

Abstract
Nutrients are indispensable resources that provide the macromolecular building blocks and energy requirements for sustaining cell growth and survival. Cancer cells require several key nutrients to fulfill their changing metabolic needs as they progress through stages of development. Moreover, both cell‐intrinsic and microenvironment‐influenced factors determine nutrient dependencies throughout cancer progression—for which a comprehensive characterization remains incomplete. In addition to the widely studied role of genetic alterations driving cancer metabolism, nutrient use in cancer tissue may be affected by several factors including the following: (i) diet, the primary source of bodily nutrients which influences circulating metabolite levels; (ii) tissue of origin, which can influence the tumor's reliance on specific nutrients to support cell metabolism and growth; (iii) local microenvironment, which dictates the accessibility of nutrients to tumor cells; (iv) tumor heterogeneity, which promotes metabolic plasticity and adaptation to nutrient demands; and (v) functional demand, which intensifies metabolic reprogramming to fuel the phenotypic changes required for invasion, growth, or survival. Here, we discuss the influence of these factors on nutrient metabolism and dependence during various steps of tumor development and progression.
Keywords: cancer metabolism, diet, microenvironment, nutrients, tumor heterogeneity
Subject Categories: Cancer, Metabolism
This review discusses the changes in the nutrient metabolism and dependence during various steps of tumor development and progression.
Glossary
- AIM
Apoptosis inhibitor of macrophage
- ALL
Acute lymphoblastic leukemia
- APC
Adenomatous polyposis coli
- ARG1
Arginase 1
- BCAA
Branched‐chain amino acids
- CAFs
Cancer‐associated fibroblasts
- ccRCC
Clear cell renal cell carcinoma
- CD36
Cluster of differentiation 36
- CRC
Colorectal cancer
- CTC
Circulating tumor cell
- CXCL16
Chemokine (C‐X-C motif) ligand 16
- ECM
Extracellular matrix
- EMT
Epithelial‐to-mesenchymal transition
- FABP4
Fatty acid‐binding protein 4
- FADS2
Fatty acid desaturase 2
- GLS1
Glutaminase1
- H3K27me3
Trimethylation of histone 3 lysine 27
- HCC
Hepatocellular carcinoma
- HFD
High‐fat diet
- HIF1a
Hypoxia‐inducible factor 1‐alpha
- IDO1
Indoleamine 2,3‐dioxygenase
- KRAS
Kirsten rat sarcoma oncogene
- lncRNA
Long non‐coding RNAs
- LUAD
Lung ductal adenocarcinoma
- MCT
Monocarboxylate transporter
- MDSCs
Myeloid‐derived suppressor cells
- MET
Mesenchymal‐to-epithelial transition
- MET
MET proto‐oncogene (Hepatocyte growth factor receptor)
- MITF
Microphthalmia transcription factor
- mTOR
Mammalian target of rapamycin
- MYC
MYC proto‐oncogene (Myelocytoma)
- NK
Natural killer cell
- NSCLC
Non‐small‐cell lung carcinoma
- OXPHOS
Oxidative phosphorylation
- PC
Pyruvate carboxylase
- PD‐1
Programmed cell death protein 1
- PDAC
Pancreatic ductal adenocarcinoma
- PDK1
Pyruvate dehydrogenase kinase‐1
- PDX
Patient‐derived xenograft
- PGC‐1α
PPAR‐gamma coactivator 1 alpha
- PHGDH
Phosphoglycerate dehydrogenase
- PML
Promyelocytic leukemia
- PSCs
Stroma‐associated pancreatic stellate cells
- PTEN
Phosphatase and tensin homolog
- ROS
Reactive oxygen species
- SREBP
Sterol regulatory element‐binding protein
- TCA
Tricarboxylic acid cycle
- TGF‐β
Transforming growth factor‐beta
- TIGAR
TP53‐induced glycolysis regulatory phosphatase
- VHL
von Hippel–Lindau tumor suppressor
Introduction
Over time, tumor cells need to adapt to the changing circumstances imposed by the different stages of cancer development (initiation, proliferation, invasion, and metastasis formation) and their surrounding environment to successfully sustain growth demands. One of the most fundamental aspects of this adaptation is the extensive metabolic rewiring that tumor cells undergo in order to support cell survival and proliferation (Vander Heiden & DeBerardinis, 2017). Growing evidence shows that cellular metabolism is thoroughly reprogrammed in cancer cells, enabling tumor initiation, promotion, and progression (Vazquez et al, 2016; Lunt & Fendt, 2018; Kreuzaler et al, 2019). The process of metabolic reprogramming can be highly influenced by nutrient availability to tumor cells at the local environment. Cancer cells residing in different organs have access to different nutrients and their metabolites. Both the presence and the abundance of those will determine the metabolic phenotype that cancer cells adapt to overcome different stages during the tumorigenic process (Lorendeau et al, 2015; Elia et al, 2018). In this way, identical cells in different microenvironments (including tissues, organs, or different parts of the same tumor) exhibit distinct metabolic programs that lead to a variation in phenotypes (Elia & Fendt, 2016; Rinaldi et al, 2018).
As a tumor mass grows and expands, the tumor cells within undergo fluctuations in nutrient availability. Nutrients and metabolic byproducts, such as lactate, are present in a gradient in solid tumors, from tumor edges that have greater access to oxygen and nutrients, to the tumor cores with elevated hypoxia and metabolic byproducts. Additionally, diet‐driven changes may alter the circulating nutrients and impact the function of circulating immune cells—both of which play roles in altering nutrient availability to solid tumors (Follain et al, 2020). As such, studying the nutrient and metabolite composition at the intratumoral level and in the tumor interstitial fluid can provide clues for what nutrients are available to tumor cells, what are being taken up for cellular processes, and what metabolites are secreted as a byproduct for the tumor microenvironment. Here, we review the current research on various nutrient networks that the tumor cell is capable of using during the different stages of cancer development, focusing exclusively in an in vivo setting where the complexity of interactions makes the difference in tumor nutrient dependency.
Evolution of nutrient dependence during cancer development
Nutrient availability to tumor cells is a decisive factor driving metabolic rewiring in cancer. Recent studies provide evidence that cancer cells use different metabolic processes depending on their microenvironment (da Cunha et al, 2019; Doglioni et al, 2019). For example, some tumor cells use the nutrient glutamine as a primary carbon source to feed into metabolic pathways required for in vivo growth, whereas in other tissues or malignant situations glutamine acts as a signaling molecule for tumor progression (Pan et al, 2016; Cacace et al, 2017). Currently, several in vitro models are being developed to more closely resemble in vivo cell environments in order to study metabolic dependencies (Coloff et al, 2016; Cantor et al, 2017; Elia et al, 2017; Vande Voorde et al, 2019). Although these models are useful for determining the molecular mechanisms driving tumorigenesis, the use of in vivo systems to complement these models is crucial for the translational applications of in vitro findings. These in vivo models have the advantage of presenting the most physiologically relevant contexts to study cancer, where the diversity of nutrient environments, oxygen tensions, and stromal cell populations—that are difficult to recapitulate in vitro—can be incorporated into experiments. Based on such studies, it was found that diet, oncogenic mutations, tissue of origin, and tumor microenvironment can all impact metabolic phenotypes in an in vivo situation. The role that these factors play in the metabolic adaptations that tumor cells acquire over the stages of cancer will be discussed in this section.
The first hurdle—nutrient requirements of cancer initiation and growth
Does diet play a role in tumor initiation and progression?
Clinical epidemiologic research complemented by preclinical dietary studies
Changes in whole‐body metabolism caused by diet can affect local nutrient availability in the interstitial fluid of organs. Thus, dietary nutrient intake affects the metabolites that are available to malignant cells during all stages of disease (Sullivan et al, 2019a). In humans, diet has already been implicated in affecting the risk of cancer development, primarily with dietary carbohydrates and fat being of interest. Several epidemiological studies link nutritional excess and the subsequent increase in body adiposity, weight, and obesity with increased rates and risk of various cancers in humans (Mayne et al, 2016; Goncalves et al, 2019a). The Continuous Update Project run by the World Cancer Research Fund and American Institute for Cancer Research has recently reported that overweight or obese individuals have an increased risk for a number of cancers (WCRF/AICR, 2018). Several meta‐analyses show a strong correlation between obesity and weight gain with digestive tract malignancies (liver, colon, rectal, pancreatic, and biliary cancers) and hormone‐linked cancer in women (breast and endometrial cancers) and men (prostate) (Dong et al, 2017; Kyrgiou et al, 2017; WCRF/AICR, 2018). While there is clear evidence that diet and excess nutrient intake may affect the risk of certain types of cancers, this does not necessarily mean that this translates to disease outcomes. For example, a recent meta‐analysis of 9 clinical studies suggests that triple‐negative breast cancer disease‐free progression and overall survival is unaffected by the presence of obesity (Mei et al, 2018). In brief, there appears to be a link between diet, nutrient excess, and adiposity with site‐specific cancers; however, clarifying the causal mechanisms remains an area of intense study.
Despite the importance of nutrient availability for tumor development, studying the role of diet on nutrient exposure to cancer tissues in patients is extremely challenging. There are numerous barriers to interpret nutritional interventions such as feasibility, insufficient magnitude of dietary change, nonadherence to protocols, or the design of appropriate controls (Mayne et al, 2016). In this respect, clinical observations are complemented with dietary studies in murine models to build on the mechanistic interplay between nutrient availability and cancer development, since nutritional interventions and conditions are easier to control in research animal diets (Lien & Vander Heiden, 2019). To date, a number of rodent dietary interventions have been used to study the effect of diet on cancer initiation and progression, including high‐fat diets (HFD), Western diets (high fat with carbohydrates and cholesterol), ketogenic diets (low carbohydrate), and nutrient‐restricted diets. These dietary interventions result in altered tissue and plasma nutrient profiles in animal models of disease. For example, HFD and Western diets have been shown to have the most profound effects on circulating glucose and free fatty acid concentrations throughout the exposure to modified animal feed (Lai et al, 2015; Wang et al, 2016). HFD can also influence the fate of glucose in tumors by altering the whole‐organism energy balance. Lipid‐driven perturbations in hepatic metabolism indirectly feed demanding colorectal tumor growth by stimulating hyperinsulinemia and hyperglycemia, essentially increasing growth signaling and glucose for use by the tumors in extra‐hepatic sites (Wang et al, 2018). The effect of ketogenic diets on whole‐organism physiology may benefit cancer therapy, with animals experiencing improvements in hyperglycemia and hyperinsulinemia resulting from PI3K inhibitors (Hopkins et al, 2018). Restriction diets that limit specific nutrients (such as serine‐ and glycine‐free diets) can increase survival in lymphoma and intestinal cancer and influence proliferation in melanoma and breast cancer models (Maddocks et al, 2017; Sullivan et al, 2019a). Additionally, the gut microbiome has re‐emerged as an important physiological factor in cancer development. As such, the effects of diet on the microbiome and cancer have become an area of increasing interest. The effects of these diets in tumor initiation, progression, as an intervention, and how the gut microbiome may be involved are discussed below.
Influence of diet during tumor initiation
Dietary modulations combined with well‐defined genetic or chemical induction models of murine cancers are widely used to study the role of nutrient‐induced changes in tumor initiation. High‐fat and sugar diets have been shown to accelerate tumor initiation in murine models of hepatocellular carcinoma (HCC) (Asgharpour et al, 2016; Tsuchida et al, 2018). These studies found that prolonged exposure to a Western diet (HFD with glucose, fructose, and cholesterol) provokes localized inflammation in the liver, lipotoxicity, and systemic changes in insulin levels and sensitivity, which, at a molecular level, activates cellular stress and oncogenic pathways relevant for HCC (Asgharpour et al, 2016; Tsuchida et al, 2018). Interestingly, this suggests that a Western diet may prime healthy tissue to develop tumorigenesis. In colorectal cancer (CRC), this diet was found to drive CRC initiation but not progression in adenomatous polyposis coli (APC) mutant mice—which are predisposed to develop intestinal tumors—where fat and sugar exposure increased the number but not the size of tumors. The mechanism behind this outcome may be an altered metabolism, oxidative stress, inflammation, or immune cell infiltration that accelerates the loss of heterozygosity of the Apc gene (Niku et al, 2017).
On the other hand, tumor initiation driven by sugar availability (fructose and glucose) has been reported in the presence of certain oncogenic mutations. In this respect, loss of the macrophage‐secreted apoptosis inhibitor of macrophage (AIM) in the context of a high fructose diet significantly increases HCC development, affecting insulin signaling and reactive oxygen species scavenging (Ozawa et al, 2016). In a CRC model with APC loss in intestinal stem cells, fructose supplementation by water increased tumor initiation alongside increased body weight and impaired whole‐body metabolism (Goncalves et al, 2019b). However, when the authors used oral gavage of fructose to limit weight gain and whole‐body metabolic disturbances, no impact on CRC initiation (as determined by total tumor numbers), but an impact on progression (observed by increased tumor size and grade of disease), was found (Goncalves et al, 2019b). Interestingly, the combination of dietary glucose and fructose, but not each sugar alone, significantly affected cell metabolism of CRC tumors. The presence of fructose in these tumors—even at a moderate dose—enhances glucose metabolism by depleting ATP levels, thereby activating glycolysis (via activation of the glycolytic enzyme phosphofructokinase). This metabolic rewiring led to an accelerated de novo lipogenesis which enhances intestinal tumor growth and grade, an enhancement that can be abolished by preventing fructose metabolism (Goncalves et al, 2019b). These studies suggest synergistic effects of the genetic predisposition and diet resulting in a significantly accelerated tumorigenesis, though further investigations in cancer patients are required.
Influence of diet during tumor progression
Although the role of dietary sugar and fat in cancer progression in patients remains unclear, an increased number of mouse studies provide evidence for the influence of diet on metastatic disease. In breast cancer models, dietary sugar was found to facilitate the development of secondary lung tumors (Jiang et al, 2016a; Fan et al, 2017). Moreover, it was found that human colon cancer tumor xenografts injected subcutaneously and orthotopically displayed accelerated growth and progression in diet‐induced obese and insulin‐resistant mice (O'Neill et al, 2016). Additionally, HFD has been reported to favor metastatic progression in xenograft models from cell lines of various tissue origin (Kim et al, 2011; Park et al, 2012; Pascual et al, 2017; Chen et al, 2018b). For instance, oral squamous carcinoma cells expressing the fatty acid transporter CD36 are sensitive to circulating blood fat levels resulting in increased metastatic potential triggered through HFD or palmitic acid exposure (Pascual et al, 2017). Furthermore, HFD‐induced lipid accumulation in prostate tumors enhanced the metastatic progression in mouse models with co‐deletion of phosphatase and tensin homolog (Pten) and promyelocytic leukemia (Pml) genes—two common oncomutations that cooperatively promote metastasis in human prostate cancer. The metastatic phenotype induced by dietary lipids might be driven by hyperactivation of an aberrant sterol regulatory element‐binding protein (SREBP) prometastatic lipogenic program, which is observed in metastatic human prostate cancer (Chen et al, 2018b). HDF consumption has also been shown to stimulate colon cancer progression as a consequence of small increases in fat mass in the tumor and adipose tissue which contribute to the infiltration of leukocytes in the tumor. The cross‐talk between cancer cells, leukocytes, and adipocytes within the tumor mass may promote the generation of growth factors, cytokines, and chemokines in situ, thereby stimulating tumor growth, angiogenesis, and metastasis (Park et al, 2012). Interestingly, some of these HFD‐related phenotypes can be observed in the absence of obesity and metabolic syndrome, suggesting that environmental dietary factors, rather than the development of metabolic diseases, can boost the progression of tumors.
Therapeutic window of dietary interventions
Cancer‐preventing diets are being explored for their potential to impact tumor development. Preventative effects on cancer progression and development have been suggested through consuming a Mediterranean diet, ketogenic diet (high‐fat/low‐carbohydrates), and applying caloric restriction (Lashinger et al, 2016; Hopkins et al, 2018; Piazzi et al, 2019; Castejón et al, 2020). Restriction of specific nutrients, such as serine and glycine, was found to impair colorectal cancer and lymphoma at various stages of initiation and progression, but showed a less clear effect in pancreatic cancers (Maddocks et al, 2017). In breast cancer, serine restriction inhibited tumor growth, albeit only with the loss of phosphoglycerate dehydrogenase (PHGDH), the rate‐limiting enzyme in serine biosynthesis, highlighting both the dependency of breast cancer tumor growth on serine and the metabolic adaptations that cancer cells use to sustain their growth (Sullivan et al, 2019a). Conversely, in a recent study on squamous cell carcinoma, progenitor stem cells were found to rely on serine availability, but not serine biosynthesis and PHGDH expression, to support stem cell populations and subsequent tumor initiation and burden (Baksh et al, 2020). This effect was associated with the presence of the repressive chromatin mark histone 3 lysine 27 trimethylation (H3K27me3), which controls terminal differentiation of stem cells, thus inhibiting differentiation and escape from becoming a malignant cell. H3K27me3 demethylation was discovered in tumors grown in mice fed serine‐ and glycine‐free diets, and PHGDH loss also prevented H3K27 methylation on this diet. Ultimately, PHGDH loss stimulated tumor growth in the presence of serine, by supporting tumor stem cell proliferation, but the loss of serine from the diet prevented this effect (Baksh et al, 2020). Dietary restriction of the essential amino acid methionine disrupted one‐carbon metabolism in humans, which has been shown to improve therapeutic responses of chemo‐ and radiotherapy‐resistant colorectal cancers and sarcomas in patient‐derived xenograft (PDX) models of tumor growth and progression (Gao et al, 2019). Furthermore, ketogenic diets or diets supplemented with ketones, a metabolic byproduct of fat catabolism, have been studied for potential anti‐cancer effects, with relevant translational results in glioblastoma patients (Poff et al, 2015; Martuscello et al, 2016; Feng et al, 2019). Currently, the benefit of these low‐carbohydrate (ketogenic) diets is tested in overweight women with endometrial cancers (http://www.clinicaltrials.gov/ identifier NCT03285152). Interestingly, dietary interventions administered in conjunction with anti‐cancer treatments can increase treatment efficacy. For instance, taking advantage of the insulin‐sensitizing effects of the ketogenic diets, response to PI3K inhibitors (which is dampened in the presence of high insulin levels) can be improved in multiple tumor types with varied mutational profiles in mice (Hopkins et al, 2018). Furthermore, the improvement of endocrine therapy by reducing insulin signaling via ketogenic diet is currently tested in women with estrogen receptor–positive breast cancer (http://www.clinicaltrials.gov/ identifier NCT03962647). On the other hand, growing preclinical and clinical evidence shows that short‐term fasting can enhance the efficacy of a variety of chemotherapeutic agents against multiple tumor types and can reduce side effects (Peng et al, 2012; de Groot et al, 2015; Dorff et al, 2016; Bauersfeld et al, 2018). Currently, large randomized clinical trials are validating the effect of calorie restriction and intermittent fasting on treatment side effects in cancer patients (http://www.clinicaltrials.gov/ identifiers NCT01802346, NCT00936364, NCT03162289, among others). These works highlight the importance of considering diet effects on therapy response in both, the preclinical and clinical settings.
Implications of gut microbiome in the diet‐cancer interaction
An emerging field looking at the intersection of diet and cancer involves the study of the microbiome, the commensal bacteria, and other microbial populations that live in symbiosis within the body. CRC has been especially highlighted to have interactions with the gut microbiome, the single largest home of symbiotic microbes in the body (Sender et al, 2016). Since the gut microbiome metabolizes nutrients that mammals are unable to break down for absorption and use (such as fiber), it is especially sensitive to dietary composition. Obesity in humans and HFD feeding in mice both drive population lost of diversity in the gut microbiome (Dalby et al, 2017; Foley et al, 2018). Further, the presence of colon cancer alters the microbiome populations as compared to healthy controls (Dejea et al, 2014; Feng et al, 2015). Beyond the gastrointestinal tract, interactions with the immune system have driven further research linking cancer development, immunotherapies, and the gut microbiome. Several studies have shown the complex interplay between the microbiome and cancer that brings microbiota modulation as a viable strategy for improving the clinical efficacy of anti‐cancer treatment. Ma and colleagues found that the metabolism of bile acids by microbes regulated the chemokine (C‐X‐C motif) ligand 16 (CXCL16) which attracted active natural killer (NK) T cells to control liver tumor growth (Ma et al, 2018). In humans with non‐small‐cell lung cancer (NSCLC), differential microbiome composition or antibiotic exposure negatively impacted clinical response to programmed cell death protein 1 (PD‐1) blockade (Routy et al, 2018). Similarly, microbiome composition may affect responses to anti‐PD‐1 immunotherapy in melanoma patients by modulating antigen presentation and effector T‐cell function (Gopalakrishnan et al, 2018). Recent work has begun to explore the diet‐microbiome‐cancer nexus, with HFD‐driven changes to the gut microbiome driving colon cancer development in Ras‐mutated mice (Schulz et al, 2014). In a study on multiple myeloma, major changes in human microbiomes were measured in patients versus healthy controls, and subsequent fecal microbiome transfers, specific microbe supplementation, and glutamine‐deficient diets in a mouse model of myeloma showed that microbe‐synthesized glutamine was found to be a critical driver of tumor growth (Jian et al, 2020). Further, it stands to say that many of the dietary interventions discussed above will impact the microbial flora and may play a role in cancer initiation, growth, and response to therapy, and thus, the microbiome is a critical consideration when discussing causality of diet effects on cancer (Zitvogel et al, 2017).
Taken together, it emerges that diet and environment affect circulating nutrient availability. This directly or indirectly alters nutrient availability to tumors. In the consequence, diet interventions can affect tumor growth and development at least in mouse models (Fig 1). While causality is limited in prospective and retrospective clinical studies on diet, cancer risk, and outcomes, the functional and mechanistic preclinical studies combined with ongoing large‐scale genetic and transcriptomic analysis of human tumor samples will be important to further increase our understanding of nutrient availability and its impact on cancer development.
Figure 1. Diet can determine tumor cell metabolism which may have effects on initiation and progression of different type of cancers.
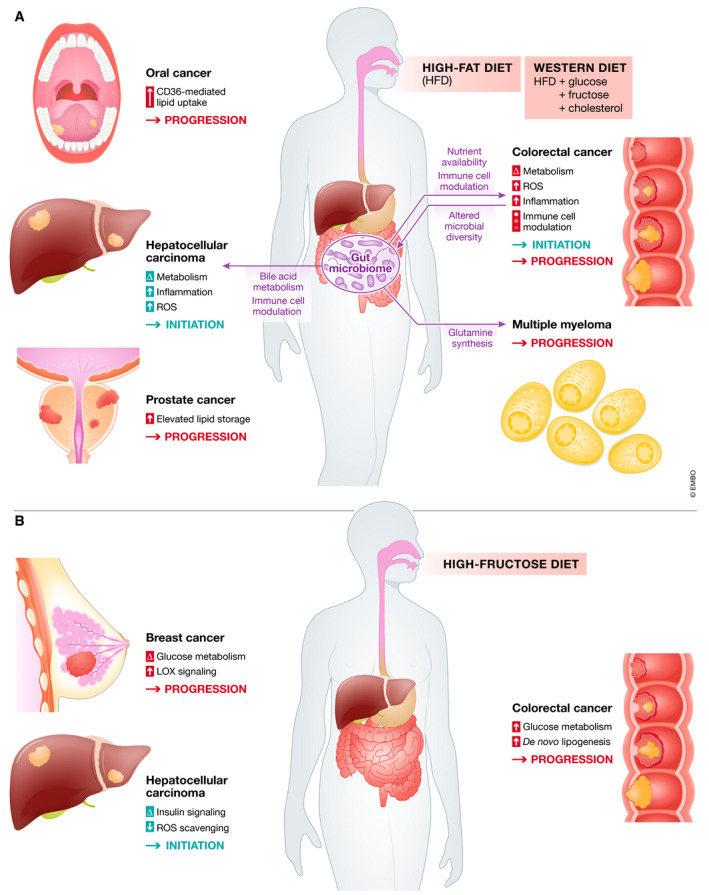
Diet intake influences nutrient availability in the body, affecting what nutrients are available during early initiation and tumor progression stages of cancer development. Various mechanisms are involved in diet‐induced cancer growth, primarily due to altered cellular metabolism, ROS production and diminished ROS scavenging, and altered immune cell infiltration or function. (A) Effects of high‐fat diet (HFD) and Western diets on initiation and progression of various cancers. Excess lipids consumed through HFD feeding can drive tumorigenesis and promote tumor growth by changing cellular metabolism to be more glycolytic, increasing levels of inflammation, increasing the production of ROS species that damage DNA and promote cellular stress mechanisms and DNA damage, and by affecting the amounts and types of immune cells that infiltrate the growing tumors. The diet also affects the composition of the gut microbiome, which impacts the growth of colorectal cancer, and also affects immune cell infiltration into tumors. Nutrients produced by microbes in the gut are also associated with promotion of cancer growth. (B) Diets high in fructose can affect initiation of hepatocellular carcinoma, but promote progression of tumors in other sites including colorectal and breast. Mechanisms involved in fructose‐driven cancer development and progression includes metabolic rewiring of cancer cells to be more glycolytic, allowing for increased de novo lipogenesis to sustain uncontrolled cell growth and altered ROS scavenging. Abbreviations and symbols: ROS (Reactive oxygen species), Δ (alterated), ↑ (high influence).
Do oncogenic mutations and tissue origin drive specific nutrient dependencies during tumor initiation?
One of the first steps in oncogenic transformation is the gain or loss of function of a gene regulating proliferative pathways (Hanahan & Weinberg, 2011). Beyond modulating proliferative signaling, a number of these genes are also involved in regulating cancer cell metabolism to support the rapid growth and division of cancer cells (Iurlaro et al, 2014). With the development and characterization of genetically modified models and in vivo metabolic tracer studies, a number of mutations have been identified that dictate nutrient requirements of malignant tissues among different organ sites and cancer types (Elia et al, 2016; Fernández‐García et al, 2020). One of the first studies showing this effect in vivo is from Yuneva and colleagues, where liver tumors—with distinct activating oncogenic mutations—exhibit unequal utilization of some nutrients such as glucose and glutamine. Here, myelocytoma (MYC)‐induced liver tumors increase lactate production from glucose and catabolism of both glucose and glutamine through the tricarboxylic (TCA) cycle, whereas hepatocyte growth factor receptor (MET)‐induced liver tumors use glucose to produce glutamine (Yuneva et al, 2012). Intriguingly, the tissue context can also affect the metabolic reprogramming outcome driven by the oncogenic mutation. In contrast to liver tumors, lung adenocarcinomas induced by MYC maintain glucose catabolism alongside elevated levels of glutamine (Yuneva et al, 2012). In comparison, Kirsten rat sarcoma oncogene (KRAS)‐driven non‐small‐cell lung carcinomas show the same increase in oxidative glucose usage to fuel the TCA cycle but minimal glutamine utilization (Davidson et al, 2016), suggesting that both, the metabolic reprogramming by MYC and KRAS, are also tissue of origin dependent (Fig 2). Another interesting example in which an oncogenic mutation and the tissue of origin can drive drastically different metabolic changes is observed in KRAS‐driven lung and pancreatic tumors. Kras mutation in lung tissue upregulates branched‐chain amino acid (BCAA) uptake and their catabolism and incorporation into proteins, whereas pancreatic tumors bearing the same mutation decrease BCAA uptake compared to adjacent normal tissues (Mayers et al, 2016). Furthermore, despite the presence of the same oncogenic mutations (KRAS activation and p53 loss), different levels of BCAA and their catabolites were found in the tumor interstitial fluid (the extracellular fluid that perfuses tumors) in lung (LUAD) and pancreatic ductal adenocarcinoma (PDAC) (Sullivan et al, 2019a). Thus, the tissue of origin is one of the major determinants of the tumor microenvironment composition and the nutrients available to tumor cells.
Figure 2. Organ‐specific tumor metabolism is influenced by the tissue of origin and oncogenic driver mutations.
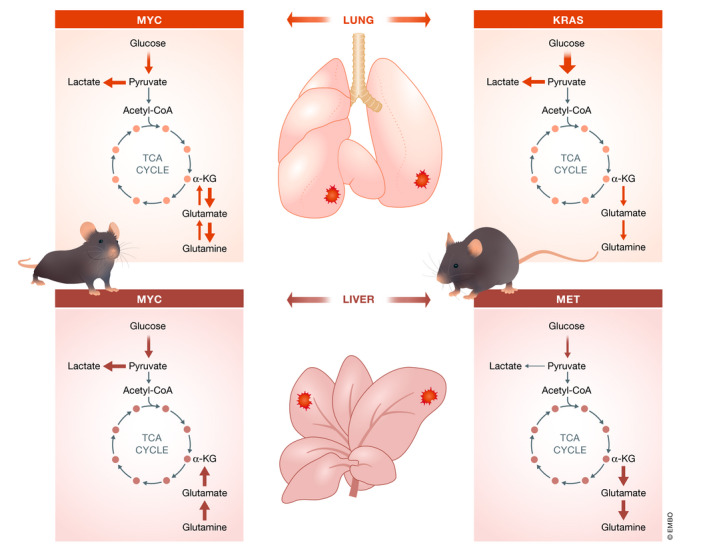
MYC‐induced lung tumor (upper left panel) shows lactate production from glucose and catabolism of glucose through the tricarboxylic cycle (TCA). While it is likely these tumors still consume glutamine, the intracellular levels are high, suggesting that glutamine synthesis exceeds consumption. KRAS‐driven lung tumor (upper right panel) resembles the increased oxidative glucose usage feeding the TCA cycle with minimal glutamine utilization. MET‐induced liver tumor (bottom right panel) mainly oxidizes glucose, with apparent net glutamine synthesis. MYC‐induced liver tumors also (bottom left panel) exhibit increased glucose uptake. However, in these tumors is observed an increased lactate production. In addition, these tumors use glutamine to fuel the citric acid cycle.
Apart from the tissue‐specific differences, there is an additional impact of variable oncogene expression on the cancer cell metabolic phenotypes. Elevated mutational burden observed in late‐stage lung tumors (specifically KRAS copy gain) has been associated with increased glucose metabolism and oxidative TCA cycle (Kerr et al, 2016). Conversely, changing nutrient availability by diet interventions has been shown to affect genetic programs. HFD fed mice bearing KRAS mutant pancreatic acinar cells enhanced KRAS activation, resulting in a metabolic shift in pancreatic tumor cells toward an aggressive glycolytic phenotype (Wang et al, 2019a). Further, the growth of KRAS‐driven tumors was not affected by the removal of serine and glycine from the diet, while APC‐ or MYC‐driven pancreatic tumors were sensitive to serine/glycine depletion resulting in reduced growth (Maddocks et al, 2017).
Nonetheless, these are only a few examples of driver mutations which—depending on the nutrient composition in the environment and the tissue of origin—activate specific metabolic pathways to support tumor cell growth. Other oncogenic mutations may have a key role in shaping metabolic needs during tumor development (Sullivan & Vander Heiden, 2019). However, the scope of how and when these changes occur is far from clear, the recent works discussed above highlight the diverse metabolic changes elicited by similar intrinsic factors (tissue of origin or genetic program). Studies using metabolite tracing approaches may be useful to clarify what global metabolic changes are happening in the clinical context of disease and whether connections can be made to disentangle oncogenic programs and their influences on metabolism in human cancer.
Fuel for established tumors—nutrients that support cancer development
Can the nutrient availability imposed by the local environment define the metabolic features of cancer cells?
Once the intrinsic features of transformed cells have activated proliferative programs, tumor growth is sustained by the interaction of cancer cells with their microenvironment. The organ environment can then redefine oncogene‐imposed metabolic dependencies of cancer cells, meaning that not only the oncogenic drivers and the tissue of origin, but also the local environment influences the development of the tumor (Lunt & Fendt, 2018; Rinaldi et al, 2018). The local environment is characterized by fluctuating metabolic properties. One of the extrinsic features that contribute to this deregulation, along with oxygen and pH, is the nutrient composition of the local environment. Diet‐driven changes in circulating nutrients (Lai et al, 2015), the function of draining lymph nodes (García Nores et al, 2016), and the tissue vasculature (Carmona‐Fontaine et al, 2017) all play roles in altering nutrient availability to solid tumors in this organ environment. Therefore, each organ has a unique metabolic profile making its local environment a particular soil for cancer cells to grow (Table 1).
Table 1.
Presence and availability of some of the most common nutrients implicated in tumor metabolism in blood and different tissues
| Nutrient | Human (concentration in μM) | Mouse (concentration in μM) | Reported location in humang | Concentration in tumor interstitial fluid (μM) | |||
|---|---|---|---|---|---|---|---|
| Intracellulara | Blooda | Intracellularb | Bloodd | Organa | Tissuea | Mousec | |
| Alpha‐ketoglutarate | 290‐350 | 0‐23 | 800 | 8‐27 | NA | NA | 8 |
| Citrate | NA | 30‐400 | 584 | 250‐600 | Prostate | All tissues | 340 |
| Dihydroxyacetone phosphate (DHAP) | 60‐220 | 16 | 1630 | 20‐300 | NA | NA | 750 |
| Fumarate | NA | NA | 485 | NA | Prostate | NA | 2.5 |
| Glucose | NA | 3000‐6000 | NA | 5000‐11000 | Lung (5600e), brain (1000), abdomen (8000‐10000f), bladder, Liver, prostate, pancreas, kidney | Epithelial, muscle, adipose | 2700‐4500f |
| Glucose 6‐phosphate | 26‐50 | 5‐30 | 675 | 2‐15 | Liver, kidney | Muscle (smooth and skeletal), adipose | 107 |
| Glyceraldehyde 3‐phosphate | 5.70‐7.70 | 5 | 141 | 30‐100 | Kidney | Smooth muscle | 160 |
| Lactate | 600‐3,500 | 500‐2,000 | NA | 3,000‐8,000 | Lung, abdomen (5,600f) | All tissues | 8900‐16700f |
| Malate | 2,800‐3,600 | 3‐21 | 1,390 | 20‐100 | Prostate | NA | 155 |
| Phosphoenolpyruvate | 15‐19 | 14.5 | 12 | 0.1‐2.5 | Prostate | NA | 15 |
| 3‐Phosphoglycerate | 45 | 47 | NA | 1‐2.3 | Prostate | NA | 30 |
| Pyruvate | 27‐127 | 20‐250 | 5880 | 40‐200 | Testicle, spleen, brain, liver, kidney, pancreas | Muscle (smooth, skeletal and cardiac), adipose | 40 |
| Succinate | NA | 6‐16 | 352 | 100‐500 | Spleen, brain, liver, prostate, kidney, pancreas | Muscle (smooth and skeletal), adipose | 110 |
| Alanine | NA | 200‐300 | 6980 | 500‐1000 | Prostate | All tissues | 1100 |
| Arginine | NA | 60‐130 | 255 | 110‐200 | Intestine, spleen, bladder, liver, testicle, prostate, kidney, pancreas | Muscle (smooth and skeletal), epithelial, adipose | 2 |
| Asparagine | NA | 30‐80 | 215 | 50‐120 | Prostate | All tissues | 115 |
| Aspartate | NA | 6‐20 | 14900 | 15‐70 | Prostate | All tissues | 400 |
| Cysteine | 30‐300 | 84 | NA | Intestine, spleen, liver, testicle, prostate, kidney | Muscle (smooth and skeletal), epithelial | NA | |
| Cystine | 60‐120 | 60‐120 | NA | 30‐135 | NA | All tissues | 50 |
| Glutamate | 1200‐1600 | 20‐145 | 63800 | 50‐170 | Intestine, spleen, prostate, kidney, pancreas | Muscle (smooth and skeletal), epithelial, adipose | 1050 |
| Glutamine | NA | 390‐900 | 17200 | 500‐900 | Intestine, testicle, spleen, prostate, kidney, pancreas | NA | 800e |
| Glycine | NA | 120‐325 | 3710 | 80‐350 | Intestine, spleen, bladder, brain, prostate, kidney, pancreas | Epithelial | 490 |
| Histidine | NA | 80‐240 | 410 | 45‐165 | Prostate | All tissues | 95 |
| Hydroxyproline | NA | 3‐30 | NA | NA | Spleen, liver, skin, prostate, kidney, pancreas | Muscle (smooth and skeletal) | NA |
| Methionine | NA | 10‐45 | 639 | 60‐350 | Spleen, liver, prostate, kidney, pancreas | Smooth muscle | 75 |
| Proline | NA | 100‐350 | 1230 | 45‐135 | Prostate | All tissues | 120 |
| Serine | NA | 50‐200 | 4860 | 10‐50 | Prostate | All tissues | 90 |
NA, Not available.
Wishart et al (2018). Estimated concentrations in μM from studies in healthy adult compiled in the Human Metabolome Database.
Park et al (2016). Measurements of absolute concentrations (μM) in immortalized baby mouse kidney cells (iBMK cells).
Sullivan et al (2019a). Microenvironmental metabolites in murine pancreatic adenocarcinoma interstitial fluid.
Sullivan et al (2019a). Plasma concentration (μM) in control (healthy) mice.
Fisher (1984). Measurements in isolated perfused rat lung.
Ann Burgess & Sylan (1962). Measurements in mouse tumor tissues.
Healthy organs and tissues in which nutrients have been verified and their concentration (when known) in normal conditions.
Adaptations to lung environment
For most healthy tissues, glucose serves as a primary source of energy meaning that this metabolite is highly available in most organs. For example, glucose oxidation in the lung has been estimated to be comparable to most other metabolically active organs (O'Neil & Tierney, 1974; Fisher, 1984). Although the lung nutrient environment has been primarily studied in murine models, there are strong indications of remarkable dependence on oxidative glucose metabolism in different human lung tumors (Fan et al, 2009; Davidson et al, 2016; Hensley et al, 2016). Interestingly, lung cancer cells grown in vitro rely on glutamine as the preferred anaplerotic carbon source to support the TCA cycle, but upon returning to the in vivo lung environment, glucose metabolism supports TCA anaplerosis (Davidson et al, 2016). In addition, even under well‐oxygenated conditions, lactate production in the lung is elevated when compared to many other tissues (Fisher, 1984) presumably to minimize local oxygen consumption, thereby enhancing overall oxygen delivery to other tissues (Liu & Summer, 2019). This makes lactate highly available in the local environment, allowing it to be used as a carbon source to support the growth of human non‐small‐cell lung cancers (Hensley et al, 2016; Faubert et al, 2017). Similarly, elevated pyruvate availability (compared to blood) in the lung environment favors pyruvate carboxylase (PC)‐dependent anaplerosis to fuel the TCA cycle, which is required for tumor survival and proliferation (Christen et al, 2016) (Fig 3A). NSCLC patients show PC upregulation and enhanced PC activity (compared to Glutaminase1 (GLS1)) in the primary tumors compared to normal adjacent tissues (Sellers et al, 2015). Interestingly, breast cancer cells growing in the lung as secondary tumors also increase PC‐dependent anaplerosis compared to corresponding primary tumors (Christen et al, 2016). While primary triple‐negative breast cancers often use glutamine anaplerosis and are consequently susceptible to GLS1 inhibitors (Elia et al, 2016), different nutrient availability in the lung microenvironment may rewire anaplerotic pathways in cells with the same origin, losing this drug susceptibility. This switch between GLS1‐dependent anaplerosis and PC‐dependent anaplerosis can be recapitulated when growing breast cancer cells in vitro in the presence or absence of pyruvate (Christen et al, 2016). This suggests that, although tumor cells maintain some metabolic features of their tissue of origin (Gaude & Frezza, 2016), different in vivo organ microenvironments can drive different metabolic adaptations, even among cancer cells of the same origin.
Figure 3. Tumors have the ability to consume multiple nutrients to fuel the central carbon metabolism.
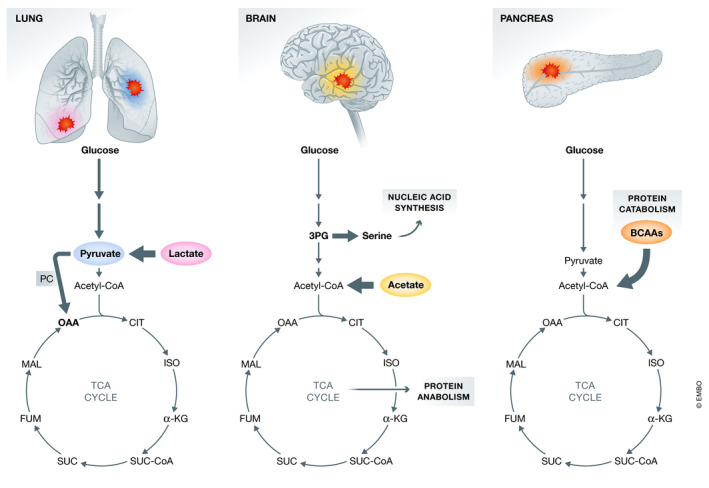
The plasticity of tumor cells allows them to adapt to the unique local environment of each organ, as noted in lung, brain, and pancreas. This flexibility allows them to take advantage of the available nutrients to fulfill energetic and biosynthetic demands promoting tumor growth.
Adaptations to brain environment
The human brain uses about 20% of the body's daily supply of glucose—making it the largest consumer of glucose of all body tissues (Erbslöh et al, 1958; Mergenthaler et al, 2013). Due to the lack of fuel storage, brain tissue requires a continuous supply of glucose and, under normal conditions, the supply of this metabolite from the blood is high in the brain microenvironment. Hence, genetically diverse human glioblastoma tumors (orthotopically transplanted into mice) utilize mitochondrial glucose oxidation to replenish biosynthetic intermediates of the TCA cycle and to contribute to a large glutamine pool during tumor growth (Marin‐Valencia et al, 2012). Apart from its mitochondrial fate, glucose has been shown to support protein and nucleic acid synthesis by preferentially supplying pools of macromolecular precursors such as glutamine, glutamate, and glycine in in situ studies of human brain tumor metabolism (Fig 3B) (Maher et al, 2012; Tardito et al, 2015). In fact, the limitation of serine and glycine in the brain environment restricts the growth of metastatic cells colonizing this organ. To overcome this metabolic constrain, disseminated cancer cells enhance de novo serine synthesis necessary for nucleotide production and cell proliferation, leading to a strong dependency on this biosynthetic pathway in metastatic brain tumors (Ngo et al, 2020). Additionally, brain cells can metabolically respond to physiological changes such as starvation or altered neuronal activity. Brain tissue can produce substantial amounts of acetate (Jang et al, 2019), which can be used (among others, such as ketone bodies or short and medium‐chain fatty acids) as an alternative fuel when blood glucose is low (Ebert et al, 2003; Mason et al, 2006; Deelchand et al, 2009). This may benefit brain‐resident tumors, as patient‐derived glioblastoma (but not normal brain tissue) has been shown to avidly consume available acetate, as opposed to glucose, to fuel the TCA cycle (Fig 3B) (Mashimo et al, 2014). Interestingly, brain metastases from different origins, even with established glucose oxidation phenotypes, can adapt to use acetate as an alternative energy source in the new environment. These observations suggest that the unique brain microenvironment may favor tumors of diverse origins that can utilize acetate as a main biosynthetic substrate (Mashimo et al, 2014) or efficiently synthesize serine for cell growth (Ngo et al, 2020).
Adaptations to other environments
Further metabolic plasticity in tumors has been reported in other organs such as the pancreas and liver. Poorly vascularized pancreatic tumors experience deficient nutrient availability due to perfusion limitations, causing extracellular protein catabolism to become a source for metabolic intermediates. In this organ, KRAS‐driven PDAC tumors, which are low in upper glycolytic intermediates, glutamine, and serine, can obtain sufficient amino acids via extracellular protein scavenging to fuel their metabolic requirements (Kamphorst et al, 2015; Davidson et al, 2017) (Fig 3C). The fact that PDAC tumors may present permeable and leaky blood vessels due to the high interstitial pressure and lymphatic deficiency may result in plasma protein accumulation in the microenvironment of these tumors which boosts protein scavenging (Kamphorst et al, 2015).
Under physiological conditions, the liver produces, stores, and releases glucose to maintain body homeostasis. This may make glucose highly available in this environment, and, contrary to lung tumors, the liver microenvironment is naturally more conducive to cells that display a high glycolytic profile. Along this line, mouse models of liver cancer show elevations of glycolytic flux profiles, lactate levels, and high anaplerotic glutamine utilization (Hu et al, 2011; Yuneva et al, 2012; Muir et al, 2017). Changes in glucose metabolism among distinct organs have been also found in cancer patients. Contrary to human lung (Hensley et al, 2016) and brain tumors (Maher et al, 2012), which show significant levels of glucose oxidation, tumors growing in the kidney display enhanced glycolytic flux with minimal glucose oxidation and turnover of the TCA cycle (Courtney et al, 2018). Cancer cells growing in the liver can also rewire palmitate metabolism by desaturating palmitate using fatty acid desaturase 2 (FADS2) to the unusual fatty acid sapienate which can potentially influence oncogenic lipid‐signaling networks (Vriens et al, 2019; Triki et al, 2020), suggesting an alternative mechanism of metabolic plasticity triggered by fatty acid metabolism.
Interestingly, tissues differ substantially in their susceptibility toward specific oncogenic events, meaning that some tumors display a predominance of mutations that affect metabolic networks based on their site of origin (Schneider et al, 2017). For example, von Hippel–Lindau tumor suppressor (VHL) is inactivated in clear cell renal cell carcinoma (ccRCC), whereas it is only rarely mutated in other tissues. VHL loss drives a pseudo‐hypoxic state by preventing the degradation of hypoxia‐inducible factor‐α (HIF‐α). This state is associated with increased glycolysis and suppressed glucose oxidation, which had been observed in ccRCC tumors compared to the adjacent kidney tissue (Courtney et al, 2018). Similarly, tissue‐specific co‐occurring mutations in the serine‐threonine kinase 11 (STK11) are a common feature of KRAS‐driven NSCLC. The co‐mutation of both genes influence glycolysis and enhanced oxidative phosphorylation, a reprogrammed glucose metabolism that has been reported in lung cancer patients (Faubert et al, 2017; Caiola et al, 2018). In contrast, drivers that affect metabolic networks in the liver (such as TP53) are less specific and the consequences of these mutations depend on the developmental context (Schneider et al, 2017).
Although the main evidence in humans has been shown in glucose enriched‐environments, emerging studies are pointing that nutrients may have an influential role in the tissue environment in modulating in vivo cancer metabolism. The flexibility that cancer cells exhibit may create opportunities for tumor cells to adapt to growth in new environments that initially seem hostile due to the nutrient milieu. Therefore, the environment dictates the nutrient availability in the tumor niche and only cancer cells with sufficient metabolic flexibility are able not only to adapt to these environmental conditions but can also rewire their metabolism to promote tumor growth.
Can tumor heterogeneity provide an advantageous situation even during nutrient fluctuations?
As discussed, the metabolic flexibility exhibited by cancer cells is advantageous for the adaptation to nutrient variability in the organ environment. However, the question remains: Is metabolic flexibility also important for cells to respond to changes within the tumor? This seems plausible, considering an increasing number of works that describe pronounced heterogeneity and metabolic flexibility within distinct regions of solid tumors (Hensley et al, 2016; Xiao et al, 2019; Tasdogan et al, 2020; Vivas‐García et al, 2020). This intratumoral metabolic heterogeneity is imposed by (i) intrinsic factors of the tumor cells, such as genetic alterations, or (ii) by local pathophysiological conditions, such as nutrient deprivation.
Intrinsic factors triggering tumor heterogeneity
Although tumorigenesis may begin with a homogeneous genetic background, it concludes with billions of malignant cells that are mutationally diverse and amplified by clonal selection (Marusyk & Polyak, 2010). Clonal heterogeneity within a tumor has been reported for a variety of malignancies (Marusyk & Polyak, 2010) and recent advances in single‐cell sequencing technology have further uncovered individual tumor heterogeneity in clinical samples, revealing the existence of multiple tumor cell states (Liu et al, 2017; Puram et al, 2017; Wang et al, 2019b). Interestingly, distinct tumor subpopulations exhibit striking differences in the metabolic features of these distinct cell states. For instance, high metabolic heterogeneity in the TCA cycle and oxidative phosphorylation (OXPHOS) has been observed at the single‐cell level in human melanomas and head and neck squamous cell carcinoma, suggesting that variation in mitochondrial activity may be the major contributor to intratumoral metabolic heterogeneity in these tumors (Xiao et al, 2019). In fact, an increased mitochondrial activity in epidermal stem cell populations can drive inhibition of de novo serine synthesis pathway, which in turn allows α‐ketoglutarate accumulation and the subsequently demethylation repression—beneficial for squamous cell carcinoma initiation (Baksh et al, 2020). In the context of therapy resistance, clusters of drug‐tolerant cells were found to be defined by the oncogene MITF (microphthalmia transcription factor) and a decreased proliferative gene expression signature in PDX melanoma models (Rambow et al, 2018). In squamous cell carcinoma, a subpopulation of slow‐cycling stem cells in the vicinity of tumor‐vasculature displayed activated transforming growth factor‐beta (TGF‐β) signaling, enhancing glutathione metabolism, which was associated with the development of therapy resistance (Oshimori et al, 2015). In both situations, the resistant cell subpopulations (with different metabolic features) were associated with disease relapse. In fact, subpopulations of stem cell‐like tumor cells are reported to support metabolic adaptations, protecting tumor cells from fluctuations in essential nutrients for tumor development (Ahmed et al, 2018; De Francesco et al, 2018). This would suggest that individual cells could become more or less stem‐like, impacting the metabolic profile and hence allow the tumor cells the flexibility to respond to changing microenvironments.
Extrinsic factors triggering tumor heterogeneity
The microenvironment within a tumor is not completely homogeneous, having regions that present different vasculature and different infiltrating normal cell populations. This spatial heterogeneity affects the nutrient composition that tumor cells are exposed to. Most tumors have partially structured habitats in which individual cells have varying proximity to the vasculature and hence different levels of oxygen and metabolite accessibility (Hoogsteen et al, 2007; Da Ponte et al, 2017). Interestingly, cancer cells engage into a metabolic symbiosis across different regions of the same tumor. That is, vessel‐distant (or hypoxic) tumor areas are characterized by increased expression of the lactate monocarboxylate transporter (MCT4) and release lactate. In contrast, tumor areas relatively distant from hypoxic areas preferentially express MCT1, which facilitates lactate uptake from the microenvironment resulting in oxidative metabolism (Allen et al, 2016; Jiménez‐Valerio et al, 2016; Pisarsky et al, 2016) (Fig 4). This metabolic symbiosis between lactate‐generating and lactate‐consuming cells is associated with adaptive resistance to anti‐angiogenic therapy in mouse models of breast cancer (Pisarsky et al, 2016), renal cell carcinoma (Jiménez‐Valerio et al, 2016), and pancreatic neuroendocrine tumors (Allen et al, 2016). Similarly, the level of perfusion within different tumors and even different tumor areas can determine glucose utilization as observed in lung cancer patients. Metabolic heterogeneous regions observed in the tumors of these patients suggest a metabolic symbiosis, in which tumor tissue with poorly perfused regions preferentially oxidize glucose whereas highly perfused tumor areas can rely on nutrients other than glucose (e.g., lactate) to support their metabolism (Hensley et al, 2016). These studies suggest that tumor zmetabolism heterogeneity observed in vivo is strongly influenced by the microenvironment and might confer an advantage for cells undergoing different situations.
Figure 4. Model of metabolic symbiosis within a tumor mass sustains metabolic limitations due to local variations in nutrient levels.
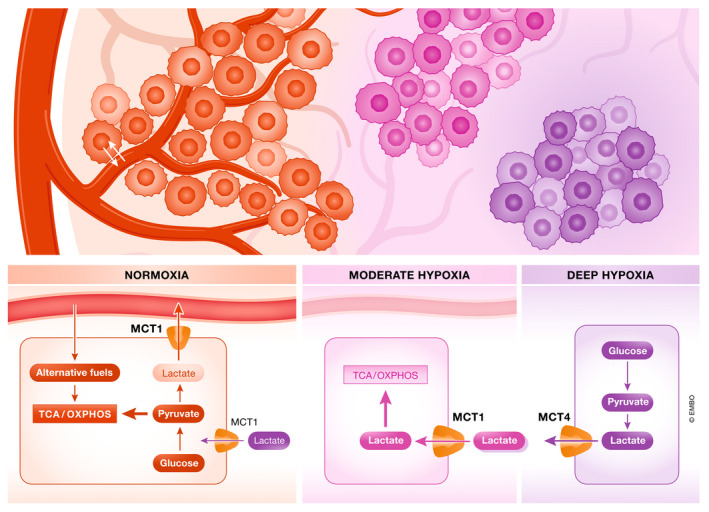
Tumor heterogeneity showed in three different regions within the tumor mass: in regions of deep hypoxia (blood vessels‐distant, in purple), oxidative phosphorylation (OXPHOS) is prevented and substrate availability is reduced. Cancer cells are highly dependent on glycolysis and thus release lactate through monocarboxylate transporter 4 (MCT4). In a moderate hypoxic environment (in pink), various substrates including lactate (exported from highly glycolytic cancer cells) may contribute to fuel the tricarboxylic acid (TCA) cycle and OXPHOS. These cancer cells are characterized by a high expression of MCT1. Under normoxia (surrounding tumor blood vessels, in red), cancer cells can easily exchange nutrients and oxygen into the bloodstream. In this situation, aerobic glycolysis and glucose oxidation in the mitochondria are fully active, and different nutrients (glucose, lactate) can fuel TCA cycle.
Tumor‐associated stromal cells reside in the tumor microenvironment and contribute to the nutrient composition in the milieu around them. Cancer cells can influence the stroma behavior to benefit from nutrients released by these tumor‐associated stromal components to overcome metabolic constraints within the tumor. For example, in ovarian cancer, tumor cell‐secreted metabolites such as lactate are posited to induce the upregulation of glutamine anabolic pathways in cancer‐associated fibroblasts (CAFs). This metabolic cross‐talk allows CAFs to harness carbon and nitrogen from noncanonical sources to synthesize glutamine that sustains tumor growth in nutrient‐deprived conditions (Yang et al, 2016). Conversely, in prostate cancer, CAFs, influenced by tumor HIF1α signaling, release lactate via MCT4 and pancreatic tumor cells take it up via MCT1, being this metabolic interaction clinically associated with poor prognosis (Fiaschi et al, 2012; Pértega‐Gomes et al, 2014). In the pancreatic tumor environment where glucose and serum‐derived nutrients are limited, cancer cells stimulate alanine secretion by induction of autophagy in stroma‐associated pancreatic stellate cells (PSCs) and selectively consume the PSC‐released alanine, using this carbon source in the TCA cycle (Sousa et al, 2016). These observations suggest that the tissue of origin may influence the behavior of tumor‐associated stromal cells residing in the milieu.
While robust data reveal how heterogeneity becomes particularly beneficial when tumors face strong selective pressures, such as chemotherapy (Ding et al, 2012; Kreso et al, 2013) or metastatic barriers (Wu et al, 2012; Casasent et al, 2018), further studies are needed to decipher the advantage of tumor heterogeneity during nutrient limitation. These initial studies suggest that metabolic heterogeneity within tumors allows cells to sustain cell survival in fluctuating nutrient environments, which might influence therapeutic vulnerabilities.
Traversing a perilous environment—the road to metastasis
What metabolic inputs support tumor cell survival in circulation?
Primary tumors may acquire functional mutations during tumor development that initiate the invasion‐metastatic cascade whereby cancer cells adopt an epithelial‐to‐mesenchymal (EMT) phenotype to disseminate from primary lesions into circulation (Lu & Kang, 2019; Vivas‐García et al, 2020). Once cancer cells leave the primary tumor and embark on this journey, they must undoubtedly adapt their metabolism to overcome the stresses and dangers they face in circulation.
To leave the primary tumor, cancer cells must develop a more motile and invasive cellular phenotype, which requires drastic transcriptional and metabolic changes. The process of primary tumor detachment causes a spike in reactive oxygen species (ROS) that circulating tumor cells (CTCs) need to counteract to survive (Friesen et al, 2004; Schafer et al, 2009). However, a non‐excessive amount of ROS can actually be beneficial since it boosts the activation of antioxidant programs, providing protection from cellular stress. In fact, detachment‐induced ROS may serve a protective role by priming cancer cells with enhanced oxidative defenses that may also aid in drug resistance (Fig 5A) (Gong et al, 2015; Elia et al, 2018; Cheung et al, 2020). In mouse models of breast and colon cancer metastasis, mitochondrial ROS (induced by chemotherapy agents such as doxorubicin) contribute to high baseline DNA damage in CTCs. Evidence suggests that this primes DNA repair pathways and antioxidant protein defenses and, hence, enhances chemotherapy resistance (Gong et al, 2015; Alix‐Panabières et al, 2020). However, a recent study in PDAC highlights the context‐dependent importance of ROS, where suppressing ROS supports tumor development while increased ROS after dissemination promotes metastasis in particular cancer settings (Cheung et al, 2020; Fendt & Lunt, 2020). This low‐to‐high shift in ROS regulation was mediated by expression of the pentose phosphate pathway‐promoting enzyme TIGAR (TP53‐induced glycolysis regulatory phosphatase) and is contrary to what has been shown to drive skin, melanoma, and lung cancer metastasis (Gal et al, 2015; Lignitto et al, 2019). The dynamic regulation of ROS has also been implicated in myeloid state cycling of hematopoietic stem cells, where low ROS maintains quiescence and self‐renewal while high ROS initiates their differentiation and migration (Ludin et al, 2014). Since stem‐like CTCs have been characterized as having more aggressive propensities for metastasis, it is likely this balance in redox state plays a key role in regulating metastasis at secondary sites. However, as exemplified by tumor dependent on TIGAR activity, the balance between excessive ROS and “just enough” to drive the phenotypic requirements of cancer progression is also dependent on genetic background and cancer type.
Figure 5. Cancer cell metabolism influences the tumor microenvironment and can support metastasis formation.
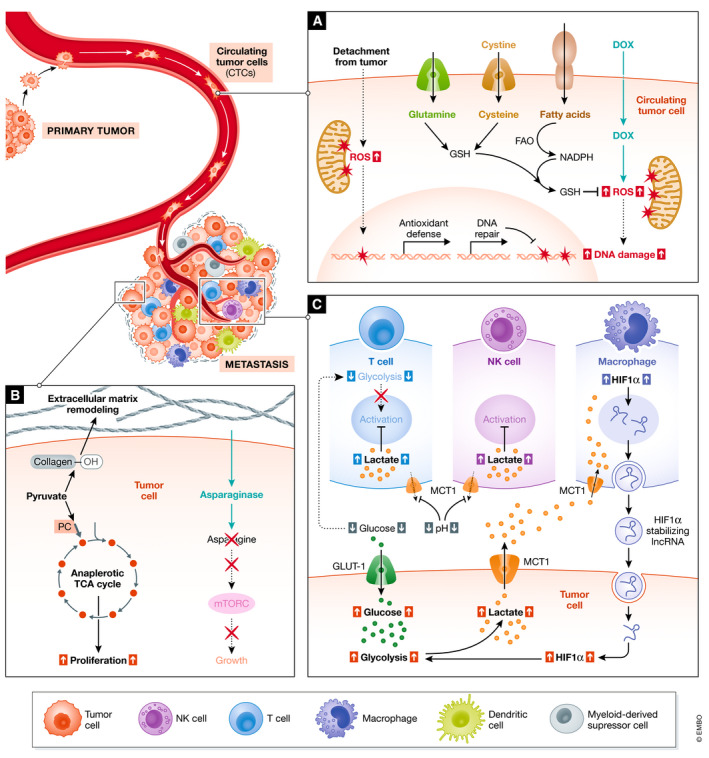
Cell types are indicated with respect to their location in the tumor microenvironment (labeled in the bottom edge), plus nutrients that have been shown to influence their activity in vivo. (A) The mechanistic consequences of tumor detachment and response to doxorubicin therapy can drive gene expression changes regulating nutrient utilization in circulating tumor cells. Abbreviations: Doxorubicin (DOX), GSH (glutathione), FAO (fatty acid oxidation), ROS (reactive oxygen species). (B) Immunoregulatory interactions can be elicited by metabolic byproducts of the intratumoral metabolism, which can act as immune regulators in the metastatic niche. (C) Nutrients can influence metastatic seeding and outgrowth by modulating immune responses and effector functions. Abbreviations: TCA (Tricarboxylic acid cycle), mTOR (mammalian target of rapamycin).
These studies suggest that metabolic pathways involved in protection from ROS support metastasis formation. Thus, antioxidant promoting metabolites such as cysteine and glutamine may be important to maintain ROS in CTCs and combat oxidative damage (Fig 5A) (Friesen et al, 2004; Knott et al, 2018; Combs & DeNicola, 2019). Cysteine and glutamine are metabolic precursors for the synthesis of glutathione, which maintains redox balance and keeps intracellular ROS at a relatively low level. Studies in mouse models of metastasis and tumorigenesis have elucidated the roles of glutamine and cysteine in regulating oxidative stress to promote survival. Glutamine can promote a stem cell phenotype by maintaining levels of ROS that regulate the β‐catenin pathway through dephosphorylation (Liao et al, 2017). In addition, oncogenic RAS transformation can redirect glutamine to maintain cellular redox balance by stimulating the transcription of cystine/glutamate antiporters to enhance glutathione levels, ultimately contributing to tumor progression (Lim et al, 2019). Reductive glutamine metabolism can also regulate oxidative stress by supporting NADPH production within the mitochondria to quench ROS via glutathione generation (Jiang et al, 2016b). Hence, dependence of cancer cells on fatty acid oxidation in vivo has been linked to the promotion of antioxidant defense via NADPH production as well as enhanced drug resistance in glioblastoma and gastric cancer (Pike et al, 2011; He et al, 2019).
These nutrients can provide defense not only against chemotherapy and detachment‐induced stress but also against the mechanical damage induced by circulatory shear stress. Using a model of metastatic breast cancer, Fu and colleagues show that ROS buffering via high manganese superoxide dismutase activity enhances in vivo breast cancer CTC resiliency to circulatory shear stress, increasing the number of cells that successfully metastasize to the lung (Fu et al, 2016). Interestingly, a microfluidics assay of breast, lung, and ovarian CTCs revealed that the high shear stress experienced in arteries during exercise is enough to cause ROS‐driven necrosis of nearly 90% of CTCs (Regmi et al, 2017). Taken together, these studies suggest the enhancement of antioxidant pathways after dissemination in CTCs may initially serve to combat the physical strains of circulatory travel across different physiological states, but then provide an additional benefit toward resisting chemotherapies with oxidative damage‐driven mechanisms. Hence, targeting the regulation of nutrients influencing ROS maintenance can improve current treatments by influencing the survival and drug‐resistant properties of CTCs.
How does the local environment of secondary organs influence the metabolic phenotypes of metastases?
Despite our understanding of metabolic demands and plasticity in primary tumors, the role of nutritional environments once CTCs reach secondary sites of metastasis is less clear. In the metastatic sites, disseminated cancer cells may adapt their metabolism to promote metastatic seeding and colonization. Cancer cells that seed in distant tissues can transition between quiescent and proliferative cell states, depending on the presence or absence of optimal growth signals and conditions (Wells et al, 2013; Lambert et al, 2017). For proliferative cancer cells, glutamine and asparagine have been shown to be instrumental in fueling the mesenchymal‐to‐epithelial (MET) switch in metastasis (Krall et al, 2016; Brabletz et al, 2018; Luo et al, 2018). In the 1960s, asparagine was first identified as a metabolic vulnerability for acute lymphoblastic leukemia (ALL)—whereby treatment of patients with the enzyme L‐asparaginase depletes asparagine from the plasma by converting it to aspartic acid and ammonia (Broome, 1961; Egler et al, 2016). More recently, roles for asparagine in metastasis have been uncovered. L‐asparagine content is selectively elevated in proteins that drive EMT during the metastatic invasion, and limiting de novo asparagine synthesis via asparaginase treatment significantly reduced the capacity for in vivo colonization in the 4T1‐T mouse model (Fig 5B) (Knott et al, 2018). This may be due to asparagine's role in stimulating the mammalian target of rapamycin (mTOR)‐mediated synthesis of proteins necessary for EMT during invasion, even in glutamine‐deprived secondary sites (Pavlova et al, 2018). In contrast to the nutrients that are involved in CTC seeding, much less attention has been given to the environmental cues that trigger dormant states in cancer (Aguirre‐Ghiso & Sosa, 2018). Investigating these influences may lead to further understanding of why some metastatic cancers can remain dormant for long periods before relapse.
Growing evidence indicates that primary tumors and their metastasis are metabolically different, suggesting that the local environment affects fitness for metastatic outgrowth. One of these adaptations has been observed during breast‐derived lung metastasis where, once early metastatic lesions are established, pyruvate further drives PC activity, which supports the TCA cycle anaplerosis needed for proliferation (Christen et al, 2016). Accordingly, genetic deletion of PC impaired pulmonary but not extra‐pulmonary metastasis—suggesting that PC activity could be important for organ tropism (Shinde et al, 2018). The metabolic changes induced by the organ microenvironment were also found to be reversible with the observation that transplanting liver metastasis‐derived cancer cells back into the primary subcutaneous site induced a parental metabolic state (Piskounova et al, 2015). This suggests that metastases of different tumors growing in the same organ might be metabolically more similar than metastases of the same primary tumor growing in different organs. Overall, oncogenic mutations drive nutritional requirements during (primary) tumor initiation and formation, whereas the nutrient status of the tumor microenvironment in distant organs is a key contributing factor supporting metastatic growth of the (secondary) tumor. Further work is needed in this area, particularly to establish specific nutrient content of different human organs and to understand how diet affects nutrient fluctuation and availability in the tumor microenvironment.
Does the phenotype of disseminated cancer cells drive the metabolic dependencies during metastasis?
Metabolic phenotypes of cancer cells can delimit the fate of those cells that are spread from the primary tumor to other tissues. For example, it has been shown that CTCs rely on lactate uptake by MCT1 to manage oxidative stress in PDX models of melanoma. MCT1 phenotype increases the chances of successful seeding by managing more surviving disseminated cancer cells and thereby increases metastasis formation. Interestingly, MCT1 inhibition showed little effect on primary subcutaneous tumors but depleted circulating melanoma cells and reduced metastatic burden (Tasdogan et al, 2020). This highlights an important consideration: The circulatory environment (as opposed to the primary tumor) may select for those CTC populations with a metabolism that sustains the unique demands of circulatory travel, and phenotypic heterogeneity in CTC fitness confers differences in metastatic potential. Once CTCs successfully extravasate and arrive at a distant organ, their phenotype can also dictate the nutrient requirements of these cells to successfully colonize the new tissue. For example, breast cancer cells colonizing lung tissue rely on pyruvate to remodel the extracellular matrix (ECM) of the metastatic niche (Elia et al, 2019), which benefits metastatic outgrowth. As a result, inhibiting the monocarboxylate transporter for pyruvate (MCT2) or mitochondrial alanine aminotransferase enzyme (involved in pyruvate metabolism) is sufficient to impair lung metastasis formation in an in vivo model of breast cancer metastasis (Elia et al, 2019).
The metabolic phenotypes of primary tumor subclones may also predict which secondary sites they will be able to colonize. For instance, depending on the site of metastasis, metastatic breast cancer cells display a different PPAR‐gamma coactivator 1 alpha (PGC‐1α) expression likely acquired from a heterogeneous primary tumor. Unlike liver or bone, in lung tissues, a PGC‐1α‐dependent breast metastatic phenotype drives bioenergetic flexibility in colonizing cells by promoting both the glycolytic and mitochondrial functions of cancer cells, features likely required to colonize this organ (Andrzejewski et al, 2017). In contrast, liver metastasis from breast cancer tumors engages distinct metabolic programs, characterized by an increase in aerobic glycolysis and a concomitant reduction in mitochondrial metabolism. This phenotype is driven by expression of HIF‐1α‐target Pyruvate dehydrogenase kinase‐1 (PDK1), whose activity is required for efficient liver metastasis (Dupuy et al, 2015). Although the metabolic plasticity of tumor cells may enhance their propensity for metastatic seeding (McGuirk et al, 2020), colonizing specific secondary sites may demand phenotypes that impose strict metabolic and nutritional requirements on CTCs. Consequently, different populations of cells leaving a heterogeneous primary tumor may be selected for upon reaching secondary sites based on the ability of their distinct metabolic programs to facilitate the requirements for tissue‐specific colonization.
Are there additional external factors that affect nutrient availability in the secondary organ?
It becomes apparent that the metabolic traits of secondary tumors are, in part, nutrient environment‐dependent. Additionally, there are complementary levels of manipulating the nutrient environment that influence the adaptation of metastatic cells to a new organ. One of these additional factors is the diet, which can affect nutrient availability to both the primary tumor as well as the tissues for potential secondary tumor establishment. Thus, increased dietary asparagine intake promotes metastatic progression and, more interestingly, asparagine restriction reduces metastasis without affecting the growth of the primary tumor (Knott et al, 2018). Another important regulator of the tumorigenic process is the tumor secretome. The secretome consists of key regulatory molecules, derived from the primary tumor or tumor‐associated cells, which influence the microenvironment of secondary organs—priming them for the cultivation of secondary tumors (Shinde et al, 2018). In this regard, recent metabolomics analysis of the in vivo secretome of lung cancer models revealed that increased succinate secretion into plasma by cancer cells induces macrophage‐dependent cytokine signaling in the metastatic niche (Wu et al, 2019). This signaling stimulates cancer cell migration and EMT, enhancing cancer metastasis. Interestingly, serum succinate has been found to be significantly higher in 97 lung cancer patients compared to 21 healthy patients and warrants further study to understand the effect on disease progression (Wu et al, 2019). Furthermore, the secretome can influence distal stromal cells to cultivate a more permissive tumor microenvironment for metastasis. In metastatic breast cancer, primary tumor‐secreted miRNA‐122 suppresses glucose utilization in stromal cells to accommodate the elevated energy demand of cancer cells during metastatic growth (Fong et al, 2015). In other words, these factors can support metastatic cells in the competition for limited nutrient supplies.
Another key component of both primary and metastatic tumor growth is the manipulation of the host immune system (Fig 5C). Immune avoidance is a well‐documented hallmark of cancer progression, and recent work has helped clarify the interplay between nutrient availability and tumor immunogenicity (Doglioni et al, 2019). For example, monocytes have been implicated in fueling proliferative cancer metabolism (Mondanelli et al, 2019; Vitale et al, 2019). Tumor cell metabolism can also deplete local nutrient levels, driving immune suppression, and immune cell‐tumor interactions that allow for more favorable tumor growth. An inverse relationship between T‐cell infiltration and glucose metabolism in squamous cell carcinoma has been described (Ottensmeier et al, 2016). This was attributed to the consumption of local glucose by tumors as a mechanism to exhaust glycolysis‐dependent T‐cell infiltrates into a quiescent‐like state. In addition to depleting resources, cancer cells can also stimulate immunoregulatory pathways in local lymphocytes to avoid cytotoxicity. Highly glycolytic tumors cultivate a tumor microenvironment with low pH and by secreting high concentrations of lactate, which suppresses T‐cell activation and NK cell function in the metastatic niche (Huber et al, 2017; Payen et al, 2019). In contrast to the direct depletion of the glycolytic substrate (glucose) from activating T cells, acidic tumor microenvironments halt T‐cell expansion by inhibiting the export of lactate by MCT1. This transporter requires a favorable concentration gradient for co‐transport of H+/lactate into the extracellular space; thus, acidic microenvironments with high lactate do not favor lactate export from T cells and result in suppression. In addition, lactate release from glycolytic tumors can stabilize HIF1α in tumor‐associated macrophages. This HIF1α stabilization in macrophages promotes the expression of a HIF1α‐stabilizing lncRNA that is delivered back to tumor cells through extracellular vesicles—fueling tumor growth, lactate production, and T‐cell/NK cell suppression in a positive feedback loop (Chen et al, 2018a; Vitale et al, 2019). Similar immunoregulatory mechanisms have been described for macrophages, myeloid‐derived suppressor cells (MDSCs), and dendritic cells in the in vivo tumor microenvironment. Tumors can stimulate these cells to upregulate key catabolic enzymes, such as arginase 1 (ARG1) or indoleamine 2,3‐dioxygenase 1 (IDO1), depleting local arginine and tryptophan which are essential for T‐cell proliferation and differentiation in vivo (Mondanelli et al, 2019). Thus, there is an emerging role for tumor‐associated immune cells in regulating the local nutrient microenvironment to promote anti‐inflammatory responses in cancer. Cancer cells can also interact with other stroma cells to shape the metabolic requirements to promote metastasis. For instance, adipocytes can transfer fatty acids to disseminated cancer cells arriving at organs exhibiting adipocyte‐rich environments for fueling tumor growth. It has been shown that primary human omental adipocytes promote homing, migration, and invasion of ovarian cancer cells by upregulation of fatty acid‐binding protein 4 (FABP4) on the adipocyte‐tumor cell interface (Nieman et al, 2011).
The commonly accepted “seed and soil” hypothesis of metastasis (Mathot & Stenninger, 2012) acknowledges the nutritional suitability of the soil (tissue) to promote seeding (metastasis). However, recent data suggest this concept needs to incorporate the “composition” of the soil, that is, the other physical components of secondary sites (such as vascular/mesothelial cell density, EMT signals, ECM composition) that affect the ability of seeds to penetrate the soil and surface. Cancer cells in secondary sites must receive extracellular invasion signals to trigger EMT, and cells must adapt their metabolism to support this invasion phenotype. ECM modifications are also necessary during metastatic seeding, where metabolic reliance on pyruvate in the lung is shaped by a phenotypic need for breast cancer cells to remodel ECM—rather than to fuel growth (Elia et al, 2019). Endothelial transmigration also depends on “soil density” and the ability of CTCs to process ECM proteins to navigate through dense tissues (Jiang et al, 2015). Some studies in murine models have also investigated the influence of tissue vasculature and endothelial surface molecules in determining metastatic potential (Ruoslahti & Rajotte, 2000; Bugyik et al, 2016). This “soil composition” seems to be therapeutically exploitable, where recent targeting of angiogenic tissue vasculature by a synthetic peptide disrupted the permeability of pre‐metastatic lung vasculature, preventing metastatic seeding in mouse models of lung cancer and melanoma (He et al, 2020). Thus, not only nutrient availability but phenotypic requirements of cancer cells in secondary sites dictate metabolic needs, as these necessitate changes in metabolism to support successful metastasis.
Concluding remarks
Due to the high relevance of the nutrient dependence in cancer cells to support continued growth, there is a significant therapeutic window for metabolism‐based cancer treatment. Although currently under development, targeting the nutrient requirements of cancer is emerging as an effective method of improving existing therapy approaches (Garcia‐Bermudez et al, 2020). Perhaps one of the earliest methods of targeting nutrient dependencies in blood cancers was the reduction of serum L‐asparagine via guinea‐pig‐isolated L‐asparaginase treatment to treat acute lymphoblastic leukemia (Dolowy et al, 1966). Since this landmark discovery, other means of “nutrient‐based metabolic therapy” have been explored. Similar to L‐asparaginase, an engineered cystathionine gamma‐lyase enzyme with a higher affinity for L‐cysteine than L‐cystathionine was designed to deplete serum cystine (Cramer et al, 2017). By systemically depleting cystine in prostate cancer xenograft mouse models, treated animals showed a marked impairment in tumor growth. In addition, since the influence of the diet on nutrient availability is a self‐evident fact, dietary interventions are emerging as synergistic with traditional treatments to improve the efficacy of anti‐cancer therapies (Lévesque et al, 2019). Alternatively, combining metabolic drugs with dietary interventions (Hopkins et al, 2018) and anti‐angiogenic therapies (Pisarsky et al, 2016) or targeting those nutrient transporters that are enriched in tumor cells hold great promise for diagnosis (Zhang & Wang, 2020) and therapeutics (Arensman et al, 2019; Elia et al, 2019). Thus, understanding the adaptations of tumor cells to nutrient fluctuations will open a new therapeutic window to improve the clinical outcome of cancer patients.
However, it still remains a significant challenge to successfully translate the nutritional dependencies that are found in cancer into patient‐relevant interventions. This is in part due to the stark contrast between broad nutrient composition available to tumors in various tissues in vivo compared to the selective nutrient availability in standard in vitro culture medium. Additionally, the inability to fully recapitulate the physiological complexity of the tumor microenvironment in vitro (i.e., nutrient gradients, stromal populations, tissue‐resident lymphocytes, varying diet in patients) to accurately recapitulate disease states merits consideration. For many nutrients, physiological concentrations in organs have been estimated in animal models. It remains to be seen whether the nutrients levels found in mouse tumors are the same as those in humans. This may drive discrepancies in drug response between tumor cells in vivo and cells in culture (Muir & Vander Heiden, 2018) or variable drug sensitivity within tumor due to the metabolic heterogeneity that exists between and within human tumors (Faubert & DeBerardinis, 2017). In fact, combination therapies that address intratumor heterogeneity need to be further considered to improve the outcome of some of the current metabolic‐based cancer treatments.
Unfortunately, as cancer recurrence is common for many patients, addressing the role of nutrient utilization and metabolism in persisting cells in different tissues is of substantial interest. Since recurrence often occurs in sites distinct from primary cancers, comprehensive investigations of the metabolic dependencies in various tissues or environments are desperately needed. As outlined, various populations of cells may arise during cancer progression—each “giving” and “taking” certain nutrients or soluble factors in an exchange with the local environment. Thus, cancer cells play an integral part in shaping the local microenvironment where subsequent daughter cells will need to adapt. To better understand the dynamic changes that occur during this cancer development, single‐cell RNA sequencing technology provides the power to analyze even rare populations of circulating and solid tumor cells for distinct metabolic signatures that develop under various contexts (Lambrechts et al, 2018; Rambow et al, 2018; Xiao et al, 2019). Leveraging the ability of single‐cell technology to unmask heterogenous cell populations with high throughput methods of cellular metabolomics represents the next major step for identification of metabolic signatures among cancer cell populations. This information may contribute to nutrient‐targeting therapies that can supplement common chemotherapeutics or elucidate novel targets for personalized cancer therapy.
In need of answers.
Could cancer type‐specific nutrient dependencies be translated into precision medicine targets? Could circulating nutrient levels be used to predict response to specific cancer treatments?
Can dietary interventions modify the local environment to be able to control the nutrient availability in tumor niches? Can these dietary interventions be combined with metabolic therapies to improve patient response? Is this a therapeutic window to translate dietary interventions to prevent metastatic diseases?
Since the role of diet has been shown to influence cancer progression, can dietary interventions modify circulatory nutrient levels that are supporting the requirements of CTCs? Can an altered nutrient intake prevent seeding of these disseminated cancer cells or the awakening of dormant cells?
Is there a specific nutrient that can stimulate metastatic cells to shift from the quiescent state to proliferative? Could this present an option to target cellular metabolism to prevent the growth of secondary tumors?
What factors determine which cells in a heterogeneous tumor are more likely to disseminate and enter the circulation? Is there a specific nutrient component allowing this population of cells to be more fit in a specific secondary organ?
Conflict of interest
S.‐M.F. has received funding from Bayer, Merck, and Black Belt Therapeutics and has consulted for Fund+. The other authors have no conflict of interest to declare.
Acknowledgements
We thank Matteo Rossi and Oskar Marín‐Béjar for their feedback on the manuscript. PAM is supported by a Marie Skłodowska‐Curie Actions individual fellowship. AMC is supported by a Boehringer Ingelheim Fonds PhD Fellowship. SMF acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement n. 771486–MetaRegulation, FWO Projects (G098120N and G088318N), Fonds Baillet Latour and KU Leuven Methusalem Co‐funding.
EMBO Reports (2020) 21: e50635
See the Glossary for abbreviations used in this article.
References
- Aguirre‐Ghiso JA, Sosa MS (2018) Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Annu Rev Cancer Biol 2: 377–393 [Google Scholar]
- Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G (2018) Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin Cancer Biol 53: 265–281 [DOI] [PubMed] [Google Scholar]
- Alix‐Panabières C, Cayrefourcq L, Mazard T, Maudelonde T, Assenat E, Assou S (2020) Molecular Portrait of Metastasis‐Competent Circulating Tumor Cells in Colon Cancer Reveals the Crucial Role of Genes Regulating Energy Metabolism and DNA Repair. Clin Chem 63: 700–713 [DOI] [PubMed] [Google Scholar]
- Allen E, Miéville P, Warren CM, Saghafinia S, Li L, Peng MW, Hanahan D (2016) Metabolic symbiosis enables adaptive resistance to anti‐angiogenic therapy that is dependent on mTOR signaling. Cell Rep 15: 1144–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrzejewski S, Klimcakova E, Johnson RM, Tabariès S, Annis MG, McGuirk S, Northey JJ, Chénard V, Sriram U, Papadopoli DJ et al (2017) PGC‐1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab 26: 778–787.e5 [DOI] [PubMed] [Google Scholar]
- Ann Burgess E, Sylan A (1962) Glucose, lactate, and lactic dehydrogenase activity in normal intersririal fluid and thar of solid mouse tumors* [PubMed]
- Arensman MD, Yang XS, Leahy DM, Toral‐Barza L, Mileski M, Rosfjord EC, Wang F, Deng S, Myers JS, Abraham RT et al (2019) Cystine–glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci USA 116: 9533–9542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F et al (2016) A diet‐induced animal model of non‐alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 65: 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baksh SC, Todorova PK, Gur‐Cohen S, Hurwitz B, Ge Y, Novak JSS, Tierney MT, Dela Cruz‐Racelis J, Fuchs E, Finley LWS (2020) Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat Cell Biol 22: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersfeld SP, Kessler CS, Wischnewsky M, Jaensch A, Steckhan N, Stange R, Kunz B, Brückner B, Sehouli J, Michalsen A (2018) The effects of short‐term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: a randomized cross‐over pilot study. BMC Cancer 18: 476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA (2018) EMT in cancer. Nat Rev Cancer 128–134 [DOI] [PubMed] [Google Scholar]
- Broome JD (1961) Evidence that the L‐asparaginase activity of guinea pig serum is responsible for its antilymphoma effects. Nature 191: 1114–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugyik E, Renyi‐Vamos F, Szabo V, Dezso K, Ecker N, Rokusz A, Nagy P, Dome B, Paku S (2016) Mechanisms of vascularization in murine models of primary and metastatic tumor growth. Chin J Cancer 35: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace A, Sboarina M, Vazeille T, Sonveaux P (2017) Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene 36: 2074–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiola E, Falcetta F, Giordano S, Marabese M, Garassino MC, Broggini M, Pastorelli R, Brunelli L (2018) Co‐occurring KRAS mutation/LKB1 loss in non‐small cell lung cancer cells results in enhanced metabolic activity susceptible to caloric restriction: an in vitro integrated multilevel approach. J Exp Clin Cancer Res 37: 302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu‐Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, Sabatini DM (2017) Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169: 258–272.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona‐Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB (2017) Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci USA 114: 2934–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, Casasent T, Meric‐Bernstam F, Edgerton ME, Navin NE (2018) Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 172: 205–217.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castejón M, Plaza A, Martinez‐Romero J, Fernandez‐Marcos PJ, de Cabo R, Diaz‐Ruiz A (2020) Energy restriction and colorectal cancer: a call for additional research. Nutrients 12: 144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Cao S, Situ B, Zhong J, Hu Y, Li S, Huang J, Xu J, Wu S, Lin J et al (2018a) Metabolic reprogramming‐based characterization of circulating tumor cells in prostate cancer. J Exp Clin Cancer Res 37: 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez‐Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM et al (2018b) An aberrant SREBP‐dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 50: 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, Vousden KH (2020) Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37: 168–182.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, Elia I, Buescher JM, Orth MF, Davidson SM et al (2016) Breast cancer‐derived lung metastases show increased pyruvate carboxylase‐dependent anaplerosis. Cell Rep 17: 837–848 [DOI] [PubMed] [Google Scholar]
- Coloff JL, Murphy JP, Braun CR, Gygi SP, Selfors LM, Brugge Correspondence JS (2016) Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab 23: 867–880 [DOI] [PubMed] [Google Scholar]
- Combs JA, DeNicola GM (2019) The non‐essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers 11: 678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y et al (2018) Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo . Cell Metab 28: 793–800.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S et al (2017) Systemic depletion of L‐cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 23: 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha BR, Domingos C, Buzzo Stefanini AC, Henrique T, Polachini GM, Castelo‐Branco P, Tajara EH (2019) Cellular interactions in the tumor microenvironment: the role of secretome. J Cancer 10: 4574–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Ponte KF, Berro DH, Collet S, Constans JM, Emery E, Valable S, Guillamo JS (2017) In vivo relationship between hypoxia and angiogenesis in human glioblastoma: a multimodal imaging study. J Nucl Med 58: 1574–1579 [DOI] [PubMed] [Google Scholar]
- Dalby MJ, Ross AW, Walker AW, Morgan PJ (2017) Dietary uncoupling of gut microbiota and energy harvesting from obesity and glucose tolerance in mice. Cell Rep 21: 1521–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O'Brien JP, Pierce KA et al (2016) Environment impacts the metabolic dependencies of ras‐driven non‐small cell lung cancer. Cell Metab 23: 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR et al (2017) Direct evidence for cancer‐cell‐autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23: 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco EM, Sotgia F, Lisanti MP (2018) Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J 475: 1611–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelchand DK, Shestov AA, Koski DM, Uğurbil K, Henry P‐G (2009) Acetate transport and utilization in the rat brain. J Neurochem 109(Suppl 1): 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M et al (2014) Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci 111: 18321–18326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD et al (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature 481: 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doglioni G, Parik S, Fendt SM (2019) Interactions in the (pre)metastatic niche support metastasis formation. Front Oncol 9: 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolowy WC, Henson D, Cornet J, Sellin H (1966) Toxic and antineoplastic effects of l‐asparaginase: study of mice with lymphoma and normal monkeys and report on a child with leukemia. Cancer 19: 1813–1819 [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhou J, Zhu Y, Luo L, He T, Hu H, Liu H, Zhang Y, Luo D, Xu S et al (2017) Abdominal obesity and colorectal cancer risk: systematic review and meta‐analysis of prospective studies. Biosci Rep 37: BSR20170945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorff TB, Groshen S, Garcia A, Shah M, Tsao‐Wei D, Pham H, Cheng C‐W, Brandhorst S, Cohen P, Wei M et al (2016) Safety and feasibility of fasting in combination with platinum‐based chemotherapy [DOI] [PMC free article] [PubMed]
- Dupuy F, Tabariès S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S (2015) PDK1‐dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab 22: 577–589 [DOI] [PubMed] [Google Scholar]
- Ebert D, Haller RG, Walton ME (2003) Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23: 5928–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egler R, Ahuja S, Matloub Y (2016) L‐asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother 7: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia I, Fendt SM (2016) In vivo cancer metabolism is defined by the nutrient microenvironment. Transl Cancer Res 5: S1284–S1287 [Google Scholar]
- Elia I, Schmieder R, Christen S, Fendt SM (2016) Organ‐specific cancer metabolism and its potential for therapy Handbook of experimental pharmacology, pp 321–353. New York LLC: Springer; [DOI] [PubMed] [Google Scholar]
- Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grünewald TGP, Fendt SM (2017) Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia I, Doglioni G, Fendt SM (2018) Metabolic hallmarks of metastasis formation. Trends Cell Biol 28: 673–684 [DOI] [PubMed] [Google Scholar]
- Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, Boon R, Escalona‐Noguero C, Torrekens S, Verfaillie C et al (2019) Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbslöh F, Bernsmeier A, Hillesheim HR (1958) Der Glucoseverbrauch des Gehirns und seine Abhängigkeit von der Leber. Archiv für Psychiatrie und Nervenkrankheiten Vereinigt mit Zeitschrift für die Gesamte Neurologie und Psychiatrie 196: 611–626 [DOI] [PubMed] [Google Scholar]
- Fan TWM, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM (2009) Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope‐resolved metabolomics (SIRM). Mol Cancer 8: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Liu H, Liu M, Wang Y, Qiu L, Cui Y (2017) Increased utilization of fructose has a positive effect on the development of breast cancer. PeerJ 5: e3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, DeBerardinis RJ (2017) Analyzing tumor metabolism in vivo . Annu Rev Cancer Biol 1: 99–117 [Google Scholar]
- Faubert B, Li KY, Cai L, Young JD, Kernstine K, Deberardinis Correspondence RJ (2017) Lactate metabolism in human lung tumors. Cell 171: 358–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Lunt SY (2020) Dynamic ROS regulation by TIGAR: balancing anti‐cancer and pro‐metastasis effects. Cancer Cell 37: 141–142 [DOI] [PubMed] [Google Scholar]
- Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z et al (2015) Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat Commun 6: 6528 [DOI] [PubMed] [Google Scholar]
- Feng S, Wang H, Liu J, Aa J, Zhou F, Wang G (2019) Multi‐dimensional roles of ketone bodies in cancer biology: opportunities for cancer therapy. Pharmacol Res 150: 104500 [DOI] [PubMed] [Google Scholar]
- Fernández‐García J, Altea‐Manzano P, Pranzini E, Fendt SM (2020) Stable isotopes for tracing mammalian‐cell metabolism in vivo . Trends Biochem Sci 45: 185–201 [DOI] [PubMed] [Google Scholar]
- Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P (2012) Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor‐stroma interplay. Can Res 72: 5130–5140 [DOI] [PubMed] [Google Scholar]
- Fisher AB (1984) Intermediary metabolism of the lung. Environ Health Perspect 55: 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley KP, Zlitni S, Denou E, Duggan BM, Chan RW, Stearns JC, Schertzer JD (2018) Long term but not short term exposure to obesity related microbiota promotes host insulin resistance. Nat Commun 9: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follain G, Herrmann D, Harlepp S, Hyenne V, Osmani N, Warren SC, Timpson P, Goetz JG (2020) Fluids and their mechanics in tumour transit: shaping metastasis. Nat Rev Cancer 20: 107–124 [DOI] [PubMed] [Google Scholar]
- Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O'Connor STF, Li S, Chin AR et al (2015) Breast‐cancer‐secreted miR‐122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17: 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen C, Kiess Y, Debatin KM (2004) A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ 11: S73–S85 [DOI] [PubMed] [Google Scholar]
- Fu A, Ma S, Wei N, Tan BXX, Tan EY, Luo KQ (2016) High expression of MnSOD promotes survival of circulating breast cancer cells and increases their resistance to doxorubicin. Oncotarget 7: 50239–50257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal K Le, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyürek LM, Lindahl P, Nilsson J et al (2015) Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 7: 308re8 [DOI] [PubMed] [Google Scholar]
- Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, Richie JP, Ciccarella A, Calcagnotto A, Mikhael PG et al (2019) Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572: 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Nores GD, Cuzzone DA, Albano NJ, Hespe GE, Kataru RP, Torrisi JS, Gardenier JC, Savetsky IL, Aschen SZ, Nitti MD et al (2016) Obesity but not high‐fat diet impairs lymphatic function. Int J Obes 40: 1582–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Bermudez J, Williams RT, Guarecuco R, Birsoy K (2020) Targeting extracellular nutrient dependencies of cancer cells. Mol Metab 33: 67–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, Frezza C (2016) Tissue‐specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 7: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves MD, Hopkins BD, Cantley LC (2019a) Dietary fat and sugar in promoting cancer development and progression. Annu Rev Cancer Biol 3: 255–273 [Google Scholar]
- Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K et al (2019b) High‐fructose corn syrup enhances intestinal tumor growth in mice. Science 363: 1345–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Liu B, Yao Y, Qu S, Luo W, Tan W, Liu Q, Yao H, Zou L, Su F et al (2015) Potentiated DNA damage response in circulating breast tumor cells confers resistance to chemotherapy. J Biol Chem 290: 14811–14825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC et al (2018) Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 359: 97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot S, Vreeswijk MP, Welters MJ, Gravesteijn G, Boei JJ, Jochems A, Houtsma D, Putter H, van der Hoeven JJ, Nortier JW et al (2015) The effects of short‐term fasting on tolerance to (neo) adjuvant chemotherapy in HER2‐negative breast cancer patients: a randomized pilot study. BMC Cancer 15: 652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, Liu Z, Yao Z, Wu Q, Liao W et al (2019) MSC‐regulated lncRNA MACC1‐AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 38: 4637–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Johansson‐Percival A, Backhouse J, Li J, Lee GYF, Hamzah J, Ganss R (2020) Remodeling of metastatic vasculature reduces lung colonization and sensitizes overt metastases to immunotherapy. Cell Rep 30: 714–724.e5 [DOI] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev‐Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L et al (2016) Metabolic heterogeneity in human lung tumors. Cell 164: 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogsteen IJ, Marres HAM, van der Kogel AJ, Kaanders JHAM (2007) The hypoxic tumour microenvironment, patient selection and hypoxia‐modifying treatments. Clin Oncol 19: 385–396 [DOI] [PubMed] [Google Scholar]
- Hopkins BD, Pauli C, Xing D, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR et al (2018) Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PEZ, Nelson SJ, Kurhanewicz J, Vigneron DB, Goga A (2011) 13C‐pyruvate imaging reveals alterations in glycolysis that precede c‐Myc‐induced tumor formation and regression. Cell Metab 14: 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C, Rivoltini L (2017) Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. New Anticancer Paradigm Oncol Med 43: 74–89 [DOI] [PubMed] [Google Scholar]
- Iurlaro R, León‐Annicchiarico CL, Muñoz‐Pinedo C (2014) Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol 542: 59–80 [DOI] [PubMed] [Google Scholar]
- Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, Morscher RJ, Reyes J, Frezza C, Hwang HY et al (2019) Metabolite exchange between mammalian organs quantified in pigs. Cell Metab 07/80: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Zhu Y, Ouyang J, Wang Y, Lei Q, Xia J, Guan Y, Zhang J, Guo J, He Y et al (2020) Alterations of gut microbiome accelerate multiple myeloma progression by increasing the relative abundances of nitrogen‐recycling bacteria. Microbiome 8: 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P et al (2015) Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol 00: S244–S275 [DOI] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B et al (2016a) Reductive carboxylation supports redox homeostasis during anchorage‐independent growth. Nature 532: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Pan Y, Rhea PR, Tan L, Gagea M, Cohen L, Fischer SM, Yang P (2016b) A sucrose‐enriched diet promotes tumorigenesis in mammary gland in part through the 12‐lipoxygenase pathway. Can Res 76: 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez‐Valerio G, Martínez‐Lozano M, Bassani N, Vidal A, Ochoa‐de-Olza M, Suárez C, García‐del-Muro X, Carles J, Viñals F, Graupera M et al (2016) Resistance to antiangiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep 15: 1134–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar‐Sagi D et al (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Can Res 75: 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP (2016) Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531: 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Choi MR, Park H, Kim M, Hong JE, Lee JY, Chun HS, Lee KW, Yoon Park JH (2011) Dietary fat increases solid tumor growth and metastasis of 4T1 murine mammary carcinoma cells and mortality in obesity‐resistant BALB/c mice. Breast Cancer Res 3: R78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL et al (2018) Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554: 378–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7: 11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, O'Brien CA, Van Galen P, Gan OI, Notta F, Brown AMK, Ng K, Jing M, Wienholds E, Dunant C et al (2013) Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 339: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzaler P, Panina Y, Segal J, Yuneva M (2019) Adapt and conquer: metabolic flexibility in cancer growth, invasion and evasion. Mol Metab 33: 83–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrgiou M, Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, Martin‐Hirsch P, Tsilidis KK (2017) Adiposity and cancer at major anatomical sites: umbrella review of the literature. BMJ 356: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YS, Chen WC, Kuo TC, Ho CT, Kuo CH, Tseng YJ, Lu KH, Lin SH, Panyod S, Sheen LY (2015) Mass‐spectrometry‐based serum metabolomics of a C57BL/6J mouse model of high‐fat‐diet‐induced non‐alcoholic fatty liver disease development. J Agricult Food Chem 63: 7873–7884 [DOI] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, Weinberg RA (2017) Emerging biological principles of metastasis. Cell 168: 670–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde K et al (2018) Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24: 1277–1289 [DOI] [PubMed] [Google Scholar]
- Lashinger L. M., OFlanagan C. H., Dunlap S. M., Rasmussen A. J., Sweeney S., Guo J. Y., Lodi A., Tiziani S., White E., Hursting S. D. (2016) Starving cancer from the outside and inside: separate and combined effects of calorie restriction and autophagy inhibition on Ras‐driven tumors. Cancer Metab 4: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque S, Pol JG, Ferrere G, Galluzzi L, Zitvogel L, Kroemer G (2019) Trial watch: dietary interventions for cancer therapy. OncoImmunology 8: 1591878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Liu PP, Hou G, Shao J, Yang J, Liu K, Lu W, Wen S, Hu Y, Huang P (2017) Regulation of stem‐like cancer cells by glutamine through β‐catenin pathway mediated by redox signaling. Mol Cancer 16: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EC, Vander Heiden MG (2019) A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer 19: 651–661 [DOI] [PubMed] [Google Scholar]
- Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B et al (2019) Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178: 316–329.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P et al (2019) Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci USA 116: 9433–9442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Summer R (2019) Cellular metabolism in lung health and disease. Annu Rev Physiol 81: 403–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu Y, Di J, Su Z, Yang H, Jiang B, Wang Z, Zhuang M, Bai F, Su X (2017) Multi‐region and single‐cell sequencing reveal variable genomic heterogeneity in rectal cancer. BMC Cancer 17: 787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorendeau D, Christen S, Rinaldi G, Fendt SM (2015) Metabolic control of signalling pathways and metabolic auto‐regulation. Biol Cell 107: 251–272 [DOI] [PubMed] [Google Scholar]
- Lu W, Kang Y (2019) Epithelial‐mesenchymal plasticity in cancer progression and metastasis. Dev Cell 49: 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludin A, Gur‐Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, Lapidot T (2014) Reactive oxygen species regulate hematopoietic stem cell self‐renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal 21: 1605–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Fendt SM (2018) Metabolism – A cornerstone of cancer initiation, progression, immune evasion and treatment response. Curr Opin Syst Biol 8: 67–72 [Google Scholar]
- Luo M, Brooks M, Wicha MS (2018) Asparagine and glutamine: co‐conspirators fueling metastasis. Cell Metab 27: 947–949 [DOI] [PubMed] [Google Scholar]
- Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V et al (2018) Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360: 858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, Van Den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F et al (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544: 372–376 [DOI] [PubMed] [Google Scholar]
- Maher EA, Marin‐Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A , Jeffrey FM, Choi C, Madden C et al (2012) Metabolism of [U‐ 13 C]glucose in human brain tumors in vivo . NMR Biomed 25: 1234–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z et al (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo . Cell Metab 15: 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuscello RT, Vedam‐Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov O, Deleyrolle LP et al (2016) A supplemented high‐fat low‐carbohydrate diet for the treatment of glioblastoma. Clin Cancer Res 22: 2482–2495 [DOI] [PubMed] [Google Scholar]
- Marusyk A, Polyak K (2010) Tumor heterogeneity: causes and consequences. Biochim Biophys Acta Rev Cancer 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C et al (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159: 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI (2006) Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes 55: 929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathot L, Stenninger J (2012) Behavior of seeds and soil in the mechanism of metastasis: a deeper understanding. Cancer Sci 103: 626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM et al (2016) Tissue of origin dictates branched‐chain amino acid metabolism in mutant Kras‐driven cancers. Science 353: 1161–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne ST, Playdon MC, Rock CL (2016) Diet, nutrition, and cancer: past, present and future. Nat Rev Clin Oncol 13: 504–515 [DOI] [PubMed] [Google Scholar]
- McGuirk S, Audet‐Delage Y, St‐Pierre J (2020) Metabolic fitness and plasticity in cancer progression. Trends Cancer 6: 49–61 [DOI] [PubMed] [Google Scholar]
- Mei L, He L, Song Y, Lv Y, Zhang L, Hao F, Xu M (2018) Association between obesity with disease‐free survival and overall survival in triple‐negative breast cancer A meta‐analysis. Medicine 97: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36: 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondanelli G, Iacono A, Allegrucci M, Puccetti P, Grohmann U (2019) Immunoregulatory interplay between arginine and tryptophan metabolism in health and disease. Front Immunol 10: 1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Vander Heiden MG (2018) The nutrient environment affects therapy. Science 360: 962–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. ELife 6: e27713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo B, Kim E, Osorio‐Vasquez V, Doll S, Bustraan S, Liang RJ, Luengo A, Davidson SM, Ali A, Ferraro GB et al (2020) Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition [DOI] [PMC free article] [PubMed]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell‐Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS et al (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17: 1498–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niku M, Pajari AM, Sarantaus L, Päivärinta E, Storvik M, Heiman‐Lindh A, Suokas S, Nyström M, Mutanen M (2017) Western diet enhances intestinal tumorigenesis in Min/+ mice, associating with mucosal metabolic and inflammatory stress and loss of Apc heterozygosity. J Nutr Biochem 39: 126–133 [DOI] [PubMed] [Google Scholar]
- O'Neil JJ, Tierney DF (1974) Rat lung metabolism: glucose utilization by isolated perfused lungs and tissue slices. Am J Physiol, 226: 867–873 [DOI] [PubMed] [Google Scholar]
- O'Neill AM, Burrington CM, Gillaspie EA, Lynch DT, Horsman MJ, Greene MW (2016) High‐fat Western diet–induced obesity contributes to increased tumor growth in mouse models of human colon cancer. Nutr Res 36: 1325–1334 [DOI] [PubMed] [Google Scholar]
- Oshimori N, Oristian D, Fuchs E (2015) TGF‐β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160: 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottensmeier CH, Perry KL, Harden EL, Stasakova J, Jenei V, Fleming J, Wood O, Woo J, Woelk CH, Thomas GJ et al (2016) Upregulated glucose metabolism correlates inversely with CD8+ T‐cell infiltration and survival in squamous cell carcinoma. Can Res 76: 4136 [DOI] [PubMed] [Google Scholar]
- Ozawa T, Maehara N, Kai T, Arai S, Miyazaki T (2016) Dietary fructose‐induced hepatocellular carcinoma development manifested in mice lacking apoptosis inhibitor of macrophage (AIM). Genes Cells 21: 1320–1332 [DOI] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez‐Davies JE, Rosales KK, Li H et al (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18: 1090–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kim M, Kwon GT, Lim DY, Yu R, Sung M‐K, Lee KW, Daily JW, Park JHY (2012) A high‐fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity‐resistant BALB/c mice. Mol Carcinog 51: 869–880 [DOI] [PubMed] [Google Scholar]
- Park JO, Rubin SA, Xu YF, Amador‐Noguez D, Fan J, Shlomi T, Rabinowitz JD (2016) Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol 12: 482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CSO, Berenguer A, Prats N, Toll A, Hueto JA et al (2017) Targeting metastasis‐initiating cells through the fatty acid receptor CD36. Nature 541: 41–45 [DOI] [PubMed] [Google Scholar]
- Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB, Zhang J (2018) as extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab 27: 428–438.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P (2019) Monocarboxylate transporters in cancer. Mol Metab 33: 48–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Robertson L, Gallinetti J, Mejia P, Vose S, Charlip A, Chu T, Mitchell JR (2012) Surgical stress resistance induced by single amino acid deprivation requires Gcn2 in mice. Sci Transl Med 4: 118ra11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pértega‐Gomes N, Vizcaíno JR, Attig J, Jurmeister S, Lopes C, Baltazar F (2014) A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 14: 352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazzi G, Prossomariti A, Baldassarre M, Montagna C, Vitaglione P, Fogliano V, Biagi E, Candela M, Brigidi P, Balbi T et al (2019) A mediterranean diet mix has chemopreventive effects in a murine model of colorectal cancer modulating apoptosis and the gut microbiota. Front Oncol 9: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M (2011) Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Bioenerget Cancer 1807: 726–734 [DOI] [PubMed] [Google Scholar]
- Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann J, Hess C, Christofori G (2016) Targeting metabolic symbiosis to overcome resistance to anti‐angiogenic therapy. Cell Rep 15: 1161–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527: 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poff AM, Ward N, Seyfried TN, Arnold P, D'Agostino DP (2015) Non‐toxic metabolic management of metastatic cancer in VM mice: novel combination of ketogenic diet, ketone supplementation, and hyperbaric oxygen therapy. PLoS ONE 10: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS et al (2017) Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171: 1611–1624.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin‐Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec‐Rychter M et al (2018) Toward minimal residual disease‐directed therapy in melanoma. Cell 174: 843–855.e19 [DOI] [PubMed] [Google Scholar]
- Regmi S, Fu A, Luo KQ (2017) High shear stresses under exercise condition destroy circulating tumor cells in a microfluidic system. Sci Rep 7: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi G, Rossi M, Fendt SM (2018) Metabolic interactions in cancer: cellular metabolism at the interface between the microenvironment, the cancer cell phenotype and the epigenetic landscape. Wiley Interdiscip Rev Syst Biol Med 10: e1397 [DOI] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP et al (2018) Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science 359: 91–97 [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Rajotte D (2000) An address system in the vasculature of normal tissues and tumors. Annu Rev Immunol 18: 813–827 [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart‐Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G, Schmidt‐Supprian M, Rad R, Saur D (2017) Tissue‐specific tumorigenesis: context matters. Nat Rev Cancer 17: 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz MD, Atay Ç, Heringer J, Romrig FK, Schwitalla S, Aydin B, Ziegler PK, Varga J, Reindl W, Pommerenke C et al (2014) High‐fat‐diet‐mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514: 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Ii MB, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R et al (2015) Pyruvate carboxylase is critical for non‐small‐cell lung cancer proliferation. J Clin Invest 125: 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde A, Wilmanski T, Chen H, Teegarden D, Wendt MK (2018) Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res 20: 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H et al (2016) Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536: 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Vander Heiden MG (2019) Determinants of nutrient limitation in cancer. Crit Rev Biochem Mol Biol 54: 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Heiden MGV, Muir A (2019a) Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8: 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, Meeth K, Muir A, Darnell AM, Bosenberg MW et al (2019b) Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab 29: 1410–1421.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PØ, Weinstock A, Wagner A et al (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine‐restricted glioblastoma. Nat Cell Biol 17: 1556–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M et al (2020) Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577: 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triki M, Rinaldi G, Planque M, Broekaert D, Winkelkotte AM, Maier CR, Raman SJ, Vandekeere A, Van Elsen J, Orth MF et al (2020) mTOR signaling and SREBP activity increase FADS2 expression and can activate sapienate biosynthesis. Cell Rep 31: 107806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Lee Y, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel M, Goossens N, Chou H, Hoshida Y et al (2018) A simple diet‐ and chemical‐induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69: 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, Tardito S (2019) Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv 5: eaau7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168: 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Kamphorst JJ, Markert EK, Schug ZT, Tardito S, Gottlieb E (2016) Cancer metabolism at a glance. J Cell Sci 129: 3367–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L (2019) Macrophages and metabolism in the tumor microenvironment. Cell Metab 30: 36–50 [DOI] [PubMed] [Google Scholar]
- Vivas‐García Y, Falletta P, Liebing J, Louphrasitthiphol P, Feng Y, Chauhan J, Scott DA, Glodde N, Chocarro‐Calvo A, Bonham S et al (2020) Lineage‐restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol Cell 77: 120–137.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona‐Noguero C, Schmieder R, Cornfield T et al (2019) Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 00: 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang J, Yang H, Sanidad KZ, Hammock BD, Kim D, Zhang G (2016) Effects of high‐fat diet on plasma profiles of eicosanoid metabolites in mice. Prostaglandins Other Lipid Mediat 127: 9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nasiri AR, Damsky WE, Perry CJ, Zhang X‐M, Rabin‐Court A, Pollak MN, Shulman GI, Perry RJ (2018) Uncoupling hepatic oxidative phosphorylation reduces tumor growth in two murine models of colon cancer. Cell Rep 24: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Bi Y, Hu L, Luo Y, Ji J, Mao AZ, Logsdon CD, Li E, Abbruzzese JL, Li Z et al (2019a) Obesogenic high‐fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun Signal 17: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Babikir H, Müller S, Yagnik G, Shamardani K, Catalan F, Kohanbash G, Alvarado B, Di Lullo E, Kriegstein A et al (2019b) The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov 9: 1708–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WCRF/AICR (2018) World Cancer Research Fund/American Institute for Cancer Research. Continuous Update Project Expert Report 2018. Body fatness and weight gain and the risk of cancer., Continuous Update Project Expert Report 2018
- Wells A, Griffith L, Wells JZ, Taylor DP (2013) The dormancy dilemma: quiescence versus balanced proliferation. Can Res 73: 3811–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez‐Fresno R, Sajed T, Johnson D, Li C, Karu N (2018) HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res 46(D1): D608–D617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJH, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG et al (2012) Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482: 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J‐Y, Huang T, Hsieh Y, Wang Y, Yen C, Lee G, Yeh C, Peng Y, Kuo Y‐Y, Wen H et al (2019) Cancer‐derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell 77: 213–227 [DOI] [PubMed] [Google Scholar]
- Xiao Z, Dai Z, Locasale JW (2019) Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun 10: 3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R et al (2016) Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment‐regulated cancer cell growth. Cell Metab 24: 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TWM, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y et al (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15: 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J (2020) Targeting uptake transporters for cancer imaging and treatment. Acta Pharmaceutica Sinica B 00: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Daillère R, Roberti MP, Routy B, Kroemer G (2017) Anticancer effects of the microbiome and its products. Nat Rev Microbiol 15: 465–478 [DOI] [PubMed] [Google Scholar]