

Abstract
Protein-tyrosine phosphatase 1B (PTP1B) is the canonical enzyme for investigating how distinct structural elements influence enzyme catalytic activity. Although it is recognized that dynamics are essential for PTP1B function, the data collected thus far have not resolved whether distinct elements are dynamically coordinated or, alternatively, whether they fulfill their respective functions independently. To answer this question, we performed a comprehensive 13C-methyl relaxation study of Ile, Leu, and Val (ILV) residues of PTP1B, which, because of its substantially increased sensitivity, provides a comprehensive understanding of the influence of protein motions on different time scales for enzyme function. We discovered that PTP1B exhibits dynamics at three distinct time scales. First, it undergoes a distinctive slow motion that allows for the dynamic binding and release of its two most N-terminal helices from the catalytic core. Second, we showed that PTP1B 13C-methyl group side chain fast time-scale dynamics and 15N backbone fast time-scale dynamics are fully consistent, demonstrating that fast fluctuations are essential for the allosteric control of PTP1B activity. Third, and most importantly, using 13C ILV constant-time Carr–Purcell–Meiboom–Gill relaxation measurements experiments, we demonstrated that all four catalytically important loops—the WPD, Q, E, and substrate-binding loops—work in dynamic unity throughout the catalytic cycle of PTP1B. Thus, these data show that PTP1B activity is not controlled by a single functional element, but instead all key elements are dynamically coordinated. Together, these data provide the first fully comprehensive picture on how the validated drug target PTP1B functions.
Keywords: protein-tyrosine phosphatase, PTP1B, enzyme, NMR spectroscopy, 13C methyl ILV dynamics, ct-CPMG, nuclear magnetic resonance (NMR), protein dynamic, enzyme catalysis, enzyme mechanism
Protein-tyrosine phosphatase 1B (PTP1B, PTPN1) was the first non–receptor-bound protein-tyrosine phosphatase (PTP) isolated (1). Not surprisingly, it is also the best-studied member of the human PTP family (2). Since its discovery, PTP1B has been shown to have diverse roles in multiple cellular processes, especially glucose uptake, body mass regulation, motility, and proliferation. As a consequence, PTP1B is a validated target for multiple diseases, especially diabetes and cancer (3).
PTP1B catalyzes the hydrolysis of phosphorylated tyrosine residues (4). The catalytic site is defined by: 1) the PTP loop ((I/V)HCXXGXXR(S/T)G), which includes Cys215, which functions as the catalytic nucleophile in the first step of hydrolysis; 2) the WPD loop, 179WPD181, which contains Asp181, which functions as the proton donor and acceptor during phosphoryl transfer; 3) the E loop, which plays a role in substrate recruitment; 4) the substrate-binding loop (SBL), which restricts dephosphorylation to tyrosine residues; and 5) the Q loop (residues 261–265), which includes the key residue Gln262, which is responsible for coordinating a nucleophilic water, ensuring that PTPs do not function as phosphotransferases (Fig. 1A) (5). Upon substrate binding, it is the WPD loop that undergoes the largest structural change, moving from an open (hydrolysis incompetent) to a closed (hydrolysis competent) position. Finally, PTP1B was also shown nearly 20 years ago to contain an allosteric binding pocket that is ∼20 Å away from the catalytic site, at the intersection of helices α3, α6, and α7 (6).
Figure 1.
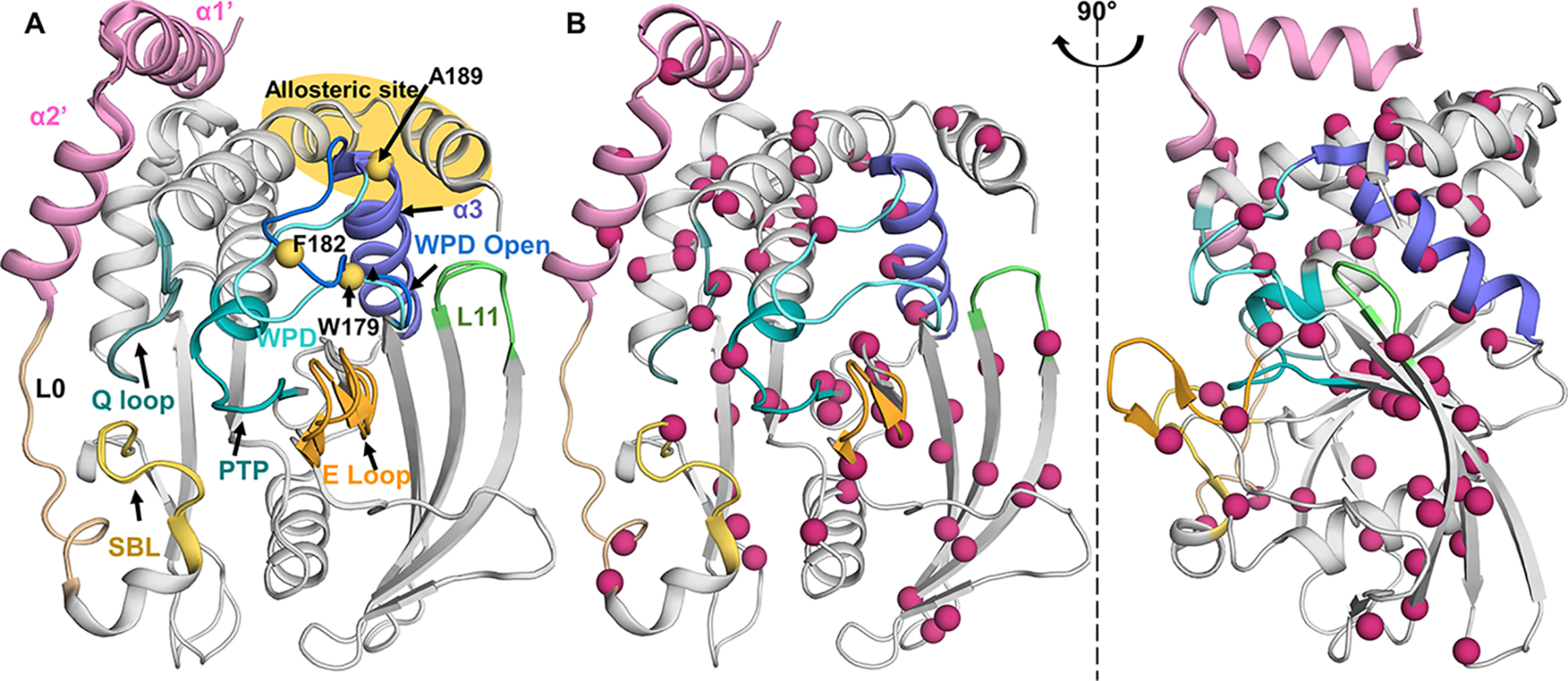
13C ILV methyl groups are well-dispersed throughout PTP1B. A, overlay of PTP1B (open state, PDB 5K9V; closed state, PDB 5K9W); active site: PTP and Q loop (teal), WPD loop (open, blue; closed, cyan); substrate recruitment: E and SBL loop (yellow/orange). Purple, helix α3; green, loop L11. The known allosteric site is highlighted in yellow at the intersection of helices α3, α6, and α7. Residues previously studied by 15N ct-CPMG relaxation measurements are shown as yellow spheres. B, PTP1B (PDB 5K9W) Ile, Leu, and Val residues depicted as pink spheres; well-distributed throughout PTP1B.
Recently, a number of reports have shown that dynamics play a critical role in PTP1B enzyme activity and allostery (7–9). In addition to dynamics, these reports also highlighted the importance of structural rigidity in the extended WPD loop, particularly for proline residue Pro185 (7). This proline is essential for PTP1B activity because it controls an indispensable CH/π switch that associates either with Trp179 within the WPD loop (closed state) or Phe269 from helix α6 (open state; Fig. S1). This CH/π switch controls WPD motion (7, 10). Further, PTP1B helix α3 was identified as the mechanical/dynamics support that drives the transition between the open and closed states of the WPD loop and serves as the connector between the WPD loop and the allosteric pocket and helix α7 (7).
Together, these structural and dynamics data have revealed many key aspects of PTP1B activity and regulation. However, there remains a key unresolved question: whether the observed dynamics for distinct structural elements are independent of one another, or, alternatively, if their dynamics are coordinated so that all catalytically critical residues work in dynamic unity throughout the PTP1B catalytic cycle. The previously reported data were primarily based on 15N-based protein backbone NMR measurements (7, 11), which are excellent reporters on many aspects of protein function. However, when working with larger proteins such as PTP1B, some of these experiments have low sensitivity, decreasing measurement accuracy and making the data statistically unreliable.
Thus, to answer this question, we used 13C-methyl relaxation studies of Ile, Leu, and Val (ILV) residues in PTP1B (12–14). The fast rotation of methyl groups in ILV residues ensures that these NMR experiments have high sensitivity, allowing the dynamics of PTP1B to be determined at three distinct time scales: fast (ps/ns), intermediate (µs/ms), and slow (ms/s). Our data confirm our previous discovery (using only 15N-based protein backbone NMR measurements) that fast motions are critical for the regulation of allostery in PTP1B. However, we also demonstrate that intermediate time-scale motions regulate PTP1B activity. Critically, the motions observed in our 13C ILV data extend far beyond the previously reported movement of the WPD loop and have revealed that PTP1B substrate recruitment and dephosphorylation function in dynamic unison. Such behavior has also been seen for other enzymes, including the enzymes dihydrofolate reductase (15, 16), cyclophilin A (17), and the kinases p38 (18) and extracellular signal–regulated kinase 2 (19). Together, this work lays the foundation for a comprehensive understanding of conformation and dynamics for the entire PTP family specifically and enzyme function and protein allostery generally.
Results
13C ILV methyl assignment of PTP1B identifies reporters distributed throughout the protein
Fundamental insights into the dynamics and the function of a protein can be obtained by studying the 15N backbone and/or the 13C side chain dynamics, especially of Ile, Leu, and Val residues. We recently reported the 15N-based sequence specific backbone assignment of the folded catalytic domain of PTP1B (∼35 kDa; residues 1–301; hereafter referred to as PTP1B) and leveraged these data to define how 15N fast time-scale dynamics contributes to PTP1B activity, especially allostery (20). However, the relatively large size of PTP1B, together with the inability to concentrate the protein to ≥250 μm, made 15N constant-time Carr–Purcell–Meiboom–Gill (ct-CPMG) data analysis, which reports on intermediate (µs to ms) events, statistically inaccurate and thus unable to provide insights into the role of intermediate time-scale dynamics for PTP1B function. The only intermediate (µs to ms) time-scale experiments previously performed on PTP1B used three reporters (Trp179 and Phe182 in the WPD loop and Ala189 in helix α3; Fig. 1A) in free PTP1B and a single reporter (Ala189 in helix α3) in a peptide-bound PTP1B complex (11). These limited data suggested that intermediate time-scale motions control the WPD loop movement and subsequently its catalytic activity. However, because of the very limited number of reporters, how intermediate time-scale motions from other regions of PTP1B also contribute to its catalytic function is completely unknown. Because a few reports have recently shown that in other signaling enzymes (kinases and phosphatases) the intermediate time-scale dynamics of distinct functional regions typically work in concert for full functionality, additional studies of PTP1B that report on all functional regions are needed to understand how PTP1B intermediate time-scale dynamics control PTP1B substrate binding and activity.
PTP1B contains 60 ILV residues that are uniformly distributed throughout its sequence and structure (16 Ile, 28 Leu, and 16 Val; Fig. 1B). A suite of complementary experiments (21), which included NOE experiments and measurements on 16 single amino acid variants, enabled us to complete the PTP1B 13C ILV methyl group assignment in both its free (Fig. S2A; open; 98% assigned; only Leu88 is missing because of overlap) and active-site inhibitor TCS-401–bound forms (Fig. S2B; closed; 96% assigned; Leu88 and Val49Cδ2 are overlapped; Ile219 is either broadened beyond detectability or overlapped). Our complete ILV assignment also correlates well with a partial assignment of the Ile-only region of PTP1B that was recently reported (10).
NMR 15N-based sequence-specific backbone assignments of large proteins, such as PTP1B, are often incomplete because of either slow hydrogen/deuterium (H/D) back exchange after protein expression in D2O-based medium or intermediate conformational exchange, which broadens peaks beyond detection (22). These are not issues for 13C ILV methyl group assignment. Nevertheless, when counting the number of peaks in the Ile Cδ region of the spectrum, we observed 4 more peaks than expected (16 expected; 20 observed; Fig. S3A). The assignment showed that these peaks belong to a minor conformation of Ile10, Ile19, Ile23, and Ile246, which form the interface between the catalytic PTP1B domain and helices α1′ and α2′ (Fig. S3B). Thus, it is likely that helices α1′ and α2′ disengage from the core protein in a slow dynamics event (pB = ∼25%), because they are only loosely connected to the catalytic domain via L0, a 10–amino acid–long linker (PTP1B residues 28–37).
PTP1B fast time-scale 13C ILV relaxation data correlate with PTP1B 15N backbone relaxation data
To define the PTP1B motions at different time scales, we recorded T1 and T1ρ/T2 fast time scale 13C ILV side-chain relaxation data (Fig. 2A and Fig. S4). The values of these parameters typically correlate with the distance of the 13C-methyl group from the backbone, i.e. the further the distance, the higher the flexibility. The fast time-scale 13C ILV relaxation data show this is also true for PTP1B, in which on average the methyl groups of valine residues rotate the slowest, followed by leucine and isoleucine (Fig. S5, A and B). Plotting the PTP1B T2 13C ILV side chain relaxation data against the protein sequence and comparing it directly with our previously reported 15N backbone relaxation data (T1, T2) show an overall similar behavior. This includes a sharp increase in overall dynamics of helix α7, which we previously established as the central modulator of allostery in PTP1B (Fig. 2, A and B). The only region in which the T2 13C ILV side-chain and 15N backbone relaxation data differ is for PTP1B helix α3, which modulates WPD motion and subsequently PTP1B activity; helix α3 did not show the increased dynamics identified in the 15N backbone relaxation data (7).
Figure 2.

Change in fast time-scale 13C ILV relaxation dynamics of PTP1B upon active-site interaction. A, 15N T2 relaxation and 13C-methyl transverse T2 rates for PTP1B (black) and PTP1B:TCS401 (red). The regions of secondary structures are illustrated at the top of the figure. B, Val287 and Leu294 (yellow) show a large change in dynamics as helix α7 becomes less flexible when inhibitor binds and becomes part of the allosteric site (blue). Val155 (yellow) interacts with key allosteric residues Tyr152 and Tyr153 to communicate to the WPD loop (green). The panel also shows a dramatic decrease in fast time-scale dynamics.
13C ILV assignment and side chain dynamics of closed PTP1B
Next, we determined the consequences of active-site inhibitor binding on PTP1B chemical shifts and dynamics. TCS401 is a small (306 Da) active-site inhibitor that binds PTP1B with a KD of ∼26 ± 2 μm (23). We and others have shown that the binding of small molecule inhibitors and/or substrate peptides leads to the closure of the WPD loop and a rearrangement of PTP1B helix α3, which rigidifies helix α7. As expected, direct comparison of the 15N-backbone chemical shift perturbation data with that of 13C ILV shows excellent overlap (Fig. S6). Notably, PTP1B helices α1′ and α2′ still disengage from the core enzyme (pB = ∼29%) in TCS401-saturated PTP1B, showing that binding of TCS-401 does not influence this slow dynamics event (Fig. S3C).
To determine whether altered dynamics accompanies active-site inhibitor binding, we repeated the 13C ILV side chain relaxation dynamics measurements with TCS401-saturated PTP1B. The most significant changes were observed in helix α7, in which the dynamics detected in the TCS401-free state were quenched in the TCS401-saturated state. This is identical to what was observed in the 15N relaxation data (R1, R2) that probe fast time-scale backbone dynamics (Fig. 2A) (7). Val287 and Leu294 show the largest changes, which define the core of the PTP1B allosteric site, at the intersection of helices α3, α6, and α7 (Fig. 2B). Thus, these results further highlight the critical role of helix α7 and its intrinsic dynamics for the PTP1B allosteric network. Finally, consistent with the PTP1B 15N backbone relaxation data and the 13C ILV chemical shift perturbation data, Val155 also has a statistically significant reduction in 13C ILV side-chain fast time-scale dynamics (Fig. 2A). Val155 abuts the L11 loop, which includes Tyr152 and Tyr153, two residues that are important for the allosteric pathway in PTP1B. Together, these data show that the fast time-scale dynamics results between the 15N backbone relaxation and the 13C ILV side chain relaxation data are consistent, a critical step before evaluating the 13C ILV side-chain ct-CPMG data.
13C ILV intermediate time-scale dynamics of PTP1B
Next, we measured 13C ILV ct-CPMG side-chain relaxation data (all 13C ILV ct-CPMG for the studies reported here are recorded at two magnetic fields; 14.1 and 18.8 T), which reports on µs-to-ms motions, for free PTP1B. Enzymatic reactions for signaling enzymes such as PTP1B often have catalytic turnover rates (PTP1B 15–60 s−1) that can be correlated with protein dynamics in the μs-to-ms time scale. In ct-CPMG measurements, residues undergoing μs-to-ms conformational exchange dynamics show changes in the effective relaxation rate R2 (R2,eff), which are measured as a function of the repetition frequency (νCPMG). Plots of R2,eff versus νCPMG are curved for those residues experiencing μs-to-ms time-scale dynamics. Fitting these curves to a two-state model (Carver–Richards) allows the populations (pA and pB) and the exchange rates between (kex) these populations to be extracted. A significant number of residues in free (38 residues) and TCS401-saturated (23 residues) PTP1B exhibited μs-to-ms conformational exchange dynamics. Using an approach that we used previously to analyze p38 mitogen-activated protein kinase dynamics (18), we next identified residues that experience similar exchange dynamics. Briefly, this approach identifies groups of residues with similar fluctuations and thus provides increased statistical significance when evaluating ct-CPMG data.
In free PTP1B, we identified two groups of residues with uniform μs-to-ms conformational exchange dynamics (Table 1). The two clusters showed fast μs exchange dynamics with kex of 3550–6160 s−1. We identified a small cluster of three residues (group 1) with very fast exchange dynamics (kex = 6160 ± 360 s−1 and pB = 1.8 ± 1.0%). This cluster exclusively includes residues on the surface of PTP1B, and thus these dynamics likely reflect interactions with bulk solvent (Fig. 3A). In contrast, a large cluster (33 residues; group 2) shows exchange dynamics of kex = 3550 ± 70 s−1 and pB = 3.2 ± 0.4% (pA open PTP1B form; pB closed or similar to the PTP1B closed form) and includes residues from most secondary structure elements of PTP1B, including the WPD loop and all other regions of PTP1B that have identified functions in the catalytic activity and allostery of PTP1B. This demonstrates that, in its free form, PTP1B exhibits largely uniform exchange dynamics (Fig. 3A).
Table 1.
PTP1B residues: specific intermediate exchange group
| kex (s−1) | pB (%) | Residues |
|---|---|---|
| 3550 ± 70 | 3.2 ± 0.4 | Ile19, Val49 (Cγ1 and Cγ2), Ile72, Leu83 (Cδ1 and Cδ2), Val107 (Cγ1), Val113 (Cγ1 and Cγ2), Leu119 (Cδ1 and Cδ2), Ile134, Leu142 (Cδ1), Leu144 (Cδ1 and Cδ2), Val155 (Cγ1 and Cγ2), Leu160 (Cδ1 and Cδ2), Leu163 (Cδ1 and Cδ2), Ile171, Leu172 (Cδ1), Val184 (Cγ1), Leu195 (Cδ1 and Cδ2), Val212 (Cγ1), Ile219, Leu227 (Cδ1 and Cδ2), Leu232 (Cδ1), Leu233 (Cδ1 and Cδ2), Leu234 (Cδ1), Val249 (Cγ1 and Cγ2), Leu250 (Cδ1 and Cδ2), Leu260 (Cδ1), Leu261 (Cδ1), Leu267 (Cδ1), Leu272 (Cδ1), Val274 (Cγ1 and Cγ2), Leu275 (Cδ1), Val287 (Cγ1), Leu294 (Cδ1 and Cδ2) |
| 6160 ± 360 | 1.8 ± 1.0 | Leu110 (Cδ1 and Cδ2), Leu140 (Cδ1 and Cδ2), Ile281 |
Figure 3.

Change in intermediate time-scale 13C ILV relaxation dynamics of PTP1B upon active-site interaction. A, PTP1B (open confirmation; PDB 5K9V): group 1 (kex = 6160 s−1; orange); group 2 (kex = 3550 s−1; teal) are shown. B, PTP1B (closed confirmation; PDB 5K9W): TCS401 shown as sticks (cyan). Group 1 (kex = 2100 s−1; blue), group 2 (kex = 780 s−1; yellow), and group 3 (kex = 550 s−1; raspberry) are shown. C, detailed analysis of group 3 residues. Residues Val49 (SBL), Val113, and Leu119 (E loop) are important for substrate recruitment (yellow box) and surround the PTP1B active site. Residues Val184 (WPD loop) and Ile261 (Q loop) are essential for substrate hydrolysis and enzyme regeneration (teal box).
Comparing our 13C ILV ct-CPMG side-chain relaxation data (38 residues) with previously reported 15N backbone ct-CPMG data (three residues; fit with a kex = 900 s−1 and pB = 2.3%; Fig. 1A) shows similarity (11). Our pB values are similar to the ones resulting from the 15N backbone ct-CPMG data evaluation (13C ILV ct-CPMG pB values, 3.2%; 15N backbone ct-CPMG pB, 2.3%). The populations extracted from our 13C ILV ct-CPMG side chain relaxation data have larger errors, because of the 4-fold increase in kex, which makes fitting accurate populations more difficult. This 4-fold faster 13C kex, most likely represents a fast (probably side chain–associated) motion that drives an underlying slower (backbone-associated) motion that was detected in the 15N data. Together, these data show that free PTP1B exhibits mostly fast exchange dynamics and that these can be partitioned into two statistically significant groups. However, the largest, most significant group encompasses most of the protein and highlights the largely uniform exchange behavior throughout free PTP1B.
Active-site binding alters 13C ILV intermediate time-scale dynamics of PTP1B
Next, we evaluated the 13C ILV ct-CPMG side-chain relaxation data for TCS401-saturated (inhibitor bound) PTP1B (WPD loop is closed; helix α7 becomes rigidified). The data show a large reduction in the overall exchange dynamics of PTP1B when the active site is occupied. We were able to fit three statistically relevant residue groups (Table 2). Group 1 (3 residues), which exhibits the fastest exchange dynamics (kex = 2100 ± 240 s−1; pB = 0.4 ± 0.03%), is a small group and includes only residues in β-strands 8 and 9, which are distant from the PTP1B active site. The exchange dynamics for group 2, which is the largest group (13 residues), are significantly slower (kex = 780 ± 50 s−1; pB = 2.1 ± 0.2%) and composed of residues surrounding the PTP1B active site and residues present within helices α3/4/6 and β-strands 4/9/10/11. Finally, group 3 (7 residues) exhibits the slowest exchange dynamics (kex = 550 ± 40 s−1; pB = 7.8 ± 0.6%). All of these residues belong to structural elements that are critical for PTP1B substrate recruitment and activity, including Val184 in the WPD loop (activity), Val113 and Leu119 of the E loop (substrate recruitment), Val49 of the substrate recruitment loop (substrate recruitment), and Ile261 of the Q loop (activity; Fig. 3, B and C). The linked dynamics of these disparate sites in the TCS401-bound state strongly suggest that motions within the active site become synchronized upon binding of inhibitors/substrates.
Table 2.
PTP1B residues in complex with TCS401; specific intermediate exchange group
| kex (s−1) | pB (%) | Residues |
|---|---|---|
| 550 ± 40 | 7.8 ± 0.6 | Val49 (Cγ1), Val107 (Cγ1), Val113 (Cγ1 and Cγ2), Leu119 (Cδ1), Val184 (Cγ1), Ile261, Ile281 |
| 780 ± 50 | 2.1 ± 0.1 | Leu83 (Cδ1), Leu110 (Cδ1 and Cδ2), Val155 (Cγ1), Leu171 (Cδ1), Leu195 (Cδ1 and Cδ2), Leu204 (Cδ1), Val211 (Cγ1), Val213 (Cγ1), Leu227 (Cδ1 and Cδ2), Leu250 (Cδ1 and Cδ2), Leu260 (Cδ1), Leu272 (Cδ1), Val287 (Cγ1) |
| 2100 ± 240 | 0.40 ± 0.03 | Leu140 (Cδ1 and Cδ2), Leu142 (Cδ1 and Cδ2), Leu160 (Cδ1) |
Only a limited comparison can be made between our 13C ILV ct-CPMG data and the previous 15N backbone ct-CPMG data of closed PTP1B, because the latter analysis is based on a single residue (Ala198 in helix α3) and was recorded using a peptide with a nonhydrolyzable tyrosine analog (11). Our data show that the population of the PTP1B open state in TCS401-saturated PTP1B (pB) is 7.8%, compared with 14% for peptide-saturated PTP1B. This difference in population either reflects the less optimal fit of a single residue or simply reflects the higher affinity of TCS401 for PTP1B versus a substrate-like peptide (low µm versus high µm to low mm), leading to a higher population of the closed state of PTP1B in our data.
Discussion
Protein dynamics at multiple time scales are essential for protein function and regulation (24–26). NMR spectroscopy is distinctively qualified to report on protein dynamics on multiple time scales, from ps to h. 15N-backbone dynamics, which report on the dynamics of the amide 15N-HN vector, is the most widely used technique because it has one reporter for each amino acid (except for proline). However, this approach is of limited applicability for larger proteins (≥35 kDa) because their increased overall correlation time τc limits sensitivity and resolution. Thus, for these systems the use of 13C-methyl groups for dynamics measurements, especially from ILV residues, has become a routine approach. 13C-H vectors in ILV methyl groups rotate rapidly and thus provide high sensitivity and sharp lines even for very large proteins. Here, we have used 13C ILV relaxation measurements to define the dynamics of PTP1B at different time scales to understand how PTP1B dynamics correlates with enzymatic function. In particular, although it is well-accepted that multiple structural elements of PTP1B are essential for catalysis (WPD-, SBL-, and Q loops), we set out to determine whether and how these elements are dynamically coordinated, or, alternatively, whether they fulfill their respective functions independently of one another.
Using 13C-methyl group dynamics relaxation experiments, we discovered that PTP1B exhibits dynamics at three distinct time scales. First, and somewhat unexpectedly, we discovered that PTP1B undergoes a distinctive slow motion that allows for the dynamic binding and release of helices α1′ and α2′ from the core catalytic domain. Among the nonreceptor PTP family, these helices are unique to PTP1B and its most closely related homolog, TCPTP/PTPN2 (2), suggesting they might have a specific function. If and how these helices contribute to PTP1B (and TCPTP) activity is unknown; however, an intriguing possibility is that the dynamic binding and release of these helices may facilitate substrate recruitment. Second, we confirmed that, in PTP1B, both PTP1B 13C-methyl group side chain fast time-scale dynamics (this work) and 15N backbone fast time-scale dynamics in elements distal from the active site, particularly helix α7 (7), are essential for the allosteric control of PTP1B activity. Thus, these data confirm that, in PTP1B, allostery is governed by local fast time-scale dynamics, which modulates intermediate time-scale dynamics that controls the catalytic activity of PTP1B.
Third, we performed 13C ILV ct-CPMG relaxation measurements to measure µs-to-ms exchange dynamics, because this time scale is most often correlated with enzymatic function. In agreement to a previous report, we see a significant 6.5-fold reduction of the WPD loop motion when the active site is occupied (open, kex = 3550 ± 70 s−1; closed, kex = 550 ± 40 s−1). However, our comprehensive relaxation data, which reports on dynamics throughout PTP1B (up to 56 distinct residues), significantly expands this observation. Namely, the data show that the functionally critical elements of PTP1B structure fluctuate coherently and distinctly from the rest of the protein when a substrate analog/inhibitor binds the PTP1B active site. This includes residues from the WPD loop, the substrate recruitment E loop, the SBL, and the Q loop. This shows that all catalytically critical residues work in dynamic unity throughout the catalytic cycle of PTP1B.
It is well-recognized that conformational dynamics/plasticity can have an essential role in the catalytic cycle of multiple enzymes. For instance, in cyclophilin A, the observed rate-limiting dynamics reflect coordinated motions across its active site, even in the absence of obvious backbone changes in the corresponding crystal structures (17). However, in the case of PTP1B and other PTPN family members, crystal structures show clearly defined changes in the position of the WPD loop (open/closed) that, in principle, might depict the full range of conformational heterogeneity that allow for the catalytic activity (27). Indeed, similar limited motions that are rate-limiting have been reported for the enzyme triosephosphate isomerase (28). Furthermore, previous results indicated that motions of the PTP1B WPD loop were apparently correlated to the catalytic rate (11), suggesting that this limited model of conformational heterogeneity (WPD loop open/closed) was likely appropriate.
However, further experiments demonstrated that the rate of catalysis of PTP1B is curiously disconnected from the WPD loop's dynamic equilibrium and suggested the involvement of adjacent structural elements in rate-limiting dynamics and thus catalytic activity (7, 10). Indeed, crystallographic evidence from the related protein-tyrosine phosphatase PTPN7 (HePTP) showed coordinated motions of the E loop and WPD loop in response to depletion of a phosphate analog from the binding site (29). Thus, it is likely that coherent/coordinated fluctuations within the active site, either in response to loop opening or in a completely closed state, control catalysis. Our new data add significant support to this latter view. Critically, the greater density of probes provided by uniform ILV labeling allows for the comprehensive observation of dynamic effects throughout the PTP1B active site and shows that all catalytically significant structural elements/loop are engaged in a coherent fluctuation in the presence of an inhibitor, which functioned as a proxy substrate in our analysis. Thus, although the WPD loop mobility is important, it is clearly not the sole contributor to PTP1B dynamics and function at the PTP1B active site.
Taken together, these data suggest that in PTP1B, intermediate dynamics are important for substrate binding and product release, whereas fast dynamics, in elements distal from the chemical reaction, are important for the allosteric control of the overall function of PTP1B. Because these structural features are highly conserved within the PTP family of proteins, it is likely that this pattern of dynamic influence is conserved throughout the PTP family (30). Furthermore, it is likely that sites within these networks can be targeted by new allosteric approaches to modulate PTP function.
Materials and methods
Protein expression
DNA coding the human PTP1B catalytic domain (Uniprot P18031; residues 1–301) was used as previously described (20, 31). For the assignment of ILV residues, the following PTP1B variants were created (QuikChange (Agilent) site-directed mutagenesis): V34A, L37I, L88I, V108A, L172I, L192A, L195A, L199I, L204A, V213A, I219V, L233I, V244I, I261V, L294A, and L299I. Furthermore, also the deletion variant PTP1BΔ7 (PTP1B residues 1–284) was used. For protein expression, plasmid DNA was transformed into Escherichia coli BL21 (DE3) RIL cells (Agilent). The cells were grown in M9 minimal medium in the presence of selective antibiotics at 37 °C to an A600 of ∼0.6, upon amino acid precursors (type depending on labeling scheme) were added. The cells continued to grow at 37 °C until an A600 of ∼0.8 was reached, and expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside. Induction proceeded for ∼20 h at 18 °C prior to harvesting by centrifugation at 8,000 × g. Cell pellets were stored at −80°C until purification. All PTP1B expression in D2O (22) required careful adaptation (successively in medium containing 25%, 50%, 75%, 90%, and 100% D2O) to optimize the PTP1B yields.
Multiple labeling schemes were used for different measurements. For the assignment of PTP1B ILV, PTP1B was expressed with 1 g/liter 15NH4Cl, 4 g/liter [2H,13C]-d-glucose, [13C5,3-2H1] 120 mg/liter α-ketoisovaleric acid sodium salt (CDLM 4418) and 60 mg/liter [13C4;3,3-2H2]α-ketobutyric acid sodium salt (CDLM 4611) in 100% D2O (scheme 1). To assist in the assignment, PTP1B variants were expressed in M9 medium and labeled with 120 mg/liter [13C]α-ketoisovaleric acid (CDLM 6821) and 60 mg/liter [13C2]α-ketobutyric acid (CDLM 6820) in 100% H2O (scheme 2). For NOE-based assignment spectra, PTP1B was expressed with 1 g/liter 15NH4Cl, 4 g/liter [2H,12C]-d-glucose, 120 mg/liter [13C5,3-2H1]α-ketoisovaleric acid (CDLM 7318) and 60 mg/liter [13C4,3,3-2H2]α-ketobutyric acid (CDLM 7317) in 100% D2O (scheme 3). Lastly, for relaxation measurements, PTP1B was expressed with 4 g/liter [2H,12C]-d-glucose, 120 mg/liter [3-13C,3-methyl-2H2, 3,4,4,4-2H4]α-ketoisovaleric acid (CDLM 7354) and 60 mg/liter [4-13C, 4-2H2, 3-2H2]α-ketobutyric acid (CDLM 7353) in 100% D2O (scheme 4).
Protein purification
PTP1B was purified as previously described into NMR Buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 5 mm DTT) (7, 20). Purified protein was either used immediately or flash-frozen in liquid nitrogen for storage at −80°C. Samples for NMR dynamics experiments were aliquoted into 550-μl aliquots, flash-frozen in liquid nitrogen, and lyophilized for 24 h. The protein was then resuspended in 550 µl of D2O. Typical PTP1B protein yields are ∼55 mg/liter in D2O-based M9 minimal medium, and variant yields are ∼21–48 mg/liter in M9 minimal medium at 98% purity.
NMR measurements
All PTP1B 13C-methyl ILV assignment NMR measurements were performed using a Bruker Advance Neo 800 MHz NMR spectrometer with a TCI HCN-active z-gradient cryoprobe at 298 K. A 3D (13C,13C,1H)HMCM(CG)CBCA experiment functioned as the initial assignment step using PTP1B produced following scheme 1 (32). A 3D (13C,13C,1H) HSQC-NOESY-HSQC (τm = 200 ms) and a 3D (13C,15N,1H)HSQC-NOESY-Heteronuclear multiple quantum coherence (HMQC) (τm = 400 ms) spectra were recorded using PTP1B produced following scheme 3. The final concentration of PTP1B for these measurements was 0.35 mm in NMR buffer containing 10% D2O. All data were processed using NMRPipe (33) or Topspin 4.0.5 and analyzed using NMRFAM-SPARKY (34).
13C-Methyl relaxation NMR measurements (13CHD2) were performed on Bruker Advance Neo 600- and 800-MHz spectrometers equipped with TCI HCN-active z-gradient cryoprobes at 298 K. Data were recorded on 2H,12C,15N-labeled PTP1B with 13CHD2-labeled ILV methyl groups (scheme 4), either free or inhibitor/TCS401-saturated at a final protein concentration of 0.25 mm in NMR buffer and 100% D2O. Sample concentration was tightly monitored to ensure no effect on τc and thus T1ρ measurements. TCS401 inhibitor was carefully titrated to achieve full saturation. Upon saturation (chemical shifts of interacting residues stopped changing; usually at 1:3 ratio), additional TCS401 (to 1:6 ratio) was added to ensure that all experiments were performed under fully inhibitor-saturated conditions and thus that the observed CPMG dispersions are independent of ligand on/off exchange events. All relaxation data were recorded as a pseudo-3D in a fully interleaved manner.
T1 relaxation delays were 20, 500, 1000, 1200, 1400, 1600, 2000, 4000, and 5500 ms (D1 (recycle delay) of 4.5 s; 800 MHz; 1200 and 4000 ms were repeated for measurement error assessment) and 20, 800, 1000, 1200, 1400, 1600, 2000, 2400, 3000, 3200, and 4000 ms (D1 of 4.2 s; 600 MHz; 1200 ms was repeated for measurement error assessment). T1ρ relaxation delays were 5, 30, 50, 60, 90, 100, 120, 150, and 180 (D1 of 2.5 s; 800 MHz; 50 and 150 ms was repeated for measurement error assessment) and 5, 30, 50, 70, 80, 90, 100, 160, 170, and 200 (D1 of 3.2 s; 600 MHz; 70 and 130 ms were repeated for measurement error assessment). The measurement errors between repeat measurements were 2.8% at 18.8T and 2.6% at 14.1T.
ct-CPMG relaxation dispersion experiments were performed at 298 K at two magnetic field strengths (14.1 and 18.8 T). A constant time of 40 ms between 15N refocusing pulses and 10 different delay times corresponding to the following CPMG frequencies 50, 100, 250, 400, 600, 800, 1000, 1200, 1600, and 2000 Hz were used (100 and 800 Hz were repeated for measurement error assessment; which was ∼2.8%). D1 was set to 3.2 and 3.8 s for experiments performed on 14.1T and 18.8T magnetic field strengths, respectively.
Relaxation analysis
T1 and T1ρ values were calculated using NMRviewJ using the peak intensities (jitter function) and exponential decay fitting function. Errors were also determined via relaxation curve fitting. T2 was extracted from T1ρ by: , where β is the effective rotation angle for each 15N nucleus as determined by the strength of the spin-lock field and the chemical shift offset of the nucleus from the spin-lock frequency.
ct-CPMG relaxation dispersion intensity measurements were performed in NMRviewJ (jitter function) and converted to R2eff by: . PTP1B residues were fit individually to the Carver–Richards equation for a system in two-state exchange using a Levenberg–Marquardt algorithm. Attempts were then made to fit residues into groups. The quality of the group fit was evaluated using the Bayesian information criterion (BIC) to compare the group fit to the results of the individual fits using ΔBIC = BICgroup − BICindividual. The more negative the ΔBIC, the better the fit. Groups were refined through several rounds. A residue was kept in a specific group if the BIC was less than −1, and the residuals were randomly distributed.
NMR analysis of TCS401 inhibitor binding
TCS401 was titrated into 0.25 mm PTP1B at molar ratios of 0:1, 0.5:1, 1:1, 2:1, and 3:1 (TCS401:PTP1B). 2D [1H,13C] HSQC spectra were recorded for each titration point. TCS401 was solubilized in d6-DMSO (25 mm). No chemical shift differences were identified in the PTP1B 2D [1H,15N] Transverse Relaxation Optimized Spectroscopy (TROSY) or 2D [1H,13C]HSQC spectrum upon the addition of d6-DMSO. Chemical shift perturbations (Δδ) between apo PTP1B and inhibitor-bound PTP1B spectra were calculated using: .
Supplementary Material
This article contains supporting information.
Author contributions—K. R. T., M. W. C., and G. S. K. data curation; K. R. T., M. W. C., and G. S. K. formal analysis; K. R. T. investigation; K. R. T. and M. W. C. methodology; K. R. T., M. W. C., G. S. K., R. P., and W. P. writing-review and editing; M. W. C. software; R. P. and W. P. conceptualization; R. P. and W. P. supervision; R. P. and W. P. funding acquisition; W. P. resources; W. P. writing-original draft; W. P. project administration.
Funding and additional information—This work was supported by the American Diabetes Association Pathway to Stop Diabetes Grant 1-14-ACN-31 (to W. P.) and by National Institutes of Health Grants and R01GM098482 (to R. P.) and T32GM008804 (to K. R. T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PTP
- protein-tyrosine phosphatase
- PDB
- Protein Data Bank
- SBL
- substrate-binding loop
- ct-CPMG
- constant-time Carr–Purcell–Meiboom–Gill
- HSQC
- heteronuclear single quantum coherence
- BIC
- Bayesian information criterion.
References
- 1. Tonks N. K., Diltz C. D., and Fischer E. H. (1988) Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 263, 6722–6730 [PubMed] [Google Scholar]
- 2. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., and Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 10.1016/j.cell.2004.05.018 [DOI] [PubMed] [Google Scholar]
- 3. Feldhammer M., Uetani N., Miranda-Saavedra D., and Tremblay M. L. (2013) PTP1B: a simple enzyme for a complex world. Crit. Rev. Biochem. Mol. Biol. 48, 430–445 10.3109/10409238.2013.819830 [DOI] [PubMed] [Google Scholar]
- 4. Pannifer A. D., Flint A. J., Tonks N. K., and Barford D. (1998) Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 273, 10454–10462 10.1074/jbc.273.17.10454 [DOI] [PubMed] [Google Scholar]
- 5. Andersen J. N., Mortensen O. H., Peters G. H., Drake P. G., Iversen L. F., Olsen O. H., Jansen P. G., Andersen H. S., Tonks N. K., and Møller N. P. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell Biol. 21, 7117–7136 10.1128/MCB.21.21.7117-7136.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wiesmann C., Barr K. J., Kung J., Zhu J., Erlanson D. A., Shen W., Fahr B. J., Zhong M., Taylor L., Randal M., McDowell R. S., and Hansen S. K. (2004) Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 11, 730–737 10.1038/nsmb803 [DOI] [PubMed] [Google Scholar]
- 7. Choy M. S., Li Y., Machado L. E. S. F., Kunze M. B. A., Connors C. R., Wei X., Lindorff-Larsen K., Page R., and Peti W. (2017) Conformational rigidity and protein dynamics at distinct timescales regulate PTP1B activity and allostery. Mol. Cell 65, 644–658.e5 10.1016/j.molcel.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krishnan N., Koveal D., Miller D. H., Xue B., Akshinthala S. D., Kragelj J., Jensen M. R., Gauss C.-M., Page R., Blackledge M., Muthuswamy S. K., Peti W., and Tonks N. K. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 10.1038/nchembio.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keedy D. A., Hill Z. B., Biel J. T., Kang E., Rettenmaier T. J., Brandão-Neto J., Pearce N. M., von Delft F., Wells J. A., and Fraser J. S. (2018) An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife 7, e36307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cui D. S., Lipchock J. M., Brookner D., and Loria J. P. (2019) Uncovering the molecular interactions in the catalytic loop that modulate the conformational dynamics in protein tyrosine phosphatase 1B. J. Am. Chem. Soc. 141, 12634–12647 10.1021/jacs.9b04470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whittier S. K., Hengge A. C., and Loria J. P. (2013) Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 341, 899–903 10.1126/science.1241735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruschak A. M., and Kay L. E. (2010) Methyl groups as probes of supra-molecular structure, dynamics and function. J. Biomol. NMR 46, 75–87 10.1007/s10858-009-9376-1 [DOI] [PubMed] [Google Scholar]
- 13. Tzeng S.-R., and Kalodimos C. G. (2011) Protein dynamics and allostery: an NMR view. Curr. Opin. Struct. Biol. 21, 62–67 10.1016/j.sbi.2010.10.007 [DOI] [PubMed] [Google Scholar]
- 14. Rosenzweig R., and Kay L. E. (2014) Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem. 83, 291–315 10.1146/annurev-biochem-060713-035829 [DOI] [PubMed] [Google Scholar]
- 15. Boehr D. D., McElheny D., Dyson H. J., and Wright P. E. (2006) The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 10.1126/science.1130258 [DOI] [PubMed] [Google Scholar]
- 16. Bhabha G., Ekiert D. C., Jennewein M., Zmasek C. M., Tuttle L. M., Kroon G., Dyson H. J., Godzik A., Wilson I. A., and Wright P. E. (2013) Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 20, 1243–1249 10.1038/nsmb.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Otten R., Liu L., Kenner L. R., Clarkson M. W., Mavor D., Tawfik D. S., Kern D., and Fraser J. S. (2018) Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 9, 1314 10.1038/s41467-018-03562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar G. S., Clarkson M. W., Kunze M. B. A., Granata D., Wand A. J., Lindorff-Larsen K., Page R., and Peti W. (2018) Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl. Acad. Sci. U.S.A. 115, 4655–4660 10.1073/pnas.1721441115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iverson D. B., Xiao Y., Jones D. N., Eisenmesser E. Z., and Ahn N. G. (2020) Activation loop dynamics are coupled to core motions in extracellular signal-regulated kinase-2. Biochemistry 59, 2698–2706 10.1021/acs.biochem.0c00485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krishnan N., Krishnan K., Connors C. R., Choy M. S., Page R., Peti W., Van Aelst L., Shea S. D., and Tonks N. K. (2015) PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J. Clin. Invest. 125, 3163–3177 10.1172/JCI80323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tugarinov V., and Kay L. E. (2003) Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 125, 13868–13878 10.1021/ja030345s [DOI] [PubMed] [Google Scholar]
- 22. Peti W., and Page R. (2016) NMR spectroscopy to study MAP kinase binding to MAP kinase phosphatases. Methods Mol. Biol. 1447, 181–196 10.1007/978-1-4939-3746-2_11 [DOI] [PubMed] [Google Scholar]
- 23. Iversen L. F., Andersen H. S., Branner S., Mortensen S. B., Peters G. H., Norris K., Olsen O. H., Jeppesen C. B., Lundt B. F., Ripka W., Møller K. B., and Møller N. P. (2000) Structure-based design of a low molecular weight, nonphosphorus, nonpeptide, and highly selective inhibitor of protein-tyrosine phosphatase 1B. J. Biol. Chem. 275, 10300–10307 10.1074/jbc.275.14.10300 [DOI] [PubMed] [Google Scholar]
- 24. Tzeng S.-R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 10.1038/nature11271 [DOI] [PubMed] [Google Scholar]
- 25. Henzler-Wildman K. A., Lei M., Thai V., Kerns S. J., Karplus M., and Kern D. (2007) A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916 10.1038/nature06407 [DOI] [PubMed] [Google Scholar]
- 26. Lisi G. P., and Loria J. P. (2016) Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function. Prog. Nucl. Magn. Reson. Spectrosc. 92–93, 1–17 10.1016/j.pnmrs.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barr A. J., Ugochukwu E., Lee W. H., King O. N. F., Filippakopoulos P., Alfano I., Savitsky P., Burgess-Brown N. A., Müller S., and Knapp S. (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–363 10.1016/j.cell.2008.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Massi F., Wang C., and Palmer A. G. (2006) Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry 45, 10787–10794 10.1021/bi060764c [DOI] [PubMed] [Google Scholar]
- 29. Critton D. A., Tautz L., and Page R. (2011) Visualizing active-site dynamics in single crystals of HePTP: opening of the WPD loop involves coordinated movement of the E loop. J. Mol. Biol. 405, 619–629 10.1016/j.jmb.2010.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hjortness M. K., Riccardi L., Hongdusit A., Zwart P. H., Sankaran B., De Vivo M., and Fox J. M. (2018) Evolutionarily conserved allosteric communication in protein tyrosine phosphatases. Biochemistry 57, 6443–6451 10.1021/acs.biochem.8b00656 [DOI] [PubMed] [Google Scholar]
- 31. Peti W., and Page R. (2007) Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr. Purif. 51, 1–10 10.1016/j.pep.2006.06.024 [DOI] [PubMed] [Google Scholar]
- 32. Krejcirikova A., and Tugarinov V. (2012) 3D-TROSY-based backbone and ILV-methyl resonance assignments of a 319-residue homodimer from a single protein sample. J. Biomol. NMR 54, 135–143 10.1007/s10858-012-9667-9 [DOI] [PubMed] [Google Scholar]
- 33. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- 34. Lee W., Tonelli M., and Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.