had no significant effects against the solid tumor cells (Figures 1A and S1D). The effect of SIRT3 depletion was partially rescued by co-expressing an shRNA-resistant form of SIRT3 (Figure S1E). Full rescue was not achieved, perhaps because we could not precisely titrate levels of exogenous SIRT3. However, a catalytically dead mutant (SIRT3H248Y) failed to rescue, indicating that the enzymatic function of SIRT3 is required for its function in DLBCL (Figure S1E).
Figure 1. SIRT3 is Highly Expressed in DLBCL and Linked to Unfavorable Outcome.
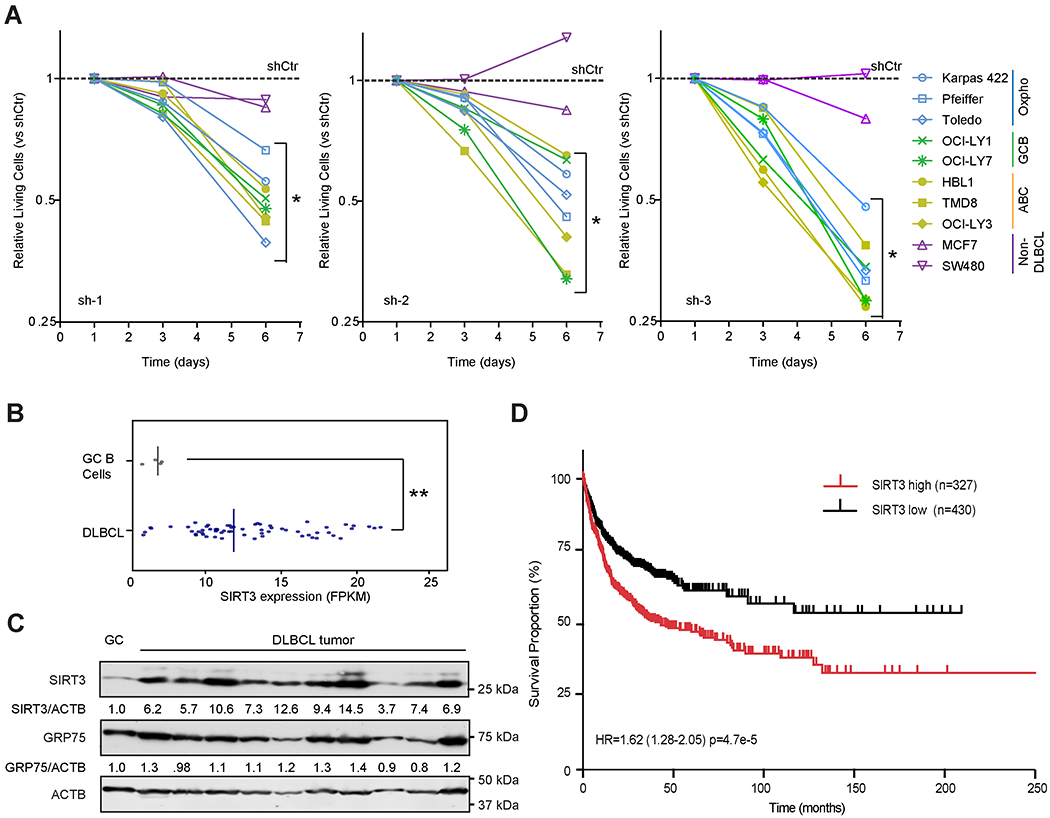
(A) Effect of SIRT3 shRNAs on the proliferation of DLBCL and non-DLBCL cell lines. Each cell line was infected with lentivirus expressing control or SIRT3 shRNA and YFP. YFP+ viable (DAPI−) cells were monitored by flow-cytometry for 6 days. Data were normalized to cells transduced with control shRNA (dashed line).
(B) SIRT3 mRNA expression detected by RNA-seq and indicated by FPKM (Fragment Per Kilobase Million) value in GC B cells (n=4) and primary DLBCL tumor samples (n=56). The median values of SIRT3 mRNA expression (the vertical line) were shown in the plot.
(C) Immunobloting for SIRT3 in primary DLBCL samples and purified human tonsillar GC B cells. GRP75 and ACTB were used as loading controls for mitochondrial and total protein, respectively. Densitometry results are indicated below the respective images.
(D) Kaplan-Meier overall survival analysis of DLBCL patients whose tumors express high (n=327) or low (n=430) SIRT3.
*p value<0.05, **p value<0.01, ***p value <0.001. Error bars represent the mean +/− SD of three or more replicates. See also Figure S1.
SIRT3 is Overexpressed in DLBCLs and Linked to Inferior Clinical Outcome
We compared and contrasted the level of SIRT3 expression in primary DLBCL specimens with normal GC B cells, which are the cell of origin of DLBCLs. Analysis of the RNA-seq data from a cohort of 56 DLBCL cases and four normal purified human tonsil GC B-cell (CD20+lgD−CD77+) specimens, indicated that SIRT3 mRNA was significantly elevated in DLBCLs compared to GC B cells (Figure 1B). We also observed that SIRT3 protein was consistently more abundant in primary DLBCL specimens and DLBCL cell lines than in GC B cells (Figures 1C and S1F).
We next evaluated the association of the expression of SIRT3 with clinical outcome in 757 patients corresponding to four publicly available DLBCL gene expression profiling cohorts (Hummel et al., 2006; Jais et al., 2008; Lenz et al., 2008a; Shaknovich et al., 2010). The optimal cutoff for SIRT3 expression was defined as the point with the most significant log-rank p value split (Budczies et al., 2012). We observed that higher SIRT3 expression was significantly associated with inferior overall survival (Figure 1D). We lacked statistical power for a multivariate analysis since the annotation of clinical features was not concordant between these cohorts. No other sirtuin family members whose expression was consistently linked to inferior outcome (not shown).
SIRT3 Maintains DLBCL Proliferation, Survival and Self-renewal
To understand the nature of SIRT3 dependency, we performed a series of phenotypic assays in DLBCL cells. Loss of SIRT3 significantly inhibited cell proliferation compared to control cells as determined by cell counts (Figure 2A). Tracking intravital dye dilution over 5 days, we observed that YFP+ cells with SIRT3 shRNAs but not control shRNA retained more dye compared to YFP-(uninfected) cells (Figure S2A), indicating inhibition of cell proliferation. In cell cycle progression assays using BrdU labeling, SIRT3 shRNAs caused a consistent reduction of YFP+S-phase cells but expansion of cells in G0/G1 as compared to YFP− cells (Figures 2B–2C). To study effects on cell death, SIRT3 shRNA transduced cells were stained with Annexin V and DAPI and analyzed by flow-cytometry at two timepoints. There was no induction of cell death 3 days after shRNA transduction. However, by day 10, we observed 2-4 folds increase in apoptosis in SIRT3 depleted vs control cells (Figure S2B). Finally, we observed significant loss self-renewal ability in colony-forming assay after SIRT3 shRNA transduction (Figures 2D and S2C).
Figure 2. SIRT3 Expression Sustains DLBCL Proliferation and Survival In vitro and In vivo.
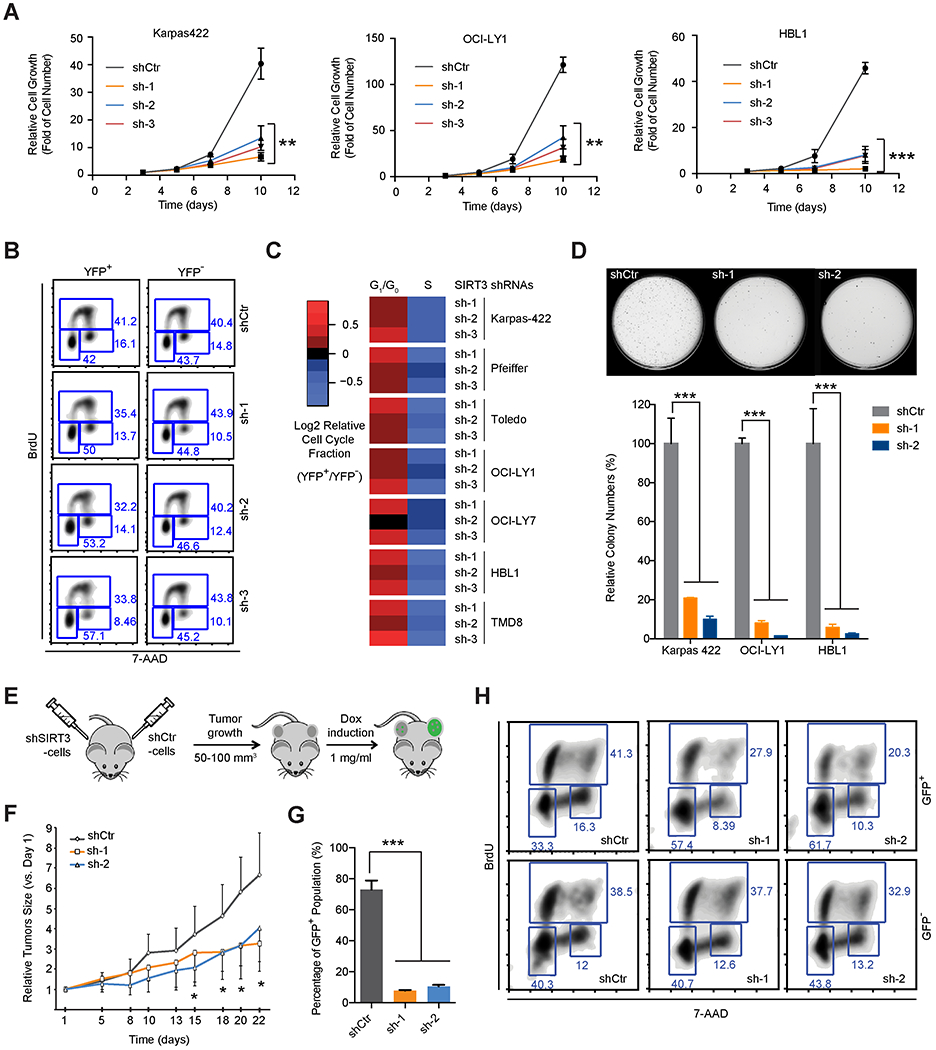
(A) Growth curves of DLBCL cell lines with SIRT3 or control shRNAs. Relative cell growth represents the fold increase of cell numbers normalized to that from first time point (the third day post transduction).
(B) Cell cycle analysis of Karpas 422 cells with SIRT3 or control shRNAs using BrdU and 7AAD staining. YFP+ and YFP− cells represent infected and non-infected cells respectively co-cultured in the same flask.
(C) Heatmap summarizing cell cycle changes induced by SIRT3 shRNAs in DLBCL cell lines. The color scale reflects the Log2 relative cell cycle fractions (YFP+/YFP−), calculated by comparing the cell cycle profiles of YFP+ and YFP− in the same flasks.
(D) Photomicrographs (top) and quantification (bottom) of colony formation in SIRT3 or control shRNA transduced Karpas 422 cells after 14 days in culture.
(E) Schema of xenograft studies with inducible SIRT3 or control shRNA expressing cells.
(F) Growth curves of xenografted tumors (Karpas 422) after induction of control or SIRT3 shRNAs expression with doxycycline in vivo. Relative tumor volumes were calculated by normalizing against the tumor volume at day one following doxycycline administration.
(G) Percentage of GFP+ cells in xenografted tumors. Xenografted tumor cells were harvested at 3 weeks post-induction with doxycycline and analyzed by flow-cytometry.
(F) Cell cycle analyses performed after in vivo BrdU incorporation in xenografted tumors following induction of SIRT3 or control shRNA expression.
*p value<0.05, **p value<0.01, ***p value <0.001. Error bars represent the mean +/− SD of three or more replicates. See also Figure S2.
SIRT3 is Required to Maintain the Growth of Lymphomas In vivo
To determine whether SIRT3 is required for the growth of DLBCL in vivo, we used a lentivirus vector expressing doxycycline inducible shRNA and GFP, which indicates the cells with shRNA expression after doxycycline induction. Karpas 422 cells were transduced with the inducible vectors expressing control or two independent SIRT3 shRNAs, then implanted in mice. shRNA expression was induced when tumors grew to 50-100 mm3 (Figure 2E). We observed that the expression of SIRT3 shRNAs significantly suppressed tumor growth as compared to controls (Figure 2F). Although all tumors started with around 70% GFP+ cells, tumors expression SIRT3 shRNA, but not control, were severely depleted of GFP+ cells after doxycycline exposure (Figure 2G). We administered BrdU to these mice prior to euthanasia and observed that viable GFP+ cells with SIRT3 vs control shRNA contained reduced proportion of S phase and increased fraction of G0/G1 phase populations, whereas GFP− cells exhibited no perturbation (Figure 2H).
SIRT3 is Dispensable for GC B cell Formation In vivo
Many lymphoma oncoproteins (e.g. BCL6 and EZH2) also play essential roles in GC formation (Beguelin et al., 2013; Dent et al., 1997). Therefore, we evaluated the role of Sirt3 in GC formation in constitutive Sirt3−/− mice, which are viable and healthy under normal conditions (Hirschey et al., 2010). GC formation was induced in Sirt3−/− and wild-type control mice by immunization with a T-cell dependent antigen. Mice were sacrificed 10 days later at the peak of the GC reaction and spleens examined by immunohistochemistry (IHC) and flow-cytometry. The splenic architecture of Sirt3−/− mice was unperturbed as compared with wild-type mice. Staining of spleen sections using the GC B-cell specific lectin peanut agglutinin (PNA) (Figure 3A) showed no difference in the number of GCs (Figure 3B), the total spleen area occupied by GCs (Figure 3C) or the average area of each GC (Figure 3D). Immunophenotyping likewise yielded similar percentage of GC B cells (B220+FAS+CD38low) in Sirt3−/− vs wild-type mice (Figures 3E–3F and S3A). No significant changes were observed among follicular B and marginal zone B cells (Figures 3E–3F and S3B). SIRT3 is thus dispensable for GC formation and is an acquired adaptation associated with malignant lymphomas.
Figure 3. SIRT3 is Dispensable for GC B-cell Function In vivo.
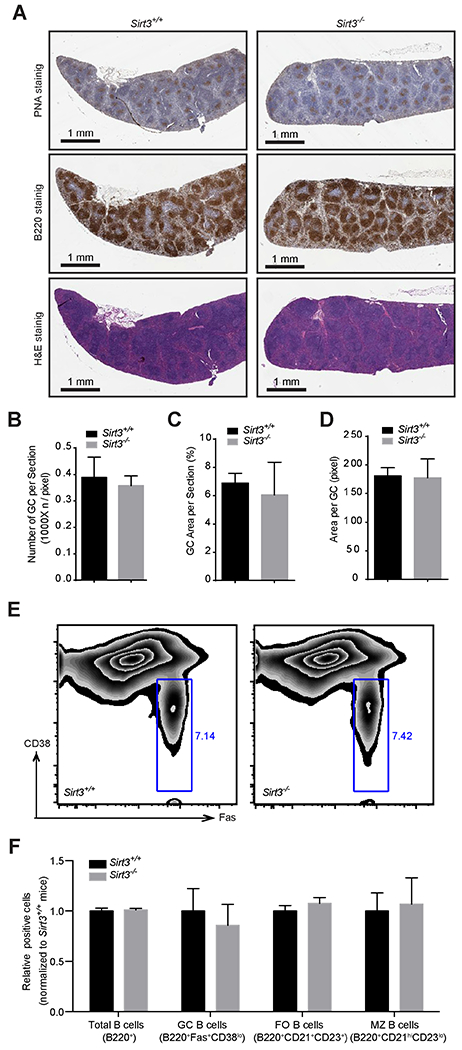
(A) PNA (top), B220 (middle) and hematoxylin and Eosin (FI&E) (bottom) staining of spleen sections derived from Sirt3+/+ and Sirt3−/− mice.
(B) Numbers of GCs per spleen sections derived from Sirt3+/+ and Sirt3−/− mice.
(C) Surface area of splenic sections occupied by GCs from Sirt3+/+ and Sirt3−/− mice.
(D) Surface area of individual GCs in spleen sections from Sirt3+/+ and Sirt3−/− mice.
(E) Flow cytometry to determine the relative abundance of GC B cells (CD38lowFAS+, indicated by gating) in DAPI−B220+ Sirt3+/+ and Sirt3−/− splenocytes.
(F) Relative percentage total B cells, GC B cells, follicular (FO) B cells, and marginal zone (MZ) B cells from Sirt3+/+ and Sirt3−/− splenocytes detected by immunophenotyping with the indicated stains.
Error bars represent the mean +/− SD of three or more replicates. See also Figure S3.
SIRT3 is Required to Maintain the TCA cycle in DLBCLs
Cancer cells undergo metabolic reprogramming to meet their energetic and biosynthetic demands (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). SIRT3 localizes in mitochondria and deacetylates various metabolic enzymes (Baeza et al., 2016; Hebert et al., 2013; Yang et al., 2016). Thus, it is plausible that DLBCL dependency on SIRT3 is linked to effects on mitochondrial metabolic pathways. To understand SIRT3 mechanism of action in DLBCL, we performed metabolomic profiling in Karpas 422 cells after transduction with SIRT3 or control shRNA. Unsupervised hierarchical clustering analysis indicated a clear distinction between metabolic profiles in SIRT3 vs control shRNA DLBCL cells (Figure 4A).
Figure 4. Loss of SIRT3 Impairs the TCA Cycle Metabolism.
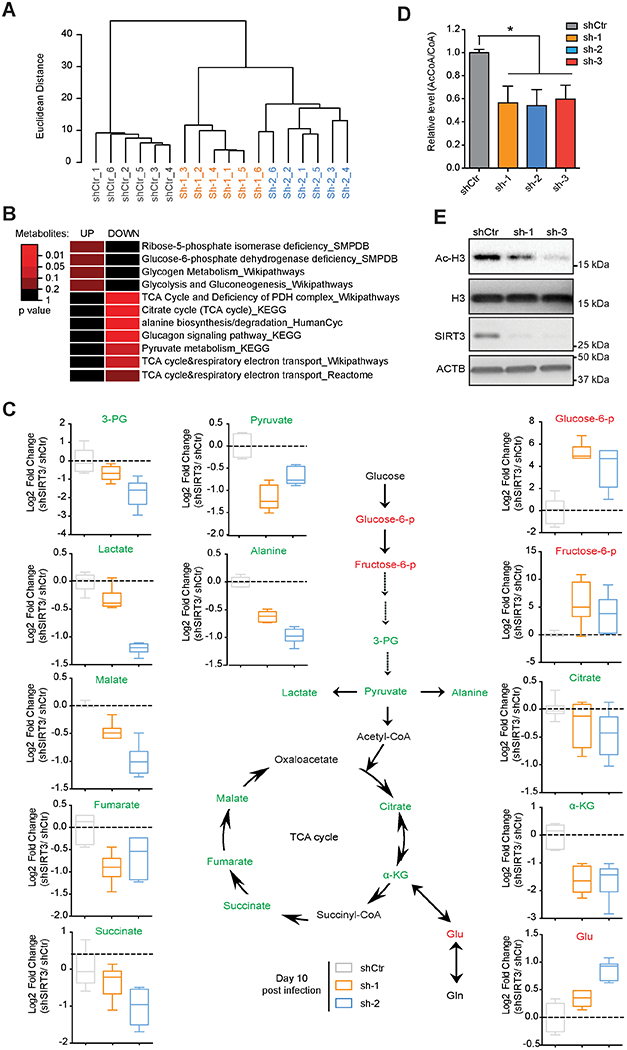
(A) Unsupervised hierarchical clustering of metabolomic profiles performed in SIRT3 or control shRNA transduced Karpas 422 at day 10 post-infection.
(B) Fleatmap showing significantly over-represented metabolic pathways among differential metabolites in Karpas 422 cells with SIRT3 shRNAs. The color key represents p values of metabolites enriched in various metabolic pathways (Wilcoxon test).
(C) Relative abundance of the indicated metabolites (z score, normalized to control) detected by metabolic profiling from Karpas 422 cells transduced with SIRT3 or control shRNAs. Green, red and black text is used to indicate decreased, increased and no detection/change, respectively. Box plot represents lower quartile; median and upper quartile and whiskers show observed minimum and maximum values.
(D) AcCoA/total CoA ratios determined by LC/MS/MC in Karpas 422 cells expressing SIRT3 or control shRNAs. *p value<0.05. Error bars represent the mean +/− SD of three or more replicates.
(E) Western blot performed with Ac-H3 antibodies in Karpas 422 cells transduced with SIRT3 or control shRNAs. Total H3 and ACTB immunoblots were also performed as loading controls for histone and total protein respectively.
See also Figure S4.
To determine which metabolic pathways were affected, we performed a pathway enrichment analysis using >3000 annotated pathways from 11 databases (Kamburov et al., 2011). This analysis indicated major reduction of TCA metabolites after SIRT3 knockdown, as well as reduction of pyruvate and alanine metabolic pathways, whereas glycolysis and glucose metabolism were induced (Figure 4B). We observed depletion of key TCA cycle metabolites such as citrate, α-ketoglutarate (αKG), succinate, fumarate, and malate (Figure 4C). We also observed accumulation of glutamate, reduction in downstream glycolytic metabolites (3-phosphoglyceric acid, pyruvate, alanine and lactate) and increase of upstream glycolysis metabolites (glucose 6-phophate and fructose 6-phosphate) (Figure 4C). TCA cycle defects are generally accompanied with decrease of acetyl-coenzyme A (AcCoA) production (Eisenberg et al., 2014; Martinez-Reyes et al., 2016; Schroeder et al., 2014). Indeed, we observed significant reduction of AcCoA vs total CoA after SIRT3 depletion (Figure 4D). The impact of acetyl-CoA reduction was underlined by the marked decrease in acetylated histone 3 (Ac-H3) (Figure 4E). These effects were generally reproducible in a second DLBCL cell line (Figures S4A–C).
Perturbation of the TCA Cycle by SIRT3 shRNAs Activates Autophagy
Impairment of the TCA cycle and AcCoA levels is reported to induce autophagy (Eisenberg et al., 2014; Martinez-Reyes et al., 2016), perhaps because cells must obtain metabolic precursors from alternative sources. Upon induction of autophagy, LC3I is conjugated to phosphatidylethanolamine to form LC3II, resulting in increased LC3II/LC3I ratios (Klionsky et al., 2016; Noda and Inagaki, 2015). We observed that SIRT3 depletion caused increased LC3II/LC3I ratios and reduction of p62 protein, which is degraded upon induction of autophagy (Klionsky et al., 2016; Noda and Inagaki, 2015), in DLBCL cell lines (Figure 5A). We also used a fluorescent reporter (mCherry-EGFP-LC3B) to quantify autophagic flux, whereby autophagy active cells manifest EGFPlow and mCherryhigh signals (Gump et al., 2014; Klionsky et al., 2016; Noda and Inagaki, 2015). SIRT3 knockdown enhanced abundance of EGFPlowmCherryhigh cells (Figure 5B) and autophagy active cells showed higher mCherry/EGFP signal across multiple cell lines (Figures 5C–5E and S5A). Activation of autophagy by SIRT3 depletion was partially rescued by re-expression of SIRT3, but not SIRT3H248Y (Figure S5B). Therefore, SIRT3 suppresses autophagy through its protein deacetylase activity.
Figure 5. Loss of SIRT3 Activates Autophagy in DLBCL Cells.
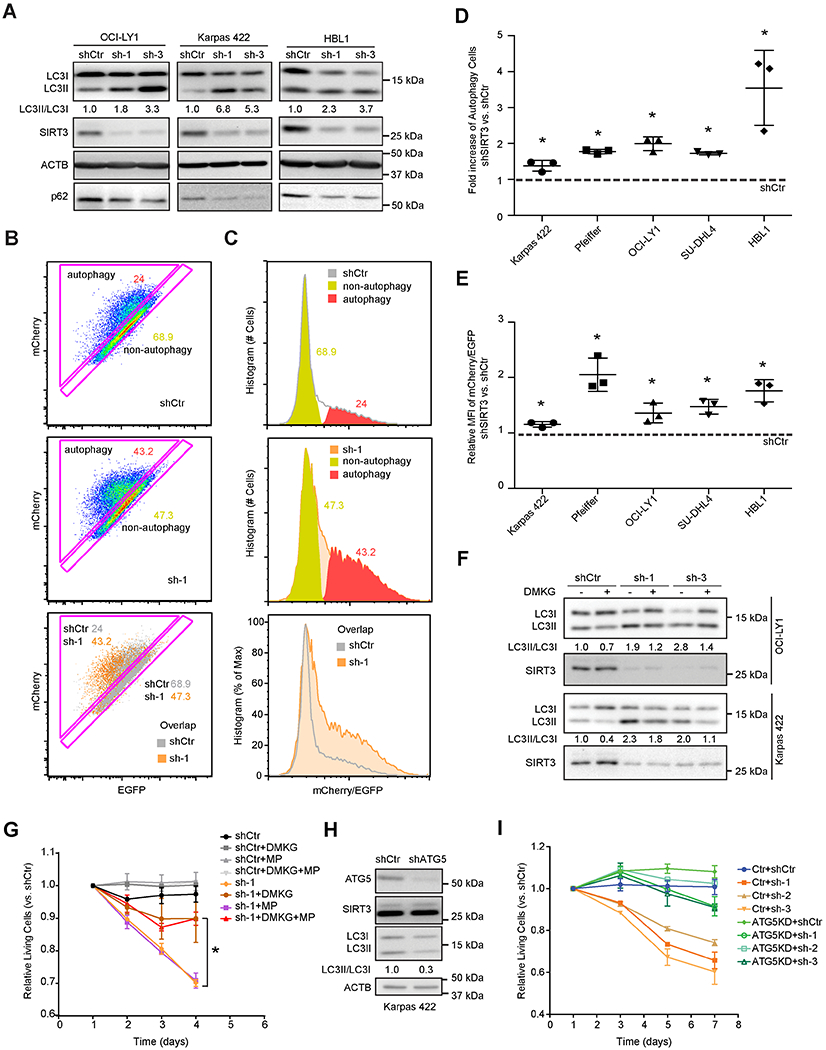
(A) Autophagy levels by detecting the ratio of LC3II/LC3I and p62 level in DLBCL cell lines with SIRT3 or control shRNAs. LC3II/LC3I ratios were quantified by densitometry and normalized to control shRNA (shCtr).
(B) Flow-cytometric analysis of DLBCL cells with a mCherry-EGFP-LC3 reporter showing autophagy activated (triangle) vs autophagy inactivated (along the diagonal) cells. Scatter plots of DLBCL cells transduced with shCtr (top), shSIRT3-sh-1 (middle), and their overlay (bottom) were shown.
(C) Representative histograms showing mCherry/EGFP signal from DLBCL cells transduced with shCtr (top), shSIRT3-sh-1 (middle) and their overlay (bottom). Red and yellow areas represent the signal intensities of mCherry/EGFP signals of autophagy active or inactive populations in Figure 5B, respectively.
(D) Fold changes of autophagy active cells in different DLBCL cell lines with SIRT3 shRNA vs control. The percentage of autophagy active populations, gated as Figure 5B, were used to calculate the fold increase of autophagy active cells in SIRT3 depleted vs controls cells (shSIRT3/shCtr).
(E) Fold changes of autophagy levels by comparing the mean fluorescence intensity (MFI) of mCherry/EGFP signal (calculated with histogram as in Figure 5C) in DLBCL cell lines with SIRT3 shRNA vs control (shSIRT3/shCtr).
(F) Effects of DMKG on autophagy (LC3II/LC3I) in DLBCL cells with SIRT3 or control shRNAs. Cells were incubated with DMKG (4 mM) for 3-4 hr before collecting for Western blot using LC3 or SIRT3 antibodies. LC3II/I ratios were calculated by densitometry and normalized to that of control cells.
(G) Effect of a 4-day exposure to DMKG, MP and both on the proliferation of Karpas 422 cells transduced with SIRT3 or control shRNA. The abundance of YFP+ vs YFP− (transduced vs non-transduced) cell populations were measured and normalized to the first time point.
(H) Karpas 422 cells were transduced with ATG5 or control shRNA and western blots performed using antibodies for ATG5, SIRT3, LC3, along with ACTB as loading controls. Densitometry was performed to determine the LC3II/LC3I ratios and normalized to that of control cells.
(I) Karpas 422 cells stable transfected with an ATG5 shRNA or control shRNA and selected by puromycin, then transduced with SIRT3 or control shRNA. Cell proliferation showed the relative abundance of YFP+ vs YFP− populations and normalized to the first time point
*p value<0.05. Error bars represent the mean +/− SD of three or more replicates. See also Figure S5.
To test for actual link of autophagy with impaired TCA cycle induced by SIRT3 depletion, we used dimethyl-αKG (DMKG) to replenish TCA cycle metabolism (Eisenberg et al., 2014). We observed that DMKG rescued LC3II/LC3I ratios (i.e. autophagy) in DLBCL cells (Figure 5F). In addition, restoring the TCA cycle with DMKG and methyl-pyruvate (MP) (Herranz et al., 2015; Son et al., 2013) rescued proliferation arrest caused by SIRT3 depletion (Figures 5G, S5C–S5D). Whereas DMKG alone rescued SIRT3 depleted Karpas 422 (Figure 5G) and OCI-LY1 (Figure S5C) cells, HBL1 cells required both DMKG and MP (Figure S5D) pointing to a partial dependency of HBL1 cells on the pyruvate pathway. To test if autophagy contributes to proliferation arrest caused by SIRT3 depletion, we knocked down ATG5, which is required for formation of autophagosomes (Noda and Inagaki, 2015; Takamura et al., 2011). As expected, ATG5 shRNA impaired autophagy in DLBCL cells (Figure 5H). However, we also observed that ATG5 depleted cells manifested resistance to SIRT3 knockdown (Figure 5I). Hence proliferation arrest induced by SIRT3 depletion in DLBCL is at least in part caused by autophagy that is triggered by impairment of the TCA cycle.
These data suggest that autophagy has a tumor suppressor effect in DLBCL. Indeed by examining the genetic profiles of 694 DLBCL samples (Green et al., 2014) we observed that 6q deletion, which contains the ATG5 locus, was one of most highly recurrent genetic alterations (17% of cases, Figure S5E). We confirmed this observation in an independent cohort of patients (Chapuy et al., 2018). Strikingly, ATG5 is within the 6q21 region that is most reproducibly deleted. Analysis of gene expression in the same patients revealed that ATG5 deletion is accompanied by significant reduction of ATG5 transcript abundance (Figure S5F), providing genetic evidence that autophagy is a tumor suppressive in the DLBCL context.
SIRT3 Drives Anaplerotic Glutaminolysis to Power the TCA cycle in DLBCL Cells
B-cell lymphomas use both glucose and glutamine to power the TCA cycle to generate energy and metabolic precursors (Caro et al., 2012; Le et al., 2012). Hence, we considered two possible explanations for loss of TCA cycle function: i) impaired glycolysis, or ii) block of anaplerotic reactions for glutamate/glutamine usage. To determine which is occurring, we performed metabolic tracing studies using 13C-labeled glutamine ([U-13C5] glutamine) and glucose ([U-13C6] glucose). Glutamine is used in TCA cycle after being transformed into glutamate, which is then converted to αKG by glutamate dehydrogenase (GDH). In the TCA cycle, αKG can be used through de-carboxylation to produce succinate (forward) or carboxylation to form citrate (reverse) (Figure 6A). Analysis of 13C glutamine labeled metabolites after SIRT3 knockdown showed decreased contributions of 13C from glutamine to both forward (M+4-succinate, fumarate, malate and aspartate) and reverse TCA cycle (M+5-citrate, M+3-malate and -aspartate, Figures 6A–6B). In contrast, 13C glucose labeling experiments revealed increased 13C incorporation into the TCA cycle metabolites after SIRT3 knockdown (Figures S6A–S6B). At the same time, there was reduction of 13C incorporation to alternative glycolysis pathway metabolites including 3-PG, serine, alanine and lactate (Figures S6A–S6B). The results were further validated in13C-glutamine and 13C-glucose tracing studies in OCI-LY1 cells, confirming that SIRT3 depletion impairs glutamine entry into the TCA cycle and increases utilization of glucose (Figures S6C–S6D).
Figure 6. Glutamine Dehydrogenase is a Key Downstream Target of SIRT3 in DLBCL cells.
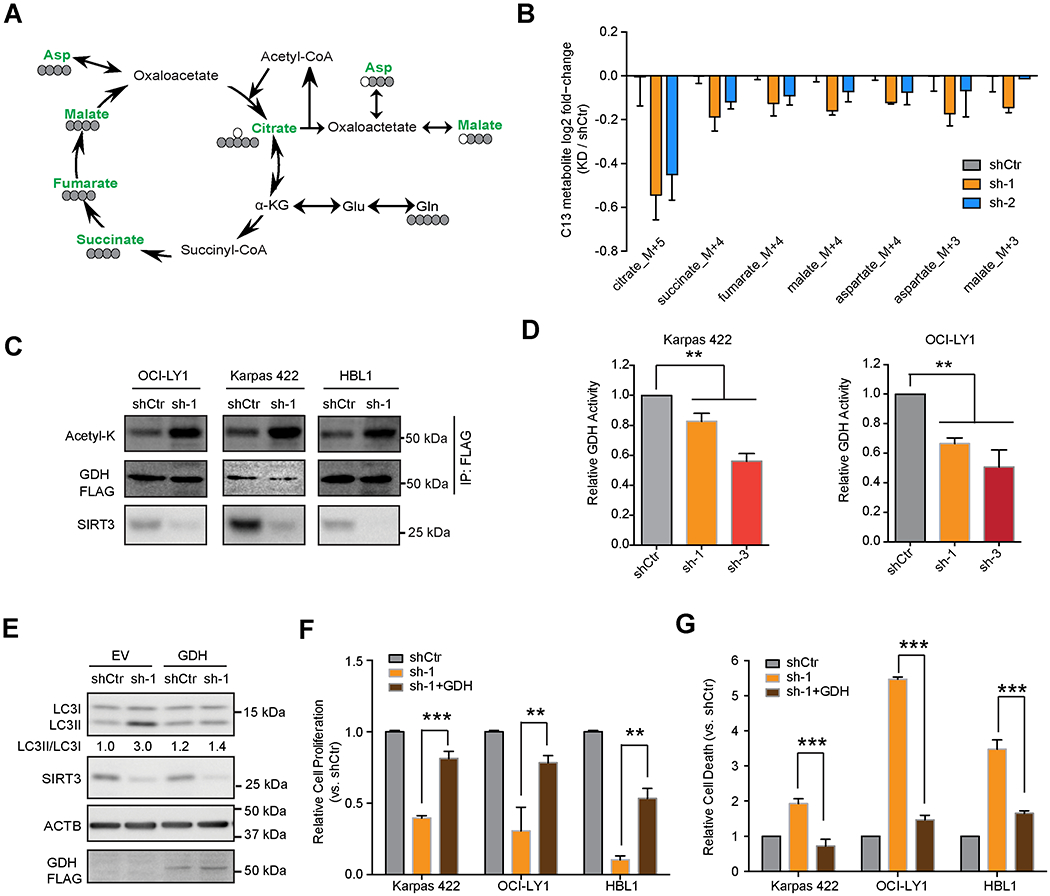
(A) Karpas 422 cells were transduced with SIRT3 or control shRNA followed by exposure to [U-13C5] glutamine. Color green indicates reduction and black indicates no change/detection. Carbon atom (13C in grey and 12C in white circles) transitions and tracers were used to detect glutamine metabolism.
(B) Relative incorporation of 13C glutamine in downstream metabolites in Karpas 422 cells. The Y-axis indicates log fold changes in 13C-metabolites in cells transduced SIRT3 shRNA vs control.
(C) Acetylation levels of GDFI-FLAG in the indicated DLBCL cell lines with SIRT3 vs control shRNA. Immunoprecipitations followed by Western blots for acetyl-lysine (acetyl-K), GDH-FLAG (anti-FLAG).
(D) GDH activity was measured by GDH activity kit with cell lysates of Karpas 422 (left) and OCI-LY1 (right) DLBCL cells with SIRT3 or control shRNA. The Y axis shows the relative GDH activity normalized to that in control cells.
(E) Autophagy level (LC3II/LC3I) caused by SIRT3 shRNA (sh-1) in control (empty vector, EV) or exogenous GDH expressing cells (GDH). Densitometry results (LC3II/LC3I) were normalized to that in control cells (EV, shCtr cells).
(F) Relative cell numbers of control and SIRT3 depleted cells with or without co-expression of GDH. Results were normalized to cell numbers of controls (with control shRNA without GDH expression) at day 10 post-infection.
(G) Relative cell death in control or SIRT3 depleted cells with or without co-expression of GDH. The percentage of DAPI+ cells were gated and normalized to controls (with control shRNA without GDH expression) at day 10 post-infection.
*p value<0.05, **p value<0.01, ***p value <0.001. Error bars represent the mean +/− SD of three or more replicates. See also Figure S6.
Glutamine Dehydrogenase is a Key Downstream Target of SIRT3 in DLBCL Cells
We next explored the direct mechanism through which SIRT3 drives glutamine entry to the TCA cycle. Protein acetylation plays an important role in regulating the activity of metabolic pathways (Baeza et al., 2016; Hebert et al., 2013; Yang et al., 2016). As expected, SIRT3 depletion caused an increase in mitochondrial protein acetylation in DLBCL cells (Figure S6E). We then tested whether GDH, which converts glutamate to αKG to enter the TCA cycle, could be a key target of SIRT3 deacetylation (Lombard et al., 2007). For this, we expressed FLAG tagged GDH in three DLBCL cell lines with SIRT3 or control shRNAs then performed FLAG-IP in these cells followed by acetyl-lysine immunoblot. In all cases, SIRT3 depletion induced GDH hyperacetylation (Figure 6C). We also performed GDH enzymatic activity assays with DLBCL cell lysates after SIRT3 knockdown and observed decreased GDH activity in accordance with the notion that acetylation impairs its enzymatic activity (Figures 6D and S6F). To determine whether GDH activity is critical to the SIRT3 phenotype, we overexpressed GDH in SIRT3 depleted cells and found that it prevented induction of autophagy after SIRT3 knockdown (LC3II/LC3I ratios, Figure 6E). GDH overexpression also significantly rescued SIRT3 cell proliferation and survival effects (Figures 6F–6G and S6G). Reciprocally, GDH knockdown induced proliferation arrest and activation of autophagy in DLBCL cells, similar to what we observe with SIRT3 knockdown (Figures S6H–S6I). Therefore, deacetylation and induction of GDH activity is critical to the effect of SIRT3 in DLBCL cells.
SIRT3 is Required for Lymphomagenesis In vivo
To determine whether SIRT3 might be required to support the malignant transformation of GC B cells into lymphomas and not just maintaining proliferation of established DLBCL cells, we crossed Sirt3−/− mice with VavP-Bcl2 transgenic mice, which develop GC derived lymphomas (Egle et al., 2004). VavP-Bcl2;Sirt3+/+ mice developed lymphoma by 6 months of age, manifesting as gross enlargement and distortion of lymphoid follicles and aberrant GC structures in PNA and B220 IHC staining (Figure S7A). In contrast, VavP-Bcl2;Sirt3−/− mice of similar age still maintained follicle structures with less invasive outgrowth of B cells (Figure S7A). An additional cohort of mice generated by transplanting bone marrow cells from VavP-Bcl2;Sirt3−/− and VavP-Bcl2;Sirt3+/+ mice into lethally irradiated recipients were sacrificed ~110 days after transplant to determine whether incipient lymphomas were developing. Mice transplanted with VavP-Bcl2;Sirt3−/− cells displayed significantly less splenomegaly than (Figure 7A), but similar body weight to (Figure S7B), those transplanted with VavP-Bcl2;Sirt3+/+ cells. As expected, mice transplanted with VavP-Bcl2;Sirt3−/− cells developed systemic lymphoma with infiltration of highly proliferative B cells consistent with DLBCL in multiple organs including lung and liver (Figures 7B and S7C). In marked contrast, mice transplanted with VavP-Bcl2;Sirt3−/− cells had no evidence of systemic lymphoma (Figures 7B and S7C) and manifested significantly improved survival vs those transplanted with VavP-Bcl2;Sirt3+/+ cells (Figure 7C). Hence SIRT3 contributes to lymphomagenesis in vivo, underlining a critical role for its metabolic effects in B-cell transformation.
Figure 7. SIRT3 is Required for Lymphomagenesis.
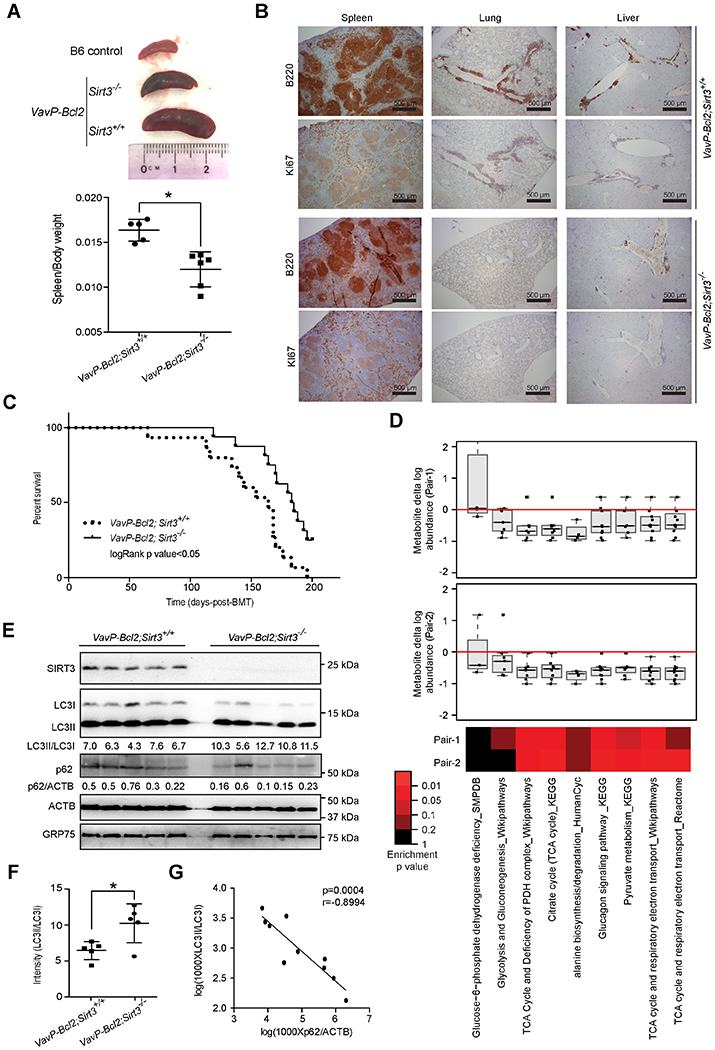
(A) Representative images of spleens from control C57B6/J(B6) mouse or recipient mice with VavP-Bcl2;Sirt3+/+ or VavP-Bcl2;Sirt3−/− bone marrow cells at 110 days post bone marrow transplantation (top) and comparison of spleen/body weight ratios from mice with VavP-Bcl2;Sirt3+/+ or VavP-Bcl2;Sirt3−/− bone marrow cells (bottom).
(B) H&E, B220 ,and Ki67 staining of spleen, lung and liver sections from mice transplanted with VavP-Bcl2;Sirt3+/+ and VavP-Bcl2;Sirt3−/− bone marrow cells.
(C) Kaplan-Meier curve showing the overall survival of mice transplanted with VavP-Bcl2;Sirt3+/+ (n=14) or VavP-Bcl2;Sirt3−/− (n=16) bone marrow cells.
(D) The box plots show relative change in metabolite abundance in Sirt3−/− vs Sirt3+/+ lymphomas, and the heatmap shows statistical significance of these changes as determined by Wilcoxon test. Box plot represents lower quartile; median and upper quartile and whiskers show observed minimum and maximum values.
(E) Protein levels of SIRT3, LC3, p62 in splenocytes from VavP-Bcl2;Sirt3+/+ or VavP-Bcl2;Sirt3−/− mice, GRP75 and ACTB were blotted as loading controls for mitochondrial and total protein respectively. The ratios of LC3II/LC3I and p62/ACTB were calculated with densitometry results.
(F) Quantification of LC3II/LC3I ratios in splenocytes from VavP-Bcl2;Sirt3+/+ or VavP-Bcl2;Sirt3−/− mice based on densitometry results in (E).
(G) A correlation plot of p62/ACTB and LC3II/LC3I ratios in splenocytes of VavP-Bcl2;Sirt3+/+ and VavP-Bcl2;Sirt3−/− mice, based on densitometry results in (E).
*p value<0.05. Error bars represent the mean +/− SD of three or more replicates. See also Figure S7.
Loss of SIRT3 Impaired the TCA cycle Metabolism and Induced Autophagy in Primary Lymphomas In vivo
To determine whether SIRT3 mediated similar effects on metabolism in primary lymphomas in vivo, we enriched for B220+ cells from spleens of VavP-Bcl2;Sirt3+/+ and VavP-Bcl2;Sirt3−/− mice for metabolic profiling, matching mice for gender and age. Similar to human cell lines, absence of SIRT3 caused a reduction in TCA metabolites, pyruvate metabolism, and glucagon signaling (Figure 7D). Because direct measurement of AcCoA was impossible in these specimens, we examined Ac-H3, as a surrogate, and observed significantly lower abundance of in VavP-Bcl2;Sirt3−/− splenocytes (Figure S7E). We accordingly also observed higher ratio of LC3II/LC3I in VavP-Bcl2;Sirt3−/− splenocytes (Figures 7E–7F), which was inversely correlated to p62 abundance (Figure 7G). Moreover, spleen/body weight were negatively correlated with autophagy (Figure S7F), which is in accordance with the proposed tumor suppressor function of autophagy (Kenific and Debnath, 2015), suggesting that SIRT3 overcomes this mechanism to support the growth of B-cell lymphomas.
Therapeutic Targeting of DLBCL with Small Molecule SIRT3 Inhibitors
Given its important role in proliferation and survival of DLBCL cells but being dispensable to normal cells, SIRT3 could serve as an attractive drug target. We developed a series of small molecules including a SIRT2 selective thiomyristoyl lysine compound called TM (Jing et al., 2016) and Biotin-TM3 (selective to SIRT1 and SIRT2). To improve SIRT3 inhibition, we developed a compound called JH-T4 that can inhibit all three Class I sirtuins at micro- to nanomolar concentrations in biochemical enzymatic assays (Figures 8A–8B and S8A). To achieve superior penetration into mitochondria, JH-T4 was modified by replacing the benzyl carbamoyl group with a triphenylphosphonium (TPP) mitochondrial targeting moiety to yield the compound, YC8-02 (Figures 8A–8B and S8B–S8C). Compared to JH-T4, YC8-02 manifested increased ratio of mitochondria to total cell concentration, as measured by mass spectrometry in purified mitochondria extracts and total cell lysates (Figure S8D). YC8-02 was also unexpectedly more efficient than JH-T4 at inhibiting SIRT3 in in vitro biochemical assays (Figures 8B and S8C).
Figure. 8. Effects of SIRT3 inhibitor on the Metabolic Phenotype of DLBCL cells.
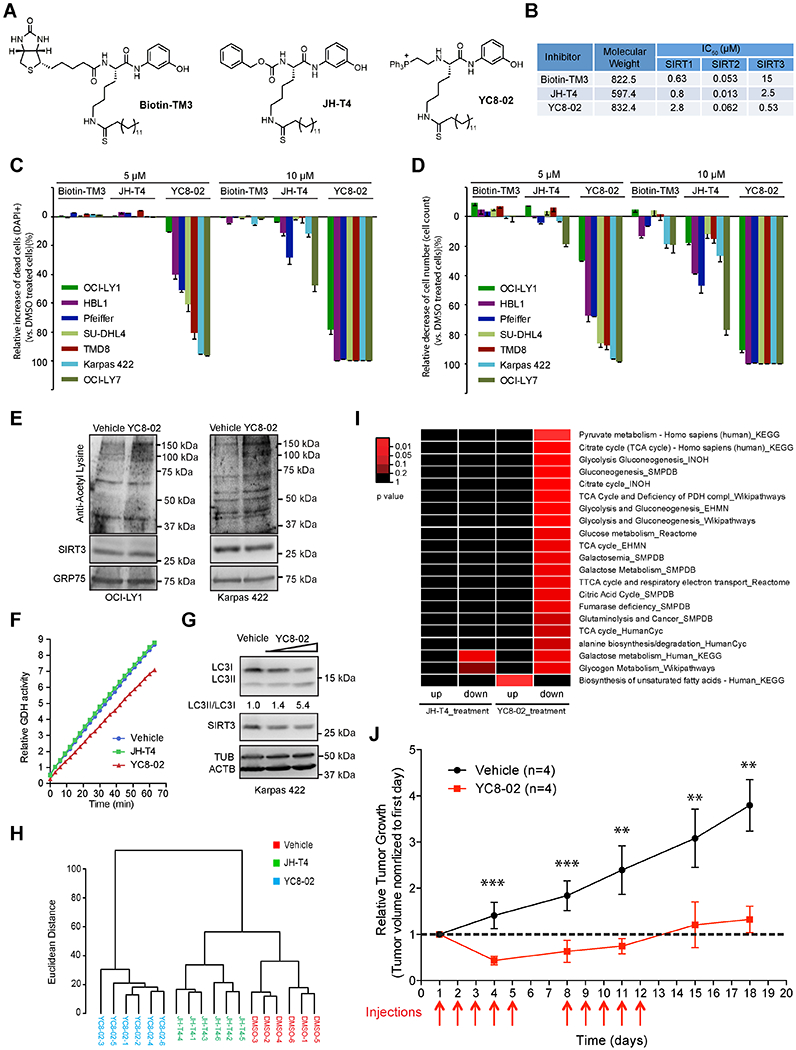
(A) Chemical structures of Biotin-TM3, JH-T4, and YC8-02.
(B) Percentage of dead cells (DAPI+) of DLBCL cell exposed to Biotin-TM3, JH-T4, and YC8-02 at 5 μM or 10 μM concentrations for 48 hr. Data were normalized to vehicle treated controls.
(C) Biochemical ICso values for Biotin-TM3, JH-T4, and YC8-02 were determined based on inhibition of deacetylase activities of SIRT1, SIR2 and SIRT3in vitro.
(D) Cell numbers changes of DLBCL cells treated with Biotin-TM3, JH-T4, and YC8-02 at 5 μM or 10 μM for 48 hr. Data were normalized to vehicle treated controls.
(E) Mitochondrial protein acetylation in OCI-LY1 and Karpas 422 cells (1 million/ml) treated with vehicle (DMSO) or YC8-02 at 7.5 μM and 5 μM, respectively. The mitochondrial protein fractions were enriched at 6 and 12 hr post treatment for Western blot using acetyl-lysine antibodies.
(F) GDH activity from cell lysates of Karpas 422 cells treated with 5 μM YC8-02, JH-T4 or vehicle for 48 hr. The Y axis shows the GDH activity in a time-lapse measurement.
(G) Autophagy levels (LC3II/LC3I) in Karpas 422 cells exposed to YC8-02 at 3 μM and 5 μM or vehicle for 40 hr. Densitometry was used to calculate LCII/I ratios normalized to that from vehicle treated control cells.
(H) Unsupervised hierarchical clustering of metabolomic profiles in Karpas 422 cells treated with vehicle (DMSO), JH-T4 or YC8-02.
(I) Heatmap showing significantly up or down-regulated metabolic pathways in Karpas 422 cells treated with JH-T4or YC8-02 vs vehicle controls. The color key indicates statistical significance based on the enrichment of metabolites in various pathways (Wilcoxon test).
(J) In vivo activity of YC8-02 against DLBCL xenografts. Treatments were done once per day for five days (weekdays) for two weeks (red arrows below x axis). Relative tumor volumes were calculated at the indicated timepoints by normalizing to the tumor volume of the first day prior to initiating treatment.
**p value < 0.01; ***p value< 0.001. Error bars represent the mean +/− SD of three or more replicates. See also Figure S8.
We next evaluated the effect of these compounds on the viability and the proliferation of DLBCL cell lines. YC8-02 was the most effective one (Figures 8C–8D, S8E–S8F), JH-T4 lacks the TPP motif and was much less active, Biotin-TM3 does not target SIRT3 and was inactive in these assays. Therefore, mitochondrial targeting of SIRT3 was critical to the YC8-02 effect in DLBCLs. Moreover, YC8-02 effects were selective to DLBCL vs human solid tumor cell lines or cord blood cells (Figures S8G–S8H).
To validate that the effects of YC8-02 were linked to suppression of SIRT3, we first observed that treatment of DLBCL cells with YC8-02 increased mitochondrial protein acetylation (Figure 8E). Moreover, YC8-02, but not JH-T4, inhibited GDH activity (Figure 8F) and induced autophagy (Figure 8G) in DLBCL cells. Finally, we performed metabolomic profiling in Karpas 422 cells treated with YC8-02, JH-T4 or vehicle. YC8-02 manifested a distinct profile as compared to JH-T4 and vehicle (Figure 8H). YC8-02 induced greater metabolic perturbation (95 metabolites down and 93 up, p<0.1, logFC>0.3) than JH-T4 (34 down and 35 up, p<0.1, logFC>0.3). YC8-02 but not JH-T4 treatment caused reduction of TCA cycle metabolites as well as pyruvate metabolism, alanine biosynthesis and glutaminolysis (Figure 8I). Using an additional quantitative approach, YC8-02 but not JH-T4 enriched for metabolic pathways suppressed by SIRT3 depletion, indicating that the active compound could phenocopy loss of SIRT3 (Figure S8I). We next tested if YC8-02 could suppress lymphoma growth in vivo. We first assayed toxicity by treating C57BL/6 mice with 30 mg/kg of YC8-02 or vehicle over five days and observed no evidence of discomfort or weight loss (Figure S8J). The same dose of YC8-02 was then used to treat NOD/SCID mice bearing DLBCL xenografts from Karpas 422 cells, in two five-day pulses. We observed that YC8-02 caused regression of the tumors during the time mice were receiving drug. The suppressive effects persisted for several days after the treatments until the mice were sacrificed (Figure 8J). Lastly, we tested another reported SIRT3 selective inhibitor, compound#8 (Mahajan et al., 2014), which although is less potent than YC8-02 is relatively selective to SIRT3 in vitro (Figure S8K). Compound#8 also reduced DLBCL cell viability (Figure S8L) and triggered activation of autophagy (Figure S8M). Collectively, these data indicate that SIRT3 is a therapeutic target in DLBCL and support the rationale for development of clinical grade SIRT3 selective inhibitors.
Discussion
Herein, we provide evidence that SIRT3 is a metabolic, non-oncogene dependency in DLBCL where it is required to maintain the accelerated and sustained metabolic needs of these rapidly dividing and growing tumors. SIRT3 dependency is independent of genetic background and DLBCL subtype, indicating that this is a more generalized biological feature of this disease. Pro-oncogenic effects of SIRT3 were also noted in certain tumor cell lines such as oral and bladder cancers (Chen et al., 2014). Two other studies explored SIRT3 knockdown in B-cell lymphoma cell lines but did not note its pro-oncogenic role (Yu et al., 2016; Yuan et al., 2017). The most likely explanation for this is the use of transient siRNA by one report (Yu et al., 2016) and the use of antibiotic selection in the other (Yuan et al., 2017), which can yield selection of resistant clones. Other studies have reported a tumor suppressor function for SIRT3 for example in breast and ovarian cancers (Bell et al., 2011; Finley et al., 2011), where loss of SIRT3 stabilized HIF1α (hypoxia inducible factor 1α), leading to transcription of glycolytic genes and induction of the Warburg effect. Hence the putative oncogenic and tumor-suppressive roles of SIRT3 are context-specific and may be linked to how its downstream functions play into malignant transformation in different tissues.
Along these lines, it is worth noting that SIRT3 was dispensable for the normal GC B cells from which DLBCLs arise. Like DLBCLs, normal GC B cells also have extreme metabolic needs given their rapid proliferation (Boothby and Rickert, 2017; Ersching et al., 2017; Jellusova et al., 2017). Yet in GC reactions, proliferation is self-limited. When B cells are activated to enter the GC proliferative zone, they first undergo a brief metabolic charging phase when they accumulate metabolic precursors but do not replicate (Boothby and Rickert, 2017; Ersching et al., 2017; Jellusova et al., 2017; Mesin et al., 2016). This anabolic program is shut down during the GC B cell proliferative burst, and depletion of nutrients is believed to limit GC B cell proliferation (Boothby and Rickert, 2017; Jellusova et al., 2017; Mesin et al., 2016). Strictly compartmentalizing anabolic growth from rapid proliferation in the GC may help to limit malignant transformation. Hence, it is tempting to speculate that upregulation of SIRT3 contributes to lymphomagenesis by reprogramming GC B cells to overcome this barrier between metabolic charging and proliferation by enabling B cells to more efficiently metabolize nutrients through anaplerotic pathways.
We find that SIRT3 drives anaplerotic glutaminolysis to fuel the TCA cycle in DLBCL cells independent of whether DLBCLs are of the GCB or ABC subtypes (Alizadeh et al., 2000) or the OxPhos or B cell receptor (BCR) dependent categories (Monti et al., 2005). The OxPhos-DLBCLs can use fatty acids as a source for the TCA cycle and oxidative phosphorylation while the BCR-DLBCLs are more efficient at glycolysis (Caro et al., 2012). Despite these nuances, both the OxPhos and BCR subtypes use glutamine to fuel the TCA cycle (Caro et al., 2012). Hence our results provide the basis to understand the overarching glutamine dependency across DLBCL subtypes, which is mediated in large part through SIRT3 deacetylation of GDH. SIRT3 is thus a critical upstream enzyme in this metabolic cascade. Although our studies point to GDH as a critical target of SIRT3 in DLBCL, these results do not rule out that other SIRT3 TCA substrates downstream of GDH could contribute to its actions (Hebert et al., 2013; Yang et al., 2016). Moreover, SIRT3 may contribute to other biological features of DLBCL through other mitochondrial pathways (Tao et al., 2010). Such considerations warrant further studies.
We present genetic and mechanistic evidence that autophagy is generally a tumor suppressor in DLBCL, downstream of SIRT3. Our results are consistent with data showing autophagy activation under metabolic stress conditions, such as reduction in the TCA cycle activity with decreased AcCoA production (Eisenberg et al., 2014; Marino et al., 2014; Martinez-Reyes et al., 2016). Indeed, autophagy was initially characterized as a tumor suppressing mechanism and mosaic deletion of Atg5 caused liver tumors in mice (Liu et al., 2017; Pietrocola et al., 2016; Takamura et al., 2011). In contrast, autophagy can also facilitate survival of cancer cells in response to stress by mitigating cellular damage and recycling nutrients to sustain tumor metabolism (Kenific and Debnath, 2015; Kimmelman and White, 2017). Autophagy thus has context-specific roles in cancer, which may depend on the identity and type of proteins that undergo autophagic protein degradation. For example, autophagy may impair DNA replication and metabolic pathways when it induces degradation of proteins important to these functions (Mathew et al., 2014), and can either promote or inhibit apoptosis by degrading particular proteins in response to death stimuli (Gump et al., 2014; Kenific and Debnath, 2015).
The requirement of SIRT3 to maintain the growth and survival of DLBCL cells points toward targeting SIRT3 as a potential therapeutic approach. As all sirtuins require NAD+ for their enzymatic activity, niacinamide and AP0866 (a NAD+ biosynthesis inhibitor) were suggested to have therapeutic activity as pan-sirtuin inhibitors in lymphoma patients (Amengual et al., 2013; Ginet et al., 2014; Nahimana et al., 2009). SIRT3 expression rescued NAD+depletion induced cell death (Yang et al., 2007). The importance of SIRT3 in DLBCL inspired us to develop the mitochondrial targeted Class I sirtuin inhibitor YC8-02 as a DLBCL therapeutic agent, relevant to a broad cross section of patients including those with inferior clinical outcomes based on current standard of care. Given our finding of autophagy as a tumor suppressor mechanism in DLBCL that is suppressed by SIRT3, it will be important to consider the presence of ATG5 deletion as a potential source of resistance to SIRT3 inhibitors to aid in patient selection for future clinical trials.
STAR*METHODS
CONTACTS FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ari Melnick (amm2014@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Tonsil and DLBCL samples
Primary human de-identified leftover tonsil samples and DLBCL samples were obtained and processed under the Institutional Review Board protocols of Weill Cornell Medicine (#0804009762) and the University of Turin (#0081521) after written informed consent, in accordance to the Declaration of Helsinki protocol. Human B cell populations were affinity-purified using standard protocols and GC B cell purity was determined by flow cytometry analysis of surface logD (BD Pharmingen), CD77 (AbD Serotech) and CD38 (BD Pharmingen) (Beguelin et al., 2013).
Mice
The Research Animal Resource Center of the Weill Cornell Medicine approved all mouse procedures. All mice were bred and housed on a 12 hr light/dark cycle. Sirt3−/− and 129S1 mice were purchased from Jackson laboratory. 8-14 weeks old female SCID, NOD/SCID and C57BL/6J Cd45.1 mice, purchased from the Jackson lab, were used as recipient mice in xenograft and bone marrow transplantation experiments. VavP-Bcl2(Egle et al., 2004) and Sirt3−/− mice were crossed to generate VavP-Bcl2;Sirt3−/− and VavP-Bcl2;Sirt3+/+ mice as donor mice for bone marrow transplantation. All mice included in the survival analysis were euthanized when tumors reached maximum size allowed by the animal protocol.
Cell Lines
The DLBCL cell lines, OCI-Ly1 and OCI-Ly7, were grown in Iscove’s medium supplemented with 10% FBS and penicillin G/streptomycin; HBL-1, Karpas 422, OCI-LY3, Pfeiffer, SU-DHL4, TMD8 and Toledo were grown in RPMI medium supplemented with 10% FBS, penicillin G/streptomycin, L-glutamine, and HEPES. MCF7, SW480 and HEK-293T cell lines were cultured in DMEM medium supplemented with 10% FBS, 1% penicillin G/streptomycin.
METHOD DETAILS
Generation of lentiviruses
pLKO.1 lentiviral expression vector containing the puromycin resistance gene was used to co-express individual shRNAs with yellow fluorescent protein (YFP). pLKO.1 was further modified for rescue experiments by inserting sequences containing SIRT3−, SIRT3H248Yor GDH. For inducible expression of SIRT3 shRNAs in tumor xenograft studies, we used pLVUTH-KRAB-KM vector with tet-inducible promotor(Szulc et al., 2006). Autophagy reporter plasmids were derived from control or shSIRT3 pLKO.1 plasmid by adding mCherry-EGFP-LC3 and puromycin resistant genes. The lentiviruses were generated by co-expressing VSV-G and delta-8.9 in HEK-293T cells, and then concentrated using PEG-it (System Biosciences). Cells were infected and cultured for at least three days before adding puromycin for selection and various biological assays.
Cell lysis, immunoblotting and immunoprecipitation
Normal germinal center (GC) B-cells, DLBCL cell lines or primary DLBCL specimens were lysed using RIPA lysis buffer containing complete protease inhibitor cocktail to prepare whole cell lysates or for immunoprecipitation (IP). Whole cell lysates or IP products were resolved by SDS-PAGE, transferred to PVDF membrane (Bio-Rad), and probed with the indicated primary antibodies: anti-SIRT3, anti-GRP75, anti-Acetylated histone 3, anti-Acetylated lysine, anti-LC3 andanti-p62 were from purchased from Cell signaling technology; anti-ACTB, anti-histone 3, and anti-GDH antibodies were purchased from Sigma, Abeam and Proteintech Group respectively. Membranes were then incubated with a corresponding peroxidase-conjugated secondary antibody. Protein signals were detected using enhanced chemiluminescence. Densitometry values were obtained by using ImageJ 1.44o.
RNA-sequencing and analyses
Total RNA was extracted from normal GC B cells (from human tonsils) and DLBCL tumors using Trizol (LifeTechnologies) and RNeasy isolation Kit (Qiagen). RNA concentration was determined using Qubit (LifeTechnologies) and integrity was verified using Agilent 2100 Bioanalyzer (Agilent Technologies). Libraries were generated using the TruSeq RNA sample kit (Illumina). First-strand synthesis was performed using random oligos and SuperscriptIII (Invitrogen). After second-strand synthesis, a 200-bp paired-end library was prepared following the Illumina paired-end library preparation protocol. Pair-end sequencing (PE50) was performed on Illumina HiSeq2000. RNA sequencing results were aligned to hg19, respectively, using STAR (Dobin et al., 2013)and annotated to RefSeq using the Rsubread package(Liao et al., 2013).
Cell proliferation and cell cycle analyses
To evaluate the effects of SIRT3 knockdown on DLBCL cell proliferation, DLBCL and non-DLBCL cell lines were infected with lentiviruses carrying control or SIRT3 shRNAs with yellow fluorescent protein (YFP). Viable YFP+ cells in control and SIRT3 depleted cells were quantified by flow-cytometry. The change in the percentage of YFP+ population was calculated by normalizing to first time point (5 days post infection). For growth curves, Karpas 422, OCI-LYI and HBL-1 cells expressing control or SIRT3 shRNAs were counted over a period of 7 days by trypan blue exclusion. For assessment of cell proliferation using the intravital fluorescent proliferation dye, infected (YFP+, with shRNA) or non-infected (YFP−) Karpas 422 and OCI-LY1 cells were stained with eFIuor 670 (Invitrogen).YFP+ and YFP− cells were co-cultured and the dilution of the fluorescent dye in YFP+ and YFP− cells was monitored after 5 days by flow-cytometry. For cell cycle analyses, infected (YFP+) and non-infected (YFP−) cells were analyzed 7-10 days post infection using the BrdU Flow kit (BD Pharmingen™).The data was analyzed using FlowJo software.
Colony-forming assay
Karpas 422, OCI-LY1 and HBL1 cells transduced with control or SIRT3 shRNAs were plated in triplicates in six well plates containing MethoCult™ FI4230 (Stem cell technology). The numbers of colonies were counted from each well after 10-14 days. Colony numbers from SIRT3 depleted cells were normalized to control cells. Data were plotted as an average from three independent experiments. Photographs were taken using a Zeiss Axioskop imaging microscope.
Cell viability assay
Control and SIRT3 shRNA transduced HBL1, Karpas 422 and OCI-LY1 cell lines were washed with PBS and resuspended in staining buffer containing Annexin V-APC (BD biosciences) and DAPI (1μg/ml). The percentage of apoptotic cells was determined by flow-cytometric analyses of Annexin V and DAPI positive cells at days 3 and 10. For cell viability studies using Class I sirtuin inhibitors, DLBCL cells were stained with DAPI (1μg/ml) and percentages of viable cells (DAPI−) were measured by flow-cytometry. The data was analyzed using FlowJo software.
Mice xenograft and in vivo cell cycle analyses
Karpas 422 cells infected with pLVUTFI-KRAB lentiviruses with inducible control or SIRT3 shRNAs (infection efficiency was around 70%) were used for the xenograft experiment. Eight-week old female SCID mice housed in barrier environment were subcutaneously injected in the left and right flank with 107Karpas 422 cells with control and SIRT3 shRNA respectively. After about 2 weeks when the tumor volumes were around 50-100 mm3, engrafted mice were given doxycycline in drinking water (1 mg/ml) to induce shRNA and EGFP expression. Tumor volumes were monitored every 2-3 days using electronic digital calipers in 2 dimensions. Tumor volume was calculated using the following formula: tumor volume (mm3) = (smallest diameter2 × largest diameter)/2. The mice were sacrificed after three weeks and tumors were harvested. One day before euthanasia, mice were injected with BrdU (50mg/kg) by tail vein injection to label tumor cells for in vivo assessment of cell cycle distribution. Tumors were homogenized and ficolled to get single cell suspension for flow-cytometry.
Germinal center assessment
Control Sirt3+/+ and Sirt3−/− mice were immunized intraperitoneally at 8 to 12 weeks of age with 0.5 ml suspension of 2% sheep red blood cell (SRBC) in PBS (Cocalico Biologicals) to induce GC formation. Mice were sacrificed after 10 days and spleens were isolated from control and SIRT3 knockout mice. The spleen sections derived from Sirt3+/+ and Sirt3−/−mice were stained by hematoxylin and eosin (FI&E) and peanut agglutinin (PNA) using standard procedures. The number of GCs, the total spleen area occupied by GCs and the average area occupied by the GCs were quantified using ImageJ 1,44o (NIH) software. To determine the percentage of GC B-cell population, single-cell suspensions from spleens derived from Sirt3+/+ and Sirt3−/− mice were stained using the following fluorescent-labeled anti-mouse antibodies: FITC conjugated anti-B220, PE conjugated anti-FAS, APC conjugated anti-CD38 from BD biosciences and analyzed by flow-cytometry. To evaluate follicular and marginal zone B-cell populations, splenic B cells were stained with APC conjugated anti-B220, FITC conjugated anti-CD21 and PE conjugated anti-CD38 from BD biosciences and then analyzed by flow-cytometry. DAPI was used for the exclusion of dead cells. The data was analyzed by FlowJo software.
Metabolomic profiling
The metabolomic studies were done with Karpas 422 and OCI-LY1 cells with control or SIRT3 shRNAs from day 10 post infections after puromycin selection. In vivo metabolic profiling was done using B220+ splenocytes from VavP-Bcl2;Sirt3+/+ and VavP-Bcl2;Sirt3+/+ mice. B220+ cells were enriched (around 90%) by CD43 negative selection. 4-6 replicates with 107 cells per replicate were used in each study. Cell pellets were flash frozen by dry ice and shipped to Metabolon™ for non-targeted metabolomic profiling.
Mass spectrometry analysis of acetyl-CoA and CoA
Karpas 422 cells transduced with control or SIRT3 shRNAs were cultured for 10 days and collected (7-8 million cells per condition) for quantification of AcCoA and total CoA. Cell pellets were lysed with pre-chilled 10% TCA (trichloroacetic acid) and then flash frozen using liquid N2. Samples were vortexed and centrifuged at 14,000 rpm (20000 x g) for 15 minutes at 4°C to remove protein. Acyl-CoAs were purified using an OASIS HLB 96-well plate solid phase extraction (SPE) plate (30 mg) and vacuum manifold. SPE wells were first conditioned with 1 ml of methanol, then equilibrated with 2 x 1 ml of water. Supernatants from cell extracts were then applied to the SPE column. After washing, acyl-CoA was eluted using three successive applications of 0.5 ml of methanol containing 25 mM ammonium acetate. Eluted acyl-CoA was dried for 5 hr in a Genevac EZ-2 Elite using the program FILPC1. Dried samples were resuspended into 110 μL of 50mM Ammonium Acetate pH 4.5 for LC-MS analysis. Acyl-CoA levels were detected using an Agilent 6550 iFunnel Q-TOF LC/MS in positive ionization mode with an Agilent Dual Jet Stream Source.
Measurement of autophagic flux
DLBCL cell lines (HBL1, Karpas 422, 0CI-LY1, Pfeiffer and SU-DHL4) transduced with viruses containing control or SIRT3 shRNA, autophagy reporter plasmids (mCherry-EGFP-LC3) and puromycin resistant gene. Infected cells were first selected with puromycin for at least four days and cells with dual-color florescence were analyzed by flow-cytometry. Cells with high levels of autophagy were defined as those having a high mCherry/EGFP fluorescence ratio as previously describe (Gump et al., 2014).
Metabolic flux assay
For steady state metabolomic analyses, DLBCL cells (Karpas 422 and OCI-LY1) infected with lentiviruses containing control or SIRT3 shRNA were selected with puromycin (1pg/ml) for 7 days, and then grown in a complete media (RPMI, 2 mM glutamine, 10 mM glucose, 10% FBS) for 24 hr. The complete media was changed to 13C labeled medium. To trace glutamine or glucose metabolism in the flux analyses, cells were grown as above and then transferred into glutamine- or glucose-free RPMI containing 10% dialyzed FBS and 2 mM [U-13C5]-glutamine or 10mM [U-13C6]-glucose (Cambridge Isotope Labs) overnight for steady state labeling. Cells were collected by spinning at 300 g for 5 minutes and metabolite extractions were done by adding pre-chilled (−80°C) 80% methanol solution. Each replicate was from extracts of five million cells. Metabolite fractions were collected and analyzed by targeted LC-MS/MS via selected reaction monitoring (SRM) as described (Son et al., 2013).
Measurement of GDH Activity
The GDH activity in DLBCL cells with control or SIRT3 shRNAs was measured using the GDH activity assay kit (Biovision) according to the instructions of the manufacturer.
Immunohistology
Mice organs were fixed in 4% formaldehyde and embedded in paraffin. Deparaffinized slides were antigen retrieved in citrate buffer pH 6.4 and endogenous peroxidase (HRP) activity was blocked by treating the sections with 3% hydrogen peroxide in methanol. Indirect immunohistochemistry was performed with anti-species specific biotinylated secondary antibodies followed by avidin-horseradish peroxidase or avidin-AP and developed by Vector Blue or DAB color substrates (Vector Laboratories). Sections were counterstained with hematoxylin if necessary. The following antibodies were used: biotin-conjugated anti-PNA (Vector Laboratories), biotin-conjugated anti-B220 (Invitrogen RM2615), Ki67 (Vector VPK451). Slides were scanned using a Zeiss Mirax Slide Scanner and photomicrographs were examined using Pannoramic Viewer software. ImageJ 1,44o software (NIH) was used to quantify GC areas.
General methods and materials for class I sirtuin inhibitors
Reagents were obtained from AlfaAesar and Sigma-Aldrich in at the highest purity available and used as supplied. 1H NMR were performed on INOVA 400 spectrometers. Analytic HPLC analysis was carried out on a SHIMADZU LC with Kinetex 5u EVO C18 100A column (100 mm × 4.60 mm, 5 μm, Phenomenex) monitoring at 215 nm and 326 nm. Solvents for analytical HPLC were water with 0.1% trifluoroacetic acid (TFA) as solvent A and acetonitrile with 0.1% TFA as solvent B. Compounds were analyzed at a flow rate of 0.5 ml/min. LC-MS experiments were carried out on a Shimadzu HPLC LC20-AD and ThermoScientific LCQ Fleet with a Kinetex 5 μm EVO C18 100A column (30 × 2.1 mm, 5 μm, Phenomenex) monitoring at 215 and 260 nm with positive mode for detection of mass-to-charge ratio of ions. Solvents for LC-MS were water with 0.1% acetic acid (solvent A) and acetonitrile with 0.1% acetic acid (solvent B) at a flow rate of 0.3 ml/min.
Synthesis of JH-T4
As in Figure S8A, compound 1 was synthesized following a procedure described in the literature (Jing et al., 2016). To a solution of compound 1 (86 mg, 0.17 mmol) and N-methylmorpholine (37.0 μl, 0.34 mmol) in dry dichloromethane (1.7 ml), isobutylchloroformate (38.0 μl, 0.27 mmol) was added dropwisely at 0°C. The reaction mixture was stirred for 30 min at 0°C. 3-Aminophenol (24 mg, 0.22 mmol) was then added at 0°C and the reaction mixture was stirred for 16 hr at room temperature. The solvent was removed under reduced pressure and the resulting residue was dissolved with EtOAc (40 ml) and washed with brine (20 ml). The organic layer was dried over anhydrous sodium sulfate. After removal of the solvents in vacuum, the residue was purified by flash chromatography on silica gel (hexanes:EtOAc = 3:2) to afford the expected compoundT4 (JH-T4) (48 mg, 47 % yield) as a white solid. 1H NMR (400 MHz, CDCb): δ 8.53 (s, 1H), 7.68 (s, 1H), 7.39 (m, 1H), 7.32 (m, 6H), 7.12 (t, J = 8.1 Hz, 1H), 6.86 (d, J = 7.9 Hz, 1H), 6.61 (dd, J = 8.3, 2.3 Hz, 1H), 5.65 (d, J = 7.9 Hz, 1H), 5.28 – 4.89 (m, 2H), 4.35 (m, 1H), 3.61 (m, 2H), 2.57 (t, J= 7.5 Hz, 2H), 1,90(m, 1H), 1.67 (m, 5H), 1.43 (m, 2H), 1.22 (m, 21H), 0.86 (t, J = 6.7 Hz, 3H). LCMS (ESI) calcd.ForC34H52N304S [M+H]+598.36, obsd. 598.25
Synthesis of YC8-02
As in Figure S8B, compound 2 was synthesized following a procedure described in the literature(He et al., 2014).To a solution of compound 2(110 mg, 0.185mmol) and N-methylmorpholine (24.0 μl, 0.22 mmol) in dry dichloromethane (1.85 ml), isobutylchloroformate (31.0 μl, 0.22 mmol) was added dropwisely at 0°C. The reaction mixture was stirred for 30 min at 0°C. 3-Aminophenol (24 mg, 0.22mmol) was then added at 0°C and the reaction mixture was stirred for 16 h at room temperature. The solvent was removed under reduced pressure and the resulting residue was dissolved with EtOAc (40 ml) and washed with brine (20 ml). The organic layer was dried over anhydrous sodium sulfate. After removal of the solvents in vacuum, the residue was purified by flash chromatography on silica gel (hexanes:EtOAc = 1:1) to afford the expected compound3(67 mg, 53 % yield).Compound 3 was deprotected using 20 % piperidine in DCM (1 ml) for 2 h at room temperature. The solvent was removed under reduced pressure and the resulting residue was dissolved with EtOAc (40 ml) and washed with brine (20 ml). The organic layer was dried over anhydrous sodium sulfate. After removal of the solvents in vacuum, the residue was purified by flash chromatography on silica gel (DCM:MeOH = 85:15) to afford deprotected compound. To a solution of the resulting deprotected compound in DMF (0.5 ml) was added diisopropylethylamine (28.0μl, 0.2 mmol) and (2-Bromoethyl) triphenylphosphonium bromide (54 mg, 0.12mmol). The reaction mixture was stirred for 2 h at room temperature. The mixture was extracted with EtOAc (40 ml) and washed with brine (20 ml). The organic layer was dried over anhydrous sodium sulfate. After removal of the solvents in vacuum, the residue was purified by flash chromatography on silica gel (DCM:MeOH = 9:1) to afford the expected compound YC8-02 (13 mg, 17 % yield). 1H NMR (400 MHz, CDCb): δ 10.38 (s, 1H), 9.47 (s, 1H), 7.93 – 7.39 (m, 18H), 7.13 (d, J = 8.0 Hz, 1H), 6.95 (t, J = 8.0 Hz, 1H), 6.57 (dd, J = 8.1,2.1 Hz, 1H), 4.04 – 3.79 (m, 1H), 3.61 (s, 2H), 3.53 – 3.43 (m, 2H), 3.04 – 2.78 (m, 2H), 2.70 (dd, J = 9.0, 6.4 Hz, 2H), 1.82 – 1.43 (m, 4H), 1.37 – 1.09 (m, 24H), 0.84 (t, J = 6.7 Hz, 3H).LCMS (ESI) calcd. ForC46H63N302PS+ [M]+752.44, obsd. 752.51.
HPLC-based inhibition assay of sirtuins
Different concentrations (0.686, 2.058, 6.173, 18.519, 55.556, 166.667μM) of small molecule inhibitors were pre-incubated with 0.3 μM sirtuins (SIRT1, SIRT2, and SIRT3) and 1 mM NAD in buffer (20 mM Tris-HCI, pH 8.0, 1 mM DTT) at 37 °C for 10 min. Then 10 μM of acyl peptide (KQTARK(Ac)STGGWW)was added to initiate the reactions. The reactions were then incubated at 37°C in a total volume of 60 μl (5 min for SIRT1 and SIRT2, 20 min for SIRT3). The conversion of each reaction was less than 20%.The reactions were stopped by adding 60 μl of an aqueous solution of 50% methanol containing 200 mM HCI and 320 mM acetic acid. The reaction mixtures were then centrifuged at 17000g for 10 min and analyzed on an analytical HPLC with Kinetex 5u EVO C18 100A column (100 mm × 4.60 mm, 5.0 μm, Phenomenex). The gradient was: 0% B for 2 min, 0 to 30% B in 13 min, and then 30% to 100% B in 10 min at a flow rate of 0.5 ml/min. All reactions were done in duplicate. The product and the substrate peaks were quantified using HPLC UV absorption traces at 280 nm.
Drug uptake in mitochondria
Karpas 422 cells (1 million/ml) were treated with 3μM or 5μM JH-T4 or YC8-02 for 1 hour. 3 and 15 million cells were used for whole cell or mitochondria extraction respectively. Mitochondria were extracted using Qproteome Mitochondria Isolation Kit (Qiagen). Whole cells or mitochondria pellets were lysed with ACN solution (Acetonitrile:H2O, 2:1). 50 μL total cell lysates or mitochondria extracts were added to 50 μL acetonitrile and centrifuged at 17000g for 10 min to remove precipitated proteins. The supernatants were added to amide analogues for JH-T4 and YC8-02 as internal standards. The final concentration of internal standards was 40 nM and analyzed on LC-MS with positive mode for detection of mass-to-charge ratio of ions. The compound concentration was quantified by comparing the ion peak area of the inhibitors and corresponding internal standards. All solutions were done in duplicates.
Drug toxicity study and treatment in mice
YC8-02 was dissolved into DMSO at concentration of 36 mM as stock solution for in vivo toxicity and treatment study. To make drug solutions for intraperitoneal (IP) injection, we first mix drug stock solution with the same volume of Kolliphor® EL (Sigma-Aldrich), then add 8 volumes of PBS and mix well to make the final solutions for injection. The toxicity study was done with female C57BL/6J mice (3 months old). We set up 2 groups of mice to be injected with vehicle (DMSO, Kolliphor, PBS solution) and YC8-02 at 30 mg/kg. Each group had 3 mice. Each mouse was treated once per day for 5 days, and all mice were weighted every time before injections. Toxicity study stopped at day 8 (3 days after the last injection) after the last measurement of mice body weight. Mice body weight was used to evaluate the weight changes during and after treatment as shown in Figure S8J. In vivo DLBCL treatment study was carried out in NOD/SCID mice bearing xenografted DLBCL tumors. Tumor xenografts were done as describe above. Ten-week old female NOD/SCID mice housed in barrier environment were subcutaneously injected in the right flank with 7X106 Karpas 422 cells. After about 2 and half weeks when the tumor volumes were around 100-200 mm3, we randomly separate mice into two groups (each group n=4) for treatment via IP injection with vehicle and YC8-02 solution (30 mg/kg). Mice were treated once per day during week days for two weeks, and tumor volumes were monitored twice a week using electronic digital calipers in 2 dimensions. Tumor volume was calculated using the following formula: tumor volume (mm3) = (smallest diameter2 × largest diameter)/2. The experiment was stopped at 3 weeks after the first injection when control mice were reaching the maximal allowed tumor burden and tumor volumes used to generate Figure 8J.
QUANTIFICATION AND STATISTICAL ANALYSES
Survival analyses
Raw microarray data were downloaded from GEO at NCBI and annotated with Brainarray CDF annotation file(Dai et al., 2005). Different microarray data sets were combined after RMA normalization and the batch effects were adjusted with ComBat method(Johnson et al., 2007). The survival probability was estimated by Kaplan-Meier method and its association with gene expression was tested by log-rank test. Cox proportional hazard regression was used to calculate hazard ratio and to test association with gene expression while adjusting for clinical features. All analyses were performed using R packages from Bioconductor and statistical software SAS 9.4 (SAS Institute, Cary, NC).
Metabolomic analyses
Metabolomic data was obtained from Metabolon™ through untargeted metabolomic profiling using DLBCL cell lines or B220+ cells isolated from mice. Data was preprocessed to exclude metabolites that contain missing values for ≥ 20% and metabolite abundances were log-transformed and quantile normalized. Differentially abundant metabolites were determined using a linear model fit with moderated t-statistics from the metabolomics R package using a fold-change threshold of 1.2 and Benjamini-Hochberg adjusted p value <0.1 (De Livera et al., 2012; De Livera et al., 2015). Hierarchical clustering was performed on log-transformed metabolite data using Euclidean distance and Ward’s method. Biological significance of the metabolomics signatures was assessed via over-representation or Wilcoxon enrichment analyses using Integrated Molecular Pathway Level Analysis (IMPaLA) (ref; http://impala.molgen.mpg.de).
DLBCL DNA copy number
Single nucleotide polymorphism (SNP) microarrays (Affymetrix 250k Sty; Affymetrix 250k Nsp; Affymetrix SNP6.0) and array comparative genome hybridization (CGH; Agilent 244k) arrays from previously studies(Bodker et al., 2013; Challa-Malladi et al., 2011; Compagno et al., 2009; Green et al., 2011; Kato et al., 2009; Lenz et al., 2008b; Monti et al., 2012)were downloaded from the gene expression omnibus (accessions, GSE34171, GSE12906, GSE15127, GSE37977, GSE22082, GSE11318;www.ncbi.nlm.nih.gov/geo/) or shared by the authors of the originating publication. All raw data were uniformly processed to generate Log2 copy number as previously described(Green et al., 2014), then segmented using the circular binary segmentation (CBS) module in GenePattern (Reich et al., 2006). Significant DNA copy number gains and losses were calculated using GISTIC2(Mermel et al., 2011) in GenePattern, with a DNA copy number change threshold of 0.1.
DLBCL gene expression analysis
Raw cel files for Affymetrix U133 plus 2.0 gene expression microarrays were obtained for 249 DLBCL tumors from previously published studies(Lenz et al., 2008a; Monti et al., 2012) via the gene expression gene expression omnibus (accessions GSE34171 and GSE10846; www.ncbi.nlm.nih.gov/qeo/). The data were RMA normalized using the ExpressionFileCreator module and batch corrected using the Com Bat module of GenePattern(Reich et al., 2006) (https://cloud.genepattern.org). The expression of genes was compared between diploid and 6q21 deleted cases using the average probe intensity and a Student’s T-test.
Statistical analyses
A two-tailed Student’s t test was used to determine the statistical significance of the results, p values ≤0.05 were considered statistically significant. Correlations were determined using Pearson correlation coefficients using GraphPad Prism. Log rank tests≤0.05 were standard for significance between subgroup of patients or animals.
DATA and SOFTWARE AVAILABILITY
RNA sequencing data from this paper have been deposited in the Gene Expression Omnibus (GEO) database. The accession numbers for the RNA sequencing data of GC B cells and DLBCL tumor cells are GSE45982 and GSE130751, respectively.
Supplementary Material
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SIRT3 antibody | Cell Signaling Technology | Cat#5940S |
| Beta-Actin antibody | Sigma-Aldrich | Cat#A5441-.2ML |
| B220 antibody for IHC | BD Bioscience | Cat#550286 |
| KI67 antibody for IHC | Vector Laboratories | Cat#VP-K451 |
| B220 antibody (FITC) for flow | BD Bioscience | Cat#553087 |
| CD38 antibody (APC) for flow | ThermoFisher Scientific | Cat#17-0381-81 |
| FAS/CD95 antibody (PE) for flow | BD Bioscience | Cat#557653 |
| CD21 antibody (FITC) for flow | ThermoFisher Scientific | Cat#11-0212-81 |
| CD23 antibody (PE) for flow | ThermoFisher Scientific | Cat#50-102-71 |
| H3 Ac antibody | Cell Signaling Technology | Cat#8848S |
| H3 antibody | Millipore (Upstate) | Cat#09-838 |
| LC3 antibody | Cell Signaling Technology | Cat#12741S |
| P62 antibody | Cell Signaling Technology | Cat#8025S |
| ATG5 antibody | Cell Signaling Technology | Cat#12994S |
| Ac-K antibody | Cell Signaling Technology; ImmuneChem | Cat#9441S; Cat#ICP0380 |
| FLAG antibody | Sigma-Aldrich | Cat#F3165-MG |
| Grp75 antibody | Cell Signaling Technology | Cat#3593S |
| Beta-Tubulin antibody | Sigma-Aldrich | Cat#T6199-200UL |
| SIRT1 antibody | Cell Signaling Technology | Cat#9475P |
| SIRT2 antibody | Cell Signaling Technology | Cat#12650P |
| SIRT5 antibody | Cell Signaling Technology | Cat#8782P |
| SIRT6 antibody | Cell Signaling Technology | Cat#2590S |
| SIRT7 antibody | Cell Signaling Technology | Cat#5360P |
| GDH antibody | ProteinTech | Cat#14299-1-AP |
| Secondary antibody anti rabbit | Santa Cruz Biotechnology; Cell Signaling Technology | Cat#sc-2004; Cat#7074S |
| Secondary antibody anti mouse | Santa Cruz Biotechnology; Cell Signaling Technology | Cat#sc-2005; Cat#7076S |
| Anti-FLAG-beads | Sigma-Aldrich | Cat#A2220-5ML |
| Bacterial and Virus Strains | ||
| pLKO.1-shScramble-puro/YFP | This paper | N/A |
| pLKO.1-shSIRT3-1-puro/YFP | MSKCC GES Core/this paper | TRCN0000038892 |
| pLKO.1-shSIRT3-2-puro/YFP | This paper | N/A |
| pLKO.1-shSIRT3-3-puro/YFP | This paper | N/A |
| pLKO.1-shSIRT1-puro/YFP | MSKCC GES Core/this paper | TRCN0000018981 |
| pLKO.1-shSIRT2-puro/YFP | MSKCC GES Core/this paper | TRCN0000040218 |
| pLKO.1-shSIRT4-puro/YFP | MSKCC GES Core/this paper | TRCN0000018944 |
| pLKO.1-shSIRT5-puro/YFP | MSKCC GES Core/this paper | TRCN0000018547 |
| pLKO.1-shSIRT6-puro/YFP | MSKCC GES Core/this paper | TRCN0000050476 |
| pLKO.1-shSIRT7-puro/YFP | MSKCC GES Core/this paper | TRCN0000020256 |
| pLKO.1-shATG5-puro | MSKCC GES Core | TRCN0000151963 |
| pLKO.1-shGDH-1-YFP | This paper | N/A |
| pLKO.1-shGDH-2-YFP | This paper | N/A |
| pLKO.1-shScramble-SIRT3m | This paper | N/A |
| pLKO.1-shScramble-SIRT3HYm | This paper | N/A |
| pLKO.1-shSIRT3-1-SIRT3m | This paper | N/A |
| pLKO.1-shSIRT3-2-SIRT3m | This paper | N/A |
| pLKO.1-shSIRT3-1-SIRT3HYm | This paper | N/A |
| pLKO.1-shSIRT3-2-SIRT3HYm | This paper | N/A |
| pLVUTH-KRAB-KM-shScramble | This paper | N/A |
| pLVUTH-KRAB-KM-shSIRT3-1 | This paper | N/A |
| pLVUTH-KRAB-KM-shSIRT3-2 | This paper | N/A |
| pLKO.1-shScramble-mCherry-GFP-LC3B-EF1-puro | This paper | N/A |
| pLKO.1-shSIRT3-mCherry-GFP-LC3B-EF1-puro | This paper | N/A |
| pLKO.1-shSramble-GDH1 | This paper | N/A |
| pLKO.1-shSIRT3-1-GDH1 | This paper | N/A |
| Biological Samples | ||
| DLBCL tumor protein samples | Weill Cornell Medicine, Department of Pathology | N/A |
| Tonsils for B cell isolations | Weill Cornell Medicine, Department of Pathology | N/A |
| umbilical cord blood-derived | New York Blood Center (NYBC) | N/A |
| HSPCs (deidentified) | ||
| Sheeps red blood in Alsevers (SRBC) | Cocalico Biological | Cat# 20-1334A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Peanut Agglutinin Biotiny (PNA) | Vector Laboratories | Cat#B-1075 |
| Hematoxylin Eosin | Vector Laboratories | Cat#H-3502 |
| DAPI | Life Technologies | Cat#D3571 |
| D-Glucose (U13C6, 99%) | Cambridge Isotope Laboratories, Inc. | CLM-1396-25-1 |
| L-Glutamine (13C5, 99%) | Cambridge Isotope Laboratories, Inc. | CLM-1822-H-0.1 |
| Dimethyl 2-oxoglutarate 96% (DMKG) | Sigma Aldrich | Cat#349631-5G |
| Methyl pyruvate 90%, technical grade (MP) | Sigma Aldrich | Cat#371173-25G |
| Biotin-TM3 | This paper | N/A |
| JH-T4 | This paper | N/A |
| YC8-02 | This paper | N/A |
| Fetal Bovine Serum, Dialyzed | Life Technologies | Cat## 26400044 |
| Kolliphor™ EL | Sigma Aldrich | Cat#C5135-500G |
| Recombinant SIRT1, SIRT2 and SIRT3 protein | This paper | N/A |
| In vitro assay substrate (AcH3K9 peptide) | This paper | N/A |
| NAD | VWR life Science | VWR 0455 |
| Critical Commercial Assays | ||
| MethoCult H4230 | StemCell Technologies Inc. | Cat# 04230 |
| BrdU staining kit | BD Biosciences | Cat#552598 |
| GDH activity assay kit | BioVision | Cat#K729-100 |
| Mitochondrial isolation kit | Qiagen; MilliporeSigma™ | Cat#37612; Cat#QIA88-1KIT |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7573 |
| CELL PROLIF DYE EF670 | Thermo Fisher Scientific | Cat#65-0840-90 |
| APC-Annexin V | BioLegend | Cat##640919 |
| Deposited Data | ||
| Raw data of gene expression and clinical information for survival analysis | Hummel et al., 2006 | GEO:GSE4475 |
| Raw data of gene expression and clinical information for survival analysis | Jais et al., 2008 | ArrayExpress: E-TABM-346 |
| Raw data of gene expression and clinical information for survival analysis | Lenz et al., 2008 | GEO:GSE10846 |
| Raw data of gene expression and clinical information for survival analysis | Shaknovich et al., 2010 | GEO:GSE23501 |
| Raw data for DNA copy number analysis | Monti et al., 2012 | GEO:GSE34171 |
| Raw data for DNA copy number analysis | Kato et al., 2009 | GEO:GSE12906 |
| Raw data for DNA copy number analysis | Compagno et al., 2009 | GEO:GSE15127 |
| Raw data for DNA copy number analysis | Bødker et al., 2013 | GEO:GSE37977 |
| Raw data for DNA copy number analysis | Green et al., 2011 | GEO:GSE22082 |
| Raw data for DNA copy number analysis | Lenz et al., 2008 | GEO:GSE11318 |
| Raw data of gene expression of GCB cells | Beguelin et al., 2013 | GEO:GSE45982 |
| Raw data of gene expression of DLBCL tumor cells | This paper | GEO:GSE130751 |
| Experimental Models: Cell Lines | ||
| OCI-LY1 | Ontario Cancer Institute (OCI) | N/A |
| OCI-LY7 | O Ontario Cancer Institute (OCI) | N/A |
| SUDHL-4 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | ACC-495 |
| Karpas 422 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | ACC-32 |
| Pfeiffer | American Type Culture Collection (ATCC) | CRL-2632 |
| Toledo | American Type Culture Collection (ATCC) | CRL-2631 |
| HBL1 | Jose A. Martinez-Climent (Universidad de Navarra, Pamplona, Spain) | N/A |
| OCI-LY3 | Ontario Cancer Institute (OCI) | N/A |
| TMD8 | Louis M. Staudt (National Cancer Institute, Bethesda, Maryland, USA) | N/A |
| HEK-293T | American Type Culture Collection (ATCC) | CRL-1573 |
| MCF7 | American Type Culture Collection (ATCC) | HTB-22 |
| SW480 | American Type Culture Collection (ATCC) | CCL-228 |
| Experimental Models: Organisms/Strains | ||
| 129S1 | Jackson Laboratory | Stock No:002448 |
| Sirt3 KO | Jackson Laboratory | Stock No:012755 |
| SCID mouse | Jackson Laboratory | Stock No:001303 |
| C57BL/6 (B6) | Jackson Laboratory | Stock No:000664 |
| C57BL/6 (B6) CD45.1 | Jackson Laboratory | Stock No:002014 |
| VavP-Bcl2 | Egle et al., 2004 | N/A |
| VavP-Bcl2-sirt3+/+ | This paper | N/A |
| VavP-Bcl2-sirt3−/− | This paper | N/A |
| Oligonucleotides | ||
| shScramble:CAACAAGATGAAGAGCACCAA | This paper | N/A |
| shSIRT3-2:TTGCTGCATGTGGTTGAIIIC | This paper | N/A |
| shSIRT3-3:GGTGGAAGAAGGTCCATATCT | This paper | N/A |
| shGDH-1:GCCCTATGAAGGAAGCATCTT | This paper | N/A |
| shGDH-2:GCTGCCTATGTCAATGCCATT | This paper | N/A |
| Recombinant DNA | ||
| pLVUTH-KRAB-KM | Szulc et al., 2006 | N/A |
| pLKO.I-TRC vector | Addgene | 10878 |
| Software and Algorithms | ||
| STAR | Dobin et al., 2013 | http://github.com/alexdobin/STAR |
| Rsubread | Liao et al., 2013 | http://bioconductor.org/packages/release/bioc/html/Rsebread.html |
| metabolomics | De Livera et al., 2012; De Livera et al., 2015 | http://cran.r-proiect.org/packages=metabolomics |
| IMPaLA | Kamburov et al., 2011 | http://impala.molgen.mpg.de |
| GenePattern | Reich et al., 2006 | http://cloud.genepattern.org |
| Brainarray CDF | Dai et al., 2005 | http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/genomic_curated_CDF.asp |
| Combat | Johnson et al., 2007 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Cutoff finder | Budczies et al., 2012 | http://molpath.charite.de/cutoff/ |
| Flowjo | Flowjo | https://www.flowjo.com |
| ImageJ | ImageJ | https://imagei.nih.gov/ij/ |
| GraphPad Prism 6.0 | GraphPad Software | https://www.graphpad.com/ |
Significance.
We still lack knowledge and targets to treat highly aggressive and heterogeneous DLBLCs. In this study, we discovered that reduced SIRT3 expression inhibited cell proliferation in different subtypes of DLBCLs cells, impaired lymphomagenesis in VavP-Bcl2 mouse lymphoma model and was associated with better survival in human DLBCL patients. Though metabolomic profiling and metabolic tracing study, we found that SIRT3 maintained DLBCLs’ metabolism by potentiating the TCA cycle through anaplerotic glutaminolysis. These findings prompted us to develop a mitochondria-targeting SIRT3 inhibitor, YC8-02, to treat diseases like DLBCLs, since SIRT3 expression is dispensable for tissue homeostasis under normal conditions.
ACKNOWLEDGMENTS
We thank the Melnick lab for helpful discussions and suggestions, the Tri-lnstitutional Therapeutic Discovery Institute for help with the synthesis of small molecules. A.M. is supported by LLS SCOR 7012-16, a Falk Medical Research Catalyst Award, the Chemotherapy Foundation, and a TDI Institute Award and the Follicular Lymphoma Consortium.
Footnotes
DECLARATION OF INTEREST
A M. receives research funding from Janssen and has consulted for Epizyme and Constellation.
REFERENCES
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Amengual JE, Clark-Garvey S, Kalac M, Scotto L, Marchi E, Neylon E, Johannet R, Wei Y, Zain J, and O’Connor OA (2013). Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood 122, 2104–2113. [DOI] [PubMed] [Google Scholar]
- Baeza J, Smallegan MJ, and Denu JM (2016). Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem Sci 41, 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, and Dalla-Favera R (2015). Germinal centres and B cell lymphomagenesis. Nat Rev Immunol 15,172–184. [DOI] [PubMed] [Google Scholar]
- Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Emerling BM, Ricoult SJ, and Guarente L (2011). SirT3 suppresses hypoxia inducible factor lalpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30, 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereshchenko OR, Gu W, and Dalla-Favera R (2002). Acetylation inactivates the transcriptional repressor BCL6. Nat Genet 32, 606–613. [DOI] [PubMed] [Google Scholar]
- Bodker JS, Gyrup C, Johansen R, Schmitz A, Madsen J, Johnsen HE, Bogsted M, Dybkaer K, and Nyegaard M (2013). Performance comparison of Affymetrix SNP6.0 and cytogenetic 2.7M whole-genome microarrays in complex cancer samples. Cytogenet Genome Res 139, 80–87. [DOI] [PubMed] [Google Scholar]
- Boothby M, and Rickert RC (2017). Metabolic Regulation of the Immune Humoral Response. Immunity 46, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budczies J, Klauschen E, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, and Denkert C (2012). Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 7, e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro R, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, et al. (2012). Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, and Guarente L (2015). The multifaceted functions of sirtuins in cancer. Nat Rev Cancer 15, 608–624. [DOI] [PubMed] [Google Scholar]
- Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, and Dalla-Favera R (2011). Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 20, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. (2018). Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, and Huang J (2014). Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis 5, e1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni E, Ponzoni M, Scandurra M, Califano A, et al. (2009). Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Rostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. (2005). Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Livera AM, Dias DA, De Souza D, Rupasinghe T, Pyke J, Tull D, Roessner U, McConville M, and Speed TP (2012). Normalizing and integrating metabolomics data. Anal Chem 84, 10768–10776. [DOI] [PubMed] [Google Scholar]
- De Livera AM, Sysi-Aho M, Jacob L, Gagnon-Bartsch JA, Castillo S, Simpson JA, and Speed TP (2015). Statistical methods for handling unwanted variation in metabolomics data. Anal Chem 87, 3606–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, and Staudt LM (1997). Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 276, 589–592. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger E, Drenkow J, Zaleski C, Jha S, Batut R, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29,15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bath ML, O’Reilly L, and Coiy S (2004). VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 103, 2276–2283. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Schroeder S, Andiyushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola E, Harger A, Zimmermann A, et al. (2014). Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab 19, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, Dominguez-Sola D, Sabatini DM, and Victora GD (2017). Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORCl Kinase. Immunity 46,1045–1058 e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, et al. (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginet V, Puyal J, Rummel C, Aubry D, Breton C, Cloux AJ, Majjigapu SR, Sordat B, Vogel R, Bruzzone S, et al. (2014). A critical role of autophagy in antileukemia/lymphoma effects of AP0866, an inhibitor of NAD biosynthesis. Autophagy 10, 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Aya-Bonilla C, Gandhi MK, Lea RA, Wellwood J, Wood P, Marlton P, and Griffiths LR (2011). Integrative genomic profiling reveals conserved genetic mechanisms for tumorigenesis in common entities of non-Hodgkin’s lymphoma. Genes Chromosomes Cancer 50, 313–326. [DOI] [PubMed] [Google Scholar]
- Green MR, Vicente-Duenas C, Romero-Camarero I, Long Liu C, Dai B, Gonzalez-Herrero I, Garcia-Ramirez I, Alonso-Escudero E, Iqbal J, Chan WC, et al. (2014). Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat Commun 5, 3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, and Thorburn A (2014). Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 16, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Deng CX, Finley LW, Kim HS, and Gius D (2012). SIRT3 is a mitochondrial tumor suppressor: a scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res 72, 2468–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144,646–674. [DOI] [PubMed] [Google Scholar]
- He B, Hu J, Zhang X, and Lin H (2014). Thiomyristoyl peptides as cell-permeable Sirt6 inhibitors. Org Biomol Chem 12, 7498–7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. (2013). Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell 49,186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, and Bedalov A (2006). Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 66, 4368–4377. [DOI] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, et al. (2015). Metabolic reprogramming induces resistance to anti-NOTCHl therapies in T cell acute lymphoblastic leukemia. Nat Med 21,1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464,121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, and Auwerx J (2012). Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TE, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, et al. (2006). A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 354, 2419–2430. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, and Younes A (2014). Precision therapy for lymphoma-current state and future directions. Nat Rev Clin Oncol 11, 585–596. [DOI] [PubMed] [Google Scholar]
- Jais JP, Haioun C, Molina TJ, Rickman DS, de Reynies A, Berger F, Gisselbrecht C, Briere J, Reyes F, Gaulard P, et al. (2008). The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia 22,1917–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung CR, Chen C, Richardson AD, Conner EM, Benschop RJ, Woodgett JR, and Rickert RC (2017). Gsk3 is a metabolic checkpoint regulator in B cells. Nat Immunol 18, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Hu J, He B, Negron Abril YL, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, et al. (2016). A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 29,767–768. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8,118–127. [DOI] [PubMed] [Google Scholar]
- Kamburov A, Cavill R, Ebbels TM, Herwig R, and Keun HC (2011). Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918. [DOI] [PubMed] [Google Scholar]
- Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, et al. (2009). Frequent inactivation of A20 in B-cell lymphomas. Nature 459, 712–716. [DOI] [PubMed] [Google Scholar]
- Kenific CM, and Debnath J (2015). Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol 25, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, and White E (2017). Autophagy and Tumor Metabolism. Cell Metab 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12,1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15,110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, et al. (2008a). Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 359, 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. (2008b). Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A 105,13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, Cho C, Machida K, Chen D, and Ou JJ (2017). Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell 68, 281–292 e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan SS, Scian M, Sripathy S, Posakony J, Lao U, Loe TK, Leko V, Thalhofer A, Schuler AD, Bedalov A, and Simon JA (2014). Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J Med Chem 57, 3283–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andiyushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, et al. (2014). Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell 53, 710–725. [DOI] [PubMed] [Google Scholar]
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, et al. (2016). TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell 61,199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, and White E (2014). Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 55, 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, Inguilizian H, Mermel C, Currie T, Dogan A, et al. (2012). Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 22, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, et al. (2005). Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 105,1851–1861. [DOI] [PubMed] [Google Scholar]
- Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, Tjornelund J, Dawson KM, Dupuis M, and Duchosal MA (2009). The NAD biosynthesis inhibitor AP0866 has potent antitumor activity against hematologic malignancies. Blood 113, 3276–3286. [DOI] [PubMed] [Google Scholar]
- Noda NN, and Inagaki F (2015). Mechanisms of Autophagy. Annu Rev Biophys 44,101–122. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer R, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch R, et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola E, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi E, Jacquelot N, Yamazaki T, et al. (2016). Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 30,147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo R, and Mesirov JP (2006). GenePattern 2.0. Nat Genet 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Schroeder S, Pendl T, Zimmermann A, Eisenberg T, Carmona-Gutierrez D, Ruckenstuhl C, Marino G, Pietrocola E, Harger A, Magnes C, et al. (2014). Acetyl-coenzyme A: a metabolic master regulator of autophagy and longevity. Autophagy 10,1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, Gascoyne RD, Elemento O, and Melnick A (2010). DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 116, e81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496,101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc J, Wiznerowicz M, Sauvain MO, Trono D, and Aebischer P (2006). A versatile tool for conditional gene expression and knockdown. Nat Methods 3,109–116. [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, and Mizushima N (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. (2007). Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130,1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MR, Vyas FS, Zaganjor E, et al. (2016). Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 167, 985–1000 el021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Denu RA, Krautkramer KA, Grindle KM, Yang DT, Asimakopoulos E, Hematti P, and Denu JM (2016). Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J Biol Chem 291, 3268–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, He M, Cheng F, Bai R, da Silva SR, Aguiar RC, and Gao SJ (2017). Tenovin-6 inhibits proliferation and survival of diffuse large B-cell lymphoma cells by blocking autophagy. Oncotarget 8,14912–14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data from this paper have been deposited in the Gene Expression Omnibus (GEO) database. The accession numbers for the RNA sequencing data of GC B cells and DLBCL tumor cells are GSE45982 and GSE130751, respectively.