

Summary
Newborn screening (NBS) was established as a public health program in the 1960s and is crucial for facilitating detection of certain medical conditions in which early intervention can prevent serious, life-threatening health problems. Genomic sequencing can potentially expand the screening for rare hereditary disorders, but many questions surround its possible use for this purpose. We examined the use of exome sequencing (ES) for NBS in the North Carolina Newborn Exome Sequencing for Universal Screening (NC NEXUS) project, comparing the yield from ES used in a screening versus a diagnostic context. We enrolled healthy newborns and children with metabolic diseases or hearing loss (106 participants total). ES confirmed the participant’s underlying diagnosis in 15 out of 17 (88%) children with metabolic disorders and in 5 out of 28 (∼18%) children with hearing loss. We discovered actionable findings in four participants that would not have been detected by standard NBS. A subset of parents was eligible to receive additional information for their child about childhood-onset conditions with low or no clinical actionability, clinically actionable adult-onset conditions, and carrier status for autosomal-recessive conditions. We found pathogenic variants associated with hereditary breast and/or ovarian cancer in two children, a likely pathogenic variant in the gene associated with Lowe syndrome in one child, and an average of 1.8 reportable variants per child for carrier results. These results highlight the benefits and limitations of using genomic sequencing for NBS and the challenges of using such technology in future precision medicine approaches.
Keywords: newborn screening, exome sequencing, inborn errors of metabolism, hearing loss, pathogenic
Introduction
Newborn screening was established in the early 1960s, beginning with phenylketonuria1 (PKU) (MIM: 261600), for which early intervention can prevent permanent intellectual disability. In the 1990s, tandem mass spectrometry (TMS) was employed to efficiently screen neonates for numerous metabolites, which resulted in an increase in the number of genetic conditions screened.2, 3, 4 As of July 2018, The American College of Medical Genetics and Genomics and the United States federal Advisory Committee on Heritable Disorders in Newborns and Children have identified 35 core conditions and 26 secondary conditions to be included on the recommended uniform screening panel (RUSP, recommended by the Secretary of the Department of Health and Human Services), including specific disorders of organic acids, fatty acid oxidation, amino acids, as well as endocrine and hemoglobin disorders.5, 6, 7 Most infants in the United States also receive point-of-care newborn screening for hearing and critical congenital heart disease.
Advances in genomic sequencing technologies (next-generation sequencing, or NGS), such as exome and genome sequencing (ES or GS, respectively), have emerged as powerful tools for identifying an individual’s genomic changes at the DNA base level.8 These technologies have the potential to dramatically increase the ability to screen newborns for rare hereditary disorders that would not be detected using traditional screening methods. Given this potential, the National Institute of Child Health and Human Development (NICHD) and the National Human Genome Research Institute (NHGRI) sponsored a cohort of studies, the Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) consortium, to investigate the use of this technology in newborns.9, 10, 11, 12 One of these studies recently reported on the use of NGS in healthy newborns and reported additional findings that would have been missed by existing newborn screening modalities.10 Thus, for some cases, genomic sequencing may clarify the underlying genetic cause of an individual’s disease and directly impact a patient’s diagnosis, intervention, and/or treatment. In addition, genomic sequencing may also be useful for screening asymptomatic individuals.
In the North Carolina Newborn Exome Sequencing for Universal Screening (NC NEXUS) study, we explored the possible use of ES in the context of newborn screening and studied the responses of parents to this new technology. The study design13 included two cohorts of infants and children: (1) healthy newborns whose parents were approached for participation in the study prenatally; and (2) infants and children (<5 years of age) already clinically diagnosed with conditions detected with current newborn screening methods (inborn errors of metabolism and hearing loss).
In the current manuscript, we report the molecular analysis results from the cohort of 106 children who were enrolled in the study. Molecular analysts examined ES data using a next-generation sequencing newborn screen (NGS-NBS) panel of 466 genes,14 while being blinded to the identity of the participant’s cohort (well-child, metabolic, or hearing loss). In our analysis strategy, we defined strict criteria for assessing the screen as abnormal or “positive,” which allowed us to examine the sensitivity of ES when used as a screening test to detect a genetic alteration in the context of a theoretical newborn screen. For those participants in the metabolic cohort or the hearing loss cohort, we then performed a second, indication-based, diagnostic analysis assessing the ES results for additional genes that were likely associated with the participant’s phenotype. In addition, parents were randomized to one of two study arms. In one study arm, parents could elect to learn additional findings from their child’s ES. The results of the initial NGS-NBS screening, indication-based analyses, and additional genomic findings are presented in this work.
Subjects and Methods
Recruitment and Sample Collection
The study protocol was approved by the University of North Carolina Institutional Review Board and all participants were consented to the study by a certified genetic counselor. The NC NEXUS randomized controlled trial protocol and study design were previously described.13 As part of that protocol, 17 children previously diagnosed with inborn errors of metabolism and 28 children previously diagnosed with hearing loss were recruited into the study. Additionally, we recruited parents in the prenatal period (pregnancies of 18 weeks or longer) in which the fetus did not have a positive or pending chromosomal abnormality or congenital malformation diagnostic test result. The mothers subsequently gave birth to 61 infants; these babies were classified into the well-child cohort. Participant demographic information is reported in Table S1.
Decision Aids
As part of the overall study, we previously described a decision aid for parents who would be offered ES for their child.13,15 This web-based electronic decision aid was developed to enhance parents’ abilities to make decisions about learning genomic information for their child; the aid included both an educational component and a values clarification exercise. The educational component included information about newborn screening, genomic sequencing, and the process of using genomic sequencing to identify genetic variants associated with specific diseases. This educational component was followed by a values clarification exercise in which parents classified five reasons for and five reasons against having genomic sequencing for their child by level of importance.15, 16, 17 After viewing the decision aid, parents met face to face with a genetic counselor for additional clarification before being asked to decide whether to consent to ES for their child.
Age-Based Semiquantitative Metric
As previously described,14 we determined actionability of gene-disease pairs using the age-based semiquantitative metric (ASQM). Conditions were placed into one of four categories: category 1, pediatric conditions with high medical actionability; category 2, pediatric conditions with low or no medical actionability; category 3, adult conditions with high medical actionability; and category 4, adult conditions with low or no medical actionability. In total, 822 gene-disease pairs were assessed, enriched for those with pediatric onset of disease and suspected actionability. Category 1 (466 gene-disease pairs) included many core RUSP conditions, as well as other disorders with onset in infancy or in childhood that have treatment, monitoring, and/or medical management that can potentially improve clinical outcomes; these conditions are comparable to those detected by current NBS and therefore all NC NEXUS study participants were eligible for disclosure of abnormal, positive findings.
Randomization for Additional Findings
Parents were randomized to one of two study arms. One arm was the “control arm” (1/3 of participants) in which parents learned findings from the NGS-NBS panel and, if applicable, indication-based analysis. The second arm was the “decision arm” (2/3 of participants) in which parents learned findings from the NGS-NBS (and indication-based analysis if applicable) and were also asked to decide whether or not they wanted to learn about additional types of genomic findings from up to three categories. These three categories included: childhood-onset conditions with low or no clinical actionability, adult-onset conditions with high actionability, and carrier status for autosomal-recessive disorders.
Exome Sequencing
The BioSpecimen Processing Facility at the University of North Carolina at Chapel Hill isolated genomic DNA from saliva and oral epithelial cell samples using PureGene chemistry. Exome libraries, including molecular barcoding and exome capture, were prepared using Agilent SureSelect XT kits (Human All Exon V6 probes) according to the manufacturer’s low input guidelines. The University of North Carolina High-Throughput Sequencing Facility performed exome sequencing on an Illumina HiSeq2500 with minimum depth of 40× coverage. Raw sequence reads were mapped to the human genome reference GRCh38.p7 using BWA-MEM version 0.7.12,18 duplicate reads were marked using Picard MarkDuplicates (v.1.130), and variants were called using FreeBayes, v.1.1.0.19
Bioinformatics and Molecular Analysis
Variants were prioritized based on previous classification as pathogenic (P) or likely pathogenic (LP) in a ClinVar submission,20 minor allele frequency in a reference database,21,22 and predicted effect of the variant on the protein (frameshift, nonsense, canonical splice-site, missense, synonymous, intronic variants). All rare possibly damaging variants in 466 genes in category 1 were reviewed by a molecular analyst for each of the 106 participants. For the initial NGS-NBS analysis, molecular analysts were blinded to the participant’s cohort (metabolic, hearing loss, or well-child). Variants were classified based on ACMG/AMP interpretation guidelines23,24 using collected evidence from population databases,21,22 ClinVar,20 and the primary literature. Pathogenic and/or LP variants that were considered an abnormal “positive screen” (heterozygous P/LP variants in a gene associated with a dominant condition; two or more P/LP variants in a gene associated with a recessive condition) were flagged for further review by a committee of molecular geneticists and physicians, genetic counselors, and researchers. Variants of uncertain significance (VUS) were not returned in the NGS-NBS screen. After completion of the first NGS-NBS analysis, the participant’s cohort was revealed to the molecular analyst. If the participant was part of the metabolic cohort or hearing loss cohort, ES data were further analyzed by filtering for variants in a subset of genes within indication-based “diagnostic lists,” containing genes associated with the relevant phenotypes. For the indication-based diagnostic analyses, P, LP, and VUS were reported. All variants were confirmed in a duplicate sample using an orthogonal method performed in the UNC Hospitals Molecular Genetics CLIA-certified laboratory. Copy number variant analysis was not conducted due to restrictions based on the study’s Investigational Device Exemption approved by the FDA.13
For participants who decided to learn some or all of the additional findings categories, ES data were analyzed to identify P/LP variants in 22 genes associated with adult-onset actionable conditions, 234 genes associated with childhood-onset nonactionable conditions, and/or 871 genes associated with carrier findings for autosomal-recessive conditions.
All parents were seen by a genetic counselor and/or clinical geneticist for return of any positive results in diagnostic, NGS-NBS, childhood-onset medically non-actionable, and adult-onset medically actionable categories. Carrier status results were conveyed by a genetic counselor only by phone. Recommendations and options for further testing or evaluation were provided but not part of the study. Parents were given a copy of the CLIA-confirmed result report for their child and were given the option of having the results placed in their child’s electronic health record.
Results
Molecular analysis was completed for 106 participants: 61 in the healthy/well-child cohort, 17 in the metabolic cohort, and 28 in the hearing loss cohort. A total of 43 out of 46 variants detected by ES were confirmed by orthogonal testing in the CLIA-certified laboratory prior to reporting. Additionally, 5 variants that were previously identified by clinical molecular testing were also detected by ES.
Metabolic Cohort
NGS-NBS analysis was deemed to represent an abnormal positive screen result in 15 of 17 participants (88%) in the metabolic cohort (Tables 1 and 2). These individuals had a combination of pathogenic and/or likely pathogenic variants in genes implicated in metabolic conditions, that, when blinded to the participants’ cohort, were determined to be reportable in a screening setting.
Table 1.
Summary of NGS-NBS and Diagnostic Findings in the NC NEXUS Study
| NC NEXUS Cohort | Total Number of Participants in Cohort | NGS-NBS Abnormal Positive Screen | NGS-NBS Normal Negative Screen | Diagnostic Positive | Diagnostic Inconclusive | Diagnostic Negative |
|---|---|---|---|---|---|---|
| Metabolic | 17 | 15 | 2 | 15 | 2 | 0 |
| Hearing loss | 28 | 7∗ | 21 | 5 | 5 | 18 |
| Healthy/well-child | 61 | 1∗ | 60 | 0 | 0 | 0 |
| Sum | ||||||
| Unanticipated findings | 106 | 4 | N/A | N/A | N/A | N/A |
An abnormal NGS-NBS, or positive screen, was indicated by observing likely pathogenic and/or pathogenic variants in genes associated with pediatric conditions with high medical actionability. A normal NGS-NBS, or negative screen, was defined by the absence of likely pathogenic or pathogenic variants found in these genes. Diagnostic positive indicates the presence of likely pathogenic or pathogenic variants found in gene(s) on the metabolic or hearing loss diagnostic list that are consistent with the participant’s disorder. Diagnostic inconclusive indicates an inconclusive result (i.e., a single heterozygous variant found in a gene associated with an autosomal-recessive condition and/or variants of uncertain significance [VUS] in genes on the diagnostic list). A negative diagnostic result indicated that we did not detect any pathogenic or likely pathogenic variants or any VUS on the diagnostic gene lists. Numbers with asterisks include two participants in the hearing loss cohort with an abnormal NGS-NBS due to likely pathogenic variants in DSC2 or F11 plus one participant in the healthy/well-child cohort that was determined to have an abnormal NGS-NBS due to a pathogenic variant in LDLR associated with familial hypercholesterolemia. NGS-NBS, next-generation sequencing newborn screen.
Table 2.
NGS-NBS and Diagnostic Findings in the NC NEXUS Inborn Errors of Metabolism Cohort
| NC NEXUS Participant | NGS-NBS Result | Diagnostic (Dx) Result | Gene(s) | Disease Association with Gene (Inheritance) | Variant and (Predicted Protein Change) | Zygosity in NC NEXUS Participant |
|---|---|---|---|---|---|---|
| M001 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.799G>A (p.Gly267Arg); c.985A>G (p.Lys329Glu) | heterozygous for both variants |
| M002 | abnormal | positive | PAH | PKU (AR) | c.1315+1G>A (p.?); c.782G>A (p.Arg261Gln) | heterozygous for both variants |
| M003 | abnormal | positive | PAH,OTC | PKU (AR), OTC deficiency (XL) | PAH c.1222C>T (p.Arg408Trp) and c.896T>G (p.Phe299Cys); OTC c.1061T>G (p.Phe354Cys) | heterozygous for PAH and OTC variants |
| M004 | abnormal | positive | PAH | PKU (AR) | c.1315+1G>A (p.?); c.284_286del (p.Ile95del) | heterozygous for both variants |
| M005 | abnormal | positive | PAH | PKU (AR) | c.194T>C (p.Ile65Thr); c.814G>T (p.Gly272∗) | heterozygous for both variants |
| M006 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu) | homozygous |
| M007 | abnormal | positive | PAH | PKU (AR) | c.1315+1G>A (p.?); c.842C>T (p.Pro281Leu) | heterozygous for both variants |
| M008 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu) | homozygous |
| M009 | normal | inconclusive (VUS) | MLYCD | malonyl-coA decarboxylase deficiency (AR) | c.1013T>C (p.Leu338Pro) | homozygous |
| M010 | abnormal | positive | PAH | PKU (AR) | c.117C>G (p.Phe39Leu); c.842C>T (p.Pro281Leu) | heterozygous for both variants |
| M011 | abnormal | positive | PAH | PKU (AR) | c.194T>C (p.Ile65Thr) | homozygous |
| M012 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu) | homozygous |
| M013 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu) | homozygous |
| M014 | abnormal | positive | SLC22A5 | renal carnitine transport deficiency (AR) | c.506G>A (p.Arg169Gln) | homozygous |
| M015 | normal | inconclusive (heterozygous variant) | BCKDHA | MSUD type 1A (AR) | c.1312T>A (p.Tyr438Asn) | heterozygous |
| M016 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu) | homozygous |
| M017 | abnormal | positive | ACADM | MCAD deficiency (AR) | c.985A>G (p.Lys329Glu); c.199T>C (p.Tyr67His) | heterozygous for both variants |
The NC NEXUS participant column includes “M” (indicating inborn errors of metabolism cohort) followed by the participant number. Each row in the table represents one NC NEXUS participant. An abnormal NGS-NBS result was indicated by a likely pathogenic or pathogenic variant found in the newborn screen gene list. A positive diagnostic result was indicated by a likely pathogenic or pathogenic variant found in a gene on the inborn errors of metabolism diagnostic list. An inconclusive diagnostic result was indicated by any variant of uncertain significance (VUS) finding or a single heterozygous variant found in a gene associated with autosomal-recessive inborn errors of metabolism. A negative diagnostic result indicated we did not detect any pathogenic or likely pathogenic variants or any VUS on the inborn errors of metabolism gene list. Gene symbols are italicized. NGS-NBS, next-generation sequencing newborn screen; AR, autosomal-recessive pattern of inheritance; XL, X-linked pattern of inheritance; PKU, phenylketonuria; MCAD deficiency, medium-chain acyl-coA dehydrogenase deficiency; MSUD, maple syrup urine disease.
Seven participants previously diagnosed with phenylketonuria (PKU) by newborn screening had pathogenic variants in PAH (MIM: 612349); six of them were likely compound heterozygous for two different variants (ranging from missense, canonical splice-site, nonsense, and/or deletion variants). In view of their previously established diagnosis of PKU, we did not routinely perform parental testing to confirm the phase of these PAH variants. The seventh participant was likely homozygous for a pathogenic missense variant in PAH (c.194T>C [p.Ile65Thr]), although we did not rule out the possibility of a deletion in trans.
Seven participants previously diagnosed with medium-chain acyl-coA dehydrogenase (MCAD) deficiency (MIM: 201450) by newborn screening had pathogenic variants in ACADM (MIM: 607008); five of these seven were homozygous for a well-known pathogenic missense variant (c.985A>G [p.Lys329Glu]) reported previously in individuals with MCAD deficiency.25, 26, 27, 28 Two of these seven were likely compound heterozygous for two different variants in the ACADM gene: c.985A>G (p.Lys329Glu) and c.799G>A (p.Gly267Arg); c.985A>G (p.Lys329Glu) and c.199T>C (p.Tyr67His).
One participant was previously diagnosed with primary carnitine deficiency (MIM: 212140) by newborn screening due to a borderline low free carnitine level (C0) found by TMS. This individual was homozygous for a pathogenic SLC22A5 (MIM: 603377) missense variant (c.506G>A [p.Arg169Gln]) consistent with his clinical diagnosis (see GeneReviews by El-Hattab in Web Resources).29, 30, 31, 32, 33, 34
NGS-NBS analysis was deemed normal, or “negative,” in 2 of 17 participants (12%) in the metabolic cohort. However, after unblinding, diagnostic analysis revealed suggestive but inconclusive findings (Tables 1 and 2). One participant previously diagnosed with maple syrup urine disease (MSUD [MIM: 248600]) was heterozygous for a pathogenic missense variant in the BCKDHA (MIM: 608348) gene (c.1312T>A [p.Tyr438Asn]) associated with classic, autosomal-recessive MSUD.35, 36, 37, 38, 39 Since we did not identify any additional variants in BCKDHA that might explain a genetic etiology for this participant’s disease, we classified this result as inconclusive. Another participant previously diagnosed with malonyl-CoA decarboxylase deficiency (MIM: 248360) was homozygous for a variant (c.1013T>C [p.Leu338Pro]) in MLYCD (MIM: 606761), the gene that encodes the malonyl-CoA decarboxylase enzyme. This variant is present in population databases at low frequency (Genome Aggregation Database total allele frequency: 1 allele/249,584 alleles) and is located in a well-conserved catalytic domain of the encoded protein, with other reported benign variants observed nearby.20, 21, 22,40 The variant was not previously reported in the clinical literature, and many of the other reported pathogenic variants in this gene are putative loss of function.41, 42, 43, 44 Importantly, this infant was born prematurely at 23 weeks gestation and required several repeat newborn screens due to inconclusive findings prior to eventual confirmation of malonyl-CoA decarboxylase deficiency at 7 weeks of age. It is worth noting that a ClinVar20 entry is available for this variant (ClinVar: VCV000432061.2) with a “likely pathogenic” interpretation. Based on clinical testing (not known to the research team at the time of the analysis), we believe this observation to be the same as the NC NEXUS participant described here. The discordance between our interpretation and GeneDx’s interpretation is likely due to our more conservative use of evidence codes related to in silico predictors and the location of the variant within a catalytic domain. Considering there was no available in vivo or in vitro functional evidence to confirm a loss-of-function effect for this variant, we continue to interpret this rare novel missense variant as a VUS due to insufficient evidence. In the context of the participant’s phenotypic information, the MLYCD variant provides a suggestive but not definitive explanation for the genetic cause of the participant’s disorder.
Hearing Loss Cohort
NGS-NBS analysis was deemed to represent an abnormal positive screen result in 5 of 28 participants (18%) in the hearing loss cohort (Tables 1 and 3). These individuals had a combination of pathogenic and/or likely pathogenic variants in genes implicated in hearing loss that, when blinded to cohort, were determined to be reportable in a screening setting. Two participants were each presumed to be compound heterozygous for two variants in an Usher syndrome gene, USH2A (MIM: 608400) based on ES data (c.1256G>T [p.Cys419Phe] and c.3686T>G [p.Leu1229Ter]; c.4338_4339del [p.Cys1447fs] and c.2299del [p.Glu767fs]). In both cases, we subsequently confirmed that the variants were in trans by parental testing. A clinical geneticist and genetic counselor discussed the implications of this diagnosis, including the likelihood of vision loss due to retinitis pigmentosa, with the parents of these children during the in-person return of results visits. In addition, the geneticist and genetic counselor provided information regarding clinical trials for treatment of retinitis pigmentosa to the parents. One participant was homozygous for a 1-bp frameshift deletion (c.35del [p.Gly12fs]) in GJB2 ([MIM: 121011]; encoding connexin 26), a common pathogenic variant associated with DFNB1 nonsyndromic deafness (MIM: 220290).45, 46, 47, 48 One participant was compound heterozygous for two pathogenic missense variants (c.626G>T [p.Gly209Val] and c.1151A>G [p.Glu384Gly]) in SLC26A4 (MIM: 605646), a gene associated with Pendred syndrome (MIM: 274600), an autosomal-recessive form of syndromic hearing loss.49, 50, 51, 52, 53 Finally, one participant was heterozygous for a pathogenic variant (c.5597C>T [p.Thr1866Met]) in TECTA (MIM: 602574), a gene associated with autosomal-dominant hearing loss (MIM: 601543).54, 55, 56 Of note, this infant passed her newborn hearing screen, but presented at 1 year of age with unilateral hearing loss that progressed to both ears. She was included in the study after referral by her audiologist, despite not fitting the original enrollment criteria, as her hearing loss was not detected by newborn screening.
Table 3.
Participants in the NC NEXUS Hearing Loss Cohort with Positive or Inconclusive Findings
| NC NEXUS Participant | NGS-NBS Result | Diagnostic (Dx) Result | Gene(s) | Disease Association with Gene (Inheritance) | Variant and (Predicted Protein Change) | Zygosity in NC NEXUS Participant |
|---|---|---|---|---|---|---|
| HL003 | abnormal | positive | SLC26A4 | Pendred syndrome (AR) | c.626G>T (p.Gly209Val); c.1151A>G (p.Glu384Gly) | heterozygous for both variants |
| HL004 | normal | inconclusive VUS | MARVELD2 MYO7A | deafness 49 (AR); deafness 2 (AR); Usher type 1B (AR) | MARVELD2 c.1183−1G>A (p.?); MYO7A c.5824G>A (p.Gly1942Arg) | heterozygous for MARVELD2 variant; homozygous for MYO7A variant |
| HL010 | abnormal | positive | USH2A | Usher type 2A (AR) | c.1256G>T (p.Cys419Phe); c.3686T>G (p.Leu1229∗) | heterozygous for both variants |
| HL013 | abnormal | negative | F11 | factor XI deficiency (AR) | c.1489C>T (p.Arg497∗); c.1608G>C (p.Lys536Asn) | heterozygous for both variants |
| HL014 | abnormal | positive | GJB2 | deafness 1A (AR) | c.35del (p.Gly12fs) | homozygous |
| HL016 | abnormal | positive | USH2A | Usher type 2A (AR) | c.4338_4339del (p.Cys1447fs); c.2299del (p.Glu767fs) | heterozygous for both variants |
| HL017 | abnormal | positive | TECTA | deafness 8/12 (AD) | c.5597C>T (p.Thr1866Met) | heterozygous |
| HL021 | abnormal | negative | DSC2 | arrhythmogenic right ventricular dysplasia (ARVD11) (AD) | c.631−2A>G (p.?) | heterozygous |
| HL022 | normal | inconclusive VUS | TMPRSS3 | nonsyndromic hearing loss and deafness (AR) | c.208del (p.His70fs); c.1151T>A (p.Met384Lys) | heterozygous for both variants |
| HL025 | normal | inconclusive (heterozygous variant) | GJB2 | deafness (AR) | c.35del (p.Gly12fs) | heterozygous |
| HL026 | normal | inconclusive VUS | LOXHD1; CEMIP; MYH14 | hearing loss (AR or AD) | LOXHD1 c.2914G>A (p.Glu972Lys); LOXHD1 c.3161C>T (p.Thr1054Met); CEMIP/KIAA1199 c.58C>T (p.Leu20Phe); MYH14 c.5942G>C (p.Gly1981Ala) | heterozygous for all variants |
| HL027 | normal | inconclusive VUS | SOX10 | Waardenburg syndrome (AD) | c.482G>A (p.Arg161His) | heterozygous |
The NC NEXUS participant column includes “HL” (indicating hearing loss cohort) followed by the participant number. Each row in the table represents one NC NEXUS participant. An abnormal NGS-NBS result was indicated by a likely pathogenic or pathogenic variant found in the newborn screen gene list. A positive diagnostic result was indicated by a likely pathogenic or pathogenic variant found in a gene on the hearing loss list. An inconclusive diagnostic result was indicated by any variant of uncertain significance (VUS) finding or a heterozygous variant found in a gene associated with an autosomal-recessive form of hearing loss. A negative diagnostic result indicated we did not detect any pathogenic or likely pathogenic variants or any VUS on the hearing loss gene list. Gene symbols are italicized. NGS-NBS, next-generation sequencing newborn screen; AR, autosomal-recessive pattern of inheritance; AD, autosomal-dominant pattern of inheritance.
In 5 of 28 participants in the hearing loss cohort, NGS-NBS screening analysis was deemed normal, or “negative.” However, after unblinding, diagnostic analysis identified five “inconclusive” results that may provide a possible explanation for their hearing loss (Tables 1 and 3). One participant was heterozygous for the pathogenic c.35del (p.Gly12fs) deletion in GJB2. No additional pathogenic variants were detected in GJB2, and analysis in the CLIA-certified laboratory for intragenic GJB6 (MIM: 604418) deletions was negative; however, we cannot rule out the presence of a second pathogenic variant in GJB2 not detected by ES, such as a partial gene deletion or cryptic splice alteration caused by a non-coding variant.
Other “inconclusive” findings were reported that did not provide definitive diagnostic results. One participant was heterozygous for four different missense VUSs in genes associated with hearing loss: two variants in LOXHD1 (MIM: 613072) (c.2914G>A [p.Glu972Lys] and c.3161C>T [p.Thr1054Met]), one variant in CEMIP, also known as KIAA1199 (MIM: 608366) (c.58C>T [p.Leu20Phe]), and one variant in MYH14 (MIM: 608568) (c.5942G>C [p.Gly1981Ala]). One participant was heterozygous for a missense VUS in SOX10 (MIM: 602229) (c.482G>A [p.Arg161His]), a gene associated with autosomal-dominant Waardenburg syndrome57 (MIM: 611584, 613266). One participant was homozygous for a missense VUS in MYO7A (MIM: 276903) (c.5824G>A [p.Gly1942Arg]), a gene associated with autosomal-recessive syndromic and non-syndromic hearing loss58,59 (MIM: 276900, 600060). One participant was compound heterozygous for one likely pathogenic variant and one VUS in TMPRSS3 (MIM: 605511): c.208del (p.His70fs) and c.1151T>A (p.Met384Lys); parental testing indicated that each variant was in trans.
We did not identify any P or LP variants on the diagnostic gene lists for 18 out of 28 (64%) participants in the hearing loss cohort; therefore, diagnostic analyses were deemed “negative” for identifying a genetic cause of hearing loss in these individuals (Table 1). Interestingly, one participant referred by audiologists to the hearing loss cohort had a rare syndromic diagnosis, Warsaw breakage syndrome (MIM: 613398), which is characterized by severe microcephaly and intellectual disability, growth restriction, and sensorineural hearing loss due to cochlear hypoplasia (see GeneReviews by Alkhunaisi et al. in Web Resources).60 Clinical quad exome sequencing of the similarly affected sibling, this participant, and parents revealed a homozygous pathogenic variant in exon 13 of the DDX11 (MIM: 601150) gene (GenBank: NM_030653.4; c.1403dup [p.Ser469fs]) in the participant and his sibling. This DDX11 c.1403dup (p.Ser469fs) variant was present in the NC NEXUS research ES data but was not examined because DDX11 was not included in either the NGS-NBS list or the hearing loss diagnostic list. If additional phenotypic information had been available at the time of molecular analysis, it is likely this variant would have been recognized as having diagnostic significance.
NGS-NBS Conditions
An abnormal, positive screen NGS-NBS result was also defined as a finding that predicted a childhood-onset actionable condition in any individual in the well-child cohort, or a condition unrelated to the indicated diagnosis of a member of one of the affected cohorts. NGS-NBS analysis was deemed abnormal, or positive in 4 of 106 participants (Table 4). One participant in the well-child cohort was heterozygous for a missense LDLR (MIM: 606945) variant (c.502G>A [p.Asp168Asn]) previously reported in autosomal-dominant familial hypercholesterolemia61, 62, 63, 64, 65, 66, 67 (MIM: 143890). After disclosure of this finding, the parents stated that they were aware of a family history of hypercholesterolemia.
Table 4.
Other Actionable Findings in the NC NEXUS NGS-NBS
| NC NEXUS Participant | NGS-NBS Result | Gene | Disease Association with Gene (Inheritance) | Variant and (Predicted Protein Change) | Zygosity in NC NEXUS Participant |
|---|---|---|---|---|---|
| M003 | abnormal | OTC | OTC deficiency (XL) | c.1061T>G (p.Phe354Cys) | heterozygous |
| NB012 | abnormal | LDLR | familial hypercholesterolemia (AD or AR) | c.502G>A (p.Asp168Asn) | heterozygous |
| HL013 | abnormal | F11 | factor XI deficiency (AR) | c.1489C>T (p.Arg497∗), c.1608G>C (p.Lys536Asn) | heterozygous for both variants |
| HL021 | abnormal | DSC2 | arrhythmogenic right ventricular dysplasia (ARVD11) (AD) | c.631−2A>G (p.?) | heterozygous |
The NC NEXUS participant column includes “M” (indicating inborn errors of metabolism cohort), “HL” (indicating hearing loss cohort) or “NB” (indicating well-child cohort) followed by the participant number. Each row in the table represents one NC NEXUS participant. Other actionable findings were defined by pathogenic or likely pathogenic variants found in the NGS-NBS that are associated with a disorder not previously known in the NC NEXUS participant. Gene symbols are italicized. NGS-NBS, next-generation sequencing newborn screen; XL, X-linked inheritance pattern; AR, autosomal-recessive pattern of inheritance; AD, autosomal-dominant pattern of inheritance.
One female participant in the metabolic cohort, diagnosed with PKU, was a carrier of a missense variant (c.1061T>G [p.Phe354Cys]) in the ornithine transcarbamylase (OTC [MIM: 300461]) gene. This variant was previously reported in mild OTC deficiency68,69 (MIM: 311250), an X-linked disease with variable expressivity in carrier females. Although this participant was diagnosed with PKU by the standard newborn screen at birth, the OTC deficiency was an unexpected finding arising from the ES analysis. Her ammonia levels are normal, and the variant was also found in her unaffected grandfather. Her younger male sibling was prenatally diagnosed with the variant, which enabled early measurement of ammonia level immediately after delivery and appropriate ongoing health supervision. So far, he has been asymptomatic. One hearing loss cohort participant was heterozygous for a canonical splice-site variant in DSC2 (MIM: 125645) (c.631−2A>G [p.?]). This variant has been reported previously in individuals with autosomal-dominant arrhythmogenic right ventricular dysplasia70 (MIM: 610476). This child has had a normal echo and it was recommended that the child be followed by a cardiologist. Another hearing loss cohort participant had two variants in F11 (MIM: 264900) (nonsense variant c.1489C>T [p.Arg497Ter] and missense variant c.1608G>C [p.Lys536Asn]). Both variants have been reported previously in individuals with autosomal-recessive factor XI deficiency71, 72, 73 (MIM: 612416). This child has a history of frequent episodes of epistaxis (nosebleeds), one requiring cauterization. It was recommended that she be evaluated by a hematologist, and it was also recommended that precautions are taken prior to any surgical procedures.
Additional Genomic Findings
A total of 65 participants were randomized into the experimental decision arm of the study, viewed the decision aid, and attended the study visit with a genetic counselor. More than 90% of these parents chose to learn at least one category of additional genomic information: 47 (72.3%) requested all three categories of additional information, 7 requested additional findings for both adult-onset actionable conditions and carrier status, 2 requested additional findings for the adult-onset actionable category only, 1 requested carrier status only, 1 requested findings for the adult-onset actionable and childhood-onset nonactionable conditions categories, and 1 requested the childhood-onset nonactionable category only. The parents of 6 participants (9.2%) elected not to receive any of the additional information categories. Details about the factors associated with parental decision making will be reported in a separate manuscript.
Two participants had additional findings in the adult-onset actionable category (Table S2). A participant in the metabolic cohort (M007) was heterozygous for a pathogenic deletion RAD51C (MIM: 602774) variant (c.905−2_905−1del [p.?]), which is predicted to alter a splice site and has been reported previously in autosomal-dominant familial ovarian cancer74,75 (MIM: 613399). This participant had a known family history of breast cancer but no known family history of ovarian cancer. A participant in the well-child cohort (NB040) was heterozygous for a nonsense c.7480C>T (p.Arg2494Ter) variant in BRCA2 (MIM: 600185) that is a known pathogenic variant associated with autosomal-dominant familial breast and ovarian cancer76, 77, 78, 79, 80, 81, 82, 83 (MIM: 612555) This participant’s parents reported a maternal family history of breast and pancreatic cancer. The child’s mother was referred to adult genetics.
One participant in the hearing loss cohort (HL015) had an additional finding for the childhood-onset nonactionable conditions category (Table S2). This participant was hemizygous for a likely pathogenic nonsense variant (c.741G>A [p.Trp247Ter]) in OCRL (MIM: 300535). This gene is associated with Lowe syndrome84 (MIM: 309000), and further correlation with clinical findings indicated that the participant has multiple features consistent with Lowe syndrome, including congenital cataracts with severe vision loss, intellectual disability, and renal impairment. Indeed, we subsequently learned that the patient had been seen in clinical genetics and that clinical exome sequencing was considered but not performed due to concerns about cost. Although hearing loss is not a prominent feature of Lowe syndrome, there is at least one previous case report85 involving an individual with Lowe syndrome who also had hearing loss, suggesting that the finding may provide a comprehensive diagnostic result for this participant’s symptoms. The mother of this patient is a carrier of the variant and cascade testing of other family members was negative.
The number of carrier findings in children whose parents requested this category ranged from 0 to 7 variants, with an average of 1.8 reportable carrier findings per individual (Figure 1, Table S2). A total of 100 out of 109 (92%) potential carrier findings detected by ES were confirmed by orthogonal testing in a CLIA-certified laboratory and reported. The nine variants that failed to be confirmed by orthogonal testing had low read depth for both alleles in the ES data. We observed several recurrent carrier findings; the most common were variants in the genes HFE (MIM: 613609), GALT (MIM:606999), SERPINA1 (MIM:107400), and RBM8A (MIM: 605313) (Figure 2). A total of 11 participants carried the HFE c.187C>G (p.His63Asp) variant, while 4 carried the HFE c.845G>A (p.Cys282Tyr) variant, both associated with hereditary hemochromatosis (see GeneReviews by Barton and Edwards in Web Resources)86 (MIM: 235200). Four participants each carried the SERPINA1 S or Z alleles (c.863A>T [p.Glu288Val] and c.1096G>A [p.Glu366Lys]), respectively) associated with alpha-1-antitrypsin deficiency (see GeneReviews by Stoller et al. in Web Resources) (MIM: 613490). Five participants were carriers for variants in GALT that are associated with the Duarte 2 allele87, 88, 89 (including one missense variant and three intronic variants), while two participants were carriers for a variant associated with classic galactosemia90,91 (MIM: 230400), c.563A>G (p.Gln188Arg). Five participants carried a hypomorphic variant located in the 5′ untranslated region (UTR) in RBM8A (c.−21G>A [p.?]), associated with thrombocytopenia-absent radius (TAR) syndrome92,93 (MIM: 274000). Additionally, we identified three carriers of pathogenic CFTR (MIM: 602421) variants associated with cystic fibrosis94, 95, 96, 97 (MIM: 219700); two participants carried the CFTR c.1521_1523del (p.Phe508del) founder variant and one participant carried the CFTR c.1519_1521del (p.Ile507del) variant (Table S2). All variants reported in the NC NEXUS study are listed in Table S3.
Figure 1.
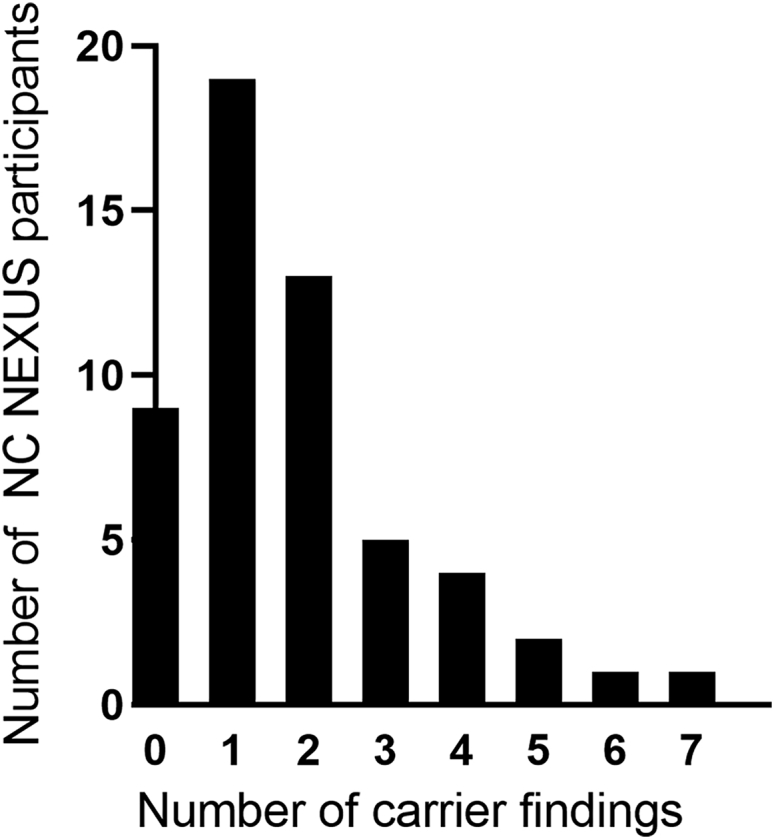
Number of Carrier Findings for NC NEXUS Participants that Were Randomized into the Experimental Group and Elected to Receive Carrier Findings
Number of carrier findings is represented on the x axis and the number of NC NEXUS participants is represented on the y axis.
Figure 2.

Variants in HFE, SERPINA1, GALT, and RBM8A Were the Most Frequently Observed Carrier Findings
Variants/predicted protein changes and genes in which they are located are represented on the x axis. Total number of carrier findings in the NC NEXUS study is represented on the y axis.
Discussion
The potential use of genome-scale sequencing to screen healthy individuals for monogenic disorders—before symptoms manifest—promises the tantalizing possibility of ameliorating these conditions, but also raises numerous challenges. Among the barriers to the use of genomic sequencing in any screening endeavor is the performance of this technology and our ability to interpret variants in the absence of phenotypic indications. Other critical issues include the kinds of genomic information that should be offered, how parental informed consent can be obtained, and how the overall process and the potential results will impact families. To address these questions, the NC NEXUS study enrolled a cohort of 106 children: 17 with inborn errors of metabolism, 28 with hearing loss, and 61 healthy newborns. We explored the analytical performance of ES in the context of newborn screening, both from a “screening” standpoint as well as in an “indication-based” interpretation mode.
Analytic Performance
In order to assess how well ES would perform for predictive purposes, molecular analysts were initially blinded to the cohort status of the individual, enabling us to simulate the conditions of a true screening test, in which the individual is assumed to be at population risk. We did not perform trio analyses, which can be helpful for identifying de novo and compound heterozygous variants, although we did conduct follow-up parental sequencing whenever possible where compound heterozygosity was suspected. From the standpoint of implementation workflows, it is reasonable to attempt to obtain trio samples in order to maximize the diagnostic yield of clinical diagnostic sequencing; however, comprehensive trio sample collection would be impractical within current NBS practices.
Screening is unlike indication-based diagnostic testing, in which the analyst can utilize phenotypic information to judge which variants are most likely to be relevant, and where the disclosure of inconclusive results (variants of uncertain significance, single heterozygous variants in genes associated with recessive conditions, etc.) can still be informative and allow for more detailed clinical follow-up. However, in a population at low risk for any given monogenic disorder, disclosure of genomic findings with lower certainty will inevitably lead to an extremely high false positive rate, and we therefore set relatively stringent thresholds for the results to qualify as an abnormal positive screen. This decision reflects the inevitable balance between increasing sensitivity to maximize case finding versus establishing stringent thresholds to reduce false positives. As expected, a small number of participants (4 out of the total 106) had abnormal positive screen NGS-NBS results (beyond those that would be expected for those in the affected cohorts), but the results have implications for their health supervision. These findings exemplify the expansion of the possible range of conditions that could be identified in newborns and thereby influence future health care and screening to ultimately improve clinical outcomes. These findings had implications for other family members, but longitudinal follow-up of a larger number of screened individuals will be required to assess outcomes, clinical utility, and the economic value of NGS-NBS. Among the affected cohorts, the screening analysis predicted the presence of inborn errors of metabolism in 15 out of 17 individuals and hearing loss in 5 out of 28 individuals. Thus, at a population level, it is unlikely that genomic sequencing could replace current screening based on biochemical measurements or audiology testing.
Rather than being used purely in a screening mode of analysis, genomic information could also be used in conjunction with phenotypic information (such as a metabolite levels) to perform a “secondary” or “indication-based” analysis that may improve the sensitivity and specificity of newborn screening for inborn errors of metabolism. In our study, after determining the “screening” results for a given individual, the analyst was unblinded to the cohort that the individual was from, and a more sensitive diagnostic analysis was conducted. The yield of sequencing improved somewhat in the context of indication-based analysis: the two remaining individuals in the metabolic cohort and several members of the hearing loss cohort had highly suggestive but “inconclusive” results (e.g., a single heterozygous pathogenic variant, a homozygous missense VUS in a gene associated with an autosomal-recessive disease). In the case of the participant with malonyl-CoA decarboxylase deficiency, this phenotype information was valuable for detecting a possible explanation for the child’s deficiency in the indication-based analysis (homozygous MLYCD c.1013T>C [p.Leu338Pro] variant). Genomic analysis may also improve the specificity of newborn screening. For example, low total carnitine levels found by TMS may be indicative of primary carnitine deficiency, a condition that can result in encephalopathy, cardiomyopathy, and hypoglycemia; however, carnitine levels may also be influenced by other factors, such as prematurity and/or drug interactions (see GeneReviews by El-Hattab in Web Resources).33,34 Detecting pathogenic variants in SLC22A5 provides an unambiguous molecular diagnosis. These findings suggest that sequencing might be useful as an adjunct to traditional NBS methods, and that with improved detection of variants (such as non-canonical splicing variants, copy number variants, or rearrangements) and more extensive interpretive databases, the positive predictive value of genomic screening may improve.
A potential explanation for the relatively low diagnostic yield of ES for participants in the hearing loss cohort is that numerous factors98, 99, 100, 101, 102 contribute to hearing loss, including non-genetic factors (i.e., environmental, infections, premature birth, etc.) that are estimated to account for approximately 20% of cases of prelingual hearing loss (see GeneReviews by Shearer et al. in Web Resources). The diagnostic rate in our study likely reflects the genetic heterogeneity of the monogenic forms of hearing loss103,104 as well as the incomplete knowledge of pathogenic variants. Ascertainment bias also may have led to a larger fraction of the hearing loss cohort having complex syndromic conditions that led the referring providers to consider exome sequencing as a useful diagnostic test. Other hearing loss patients may already have had panel testing, thus depleting the enrolled study population of the more commonly identified nonsyndromic hearing loss conditions. In addition, certain genetic forms of hearing loss are caused by copy number variants or mitochondrial DNA variants (see GeneReviews by Shearer et al. in Web Resources),105 which were not assessed in this study. Due to the heterogeneity of the etiologies of hearing loss, it seems clear that phenotypic screening (at birth and during childhood) will remain necessary, even if detection of the genetic mechanisms improves.
On the other hand, many types of hearing loss that would be detectable with genomic sequencing (for example, hearing loss due to pathogenic or likely pathogenic variants in TECTA as shown in one NC NEXUS subject) do not have congenital onset and would therefore be missed at birth. The pathogenic TECTA variant finding demonstrates the potential for NGS-NBS to detect hearing loss risk in children who are not identified through traditional newborn hearing screening. The identification of a genetic risk for hearing loss in this setting could be followed up with more aggressive audiology evaluations during infancy and early childhood in order to facilitate interventions and reduce the chance of language impairment. In other cases, hearing loss is associated with a broader syndromic condition, for example the NC NEXUS subjects with Warsaw breakage syndrome or Lowe syndrome. These examples raise intriguing points regarding the implementation of genomic screening in newborns, where testing in the context of a specific phenotype can be a powerful diagnostic tool enabling families and clinicians to better manage an individual’s health condition. However, it is unclear whether routine genomic screening of all healthy newborns for such conditions would be useful in a broader context without clinical interventions to ameliorate symptoms; in such cases, parental input will be critical.
Additional Genomic Findings
One question raised by the prospect of genomic sequencing in newborns is how parents will be engaged in the process of informed decision making. A subsequent manuscript will describe in detail how parents used an online decision aid, factors associated with decisions about whether or not to learn additional genomic findings, and parents’ reactions to learning the results of these additional analyses. From a molecular analysis standpoint, carrier findings reported in this study were similar to other studies.10,106, 107, 108, 109 Interestingly, many of the variants we observed are milder or hypomorphic variants; in order to cause disease, these alleles must be in trans with a more severe variant in the same gene (e.g., the RBM8A c.−21G>A [p.?] variant and the HFE c.187C>G [p.His63Asp] variant). These hypomorphic variants complicate the analysis and the reporting of carrier findings. In addition, identifying carrier status in a newborn is not fully informative with regards to carrier status in the parents or siblings without further analysis, which decreases the clinical utility of reporting carrier findings in infants.
Other molecular findings predictive of a current or future clinical diagnosis in the patient, which we categorized in this study as childhood-onset nonactionable and adult-onset actionable conditions, can provide clinically relevant information, yet it is unclear whether this information should be sought routinely in the course of newborn screening. In the context of an otherwise healthy newborn, this information could potentially allow parents and providers to prepare for future eventualities, to avoid a “diagnostic odyssey,” or even to alert other family members that they could also have a disease-causing variant that may be immediately relevant to their health, as in the case of hereditary cancer predisposition. However, the long-term impact of this information on clinical outcomes and family psychosocial dynamics is unknown. We also recognize that the screening of healthy adults for medically actionable conditions such as cancer predisposition is becoming increasingly available, thereby reducing the imperative to discover these findings in an infant.
Conclusions and Future Directions
Clinical applications of genomic sequencing technologies offer great opportunities in both diagnostic testing and screening. In the context of NBS, sequencing-based approaches cannot fully replace biochemical or phenotypic screens because of etiologic heterogeneity and the challenges of variant interpretation. However, sequencing approaches have the advantage of being able to identify virtually any condition with a known genetic cause. Thus, augmentation of newborn screening with some form of genomic sequencing seems inevitable. In the near term, sequencing of genes already associated with conditions that are screened for in traditional NBS may help to improve clinical sensitivity and specificity. Expanding the current NBS panel to include other actionable conditions detectable only by sequencing could further enhance the public health benefits of NBS, if interpretive challenges can be overcome in order to balance case detection versus false positives. Providing additional genomic information beyond the most actionable conditions, while potentially of interest to many parents, may increase the complexity of informed consent and thereby serve to distract from the primary health benefits of NGS-NBS. There will be significant costs, ethical considerations, and implementation challenges involved in conducting genome-scale sequencing in healthy newborns. These challenges may limit broad application across the entire population, thus failing to deliver on the promise of the Human Genome Project. We should therefore continue to explore innovative approaches to NGS-NBS that will enable the most comprehensive adoption in the general population, improve outcomes to the greatest extent possible, and maximize societal benefits in a cost-effective manner.
Declaration of Interests
D.B.B. is involved in an unrelated research study that receives contributed equipment and reagents from Asuragen and an unrelated research study that receives partial funding from Sarepta Therapeutics. B.C.P. is an investigator on a different research study that receives in-kind support (reagents and sequencing consumables) from Illumina (San Diego, CA). K.E.W. is President and Chair of the Executive Committee and Board of Directors for the Association for Molecular Pathology, a member of the Advisory Committee for the US FDA Medical Devices Molecular and Clinical Genetics Devices Panel, and a member of the Consultant Advisory Panel for BlueCross BlueShield of North Carolina.
Acknowledgments
We thank the families who participated in the study. This work was funded by the National Human Genome Research Institute and the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health as part of the Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) Program, U19 HD077632. We gratefully acknowledge the UNC BioSpecimen Processing Facility and the UNC High-Throughput Sequencing Facility for their work in DNA extraction, sample preparation and exome sequencing, and Manyu Li and the UNC Hospitals Clinical Molecular Genetics Laboratory. We also thank Jason Reilly, Ian Wilhelmsen, Kirk Wilhelmsen, Dylan Young, and the Renaissance Computing Institute (RENCI) at UNC for bioinformatics analysis and expertise; Alexandra Arreola and Claire Edgerly for contributions to molecular analysis; and Lonna Mollison and Kathleen Wallace for their contributions to curating the NGS-NBS and additional findings gene lists.
Published: August 26, 2020
Footnotes
Supplemental Data can be found online at https://doi.org/10.1016/j.ajhg.2020.08.001.
Web Resources
ClinGen Allele Registry, http://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/landing
ClinGen Variant Pathogenicity Curation Interface, https://curation.clinicalgenome.org/
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/submitters/506663
ExAC Browser, http://exac.broadinstitute.org/
GeneReviews, Alkhunaizi, E., Brosh, R.M., Alkuraya, F.S., and Chitayat, D. (2019). Warsaw syndrome. https://www.ncbi.nlm.nih.gov/books/NBK541972/
GeneReviews, Barton, J.C., and Edwards, C.Q. (2018). HFE hemochromatosis. https://www.ncbi.nlm.nih.gov/books/NBK1440/
GeneReviews, El-Hattab, A.W. (2016). Systemic primary carnitine deficiency, https://www.ncbi.nlm.nih.gov/books/NBK84551/
GeneReviews, Stoller, J.K., Hupertz, V., and Aboussouan, L.S. (2020). Alpha-1 antitrypsin deficiency: gene review. https://www.ncbi.nlm.nih.gov/books/NBK1519/
GeneReviews, Shearer, A.E., Hildebrand, M.S., and Smith, R.J. (2017). Hereditary hearing loss and deafness overview. https://www.ncbi.nlm.nih.gov/books/NBK1434/
gnomAD Browser, https://gnomad.broadinstitute.org/
Mutalyzer, https://mutalyzer.nl/index
OMIM, https://www.omim.org/
UCSC Genome Browser, https://genome.ucsc.edu
Data and Code Availability
The variants discussed in the paper were submitted to ClinVar and variant accession numbers can be found in Table S3. ClinicalTrials.gov Identifier: NCT02826694.
Supplemental Data
References
- 1.Guthrie R., Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- 2.Frazier D.M., Millington D.S., McCandless S.E., Koeberl D.D., Weavil S.D., Chaing S.H., Muenzer J. The tandem mass spectrometry newborn screening experience in North Carolina: 1997-2005. J. Inherit. Metab. Dis. 2006;29:76–85. doi: 10.1007/s10545-006-0228-9. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC) Impact of expanded newborn screening--United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 2008;57:1012–1015. [PubMed] [Google Scholar]
- 4.Hammett-Stabler C.A., Garg U. Humana Press; 2010. Clinical Applications of Mass Spectrometry Methods and Protocols. [DOI] [PubMed] [Google Scholar]
- 5.Watson M.S., Mann M.Y., Lloyd-Puryear M.A., Rinaldo P., Howell R.R., Cordero J., Edwards E.S., Howse J.L., Mullaley T., Van Dyck P., American College of Medical Genetics Newborn Screening Expert Group Newborn screening: toward a uniform screening panel and system--executive summary. Pediatrics. 2006;117:S296–S307. doi: 10.1542/peds.2005-2633I. [DOI] [PubMed] [Google Scholar]
- 6.Sweetman L., Millington D.S., Therrell B.L., Hannon W.H., Popovich B., Watson M.S., Mann M.Y., Lloyd-Puryear M.A., van Dyck P.C. Naming and counting disorders (conditions) included in newborn screening panels. Pediatrics. 2006;117:S308–S314. doi: 10.1542/peds.2005-2633J. [DOI] [PubMed] [Google Scholar]
- 7.Watson M.S., Mann M.Y., Lloyd-Puryear M.A., Rinaldo P., Howell R.R. Executive Summary. Genet. Med. 2006;8:1S–11S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloğlu A., Ozen S., Sanjad S. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg J.S., Agrawal P.B., Bailey D.B., Jr., Beggs A.H., Brenner S.E., Brower A.M., Cakici J.A., Ceyhan-Birsoy O., Chan K., Chen F. Newborn sequencing in genomic medicine and public health. Pediatrics. 2017;139 doi: 10.1542/peds.2016-2252. e20162252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceyhan-Birsoy O., Murry J.B., Machini K., Lebo M.S., Yu T.W., Fayer S., Genetti C.A., Schwartz T.S., Agrawal P.B., Parad R.B., BabySeq Project Team Interpretation of genomic sequencing results in healthy and ill newborns: Results from the BabySeq project. Am. J. Hum. Genet. 2019;104:76–93. doi: 10.1016/j.ajhg.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingsmore S.F., Cakici J.A., Clark M.M., Gaughran M., Feddock M., Batalov S., Bainbridge M.N., Carroll J., Caylor S.A., Clarke C., RCIGM Investigators A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am. J. Hum. Genet. 2019;105:719–733. doi: 10.1016/j.ajhg.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bassaganyas L., Freedman G., Vaka D., Wan E., Lao R., Chen F., Kvale M., Currier R.J., Puck J.M., Kwok P.Y. Whole exome and whole genome sequencing with dried blood spot DNA without whole genome amplification. Hum. Mutat. 2018;39:167–171. doi: 10.1002/humu.23356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milko L.V., Rini C., Lewis M.A., Butterfield R.M., Lin F.C., Paquin R.S., Powell B.C., Roche M.I., Souris K.J., Bailey D.B., Jr. Evaluating parents’ decisions about next-generation sequencing for their child in the NC NEXUS (North Carolina Newborn Exome Sequencing for Universal Screening) study: a randomized controlled trial protocol. Trials. 2018;19:344. doi: 10.1186/s13063-018-2686-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milko L.V., O’Daniel J.M., DeCristo D.M., Crowley S.B., Foreman A.K.M., Wallace K.E., Mollison L.F., Strande N.T., Girnary Z.S., Boshe L.J. An age-based framework for evaluating genome-scale sequencing results in newborn screening. J. Pediatr. 2019;209:68–76. doi: 10.1016/j.jpeds.2018.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis M.A., Paquin R.S., Roche M.I., Furberg R.D., Rini C., Berg J.S., Powell C.M., Bailey D.B., Jr. Supporting parental decisions about genomic sequencing for newborn screening: The NC NEXUS decision aid. Pediatrics. 2016;137(Suppl 1):S16–S23. doi: 10.1542/peds.2015-3731E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis M.A., Stine A., Paquin R.S., Mansfield C., Wood D., Rini C., Roche M.I., Powell C.M., Berg J.S., Bailey D.B., Jr. Parental preferences toward genomic sequencing for non-medically actionable conditions in children: a discrete-choice experiment. Genet. Med. 2018;20:181–189. doi: 10.1038/gim.2017.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paquin R.S., Peinado S., Lewis M.A., Biesecker B.B., Rini C., Roche M. A behavior-theoretic evaluation of values clarification on parental beliefs and intentions toward genomic sequencing for newborns. Soc. Sci. Med. 2018;112037:30651–30658. doi: 10.1016/j.socscimed.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 1303.3997v1. [Google Scholar]
- 19.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXIv. 2012 1207.3907. [Google Scholar]
- 20.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., Genome Aggregation Database Consortium The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oza A.M., DiStefano M.T., Hemphill S.E., Cushman B.J., Grant A.R., Siegert R.K., Shen J., Chapin A., Boczek N.J., Schimmenti L.A., ClinGen Hearing Loss Clinical Domain Working Group Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018;39:1593–1613. doi: 10.1002/humu.23630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier E.M., Liebl B., Röschinger W., Nennstiel-Ratzel U., Fingerhut R., Olgemöller B., Busch U., Krone N., v Kries R., Roscher A.A. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum. Mutat. 2005;25:443–452. doi: 10.1002/humu.20163. [DOI] [PubMed] [Google Scholar]
- 26.Gregersen N., Andresen B.S., Bross P., Winter V., Rüdiger N., Engst S., Christensen E., Kelly D., Strauss A.W., Kølvraa S. Molecular characterization of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: identification of a lys329 to glu mutation in the MCAD gene, and expression of inactive mutant enzyme protein in E. coli. Hum. Genet. 1991;86:545–551. doi: 10.1007/BF00201539. [DOI] [PubMed] [Google Scholar]
- 27.Andresen B.S., Dobrowolski S.F., O’Reilly L., Muenzer J., McCandless S.E., Frazier D.M., Udvari S., Bross P., Knudsen I., Banas R. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001;68:1408–1418. doi: 10.1086/320602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokota I., Coates P.M., Hale D.E., Rinaldo P., Tanaka K. Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am. J. Hum. Genet. 1991;49:1280–1291. [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon Y.A., Lee D.H., Ki C.S., Lee S.Y., Kim J.W., Lee Y.W., Park H.D. SLC22A5 mutations in a patient with systemic primary carnitine deficiency: the first Korean case confirmed by biochemical and molecular investigation. Ann. Clin. Lab. Sci. 2012;42:424–428. [PubMed] [Google Scholar]
- 30.Li F.Y., El-Hattab A.W., Bawle E.V., Boles R.G., Schmitt E.S., Scaglia F., Wong L.J. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum. Mutat. 2010;31:E1632–E1651. doi: 10.1002/humu.21311. [DOI] [PubMed] [Google Scholar]
- 31.Burwinkel B., Kreuder J., Schweitzer S., Vorgerd M., Gempel K., Gerbitz K.D., Kilimann M.W. Carnitine transporter OCTN2 mutations in systemic primary carnitine deficiency: a novel Arg169Gln mutation and a recurrent Arg282ter mutation associated with an unconventional splicing abnormality. Biochem. Biophys. Res. Commun. 1999;261:484–487. doi: 10.1006/bbrc.1999.1060. [DOI] [PubMed] [Google Scholar]
- 32.Rose E.C., di San Filippo C.A., Ndukwe Erlingsson U.C., Ardon O., Pasquali M., Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012;33:118–123. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shieh J.T.C. Genomic sequencing expansion and incomplete penetrance. Pediatrics. 2019;143(Suppl 1):S22–S26. doi: 10.1542/peds.2018-1099E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee N.C., Tang N.L.S., Chien Y.H., Chen C.A., Lin S.J., Chiu P.C., Huang A.C., Hwu W.L. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol. Genet. Metab. 2010;100:46–50. doi: 10.1016/j.ymgme.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Love-Gregory L.D., Grasela J., Hillman R.E., Phillips C.L. Evidence of common ancestry for the maple syrup urine disease (MSUD) Y438N allele in non-Mennonite MSUD patients. Mol. Genet. Metab. 2002;75:79–90. doi: 10.1006/mgme.2001.3264. [DOI] [PubMed] [Google Scholar]
- 36.Zhang B., Edenberg H.J., Crabb D.W., Harris R.A. Evidence for both a regulatory mutation and a structural mutation in a family with maple syrup urine disease. J. Clin. Invest. 1989;83:1425–1429. doi: 10.1172/JCI114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuda I., Nobukuni Y., Mitsubuchi H., Indo Y., Endo F., Asaka J., Harada A. A T-to-A substitution in the E1 α subunit gene of the branched-chain α-ketoacid dehydrogenase complex in two cell lines derived from Menonite maple syrup urine disease patients. Biochem. Biophys. Res. Commun. 1990;172:646–651. doi: 10.1016/0006-291x(90)90723-z. [DOI] [PubMed] [Google Scholar]
- 38.Fisher C.R., Chuang J.L., Cox R.P., Fisher C.W., Star R.A., Chuang D.T. Maple syrup urine disease in Mennonites. Evidence that the Y393N mutation in E1 α impedes assembly of the E1 component of branched-chain α-keto acid dehydrogenase complex. J. Clin. Invest. 1991;88:1034–1037. doi: 10.1172/JCI115363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stojiljkovic M., Klaassen K., Djordjevic M., Sarajlija A., Brasil S., Kecman B., Grkovic S., Kostic J., Rodriguez-Pombo P., Desviat L.R. Molecular and phenotypic characteristics of seven novel mutations causing branched-chain organic acidurias. Clin. Genet. 2016;90:252–257. doi: 10.1111/cge.12751. [DOI] [PubMed] [Google Scholar]
- 40.Froese D.S., Forouhar F., Tran T.H., Vollmar M., Kim Y.S., Lew S., Neely H., Seetharaman J., Shen Y., Xiao R. Crystal structures of malonyl-coenzyme A decarboxylase provide insights into its catalytic mechanism and disease-causing mutations. Structure. 2013;21:1182–1192. doi: 10.1016/j.str.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chapel-Crespo C., Gavrilov D., Sowa M., Myers J., Day-Salvatore D.L., Lynn H., Regier D., Starin D., Steenari M., Schoonderwoerd K., Abdenur J.E. Clinical, biochemical and molecular characteristics of malonyl-CoA decarboxylase deficiency and long-term follow-up of nine patients. Mol. Genet. Metab. 2019;128:113–121. doi: 10.1016/j.ymgme.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 42.Gao J., Waber L., Bennett M.J., Gibson K.M., Cohen J.C. Cloning and mutational analysis of human malonyl-coenzyme A decarboxylase. J. Lipid Res. 1999;40:178–182. [PubMed] [Google Scholar]
- 43.FitzPatrick D.R., Hill A., Tolmie J.L., Thorburn D.R., Christodoulou J. The molecular basis of malonyl-CoA decarboxylase deficiency. Am. J. Hum. Genet. 1999;65:318–326. doi: 10.1086/302492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sacksteder K.A., Morrell J.C., Wanders R.J.A., Matalon R., Gould S.J. MCD encodes peroxisomal and cytoplasmic forms of malonyl-CoA decarboxylase and is mutated in malonyl-CoA decarboxylase deficiency. J. Biol. Chem. 1999;274:24461–24468. doi: 10.1074/jbc.274.35.24461. [DOI] [PubMed] [Google Scholar]
- 45.D’Andrea P., Veronesi V., Bicego M., Melchionda S., Zelante L., Di Iorio E., Bruzzone R., Gasparini P. Hearing loss: frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002;296:685–691. doi: 10.1016/s0006-291x(02)00891-4. [DOI] [PubMed] [Google Scholar]
- 46.Zelante L., Gasparini P., Estivill X., Melchionda S., D’Agruma L., Govea N., Milá M., Monica M.D., Lutfi J., Shohat M. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997;6:1605–1609. doi: 10.1093/hmg/6.9.1605. [DOI] [PubMed] [Google Scholar]
- 47.Morell R.J., Kim H.J., Hood L.J., Goforth L., Friderici K., Fisher R., Van Camp G., Berlin C.I., Oddoux C., Ostrer H. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N. Engl. J. Med. 1998;339:1500–1505. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- 48.Carrasquillo M.M., Zlotogora J., Barges S., Chakravarti A. Two different connexin 26 mutations in an inbred kindred segregating non-syndromic recessive deafness: implications for genetic studies in isolated populations. Hum. Mol. Genet. 1997;6:2163–2172. doi: 10.1093/hmg/6.12.2163. [DOI] [PubMed] [Google Scholar]
- 49.Chattaraj P., Munjal T., Honda K., Rendtorff N.D., Ratay J.S., Muskett J.A., Risso D.S., Roux I., Gertz E.M., Schäffer A.A. A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J. Med. Genet. 2017;54:665–673. doi: 10.1136/jmedgenet-2017-104721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Hauwe P., Everett L.A., Coucke P., Scott D.A., Kraft M.L., Ris-Stalpers C., Bolder C., Otten B., de Vijlder J.J.M., Dietrich N.L. Two frequent missense mutations in Pendred syndrome. Hum. Mol. Genet. 1998;7:1099–1104. doi: 10.1093/hmg/7.7.1099. [DOI] [PubMed] [Google Scholar]
- 51.Coyle B., Reardon W., Herbrick J.A., Tsui L.C., Gausden E., Lee J., Coffey R., Grueters A., Grossman A., 4, Phelps P.D. Molecular analysis of the PDS gene in Pendred syndrome. Hum. Mol. Genet. 1998;7:1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 52.Scott D.A., Wang R., Kreman T.M., Andrews M., McDonald J.M., Bishop J.R., Smith R.J.H., Karniski L.P., Sheffield V.C. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4) Hum. Mol. Genet. 2000;9:1709–1715. doi: 10.1093/hmg/9.11.1709. [DOI] [PubMed] [Google Scholar]
- 53.Choi B.Y., Stewart A.K., Madeo A.C., Pryor S.P., Lenhard S., Kittles R., Eisenman D., Kim H.J., Niparko J., Thomsen J. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum. Mutat. 2009;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildebrand M.S., Morín M., Meyer N.C., Mayo F., Modamio-Hoybjor S., Mencía A., Olavarrieta L., Morales-Angulo C., Nishimura C.J., Workman H. DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum. Mutat. 2011;32:825–834. doi: 10.1002/humu.21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sagong B., Park R., Kim Y.H., Lee K.Y., Baek J.I., Cho H.J., Cho I.J., Kim U.K., Lee S.H. Two novel missense mutations in the TECTA gene in Korean families with autosomal dominant nonsyndromic hearing loss. Ann. Clin. Lab. Sci. 2010;40:380–385. [PubMed] [Google Scholar]
- 56.Moteki H., Nishio S.Y., Hashimoto S., Takumi Y., Iwasaki S., Takeichi N., Fukuda S., Usami S. TECTA mutations in Japanese with mid-frequency hearing loss affected by zona pellucida domain protein secretion. J. Hum. Genet. 2012;57:587–592. doi: 10.1038/jhg.2012.73. [DOI] [PubMed] [Google Scholar]
- 57.Chaoui A., Watanabe Y., Touraine R., Baral V., Goossens M., Pingault V., Bondurand N. Identification and functional analysis of SOX10 missense mutations in different subtypes of Waardenburg syndrome. Hum. Mutat. 2011;32:1436–1449. doi: 10.1002/humu.21583. [DOI] [PubMed] [Google Scholar]
- 58.Duman D., Sirmaci A., Cengiz F.B., Ozdag H., Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet. Test. Mol. Biomarkers. 2011;15:29–33. doi: 10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- 59.Bonnet C., Riahi Z., Chantot-Bastaraud S., Smagghe L., Letexier M., Marcaillou C., Lefèvre G.M., Hardelin J.P., El-Amraoui A., Singh-Estivalet A. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016;24:1730–1738. doi: 10.1038/ejhg.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van der Lelij P., Chrzanowska K.H., Godthelp B.C., Rooimans M.A., Oostra A.B., Stumm M., Zdzienicka M.Z., Joenje H., de Winter J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Day I.N.M., Whittall R.A., O’Dell S.D., Haddad L., Bolla M.K., Gudnason V., Humphries S.E. Spectrum of LDL receptor gene mutations in heterozygous familial hypercholesterolemia. Hum. Mutat. 1997;10:116–127. doi: 10.1002/(SICI)1098-1004(1997)10:2<116::AID-HUMU4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 62.Lee W.K., Haddad L., Macleod M.J., Dorrance A.M., Wilson D.J., Gaffney D., Dominiczak M.H., Packard C.J.D., Day I.N.M., Humphries S.E., Dominiczak A.F. Identification of a common low density lipoprotein receptor mutation (C163Y) in the west of Scotland. J. Med. Genet. 1998;35:573–578. doi: 10.1136/jmg.35.7.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laurie A.D., Scott R.S., George P.M. Genetic screening of patients with familial hypercholesterolaemia (FH): a New Zealand perspective. Atheroscler. Suppl. 2004;5:13–15. doi: 10.1016/j.atherosclerosissup.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Villéger L., Abifadel M., Allard D., Rabès J.P., Thiart R., Kotze M.J., Béroud C., Junien C., Boileau C., Varret M. The UMD-LDLR database: additions to the software and 490 new entries to the database. Hum. Mutat. 2002;20:81–87. doi: 10.1002/humu.10102. [DOI] [PubMed] [Google Scholar]
- 65.Etxebarria A., Benito-Vicente A., Stef M., Ostolaza H., Palacios L., Martin C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis. 2015;238:304–312. doi: 10.1016/j.atherosclerosis.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 66.Thormaehlen A.S., Schuberth C., Won H.H., Blattmann P., Joggerst-Thomalla B., Theiss S., Asselta R., Duga S., Merlini P.A., Ardissino D. Systematic cell-based phenotyping of missense alleles empowers rare variant association studies: a case for LDLR and myocardial infarction. PLoS Genet. 2015;11:e1004855. doi: 10.1371/journal.pgen.1004855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Civeira F., Jarauta E., Cenarro A., García-Otín A.L., Tejedor D., Zambón D., Mallen M., Ros E., Pocoví M. Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J. Am. Coll. Cardiol. 2008;52:1546–1553. doi: 10.1016/j.jacc.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 68.Myers J.H., Shook J.E. Vomiting, ataxia, and altered mental status in an adolescent: late-onset ornithine transcarbamylase deficiency. Am. J. Emerg. Med. 1996;14:553–557. doi: 10.1016/S0735-6757(96)90097-2. [DOI] [PubMed] [Google Scholar]
- 69.Yamaguchi S., Brailey L.L., Morizono H., Bale A.E., Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 2006;27:626–632. doi: 10.1002/humu.20339. [DOI] [PubMed] [Google Scholar]
- 70.Heuser A., Plovie E.R., Ellinor P.T., Grossmann K.S., Shin J.T., Wichter T., Basson C.T., Lerman B.B., Sasse-Klaassen S., Thierfelder L. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mitchell M., Mountford R., Butler R., Alhaq A., Dai L., Savidge G., Bolton-Maggs P.H.B. Spectrum of factor XI (F11) mutations in the UK population--116 index cases and 140 mutations. Hum. Mutat. 2006;27:829. doi: 10.1002/humu.9439. [DOI] [PubMed] [Google Scholar]
- 72.Pike G.N., Cumming A.M., Hay C.R.M., Sempasa B., Sutherland M., Thachil J., Burthem J., Bolton-Maggs P.H.B. In vitro comparison of the effect of two factor XI (FXI) concentrates on thrombin generation in major FXI deficiency. Haemophilia. 2016;22:403–410. doi: 10.1111/hae.12846. [DOI] [PubMed] [Google Scholar]
- 73.Esteban J., de la Morena-Barrio M.E., Salloum-Asfar S., Padilla J., Miñano A., Roldán V., Soria J.M., Vidal F., Corral J., Vicente V. High incidence of FXI deficiency in a Spanish town caused by 11 different mutations and the first duplication of F11: Results from the Yecla study. Haemophilia. 2017;23:e488–e496. doi: 10.1111/hae.13356. [DOI] [PubMed] [Google Scholar]
- 74.Lhota F., Zemankova P., Kleiblova P., Soukupova J., Vocka M., Stranecky V., Janatova M., Hartmannova H., Hodanova K., Kmoch S., Kleibl Z. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016;90:324–333. doi: 10.1111/cge.12748. [DOI] [PubMed] [Google Scholar]
- 75.Song H., Dicks E., Ramus S.J., Tyrer J.P., Intermaggio M.P., Hayward J., Edlund C.K., Conti D., Harrington P., Fraser L. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J. Clin. Oncol. 2015;33:2901–2907. doi: 10.1200/JCO.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Syrjäkoski K., Kuukasjärvi T., Waltering K., Haraldsson K., Auvinen A., Borg A., Kainu T., Kallioniemi O.P., Koivisto P.A. BRCA2 mutations in 154 finnish male breast cancer patients. Neoplasia. 2004;6:541–545. doi: 10.1593/neo.04193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim H., Cho D.Y., Choi D.H., Choi S.Y., Shin I., Park W., Huh S.J., Han S.H., Lee M.H., Ahn S.H., Korean Breast Cancer Study Group Characteristics and spectrum of BRCA1 and BRCA2 mutations in 3,922 Korean patients with breast and ovarian cancer. Breast Cancer Res. Treat. 2012;134:1315–1326. doi: 10.1007/s10549-012-2159-5. [DOI] [PubMed] [Google Scholar]
- 78.Sun J., Meng H., Yao L., Lv M., Bai J., Zhang J., Wang L., Ouyang T., Li J., Wang T. Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin. Cancer Res. 2017;23:6113–6119. doi: 10.1158/1078-0432.CCR-16-3227. [DOI] [PubMed] [Google Scholar]
- 79.Lim M.C., Kang S., Seo S.S., Kong S.Y., Lee B.Y., Lee S.K., Park S.Y. BRCA1 and BRCA2 germline mutations in Korean ovarian cancer patients. J. Cancer Res. Clin. Oncol. 2009;135:1593–1599. doi: 10.1007/s00432-009-0607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han S.H., Lee K.R., Lee D.G., Kim B.Y., Lee K.E., Chung W.S. Mutation analysis of BRCA1 and BRCA2 from 793 Korean patients with sporadic breast cancer. Clin. Genet. 2006;70:496–501. doi: 10.1111/j.1399-0004.2006.00717.x. [DOI] [PubMed] [Google Scholar]
- 81.Ahn S.H., Son B.H., Yoon K.S., Noh D.Y., Han W., Kim S.W., Lee E.S., Park H.L., Hong Y.J., Choi J.J. BRCA1 and BRCA2 germline mutations in Korean breast cancer patients at high risk of carrying mutations. Cancer Lett. 2007;245:90–95. doi: 10.1016/j.canlet.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 82.Seong M.W., Cho S., Noh D.Y., Han W., Kim S.W., Park C.M., Park H.W., Kim S.Y., Kim J.Y., Park S.S. Comprehensive mutational analysis of BRCA1/BRCA2 for Korean breast cancer patients: evidence of a founder mutation. Clin. Genet. 2009;76:152–160. doi: 10.1111/j.1399-0004.2009.01202.x. [DOI] [PubMed] [Google Scholar]
- 83.Choi D.H., Lee M.H., Bale A.E., Carter D., Haffty B.G. Incidence of BRCA1 and BRCA2 mutations in young Korean breast cancer patients. J. Clin. Oncol. 2004;22:1638–1645. doi: 10.1200/JCO.2004.04.179. [DOI] [PubMed] [Google Scholar]
- 84.Hichri H., Rendu J., Monnier N., Coutton C., Dorseuil O., Poussou R.V., Baujat G., Blanchard A., Nobili F., Ranchin B. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 2011;32:379–388. doi: 10.1002/humu.21391. [DOI] [PubMed] [Google Scholar]
- 85.Kim H.-K., Kim J.H., Kim Y.-M., Kim G.-H., Lee B.H., Choi J.-H., Yoo H.-W. Lowe syndrome: a single center’s experience in Korea. Korean J. Pediatr. 2014;57:140–148. doi: 10.3345/kjp.2014.57.3.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pedersen P., Milman N. Genetic screening for HFE hemochromatosis in 6,020 Danish men: penetrance of C282Y, H63D, and S65C variants. Ann. Hematol. 2009;88:775–784. doi: 10.1007/s00277-008-0679-1. [DOI] [PubMed] [Google Scholar]
- 87.Carney A.E., Sanders R.D., Garza K.R., McGaha L.A., Bean L.J.H., Coffee B.W., Thomas J.W., Cutler D.J., Kurtkaya N.L., Fridovich-Keil J.L. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1-phosphate uridylyltransferase. Hum. Mol. Genet. 2009;18:1624–1632. doi: 10.1093/hmg/ddp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kozák L., Francová H., Pijácková A., Macku J., Stastná S., Peskovová K., Martincová O., Krijt J. Presence of a deletion in the 5′ upstream region of the GALT gene in Duarte (D2) alleles. J. Med. Genet. 1999;36:576–578. [PMC free article] [PubMed] [Google Scholar]
- 89.Pyhtila B.M., Shaw K.A., Neumann S.E., Fridovich-Keil J.L. Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep. 2015;15:79–93. doi: 10.1007/8904_2014_302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fridovich-Keil J.L., Langley S.D., Mazur L.A., Lennon J.C., Dembure P.P., Elsas J.L., 2nd Identification and functional analysis of three distinct mutations in the human galactose-1-phosphate uridyltransferase gene associated with galactosemia in a single family. Am. J. Hum. Genet. 1995;56:640–646. [PMC free article] [PubMed] [Google Scholar]
- 91.Reichardt J.K.V., Packman S., Woo S.L.C. Molecular characterization of two galactosemia mutations: correlation of mutations with highly conserved domains in galactose-1-phosphate uridyl transferase. Am. J. Hum. Genet. 1991;49:860–867. [PMC free article] [PubMed] [Google Scholar]
- 92.Albers C.A., Paul D.S., Schulze H., Freson K., Stephens J.C., Smethurst P.A., Jolley J.D., Cvejic A., Kostadima M., Bertone P. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012;44:435–439, S1–S2. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bottillo I., Castori M., De Bernardo C., Fabbri R., Grammatico B., Preziosi N., Scassellati G.S., Silvestri E., Spagnuolo A., Laino L., Grammatico P. Prenatal diagnosis and post-mortem examination in a fetus with thrombocytopenia-absent radius (TAR) syndrome due to compound heterozygosity for a 1q21.1 microdeletion and a RBM8A hypomorphic allele: a case report. BMC Res. Notes. 2013;6:376. doi: 10.1186/1756-0500-6-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerem E., Corey M., Kerem B.S., Rommens J., Markiewicz D., Levison H., Tsui L.C., Durie P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508) N. Engl. J. Med. 1990;323:1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 95.Kerem B.-S., Zielenski J., Markiewicz D., Bozon D., Gazit E., Yahav J., Kennedy D., Riordan J.R., Collins F.S., Rommens J.M. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc. Natl. Acad. Sci. USA. 1990;87:8447–8451. doi: 10.1073/pnas.87.21.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Orozco L., Friedman K., Chávez M., Lezana J.L., Villarreal M.T., Carnevale A. Identification of the I507 deletion by site-directed mutagenesis. Am. J. Med. Genet. 1994;51:137–139. doi: 10.1002/ajmg.1320510210. [DOI] [PubMed] [Google Scholar]
- 97.Nelson P.V., Carey W.F., Morris C.P. Identification of a cystic fibrosis mutation: deletion of isoleucine506. Hum. Genet. 1991;86:391–393. doi: 10.1007/BF00201841. [DOI] [PubMed] [Google Scholar]
- 98.Sundstrom R.A., Van Laer L., Van Camp G., Smith R.J.H. Autosomal recessive nonsyndromic hearing loss. Am. J. Med. Genet. 1999;89:123–129. doi: 10.1002/(sici)1096-8628(19990924)89:3<123::aid-ajmg2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 99.Rehm H.L., Morton C.C. A new age in the genetics of deafness. Genet. Med. 1999;1:295–302. doi: 10.1097/00125817-199909000-00009. [DOI] [PubMed] [Google Scholar]
- 100.Korver A.M.H., Smith R.J.H., Van Camp G., Schleiss M.R., Bitner-Glindzicz M.A.K., Lustig L.R., Usami S.I., Boudewyns A.N. Congenital hearing loss. Nat. Rev. Dis. Primers. 2017;3:16094. doi: 10.1038/nrdp.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petit C., Levilliers J., Hardelin J.-P. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001;35:589–646. doi: 10.1146/annurev.genet.35.102401.091224. [DOI] [PubMed] [Google Scholar]
- 102.Petersen M.B. Non-syndromic autosomal-dominant deafness. Clin. Genet. 2002;62:1–13. doi: 10.1034/j.1399-0004.2002.620101.x. [DOI] [PubMed] [Google Scholar]
- 103.Alford R.L., Arnos K.S., Fox M., Lin J.W., Palmer C.G., Pandya A., Rehm H.L., Robin N.H., Scott D.A., Yoshinaga-Itano C., ACMG Working Group on Update of Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss. Professional Practice and Guidelines Committee American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 2014;16:347–355. doi: 10.1038/gim.2014.2. [DOI] [PubMed] [Google Scholar]
- 104.DiStefano M.T., Hemphill S.E., Oza A.M., Siegert R.K., Grant A.R., Hughes M.Y., Cushman B.J., Azaiez H., Booth K.T., Chapin A., ClinGen Hearing Loss Clinical Domain Working Group ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet. Med. 2019;21:2239–2247. doi: 10.1038/s41436-019-0487-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shearer A.E., Kolbe D.L., Azaiez H., Sloan C.M., Frees K.L., Weaver A.E., Clark E.T., Nishimura C.J., Black-Ziegelbein E.A., Smith R.J.H. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6:37. doi: 10.1186/gm554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Punj S., Akkari Y., Huang J., Yang F., Creason A., Pak C., Potter A., Dorschner M.O., Nickerson D.A., Robertson P.D. Preconception carrier screening by genome sequencing: Results from the clinical laboratory. Am. J. Hum. Genet. 2018;102:1078–1089. doi: 10.1016/j.ajhg.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Porter K.M., Kauffman T.L., Koenig B.A., Lewis K.L., Rehm H.L., Richards C.S., Strande N.T., Tabor H.K., Wolf S.M., Yang Y., members of the CSER Actionability and Return of Results Working Group Approaches to carrier testing and results disclosure in translational genomics research: The clinical sequencing exploratory research consortium experience. Mol. Genet. Genomic Med. 2018;6:898–909. doi: 10.1002/mgg3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Umstead K.L., Han P.K.J., Lewis K.L., Miller I.M., Hepler C.L., Thompson L.J., Wolfsberg T.G., Nguyen A.-D., Fredriksen M.T., Gibney G. Perceptions of uncertainties about carrier results identified by exome sequencing in a randomized controlled trial. Transl. Behav. Med. 2019;10:441–450. doi: 10.1093/tbm/ibz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Machini K., Ceyhan-Birsoy O., Azzariti D.R., Sharma H., Rossetti P., Mahanta L., Hutchinson L., McLaughlin H., Green R.C., Lebo M., Rehm H.L., MedSeq Project Analyzing and reanalyzing the genome: Findings from the MedSeq project. Am. J. Hum. Genet. 2019;105:177–188. doi: 10.1016/j.ajhg.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variants discussed in the paper were submitted to ClinVar and variant accession numbers can be found in Table S3. ClinicalTrials.gov Identifier: NCT02826694.