

Abstract
The pandemic COVID‐19 has become a global panic‐forcing life towards a compromised “new normal.” Antiviral therapy against SARS‐CoV‐2 is still lacking. Thus, development of natural inhibitors as a prophylactic measure is an attractive strategy. In this context, this work explored phytochemicals as potential inhibitors for SARS‐CoV‐2 by performing all atom molecular dynamics simulations using high performance computing for 8 rationally screened phytochemicals from Withania somnifera and Azadirachta indica and two repurposed drugs docked with the spike glycoprotein and the main protease of SARS‐CoV‐2. These phytochemicals were rationally screened from 55 Indian medicinal plants in our previous work. MM/PBSA, principal component analysis (PCA), dynamic cross correlation matrix (DCCM) plots and biological pathway enrichment analysis were performed to reveal the therapeutic efficacy of these phytochemicals. The results revealed that Withanolide R (−141.96 KJ/mol) and 2,3‐Dihydrowithaferin A (−87.60 KJ/mol) were with the lowest relative free energy of binding for main protease and the spike proteins respectively. It was also observed that the phytochemicals exhibit a remarkable multipotency with the ability to modulate various human biological pathways especially pathways in cancer. Conclusively we suggest that these compounds need further detailed in vivo experimental evaluation and clinical validation to implement them as potent therapeutic agents for combating SARS‐CoV‐2.
Keywords: India, main protease, molecular dynamics, phytochemicals, relative free binding energy, SARS‐CoV‐2, simulation, spike
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) pandemic has overwhelmed the world wide health sector (Zhu et al., 2020). As of 28th June, 2020 there have been 9,843,073 confirmed cases of COVID‐19, including 495,760 deaths (https://covid19.who.int/). Moreover, according to World Health Organization reports‐53% of the countries surveyed have partially or completely disrupted medical services for hypertension treatment; 49% for treatment for diabetes and diabetes‐related complications; 42% for cancer treatment, and 31% for cardiovascular emergencies (WHO, 2020a). Hence, this viral pandemic is forcing us to a compromised “new normal” for survival. To overcome this illness many clinical trials on vaccine development and drug repurposing are underway (WHO, 2020b). Unfortunately, till now none of the vaccines have reached the global market as the disease cure. Thus, a prophylactic treatment regime needs to be developed using natural compounds as a promising alternative therapy for containing the spread of the virus. Phytochemicals have been identified by computational drug development approaches to be effective against SARS‐CoV‐2 (Adem, Eyupoglu, Sarfraz, Rasul, & Ali, 2020; Chandel, Raj, Rathi, & Kumar, 2020; Gentile et al., 2020; Gonzalez‐Paz et al., 2020; Khaerunnisa, Kurniawan, Awaluddin, Suhartati, & Soetjipto, 2020; Khan, Zia, Ashraf, Uddin, & Ui‐Haq, 2020; Kumar, Dhanjal, Kaul, Wadhwa, & Sundar, 2020; McKee, Sternberg, Stange, Laufer, & Naujokat, 2020; Qamar ul, Alqahtani, Alamri, & Chen, 2020; Sharma & Kaur, 2020; Sun, Wong, & Guo, 2020). For example, flavonoids have been reported to be active against many viruses, and are available as natural supplements at a daily dose ranging from 100 to 500 mg (Dai et al., 2019; McKee et al., 2020; Moghaddam et al., 2014). However very few of them are undergoing clinical trials (WHO, 2020b). Therefore, looking at their potential, it can be said that more research is required to understand the efficacy of phytochemicals in the treatment of COVID‐19 by understanding their functional mechanism of inhibition by dynamically exploring their interactions within the target sites. Further, comparison of their binding modes with that of the repurposed drugs (undergoing clinical trials) will also shed light about their efficiency.
In this context, rational screening was employed in our previous work to identify effective phytochemicals from 55 Indian medicinal plants for the treatment of COVID‐19 (Parida, Paul, & Chakravorty, 2020). The phytochemicals following drug likeness properties were screened from Withania somnifera and Azadirachta indica against 8 protein targets of SARS‐CoV‐2. The phytochemicals were also observed to possess multi‐potency against various SARS CoV‐2 targets (Parida et al., 2020). This corroborates with the report by Kumar et al. (2020), where Withanone from Withania somnifera was shown to bind and stably interact with SARS‐CoV‐2 main protease.
Therefore, from our previous work and recent reports, after analysing the binding potential of these phytochemicals with multiple SARS‐CoV‐2 targets, it becomes necessary to explore their multi‐target binding affinity and stability by carrying out molecular dynamics simulations. The multi‐potential binding ability of these phytochemicals is important because it can result in overcoming multi‐drug resistance of very specific drugs designed for single targets (Kadioglu, Saeed, Johannes Greten, & Efferth, 2020). It can also be mentioned here that phytochemicals are known to bind to multiple targets (Efferth & Koch, 2011).
Among all the potential targets explored in our previous work and through literature survey, the spike glycoprotein was understood to be a lucrative target for drug development as it facilitates viral entry. The receptor to which it has the highest affinity is human angiotensin‐converting enzyme 2 (ACE2) (Y. Chen, Guo, Pan, & Zhao, 2020; Xu et al., 2020). The spike/ACE2 interface has been illustrated at the atomic level, and the efficiency of ACE2 usage was found to be a key determinant of this virus transmissibility which makes spike protein the primary target of neutralising antibody, vaccine and drug development (Li, Li, Farzan, & Harrison, 2005; Tian et al., 2020). Another important target is the SARS‐CoV‐2 main protease (Mpro). It can be targeted to prevent the virus replication within the cells. It has been reported that backbone and binding site conformations are conserved in Mpro (Joshi et al., 2020). Moreover, with its low similarity with human proteases, MPro‐ inhibitors are found to be very less cytotoxic (Anand, Ziebuhr, Wadhwani, Mesters, & Hilgenfeld, 2003; Zhang et al., 2020). Thus it can be said that SARS‐CoV‐2 spike (S) glycoprotein and main protease are crucial to the virulence and transmission of the virus. These attributes can be explored for drug development.
Therefore, in the present work, long timescale molecular dynamics simulations and calculation of free energy of binding along with biological pathway enrichment analysis were performed on the phytochemicals targeting the Spike glycoprotein and the main protease of SARS‐CoV‐2. Through this work, in light of the high infectivity rate of SARS‐CoV‐2 and lack of treatment per se, the experimental testing of phytochemicals from W. somnifera and A. indica as potential drugs against SARS‐CoV‐2 is being proposed.
2. METHODS
2.1. Computational resources
High Performance Computing resources of Amazon EC2 G4 instances with the latest generation NVIDIA T4 GPUs were employed for carrying out computational work on Amazon Web Services cloud computing platform (https://aws.amazon.com) with the help of RONIN interface (https://ronin.cloud/) as an award granted by the COVID‐19 High Performance Computing (HPC) Consortium for carrying out this work (https://covid19-hpc-consortium.org/projects). Multi GPU virtual machines with g4dn.12xlarge with 4 GPU instances and 48 virtual core CPU instances were used to carry out in total 1 μs molecular dynamics simulations. The performance obtained using this computational system configuration was 125 ns/day.
2.2. Molecular dynamics simulations
Phytochemicals from Indian medicinal plants were rationally screened by molecular docking against SARS‐CoV‐2 targets in our previous work (Parida et al., 2020). The eight top scoring phytochemicals that followed drug like properties‐ against SARS‐CoV‐2 spike glycoprotein (PDB ID: 6LZG) and the main protease (PDB ID: 6LU7) were analysed by all atom molecular dynamics (MD) simulations for 100 ns (Table 1). Two repurposed drugs that had the lowest docking score against the spike protein and the main protease were also analysed by MD simulation as a control group. All the simulations were carried out with Gromacs 2020.2 software package (Lindahl, Hess, & van der Spoel, 2020) with AMBER99SB‐ILDN force field. The force field parameters for the phytochemicals and the repurposed drugs were generated by ACPYPE (AnteChamber PYthon Parser interface) (Da Silva & Vranken, 2012). Complex charges were neutralised with sodium and chloride ions. Simulation was conducted at temperature 300 K and under a pressure of 1 bar. Each system was minimised with 5,000 steps by steepest descent algorithm. Electrostatic interactions were calculated with Particle‐Mesh‐Ewald summation (PME) (Darden, York, & Pedersen, 1993). The systems were equilibrated by 1 ns position restraint simulations of 1,000 kJ mol−1 nm−2 in the NVT and NPT ensembles. Equilibrated systems were used to simulate a 100 ns no restraint production run. Post‐MD analyses were performed, this includes root mean square deviation (RMSD), root mean square fluctuations (RMSF), the radius of gyration (Rg) and hydrogen bond occupancy.
TABLE 1.
The top scoring drug like phytochemicals used for MD simulations [Colour table can be viewed at wileyonlinelibrary.com]
| Phytochemicals | Ab | a Autodock docking score (Kcal/mol) | Source | 2D structure |
|---|---|---|---|---|
| Main protease NSP5 (PDB ID: 6lu7) | ||||
|
27‐Deoxy‐14‐hydroxywithaferin A c4 |
P1 | −10.8 | Withania somnifera |
|
|
Nimolicinol c7 |
P2 | −10.09 | Azadirachta indica |
|
|
17‐Hydroxywithaferin c6 |
P3 | −10.08 | Withanias somnifera |
|
|
Withanolide R c10 |
P4 | −9.63 | W. somnifera |
|
|
Methylprednisolone c23 |
PR5 | −8.57 | Repurposed drug |
|
| Spike protein (PDB ID: 6lzg, chain B) | ||||
|
27‐Hydroxywithanone c37 |
S1 | −8.47 | W. somnifera |
|
|
12‐Deoxywithastramonolide c40 |
S2 | −8.27 | W. somnifera |
|
|
27‐Deoxywithaferin A c36 |
S3 | −7.82 | W. somnifera |
|
|
2,3‐Dihydrowithaferin A c41 |
S4 | −7.45 | W. somnifera |
|
|
Baricitinib c56 |
SR5 | −7.62 | Repurposed drug |
|
Note: Ab: Abbreviations.
The phytochemicals (top docking scorers) were identified from a previous work to possess drug likeness properties (Parida et al., 2020).
Source: The 2D images of the phytochemicals were obtained from PubChem Database (https://pubchem.ncbi.nlm.nih.gov/).
2.3. Binding free energy calculations
Molecular Mechanics—Poisson Boltzmann Surface Area (MM‐PBSA) was applied on snapshots obtained from MD trajectory to estimate the relative binding free energy ΔG b using the GROMACS tool g_mmpbsa (Baker, Sept, Joseph, Holst, & McCammon, 2001; Kumari et al., 2014).
where b = binding free energy, v = vaccuum phase, s = solvation free energy which is the difference of solvation free energy values of complex, receptor and ligand:
where c = protein ligand complex, r = protein, l = ligand. The solvation free energy is composed of polar and non‐polar energy computed as:
where p = polar energy, np = non − polar energy. The computed free energy of binding represents the affinity and stability of the compounds with the target proteins.
2.4. Principal component analysis (PCA) and dynamics cross‐correlation matrices (DCCM) analysis
The principal component analysis (PCA) method was employed to understand the dynamical behaviour in the conformational space of SARS‐CoV‐2 spike glycoprotein and NSP5 bound to the phytochemicals and the repurposed drugs. PCA was performed using RStudio (RStudio Team, 2020) and Bio3d (Grant, Rodrigues, ElSawy, McCammon, & Caves, 2006). The eigenvectors, eigenvalues and their projection along with the first two principal components were analysed (Al‐Khafaji & Taskin Tok, 2020). PCA extracts the most variable dynamic motions in simulations, required for biological function (Amadei, Linssen, & Berendsen, 1993). It was assumed that a stable drug bound complex will undergo less dynamic fluctuations due to enhancement of rigidity of the drug bound site. Further, dynamic cross correlation matrix (DCCM) analysis was conducted to understand and compare the conformational changes of spike glycoprotein and 3CLPro upon binding of phytochemicals and the repurposed drugs. The Bio3D package was employed to perform DCCM. For both DCCM and PCA analysis we considered only Cα atomic coordinates of each residue to reduce the statistical noise.
2.5. Comparative pathway enrichment analysis between COVID‐19 and the phytochemicals protein targets
The human protein targets of the phytochemicals used in this work were predicted from SwissTargetPrediction server (Gfeller et al., 2014). The top 15 targets were retained for each phytochemical. After overlapping the targets and removal of redundancy it was possible to filter out 47 target proteins. The protein targets for human coronavirus were obtained by literature survey and a total of 172 targets were filtered after removal of redundancy (Chakrabarty, Das, Bulusu, & Roy, 2020; Zhou et al., 2020). The targets have been presented in Table S3. The gene set enrichment analysis web server, Enrichr was used for gene ontology (GO) enrichment which were summarised using REVIGO webserver (Kuleshov et al., 2016; Supek, Bošnjak, Škunca, & Šmuc, 2011). The generated networks were visualized in Cytoscape v3.8 (Shannon et al., 2003). The p‐values computed using the Fisher exact test for pathway and database enrichment were performed for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis) and VirusMint analysis using Enrichr (with p < .01). VirusMint is database of interactions between viral and human proteins (Chatr‐aryamontri et al., 2009).
3. RESULTS
3.1. Main protease (NSP5) molecular dynamics simulation analysis
Each of the best docked complexes of NSP5 with 4 phytochemicals (P1‐P4) and 1 repurposed drug (PR5), obtained from our previous work, were subjected to 100 ns MD simulations. SARS‐CoV‐2 main protease (NSP5) consists domain I (residues 8–101) and II residues (102–184) with antiparallel β‐barrel and domain III (residues 201–303) which is helical. A long loop (residues 185–200) connects domains III and II (Wu et al., 2020). The active site of NSP5 (Figure 1A) is located in between domains I and II with His41‐Cys145 as catalytic dyad (Wu et al., 2020; Yang et al., 2003). Cys145, Gly143 and Ser144 also serves as the oxyanion hole (Wu et al., 2020; Yang et al., 2003). His163, while Glu166 and Gln189 are also required for catalysis (Wu et al., 2020; Yang et al., 2003). Figure 1B illustrates stable overall RMSD over 100 ns for all the trajectories of the protein‐ligand complex (~0.2–0.45 nm). The average RMSD values considering the phytochemical bound protein complexes were P1: 0.29 nm, P2: 0.27 nm, P3: 0.25 nm, P4: 0.22 nm, the repurposed drug‐ PR5: 0.23 nm and the apo‐protein without any ligand was 0.22 nm. The average Cα, RMSD were ~0.2 nm. The average RMSD of the phytochemicals were obtained as P1: 0.24, P2: 0.31, P3: 0.2, P4: 0.18 and PR5: 0.12. This ensures that the ligands did not diffuse away from the initial binding positions and shows a stable binding of phytochemicals and the repurposed drugs making them good inhibitor candidates. Interestingly P4, PR5 RMSD distributions were overlapping with that of the apo‐protein. ΔRMSF plot (difference between RMSF of ligands and apo‐protein) revealed lesser fluctuations in the three domains for P4 and PR5 with respect to the apo‐protein, compared to the other phytochemicals (Figure 1C). Radius of gyration plots for the trajectories revealed similar structural compactness of all the ligands with the apo‐protein (average‐ 2.24 nm) in comparison to the other phytochemicals (Figure 1D).
FIGURE 1.
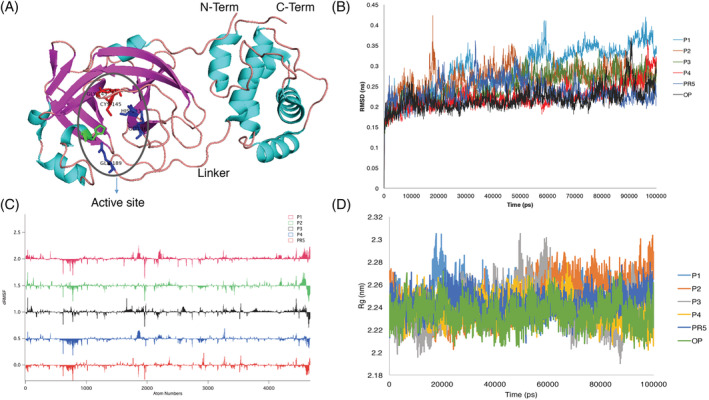
Molecular dynamics analysis of 100 ns trajectories of phytochemicals (P1‐P4) and repurposed drug (PR5) with NSP5. (A) The cartoon representation of NSP5 showing the active site, N‐terminal, C‐terminal the linker and the three domains (I, II and III). (B) Root mean square deviation (RMSD) of the 100 ns trajectories. (C) ΔRMSF stacked plot of phytochemicals (P1‐P4) and repurposed drug (PR5) with respect to NSP5 without ligand (PDB ID: 6lu7). The atom numbers are with respect to the coordinates of NSP5 (6lu7). (D) Radius of gyration over 100 ns trajectories. OP, apo‐protein. For abbreviations please refer the main text and Table 1 [Colour figure can be viewed at wileyonlinelibrary.com]
Analysis of occupancy of hydrogen bonds (distance ≤4 Å) over 100 ns of the trajectories revealed that P4 and Gln189 in the connecting loop between domain II and domain III maintained ~56% occupancy over 100 ns trajectory. Again the hydrogen bond occupancy of P4 was 28% with His41. PR5 maintained 41% with Gln189 and 18% occupancy. The hydrogen bond occupancy was higher for P4 and PR5 than other phytochemicals with the active site residues (Table S1).
Further, to extract the structural variations in details upon the binding of the phytochemicals and the repurposed drug, PCA on the Cα atom was performed. The first three Eigen vectors captured around ~50% of the motions (Figure S1). It is notable that the conformational space sampled by P4 and PR5 were very similar to the apo‐protein. Much larger conformational space was sampled by the other phytochemicals (Figures 2A and S1). The residue fluctuation (RMSF) contribution to the PCA showed reduced flexibility in PC1 and PC2 were comparable to the apo‐protein (<0.1 nm), especially in the active site regions (His41, Cys145, Gly143, Ser144, His163, Glu166, Gln189) and the domain III (residues 201‐306) for P4 and PR5 in comparison to the other phytochemicals (Figures 2B and S2).
FIGURE 2.
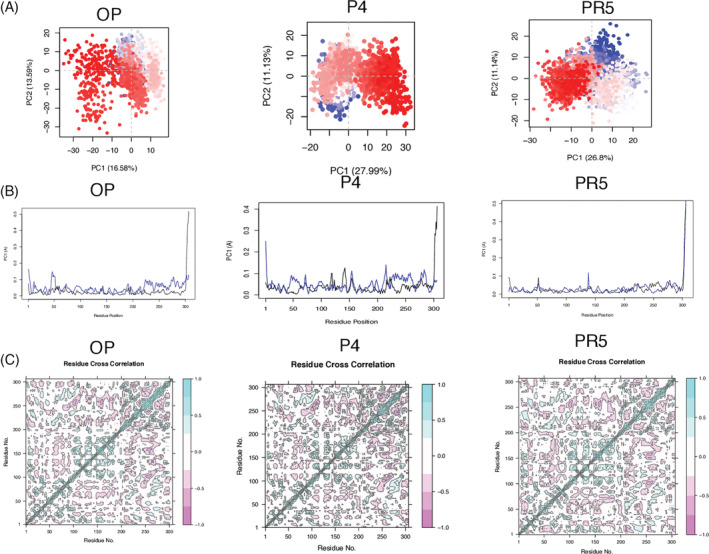
Principal component analysis (PCA) analysis of the phytochemicals (P4) and the repurposed drug (PR5) in comparison to the apo‐protein (OP). (A) The projection of PC1 on PC2. The continuous colour spectrum from blue to white to red represents simulation time. The initial timescale represented by blue, intermediate by white and final by red. (B) The RMSF of residue contribution to PCA (black: PC1, blue: PC2). (C) Dynamic cross correlation matrix (DCCM) plots for (P4, PR5 and OP). The positive value represents the positively correlated motions (cyan), while negative values represent the anti‐correlated motions (pink). For abbreviations please refer main text and Table 1 [Colour figure can be viewed at wileyonlinelibrary.com]
Furthermore, to understand the effect of phytochemicals binding on the internal dynamics of NSP5, the dynamic cross‐correlation matrix was calculated by using the coordinates of Cα atoms from the trajectories. Domain I (residues 8‐101), domain II (residues 102–184) and domain III (residues 201‐306) have been shown in Figure 2C. The DCCM plots of P4, PR5 and the apo‐protein shows overall slight increase in anti‐correlated motions on ligand binding. The other phytochemicals (P1‐P3) showed much higher increase in anti‐correlated motions amongst the three domains of NSP5 (Figure S3). In comparison to the apo‐protein correlated motions increase in the domain II (residues 102‐184) which is also the binding pocket for the ligands for the phytochemicals and the repurposed drugs. It can be said that the binding of P4 and PR5 with NSP5 creates a stable environment compared to the other phytochemicals.
3.2. Spike protein molecular dynamics simulation analysis
The four best docked complexes (S1–S4) with phytochemicals following drug likeness properties were subjected to 100 ns MD simulations along with the best docked repurposed drug (SR5). The apo‐protein of spike without ACE‐2 receptor was also subjected to 100 ns MD simulations. Figure 3A illustrates the region of spike protein that interacts with the human ACE‐2 receptor has been highlighted in red in the cartoon representation of the spike glycoprotein crystal structure. Residues important for priming with ACE2 were observed from the crystal structure to be Lys417, Gly446, Tyr449, Ala475, Asn487, Gln493, Tyr495, Gly496, Phe497, Gln498, Thr500, Asn501 and Tyr505. Figure 3B illustrates stable RMSD over 100 ns for all the trajectories (~0.2–0.4 nm). The average RMSD values for the phytochemical‐protein complexes were S1: 0.13 nm, S2: 0.15 nm, S3: 0.12 nm, S4: 0.13 nm, the repurposed drug‐ SR5: 0.15 nm and the apo‐protein without any ligand was 0.08 nm. The average Cα RMSD was ~0.15 nm. The average RMSD of the ligands were S1:0.09 nm, S2: 0.13 nm, S3: 0.05 nm, S4: 0.05 nm and SR5: 0.17 nm. This shows a stable binding of phytochemicals and the repurposed drugs making them good inhibitor candidates. ΔRMSF plot (difference between RMSF of ligands and apo‐protein) revealed lesser fluctuations in the receptor binding domain for S4 with respect to the apo‐protein, compared to the other phytochemicals (S1‐S3) and SR5 (Figure 3C). Radius of gyration plots for the trajectories revealed similar structural compactness of all the ligands with the apo‐protein (average ~ 1.8 nm) in comparison to the other phytochemicals (Figure 3D). Interestingly the compactness of SR5 decreases after 50 ns.
FIGURE 3.
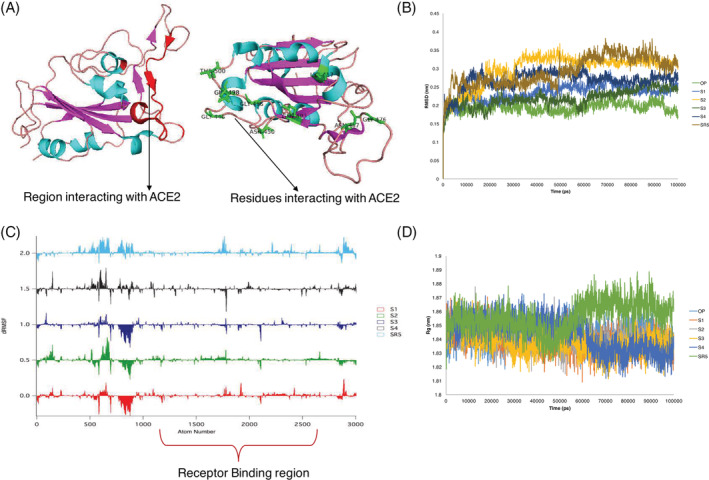
Molecular dynamics analysis of 100 ns trajectories of phytochemicals (S1‐S4) and repurposed drug (SR5) with Spike protein. (A) A cartoon representation of the receptor‐binding domain (RBD) of the spike protein (PDB ID: 6LZG). The receptor binding region is marked in red and the residues important for priming with ACE2 have been represented as sticks and labelled. (B) RMSD of the 100 ns trajectories. (C) ΔRMSF stacked plot of phytochemicals (S1‐S4) and repurposed drug (SR5) with respect to spike protein (PDB ID: 6lzg: chain B). The atom numbers are with respect to the coordinates of spike protein (6lzg: chain B). The Receptor Binding region has been highlighted in maroon parenthesis. (D) Radius of gyration over 100 ns trajectories. OP, apo‐protein. For abbreviations please refer the main text and Table 1 [Colour figure can be viewed at wileyonlinelibrary.com]
Analysis of occupancy of hydrogen bonds (distance ≤4 Å) over 100 ns of the trajectories revealed that the hydrogen bond occupancy was maximum for S4 followed by SR5 and the other phytochemicals (S1‐S3). More number of residues were involved with hydrogen bonds with S4 in comparison to S1‐S3 and SR5. Gln493, Ser494, Phe497, Tyr495, Tyr505, Asn501 of the RBD domain of spike protein were with >10% hydrogen bond occupancy with S4. Hydrogen bonding with Tyr495 was maintained for ~53% of the trajectory, following by Tyr505 with ~40% occupancy (Table S2). It can be said that the hydrogen bonding interactions of the RBD domain of spike protein was most stable with S4 compared to the other phytochemicals and the repurposed drug.
PCA on the Cα atom was performed for the apo‐protein and the ligand complex trajectories. The first five Eigen vectors captured around ~60% of the motions (Figure S4). Much larger conformational space was sampled by the phytochemicals (Figures 4A and S4). The residue fluctuation (RMSF) contribution to the PCA showed there was increased fluctuations in the loop region involving residues 280–400 of the spike protein on binding of the ligands. This can be attributed to the loops in proteins which undergo higher fluctuations compared to helices or strands. Increase in fluctuations represent conformational changes in the spike protein. Similar flexibility in PC1 and PC2 was computed for S4 in comparison to the apo‐protein. However, SR5 showed increase in flexibility in the RBD region in comparison to the apo‐protein (Figure 4B, Figure S5). S4 also showed reduced flexibility at the residues important for interacting with ACE‐2 receptor (6lzg: residue number 470‐490) compared to the other phytochemicals (S1‐S3) and SR5 showed comparatively more fluctuations.
FIGURE 4.
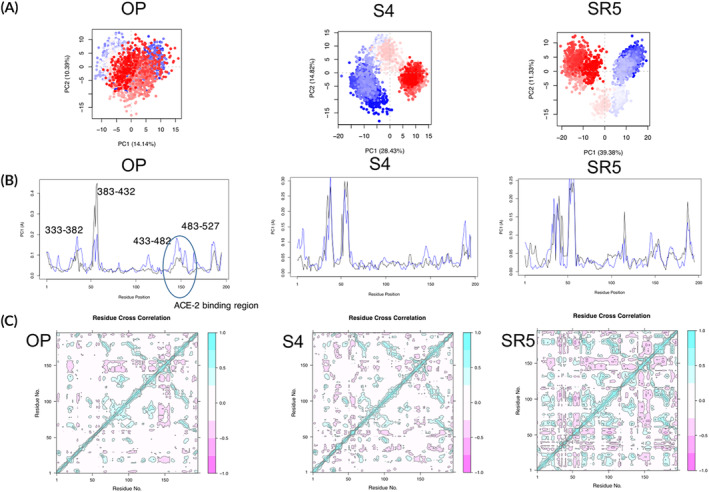
PCA analysis of the phytochemicals (S4) and the repurposed drug (SR5) in comparison to the apo‐protein (OP). (A) The projection of PC1 on PC2. The continuous colour spectrum from blue to white to red represents simulation time. The initial timescale represented by blue, intermediate by white and final by red. (B) The RMSF of residue contribution to PCA. (black: PC1, blue: PC2) (C) DCCM plots for (S4, SR5 and OP). The positive value represents the positively correlated motions (cyan), while negative values represent the anti‐correlated motions (pink). RBD domain is marked in blue circle. For abbreviations please refer main text and Table 1 [Colour figure can be viewed at wileyonlinelibrary.com]
Further, dynamic cross‐correlation matrix was calculated by using the coordinates of Cα atoms from the trajectories. The DCCM plots of S4 and the apo‐protein shows overall slight increase in anti‐correlated motions on ligand binding. The other phytochemicals (S1‐S3) and repurposed drug (SR5) showed much higher increase in anti‐correlated motions in the RBD domain of the spike protein (Figure S6). Maximum increase in anti‐correlated motions was observed for the repurposed drug. Further, in comparison to the apo‐protein correlated motions increase in the RBD domain on binding of phytochemicals and the repurposed drug. Thus it can be said that the binding of S4 with spike protein creates a stable environment compared to the other phytochemicals and the repurposed drug.
3.3. Relative binding free energy calculations for the main protease and spike protein with the phytochemicals and the repurposed drugs
The relative binding free energy were calculated for each complex of protease and spike protein using MM‐PBSA. The SEs were calculated by 500 steps of bootstrap analysis. It was observed that the relative binding free energies obtained for the complexes were in agreement with the RMSD, RMSF, PCA and DCCM calculations. Lowest relative binding energy was obtained for P4 (−141.96 KJ/mol) for the protease of SARS‐CoV‐2, lower than the repurposed drug with −108.92 KJ/mol relative binding energy. For the spike glycoprotein the lowest relative binding free energy was obtained for S4 (−87 KJ/mol) (Table 2). Interestingly the repurposed drug had a lower relative binding energy of −96.52 KJ/mol (Table 2).
TABLE 2.
Relative binding energies calculated using MM/PBSA
| Ligand | Ab | van der Waal energy (KJ/mol) | Electrostattic energy (KJ/mol) | Polar solvation energy (KJ/mol) | SASA energy (KJ/mol) | Total relative binding energy (KJ/mol) |
|---|---|---|---|---|---|---|
| Protease | ||||||
| 27‐Deoxy‐14‐hydroxywithaferin A | P1 | −92.60 (9.657) | −1.69 (3.87) | 45.88 (21.97) | −10.74 (1.97) | −59.15 (16.29) |
| 17‐Hydroxywithaferin | P2 | −147.84 (24.54) | −21.854 (12.71) | 87.99 (28.35) | −15.17 (1.86) | −96.88 (25.92) |
| Nimolicinol | P3 | −197.07 (15.26) | −13.41 (6.45) | 98.34 (11.85) | −18.93 (1.16) | −131.71 (16.37) |
| Withanolide R | P4 | −187.11 (20.48) | −14.11 (5.74) | 76.18 (8.48) | −16.91 (1.51) | −141.96 (18.39) |
| Methylprednisolone | PR4 | −158.68 (12.32) | −3.09 (1.87) | 67.80 (10.85) | −15.03 (1.08) | −108.92 (13.48) |
| Spike | ||||||
| 27‐Deoxywithaferin A | S1 | −102.52 (21.28) | −11.98 (12.31) | 57.57 (16.24) | −11.41 (2.05) | −68.34 (14.88) |
| 27‐Hydroxywithanone | S2 | −76.30 (15.09) | −77.02 (18.53) | 98.11 (48.56) | −8.78 (1.69) | −63.99 (23.93) |
| 12‐Deoxywithastramonolide | S3 | −104.85 (16.12) | −23.78 (9.81) | 72.21 (15.48) | −11.92 (1.48) | −68.34 (11.23) |
| 2,3‐Dihydrowithaferin A | S4 | −135.43 (26.270) | −25.90 (11.53) | 87.186 (22.23) | −13.45 (1.57) | −87.60 (22.68) |
| Baricitinib | SR5 | −121.08 (12.47) | −4.68 (1.49) | 40.91 (11.10) | −11.69 (1.08) | −96.52 (14.71) |
Note: Ab: Abbreviations. The 500 step bootstrap ± Standard Error have been given in parenthesis.
Further the residue contribution towards the relative free binding energy was calculated. In the case of the main protease‐P4 and PR5 complexes, the connecting loop between domain I and domain II‐ Gln189 contributed the lowest binding energy (−8.1073 KJ/mol) followed by domain II residues Met165 (−6.6762 KJ/mol), Leu167 (−2.94 KJ/mol) and Pro168 (−6.6042 KJ/mol). Domain III residues Asp187 (−2.58 KJ/mol) and Ala191 (−3.37 KJ/mol) contributed to the relative binding free energy. These residues were also observed to have lower fluctuations as analysed by the RMSF, PCA and DCCM calculations.
Further, it was observed that in the spike glycoprotein residues involved in priming to the human ACE‐2 receptor were computed to have the lowest contribution to the relative binding free energy. In the spike glycoprotein‐S4 complex Leu455 (−4.74 KJ/mol), Tyr505 (−4.13 KJ/mol) and Gln493 (−3.6 KJ/mol), were with the lowest binding free energy contributions. In the spike‐glycoprotein‐SR5 complex Leu455 (−4.96 KJ/mol), Phe456 (−11.23 KJ/mol), Tyr473 (−6.19 KJ/mol), Ala475 (−2.83 KJ/mol), Tyr489 (−8.7 KJ/mol) and Pro491(−4.30 KJ/mol) were with the lowest binding free energy contributions.
3.4. Comparative pathway enrichment analysis between COVID‐19 and the phytochemicals protein targets
The pathway network based approach is a systems level understanding of the effect on the interconnected biological processes. The top 15 human protein targets for each phytochemical were retrieved from SwissTarget Database. The SARS‐CoV‐2 human protein targets were obtained by literature survey (Chakrabarty et al., 2020; Zhou et al., 2020). Enrichr analysis of KEGG pathway enrichment for COVID‐19 and the phytochemicals revealed that AGE‐RAGE signalling pathway in diabetic complications and Human cytomegalovirus infection were common to both (Table S4). Interestingly Influenza A and pathways in cancer were the most enriched in case of the COVID‐19 and the phytochemicals respectively (Figure 5A,B). VirusMint database enrichment resulted in the proteins from both COVID‐19 and phytochemicals to be most significantly associated with Human Immunodeficiency virus I‐human protein interactions (Figure 5C,D). Further, comparative analysis of the top 100 GO terms of biological pathway enriched by the phytochemical pathway and the COVID‐19 infection pathway revealed that protein kinase activity and binding, protein homo‐dimerization activity, growth factor receptor binding, protein phosphatase binding, disordered domain specific binding, ATPase binding, ubiquitin protein ligase binding, MAP kinase activity, R‐SMAD binding, mitogen‐activated protein kinase binding (MAPK), transition metal ion binding, telomeric DNA binding, androgen receptor binding and RNA polymerase II activating transcription factor binding processes were enriched in both the cases (Figure 5, Tables S5 and S6).
FIGURE 5.
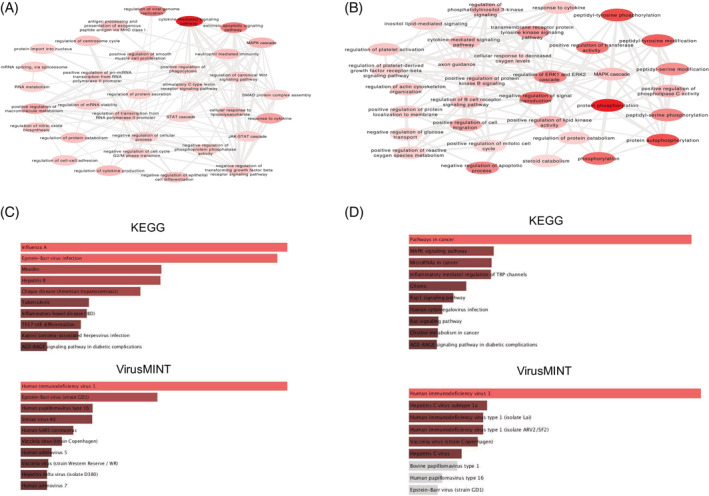
Biological processes enriched by the proteins associated with the (A) COVID‐19 and the (B) phytochemicals. The processes are coloured based on the dispensability score obtained from REVIGO where higher scores are represented by darker shades of red (Supek et al., 2011). (C) Top 10 KEGG pathway and VirusMint enrichment for COVID‐19. (D) Top 10 Kyoto Encyclopedia of Genes and Genomes (KEGG) and VirusMint enrichment for phytochemicals. The bars in panel C and D represents the p‐values computed using the Fisher exact test. The longer bars and lighter coloured bars mean that the term is more significant [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSIONS
The whole gamut of computational methods is instrumental in designing highly specific inhibitors against important viral targets‐ offering scientifically sound basis for anti‐viral drug discovery. Identification of potential inhibitors by rational screening is the first step towards the anti‐viral inhibitor discovery process. The second step involves understanding the biophysical basis of inhibitor binding mechanism through molecular dynamics simulations. Currently the viral infection, COVID‐19 is highly contagious with no effective therapy to combat the infection (Guan et al., 2020). The high infectivity is due to its spike protein of SARS‐CoV‐2 that primes with human ACE2 receptor more tightly, by a factor of 10 to 15, than SARS‐CoV spike protein (Wang et al., 2020). Another important therapeutic target is the main protease of SARS‐CoV‐2 as it is involved in maintaining the viral life cycle, being an essential enzyme for viral replication (Huynh, Wang, & Luan, 2020).
In this context previous research reported natural compounds like theaflavin, quercetin, withanone and caffeic acid phenethyl ester to be effective inhibitors of SARS‐CoV‐2 (Kumar et al., 2020; Lung et al., 2020; Zhang et al., 2020). This corroborated with our previous work were 8 phytochemicals from Withania somnifera and Azadirachta indica were found by virtual screening of 1916 phytochemicals from 55 Indian medicinal plants to dock with the main protease (NSP5) and the spike protein of SARS‐CoV‐2. Therefore, in this work these phytochemicals were subjected to 100 ns all atom molecular dynamics simulations to analyse the biochemical basis of the inhibitions. The phytochemicals docking to the SARS‐CoV‐2 targets were obtained by rational screening from our previous work (Parida et al., 2020). Two repurposed drugs‐ Methylprednisolone and Baricitinib complexes with NSP5 and spike protein respectively, with lowest docking scores, were also subjected to MD simulations. The latter was performed to compare the binding efficacy of the phytochemicals with the repurposed drugs. The parameters computed and analysed after 100 ns dynamics trajectory were RMSD, RMSF, radius of gyration (Rg), hydrogen bonding, PCA, DCCM and relative binding energies computed by MM‐PBSA. The context of performing RMSD and RMSF is because such stabilities are essential to obtain good binding affinities (J. Chen & Shen, 2009; Doniach & Eastman, 1999; Dubey, Tiwari, & Ojha, 2013). Enhancement in protein compactness on ligand binding can be elucidated by low values of radius of gyration. Again, large number of hydrogen bonds between ligand and important residues related to activity of protein signifies a strong binding affinity (Khan et al., 2020). PCA is an essential dynamics (ED) approach‐ performed to reflect overall difference in motions in the protein on binding ligands in comparison to the apo‐protein (Islam, Khan, Parves, Hossain, & Halim, 2019; Martens & Naes, 1992). Like PCA, the protein residue atomic motions correlation and non‐correlation can be computed with the help of DCCM (Hünenberger, Mark, & Van Gunsteren, 1995; McCammon, 1984).
The results showed that all the trajectories were stable with RMSD ~0.2–0.45 nm. Withanolide R and 2,3‐Dihydrowithaferin A were with the lowest relative binding energy with the main protease (−141.96 KJ/mol) and the spike protein (−87 KJ/mol) respectively. The relative binding energy of Methylprednisolone and Baricitinib were computed to be −108.92 and −96.52 KJ/mol respectively. Interestingly Baricitinib with lower binding energy than the phytochemicals. Barcitinib is an anti‐Janus kinase inhibitor and has been suggested as a potential treatment for COVID‐19 disease with high affinity for AP2‐associated protein kinase 1 (AAK1) (Richardson et al., 2020). The connecting loop between domain I and domain II‐ Gln189 in the case of protease contributed the lowest binding energy (−8.1073 KJ/mol). Similarly, for the spike protein Leu455 contributed the lowest binding energy (−4.74 KJ/mol). Thus Gln189 and Leu455 can be said to be an important inhibitor binding site in NSP5 and spike protein respectively. These residues were also found to be potent drug binding sites by previous research (Gyebi, Ogunro, Adegunloye, Ogunyemi, & Afolabi, 2020; Qamar ul et al., 2020; Shang et al., 2020). Leu455 was also reported to contribute favourably to ACE2 recognition (Shang et al., 2020). Further we identified that hydrogen bonds between Withanolide R and NSP5 were stable with residues involved in the activity of NSP5 for example‐ His41 and Gln189. Similarly, Tyr495 and Tyr505 of the ACE2 binding region were involved with hydrogen bonding with the phytochemicals in the case of spike protein. These residues were also reported to be critical in binding to the ACE2 receptor (Chikhale et al., 2020). Relative RMSF, Radius of gyration plots, PCA and DCCM analysis also showed that Withanolide R and Methylprednisolone creates a stable environment of binding compared to the other phytochemicals with NSP5. MD analysis results for spike protein showed lesser fluctuations in the ACE2 binding domain with 2,3‐Dihydrowithaferin with respect to the apo‐protein and in comparison to the other phytochemicals and Baricitinib. Hydrogen bonding interactions of the ACE2 binding domain of spike protein was also computed to be most stable with 2,3‐Dihydrowithaferin A compared to the other phytochemicals and Baricitinib. Further, PCA on the MD simulation disclosed that complexes of Withanolide R and 2,3‐Dihydrowithaferin A with the protease and the spike protein respectively have structural similarity with their apo‐form. Additional analysis of residue contribution towards PCA showed that flexibility of the active site domains of protease and the AC2 binding domain of spike protein were comparable to that of the apo‐protein on binding of these phytochemicals and the repurposed drugs. However, the flexibility in the same regions increased with the binding of other phytochemicals. Further, DCCM analysis showed that correlated motions increased in the active site region of protease and the ACE2‐binding region of the spike protein respectively. There was slight increase in overall anti‐correlated motions on binding with Withanolide R and 2,3‐Dihydrowithaferin A. The anti‐correlated motions were observed to be more with the other phytochemicals and the repurposed drugs with increase in residue fluctuations indicating inhibitor instability. Overall the phytochemicals with the lowest binding energy formed a more stable environment with the targets.
It can also be mentioned here that the therapeutic benefit of the phytochemicals is not limited to targeting spike and the main protease of SARS‐CoV‐2. In summary, these phytochemicals exhibit a remarkable multipotency with the ability to modulate various human biological processes and biochemical pathways. This was observed by the study of pathway enrichment by the phytochemicals and the SARS‐CoV‐2. Amongst many different pathways that the phytochemicals can target, enrichment of common biological pathways in COVID‐19 and by the phytochemical human target proteins, is a strong indication about their therapeutic role in treating COVID‐19 infections. Interestingly by pathway enrichment analysis it be said that these phytochemicals can be tested experimentally for elimination of the COVID‐19 infection as an alternative therapy in high risk patients with heart disease, cancer and diabetes as the pathways enriched in such conditions were also enriched in COVID‐19 and by the phytochemicals. For example, it was observed that pathways in cancer were enriched by the phytochemicals. Further, AGE‐RAGE signalling pathway in diabetic complications and human cytomegalovirus infection (CMV) pathways were enriched in both the cases. Increased levels of advanced glycation end products (AGEs) especially carboxymethyllysine (CML), have been observed to be associated with atherosclerosis, and coronary artery disease and heart failure (Hegab, Gibbons, Neyses, & Mamas, 2012). CMV is also a lung infection and HRCT findings and patterns are similar to COVID‐19 infections (Cascella, Rajnik, Cuomo, Dulebohn, & Di Napoli, 2020). The MAPK pathway enrichment can be justified because it is involved in release of pro‐inflammatory cytokines such as IL‐6 and has been implicated in acute lung injury and myocardial dysfunction (Grimes & Grimes, 2020). It was reported that activation of p38 MAPK, is involved in replication of HCoV‐229E and induction of overwhelming inflammation by SARS‐CoV‐2 (Grimes & Grimes, 2020; Kono et al., 2008). Again clinical feature associated with COVID‐19 like the SARS‐CoV infection is pulmonary fibrosis and the associated lung failure. Phytochemicals targeting the R‐SMAD binding pathway which has been reported to be involved in blocking apoptosis of SARS‐CoV‐infected host cells may result in decrease in fibrosis (Zhao, Nicholls, & Chen, 2008). Similarly, the proteins targeted by the phytochemicals and SARS‐CoV‐2 were observed to be enriched in transition metal ion binding and metal ion binding have been reported to be an integral part of HCoV‐229E viral protein (Chaturvedi & Shrivastava, 2005). The pathways involved in DNA replication and protein transcription were also enriched in SARS‐CoV‐2 infection and by the phytochemicals. Further, pathways related to androgen receptor binding were enriched in COVID‐19 and the phytochemicals. It was reported that cell entry of SARS‐CoV‐2 depends on binding of the viral spike (S) proteins to ACE2 and on S protein priming by TMPRSS2. Inhibition of TMPRSS2 (an androgen regulated gene) which is alleviated in prostate cancer patients may work to block or decrease the severity of SARS‐CoV‐2 infections (Montopoli et al., 2020). Again the ubiquitin‐proteasome system is a regulator of intracellular sorting and degradation of proteins. Inhibition of the pathway was reported to severely impair coronavirus infection. The phytochemicals were observed to enrich this pathway similar to the SARS‐CoV‐2. Conclusively it can be said that the phytochemicals can regulate COVID‐19 infection by also targeting these pathways along with the spike and the main protease of SARS‐CoV‐2.
5. CONCLUSIONS
In conclusion, this study reports the biophysical basis of interaction between eight phytochemicals and two repurposed drugs with spike protein and the main protease of SARS‐CoV‐2 by performing analysis of 100 ns molecular dynamics simulation trajectories. Overall, the simulation run confirms the stability and rigidity of the interactions of Withanolide R and 2,3‐Dihydrowithaferin A from Withania somnifera to be the most potent phytochemical inhibitor for the main protease and the spike protein respectively. The energy calculations show the binding efficiency of these phytochemicals. Further, the important residues interacting with the phytochemicals were elucidated. Conclusively through this work experimental validation is being suggested to test the efficacy of these phytochemicals for the prevention and treatment of COVID‐19.
CONFLICT OF INTEREST
No potential conflict of interest was reported by the authors.
AUTHOR CONTRIBUTIONS
All authors conceived, conducted and analysed the results of experiments equally.
Supporting information
Figure S1. Principal component analysis (PCA) analysis of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S2. Residue contribution (RMSF) to PCA of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S3. Dynamic cross correlation matrix (DCCM) plots of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S4. PCA analysis of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S5. Residue contribution (RMSF) to PCA of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S6. DCCM plots of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Table S1. Hydrogen bond occupancy with NSP5‐ P1, NSP5‐ P2, NSP5‐ P3, NSP5‐ P4, NSP5‐ PR5.
Table S2. Hydrogen bond occupancy with spike protein – S1, S2, S3, S4, SR5.
Table S3. The protein targets (HUGO symbol) for Phytochemicals and SARS‐CoV‐2 used to perform pathway enrichment analysis.
Table S4. KEGG pathway analysis of protease and spike protein.
Table S5. COVID‐19 GO enrichment obtained from Enrichr.
Table S6. Phytochemicals GO enrichment obtained from Enrichr.
ACKNOWLEDGEMENTS
This work used resources services, and support provided via the COVID‐19 HPC Consortium (https://covid19-hpc-consortium.org/), which is a unique private‐public effort to bring together government, industry, and academic leaders who are volunteering free compute time and resources in support of COVID‐19 research. We would also like to acknowledge Noor Enzymes Private Limited for providing us with the human resources needed to successfully complete this work. We would also like to specially thank Linda Hedges (HPC Solutions architect at Amazon Web Services), Tara Madhyastha (Principal research scientist at Amazon Web Services), Nathan Albrighton (Founder & CEO at RONIN) and Don Hancock (Co‐Founder at RONIN) who provided us with timely guidance and assistance to complete this work.
Parida PK, Paul D, Chakravorty D. The natural way forward: Molecular dynamics simulation analysis of phytochemicals from Indian medicinal plants as potential inhibitors of SARS‐CoV‐2 targets. Phytotherapy Research. 2020;34:3420–3433. 10.1002/ptr.6868
All the authors have contributed equally to the paper.
REFERENCES
- Adem, S. , Eyupoglu, V. , Sarfraz, I. , Rasul, A. , & Ali, M. (2020). Identification of potent COVID–19 main protease (Mpro) inhibitors from natural polyphenols: An in silico strategy unveils a hope against CORONA. Preprints. 10.20944/preprints202003.0333.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Khafaji, K. , & Taskin Tok, T. (2020). Amygdalin as multi‐target anticancer drug against targets of cell division cycle: Double docking and molecular dynamics simulation. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1742792. [published online ahead of print, 2020 Mar 24]. [DOI] [PubMed] [Google Scholar]
- Amadei, A. , Linssen, A. B. , & Berendsen, H. J. (1993). Essential dynamics of proteins. Proteins: Structure, Function, and Bioinformatics, 17(4), 412–425. [DOI] [PubMed] [Google Scholar]
- Anand, K. , Ziebuhr, J. , Wadhwani, P. , Mesters, J. R. , & Hilgenfeld, R. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti‐SARS drugs. Science, 300(5626), 1763–1767. [DOI] [PubMed] [Google Scholar]
- Baker, N. A. , Sept, D. , Joseph, S. , Holst, M. J. , & McCammon, J. A. (2001). Electrostatics of nanosystems: Application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America, 98(18), 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella, M. , Rajnik, M. , Cuomo, A. , Dulebohn, S. C. , & Di Napoli, R. (2020). Features, evaluation and treatment coronavirus (COVID‐19), Treasure Island (FL): StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK554776/ [PubMed] [Google Scholar]
- Chakrabarty, B. , Das, D. , Bulusu, G. , & Roy, A. (2020). Network‐based analysis of fatal comorbidities of COVID‐19 and potential therapeutics. ChemRxiv. 10.26434/chemrxiv.12136470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel, V. , Raj, S. , Rathi, B. , & Kumar, D. (2020). In silico identification of potent COVID–19 main protease inhibitors from FDA approved antiviral compounds and active phytochemicals through molecular docking: A drug repurposing approach. Preprints. 10.20944/preprints202003.0349.v1 [DOI] [Google Scholar]
- Chatr‐Aryamontri, A. , Ceol, A. , Peluso, D. , Nardozza, A. , Panni, S. , Sacco, F. , … Cusick, M. E. (2009). VirusMINT: A viral protein interaction database. Nucleic Acids Research, 37(suppl_1), D669–D673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi, U. C. , & Shrivastava, R. (2005). Interaction of viral proteins with metal ions: Role in maintaining the structure and functions of viruses. FEMS Immunology & Medical Microbiology, 43(2), 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , & Shen, B. (2009). Computational analysis of amino acid mutation: A proteome wide perspective. Current Proteomics, 6(4), 228–234. [Google Scholar]
- Chen, Y. , Guo, Y. , Pan, Y. , & Zhao, Z. J. (2020). Structure analysis of the receptor binding of 2019‐nCoV. Biochemical and Biophysical Research Communications, 525(1), 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale, R. V. , Sinha, S. K. , Patil, R. B. , Prasad, S. K. , Shakya, A. , Gurav, N. , … Gurav, S. S. (2020). In‐silico investigation of phytochemicals from Asparagus racemosus as plausible antiviral agent in COVID‐19. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1784289 [DOI] [PubMed] [Google Scholar]
- Da Silva, A. W. S. , & Vranken, W. F. (2012). ACPYPE ‐ Antechamber PYthon parser interfacE. BMC Research Notes, 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, W. , Bi, J. , Li, F. , Wang, S. , Huang, X. , Meng, X. , … Su, W. (2019). Antiviral efficacy of flavonoids against enterovirus 71 infection in vitro and in newborn mice. Viruses, 11(7), 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T. , York, D. , & Pedersen, L. (1993). Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092. [Google Scholar]
- Doniach, S. , & Eastman, P. (1999). Protein dynamics simulations from nanoseconds to microseconds. Current Opinion in Structural Biology, 9(2), 157–163. [DOI] [PubMed] [Google Scholar]
- Dubey, K. D. , Tiwari, R. K. , & Ojha, R. P. (2013). Recent advances in protein‐ligand interactions: Molecular dynamics simulations and binding free energy. Current Computer‐Aided Drug Design, 9(4), 518–531. [DOI] [PubMed] [Google Scholar]
- Efferth, T. , & Koch, E. (2011). Complex interactions between phytochemicals. The multi‐target therapeutic concept of phytotherapy. Current Drug Targets, 12(1), 122–132. [DOI] [PubMed] [Google Scholar]
- Gentile, D. , Patamia, V. , Scala, A. , Sciortino, M. T. , Piperno, A. , & Rescifina, A. (2020). Putative inhibitors of SARS‐CoV‐2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Marine Drugs, 18(4), 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gfeller, D. , Grosdidier, A. , Wirth, M. , Daina, A. , Michielin, O. , & Zoete, V. (2014). SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Research, 42(W1), W32–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Paz, L. A. , Lossada, C. A. , Moncayo, L. S. , Romero, F. , Paz, J. L. , Vera‐Villalobos, J. , … Alvarado, Y. J. (2020). Theoretical molecular docking study of the structural disruption of the viral 3CL‐protease of COVID19 induced by binding of capsaicin, piperine and curcumin part 1: A comparative study with chloroquine and hydrochloroquine two antimalaric drugs. Research Square. 10.21203/rs.3.rs-21206/v1 [DOI] [Google Scholar]
- Grant, B. J. , Rodrigues, A. P. , ElSawy, K. M. , McCammon, J. A. , & Caves, L. S. (2006). Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics, 22(21), 2695–2696. [DOI] [PubMed] [Google Scholar]
- Grimes, J. M. , & Grimes, K. V. (2020). p38 MAPK inhibition: A promising therapeutic approach for COVID‐19. Journal of Molecular and Cellular Cardiology, 144, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, W. J. , Ni, Z. Y. , Hu, Y. , Liang, W. H. , Ou, C. Q. , He, J. X. , … Du, B. (2020). Clinical characteristics of coronavirus disease 2019 in China. New England Journal of Medicine, 382(18), 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi, G. A. , Ogunro, O. B. , Adegunloye, A. P. , Ogunyemi, O. M. , & Afolabi, S. O. (2020). Potential inhibitors of coronavirus 3‐chymotrypsin‐like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegab, Z. , Gibbons, S. , Neyses, L. , & Mamas, M. A. (2012). Role of advanced glycation end products in cardiovascular disease. World Journal of Cardiology, 4(4), 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hünenberger, P. H. , Mark, A. E. , & Van Gunsteren, W. F. (1995). Fluctuation and cross‐correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. Journal of Molecular Biology, 252(4), 492–503. [DOI] [PubMed] [Google Scholar]
- Huynh, T. , Wang, H. , & Luan, B. (2020). In silico exploration of molecular mechanism of clinically oriented drugs for possibly inhibiting SARS‐CoV‐2's main protease. The Journal of Physical Chemistry Letters, 11(11), 4413–4420. [DOI] [PubMed] [Google Scholar]
- Islam, M. J. , Khan, A. M. , Parves, M. R. , Hossain, M. N. , & Halim, M. A. (2019). Prediction of deleterious non‐synonymous SNPs of human STK11 gene by combining algorithms, molecular docking, and molecular dynamics simulation. Scientific Reports, 9, 16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, R. S. , Jagdale, S. S. , Bansode, S. B. , Shankar, S. S. , Tellis, M. B. , Pandya, V. K. , … Kulkarni, M. J. (2020). Discovery of potential multi‐target‐directed ligands by targeting host‐specific SARS‐CoV‐2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu, O. , Saeed, M. , Johannes Greten, H. , & Efferth, T. (2020). Identification of novel compounds against three targets of SARS CoV‐2 coronavirus by combined virtual screening and supervised machine learning. Bulletin of World Health Organization. 10.2471/BLT.20.255943. [published online ahead of print, 2020 Mar 21]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaerunnisa, S. , Kurniawan, H. , Awaluddin, R. , Suhartati, S. , & Soetjipto, S. (2020). Potential inhibitor of COVID‐19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints. 10.20944/preprints202003.0226.v1 [DOI] [Google Scholar]
- Khan, S. A. , Zia, K. , Ashraf, S. , Uddin, R. , & Ui‐Haq, Z. (2020). Identification of chymotrypsin‐like protease inhibitors of SARS‐CoV‐2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Kono, M. , Tatsumi, K. , Imai, A. M. , Saito, K. , Kuriyama, T. , & Shirasawa, H. (2008). Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antiviral Research, 77(2), 150–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov, M. V. , Jones, M. R. , Rouillard, A. D. , Fernandez, N. F. , Duan, Q. , Wang, Z. , … McDermott, M. G. (2016). Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research, 44(W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, V. , Dhanjal, J. K. , Kaul, S. C. , Wadhwa, R. , & Sundar, D. (2020). Withanone and caffeic acid phenethyl ester are predicted to interact with main protease (Mpro) of SARS‐CoV‐2 and inhibit its activity. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1772108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, R. , Kumar, R. , Open Source Drug Discovery Consortium , & Lynn, A. (2014). g_mmpbsa—A GROMACS tool for high‐throughput MM‐PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. [DOI] [PubMed] [Google Scholar]
- Li, F. , Li, W. , Farzan, M. , & Harrison, S. C. (2005). Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science, 309(5742), 1864–1868. [DOI] [PubMed] [Google Scholar]
- Lindahl, A. , Hess, M. J. , & van der Spoel D.. (2020). GROMACS 2020.2 source code. Zenodo. https://zenodo.org/record/3773801#.X1MXH3kzZPY
- Lung, J. , Lin, Y. S. , Yang, Y. H. , Chou, Y. L. , Shu, L. H. , Cheng, Y. C. , … Wu, C. Y. (2020). The potential chemical structure of anti‐SARS‐CoV‐2 RNA‐dependent RNA polymerase. Journal of Medical Virology, 92, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens, H. , & Naes, T. (1992). Multivariate calibration, USA: John Wiley & Sons, Inc. [Google Scholar]
- McCammon, J. A. (1984). Protein dynamics. Reports on Progress in Physics, 47(1), 1–46. [Google Scholar]
- McKee, D. L. , Sternberg, A. , Stange, U. , Laufer, S. , & Naujokat, C. (2020). Candidate drugs against SARS‐CoV‐2 and COVID‐19. Pharmacological Research, 157, 104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam, E. , Teoh, B. T. , Sam, S. S. , Lani, R. , Hassandarvish, P. , Chik, Z. , … Zandi, K. (2014). Baicalin, a metabolite of baicalein with antiviral activity against dengue virus. Scientific Reports, 4, 5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montopoli, M. , Zumerle, S. , Vettor, R. , Rugge, M. , Zorzi, M. , Catapano, C. V. , … Prayer‐Galetti, T. (2020). Androgen‐deprivation therapies for prostate cancer and risk of infection by SARS‐CoV‐2: A population‐based study (N = 4532). Annals of Oncology, 31, 1040–1045. 10.1016/j.annonc.2020.04.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida, P. K. , Paul, D. , & Chakravorty, D. (2020). Nature to nurture‐identifying phytochemicals from Indian medicinal plants as prophylactic medicine by rational screening to be potent against multiple drug targets of SARS‐CoV‐2. ChemRxiv. 10.26434/chemrxiv.12355937 [DOI] [Google Scholar]
- Qamar ul, M. T. , Alqahtani, S. M. , Alamri, M. A. , & Chen, L. L. (2020). Structural basis of SARS‐CoV‐2 3CLpro and anti‐COVID‐19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis, 10, 313–319. 10.1016/j.jpha.2020.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, P. , Griffin, I. , Tucker, C. , Smith, D. , Oechsle, O. , Phelan, A. , & Stebbing, J. (2020). Baricitinib as potential treatment for 2019‐nCoV acute respiratory disease. Lancet, 395(10223), e30–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team . (2020). RStudio: Integrated development for R. Boston, MA: RStudio, PBC. Retrieved from https://support.rstudio.com/hc/en-us/articles/206212048-Citing-RStudio [Google Scholar]
- Shang, J. , Wan, Y. , Liu, C. , Yount, B. , Gully, K. , Yang, Y. , … Li, F. (2020). Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Pathogens, 16(3), e1008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , … Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. D. , & Kaur, I. (2020). Eucalyptol (1,8 cineole) from eucalyptus essential oil a potential inhibitor of COVID 19 corona virus infection by molecular docking studies. Preprints. 10.20944/preprints202003.0455.v1 [DOI] [Google Scholar]
- Sun, N. , Wong, W. , & Guo, J. (2020). Prediction of potential 3CLpro‐targeting anti‐SARS‐CoV‐2 compounds from Chinese medicine. Preprints. 10.20944/preprints202003.0247.v1 [DOI] [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. , & Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, X. , Li, C. , Huang, A. , Xia, S. , Lu, S. , Shi, Z. , … Ying, T. (2020). Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus‐specific human monoclonal antibody. Emerging Microbes & Infections, 9(1), 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhang, Y. , Wu, L. , Niu, S. , Song, C. , Zhang, Z. , … Wang, Q. (2020). Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell, 181(4), 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . (2020a). COVID‐19 significantly impacts health services for noncommunicable diseases. Retrieved from https://www.who.int/news‐room/detail/01‐06‐2020‐covid‐19‐significantly‐impacts‐health‐services‐for‐noncommunicable‐diseases
- World Health Organization . (2020b). International clinical trials registry platform (ICTRP). Retrieved from https://www.who.int/ictrp/en/
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y. M. , Wang, W. , Song, Z. G. , … Yuan, M. L. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Chen, P. , Wang, J. , Feng, J. , Zhou, H. , Li, X. , … Hao, P. (2020). Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences, 63(3), 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Yang, M. , Ding, Y. , Liu, Y. , Lou, Z. , Zhou, Z. , … Gao, G. F. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Lin, D. , Sun, X. , Curth, U. , Drosten, C. , Sauerhering, L. , … Hilgenfeld, R. (2020). Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science, 368(6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Nicholls, J. M. , & Chen, Y. G. (2008). Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐β signaling. Journal of Biological Chemistry, 283(6), 3272–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X. L. , Wang, X. G. , Hu, B. , Zhang, L. , Zhang, W. , … Chen, H. D. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N. , Zhang, D. , Wang, W. , Li, X. , Yang, B. , Song, J. , … Niu, P. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Principal component analysis (PCA) analysis of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S2. Residue contribution (RMSF) to PCA of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S3. Dynamic cross correlation matrix (DCCM) plots of phytochemicals and repurposed drugs for main protease (NSP5) of SARS‐CoV‐2. OP, apo‐protein. P1‐P4, phytochemicals. PR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S4. PCA analysis of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S5. Residue contribution (RMSF) to PCA of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Figure S6. DCCM plots of phytochemicals and repurposed drugs for spike protein of SARS‐CoV‐2. OP, apo‐protein. S1‐S4, phytochemicals. SR5, Repurposed drug. For abbreviations please refer Table 1 in the main manuscript.
Table S1. Hydrogen bond occupancy with NSP5‐ P1, NSP5‐ P2, NSP5‐ P3, NSP5‐ P4, NSP5‐ PR5.
Table S2. Hydrogen bond occupancy with spike protein – S1, S2, S3, S4, SR5.
Table S3. The protein targets (HUGO symbol) for Phytochemicals and SARS‐CoV‐2 used to perform pathway enrichment analysis.
Table S4. KEGG pathway analysis of protease and spike protein.
Table S5. COVID‐19 GO enrichment obtained from Enrichr.
Table S6. Phytochemicals GO enrichment obtained from Enrichr.