

Abstract
Mitogenic signals that regulate cell division often proceed through multienzyme assemblies within defined intracellular compartments. The anchoring protein Gravin restricts the action of mitotic kinases and cell-cycle effectors to defined mitotic structures. In this report we discover that genetic deletion of Gravin disrupts proper accumulation and asymmetric distribution of γ-tubulin during mitosis. We utilize a new precision pharmacology tool, Local Kinase Inhibition, to inhibit the Gravin binding partner polo-like kinase 1 at spindle poles. Using a combination of gene-editing approaches, quantitative imaging, and biochemical assays, we provide evidence that disruption of local polo-like kinase 1 signaling underlies the γ-tubulin distribution defects observed with Gravin loss. Our study uncovers a new role for Gravin in coordinating γ-tubulin recruitment during mitosis and illuminates the mechanism by which signaling enzymes regulate this process at a distinct subcellular location.
Keywords: protein kinase A anchoring protein (AKAP), protein kinase, enzyme inhibitor, protein complex, mitosis, tubulin
Spatial biology is an emerging aspect of biomedicine wherein investigators study how the subcellular location of enzymes underlies health and disease (1, 2). At the molecular level, intracellular targeting of cell-signaling enzymes is achieved through the interaction with anchoring, adaptor, or scaffolding proteins (3). Protein kinase A anchoring proteins (AKAPs) are a prototypic example of these signal-organizing proteins that are classified by their ability to anchor PKA (4–6). However, numerous studies have identified other classes of protein kinases, phosphatases, small GTPases, and effector proteins that are AKAP-binding partners (7). Thus, an important function of AKAPs is to provide a locus for the processing, integration, and termination of chemical stimuli by constraining these signaling molecules at defined subcellular locations. Recent findings highlight that AKAPs provide spatial and temporal synchronization of protein kinases that control the mammalian cell cycle. Several multivalent AKAPs such as pericentrin, AKAP450, and Gravin have been implicated in targeting PKA and other signaling enzymes to mitotic structures (8–11). For example, during mitosis Gravin anchors Aurora A and polo-like kinase 1 (Plk1) at the spindle poles (12). We previously showed that mitotic defects and cell-cycle delay ensue upon displacement of either enzyme from the Gravin scaffold. However, given the breadth of targets for these enzymes, a mechanistic dissection of their role in regulating mitotic machinery remains complex.
γ-tubulin participates in various aspects of cell division, including centrosome duplication, chromosome segregation, and mitotic progression (13–15). Importantly, γ-tubulin also initiates nucleation of microtubules at the spindle poles during mitosis (16, 17). Aberrant expression and subcellular distribution of γ-tubulin has been documented in several cancers including gliomas, medulloblastomas, breast cancer, and nonsmall cell lung cancer (18–22). Thus, gaining greater insight into how γ-tubulin is regulated is paramount to understanding its role in disease and for the development of novel therapeutics that target this protein (23, 24).
In this study, we discover that Gravin loss impairs the accumulation of γ-tubulin at the spindle poles during mitosis. We utilize a recently developed precision pharmacology tool, Local Kinase Inhibition (LoKI), to demonstrate that targeted inhibition of centrosomal Plk1 alters the accumulation and asymmetric distribution of γ-tubulin at mitotic spindle poles. Finally, we show that deletion of Gravin disrupts the formation of Nedd1/γ-tubulin sub-complexes that are necessary for tubulin ring assembly, thus illuminating a key centrosome-specific function of this anchoring protein.
Results
Gravin loss perturbs accumulation of γ-tubulin at mitotic spindle poles
Gravin organizes several signaling elements during mitosis (11, 25, 26). Conversely, removal of Gravin disrupts the localization of active kinases at mitotic centrosomes and is associated with spindle defects (12, 27). To ensure the proper assembly of mitotic spindles, γ-tubulin initiates nucleation of microtubules at the spindle poles (28). We examined whether cells with genetically ablated Gravin exhibited defects in the expression or localization of this critical spindle assembly component. First, immunoblots of mouse embryonic fibroblasts (MEFs) from WT and Gravin null (−/−) mice established that total cellular protein expression of γ-tubulin was similar for both genotypes (Fig. 1A). Next, immunofluorescence detection confirmed that γ-tubulin (magenta) decorates spindle poles in WT MEFs during mitosis (Fig. 1B). Counterstaining with α-tubulin (green) and 4′,6-diamidino-2-phenylindole (DAPI; blue) revealed the mitotic spindle and DNA, respectively (Fig. 1B). Strikingly, γ-tubulin is much less concentrated at the spindle poles in Gravin null cells (Fig. 1C). Representative heat maps highlight this phenomenon (Fig. 1, B–E). Likewise, surface plots depicting maximum intensity signals further illustrate this result (Fig. 1, D–G). Quantitative analysis using integrated intensity measurements revealed 33.4% less γ-tubulin at mitotic spindle poles in Gravin null MEFs as compared with WT cells (Fig. 1H). Complementary experiments in HeLa cells stably expressing a control or Gravin shRNA provide further evidence for this phenomenon (Fig. S1, A and B). Gravin-depleted HeLa cells display 23.6% less γ-tubulin at the spindle poles than controls (Fig. S1C). Furthermore, this defect was corrected upon rescue with murine Gravin (Fig. S1, D–F). Collectively, these findings reveal that accumulation of γ-tubulin during mitosis is disrupted in cells lacking Gravin.
Figure 1.

Loss of Gravin perturbs accumulation of γ-tubulin at mitotic spindle poles. A, immunoblot detection of Gravin (top), γ-tubulin (middle), and GAPDH (bottom) in WT and Gravin null (−/−) mouse embryonic fibroblasts (MEFs). B and C, immunofluorescence of representative WT (B) and Gravin −/− (C) mitotic cells. Composite images (left) show α-tubulin (green), DAPI (blue), and γ-tubulin (magenta). Distribution of γ-tubulin is presented in grayscale (middle) and as pseudo-color heat maps (right). Signal intensity scale (in arbitrary units; A.U.) is shown below. D and E, magnified heat maps from image insets in B and C (white boxes) highlight the γ-tubulin signal in WT (D) and Gravin −/− (E) cells. F and G, surface plots representing signal intensity of γ-tubulin in WT (F) and Gravin −/− (G) cells. H, quantification of γ-tubulin immunofluorescence at spindle poles in WT (gray) and Gravin −/− (red) MEFs. Points represent individual cells (n). Data are normalized to WT controls. WT, n = 49; Gravin −/−, n = 54; ****p < 0.0001. Experiments were conducted four times (N = 4). p-values were calculated by unpaired two-tailed Student's t test. Data are mean ± S.E.
Generation and validation of genetically engineered human Gravin KO cells
Although our initial findings postulate that Gravin loss disrupts γ-tubulin targeting to mitotic spindle poles, it is not clear whether complete deletion of Gravin in human cells produces a similar outcome. To generate Gravin KO human lines, we used CRISPR/Cas9-mediated genome editing to disrupt the AKAP12 gene on chromosome 6 in U2OS osteosarcoma cells (Fig. 2A). Targets were selected within the exon of AKAP12 that is shared by all three Gravin isoforms, α, β, and γ (Fig. 2A, middle schematic). We employed either a single guide RNA (gRNA) directed toward target 1 (Gravin KO) or a combination of two gRNAs directed toward both targets 1 and 2 (Gravin KO 2) to generate independent Gravin KO cell lines (Fig. 2A, bottom schematic).
Figure 2.
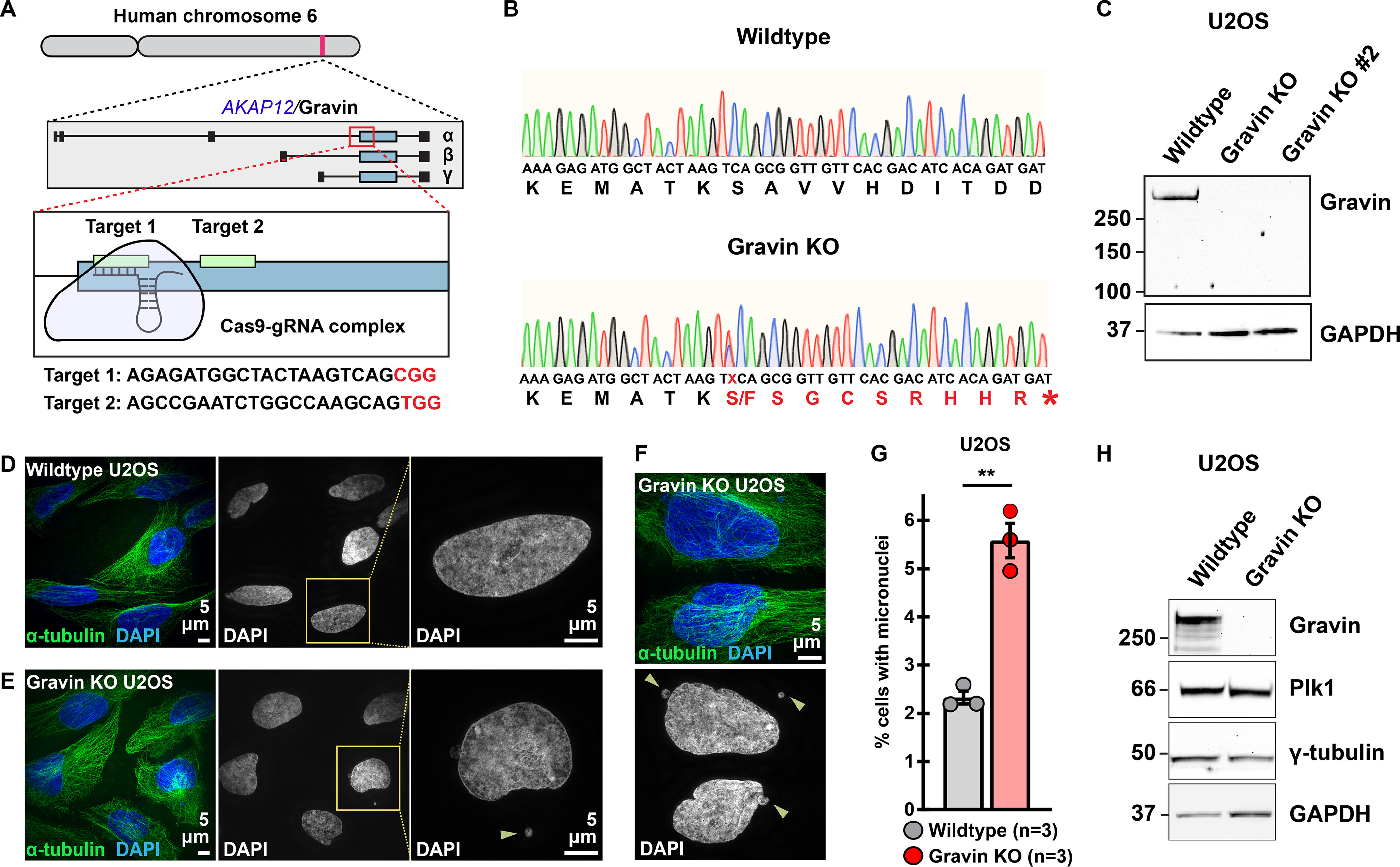
Generation of Gravin knockout U2OS cells. A, CRISPR/Cas9 gene editing of human chromosome 6 in U2OS cells to disrupt the Gravin-encoding gene, AKAP12 (top). Targets are directed to the exon that is shared by all three Gravin isoforms, α, β, and γ (middle). Two unique gRNAs were designed to targets 1 and 2 (bottom). Target sequences are presented below with protospacer adjacent motif (PAM) sites highlighted in red. B, traces depicting nucleic (small letters) and amino (large letters) acid sequences for a representative WT (top) and Gravin KO (bottom) clone. A mutation present in Gravin KO cells alters the protein coding sequence (red) to generate a premature stop codon (*) and leads to a truncated protein. Traces represent pooled allele sequences for each individual clone. Two amino acid alterations were detected (S and F). C, immunoblot detection of Gravin (top) and GAPDH (bottom) in WT and Gravin KO clonal U2OS cells. D–F, SIM images of WT (D) and Gravin KO (E and F) cells during interphase. Composite images (left) show α-tubulin (green) and DAPI (blue). DAPI stain depicted in grayscale (middle). Magnified insets (right) and yellow arrows highlight micronuclei. G, quantification of amalgamated data representing the percent of cells with micronuclei in WT (gray) and Gravin KO (red) U2OS cells. Points depict individual experiments (n): WT, n = 3; Gravin KO, n = 3; **p = 0.001. A total of 1500 cells were analyzed over three independent experiments. P-values were calculated by unpaired two-tailed Student's t test. Data are mean ± S.E. H, immunoblot detection of Gravin (blot 1), Plk1 (blot 2), γ-tubulin (blot 3), and GAPDH (blot 4) in WT and Gravin KO U2OS cells.
We validated our clonal cell lines in five stages. First, Sanger sequencing was used to confirm introduction of indels in Gravin KO clones (Fig. 2B). Sequence analysis indicates introduction of a premature stop codon that abolishes the Gravin coding region at target site 1 in our Gravin KO clone (Fig. 2B, red sequence). Second, immunoblots detected robust Gravin expression in WT cells, whereas a complete loss of the protein was observed in KO lines (Fig. 2C). Immunoblot detection of GAPDH served as a loading control (Fig. 2C). Removal of Gravin was validated with two separate antibodies against this anchoring protein (Fig. 2C and Fig. S2A). Third, we assessed our genome-edited clones for the presence of micronucleated cells. Micronuclei are structures that contain genetic material outside of the main nucleus and often result from missegregated chromosomes (29). Our experiments in Gravin-depleted HEK 293 cells demonstrate that micronuclei formation can be used as a functional readout for Gravin loss (Fig. S2B). Indeed, we observed this phenomenon in our genetically engineered human U2OS clones as well (Fig. 2, D–G). Interphase WT cells displayed a low incidence of micronuclei at baseline (Fig. 2D). On the contrary, we observed more micronucleated cells when Gravin was ablated (Fig. 2, E and F). Gravin loss resulted in a 2.4-fold increase in the number of micronucleated cells (Fig. 2G). This enrichment was further validated in an additional Gravin KO clone (Fig. S2C). Fourth, immunofluorescent detection and quantitative analyses confirmed that levels of active Plk1 (assessed by pT210–Plk1 immunofluorescence signal), a previously characterized Gravin interacting partner, were reduced at mitotic spindle poles in Gravin KO cells as compared with WT controls (Fig. S2, D–G). Finally, additional immunoblotting established that both WT and Gravin KO U2OS cell lines express normal levels of γ-tubulin and Plk1 (Fig. 2H). Together, these data provide evidence for an engineered human cell line that lacks the Gravin anchoring protein.
Gravin deletion enhances the asymmetric distribution of γ-tubulin
Gravin governs the asymmetric distribution of protein kinases at mitotic spindle poles (12). We employed super-resolution structured illumination microscopy (SIM) to examine γ-tubulin organization at each spindle pole. Accordingly, we found that this key microtubule assembly component distributes asymmetrically between the two poles in WT MEFs (Fig. 3A). This result is further illustrated by representative line plots depicting maximum intensity signals across the x-plane (Fig. 3, B and C). Quantitative analysis using integrated intensity measurements established that pole 1 clusters 19.9% more γ-tubulin than pole 2 in WT MEF cells (Fig. 3D). Parallel analyses recapitulated this phenomenon in WT U2OS cells (Fig. 3E and Fig. S3A). These additional studies demonstrated an 18.9% enrichment of γ-tubulin at pole 1 as compared with pole 2 (Fig. 3E). These data reveal the asymmetric distribution of γ-tubulin between individual spindle poles in both WT murine and human cells.
Figure 3.
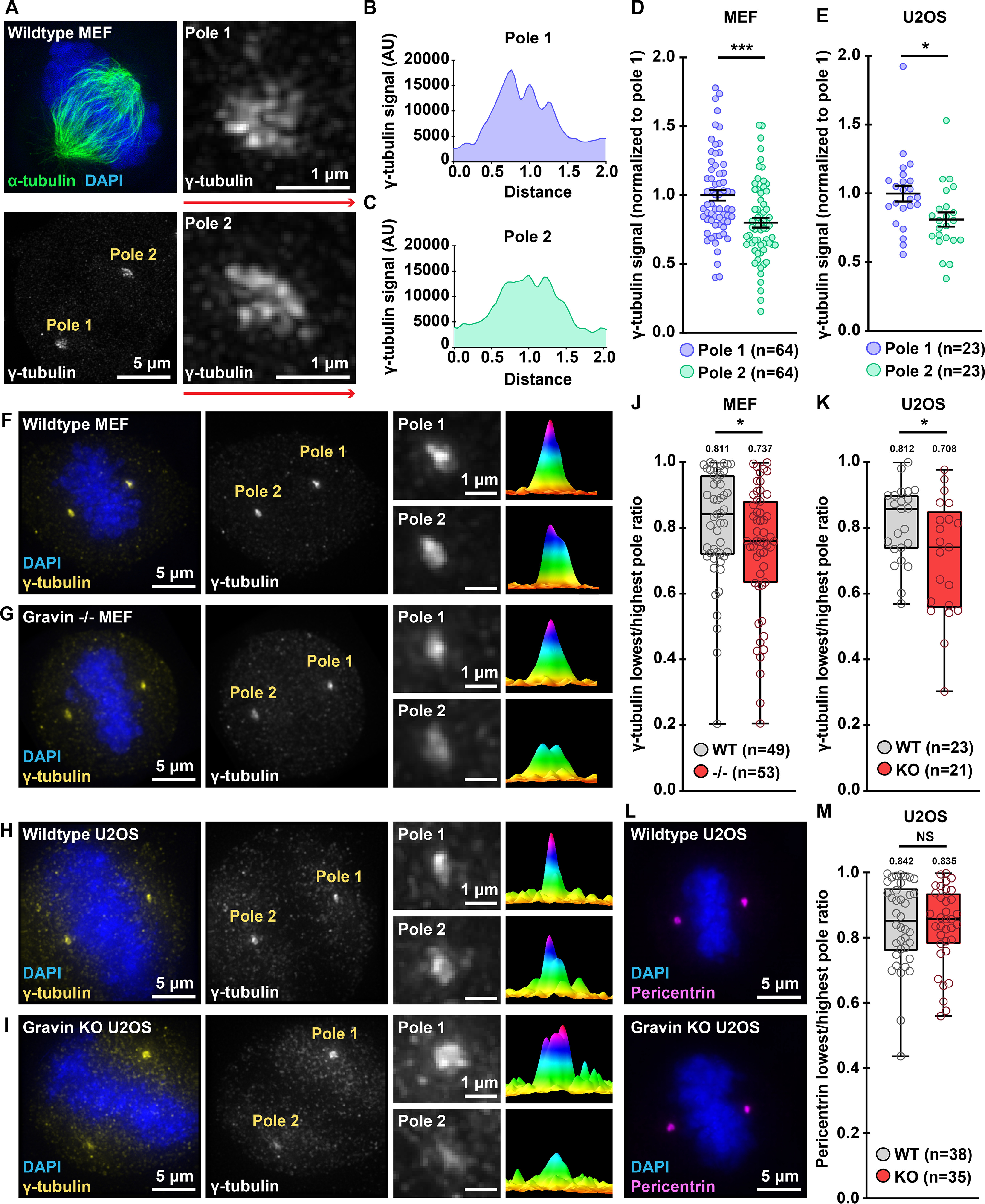
Deletion of Gravin enhances the asymmetric distribution of γ-tubulin. A, SIM micrograph of a WT MEF during mitosis. Composite images (top left) show α-tubulin (green) and DAPI (blue). Grayscale images depict γ-tubulin at both poles (bottom left). Magnified images reveal that pole 1 (top right) accumulates more γ-tubulin than pole 2 (bottom right). B and C, signal intensity (y axis) is graphed against distance (x axis) to generate plot profiles of maximum signal measurements. Distance along red lines in A were used to generate representative plot profiles for pole 1 (B) and pole 2 (C). D and E, quantification of γ-tubulin immunofluorescence at pole 1 (blue) and pole 2 (green) for MEF (D) and U2OS (E) mitotic cells; (D) pole 1, n = 64, pole 2, n = 64, ***p = 0.0002; (E) pole 1, n = 23, pole 2, n = 23, *p = 0.0181. F–I, immunofluorescence of representative WT MEF (F), Gravin −/− MEF (G), WT U2OS (H), and Gravin KO (I) mitotic cells. Composite images (far left) show DAPI (blue) and γ-tubulin (yellow). Grayscale images (middle left) depict γ-tubulin at poles. Magnified insets (middle right) show signals at individual poles. Surface plots (far right) depict signal intensity profiles of each pole. J and K, quantification of γ-tubulin immunofluorescence at each pole represented as box plots showing lowest/highest pole ratio in WT (gray) and Gravin null (red) MEF (J) and U2OS (K) mitotic cells. Central tendency line indicates median values while mean values are indicated above each plot. (J) WT, n = 49, −/−, n = 53, *p = 0.0438; (K) WT, n = 23, KO, n = 21, *p = 0.0263. L, composite immunofluorescence images of representative WT (top) and Gravin KO (bottom) U2OS mitotic cells show DAPI (blue) and pericentrin (magenta). M, quantification of pericentrin immunofluorescence at each pole represented as box blots showing lowest/highest pole ratio in WT (gray) and Gravin KO (red) U2OS mitotic cells. Central tendency line indicates median values while mean values are indicated above each plot. WT, n = 38; KO, n = 35; p = 0.8137. NS, not significant. Points in D, E, J, K, and M represent individual cells (n). Data in D and E are normalized to pole 1. Experiments were conducted at least three times (N = 3). p-values were calculated by unpaired two-tailed Student's t test. Data are mean ± S.E.
Loss of Gravin disrupts the recruitment of signaling elements to mitotic spindle poles (12, 27). Therefore, we tested whether Gravin ablation perturbs the distribution of γ-tubulin between the two poles (Fig. 3, F–K). As before, immunofluorescent staining in WT MEFs confirmed an enrichment of γ-tubulin (yellow) at one spindle pole over the other (Fig. 3F). Magnified insets and representative surface plots emphasized this result (Fig. 3F). Surprisingly, Gravin null MEFs displayed a more pronounced asymmetry in the pole-to-pole distribution of γ-tubulin (Fig. 3G). Quantitative methods were employed to further probe this observation. First, integrated intensity measurements were used to quantitate the total γ-tubulin signal at each pole. Then, the γ-tubulin signal at the pole with the lowest intensity was divided by the signal at the pole with the highest intensity, giving us the ratio of γ-tubulin between the poles (lowest/highest pole ratio). In WT MEFs, we calculated a mean lowest/highest pole ratio of 0.811 (Fig. 3J). However, in Gravin null cells we observed an average ratio of 0.737 (Fig. 3J). Parallel analyses in the CRISPR/Cas9-edited U2OS cells revealed a similar result (Fig. 3, H, I, and K). WT U2OS cells displayed a minor enrichment of γ-tubulin at one pole over the other (Fig. 3H). Quantitative analysis of multiple WT cells yielded a mean lowest/highest pole ratio of 0.812 (Fig. 3K). Consistent with our earlier findings, Gravin KO U2OS cells displayed a ratio of 0.708. This asymmetry was further validated in an additional Gravin KO clone (Fig. S3B). Immunofluorescent staining and quantitative analyses of pericentrin and centrin, two independent spindle pole markers, revealed no differences between the genotypes in the accumulation or pole-to-pole distribution of these control proteins (Fig. 3, L and M and Fig. S3, C–E). Finally, quantitative analyses of immunoblots established that total γ-tubulin expression was similar between WT and Gravin null genotypes in both MEF and U2OS cell lines (Fig. S3, F–I). Collectively, these findings suggest that Gravin deletion leads to enhanced asymmetry of γ-tubulin between mitotic spindle poles.
Precision targeting of Plk1 inhibitors to spindle poles promotes asymmetric distribution of active Plk1 and γ-tubulin
Gravin interacts with Plk1 during mitosis, and depletion of this scaffold results in loss of Gravin-mediated Plk1 anchoring and reduced pools of active kinase at the mitotic spindle poles (12, 27). Thus, it is possible that reduced Plk1 activity may contribute to the defects seen in our Gravin null U2OS cells. We previously reported that treatment with the Plk1 inhibitor BI2536 reduces the total levels of pT210-Plk1 (an index of active kinase) at centrosomes (27). Here we examined how the pT210-Plk1 pool that remains after inhibition with BI2536 distributes between the individual poles. In DMSO-treated controls, immunofluorescent detection of pT210-Plk1 revealed a mild enrichment of active kinase at one spindle pole over the other (Fig. 4A). Remarkably, inhibition of Plk1 led to greater asymmetry in the pole-to-pole distribution of pT210-Plk1 (Fig. 4A). This effect is even more pronounced with increasing concentrations of Plk1 inhibitor (Fig. 4A). Representative line plots further illustrate this result (Fig. 4B). Integrated intensity measurements were used to quantitate the total pT210-Plk1 signal at the spindle poles for each inhibitor dose (Fig. 4C). Whereas DMSO-treated cells displayed an 18.7% enrichment in pT210-Plk1 at one pole over the other, cells treated with 20 nm BI2536 showed a 67.8% difference between the two poles (Fig. 4C). These data suggest that inhibition of Plk1 perturbs the distribution of active kinase between the spindle poles.
Figure 4.
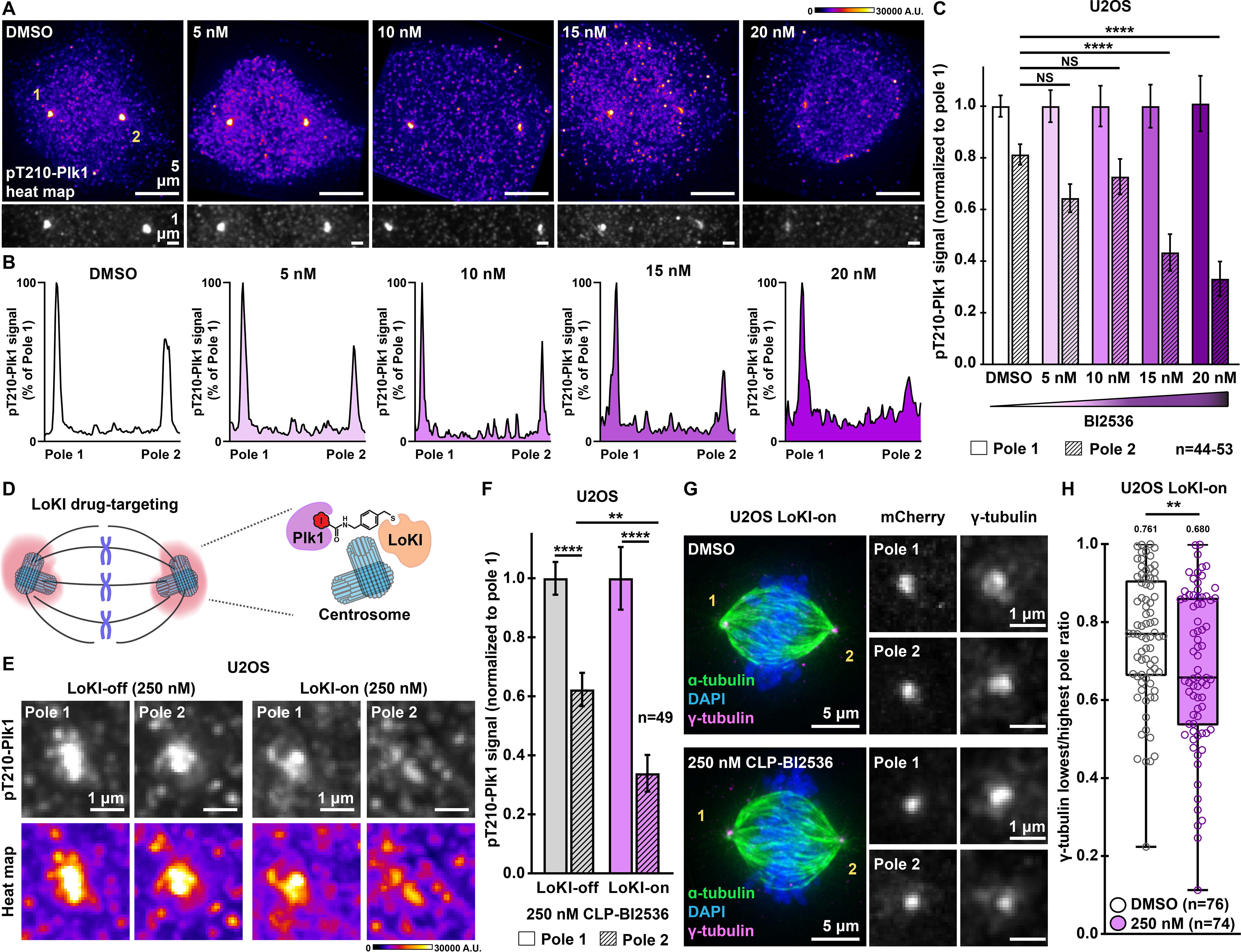
Targeting Plk1 inhibitors to spindle poles promotes a more asymmetric distribution of active Plk1 and γ-tubulin. A, immunofluorescence detection of pT210-Plk1 (an index of kinase activity) at a mitotic spindle pole in parental U2OS cells treated with DMSO or BI2536 for 4 h. Distribution of pT210-Plk1 is represented with pseudo-color heat maps (top) and in grayscale (bottom). Signal intensity scale (in arbitrary units; A.U.) is shown above. B, pT210-Plk1 signal (grayscale from A) is graphed against distance to generate plot profiles of maximum intensity signal measurements. Peaks are normalized to pole 1. BI2536 concentrations are indicated. C, quantification of amalgamated pT210-Plk1 immunofluorescence at each pole for U2OS parental cells treated with DMSO or BI2536; pole 2, DMSO, n = 53, control; 5 nm, n = 44, p = 0.1387; 10 nm, n = 44, p = 0.7016; 15 nm, n = 44, ****p < 0.0001; 20 nm, n = 44, ****p < 0.0001. D, schematic of precision drug targeting of Plk1 inhibitor to spindle poles (pink) using the Local Kinase Inhibition (LoKI) system. E, immunofluorescence of pT210-Plk1 at individual spindle poles in representative LoKI-off (left) and LoKI-on (right) U2OS cells treated with 250 nm CLP-BI2536 for 4 h. Signal of active kinase is represented in grayscale (top) and with pseudo-color heat maps (bottom). Signal intensity scale (in arbitrary units, A.U.) is shown below. F, quantification of amalgamated pT210-Plk1 immunofluorescence at each pole after 4 h of targeted BI2536 delivery; LoKI-off, pole 1, n = 49, LoKI-off, pole 2, n = 49, ****p < 0.0001; LoKI-on, pole 1, n = 49, LoKI-on, pole 2, n = 49, ****p < 0.0001; LoKI-off, pole 2, n = 49, LoKI-on, pole 2, n = 49, **p = 0.0010. G, immunofluorescence of a LoKI-on mitotic cell treated with DMSO (top) or 250 nm CLP-BI2536 (bottom) for 4 h. Composite images (left) show α-tubulin (green), DAPI (blue), and γ-tubulin (magenta). Magnified grayscale images show mCherry (middle) and γ-tubulin (right) signals at poles. H, quantification of amalgamated γ-tubulin immunofluorescence data at each pole. Data are represented as box plots showing lowest/highest pole ratio for LoKI-on cells after 4 h of treatment with DMSO (white) or 250 nm CLP-BI2536 (purple) followed by a 1-h washout. Central tendency line indicates median values while mean values are indicated above each plot. DMSO, n = 76, 250 nm, n = 74, **p = 0.0074. Values depicted in C, F, and H represent individual cells (n). Data in C and F are normalized to pole 1. Experiments were conducted at least three times (N = 3). p-values were calculated by one-way analysis of variance with Dunnett's multiple comparisons test performed with pole 2 DMSO as control (C) or unpaired two-tailed Student's t test (F and H). Data are mean ± S.E. NS, not significant.
Because Gravin anchors Plk1 at mitotic spindle poles, we assessed whether selective inhibition of Plk1 at this location disrupts the pole-to-pole distribution of active kinase. This was achieved by Local Kinase Inhibition (LoKI), a drug-targeting approach that allows us to deliver the Plk1 inhibitor chloropyrimidine (CLP)-BI2536 to centrosomes (27). This precision pharmacology tool allowed us to inhibit Plk1 specifically at spindle poles (Fig. 4D). For our studies, we treated the cells with 250 nm CLP-BI2536 for 4 h because we previously established this as the optimal drug regime to produce equivalent Plk1 inhibition as lower concentrations of nonderivatized BI2536 (27). Immunofluorescence detection of pT210-Plk1 assessed the levels of active kinase at each spindle pole (Fig. 4E and Fig. S4A). Control cells expressed a mutant construct called LoKI-off that is unable to conjugate derivatized kinase inhibitors at spindle poles. When treated with 250 nm CLP-BI2536, these cells display 37.7% less pT210-Plk1 (gray) at pole 2 as compared with pole 1 (Fig. 4, E and F). Heat plots further emphasize this result (Fig. 4E). Strikingly, cells expressing the active LoKI-on targeting platform exhibited 66.1% less pT210-Plk1 at pole 2 as compared with pole 1 when treated with the same concentration of drug (Fig. 4, E and F). Quantitative analyses established a 1.8-fold greater difference in the distribution of active kinase with spindle pole–targeted inhibitors as compared with the global drug (Fig. 4F). Additional controls were conducted in DMSO-treated U2OS cells (Fig. S4, A and B). These studies confirmed that in the absence of the drug we observe comparable distributions of pT210-Plk1 between the poles in cells expressing either LoKI targeting platform (Fig. S4, A and B). Additional validation confirmed that the enhanced asymmetry of pT210-Plk1 does not result from redistribution of total Plk1 protein (Fig. S4C). Together, these data suggest that local targeting of Plk1 inhibitor drugs to mitotic spindle poles leads to enhanced asymmetry in the distribution of active kinase at the spindle poles.
Recruitment of γ-tubulin to centrosomes relies on Plk1 activity (30, 31). Consequently, inhibition of Plk1 at this location disrupts γ-tubulin targeting to these structures (27). Moreover, in Gravin null U2OS cells we observed a trend in which the pole with higher levels of γ-tubulin corresponded to the pole with higher active kinase (Fig. S4, D and E). Thus, a logical next step was to test whether local inhibition of Plk1 enhances the asymmetrical localization of γ-tubulin between the individual spindle poles. Targeted delivery of CLP-BI2536 in LoKI-on cells promoted a more asymmetric distribution of γ-tubulin as compared with DMSO controls (Fig. 4G). Magnified insets of each pole further emphasize this result (Fig. 4G). Quantitative analysis revealed that the lowest/highest pole ratio for γ-tubulin drops from 0.761 to 0.680 when Plk1 inhibitors are targeted to the mitotic spindle poles (Fig. 4H). In contrast, a less-pronounced effect was observed when experiments were repeated in LoKI-off cells (Fig. S4, F and G). Additional control analyses used immunofluorescence of mCherry to confirm that the distribution of LoKI platform is equivalent in DMSO- and drug-treated LoKI-on cells (Fig. 4G and Fig. S4H). These precision pharmacology experiments reveal that targeted Plk1 inhibitor drugs lead to an enhanced asymmetric localization of γ-tubulin at mitotic spindle poles.
Gravin ablation diminishes interactions between γ-tubulin and upstream regulators
Nedd1, a member of the γ-tubulin ring complex (γ-TURC), coordinates γ-tubulin accumulation at centrosomes (32, 33). Furthermore, Plk1 promotes the interaction of γ-tubulin with Nedd1 to ensure proper targeting of γ-TURC during mitosis (34). We employed proximity ligation assay (PLA), a technique that marks protein-protein interactions that occur within a range of 40–60 nm, to identify protein interaction pairs in WT and Gravin null mitotic cells (Fig. 5, A–E). Immunofluorescent detection of PLA puncta (green) and DAPI (blue) uncovered Plk1/γ-tubulin interaction pairs in WT MEFs (Fig. 5A). Significantly fewer PLA puncta were detected in Gravin null cells (Fig. 5B). Quantitative analysis revealed a 24% reduction in the number of puncta observed in Gravin null MEFs as compared with WT controls (9.8 versus 12.9, respectively; Fig. 5C). Complementary experiments were carried out to assess Nedd1/γ-tubulin protein-protein interactions (Fig. 5, D–E). Again, immunofluorescent detection identified PLA puncta (yellow) and DAPI (blue) in mitotic cells (Fig. 5D). As before, Gravin null MEFs displayed fewer PLA puncta than WT control cells (Fig. 5D). Quantitation further revealed that Gravin-depleted cells displayed 34.8% less PLA puncta than WT MEFs (Fig. 5E). These findings suggest that Plk1/γ-tubulin and Nedd1/γ-tubulin interactions are reduced in cells lacking Gravin. Thus, abrogation of Plk1-mediated signaling may underlie the abnormalities in γ-tubulin accumulation observed in Gravin null cells (Fig. 5F).
Figure 5.
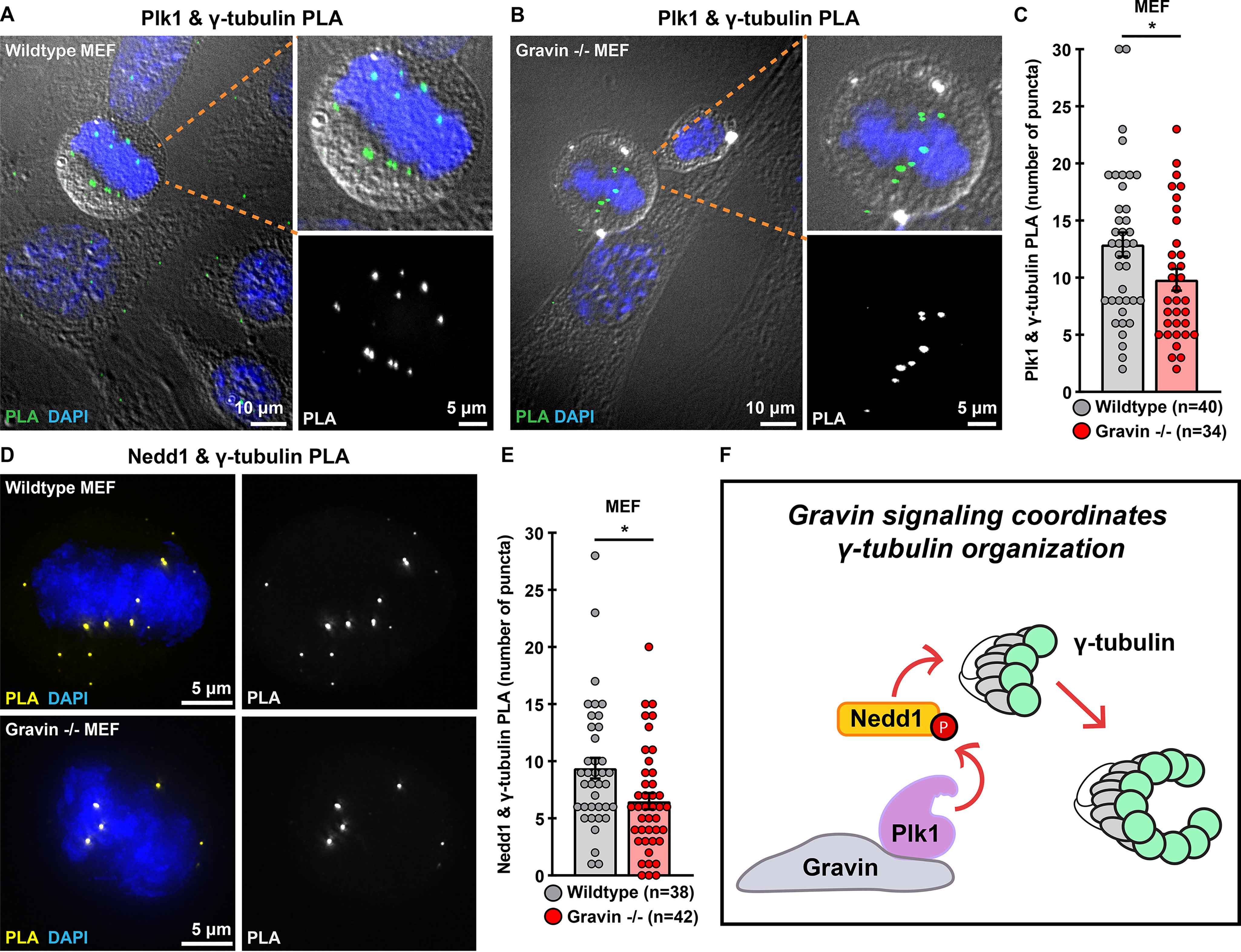
Interactions between γ-tubulin and upstream regulators are disrupted in Gravin-ablated cells. A and B, WT (A) and Gravin −/− (B) MEFs assayed with PLA reveal Plk1 and γ-tubulin interaction pairs. Composite images (left) show PLA puncta (green) and DAPI (blue). Differential interference contrast shows cell boundaries in mitotic (foreground) and interphase (background) cells. Magnified insets of composite images (top right) depict a mitotic cell. Grayscale images (bottom right) reveal PLA puncta. C, quantification of Plk1 and γ-tubulin PLA (number of puncta per cell) in WT (gray) and Gravin −/− (red) MEFs; WT, n = 40, Gravin −/−, n = 34, *p = 0.0357. D, immunofluorescence of WT (top) and Gravin −/− (bottom) MEFs assayed with PLA reveal Nedd1 and γ-tubulin interaction pairs. Composite images (left) show PLA (yellow) and DAPI (blue). Single channel images of PLA are represented in grayscale (right). E, quantification of Nedd1 and γ-tubulin PLA (number of puncta per cell) in WT (gray) and Gravin −/− (red) MEFs; WT, n = 38, Gravin −/−, n = 42, *p = 0.0131. Points in C and E represent individual cells (n). Experiments were conducted three times (N = 3). p-values were calculated by unpaired two-tailed Student's t test. Data are mean ± S.E. F, schematic depicting that Gravin organizes a signaling network that promotes accumulation of γ-tubulin (green circles) when Plk1 and Nedd1 are in proximity to γ-tubulin. “P” indicates a phosphorylated species of protein.
Discussion
Subcellular targeting and anchoring of protein kinases is a recognized molecular mechanism that enhances the precision and fidelity of protein phosphorylation events (3). Gravin regulates various aspects of mitosis, including the clustering of mitotic protein kinases to facilitate spindle formation and mitotic progression (12, 35, 36). In this study, we discover a new role for this anchoring protein in modulating the recruitment of γ-tubulin, a major regulator of microtubule nucleation. Our imaging studies in Fig. 1 reveal that loss of Gravin perturbs the accumulation of γ-tubulin at mitotic spindle poles. Elevated expression and ectopic cellular distribution of γ-tubulin has been observed in astrocytic gliomas (18). Delocalization of this key microtubule-nucleating component has also been detected in human breast cancer, with the most dramatic changes occurring in tumor cell lines that encompass the greatest metastatic potential (20). Both reports conclude that inappropriate subcellular distribution of γ-tubulin is associated with tumor progression. Our findings argue that Gravin-mediated signaling further contributes to this process. Other investigators have shown that Gravin loss is linked to defective mitoses in a variety of cancer cell lines (36, 37). Recent phosphoproteomics studies further implicate this anchoring protein as a potential prognostic biomarker for high-grade meningiomas (36, 37). Our discovery that Gravin-ablated cells display an enhanced pole-to-pole asymmetry of γ-tubulin provides additional clues into how protein scaffolds safeguard cell division events (Fig. 3, F–K). Thus, we postulate that Gravin may serve a protective role during mitosis by ensuring the proper localization of γ-tubulin at mitotic spindle poles. Collectively, these studies suggest that precise targeting of mitotic signaling components contributes to the fidelity of cell division.
Centrosomes exhibit an inherent asymmetry due, in large part, to their biogenesis from distinctive mother and daughter centrioles (38). It has been shown in many cell systems that this unequal distribution of centrosomal proteins correlates with differential cell fate (39). In addition, higher microtubule organizing activity is often a feature of the mother centriole (40). Extremes in centrosomal asymmetry must be avoided to ensure equal cell division and proper spindle assembly (41). Although the mechanistic details of this process are better understood for cell fate determination, fewer studies have investigated this process during cell division. Nonetheless, disrupting the asymmetric distribution of spindle pole–associated proteins promotes defective chromosome segregation, a hallmark of many cancers (26). Whereas previous work from our group linked centrosomal Plk1 activity in driving the localization of γ-tubulin, data in Fig. 1 and Fig. 3 provide the first evidence for a role of an AKAP in this process (27). More importantly, the present study elucidates a new role for the multifunctional kinase scaffolding protein Gravin in the maintenance of proper γ-tubulin pole-to-pole asymmetry.
Plk1 is a promising target for therapeutic intervention in cancer (42–46). This enzyme catalyzes phosphorylation events necessary for the efficient and accurate progression of cells through mitosis (47–49). Although ATP-competitive kinase inhibitors such as BI2536 are putative anticancer compounds that have entered clinical trials, conventional approaches for delivering these drugs increase off-target effects and mask the contribution of Plk1 at specified subcellular locations (42, 50, 51). With this in mind, we reasoned that manipulating molecular scaffolds such as Gravin could offer new insight into how locally constrained signaling enzymes are regulated. Moreover, pharmacological strategies that inhibit kinase activity within a specified locale permit the investigation of individual signaling events. Here, we demonstrate an application of the new precision pharmacology tool, LoKI, by targeting kinase inhibitor drugs to the subcellular location where Gravin anchors Plk1 (27, 52). The rationale for utilizing LoKI targeting was provided by previous reports that have implicated a role for Plk1 in organizing γ-tubulin at mitotic centrosomes (27, 31, 32, 53). A key advance, illustrated in Fig. 4, is that suppression of Plk1 activity at mitotic spindle poles enhances the asymmetric distribution of γ-tubulin. By taking advantage of the spatial resolution afforded by the LoKI system, we discovered that selective loss of Plk1 action at spindle poles perturbs the distribution of γ-tubulin. In addition, the data in Fig. 4, E–H uncover a link between loss of Plk1 activity at the spindle poles and an enhanced asymmetric distribution of γ-tubulin. On the basis of these results, it is possible that dampened Plk1 activity at mitotic spindle poles underlies the γ-tubulin accumulation defects observed in cells lacking Gravin. Alternatively, Gravin may facilitate phosphorylation of yet-unknown mitotic substrates that regulate γ-tubulin targeting. Irrespective of either mechanism, our findings advance the concept that Gravin-anchored kinase activity is necessary to optimally regulate the distribution of a central γ-TURC component during mitosis.
AKAPs constrain enzymes within spatially restricted signaling islands to restrict kinases and phosphatases to the immediate vicinity of select substrates (54–56). Our previous reports showed that Gravin organizes Plk1 and Aurora A kinases at spindle poles to facilitate proper spatial relay of signaling during mitosis (12, 27). A new concept emerging from this study is that cells without Gravin have lost the necessary protein-protein interactions to drive robust localization of γ-tubulin at spindle poles. This is supported by data in Fig. 5, D–E showing that γ-tubulin interaction with Nedd1, a Plk1 substrate, is disrupted in Gravin null cells. On the basis of these findings, we conclude that inappropriate targeting of Plk1 activity in Gravin-ablated cells may contribute to the loss of γ-tubulin/Nedd1 interactions. Moreover, our data are in line with a recent study that discovered that increased Plk1 mobility in Gravin-depleted cells results in aberrant phosphorylation of CEP215, a pericentriolar material component that regulates the assembly of the γ-TURC (26). In addition, these authors showed that the pole-to-pole asymmetry of this Plk1 substrate is impacted by Gravin loss (26). Because Plk1 promotes the interaction of γ-tubulin with Nedd1, it is possible that loss of this interaction in Gravin null cells drives the disruptions that we observed in γ-tubulin accumulation (34). Moreover, these interactions may be necessary for maintaining the symmetry and composition of the centrosomes. Thus, our findings in Fig. 5 offer additional mechanistic insight into how Gravin organizes signaling complexes at specific subcellular locations to ensure that kinases are positioned in close proximity to their targets.
A recognized facet of AKAPs is their ability to work within spatially restricted microenvironments to direct and insulate kinase action within a few Ångstroms of their intended targets (54, 57). Kinase anchoring is particularly effective for the modulation of a highly coordinated process such as the cell cycle (58). Thus, it is perhaps not surprising that subtle perturbations in the organization of key macromolecular complexes are responsible for certain defective signaling events that are observed in a range of cancers (6). This is evidenced by studies that have identified the AKAP12 gene, which encodes Gravin, in a deletion hotspot for a variety of human cancers (59). However, it is equally worth noting that elevated levels of Gravin in ovarian cancer have been correlated with poor prognosis (60). Thus, more studies are necessary to clarify the role of Gravin in these different pathological contexts. Another theme emerging from our work is that deploying tools that direct kinase inhibitors to specific subcellular locations provide clues into the contributions of local signaling events. This study provides a practical application of the LoKI system and demonstrates that precision pharmacology can be utilized to decipher how anchored Plk1 activity at the spindle poles drives the asymmetric distribution of γ-tubulin. Together, our findings lead us to speculate that perturbation of Gravin signaling during mitosis may underlie disease progression. However, future work is necessary to uncover additional Gravin binding partners and downstream substrates that become dysregulated when this anchoring protein is ablated in disease contexts.
Experimental procedures
Antibodies and reagents
Antibodies and reagents were obtained from the following sources: α-tubulin, clone DM1A (Sigma-Aldrich, T9026, mouse monoclonal; RRID:AB_477593); α-tubulin-FITC, clone DM1A (Sigma-Aldrich, F2168, mouse monoclonal; RRID:AB_476967); anti-Centrin, clone 20H5 (Millipore, 04-1624, mouse monoclonal, RRID:AB_10563501); FLAG M2 (Sigma-Aldrich, F3165, mouse monoclonal, RRID:AB_259529); FLAG-HRP (Sigma-Aldrich, A8592, mouse monoclonal, RRID:AB_439702); γ-tubulin (Abcam, 11317, rabbit polyclonal, RRID:AB_297921); GAPDH-HRP (Novus, NB110-40405, mouse monoclonal, RRID:AB_669249); Gravin, clone JP74 (Sigma-Aldrich, G3795, mouse monoclonal); Gravin, clone R3698 (in-house (12, 35, 36), rabbit polyclonal); Nedd1, clone H-3 (Santa Cruz Biotechnology, sc-398733, mouse monoclonal); Pericentrin (Abcam, ab4448, rabbit polyclonal; RRID:AB_304461); phospho-Plk1 Thr-210 (Biolegend, 628901, rabbit polyclonal, RRID:AB_439786); phospho-Plk1 Thr-210 clone 2A3 (Abcam, ab39068, mouse monoclonal, RRID:AB_10861033); Plk1, clone 35-206 (Millipore, 05-844, mouse monoclonal; RRID:AB_11213632); Plk1, clone F-8 (Santa Cruz Biotechnology, sc-17783, mouse monoclonal; RRID:AB_628157); SSeCKS (BD Transduction Laboratory, S63820, mouse monoclonal); donkey anti-mouse IgG, Alexa Fluor 488 (Invitrogen, A-21202); donkey anti-rabbit IgG, Alexa Fluor 647 (Invitrogen, A-31573). Amersham Biosciences ECL mouse IgG, HRP-linked F(ab′)2 fragment from sheep (GE Life Sciences, NA9310); Amersham Biosciences ECL rabbit IgG, HRP-linked F(ab′)2 fragment from donkey (GE Life Sciences, NA9340).
Plasmid constructs
Constructs for the generation of LoKI cells were generated as described in (27). In brief, LoKI constructs contain an N-terminal mCherry reporter protein, two soluble NSF attachment protein–tag domains, and a PACT (Pericentrin AKAP450 centrosomal targeting) sequence. The second soluble NSF attachment protein–tag has a varied codon sequence to simplify PCR amplification. Individual components were PCR amplified with overlapping ends and/or Gateway att sites and assembled using Gibson cloning. Constructs were subcloned into pLIX402 (a gift from David Root; Addgene plasmid 41394) using Gateway cloning. Mutant LoKI vectors were generated by performing site-directed mutagenesis with a QuikChange II XL kit (Aligent). Constructs for CRISPR/Cas9-mediated genome editing experiments expressing Cas9 and gRNAs were generated by cloning into pSpCas9(BB)-2A-Puro (PX459) vectors (Addgene plasmid 48139). Constructs were verified by Sanger sequencing.
Cell culture
U2OS cells used to generate stable cell lines (27) were purchased from ATCC and tested negative for mycoplasma contamination as assessed by the Universal Mycoplasma Detection Kit (ATCC, 30-1012K). U2OS, control and Gravin shRNA HeLa (12), control and Gravin shRNA HEK 293 (11), and immortalized MEF (generated as described in (12)) cells were maintained in DMEM, high glucose (Life Technologies) at 37 °C and 5% CO2. All media was supplemented with 10% FBS (Thermo Fisher Scientific). Infections for generation of stable knockdown in HeLa and HEK 293 cells were performed with shRNA lentiviral particles (Santa Cruz Biotechnology). Infections for generation of stable LoKI cells were performed using lentiviral particles created in-house (as described in (27)). Transient gene expression of FLAG-SSeCKS, a murine Gravin rescue construct, was performed by transfection using TransIT-LT1 reagent (Mirus).
CRISPR/Cas9 editing of Gravin
CRISPR/Cas9-mediated genome editing was used to delete the AKAP12 gene on chromosome 6 in U2OS cells. Two unique gRNAs were designed to the following targets: gRNA1, AGAGATGGCTACTAAGTCAGCGG; and gRNA 2, AGCCGAATCTGGCCAAGCAGTGG. Bold letters indicate a peptidylglycine α-amidating monooxygenase sequence. Constructs expressing Cas9 and either gRNA were generated by cloning into pSpCas9(BB)-2A-Puro (PX459) vectors (Addgene plasmid 48139). Individual vectors were transfected into U2OS cells using TransIT-LT1 reagent (Mirus) in Opti-MEM® (Life Technologies) media according to the manufacturer's instructions. Single clones were isolated using Scienceware cloning discs (Sigma-Aldrich). Clones were first screened by immunoblotting for loss of Gravin protein expression using two independent antibodies. Clones that had undetectable levels of Gravin were further checked for mutations. In brief, cells were pelleted and purification of genomic DNA was achieved by treating with 50 mm NaOH for 20 min at 95 °C followed by treatment with 1m Tris pH 8.0 and a 10 min spin at 14,000 rpm. Primers flanking the target sites were designed and the region was amplified by PCR: gRNA1-forward, TTGGACAGAGAGACTCTGAAGATGTG; gRNA1-reverse, CTTCATCTTTCTTCACAGTGAGTAGC; and gRNA2-forward, ACTGTGAAGAAAGATGAAGGGG. PCR products were cloned into vectors using the Zero BluntTM TOPOTM PCR Cloning kit (Thermo Fisher). Individual clones were isolated for plasmid DNA mini-prep and verified by Sanger sequencing.
Drug treatments
RO3306 (Sigma-Aldrich) was used to enrich mitotic cells in Gravin rescue experiments. At least 24 h post transfection, cells were treated with 10 μm RO3306 for 24 h and then incubated in complete DMEM for 25 min prior to fixation. BI2536 (AdooQ BioScience) and CLP-BI2536 (27) were used to inhibit Plk1. For LoKI experiments, cells were treated as described in (27). In brief, prior to drug treatment, cells were incubated with 1 μg/ml doxycycline hyclate (Sigma-Aldrich) in FBS-supplemented DMEM for 48–72 h to induce expression of LoKI targeting platforms. The cells were grown on 1.5 poly-d-lysine–coated coverslips (Neuvitro) for ∼16 h in complete DMEM and then treated with DMSO (Pierce) or CLP-BI2536 in serum-free DMEM for 4 h. For washout experiments (γ-tubulin data), the cells were incubated in serum-free DMEM without inhibitors for an additional 1 h. The cells were washed once with PBS prior to fixation.
Immunoblotting
The cells were lysed in radioimmune precipitation assay buffer (50 mm Tris HCl, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm NaF, 120 mm NaCl, and 5 mm β-glycerophosphate) with protease and phosphatase inhibitors (1 mm benzamidine, 1 mm AEBSF, 2 μg/ml leupeptin, and 100 nm microcystin-LR). Samples were boiled for 5 min at 95 °C in NuPAGETM LDS Sample Buffer 4× (Thermo Fisher Scientific) and 5% Eagle's Basal Medium (Sigma-Aldrich), and protein concentration was determined using a PierceTM BCA Protein Assay kit (Thermo Fisher Scientific). The samples were resolved on Bolt® 4–12% Bis-Tris Plus Gels (Invitrogen), the proteins were transferred to nitrocellulose for immunoblotting, and the membranes were probed with primary antibodies. Detection was achieved with an HRP-conjugated rabbit or mouse secondary antibody (GE Healthcare) followed by enhanced chemiluminescence with SuperSignalTM West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). Representative blots were adjusted for brightness and contrast in Fiji, and densitometry measurements were used to generate graphs.
Immunofluorescence
The cells grown on 1.5 poly-d-lysine–coated coverslips (Neuvitro) for ∼16 h were fixed in ice-cold methanol or in 4% paraformaldehyde in PBS for 10 min. The cells were permeabilized and blocked in PBS with 0.5% Triton X-100 and 1% BSA (PBSAT) for 30 min. Primary antibodies and secondary antibodies, conjugated to Alexa Fluor dyes (Invitrogen), were diluted in PBSAT, and the cells were stained for 1 h in each. Counterstaining with FITC-tubulin antibodies and/or DAPI (Thermo Fisher Scientific) was carried out for 10–45 min in PBSAT. Washes with PBSAT were carried out in one of two ways (three times for 5 min or 10 times, quick on and off) between antibody and/or dye incubation steps and prior to mounting. Coverslips were mounted on slides using ProLong® Diamond Antifade Mountant (Life Technologies, P36961).
Proximity ligation assay
The cells were methanol-fixed, permeabilized, blocked, and stained with primary antibodies as described under “Immunofluorescence.” The cells were incubated with anti-rabbit and anti-mouse probes (Duolink) and PLA was carried out according to manufacturer's instructions. Where applicable, the cells were counterstained as described above.
Microscopy
Widefield and super-resolution 3D-SIM images were acquired on a Deltavision OMX V4 (GE Healthcare) system equipped with a 60× 1.42 NA PlanApo oil immersion lens (Olympus), 405-, 488-, 568-, and 642-nm solid-state lasers, and sCMOS cameras (pco.edge). For SIM, 15 images per optical slice (three angles and five phases) were acquired. Image stacks of 4–7 μm with 0.200- (widefield) or 0.125-μm (SIM) optical thick z-sections were acquired using immersion oil with a refractive index of 1.516 or 1.518. Z-stacks were generated using the DAPI or α-tubulin channels to define the upper and lower boundaries of the plane with a 0.5–μm step size. SIM images were reconstructed using a Wiener filter setting of 0.003, and optical transfer functions measured specifically for each channel with SoftWoRx software (GE Healthcare) were used to obtain super-resolution images with a 2-fold increase in resolution both axially and laterally. Images from different color channels were registered using parameters generated from a gold grid registration slide (GE Healthcare) and SoftWoRx. Widefield images were deconvolved using SoftWoRx. For micronuclei counts and acquisition of differential interference contrast images for PLA assays, a DM16000B inverted microscope (Leica) equipped with a 63× Plan-Apocromat NA 1.4 oil objective, a CSU10 confocal spinning disk (Yokogawa), and a CoolSNAP HQ camera (Photometrics) controlled by MetaMorph 7.6.4 (Molecular Devices) was used. Representative images were adjusted for brightness and contrast in Fiji.
Image analysis
SoftWoRx (GE Healthcare) or NIH ImageJ (Fiji) software was used to generate maximum intensity projections from z-stack images. Immunofluorescence signals were measured using Fiji software. For analysis of immunofluorescence at spindle poles, sum slice 32-bit Tiff projections were generated from z-stack images. The oval selection tool in Fiji was used to draw a circle (region-of-interest) around the spindle pole and measure the signal in the 647 (γ-tubulin) channel. The area of the circle remained consistent for all experimental replicates. Using the measure function in Fiji, with “Area” and “Raw Integrated Density” predefined as measurements, values were recorded for each spindle pole and for a nearby background region.
Total signal at spindle poles: The average raw integrated density for the spindle poles was determined by adding together the raw integrated densities for each pole in a cell and dividing that value by two. The integrated density of the background was subtracted from the average integrated density of the spindle poles to yield a background-subtracted average integrated density signal. In the case of a negative value (when the background signal is higher than that at the poles), a value of zero was reported. An average spindle pole signal was calculated for each control and experimental condition. To do this, normalized average integrated densities were added together and divided by the total number of cells for a given condition. This resulted in a value representing the background-normalized average integrated density at the spindle poles. Values for Gravin null and drug-treated cells were normalized to WT or DMSO-treated controls, respectively.
Signal at individual spindle poles: The individual raw integrated densities for each spindle pole in a given cell were classified into two categories: pole 1 (the spindle pole that contains the highest fluorescence signal) and pole 2 (the pole with the lowest fluorescence signal). The background signal values were subtracted from each individual pole. As before, negative values were replaced with a zero. Values for the low pole were normalized to average high-pole values of each corresponding condition.
Lowest/highest pole ratios: Background-subtracted integrated intensity values were used to determine the total γ-tubulin signal at each pole. The immunofluorescence signal of the pole with the lowest intensity was divided by the signal of the pole with the highest intensity to determine the ratio of γ-tubulin between the poles. In cases where the value for the ratio was zero, the value was removed and not included in statistical analysis. This was to avoid misinterpretation of the data because a value of zero at both poles and a high value at pole 1 but a zero value at pole 2 would both yield the same ratio.
Micronuclei analysis: For each experimental replicate, 500 interphase cells were examined using the DAPI channel. The number of cells with micronuclei was divided by 500 to determine the percent of micronucleated cells per experiment.
PLA analysis: The number of PLA puncta per cell was quantified using the cell counter tool in Fiji.
Surface and line plots: Plots were generated in Fiji software from maximum intensity projections of representative images using the 3D Surface Plot function or the Plot Profile function. The ImageJ “fire” lookup table setting was used to generate pseudo-color images.
Statistical analysis
Statistics were performed using an unpaired two-tailed Student's t test or a one-way analysis of variance in Prism 8 software (GraphPad). All values are reported as mean ± S.E. with p-values less than 0.05 considered statistically significant. Number of independent experiments (N) and number of individual points over several experiments (n) are presented. The sample size was not statistically determined. Where applicable, n > 20 independent measurements were conducted across N ≥ 3 independent experiments. For micronuclei experiments in Gravin KO 2 (Fig. S2C), at least 1000 cells per condition over two independent experiments were assayed. Detailed analyses are presented in Table 1.
Table 1.
Statistical analyses table
| Fig. | Condition | Sample size (n) | Test | Statistics | p-value | Summary |
|---|---|---|---|---|---|---|
| 1H | WT | 49 | Unpaired two-tailed Student's t test | t = 5.855, df = 101 | <0.0001 | **** |
| Gravin −/− | 54 | |||||
| S1C | Control shRNA | 46 | Unpaired two-tailed Student's t test | t = 3.901, df = 90 | 0.0002 | *** |
| Gravin shRNA | 46 | |||||
| S1E | Control shRNA | 45 | Unpaired two-tailed Student's t test | t = 6.360, df = 86 | <0.0001 | **** |
| Gravin shRNA | 43 | |||||
| S1E | Gravin shRNA | 43 | Unpaired two-tailed Student's t test | t = 7.300, df = 86 | <0.0001 | **** |
| Gravin shRNA + rescue | 45 | |||||
| S1E | Control shRNA | 45 | Unpaired two-tailed Student's t test | t = 3.086, df = 88 | 0.0027 | ** |
| Gravin shRNA + rescue | 45 | |||||
| 2G | WT | 3 | Unpaired two-tailed Student's t test | t = 8.527, df = 4 | 0.0010 | ** |
| Gravin KO | 3 | |||||
| S2B | Control shRNA | 3 | Unpaired two-tailed Student's t test | t = 5.340, df = 4 | 0.0059 | ** |
| Gravin shRNA | 3 | |||||
| S2C | WT | 2 | Unpaired two-tailed Student's t test | t = 26.90, df = 2 | 0.0014 | ** |
| Gravin KO | 2 | |||||
| S2F | WT | 42 | Unpaired two-tailed Student's t test | t = 5.391, df = 79 | <0.0001 | **** |
| Gravin KO | 39 | |||||
| S2G | WT | 42 | Unpaired two-tailed Student's t test | t = 5.595, df = 79 | <0.0001 | **** |
| Gravin KO | 39 | |||||
| 3D | Pole 1 | 64 | Unpaired two-tailed Student's t test | t = 3.794, df = 126 | 0.0002 | *** |
| Pole2 | 64 | |||||
| 3E | Pole 1 | 23 | Unpaired two-tailed Student's t test | t = 2.454, df = 44 | 0.0181 | * |
| Pole2 | 23 | |||||
| 3J | WT | 49 | Unpaired two-tailed Student's t test | t = 2.042, df = 100 | 0.0438 | * |
| −/− | 53 | |||||
| 3K | WT | 23 | Unpaired two-tailed Student's t test | t = 2.303, df = 42 | 0.0263 | * |
| KO | 21 | |||||
| 3M | WT | 38 | Unpaired two-tailed Student's t test | t = 0.2365, df = 71 | 0.8137 | NS |
| KO | 35 | |||||
| S3B | WT | 23 | Unpaired two-tailed Student's t test | t = 2.303, df = 42 | 0.0263 | * |
| KO | 21 | |||||
| S3B | WT | 23 | Unpaired two-tailed Student's t test | t = 2.452, df = 37 | 0.0190 | * |
| KO #2 | 16 | |||||
| S3B | KO | 21 | Unpaired two-tailed Student's t test | t = 0.2261, df = 35 | 0.8225 | NS |
| KO #2 | 16 | |||||
| S3C | WT | 38 | Unpaired two-tailed Student's t test | t = 1.550, df = 71 | 0.1256 | NS |
| KO | 35 | |||||
| S3D | WT | 49 | Unpaired two-tailed Student's t test | t = 0.4315, df = 101 | 0.6671 | NS |
| KO | 54 | |||||
| S3E | WT | 49 | Unpaired two-tailed Student's t test | t = 0.2692, df = 102 | 0.7883 | NS |
| KO | 55 | |||||
| S3G | WT | 2 | Unpaired two-tailed Student's t test | t = 0.6212, df = 2 | 0.5978 | NS |
| −/− | 2 | |||||
| S3I | WT | 2 | Unpaired two-tailed Student's t test | t = 0.8504, df = 2 | 0.4847 | NS |
| KO | 2 | |||||
| 4C | DMSO (pole 2) | 53 | one-way analysis of variance with Dunnett's multiple comparisons test | F (4, 224) = 11.47 | control | - |
| 5 nm (pole 2) | 44 | 0.1387 | NS | |||
| 10 nm (pole 2) | 44 | 0.7016 | NS | |||
| 15 nm (pole 2) | 44 | <0.0001 | **** | |||
| 20 nm (pole 2) | 44 | <0.0001 | **** | |||
| 4F | LoKI-off, pole 1 | 49 | Unpaired two-tailed Student's t test | t = 4.770, df = 96 | <0.0001 | **** |
| LoKI-off, pole 2 | 49 | |||||
| 4F | LoKI-on, pole 1 | 49 | Unpaired two-tailed Student's t test | t = 5.362, df = 96 | <0.0001 | **** |
| LoKI-on, pole 2 | 49 | |||||
| 4F | LoKI-off, pole 2 | 49 | Unpaired two-tailed Student's t test | t = 3.382, df = 96 | 0.0010 | ** |
| LoKI-on, pole 2 | 49 | |||||
| 4H | DMSO | 76 | Unpaired two-tailed Student's t test | t = 2.716, df = 148 | 0.0074 | ** |
| 250 nm | 74 | |||||
| S4B | DMSO, LoKI-off (pole 2) | 44 | Unpaired two-tailed Student's t test | t = 1.163, df = 102 | 0.2477 | NS |
| DMSO, LoKI-on (pole 2) | 60 | |||||
| S4B | 100 nm, LoKI-off (pole 2) | 46 | Unpaired two-tailed Student's t test | t = 0.09352, df = 103 | 0.9257 | NS |
| 100 nm, LoKI-on (pole 2) | 59 | |||||
| S4B | 500 nm, LoKI-off (pole 2) | 53 | Unpaired two-tailed Student's t test | t = 2.612, df = 111 | 0.0102 | * |
| 500 nm, LoKI-on (pole 2) | 60 | |||||
| S4C | DMSO, LoKI-off (pole 2) | 52 | Unpaired two-tailed Student's t test | t = 0.6178, df = 109 | 0.5380 | NS |
| DMSO, LoKI-on (pole 2) | 59 | |||||
| S4C | 100 nm, LoKI-off (pole 2) | 52 | Unpaired two-tailed Student's t test | t = 0.1043, df = 108 | 0.9172 | NS |
| 100 nm, LoKI-on (pole 2) | 58 | |||||
| S4C | 250 nm, LoKI-off (pole 2) | 55 | Unpaired two-tailed Student's t test | t = 0.7968, df = 100 | 0.4275 | NS |
| 250 nm, LoKI-on (pole 2) | 47 | |||||
| S4C | 500 nm, LoKI-off (pole 2) | 52 | Unpaired two-tailed Student's t test | t = 0.2530, df = 102 | 0.8007 | NS |
| 500 nm, LoKI-on (pole 2) | 52 | |||||
| S4G | DMSO | 70 | Unpaired two-tailed Student's t test | t = 2.106, df = 135 | 0.0370 | * |
| 250 nm | 67 | |||||
| S4H | DMSO | 76 | Unpaired two-tailed Student's t test | t = 1.737, df = 146 | 0.0846 | NS |
| 250 nm | 72 | |||||
| 5C | WT | 40 | Unpaired two-tailed Student's t test | t = 2.140, df = 72 | 0.0357 | * |
| Gravin −/− | 34 | |||||
| 5E | WT | 38 | Unpaired two-tailed Student's t test | t = 2.538, df = 78 | 0.0131 | * |
| Gravin −/− | 42 |
Data availability
All raw image data can be provided upon request.
Supplementary Material
Acknowledgments
We thank members of the Scott Lab for critical discussions and feedback on the manuscript, Patrina Pellett (GE Healthcare) for technical help with super-resolution imaging techniques, and Juan-Jesus Vicente (Wordeman Laboratory, University of Washington) for help with experimental design, data analysis, and thoughtful discussion.
This article contains supporting information.
Author contributions—P. J. B. and J. D. S. conceptualization; P. J. B., I. G., R. M., and A. B. data curation; P. J. B., I. G., R. M., and A. B. formal analysis; P. J. B. and J. D. S. supervision; P. J. B., I. G., and R. M. validation; P. J. B., I. G., R. M., and A. B. investigation; P. J. B. and I. G. visualization; P. J. B. and L. W. methodology; P. J. B. and J. D. S. writing-original draft; P. J. B. and J. D. S. project administration; P. J. B., I. G., L. W., and J. D. S. writing-review and editing; L. W. resources; J. D. S. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants DK119186 and DK119192 to J.D.S. Support also from the Fibrolamellar Carcinoma Foundation (FCF). This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Plk1
- polo-like kinase 1
- LoKI
- Local Kinase Inhibition
- AKAP
- protein kinase A anchoring protein
- Nedd
- neural precursor cell expressed developmentally down-regulated protein
- MEF
- mouse embryonic fibroblast
- gRNA
- guide RNA
- pT210
- phospho-Thr-210
- SIM
- structured illumination microscopy
- CLP
- chloropyrimidine
- γ-TURC
- γ-tubulin ring complex
- AEBSF
- 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride
- LDS
- lithium dodecyl sulfate
- PBSAT
- PBS with 0.5% Triton X-100 and 1% BSA
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Scott J. D., and Pawson T. (2009) Cell signaling in space and time: where proteins come together and when they're apart. Science 326, 1220–1224 10.1126/science.1175668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lemmon M. A., Freed D. M., Schlessinger J., and Kiyatkin A. (2016) The dark side of cell signaling: positive roles for negative regulators. Cell 164, 1172–1184 10.1016/j.cell.2016.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Langeberg L. K., and Scott J. D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 10.1038/nrm3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gabrovsek L., Bucko P., Carnegie G. K., and Scott J. D. (2017) A-kinase anchoring protein (AKAP). in Encyclopedia of Signaling Molecules (Choi S. ed.), pp. 1–6, Springer, New York [Google Scholar]
- 5. Taskén K., and Aandahl E. M. (2004) Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84, 137–167 10.1152/physrev.00021.2003 [DOI] [PubMed] [Google Scholar]
- 6. Bucko P. J., and Scott J. D. (2021) Drugs that regulate local cell signaling: AKAP targeting as a therapeutic option. Annu. Rev. Pharmacol. Toxicol. 61, 10.1146/annurev-pharmtox-022420-112134 [DOI] [PubMed] [Google Scholar]
- 7. Esseltine J. L., and Scott J. D. (2013) AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 34, 648–655 10.1016/j.tips.2013.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diviani D., Langeberg L. K., Doxsey S. J., and Scott J. D. (2000) Pericentrin anchors protein kinase A at the centrosome through a newly identified RII-binding domain. Curr. Biol. 10, 417–420 10.1016/S0960-9822(00)00422-X [DOI] [PubMed] [Google Scholar]
- 9. Chen D., Purohit A., Halilovic E., Doxsey S. J., and Newton A. C. (2004) Centrosomal anchoring of protein kinase C βII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J. Biol. Chem. 279, 4829–4839 10.1074/jbc.M311196200 [DOI] [PubMed] [Google Scholar]
- 10. Witczak O., Skålhegg B. S., Keryer G., Bornens M., Taskén K., Jahnsen T., and Orstavik S. (1999) Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. EMBO J. 18, 1858–1868 10.1093/emboj/18.7.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Canton D. A., Keene C. D., Swinney K., Langeberg L. K., Nguyen V., Pelletier L., Pawson T., Wordeman L., Stella N., and Scott J. D. (2012) Gravin is a transitory effector of polo-like kinase 1 during cell division. Mol. Cell 48, 547–559 10.1016/j.molcel.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hehnly H., Canton D., Bucko P., Langeberg L. K., Ogier L., Gelman I., Santana L. F., Wordeman L., and Scott J. D. (2015) A mitotic kinase scaffold depleted in testicular seminomas impacts spindle orientation in germ line stem cells. eLife 4, e09384 10.7554/eLife.09384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alvarado-Kristensson M., Rodríguez M. J., Silió V., Valpuesta J. M., and Carrera A. C. (2009) SADB phosphorylation of γ-tubulin regulates centrosome duplication. Nat. Cell Biol. 11, 1081–1092 10.1038/ncb1921 [DOI] [PubMed] [Google Scholar]
- 14. Hendrickson T. W., Yao J., Bhadury S., Corbett A. H., and Joshi H. C. (2001) Conditional mutations in γ-tubulin reveal its involvement in chromosome segregation and cytokinesis. Mol. Biol. Cell 12, 2469–2481 10.1091/mbc.12.8.2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Müller H., Fogeron M. L., Lehmann V., Lehrach H., and Lange B. M. (2006) A centrosome-independent role for γ-TuRC proteins in the spindle assembly checkpoint. Science 314, 654–657 10.1126/science.1132834 [DOI] [PubMed] [Google Scholar]
- 16. Moritz M., Braunfeld M. B., Sedat J. W., Alberts B., and Agard D. A. (1995) Microtubule nucleation by γ-tubulin-containing rings in the centrosome. Nature 378, 638–640 10.1038/378638a0 [DOI] [PubMed] [Google Scholar]
- 17. Zheng Y., Wong M. L., Alberts B., and Mitchison T. (1995) Nucleation of microtubule assembly by a γ-tubulin-containing ring complex. Nature 378, 578–583 10.1038/378578a0 [DOI] [PubMed] [Google Scholar]
- 18. Katsetos C. D., Reddy G., Dráberová E., Smejkalová B., Del Valle L., Ashraf Q., Tadevosyan A., Yelin K., Maraziotis T., Mishra O. P., Mörk S., Legido A., Nissanov J., Baas P. W., de Chadarévian J. P., and Dráber P. (2006) Altered cellular distribution and subcellular sorting of γ-tubulin in diffuse astrocytic gliomas and human glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 65, 465–477 10.1097/01.jnen.0000229235.20995.6e [DOI] [PubMed] [Google Scholar]
- 19. Caracciolo V., D'Agostino L., Dráberová E., Sládková V., Crozier-Fitzgerald C., Agamanolis D. P., de Chadarévian J. P., Legido A., Giordano A., Dráber P., and Katsetos C. D. (2010) Differential expression and cellular distribution of γ-tubulin and βIII-tubulin in medulloblastomas and human medulloblastoma cell lines. J. Cell. Physiol. 223, 519–529 10.1002/jcp.22077 [DOI] [PubMed] [Google Scholar]
- 20. Cho E. H., Whipple R. A., Matrone M. A., Balzer E. M., and Martin S. S. (2010) Delocalization of γ-tubulin due to increased solubility in human breast cancer cell lines. Cancer Biol. Ther. 9, 66–76 10.4161/cbt.9.1.10451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niu Y., Liu T., Tse G. M., Sun B., Niu R., Li H. M., Wang H., Yang Y., Ye X., Wang Y., Yu Q., and Zhang F. (2009) Increased expression of centrosomal α, γ-tubulin in atypical ductal hyperplasia and carcinoma of the breast. Cancer Sci. 100, 580–587 10.1111/j.1349-7006.2008.01075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maounis N. F., Draberova E., Mahera E., Chorti M., Caracciolo V., Sulimenko T., Riga D., Trakas N., Emmanouilidou A., Giordano A., Draber P., and Katsetos C. D. (2012) Overexpression of γ-tubulin in non-small cell lung cancer. Histol. Histopathol. 27, 1183–1194 [DOI] [PubMed] [Google Scholar]
- 23. Friesen D. E., Barakat K. H., Semenchenko V., Perez-Pineiro R., Fenske B. W., Mane J., Wishart D. S., and Tuszynski J. A. (2012) Discovery of small molecule inhibitors that interact with γ-tubulin. Chem. Biol. Drug Des. 79, 639–652 10.1111/j.1747-0285.2012.01340.x [DOI] [PubMed] [Google Scholar]
- 24. Chinen T., Liu P., Shioda S., Pagel J., Cerikan B., Lin T. C., Gruss O., Hayashi Y., Takeno H., Shima T., Okada Y., Hayakawa I., Hayashi Y., Kigoshi H., Usui T., et al. (2015) The γ-tubulin-specific inhibitor gatastatin reveals temporal requirements of microtubule nucleation during the cell cycle. Nat. Commun. 6, 8722 10.1038/ncomms9722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Canton D. A., and Scott J. D. (2013) Anchoring proteins encounter mitotic kinases. Cell Cycle 12, 863–864 10.4161/cc.24192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colicino E. G., Garrastegui A. M., Freshour J., Santra P., Post D. E., Kotula L., and Hehnly H. (2018) Gravin regulates centrosome function through PLK1. Mol. Biol. Cell 29, 532–541 10.1091/mbc.E17-08-0524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bucko P. J., Lombard C. K., Rathbun L., Garcia I., Bhat A., Wordeman L., Smith F. D., Maly D. J., Hehnly H., and Scott J. D. (2019) Subcellular drug targeting illuminates local kinase action. eLife 8, e52220 10.7554/eLife.52220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmerman W. C., Sillibourne J., Rosa J., and Doxsey S. J. (2004) Mitosis-specific anchoring of γ tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol. Biol. Cell 15, 3642–3657 10.1091/mbc.e03-11-0796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leibowitz M. L., Zhang C. Z., and Pellman D. (2015) Chromothripsis: a new mechanism for rapid karyotype evolution. Annu. Rev. Genet. 49, 183–211 10.1146/annurev-genet-120213-092228 [DOI] [PubMed] [Google Scholar]
- 30. Haren L., Stearns T., and Lüders J. (2009) Plk1-dependent recruitment of γ-tubulin complexes to mitotic centrosomes involves multiple PCM components. PLoS ONE 4, e5976 10.1371/journal.pone.0005976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu D., and Dai W. (2011) The function of mammalian Polo-like kinase 1 in microtubule nucleation. Proc. Natl. Acad. Sci. U.S.A. 108, 11301–11302 10.1073/pnas.1107723108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haren L., Remy M. H., Bazin I., Callebaut I., Wright M., and Merdes A. (2006) NEDD1-dependent recruitment of the γ-tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J. Cell Biol. 172, 505–515 10.1083/jcb.200510028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manning J. A., Shalini S., Risk J. M., Day C. L., and Kumar S. (2010) A direct interaction with NEDD1 regulates γ-tubulin recruitment to the centrosome. PLoS ONE 5, e9618 10.1371/journal.pone.0009618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X., Chen Q., Feng J., Hou J., Yang F., Liu J., Jiang Q., and Zhang C. (2009) Sequential phosphorylation of Nedd1 by Cdk1 and Plk1 is required for targeting of the γTuRC to the centrosome. J. Cell Sci. 122, 2240–2251 10.1242/jcs.042747 [DOI] [PubMed] [Google Scholar]
- 35. Nauert J. B., Klauck T. M., Langeberg L. K., and Scott J. D. (1997) Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr. Biol. 7, 52–62 10.1016/S0960-9822(06)00027-3 [DOI] [PubMed] [Google Scholar]
- 36. Gelman I. H. (2010) Emerging roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barriergenesis. Genes Cancer 1, 1147–1156 10.1177/1947601910392984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parada C. A., Osbun J., Kaur S., Yakkioui Y., Shi M., Pan C., Busald T., Karasozen Y., Gonzalez-Cuyar L. F., Rostomily R., Zhang J., and Ferreira M. Jr (2018) Kinome and phosphoproteome of high-grade meningiomas reveal AKAP12 as a central regulator of aggressiveness and its possible role in progression. Sci. Rep. 8, 2098 10.1038/s41598-018-19308-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lattao R., Kovács L., and Glover D. M. (2017) The centrioles, centrosomes, basal bodies, and cilia of Drosophila melanogaster. Genetics 206, 33–53 10.1534/genetics.116.198168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamashita Y. M., Mahowald A. P., Perlin J. R., and Fuller M. T. (2007) Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 315, 518–521 10.1126/science.1134910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piel M., Meyer P., Khodjakov A., Rieder C. L., and Bornens M. (2000) The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J. Cell Biol. 149, 317–330 10.1083/jcb.149.2.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Siller K. H., and Doe C. Q. (2009) Spindle orientation during asymmetric cell division. Nat. Cell Biol. 11, 365–374 10.1038/ncb0409-365 [DOI] [PubMed] [Google Scholar]
- 42. Steegmaier M., Hoffmann M., Baum A., Lénárt P., Petronczki M., Krssák M., Gürtler U., Garin-Chesa P., Lieb S., Quant J., Grauert M., Adolf G. R., Kraut N., Peters J. M., and Rettig W. J. (2007) BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 17, 316–322 10.1016/j.cub.2006.12.037 [DOI] [PubMed] [Google Scholar]
- 43. Choi M., Kim W., Cheon M. G., Lee C. W., and Kim J. E. (2015) Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe following activation of the spindle assembly checkpoint in non-small cell lung cancer cells. Cancer Lett. 357, 591–601 10.1016/j.canlet.2014.12.023 [DOI] [PubMed] [Google Scholar]
- 44. Lee K. S., Burke T. R. Jr, Park J. E., Bang J. K., and Lee E. (2015) Recent advances and new strategies in targeting Plk1 for anticancer therapy. Trends Pharmacol. Sci. 36, 858–877 10.1016/j.tips.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gutteridge R. E., Ndiaye M. A., Liu X., and Ahmad N. (2016) Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol. Cancer Ther. 15, 1427–1435 10.1158/1535-7163.MCT-15-0897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng C. Y., Liu C. J., Huang Y. C., Wu S. H., Fang H. W., and Chen Y. J. (2018) BI2536 induces mitotic catastrophe and radiosensitization in human oral cancer cells. Oncotarget 9, 21231–21243 10.18632/oncotarget.25035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barr F. A., Silljé H. H., and Nigg E. A. (2004) Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–440 10.1038/nrm1401 [DOI] [PubMed] [Google Scholar]
- 48. Lens S. M., Voest E. E., and Medema R. H. (2010) Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev. Cancer 10, 825–841 10.1038/nrc2964 [DOI] [PubMed] [Google Scholar]
- 49. Combes G., Alharbi I., Braga L. G., and Elowe S. (2017) Playing polo during mitosis: PLK1 takes the lead. Oncogene 36, 4819–4827 10.1038/onc.2017.113 [DOI] [PubMed] [Google Scholar]
- 50. Lénárt P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J. J., Hoffmann M., Rettig W. J., Kraut N., and Peters J. M. (2007) The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315 10.1016/j.cub.2006.12.046 [DOI] [PubMed] [Google Scholar]
- 51. Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P. A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J., Reiter K., Qiao H., Helm D., Koch H., et al. (2017) The target landscape of clinical kinase drugs. Science 358, eaan4368 10.1126/science.aan4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kawashima A. T., and Newton A. C. (2020) Pharmacology on target. Trends Pharmacol. Sci. 41, 227–230 10.1016/j.tips.2020.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lane H. A., and Nigg E. A. (1996) Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol. 135, 1701–1713 10.1083/jcb.135.6.1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith F. D., Esseltine J. L., Nygren P. J., Veesler D., Byrne D. P., Vonderach M., Strashnov I., Eyers C. E., Eyers P. A., Langeberg L. K., and Scott J. D. (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293 10.1126/science.aaj1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Smith F. D., Reichow S. L., Esseltine J. L., Shi D., Langeberg L. K., Scott J. D., and Gonen T. (2013) Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. eLife 2, e01319 10.7554/eLife.01319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nygren P. J., Mehta S., Schweppe D. K., Langeberg L. K., Whiting J. L., Weisbrod C. R., Bruce J. E., Zhang J., Veesler D., and Scott J. D. (2017) Intrinsic disorder within AKAP79 fine-tunes anchored phosphatase activity toward substrates and drug sensitivity. eLife 6, e30872 10.7554/eLife.30872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith F. D., Omar M. H., Nygren P. J., Soughayer J., Hoshi N., Lau H. T., Snyder C. G., Branon T. C., Ghosh D., Langeberg L. K., Ting A. Y., Santana L. F., Ong S. E., Navedo M. F., and Scott J. D. (2018) Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. U.S.A 115, E11465–E11474 10.1073/pnas.1816614115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fulcher L. J., and Sapkota G. P. (2020) Mitotic kinase anchoring proteins: the navigators of cell division. Cell Cycle 19, 505–524 10.1080/15384101.2020.1728014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xia W., Unger P., Miller L., Nelson J., and Gelman I. H. (2001) The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 61, 5644–5651 [PubMed] [Google Scholar]
- 60. Bateman N. W., Jaworski E., Ao W., Wang G., Litzi T., Dubil E., Marcus C., Conrads K. A., Teng P. N., Hood B. L., Phippen N. T., Vasicek L. A., McGuire W. P., Paz K., Sidransky D., et al. (2015) Elevated AKAP12 in paclitaxel-resistant serous ovarian cancer cells is prognostic and predictive of poor survival in patients. J. Proteome Res. 14, 1900–1910 10.1021/pr5012894 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw image data can be provided upon request.