

Synopsis / Abstract:
Patients with chronic liver disease are at increased risk of developing hepatocellular carcinoma (HCC). The majority of patients diagnosed with HCC have limited treatment options and a poor overall prognosis, with a 5-year survival that is less than 15%. Thus, preventing the development of HCC represents the most important and impactful strategy to improve patient outcomes. However, current guidelines lack specific recommendations for the primary prevention of HCC. Emerging data suggest that lifestyle factors – including diabetes, obesity, alcohol consumption and an unhealthy diet – play an increasingly central role in the pathogenesis of HCC, and that primary prevention strategies focused on lifestyle modification could represent an important approach to the prevention of HCC. Moreover, both experimental and epidemiological studies have identified promising chemopreventive agents for the primary prevention of HCC, including statins, aspirin and anti-diabetic drugs. However, additional research is needed to define the optimal approach to lifestyle modification that can achieve maximal HCC risk reduction.
Keywords: Cancer prevention, hepatocellular carcinoma, chemoprevention, lifestyle, modifiable risk factor
Introduction
Hepatocellular carcinoma (HCC) represents the third leading cause of cancer-related mortality worldwide, and is a major cause of death among patients with cirrhosis1. In the United States, the incidence of HCC has tripled over the past 30 years, and mortality rates from HCC are increasing at an alarming pace2, 3. Currently, it is recommended that patients at high risk for developing HCC undergo regular surveillance ultrasonography with assessment of alpha feto-protein (AFP)4. This approach has a sensitivity of 84% (95% CI 76–92) for the detection of any-stage HCC, however the sensitivity of ultrasound for detecting early-stage HCC is only 47% (95% CI 33–61)5. Moreover, the accuracy of ultrasound varies widely with body habitus and operator expertise6, and it is underutilized among high-risk populations7. Thus, HCC is often diagnosed at a late stage, when treatment options are limited and prognosis is poor8. Despite recent advances in treatment, patients diagnosed with HCC have a five-year survival rate of less than 15%, and 70% of patients experience tumor recurrence within five years1, 3. Given these alarming trends, an urgent need remains to develop effective primary prevention strategies that improve patient outcomes by preventing the development of HCC.
HCC risk varies according to the underlying etiology of chronic liver disease, the severity of liver fibrosis, and individual clinical and demographic factors. Major risk factors for HCC include chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, alcohol-related liver disease and nonalcoholic fatty liver disease (NAFLD)9, 10. The vast majority of HCC tumors arise within cirrhotic livers, however HCC may also arise in the absence of cirrhosis, particularly in patients with chronic HBV infection and NAFLD11–13. There are also well-established disparities in the incidence of HCC, with the highest rates observed in men and in racial and ethnic minorities14, 15. Additionally, HCC cases are often clustered in areas of high poverty and unemployment, relative to the general population16. Finally, an increasing body of literature now demonstrates that environmental and lifestyle factors play a key role in the pathogenesis of HCC, including diabetes, obesity, diet and use of certain medications17–21 (Figure 1). Thus, developing comprehensive strategies for HCC prevention requires a thorough assessment of risk, based upon these diverse clinical, demographic, lifestyle and environmental factors.
Figure 1.
Overview of HCC Prevention Strategies
Given the limited treatment options and poor prognosis of HCC, strategies focused on preventing the development of HCC would likely carry the most impact. In this review, we outline recent advances in understanding of modifiable HCC risk factors, that could inform the development of much needed biomarker-based strategies for HCC prevention.
Overview of HCC Prevention Strategies
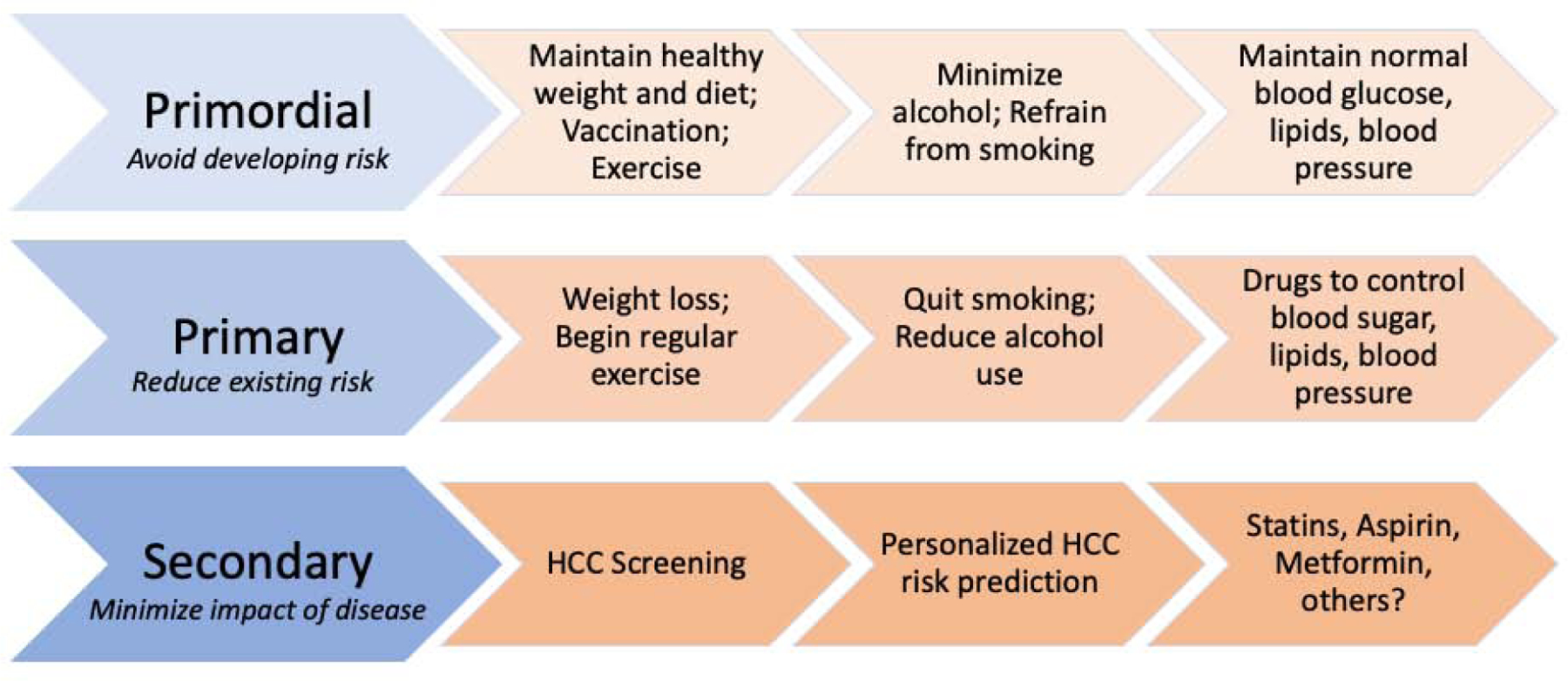
HCC prevention strategies can be applied before or during the natural history of chronic liver disease, and may be categorized as, primordial, primary, secondary and tertiary prevention strategies. Primordial prevention includes behaviors and actions that maintain overall health and thereby prevent the development of risk factors for liver disease. Primary prevention is defined as the modification of behaviors or high-risk exposures in order to reduce risk factors for liver disease. Secondary and tertiary prevention includes screening and surveillance procedures that accurately identify and diagnose existing disease, facilitate early detection and timely interventions for HCC, or which minimize risk of HCC recurrence, among patients with established disease (Figure 2).
Figure 2.

Emerging Risk Factors for Hepatocellular Carcinoma (HCC)
When this framework is applied to HCC prevention, primordial prevention involves maintenance of a healthy body weight, eating a healthy diet with minimization of alcohol use, vaccination against hepatitis B virus (HBV) infection, avoiding smoking, and maintaining normal circulating blood glucose and cholesterol levels. Primary prevention of HCC includes lifestyle and behavioral modification, including making changes to adopt a healthy diet, quit smoking or reduce alcohol consumption, weight loss, or taking medications to control or reduce risk factors, including diabetes, obesity, hypertension and/or dyslipidemia. Among patients with HBV or chronic hepatitis C virus (HCV) infection, the initiation of anti-viral therapy is also considered primary prevention, as the control of HBV DNA or the eradication of HCV infection can control these risk factors and thereby reduce long-term HCC risk. Finally, secondary prevention for patients with high-risk disease or cirrhosis includes engagement in regular HCC surveillance, every 6 months.
HCC risk can be reduced with etiology-specific treatments, which include the use of anti-viral therapy to suppress HBV DNA levels or to eradicate HCV infection, among those with chronic viral hepatitis. These etiology-specific strategies have been reviewed in detail elsewhere22. However, even with such therapies, excess HCC risk may nevertheless persist, particularly in high-risk patients or in those with cirrhosis9, 23. Furthermore, as the prevalence of lifestyle-related liver diseases grows, it is increasingly recognized that primary prevention strategies focused on lifestyle modification are likely to provide the most impactful benefits11, 22. However, to date, the optimal strategy for primary HCC prevention remains undefined.
Established and Emerging Lifestyle Risk Factors for HCC
Accumulating preclinical, clinical and epidemiological evidence demonstrate that modifiable environmental and lifestyle factors play a key role in the pathogenesis of HCC, including diet, alcohol use, obesity, type 2 diabetes and medications (Table 1). Accordingly, lifestyle modification has emerged as an important strategy for the primary prevention of HCC.
Table 1.
Summary of Prior Studies Relating Lifestyle Factors with HCC Risk
| Risk Factor | Relative Risk Estimates* | Proposed mechanisms for HCC prevention |
|---|---|---|
| Obesity | HR 1.95, 95% CI 1.46–2.46 for body mass index (BMI)>30 (vs. BMI<25)38; HR 1.59, 95% CI 1.38–1.83 for elevated waist circumference (WC) vs. normal WC193 | Hyperinsulinemia; lipotoxicity; adipokine disruption; alterations in the gut microbiome and gut-derived metabolites47, 194–196 |
| Diabetes | RR 2.01, 95% CI 1.61–2.51 for diabetes (vs. no diabetes)39 | Insulin resistance, hyperglycemia cause ROS formation, lipotoxicity and increased IGF-I and IGF-II levels, which activate Wnt signaling through PI3K/B-catenin pathways197, 198 |
| Alcohol use | HR 1.16, 95% CI 1.01–1.34 for 3 or more drinks/day (vs. non-drinking)32 | DNA adducts alter DNA repair mechanisms and change protein structure and function; induction of the CYP2E1 enzyme, mitochondrial dysfunction and ROS lead to cellular toxicity199 |
| Diet | HR 0.68, 95% CI 0.51–0.90 for the highest quintile of the Alternative Mediterranean Diet (AMED) score (vs. the lowest quintile)56 | Healthy diet reduces ROS formation and lipotoxicity, inhibits synthesis of pro-inflammatory cytokines and blocks B-catenin and COX-2 signaling pathways75, 200 |
| Coffee | RR 0.71 for consumption of >2 cups of coffee/day (vs. none)66 | Induction of UDP glucuronosyltransferase genes may have antioxidant and cytoprotective effects; caffeine inhibits CTGF and TGF-ß 201, 202 |
| Aspirin | HR 0.69, 95%CI 0.62–0.76 for low dose (<163mg/day) aspirin use (vs. non-use)166 | Inhibition of COX-2 and pro-inflammatory prostaglandins may reduce angiogenesis and tumor cell proliferation137, by blocking protein kinase 3 and NFkB pathways140, 141 |
| Statins | OR 0.63, 95%CI 0.52–0.76 for statin use (vs. non-use)120 | Blockade of diverse carcinogenic pathways governed by Myc, PI3K-Akt, integrins, Rho-dependent kinase, NFkB, and the Hippo signaling pathway102–109 |
| Metformin | OR 0.52, 95%CI 0.40–0.68170 for metformin use (vs. non-use) | AMPK-mediated inhibition of VEGF and HIF1A, preventing angiogenesis and cell signaling167, 203, and suppression of hepatic progenitor cells168 |
Abbreviations: HCC, hepatocellular carcinoma; HR, hazard ratio; RR, relative risk; OR, odds ratio; CI, confidence interval; BMI, body mass index; WC, waist circumference; ROS, reactive oxygen species; COX-2, cyclooxygenase-2; CTGF, connective tissue growth factor; NFkB, nuclear factor kappa B; VEGF, vascular endothelial growth factor; HIF1A, hypoxia-inducible factor 1-alpha; AMPK, adenosine monophosphate-activated protein kinase
Relative risk estimates were selected from meta-analyses (if available) or from the largest published cohort studies to date.
Alcohol
Alcohol use represents a major underlying etiology of HCC. Worldwide, approximately one-third of incident HCC cases are attributable to alcohol, although these rates vary markedly between regions24, 25. For example, the proportion of HCC cases attributable to alcohol is estimated to be 6% in the Middle East, 14% in Northern Africa, 20% in southern Europe, and as high as 63% in some Eastern European countries26. According to the Global Burden of Disease study, approximately 854,000 new primary liver cancers were diagnosed in 2015, and there were 815,000 liver cancer-related deaths26. Of these recorded cases of new primary liver cancer, 245,000 cases (30%) were attributable to alcohol use, with a strong male predominance (204,000 cases)26. In longitudinal cohort studies from France, Spain, Belgium and Japan, the annual incidence of HCC among adults with alcoholic cirrhosis ranged between 2.1–5.6%27–30. While prior studies have developed strategies to predict future HCC risk in this population27, 28, large-scale validation studies are still needed. This represents a research area of important unmet need, for it is projected that the proportion of HCC cases attributable to alcohol is likely to increase in the coming decades, due to the improved efficacy of anti-viral therapies for chronic viral hepatitis, to HBV vaccination strategies, and to the increasing per capita consumption of alcohol that has been recorded in regions of Northern Europe, Eastern Europe, and in the United States24, 31.
Epidemiological studies demonstrate that heavy alcohol use is independently associated with a 1.2-fold higher risk of developing incident HCC, compared to non-drinking32. In a meta-analysis of 19 cohorts and 4,445 incident cases of HCC, alcohol consumption contributed to HCC risk in a dose-dependent manner, with 46% higher risk observed with 50g of alcohol consumption/day, and 66% higher risk with 100g of alcohol consumption/day32. This excess risk is compounded in patients with underlying liver fibrosis24, 33, 34, and alcohol contributes synergistically to the development of HCC in patients with obesity, diabetes33, 34, and chronic HCV infection34. Notably, even after alcohol cessation, the excess observed HCC risk related to alcohol use appears to last for many years35. In a meta-analysis of 4 studies, HCC risk declined by approximately 6% per year with abstinence from alcohol; the authors found that for patients with cirrhosis, it would take approximately 23 years before an individual would achieve the same HCC incidence rates as a non-drinker35.
NAFLD, obesity and diabetes
Worldwide, approximately 25% of adults are affected by nonalcoholic fatty liver disease (NAFLD)17. Closely linked to obesity and diabetes, NAFLD is thought to represent the hepatic manifestation of the metabolic syndrome. While the majority of patients with NAFLD have non-progressive disease, nearly 30% of adults with NAFLD develop nonalcoholic steatohepatitis (NASH) and fibrosis, and among those cases, between 10–20% progress to cirrhosis36. NAFLD represents the most rapidly growing cause of cirrhosis in the United States, and it is also the fastest growing indication for liver transplantation, among adults with HCC37.
Obesity and diabetes are present in 51% and 23% of patients with NAFLD17, and both conditions represent independent risk factors for the development of HCC. Epidemiological studies have linked excess adiposity (defined by, total body weight, body mass index [BMI], waist circumference, etc.) to an increased risk of incident HCC18–20, and to a nearly 2-fold higher risk of HCC-related mortality38. Similarly, type 2 diabetes is significantly and independently associated with excess HCC risk20, 39–42. In a meta-analysis of 23 cohort studies, diabetes was associated with a 2-fold higher pooled relative risk of incident HCC39. Furthermore, recent evidence shows that this risk increases with longer duration of type 2 diabetes42, with additional metabolic comorbidities32, 42, and also that diabetes compounds HCC risk, among patients with chronic viral hepatitis43.
There is increasing awareness of a link between NAFLD and HCC; however, clinical data are limited and conflicting regarding the precise magnitude of this risk. In a 2011 meta-analysis, the 5–10 year risk estimates of HCC incidence ranged from 0% to 38% and displayed marked heterogeneity, due to the small sample sizes and limited numbers of cases of incident HCC, among the included studies44. This limitation was partially addressed by a 2018 retrospective cohort study of 296,707 U.S. Veterans with NAFLD and matched non-NAFLD controls, which found that a diagnosis of NAFLD was associated with a modest but statistically significant increased risk of developing HCC (incidence rate difference, 0.02 per 1000 person-years), and the highest excess risk was observed with NAFLD cirrhosis (incidence rate difference, 10.6 per 1000 person years)12. However, this cohort was primarily male (94%), with NAFLD and cirrhosis identified by administrative codes and/or by surrogate serum fibrosis scores; thus, future studies are still needed in unselected, population-based cohorts, including those with NAFLD histology, to establish more precise and generalizable estimates of HCC risk across the complete NAFLD histological spectrum.
Recent evidence has also suggested that HCC risk might be elevated in patients with NAFLD who do not have cirrhosis45. While prospective studies are still needed to fully define this relationship, it would suggest that the mechanisms that underpin NAFLD-related hepatocarcinogenesis may be less dependent upon liver fibrosis, compared to other etiologies of liver disease. It has been hypothesized that these mechanisms might relate to gut microbial dysbiosis, changes in circulating gut microbiota-derived metabolites (i.e. secondary bile acids or short chain fatty acids), oxidative stress, disruption of circadian rhythms, or dysregulation of circulating and hepatic adipokines and proinflammatory cytokines46–50. Further research is needed in animal models and in human studies, to more precisely characterize these pathways and to translate these findings to novel preventive therapies.
Dietary Patterns
A growing body of clinical and epidemiological evidence suggests that dietary patterns may influence HCC risk. Dietary patterns reflect complex combinations of nutrients and individual compounds that act synergistically within whole foods and across combinations of foods to exert biological effects, which may impact long-term health outcomes51. In one of the earliest observational studies of dietary patterns and incident HCC risk, it was demonstrated that male and female participants in the Shanghai Men’s and Women’s Health Studies who adhered to a vegetable-based dietary pattern had a significantly reduced risk of developing incident HCC52.
More recently, the National Cancer Institute launched the Dietary Patterns Methods Project, which compares validated indices of overall diet quality in relation to incident cancers53. These indices were selected based upon their established associations with cancer and cardiovascular disease54, and include the Alternative Healthy Eating Index (AHEI), the Healthy Eating Index (HEI), the Dietary Approaches to Stop Hypertension (DASH), and the Alternate Mediterranean Diet (AMED). Since that time, three observational cohort studies have evaluated index-based dietary patterns in relation to HCC incidence21, 55, 56. Two of those studies were conducted in three large, prospective U.S. cohort studies (the National Institutes of Health/AARP Diet and Health Study, the Nurses’ Health Study and the Health Professionals Follow-up Study), and found significantly lower HCC risk in participants with higher AHEI-2010 and AMED dietary scores21, 55. The third study included 169,806 adults enrolled in the prospective Multiethnic Cohort study, and found that higher AMED dietary scores were associated with significantly lower risk of incident HCC (adjusted hazard ratio for the highest quintile vs. the lowest quintile, 0.68, 95% CI 0.51–0.90)56. To date, however, published evidence is not yet sufficiently robust to recommend one particular diet for HCC primary prevention.
Individual Foods, Nutrients and Dietary Compounds
Fruit, Vegetables, Meat and Fat
Consumption of fruits and vegetables have also been studied in relation to HCC incidence. In a 2014 meta-analysis of 19 studies (1.29 million subjects and 3,912 cases of incident HCC), each 100g increase in daily vegetable intake was associated with an 8% lower risk of incident HCC, among the included cohort studies (OR 0.92, 95%CI=0.88–0.95)57. In contrast, a null association was found for fruit consumption and incident HCC risk57. Observational cohort studies and case-control studies have also evaluated the intake of red meat, white meat and fish, in relation to incident HCC. In a meta-analysis pooling results from 9 studies, the highest category of daily red meat intake was not significantly associated with increased HCC incidence, compared to the lowest category (pooled OR=1.10, 95%CI=0.85–1.42)58. In contrast, both white meat and fish consumption were significantly associated with reduced HCC risk, when the highest vs. the lowest categories of consumption were compared (pooled OR for white meat and fish=0.69 [95%CI=0.58–0.81])58.
Although human data regarding dietary fat intake and HCC risk are more limited, a notable study included 495,006 older adults enrolled in the prospective NIH-AARP cohort, and observed a higher daily intake of saturated fat at baseline was associated with a significant, 1.9-fold increased risk of incident HCC (HR 1.87, 95%CI=1.23–2.85)59. In contrast, the prospective, European EPIC cohort study did not find a significant association between saturated fats and incident HCC risk (HR 1.08, 95%CI=0.88–1.34), while monounsaturated fats were inversely associated with HCC risk (per each 5g/day, HR 0.71, 95%CI=0.55–0.92)60.
Coffee
Coffee contains well-described anti-inflammatory, antioxidant and antifibrotic properties, and it has been observed that coffee drinkers tend to have lower risk of developing advanced liver disease, including liver fibrosis61, cirrhosis and incident HCC62, 63. Both the World Cancer Research Fund and the International Agency for Research on Cancer have also published reports supporting the beneficial effects of coffee for the prevention of HCC64, 65.
A recent meta-analysis of 26 studies and 1,825 incident HCC cases demonstrated that consumption of at least 2 cups/day of coffee was associated with significantly reduced risk of incident HCC, with a pooled relative risk of 0.7166. Per each additional 2 cups of coffee consumed per day, the magnitude of observed benefit was significantly greater with caffeinated coffee (27% relative risk reduction) than with decaffeinated coffee (14% relative risk reduction)66. Overall, the strength and consistency of the epidemiological associations for coffee has led to recommendations for moderate coffee consumption for HCC prevention in the 2018 guidelines from the European Association for the Study of the Liver (EASL)67. However, several important questions remain unanswered, including the optimal “dose” and preparation of coffee (i.e. espresso vs. drip-coffee, type of coffee bean or roasting process), the optimal timing of initiation and the necessary duration of consumption during the natural history of liver disease, to achieve meaningful risk reduction. Thus, high-quality, prospective studies are needed in well-phenotyped populations, that include more specific details regarding coffee consumption.
Green Tea
Two meta-analyses have evaluated green tea consumption in relation to HCC incidence68, 69. The most recent 2016 meta-analysis included 11 Asian cohort studies of more than 460,000 individuals and 3,694 cases of liver cancer, and demonstrated a pooled relative risk for incident HCC of 0.88 (95% CI=0.81–0.97), when the highest category of green tea intake was compared to the lowest category68. In a dose-response analysis, the authors additionally found that each additional 1 cup of daily green tea was associated with a 3% reduction in HCC risk (95%CI=0.95–1.00). In contrast, data from European cohort studies has been mixed: in the European Prospective Investigation into Cancer and nutrition (EPIC) cohort, persons in the highest quintile of green tea consumption had a 59% lower risk of developing HCC (adjusted HR, 0.41 [95%CI=0.22–0.78]), compared to the lowest quintile70 while two prior Italian case-control studies found null associations71, 72.
Green tea is produced by heating or steaming fresh tea leaves at high temperatures, in processes that result in minimal oxidation and thus preservation of the polyphenols (i.e. catechins) within the tea. Between 50–75% of the primary catechins in green tea are epigallocatechin-3-gallate (EGCG), while the remainder include epigallocatechin, epicatechin-3-gallate (ECG), and epicatechin. In preclinical studies, EGCG inhibits carcinogenesis at numerous sites, including within the liver, albeit with potential risk of hepatotoxicity at high levels73. However, in an epidemiological cohort study, higher levels of urinary catechins were associated with increased HCC risk, among subjects with positive HBV surface antigens, and this excess risk was magnified in patients with low circulating retinol levels (adjusted odds ratio, 2.62, 95%CI=1.25–5.51)74. Given that green tea is the primary source of catechins, these data indicate that further research is needed to understand the relationship between green tea consumption and HCC risk, particularly among patients with chronic HBV infection.
Omega 3 Polyunsaturated Fatty Acids (PUFAs)
Preclinical data suggest that intake of the omega 3 (n-3) PUFAs, eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA) and docosahexaenoic acid (DHA), could prevent hepatocarcinogenesis by inhibiting the pro-inflammatory cyclooxygenase-2 (COX-2) enzyme, which in turn inhibits endogenous biosynthesis of prostaglandins and ß-catenin signaling pathways75, while simultaneously stimulating the endogenous biosynthesis of pro-resolution lipid mediators. When fat-1 transgenic mice (which endogenously form n-3 PUFAs) were compared to wild-type pairs, both the size and number of hepatic tumors was reduced after diethylnitrosamine (DEN) treatment, and hepatic COX-2 expression was significantly reduced, while levels of circulating n-3 PUFA-derived pro-resolution lipid mediators were significantly elevated76. These pro-resolution lipid mediators, which include lipoxins, resolvins, maresins and protectins, are also stimulated by aspirin, and have been shown in murine models to mediate anti-tumor activity (Figure 3)77.
Figure 3.
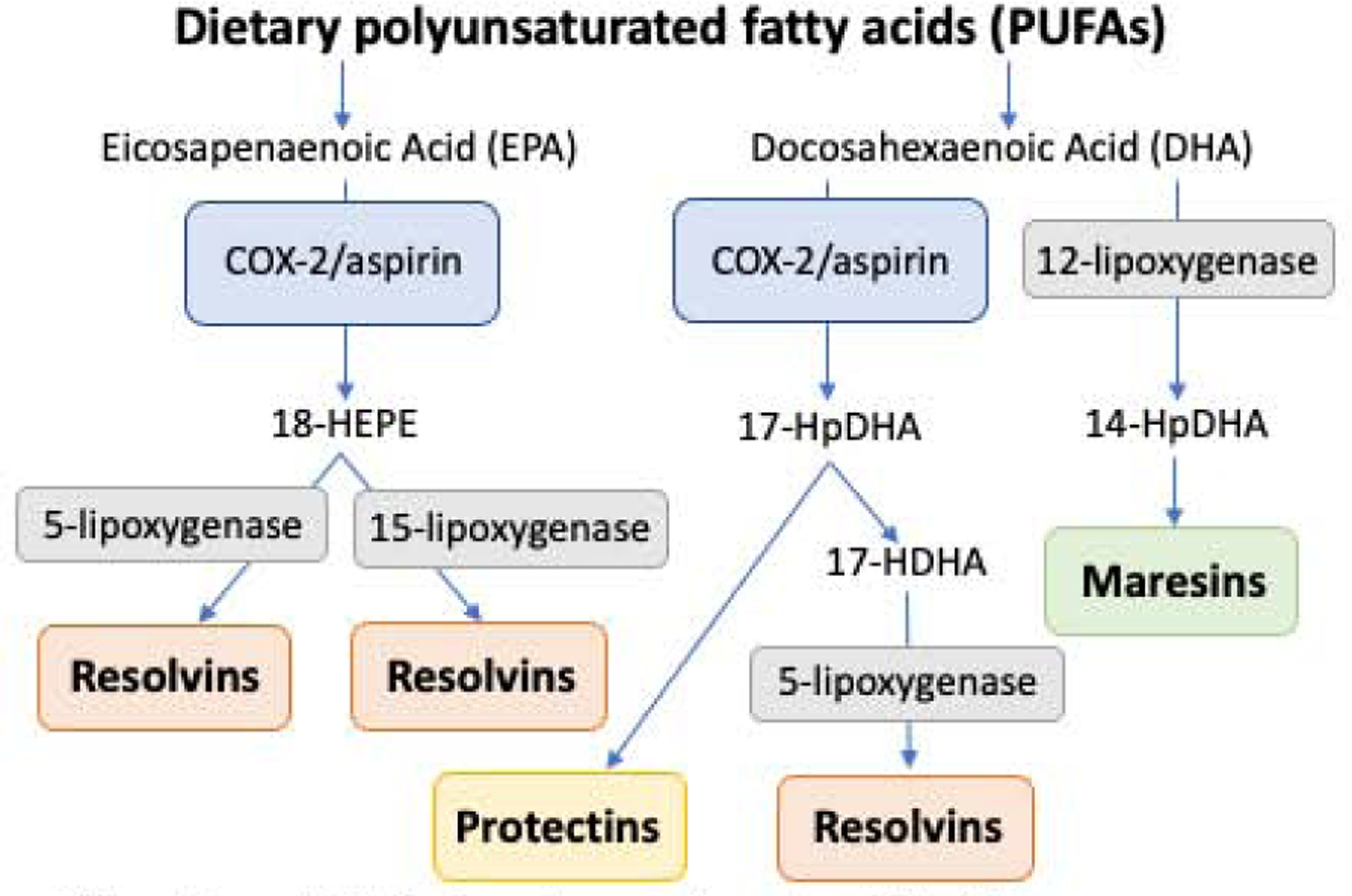
Polyunsaturated fatty acids promote the endogeneous biosynthesis of anti-inflammatory, pro-resolution lipid mediators
Abbreviations: HEPE, hydroxyeicosapentaenoic acid; HpDHA, hydroperoxydocosahexaenoic acid; HDHA, hydroxydocosahexaenoic acid
In a large, prospective cohort study of 90,296 Japanese adults consumption of an n-3 PUFA-rich diet and individual n-3 PUFA supplements were significantly and inversely associated with reduced HCC risk, in a dose-dependent manner78. Specifically, compared to the lowest quintiles of n-3 PUFA intake, the adjusted hazard ratios in the highest quintiles were, 0.64 for n-3 PUFA-rich fish, 0.56 for EPA, 0.64 for DPA, and 0.56 for DHA78. These findings are also supported by prior cohort studies that have similarly demonstrated significant, inverse associations between intake of diets rich in n-3 PUFA-rich fish or white meat, and reduced HCC risk79–81.
Vitamin D
Both preclinical and clinical studies have linked higher levels of Vitamin D, 25(OH)D to reduced HCC incidence. In vitro, administration of 1-alpha,25(OH)2D has pro-apoptotic and antiproliferative effects on numerous cancer cells82–84, and inhibits growth of HCC cell lines85, by modulating cell cycle growth via induction of p21 and p27 tumor suppressor genes and suppression of cyclins and cyclin-dependent kinases86, 87. In humans, higher vitamin D, 25(OH)D, levels have been associated with reduced HCC risk, with a relative risk of 0.5188; in contrast, low 25(OH)D3 levels have been linked to excess HCC risk in patients with chronic HBV infection (adjusted hazard ratio 1.90)89.
Several carcinogenic signaling pathways are hypothesized to be responsive to Vitamin D and its metabolites. First, 1-alpha,25(OH)2D has been shown to downregulate epidermal growth factor (EGF) receptor expression, which inhibits cell growth and promotes cell division, through mitogen-activated protein kinase (MAPK)-dependent pathways90. Second, Vitamin D3 might inhibit VEGF-mediated endothelial cell proliferation and angiogenesis91, 92. Finally, it has been posited that Vitamin D might act on insulin-like growth factor (IGF)-I and II signaling pathways93, which may in turn impact liver cancer cell proliferation.
Branched Chain Amino Acids (BCAAs)
Preclinical data suggest that elevated circulating BCAA levels might protect against hepatocarcinogenesis. In vivo, BCAA treatment enhances mTOR signaling, which reduces both liver fibrosis and HCC94, 95. In HCV-transgenic mice, BCAA administration reduces hepatic iron deposition and decreases reactive oxygen species (ROS) formation96, and in high-fat diet-fed mice with NASH, BCAA therapy represses pro-fibrogenic gene expression in hepatic stellate cells and protects hepatocytes from apoptosis97. Furthermore, in vivo, BCAA therapy suppresses expression of IL-6, IL-1b, IL-18 and tumor necrosis factor (TNF), reducing inflammation in both the liver and white adipose tissues, and inhibiting spontaneous HCC development98.
BCAA therapy has historically been used as a treatment for hepatic encephalopathy, and clinical evidence linking BCAA supplementation to HCC incidence is sparse. In a prospective study of 299 Japanese patients with cirrhosis, those provided with branched-chain amino acid (BCAA) supplementation (5.5–12.0g/day) had a significantly lower risk of developing incident HCC (RR 0.45)99, compared to controls. In a meta-analysis of 11 studies, oral BCAA supplementation in patients with established HCC was associated with improved mortality among Child-Pugh class B patients, and among those with higher levels of albumin (standardized mean difference=0.234), and lower rates of ascites (relative risk, 0.55)100. More recently, in an observational study of 166 patients undergoing evaluation for liver transplantation, reduced plasma levels of valine and the valine to phenylalanine ratio were significantly associated with increased overall mortality101.
Other Dietary Compounds
Numerous additional dietary components and phytochemicals have been examined for their potential role in HCC chemoprevention, including curcumin, resveratrol, flavonoids (including silymarin), and carotenoids. However, to date, robust clinical evidence supporting HCC preventative effects from these compounds in humans are still lacking.
Statins
Statins, or 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, are prescribed for the reduction of low-density lipoprotein (LDL) cholesterol levels. Beyond their cholesterol-lowering effects, statins also exert a diverse array of pleiotropic anti-inflammatory and anti-neoplastic effects. Both in vitro and in vivo studies demonstrate that statins block numerous carcinogenic signaling pathways, including those governed by Myc, Akt, integrins, Rho-dependent kinase, NF-kB, interleukin-6 and the Hippo pathway, among others102–109. By curtailing mevalonate synthesis, statins also inhibit downstream post-translational modification of Ras/Rho signalling proteins, which regulate cellular survival and growth, and they inhibit the cellular breakdown of p21 and p27, thereby permitting these molecules to exert potent growth-inhibitory effects106, 110.
Statins also appear to exert direct anti-fibrotic actions within the liver, which may potentiate their anti-HCC benefits. Liver fibrosis is driven by the activation of hepatic stellate cells (HSCs), which undergo a phenotypic change from a quiescent state to become proliferative myofibroblasts. In preclinical studies, statins inhibit the activation and proliferation of HSCs111–113, by upregulating Kruppel-like factor 2, a transcription factor that promotes HSC quiescence and thereby limits collagen production114. The administration of statins has also been shown to reduce pressures in the portal circulation, which may further limit hepatic fibrogenesis115, potentially through non-canonical Hedgehog signaling pathways116.
Observational studies largely support a link between statin use and reduced HCC risk. We observed a dose-dependent, inverse association between statin use and reduced risk of cirrhosis and incident HCC, among U.S. Veterans with chronic HCV infection117, and also in a post-hoc analysis of a randomized controlled trial118. Kaplan et al. studied a different cohort of U.S. veterans with new diagnoses of cirrhosis, and found significant HCC risk reduction with statin use for at least 90 days or more, compared to non-use119. These findings have been confirmed in several large meta-analyses120–122, which have demonstrated a significant, inverse association between statin use and reduced HCC risk. The observed benefits of statins were most apparent among observational studies, while in contrast, post-hoc analyses of prior randomized controlled trials (RCTs) of statins for cardiovascular disease have failed to demonstrate significant HCC risk reduction. Notably, however, those prior RCTs were not designed or powered to evaluate the long-term effect of statins on HCC incidence. Further, because they excluded patients with cirrhosis, they were comprised of study populations at very low risk for developing incident HCC120, 121.
Emerging evidence further suggests that statin class may also influence HCC risk. Specifically, statins can be broadly divided into lipophilic and hydrophilic subclasses, and it has been hypothesized that lipophilic statins (i.e. atorvastatin, simvastatin, fluvastatin and lovastatin) may confer more potent anti-HCC effects than hydrophilic statins (i.e. pravastatin, rosuvastatin). This hypothesis based upon four lines of evidence. First, in preclinical studies, lipophilic statins suppress viral replication, potentiate antiviral therapy and stimulate anti-tumor immunity to a greater degree than is observed with hydrophilic statins123–126. Second, in the setting of progressive liver fibrosis, the expression of organic anion transporter proteins on the surface of hepatocytes is markedly reduced, and this may prevent hydrophilic statins from entering hepatocytes, whereas lipophilic statins can passively diffuse across cell membranes127, 128. Third, lipophilic statins limit cell growth and promote cellular apoptosis by inducing cell cycle arrest via regulation of Ras/Raf/MEK/ERK signaling108. Further, administration of simvastatin to hepatocyte cell lines enhances expression of the proapoptotic BAX gene and suppresses expression of the antiapoptotic BCL-2 gene, indicating that lipophilic statins can induce apoptosis by acting at the pre-translational level, as well129.
Epidemiological data comparing lipophilic and hydrophilic statins is both limited and conflicting. Specifically, two prior studies did not identify differences in HCC risk with use of lipophilic compared hydrophilic statins121, 130; however, in a large, population-based cohort study of Swedish adults, we recently demonstrated significantly reduced HCC risk among lipophilic statin users, compared to non-users, while the relationship between hydrophilic statin use and incident HCC risk was null131. Although future studies are still needed to confirm these findings, such data suggest that the observed benefits previously associated with statin use in prior studies may have been driven by the unique, class-specific benefits of lipophilic statins.
To date, evidence supporting the use of statins for HCC chemoprevention is not yet sufficient to be incorporated into guidelines. First, published data derive primarily from observational studies, which lack the benefits of randomization, and are prone to selection or confounding by indication bias. Among prior studies, only a few have appropriately balanced the prevalence of underlying HCC risk factors (such as HBV and HCV infection, alcohol-related liver disease, diabetes, obesity and smoking status) between exposure groups119, 131. Such imbalances could introduce confounding by indication, particularly because physicians historically have avoided prescribing statins to patients with liver disease out of concern for hepatotoxicity. Second, high-quality, prospective data are scarce regarding the optimal statin type, necessary duration of use, or the durability of statin-related treatment response, nor is sufficient data available regarding the impact of statins in patients with NAFLD or alcohol-related liver disease. Finally, it remains unknown whether there might be potential additive benefits from the concomitant use of statins together with other medications with putative anti-HCC effects, such as aspirin or metformin. While some prior data suggests that the relationship between statin use and reduced HCC risk is not significantly modified by concurrent aspirin or antidiabetic medication use131, confirmatory studies are needed to validate these findings.
Aspirin
Preclinical evidence supports a role for aspirin in the prevention of HCC. Although the precise mechanisms remain undefined, both cyclooxygenase (COX)-dependent and COX-independent actions have been proposed. Specifically, the inducible, proinflammatory COX-2 enzyme is over-expressed in many cancers associated with obesity and chronic inflammation132, 133, including HCC134, 135, and aspirin irreversibly inhibits COX-2 expression in a dose-dependent manner136. COX-2 expression in hepatocytes promotes the spontaneous development of HCC in mice, by reducing tet methylcytosinedioxygenase 1 (TET1) expression, silencing tumor suppressor genes and activating oncogenic pathways134. Hepatocarcinogenesis has also been linked to hepatic translocation of two gut microbial metabolites, lipoleichoic acid and deoxycholic acid, which promote cellular senescence and upregulate COX-2 expression, driving the production of prostaglandin E2 and suppressing antitumor immunity47. Moreover, by stimulating production of prostaglandins, COX-2 overexpression also promotes angiogenesis and cellular proliferation137–139, by activating the pro-inflammatory protein kinase 3, mammalian target of rapamycin (MTOR) and nuclear factor k B (NFkB) signaling cascades134. In contrast, aspirin inhibits NFkB activation and protein kinase 3 signaling140–142, and in preclinical models, pharmacologic inhibition of COX-2 or prostaglandin E2 prevents the proliferation of liver cancer cells135 and promotes the resolution of liver fibrosis143–145.
The benefits of aspirin in the liver may also derive from the inhibition of platelet activity, which has been shown to limit hepatic inflammation, fibrosis and hepatocarcinogenesis146. Platelets play a central role in promoting accumulation of CD8+ T cells in the liver during chronic viral infection. They also generate platelet-derived growth factor-beta, which activates hepatic stellate cells and promotes fibrosis progression, in rodent models147. Recently, Malehmir et al demonstrated in murine models that antiplatelet therapy with aspirin prevented the development of nonalcoholic steatohepatitis (NASH) and subsequent HCC via inhibition of platelet-derived glycoprotein 1b alpha, which subsequently reduced intrahepatic platelet accumulation, activation and immune cell trafficking148. Together, these lines of evidence provide additional promising mechanistic explanations for the observed hepatoprotective effects of aspirin.
Clinical evidence regarding the impact of aspirin use on HCC incidence derives exclusively from observational studies. studies149–166. Although some have reported conflicting results, most of these observational studies have found a significant, inverse association between aspirin use and reduced risk of incident HCC (Table 2). For example, within two prospective cohorts of U.S. women and men, we demonstrated that regular aspirin use was associated with a significant, 49% lower risk of developing incident HCC (adjusted HR, 0.51, 95%CI 0.34–0.77), and these benefits were both dose- and duration-dependent165. More recently, we confirmed these associations in a nationwide, unselected population of Swedish adults with chronic HBV or HCV infection, in whom low-dose aspirin use (<163mg) was associated with significant, duration-dependent reductions in risk of developing incident HCC (adjusted HR 0.69, 95%CI 0.62–0.76) and in the risk of liver-related mortality (adjusted HR 0.73, 95%CI 0.67–0.81)166. Similarly, a pooled analysis of 10 US-based prospective cohorts (with nearly 1.1 million adults, and 679 incident HCC cases) reported a pooled HR for incident HCC of 0.68 with aspirin use, compared to non-use164. Although these lines of evidence are promising, additional prospective data are still needed to more fully characterize the potential benefits of aspirin across the complete spectrum of chronic liver disease, and also to quantify the potential risks of bleeding associated with aspirin use.
Table 2.
Observational studies of aspirin use and risk of hepatocellular carcinoma (HCC)
| STUDY (AUTHOR, YEAR) | REGION | STUDY DESIGN | HCC CASES (N) | ASPIRIN USERS (N) | TOTAL (N) | HCC RISK (OR, RR, HR; 95% CI) |
|---|---|---|---|---|---|---|
| SIMON, 2020 | Sweden | Retrospective Cohort | 1,612 | 14,205 | 50,275 | 0.69 (0.62–0.76) |
| DU, 2019 | China | Retrospective Cohort | 41 | 59 | 264 | 0.16 (0.04–0.71) |
| LEE, T, 2019 | Korea | Retrospective Cohort | 697 | 2,123 | 10, 615 | 0.70 (0.58–0.86) |
| TSOI, 2019 | Hong Kong | Retrospective Cohort | 9,370 | 204,170 | 612,509 | 0.49 (0.45–0.53) |
| HWANG, 2018 | Korea | Prospective Cohort | 2,336 | 64,782 | 460,755 | 0.87 (0.77–0.98) |
| SIMON, 2018 | USA | Prospective Cohort | 108 | 58,855 | 133,371 | 0.51 (0.34–0.77) |
| LIN, 2018 | Taiwan | Retrospective Cohort | 110 | 3,576 | 18,243 | 0.67 (0.42–1.08) |
| TSENG, 2018 | Taiwan | Retrospective Cohort | 1,750 | 23,112 | 43,800 | 0.83 (0.69–0.99) |
| LEE, M, 2017 | Korea | Retrospective Cohort* | 63 | 343 | 14,392 | 0.34 (0.15–0.77) |
| LEE, T, 2017 | Taiwan | Retrospective Cohort | NR | 5,602 | 18,080 | 0.70 (0.37–1.36) |
| KIM, 2017 | Korea | Case Control | 229 | 390 | 1,374 | 0.34 (0.15–0.78) |
| YANG, 2016 | UK | Case Control | 1,195 | 1,670 | 5,835 | 1.11 (0.86–1.44) |
| PETRICK, 2015 | USA | Prospective Cohort | 679 | 477,470 | 1,084,133 | 0.68 (0.57–0.81) |
| SAHASRABUDDHE, 2012 | USA | Prospective Cohort | 250 | 89,585 | 300,504 | 0.51 (0.35–0.75) |
| CHIU, 2011 | Taiwan | Case Control | 1,166 | 162 | 2,332 | 1.0 (0.73–1.38) |
| FRIIS, 2003 | Danish | Retrospective Cohort | 21 | 29,470 | 29,470 | 1.0 (0.60–1.50) |
| COOGAN, 2000 | USA | Case Control | 51 | 491 | 7,101 | 0.90 (0.30–2.90) |
Abbreviatons: N, number; HCC, hepatocellular carcinoma; OR, odds ratio; RR, relative risk; HR, hazard ratio; CI, confidence interval
Estimates provided are from the propensity score-matched cohort
Metformin
Anti-diabetic medications have also been explored as potential agents for HCC chemoprevention. Among them, the best studied is metformin, a biguanide derivative that blocks gluconeogenesis and enhances peripheral insulin sensitivity. Metformin exerts diverse anti-angiogenic, anti-inflammatory and anti-neoplastic effects; by activating AMPK, metformin inhibits hypoxia inducible factor 1 alpha and vascular endothelial growth factor signaling, which serve to block angiogenesis167. Metformin also suppresses hepatic progenitor cell activation168, and can inhibit cellular proliferation by suppressing NF-kB and reducing the expression of cyclin D1167. Furthermore, in murine models, metformin prevents HSC activation and attenuates fibrosis169, and it also appears to reduce HCC development, particularly when it is initiated before the development of cirrhosis168.
In humans, several prior meta-analyses have demonstrated that metformin use is associated with reduced HCC incidence. The most recent meta-analysis included 19 studies and over 550,000 patients with diabetes, and found a 48% lower risk of incident HCC with metformin use, compared to non-use (pooled OR, 0.52, 95%CI=0.40–0.68)170. Notably, the authors found no significant reduction in HCC incidence in a sub-analysis of 2 post-hoc studies of prior RCTs of metformin use among patients with diabetes (pooled OR=0.84 with metformin use vs. non-use [95%CI=0.10–6.83]); however, those 2 prior RCTs were limited by very few cases of liver cancer, and they were not designed or powered to assess HCC endpoints, thus their findings should be interpreted with caution171.
Pioglitazone, which stimulates the nuclear receptor peroxisome proliferator-activated receptor gamma, has demonstrated efficacy for reducing liver fat and inflammation, in patients with established nonalcoholic steatohepatitis (NASH)172; however, whether this translates to reduced HCC risk is still unknown. To date, one case-control study reported significantly reduced HCC risk with use of pioglitazone, compared to non-use (OR 0.83, 95%CI 0.72–0.95)173, and two studies have found significant risk reduction with use of any thiazolidinedione medication, compared to non-use174, 175, however others have demonstrated null associations176, 177. Finally, although glucagon-like peptide-1 (GLP-1) receptor agonists have demonstrated short-term efficacy for the resolution of NASH178, very little is currently known about the long-term impact of GLP-1 receptor agonists or dipeptidyl peptidase-4 (DPP-4) inhibitors on HCC incidence.
Other Potential Chemopreventive Drugs
Although published data are limited, several other medications could represent plausible agents for HCC chemoprevention, including angiotensin converting enzyme (ACE) inhibitors and menopausal hormone therapy. The renin-angiotensin axis participates in liver fibrogenesis and hepatocarcinogenesis179, and by activating NFkB, angiotensin II can promote the survival of hepatic myofibroblasts, but this effect is reversed with captopril treatment180. Moreover, telmisartan, an antiotension II type 1 receptor blocker (ARB), can prevent fibrosis and HCC development in rodents181. Finally, it is well-established that there are marked sex disparities in the incidence of HCC, with men being affected more frequently than women, leading to the hypothesis that estrogen may protect against HCC incidence. In support of this, a case-control study of 234 women with treated HCC and 282 healthy controls demonstrated that menopausal hormone therapy use was associated with reduced odds of developing HCC182. A meta-analysis of 87 studies also found that variants in the estrogen receptor 1 (ESR1) gene were associated with excess HCC risk183. Moreover, in a large consortium of prospective U.S. cohort studies, bilateral oophorectomy was significantly associated with increased HCC incidence (HR=2.67, 95%CI=1.22–5.85), after accounting for other lifestyle and clinical factors and duration of exposure to exogenous hormone therapy184. 185
The Potential Impact of Lifestyle Modification for HCC Risk Reduction
Given the growing prevalence of chronic liver disease attributable to an unhealthy lifestyle, and the significant associations between high-risk lifestyle factors and excess HCC risk, HCC prevention strategies focused on adopting a low-risk lifestyle would likely offer substantial benefits. However, in order to identify priorities for public health interventions, it is important to quantify the magnitude of contribution of lifestyle factors to HCC risk. Using two nationwide, prospective U.S. cohort studies, we recently demonstrated that over 80% of HCC cases could theoretically have been prevented, with adherence to low-risk lifestyle. Such data underscore the enormous potential impact of primary HCC prevention efforts focused on lifestyle modification. Nevertheless, important knowledge gaps still remain. In order to translate such data into meaningful recommendations, well-designed, prospective studies are needed to define the optimal approaches to lifestyle modification to achieve clinically meaningful and durable HCC risk reduction, in patients who are at high risk of developing incident HCC.
Challenges and Future Directions
Progress in the development and clinical translation of HCC prevention strategies has thus far been limited by four important barriers. First, despite promising associations between low-risk lifestyle factors and reduced HCC risk, data are lacking regarding the optimal approaches to lifestyle modification that might translate to effective and durable HCC risk reduction, in high-risk populations. Second, the molecular mechanisms of hepatocarcinogenesis remain largely uncharacterized11, 186, due to the genetic heterogeneity of HCC tumors187, and to suboptimal animal models188, which limit the ability to translate hypotheses from preclinical studies to humans. Third, research into other cancers benefits from ready access to tumor biospecimens, precursor lesions and adjacent normal tissues, which facilitates the discovery and validation of targeted, molecular chemoprevention strategies189. In contrast, access to HCC specimens is more difficult, as HCC tumors may be diagnosed without confirmatory pathological specimens. While there have been promising recent developments in molecular tools for HCC risk prediction, and in the use of liquid biopsy, the clinical utility of these approaches is not yet established11.
Finally, a major challenge has been the need for large numbers of subjects and prolonged follow-up times, in HCC chemoprevention trials. It has been hypothesized that these requirements for large populations and prolonged followed are due to the inclusion of heterogeneous study populations, which dilute potential treatment effects. For example, two large chemoprevention trials of low-dose interferon therapy for patients with advanced fibrosis or cirrhosis failed to demonstrate significant HCC risk reduction with treatment189–191. However, among patients with cirrhosis – the subgroup at highest risk of developing HCC – a significant treatment benefit was found. Thus, it is plausible that enrollment of an enriched, high-risk study population might maximize the potential to detect a treatment effect. This in turn would enable the design of more feasible and efficient clinical trials, requiring smaller numbers of patients and shorter follow-up times186.
In addition to risk-stratified enrollment, biomarker-based HCC chemoprevention trials are needed. To achieve this goal, such biomarkers must, (1) predict future risk of HCC development, (2) predict response to chemoprevention therapy, and (3) provide insight into drug pharmacokinetics. Recently, molecular biomarkers of HCC risk have been developed and are undergoing rigorous validation for these purposes11. For example, liver tissue-derived transcriptomic signatures have been validated for predicting incident HCC risk among patients with cirrhosis of any etiology, including chronic HBV or HCV infection, alcohol related liver disease, and NAFLD192. Based upon these results, enrollment was recently completed for a Phase I/II clinical trial of Erlotinib for the prevention of HCC in patients with cirrhosis (NCT02273362); this trial utilized a liver tissue transcriptomic prognostic signature as a selection factor for study enrollment and as a surrogate, biomarker-based endpoint. Demonstrating that a high-risk transcriptomic signature predicts meaningful HCC risk reduction with Erlotinib would form a strong scientific rationale for future biomarker-driven HCC chemoprevention trials that utilize molecular-based risk-stratified enrollment procedures and validated, surrogate biomarker endpoints for HCC186. Such trials would have enhanced feasibility, overcoming many of the barriers outlined above, and would therefore enable more rapid translation of preclinical discoveries to humans.
Conclusions
Given the diversity of HCC and its underlying risk factors, strategies for primordial and primary HCC prevention are likely to have broad clinical applicability for patients with chronic liver disease. Lifestyle modification or the repurposing of medications like statins, aspirin or metformin, could be readily combined with etiology-specific HCC prevention strategies, and might offer synergistic benefits. In parallel, research to better characterize the molecular determinants of HCC will help elucidate much needed prognostic biomarkers, and thereby enable the design of more efficient, biomarker-based HCC chemoprevention trials. Ultimately, combining lifestyle modification strategies with the use of safe, generic compounds and targeted biomarkers for predicting HCC risk could provide a robust and cost-effective strategy for HCC chemoprevention among at-risk patients with chronic liver disease.
Key Points:
Cancer chemoprevention approaches can include primordial, primary or secondary prevention strategies; primary prevention strategies include modification of behaviors or high-risk exposures in order to eliminate risk factors for chronic liver disease.
Epidemiological data demonstrate that modifiable lifestyle factors contribute to the pathogenesis of HCC, including an unhealthy diet, alcohol use, obesity, type 2 diabetes and non-use of certain medications, including aspirin and statins.
Lifestyle modification or the repurposing of medications used for other conditions – including statins, aspirin or metformin – represent novel and important strategies for the primary prevention of HCC.
Research to define the molecular determinants of HCC could help elucidate much-needed prognostic biomarkers and thereby facilitate the design of more efficient, biomarker-driven HCC chemoprevention trials.
Acknowledgments
Grant Support
NIH K24 DK098311 (ATC)
NIH K23 DK122104 (TGS)
Dr. Simon is supported by the Harvard University Center for AIDS Research (Career Development Award).
Dr. Chan is a Stuart and Suzanne Steele MGH Research Scholar
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures and conflicts of interest:
Dr. Chan has previously served as a consultant for Bayer Pharma AG, Janssen Pharmaceuticals, and Pfizer Inc. and Boehringer Ingelheim for unrelated work.
Dr. Simon has no disclosures and no conflicts of interest to disclose.
REFERENCES
- 1.Bertuccio P, Turati F, Carioli G, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol 2017;67:302–309. [DOI] [PubMed] [Google Scholar]
- 2.Mokdad AH, Dwyer-Lindgren L, Fitzmaurice C, et al. Trends and Patterns of Disparities in Cancer Mortality Among US Counties, 1980–2014. JAMA 2017;317:388–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–1953. [DOI] [PubMed] [Google Scholar]
- 4.Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358–380. [DOI] [PubMed] [Google Scholar]
- 5.Tzartzeva K, Obi J, Rich NE, et al. Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients With Cirrhosis: A Meta-analysis. Gastroenterology 2018;154:1706–1718 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simmons O, Fetzer DT, Yokoo T, et al. Predictors of adequate ultrasound quality for hepatocellular carcinoma surveillance in patients with cirrhosis. Aliment Pharmacol Ther 2017;45:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singal AG, Yopp AC, Gupta S, et al. Failure rates in the hepatocellular carcinoma surveillance process. Cancer Prev Res (Phila) 2012;5:1124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parikh ND, Singal AG, Hutton DW. Cost effectiveness of regorafenib as second-line therapy for patients with advanced hepatocellular carcinoma. Cancer 2017;123:3725–3731. [DOI] [PubMed] [Google Scholar]
- 9.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 2008;371:838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol 2013;47 Suppl:S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujiwara N, Friedman SL, Goossens N, et al. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol 2018;68:526–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanwal F, Kramer JR, Mapakshi S, et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018;155:1828–1837 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim J, Singal AG. Surveillance and Diagnosis of Hepatocellular Carcinoma. Clin Liver Dis (Hoboken) 2019;13:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim AK, Singal AG. Health disparities in diagnosis and treatment of hepatocellular carcinoma. Clin Liver Dis (Hoboken) 2014;4:143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu L, Sloane DA, Guo C, et al. Risk factors for primary hepatocellular carcinoma in black and white Americans in 2000. Clin Gastroenterol Hepatol 2006;4:355–60. [DOI] [PubMed] [Google Scholar]
- 16.Shebl FM, Capo-Ramos DE, Graubard BI, et al. Socioeconomic status and hepatocellular carcinoma in the United States. Cancer Epidemiol Biomarkers Prev 2012;21:1330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 18.Hagstrom H, Tynelius P, Rasmussen F. High BMI in late adolescence predicts future severe liver disease and hepatocellular carcinoma: a national, population-based cohort study in 1.2 million men. Gut 2018;67:1536–1542. [DOI] [PubMed] [Google Scholar]
- 19.Hassan MM, Abdel-Wahab R, Kaseb A, et al. Obesity Early in Adulthood Increases Risk but Does Not Affect Outcomes of Hepatocellular Carcinoma. Gastroenterology 2015;149:119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell PT, Newton CC, Freedman ND, et al. Body Mass Index, Waist Circumference, Diabetes, and Risk of Liver Cancer for U.S. Adults. Cancer Res 2016;76:6076–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y, Yang W, Simon TG, et al. Dietary Patterns and Risk of Hepatocellular Carcinoma Among U.S. Men and Women. Hepatology 2019;70:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh S, Singh PP, Roberts LR, et al. Chemopreventive strategies in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2014;11:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loomba R, Yang HI, Su J, et al. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: a prospective cohort study. Am J Epidemiol 2013;177:333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pimpin L, Cortez-Pinto H, Negro F, et al. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J Hepatol 2018;69:718–735. [DOI] [PubMed] [Google Scholar]
- 25.Schutze M, Boeing H, Pischon T, et al. Alcohol attributable burden of incidence of cancer in eight European countries based on results from prospective cohort study. BMJ 2011;342:d1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Global Burden of Disease Liver Cancer C, Akinyemiju T, Abera S, et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol 2017;3:1683–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mancebo A, Gonzalez-Dieguez ML, Cadahia V, et al. Annual incidence of hepatocellular carcinoma among patients with alcoholic cirrhosis and identification of risk groups. Clin Gastroenterol Hepatol 2013;11:95–101. [DOI] [PubMed] [Google Scholar]
- 28.Ganne-Carrie N, Chaffaut C, Bourcier V, et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J Hepatol 2018;69:1274–1283. [DOI] [PubMed] [Google Scholar]
- 29.Kodama K, Tokushige K, Hashimoto E, et al. Hepatic and extrahepatic malignancies in cirrhosis caused by nonalcoholic steatohepatitis and alcoholic liver disease. Alcohol Clin Exp Res 2013;37 Suppl 1:E247–52. [DOI] [PubMed] [Google Scholar]
- 30.Torisu Y, Ikeda K, Kobayashi M, et al. Diabetes mellitus increases the risk of hepatocarcinogenesis in patients with alcoholic cirrhosis: A preliminary report. Hepatol Res 2007;37:517–23. [DOI] [PubMed] [Google Scholar]
- 31.Haughwout Sarah P and Megan E Slater; Apparent Per Capita Alcohol Consumption: National S, and Regional Trends, 1977–2016; National Institute on Alcohol Abuse and Alcoholism, Surveillance Report #110; https://pubs.niaaa.nih.gov/publications/surveillance110/CONS16.htm; accessed 5/17/2020.
- 32.Turati F, Galeone C, Rota M, et al. Alcohol and liver cancer: a systematic review and meta-analysis of prospective studies. Ann Oncol 2014;25:1526–35. [DOI] [PubMed] [Google Scholar]
- 33.Grewal P, Viswanathen VA. Liver cancer and alcohol. Clin Liver Dis 2012;16:839–50. [DOI] [PubMed] [Google Scholar]
- 34.Hassan MM, Hwang LY, Hatten CJ, et al. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002;36:1206–13. [DOI] [PubMed] [Google Scholar]
- 35.Heckley GA, Jarl J, Asamoah BO, et al. How the risk of liver cancer changes after alcohol cessation: a review and meta-analysis of the current literature. BMC Cancer 2011;11:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274–85. [DOI] [PubMed] [Google Scholar]
- 37.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014;59:2188–95. [DOI] [PubMed] [Google Scholar]
- 38.Gupta A, Das A, Majumder K, et al. Obesity is Independently Associated With Increased Risk of Hepatocellular Cancer-related Mortality: A Systematic Review and Meta-Analysis. Am J Clin Oncol 2018;41:874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C, Wang X, Gong G, et al. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: a systematic review and meta-analysis of cohort studies. Int J Cancer 2012;130:1639–48. [DOI] [PubMed] [Google Scholar]
- 40.Lai SW, Chen PC, Liao KF, et al. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol 2012;107:46–52. [DOI] [PubMed] [Google Scholar]
- 41.Chen HF, Chen P, Li CY. Risk of malignant neoplasms of liver and biliary tract in diabetic patients with different age and sex stratifications. Hepatology 2010;52:155–63. [DOI] [PubMed] [Google Scholar]
- 42.Simon TG, King LY, Chong DQ, et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: Results from two prospective cohort studies. Hepatology 2018;67:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan Y, Zhang X, Zhang W, et al. The Influence of Metabolic Syndrome on the Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Infection in Mainland China. Cancer Epidemiol Biomarkers Prev 2019;28:2038–2046. [DOI] [PubMed] [Google Scholar]
- 44.White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012;10:1342–1359 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol 2016;14:124–31 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loo TM, Kamachi F, Watanabe Y, et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov 2017;7:522–538. [DOI] [PubMed] [Google Scholar]
- 47.Yoshimoto S, Loo TM, Atarashi K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97–101. [DOI] [PubMed] [Google Scholar]
- 48.Kettner NM, Voicu H, Finegold MJ, et al. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016;30:909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes AL, Teijeiro A, Buren S, et al. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016;30:161–175. [DOI] [PubMed] [Google Scholar]
- 51.Tapsell LC, Neale EP, Satija A, et al. Foods, Nutrients, and Dietary Patterns: Interconnections and Implications for Dietary Guidelines. Adv Nutr 2016;7:445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Xiang YB, Li HL, et al. Vegetable-based dietary pattern and liver cancer risk: results from the Shanghai women’s and men’s health studies. Cancer Sci 2013;104:1353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.National Cancer Institute. Applied Research: Cancer Control and Population Sciences Dietary Patterns Methods Project. Rockville: MNCI, 2018. [Google Scholar]
- 54.Chiuve SE, Fung TT, Rimm EB, et al. Alternative dietary indices both strongly predict risk of chronic disease. J Nutr 2012;142:1009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li WQ, Park Y, McGlynn KA, et al. Index-based dietary patterns and risk of incident hepatocellular carcinoma and mortality from chronic liver disease in a prospective study. Hepatology 2014;60:588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bogumil D, Park SY, Le Marchand L, et al. High-Quality Diets Are Associated With Reduced Risk of Hepatocellular Carcinoma and Chronic Liver Disease: The Multiethnic Cohort. Hepatol Commun 2019;3:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y, Zhang D, Feng N, et al. Increased intake of vegetables, but not fruit, reduces risk for hepatocellular carcinoma: a meta-analysis. Gastroenterology 2014;147:1031–42. [DOI] [PubMed] [Google Scholar]
- 58.Luo J, Yang Y, Liu J, et al. Systematic review with meta-analysis: meat consumption and the risk of hepatocellular carcinoma. Aliment Pharmacol Ther 2014;39:913–22. [DOI] [PubMed] [Google Scholar]
- 59.Freedman ND, Cross AJ, McGlynn KA, et al. Association of meat and fat intake with liver disease and hepatocellular carcinoma in the NIH-AARP cohort. J Natl Cancer Inst 2010;102:1354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duarte-Salles T, Fedirko V, Stepien M, et al. Dietary fat, fat subtypes and hepatocellular carcinoma in a large European cohort. Int J Cancer 2015;137:2715–28. [DOI] [PubMed] [Google Scholar]
- 61.Alferink LJM, Fittipaldi J, Kiefte-de Jong JC, et al. Coffee and herbal tea consumption is associated with lower liver stiffness in the general population: The Rotterdam study. J Hepatol 2017;67:339–348. [DOI] [PubMed] [Google Scholar]
- 62.Saab S, Mallam D, Cox GA 2nd, et al. Impact of coffee on liver diseases: a systematic review. Liver Int 2014;34:495–504. [DOI] [PubMed] [Google Scholar]
- 63.Bravi F, Tavani A, Bosetti C, et al. Coffee and the risk of hepatocellular carcinoma and chronic liver disease: a systematic review and meta-analysis of prospective studies. Eur J Cancer Prev 2017;26:368–377. [DOI] [PubMed] [Google Scholar]
- 64.Loomis D, Guyton KZ, Grosse Y, et al. Carcinogenicity of drinking coffee, mate, and very hot beverages. Lancet Oncol 2016;17:877–878. [DOI] [PubMed] [Google Scholar]
- 65.World Cancer Research Fund International/American Institute for Cancer Research. Continuous Update Project Report: diet N, Physical Activity and liver Cancer. 2015. wcrf.org/sites/default/files/Liver-Cancer-2015-Report.pdf; Accessed 05/01/2020.
- 66.Kennedy OJ, Roderick P, Buchanan R, et al. Coffee, including caffeinated and decaffeinated coffee, and the risk of hepatocellular carcinoma: a systematic review and dose-response meta-analysis. BMJ Open 2017;7:e013739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol 2018;69:182–236. [DOI] [PubMed] [Google Scholar]
- 68.Huang YQ, Lu X, Min H, et al. Green tea and liver cancer risk: A meta-analysis of prospective cohort studies in Asian populations. Nutrition 2016;32:3–8. [DOI] [PubMed] [Google Scholar]
- 69.Fon Sing M, Yang WS, Gao S, et al. Epidemiological studies of the association between tea drinking and primary liver cancer: a meta-analysis. Eur J Cancer Prev 2011;20:157–65. [DOI] [PubMed] [Google Scholar]
- 70.Bamia C, Lagiou P, Jenab M, et al. Coffee, tea and decaffeinated coffee in relation to hepatocellular carcinoma in a European population: multicentre, prospective cohort study. Int J Cancer 2015;136:1899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Montella M, Polesel J, La Vecchia C, et al. Coffee and tea consumption and risk of hepatocellular carcinoma in Italy. Int J Cancer 2007;120:1555–9. [DOI] [PubMed] [Google Scholar]
- 72.La Vecchia C, Negri E, Franceschi S, et al. Tea consumption and cancer risk. Nutr Cancer 1992;17:27–31. [DOI] [PubMed] [Google Scholar]
- 73.Lambert JD, Kennett MJ, Sang S, et al. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem Toxicol 2010;48:409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butler LM, Huang JY, Wang R, et al. Urinary biomarkers of catechins and risk of hepatocellular carcinoma in the Shanghai Cohort Study. Am J Epidemiol 2015;181:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lim K, Han C, Dai Y, et al. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol Cancer Ther 2009;8:3046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weylandt KH, Krause LF, Gomolka B, et al. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis 2011;32:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gilligan MM, Gartung A, Sulciner ML, et al. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc Natl Acad Sci U S A 2019;116:6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sawada N, Inoue M, Iwasaki M, et al. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology 2012;142:1468–75. [DOI] [PubMed] [Google Scholar]
- 79.Yu SZ, Huang XE, Koide T, et al. Hepatitis B and C viruses infection, lifestyle and genetic polymorphisms as risk factors for hepatocellular carcinoma in Haimen, China. Jpn J Cancer Res 2002;93:1287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang MP, Thomas GN, Ho SY, et al. Fish consumption and mortality in Hong Kong Chinese--the LIMOR study. Ann Epidemiol 2011;21:164–9. [DOI] [PubMed] [Google Scholar]
- 81.Talamini R, Polesel J, Montella M, et al. Food groups and risk of hepatocellular carcinoma: A multicenter case-control study in Italy. Int J Cancer 2006;119:2916–21. [DOI] [PubMed] [Google Scholar]
- 82.Chiang KC, Chen TC. Vitamin D for the prevention and treatment of pancreatic cancer. World J Gastroenterol 2009;15:3349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chiang KC, Persons KS, Istfan NW, et al. Fish oil enhances the antiproliferative effect of 1alpha,25-dihydroxyvitamin D3 on liver cancer cells. Anticancer Res 2009;29:3591–6. [PubMed] [Google Scholar]
- 84.Flanagan JN, Zheng S, Chiang KC, et al. Evaluation of 19-nor-2alpha-(3-hydroxypropyl)-1alpha,25-dihydroxyvitamin D3 as a therapeutic agent for androgen-dependent prostate cancer. Anticancer Res 2009;29:3547–53. [PubMed] [Google Scholar]
- 85.Pourgholami MH, Akhter J, Lu Y, et al. In vitro and in vivo inhibition of liver cancer cells by 1,25-dihydroxyvitamin D3. Cancer Lett 2000;151:97–102. [DOI] [PubMed] [Google Scholar]
- 86.Caputo A, Pourgholami MH, Akhter J, et al. 1,25-Dihydroxyvitamin D(3) induced cell cycle arrest in the human primary liver cancer cell line HepG2. Hepatol Res 2003;26:34–39. [DOI] [PubMed] [Google Scholar]
- 87.Hager G, Formanek M, Gedlicka C, et al. 1,25(OH)2 vitamin D3 induces elevated expression of the cell cycle-regulating genes P21 and P27 in squamous carcinoma cell lines of the head and neck. Acta Otolaryngol 2001;121:103–9. [DOI] [PubMed] [Google Scholar]
- 88.Fedirko V, Duarte-Salles T, Bamia C, et al. Prediagnostic circulating vitamin D levels and risk of hepatocellular carcinoma in European populations: a nested case-control study. Hepatology 2014;60:1222–30. [DOI] [PubMed] [Google Scholar]
- 89.Wong GL, Chan HL, Chan HY, et al. Adverse effects of vitamin D deficiency on outcomes of patients with chronic hepatitis B. Clin Gastroenterol Hepatol 2015;13:783–90 e1. [DOI] [PubMed] [Google Scholar]
- 90.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 2007;7:684–700. [DOI] [PubMed] [Google Scholar]
- 91.Chung I, Wong MK, Flynn G, et al. Differential antiproliferative effects of calcitriol on tumor-derived and matrigel-derived endothelial cells. Cancer Res 2006;66:8565–73. [DOI] [PubMed] [Google Scholar]
- 92.Iseki K, Tatsuta M, Uehara H, et al. Inhibition of angiogenesis as a mechanism for inhibition by 1alpha-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 of colon carcinogenesis induced by azoxymethane in Wistar rats. Int J Cancer 1999;81:730–3. [DOI] [PubMed] [Google Scholar]
- 93.Scharf JG, Dombrowski F, Ramadori G. The IGF axis and hepatocarcinogenesis. Mol Pathol 2001;54:138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cha JH, Bae SH, Kim HL, et al. Branched-chain amino acids ameliorate fibrosis and suppress tumor growth in a rat model of hepatocellular carcinoma with liver cirrhosis. PLoS One 2013;8:e77899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakano M, Nakashima A, Nagano T, et al. Branched-chain amino acids enhance premature senescence through mammalian target of rapamycin complex I-mediated upregulation of p21 protein. PLoS One 2013;8:e80411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korenaga M, Nishina S, Korenaga K, et al. Branched-chain amino acids reduce hepatic iron accumulation and oxidative stress in hepatitis C virus polyprotein-expressing mice. Liver Int 2015;35:1303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takegoshi K, Honda M, Okada H, et al. Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a non-alcoholic steatohepatitis mouse model. Oncotarget 2017;8:18191–18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Terakura D, Shimizu M, Iwasa J, et al. Preventive effects of branched-chain amino acid supplementation on the spontaneous development of hepatic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Carcinogenesis 2012;33:2499–506. [DOI] [PubMed] [Google Scholar]
- 99.Kawaguchi T, Shiraishi K, Ito T, et al. Branched-chain amino acids prevent hepatocarcinogenesis and prolong survival of patients with cirrhosis. Clin Gastroenterol Hepatol 2014;12:1012–8 e1. [DOI] [PubMed] [Google Scholar]
- 100.Chen L, Chen Y, Wang X, et al. Efficacy and safety of oral branched-chain amino acid supplementation in patients undergoing interventions for hepatocellular carcinoma: a meta-analysis. Nutr J 2015;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kinny-Koster B, Bartels M, Becker S, et al. Plasma Amino Acid Concentrations Predict Mortality in Patients with End-Stage Liver Disease. PLoS One 2016;11:e0159205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Higashi T, Hayashi H, Kitano Y, et al. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med Oncol 2016;33:123. [DOI] [PubMed] [Google Scholar]
- 103.Wang J, Tokoro T, Higa S, et al. Anti-inflammatory effect of pitavastatin on NF-kappaB activated by TNF-alpha in hepatocellular carcinoma cells. Biol Pharm Bull 2006;29:634–9. [DOI] [PubMed] [Google Scholar]
- 104.Relja B, Meder F, Wang M, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol 2011;38:879–85. [DOI] [PubMed] [Google Scholar]
- 105.Roudier E, Mistafa O, Stenius U. Statins induce mammalian target of rapamycin (mTOR)-mediated inhibition of Akt signaling and sensitize p53-deficient cells to cytostatic drugs. Mol Cancer Ther 2006;5:2706–15. [DOI] [PubMed] [Google Scholar]
- 106.Cao Z, Fan-Minogue H, Bellovin DI, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011;71:2286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang PM, Liu YL, Lin YC, et al. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res 2010;70:7699–709. [DOI] [PubMed] [Google Scholar]
- 108.Sutter AP, Maaser K, Hopfner M, et al. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J Hepatol 2005;43:808–16. [DOI] [PubMed] [Google Scholar]
- 109.Kah J, Wustenberg A, Keller AD, et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol Rep 2012;28:1077–83. [DOI] [PubMed] [Google Scholar]
- 110.Rao S, Porter DC, Chen X, et al. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci U S A 1999;96:7797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trebicka J, Hennenberg M, Odenthal M, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol 2010;53:702–12. [DOI] [PubMed] [Google Scholar]
- 112.Miyaki T, Nojiri S, Shinkai N, et al. Pitavastatin inhibits hepatic steatosis and fibrosis in non-alcoholic steatohepatitis model rats. Hepatol Res 2011;41:375–85. [DOI] [PubMed] [Google Scholar]
- 113.Wang W, Zhao C, Zhou J, et al. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS One 2013;8:e76538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marrone G, Maeso-Diaz R, Garcia-Cardena G, et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut 2014. [DOI] [PubMed] [Google Scholar]
- 115.Abraldes JG, Albillos A, Banares R, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651–8. [DOI] [PubMed] [Google Scholar]
- 116.Uschner FE, Ranabhat G, Choi SS, et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci Rep 2015;5:14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Simon TG, Bonilla H, Yan P, et al. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: Results from ERCHIVES. Hepatology 2016;64:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Simon TG, King LY, Zheng H, et al. Statin use is associated with a reduced risk of fibrosis progression in chronic hepatitis C. J Hepatol 2015;62:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaplan DE, Serper MA, Mehta R, et al. Effects of Hypercholesterolemia and Statin Exposure on Survival in a Large National Cohort of Patients With Cirrhosis. Gastroenterology 2019;156:1693–1706 e12. [DOI] [PubMed] [Google Scholar]
- 120.Singh S, Singh PP, Singh AG, et al. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology 2013;144:323–32. [DOI] [PubMed] [Google Scholar]
- 121.Shi M, Zheng H, Nie B, et al. Statin use and risk of liver cancer: an update meta-analysis. BMJ Open 2014;4:e005399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pradelli D, Soranna D, Scotti L, et al. Statins and primary liver cancer: a meta-analysis of observational studies. Eur J Cancer Prev 2013;22:229–34. [DOI] [PubMed] [Google Scholar]
- 123.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab 2010;21:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci U S A 2005;102:2561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xia Y, Xie Y, Yu Z, et al. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell 2018;175:1059–1073 e21. [DOI] [PubMed] [Google Scholar]
- 126.Bader T, Korba B. Simvastatin potentiates the anti-hepatitis B virus activity of FDA-approved nucleoside analogue inhibitors in vitro. Antiviral Res 2010;86:241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kato S, Smalley S, Sadarangani A, et al. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med 2010;14:1180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thakkar N, Slizgi JR, Brouwer KLR. Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci 2017;106:2282–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spampanato C, De Maria S, Sarnataro M, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol 2012;40:935–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tsan YT, Lee CH, Wang JD, et al. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J Clin Oncol 2012;30:623–30. [DOI] [PubMed] [Google Scholar]
- 131.Simon TG, Duberg AS, Aleman S, et al. Lipophilic Statins and Risk for Hepatocellular Carcinoma and Death in Patients With Chronic Viral Hepatitis: Results From a Nationwide Swedish Population. Ann Intern Med 2019;171:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med 2007;356:2131–42. [DOI] [PubMed] [Google Scholar]
- 133.Singh B, Berry JA, Shoher A, et al. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol 2005;26:1393–9. [PubMed] [Google Scholar]
- 134.Chen H, Cai W, Chu ESH, et al. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 2017;36:4415–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kern MA, Schubert D, Sahi D, et al. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002;36:885–94. [DOI] [PubMed] [Google Scholar]
- 136.Patrono C, Garcia Rodriguez LA, Landolfi R, et al. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 2005;353:2373–83. [DOI] [PubMed] [Google Scholar]
- 137.Kim AK, Dziura J, Strazzabosco M. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma: the egg of columbus or another illusion? Hepatology 2013;58:819–21. [DOI] [PubMed] [Google Scholar]
- 138.Hossain MA, Kim DH, Jang JY, et al. Aspirin enhances doxorubicin-induced apoptosis and reduces tumor growth in human hepatocellular carcinoma cells in vitro and in vivo. Int J Oncol 2012;40:1636–42. [DOI] [PubMed] [Google Scholar]
- 139.Fajardo AM, Piazza GA. Chemoprevention in gastrointestinal physiology and disease. Anti-inflammatory approaches for colorectal cancer chemoprevention. Am J Physiol Gastrointest Liver Physiol 2015;309:G59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chan TA, Morin PJ, Vogelstein B, et al. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci U S A 1998;95:681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leng J, Han C, Demetris AJ, et al. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 2003;38:756–68. [DOI] [PubMed] [Google Scholar]
- 142.Fodera D, D’Alessandro N, Cusimano A, et al. Induction of apoptosis and inhibition of cell growth in human hepatocellular carcinoma cells by COX-2 inhibitors. Ann N Y Acad Sci 2004;1028:440–9. [DOI] [PubMed] [Google Scholar]
- 143.Kanai S, Ishihara K, Kawashita E, et al. ASB14780, an Orally Active Inhibitor of Group IVA Phospholipase A2, Is a Pharmacotherapeutic Candidate for Nonalcoholic Fatty Liver Disease. J Pharmacol Exp Ther 2016;356:604–14. [DOI] [PubMed] [Google Scholar]
- 144.Hu X, Cifarelli V, Sun S, et al. Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J Lipid Res 2016;57:663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Paik YH, Kim JK, Lee JI, et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 2009;58:1517–27. [DOI] [PubMed] [Google Scholar]
- 146.Sitia G, Aiolfi R, Di Lucia P, et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci U S A 2012;109:E2165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yoshida S, Ikenaga N, Liu SB, et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014;147:1378–92. [DOI] [PubMed] [Google Scholar]
- 148.Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Simon TG, Duberg A, Aleman S, et al. Aspirin and Risk for Hepatocellular Carcinoma and Liver-Related Mortality: Results from a Nationwide Population with Chronic Viral Hepatitis. New England Journal of Medicine -pending publication, 2020. [Google Scholar]
- 150.Lee TY, Hsu YC, Tseng HC, et al. Association of Daily Aspirin Therapy With Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. JAMA Intern Med 2019;179:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tsoi KKF, Ho JMW, Chan FCH, et al. Long-term use of low-dose aspirin for cancer prevention: A 10-year population cohort study in Hong Kong. Int J Cancer 2019;145:267–273. [DOI] [PubMed] [Google Scholar]
- 152.Hwang IC, Chang J, Kim K, et al. Aspirin Use and Risk of Hepatocellular Carcinoma in a National Cohort Study of Korean Adults. Sci Rep 2018;8:4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim G, Jang SY, Han E, et al. Effect of statin on hepatocellular carcinoma in patients with type 2 diabetes: A nationwide nested case-control study. Int J Cancer 2017;140:798–806. [DOI] [PubMed] [Google Scholar]
- 154.Lin YS, Yeh CC, Huang SF, et al. Aspirin associated with risk reduction of secondary primary cancer for patients with head and neck cancer: A population-based analysis. PLoS One 2018;13:e0199014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tseng CH. Metformin and risk of hepatocellular carcinoma in patients with type 2 diabetes. Liver Int 2018;38:2018–2027. [DOI] [PubMed] [Google Scholar]
- 156.Lee TY, Wu JC, Yu SH, et al. The occurrence of hepatocellular carcinoma in different risk stratifications of clinically noncirrhotic nonalcoholic fatty liver disease. Int J Cancer 2017;141:1307–1314. [DOI] [PubMed] [Google Scholar]
- 157.Lee M, Chung GE, Lee JH, et al. Antiplatelet therapy and the risk of hepatocellular carcinoma in chronic hepatitis B patients on antiviral treatment. Hepatology 2017;66:1556–1569. [DOI] [PubMed] [Google Scholar]
- 158.Chiu HF, Ho SC, Chen CC, et al. Statin use and the risk of liver cancer: a population-based case–control study. Am J Gastroenterol 2011;106:894–8. [DOI] [PubMed] [Google Scholar]
- 159.Coogan PF, Rosenberg L, Palmer JR, et al. Nonsteroidal anti-inflammatory drugs and risk of digestive cancers at sites other than the large bowel. Cancer Epidemiol Biomarkers Prev 2000;9:119–23. [PubMed] [Google Scholar]
- 160.Friis S, Sørensen HT, McLaughlin JK, et al. A population-based cohort study of the risk of colorectal and other cancers among users of low-dose aspirin. Br J Cancer 2003;88:684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Du ZQ, Zhao JZ, Dong J, et al. Effect of low-dose aspirin administration on long-term survival of cirrhotic patients after splenectomy: A retrospective single-center study. World J Gastroenterol 2019;25:3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang B, Petrick JL, Chen J, et al. Associations of NSAID and paracetamol use with risk of primary liver cancer in the Clinical Practice Research Datalink. Cancer Epidemiol 2016;43:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sahasrabuddhe VV, Gunja MZ, Graubard BI, et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst 2012;104:1808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Petrick JL, Sahasrabuddhe VV, Chan AT, et al. NSAID Use and Risk of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma: The Liver Cancer Pooling Project. Cancer Prev Res (Phila) 2015;8:1156–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Simon TG, Ma Y, Ludvigsson JF, et al. Association Between Aspirin Use and Risk of Hepatocellular Carcinoma. JAMA Oncol 2018;4:1683–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Simon TG, Duberg AS, Aleman S, et al. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N Engl J Med 2020;382:1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zheng L, Yang W, Wu F, et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res 2013;19:5372–80. [DOI] [PubMed] [Google Scholar]
- 168.DePeralta DK, Wei L, Ghoshal S, et al. Metformin prevents hepatocellular carcinoma development by suppressing hepatic progenitor cell activation in a rat model of cirrhosis. Cancer 2016;122:1216–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shankaraiah RC, Callegari E, Guerriero P, et al. Metformin prevents liver tumourigenesis by attenuating fibrosis in a transgenic mouse model of hepatocellular carcinoma. Oncogene 2019;38:7035–7045. [DOI] [PubMed] [Google Scholar]
- 170.Ma S, Zheng Y, Xiao Y, et al. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine (Baltimore) 2017;96:e6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Home PD, Kahn SE, Jones NP, et al. Experience of malignancies with oral glucose-lowering drugs in the randomised controlled ADOPT (A Diabetes Outcome Progression Trial) and RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycaemia in Diabetes) clinical trials. Diabetologia 2010;53:1838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297–307. [DOI] [PubMed] [Google Scholar]
- 173.Chang CH, Lin JW, Wu LC, et al. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology 2012;55:1462–72. [DOI] [PubMed] [Google Scholar]
- 174.Hassan MM, Curley SA, Li D, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010;116:1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Huang MY, Chung CH, Chang WK, et al. The role of thiazolidinediones in hepatocellular carcinoma risk reduction: a population-based cohort study in Taiwan. Am J Cancer Res 2017;7:1606–1616. [PMC free article] [PubMed] [Google Scholar]
- 176.Kasmari AJ, Welch A, Liu G, et al. Independent of Cirrhosis, Hepatocellular Carcinoma Risk Is Increased with Diabetes and Metabolic Syndrome. Am J Med 2017;130:746 e1–746 e7. [DOI] [PubMed] [Google Scholar]
- 177.Kawaguchi T, Taniguchi E, Morita Y, et al. Association of exogenous insulin or sulphonylurea treatment with an increased incidence of hepatoma in patients with hepatitis C virus infection. Liver Int 2010;30:479–86. [DOI] [PubMed] [Google Scholar]
- 178.Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016;387:679–90. [DOI] [PubMed] [Google Scholar]
- 179.Hoshida Y, Fuchs BC, Tanabe KK. Prevention of hepatocellular carcinoma: potential targets, experimental models, and clinical challenges. Curr Cancer Drug Targets 2012;12:1129–59. [PMC free article] [PubMed] [Google Scholar]
- 180.Oakley F, Teoh V, Ching ASG, et al. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology 2009;136:2334–2344 e1. [DOI] [PubMed] [Google Scholar]
- 181.Tamaki Y, Nakade Y, Yamauchi T, et al. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J Gastroenterol 2013;48:491–503. [DOI] [PubMed] [Google Scholar]
- 182.Hassan MM, Botrus G, Abdel-Wahab R, et al. Estrogen Replacement Reduces Risk and Increases Survival Times of Women With Hepatocellular Carcinoma. Clin Gastroenterol Hepatol 2017;15:1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sun H, Deng Q, Pan Y, et al. Association between estrogen receptor 1 (ESR1) genetic variations and cancer risk: a meta-analysis. J BUON 2015;20:296–308. [PubMed] [Google Scholar]
- 184.McGlynn KA, Sahasrabuddhe VV, Campbell PT, et al. Reproductive factors, exogenous hormone use and risk of hepatocellular carcinoma among US women: results from the Liver Cancer Pooling Project. Br J Cancer 2015;112:1266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen HP, Shieh JJ, Chang CC, et al. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies. Gut 2013;62:606–15. [DOI] [PubMed] [Google Scholar]
- 186.Bode AM, Dong Z. Cancer prevention research - then and now. Nat Rev Cancer 2009;9:508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Le Magnen C, Dutta A, Abate-Shen C. Optimizing mouse models for precision cancer prevention. Nat Rev Cancer 2016;16:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Maresso KC, Tsai KY, Brown PH, et al. Molecular cancer prevention: Current status and future directions. CA Cancer J Clin 2015;65:345–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bruix J, Poynard T, Colombo M, et al. Maintenance therapy with peginterferon alfa-2b does not prevent hepatocellular carcinoma in cirrhotic patients with chronic hepatitis C. Gastroenterology 2011;140:1990–9. [DOI] [PubMed] [Google Scholar]
- 191.Lok AS, Everhart JE, Wright EC, et al. Maintenance peginterferon therapy and other factors associated with hepatocellular carcinoma in patients with advanced hepatitis C. Gastroenterology 2011;140:840–9; quiz e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nakagawa S, Wei L, Song WM, et al. Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 2016;30:879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rahmani J, Kord Varkaneh H, Kontogiannis V, et al. Waist Circumference and Risk of Liver Cancer: A Systematic Review and Meta-Analysis of over 2 Million Cohort Study Participants. Liver Cancer 2020;9:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Esposito K, Capuano A, Giugliano D. Metabolic syndrome and cancer: holistic or reductionist? Endocrine 2014;45:362–4. [DOI] [PubMed] [Google Scholar]
- 195.Vanni E, Bugianesi E. Obesity and liver cancer. Clin Liver Dis 2014;18:191–203. [DOI] [PubMed] [Google Scholar]
- 196.Farrell G Insulin resistance, obesity, and liver cancer. Clin Gastroenterol Hepatol 2014;12:117–9. [DOI] [PubMed] [Google Scholar]
- 197.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656–65. [DOI] [PubMed] [Google Scholar]
- 198.Gehmert S, Sadat S, Song YH, et al. The anti-apoptotic effect of IGF-1 on tissue resident stem cells is mediated via PI3-kinase dependent secreted frizzled related protein 2 (Sfrp2) release. Biochem Biophys Res Commun 2008;371:752–5. [DOI] [PubMed] [Google Scholar]
- 199.Sidharthan S, Kottilil S. Mechanisms of alcohol-induced hepatocellular carcinoma. Hepatol Int 2014;8:452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Endres S, Ghorbani R, Kelley VE, et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N Engl J Med 1989;320:265–71. [DOI] [PubMed] [Google Scholar]
- 201.Gressner OA, Lahme B, Rehbein K, et al. Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J Hepatol 2008;49:758–67. [DOI] [PubMed] [Google Scholar]
- 202.Kalthoff S, Ehmer U, Freiberg N, et al. Coffee induces expression of glucuronosyltransferases by the aryl hydrocarbon receptor and Nrf2 in liver and stomach. Gastroenterology 2010;139:1699–710, 1710 e1–2. [DOI] [PubMed] [Google Scholar]
- 203.Zhou X, Chen J, Yi G, et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016;7:873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]