

Abstract
That genome function may respond to its three-dimensional (3D) organization highlights the need for methods that can image genomes with superior coverage as well as greater genomic and optical resolution. Here, we push toward this goal by introducing OligoFISSEQ, a suite of three methods that leverage fluorescent in situ sequencing of barcoded Oligopaint probes to enable the rapid visualization of many targeted genomic regions. Applying OligoFISSEQ to human diploid fibroblast cells, we show how only four rounds of sequencing are sufficient to produce 3D maps of 66 genomic targets across 6 chromosomes in hundreds to thousands of cells. We then use OligoFISSEQ to trace chromosomes at finer resolution, following the path of the X chromosome through 46 regions, with separate studies showing compatibility of OligoFISSEQ with immunochemistry. Finally, we combined OligoFISSEQ with OligoSTORM, laying the foundation for accelerated single-molecule super-resolution imaging of large swaths of, if not entire, human genomes.
A capacity to view genomes in situ, in their entirety, and at high genomic resolution is becoming increasingly important, with one potentially enabling class of methods being fluorescent in situ hybridization (FISH)1. Indeed, it was FISH that enabled the pioneering work demonstrating chromosome territories in interphase cells2,3. Of the several methods for FISH, a number are oligomer- (oligo-) based1, one of these being Oligopaints4 (see Supplementary Note 1 for additional examples), which appends nongenomic sequences (Mainstreet and Backstreet) to enable multiple functionalities4–21. In the context of megabase-level coverage, some studies have used these functionalities to walk along contiguous megabases of the genome13,14, with others labeling up to 40 regions on single chromosomes in order to reveal chromosomal paths9,21, and still others visualizing entire, or nearly entire, genomes, one chromosome or one chromosome arm at a time15,19. Here, we demonstrate how streets enable a new technology, OligoFISSEQ, which vastly increases the number of targets that can be visualized, putting us within reach of genome-wide imaging via the visualization of a multitude of sub-chromosomal regions. As OligoFISSEQ is compatible with the single-molecule localization method of OligoSTORM5,10, it also accelerates the speed with which genomic regions can be visualized at super-resolution. Thus, OligoFISSEQ should contribute to both diffraction-limited and super-resolution views of entire genomes at high genomic resolution.
OligoFISSEQ is based on the fluorescent in situ sequencing (FISSEQ) technologies that have been honed for in situ detection of transcripts22,23 (see Supplementary Note 2 for recent iterations as well as earlier studies). Here, we present three strategies that direct the sequencing to barcodes embedded in Oligopaint streets, wherein one uses sequencing by ligation (SBL), another uses sequencing by synthesis (SBS), and a third uses sequencing by hybridization (SBH). Focusing on OligoFISSEQ with SBL, we map 66 genomic regions in human diploid PGP1 skin fibroblasts (XY; PGP1f) using only 4 rounds of sequencing. We next introduce a method to improve barcode detection and, using it in conjunction with OligoFISSEQ, trace the human X chromosome by mapping 46 regions along its length. We demonstrate that OligoFISSEQ is compatible with immunofluorescence and then conclude by combining OligoFISSEQ with OligoSTORM to achieve a much accelerated rate at which multiple genomic regions ranging in size from tens of kilobases to megabases can be visualized simultaneously at super-resolution.
Results
Principle and validation of OligoFISSEQ
FISSEQ technologies22,23 leverage next generation sequencing methods24,25 to provide in situ 3D spatial maps of transcripts that have been reverse transcribed and then amplified. As FISSEQ can also be used for in situ decoding of barcodes introduced during the generation of cDNA, we reasoned that it might be possible for FISSEQ to read barcoded Oligopaints. Furthermore, by targeting hundreds to thousands of identically barcoded Oligopaints to a genomic region, the combination of Oligopaints with FISSEQ, which we call OligoFISSEQ, could both obviate the need for target amplification, typically required by FISSEQ, while also rendering the targeted chromosomal structure amenable to imaging. Finally, as FISSEQ is carried out using diffraction-limited microscopy, we anticipated a capacity of OligoFISSEQ to image the same genomic regions in hundreds to thousands of cells and thus provide the computational and statistical power necessary for addressing cell-to-cell variability.
We began by designing an Oligopaint library that targeted 18,536 oligos to a 4.8 Mb single copy region on human chromosome 19 (Chr19–20K; Extended Data Fig. 1a) and then testing whether it could be sequenced in situ, focusing first on SBL to effect ligation-based interrogation of targets (LIT) and SBS to effect synthesis-based interrogation of targets (SIT), implementing hybridization-based interrogation of targets (HIT) only later (Fig. 1a–e). Importantly, as Oligopaint streets can accommodate multiple barcodes, we were able to design a single library to accommodate the sequencing chemistries of both LIT and SIT, with the primer binding site and barcode for LIT embedded on Mainstreet (5’ end of the Oligopaint oligo) and the primer-binding site and barcode for SIT embedded on Backstreet (3’ end) (Fig. 1a). Note that we will use LIT and SIT to refer to the steps of sequencing, per se, and OligoFISSEQ-LIT (O-LIT) and OligoFISSEQ-SIT (O-SIT) to refer to the use of LIT and SIT, respectively, in the context of OligoFISSEQ.
Fig. 1. OligoFISSEQ.
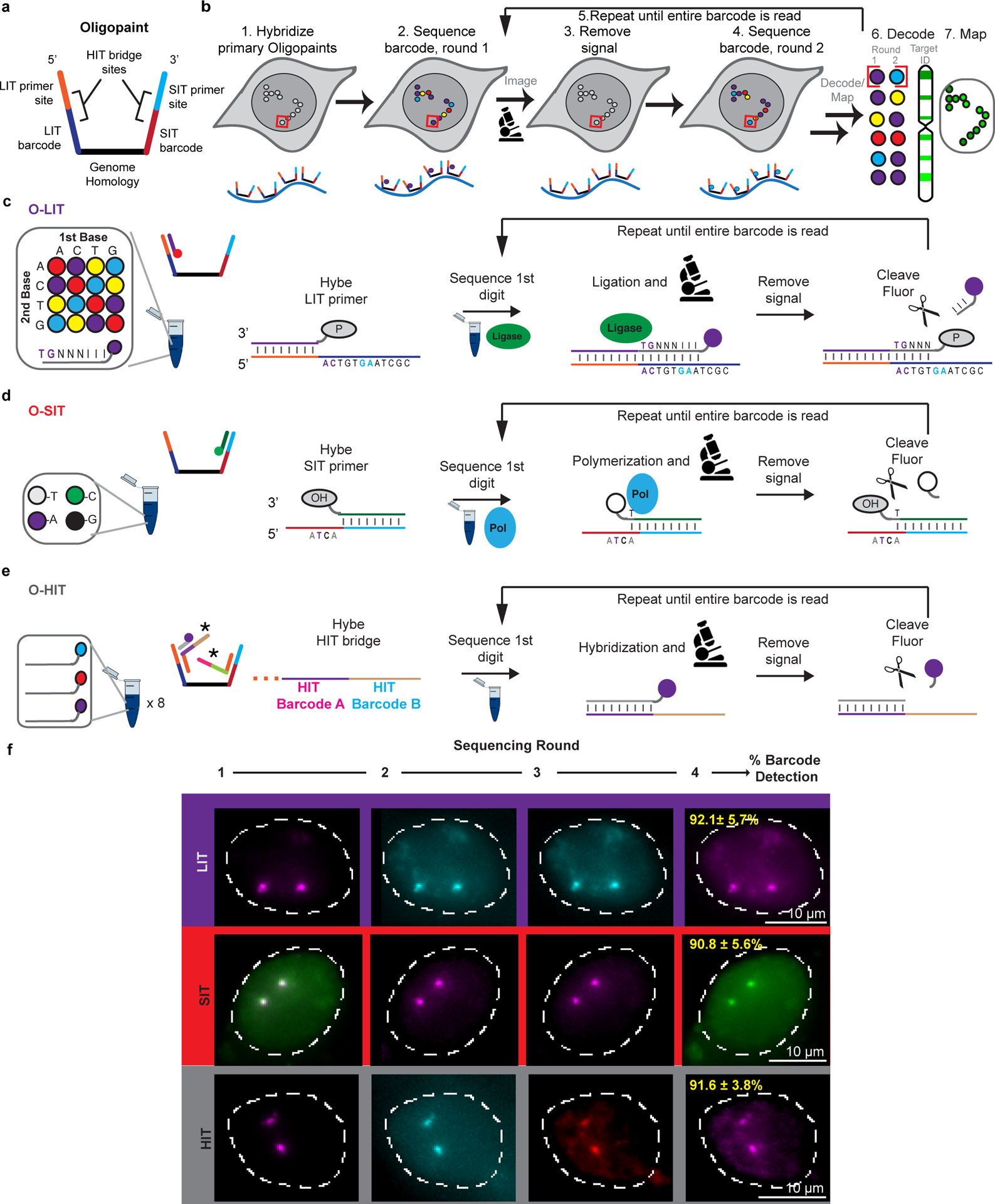
a) Oligopaint oligo used for OligoFISSEQ. Portions of the LIT and SIT primer sites and barcodes can function as binding sites for HIT bridges (panel e) as well as priming sites to amplify the Oligopaint library. LIT primers and barcodes can be encoded on both streets to increase signal. Some libraries used reverse priming sites that were chromosome-specific (common to oligos targeting the same chromosome).
b) General OligoFISSEQ workflow. In step 7, mapped targets correspond to Target ID text color in step 6 and chromosome cartoon.
c) O-LIT. After the phosphorylated LIT primer (P) is hybridized, it is ligated to an 8-mer (TGNNNIII), the first two nts of which correspond to a specific fluorophore; as Oligopaint barcodes are pre-defined, each fluorophore corresponds to only a single barcode digit. N denotes a mixture of A, C, T, or G, and I denotes deoxyinosine, a universal base.
d) O-SIT. SIT primers contain 3’hydroxyls (OH). A and C are conjugated to distinct fluorophores (shown here as purple and green, respectively), T to two fluorophores (grey), with G remaining unlabeled (black).
e) O-HIT. In this iteration using three fluorophores and 6 barcodes, each round of hybridization uses three fluorophore-labeled oligos, and two rounds suffice to identify which barcode resides at a barcode position. Given 4 barcode positions, HIT is completed within 8 rounds of hybridization. *, two bridge oligos bringing in a total of 4 HIT barcodes.
f) Representative images after 4 rounds of O-LIT, O-SIT, and O-HIT using Chr19–20K on PGP1f cells. Images represent maximum intensity z-projections. The first round of SIT identifies deoxyadenosine (labeled by a combination of purple and green and thus appears white). Mean barcode detection efficiencies with SD for LIT, SIT, and HIT represent 85, 66, and 79 total cells, respectively, from 4 replicates.
With O-LIT (Fig. 1c and Extended Data Fig. 1b), the barcode is read with SOLiD chemistry24, wherein each digit of the barcode (5-nt per digit, digit defined as the smallest unit of a code) is read by cleavable 8-mers carrying one of four fluorophores. In brief, a sequencing primer is hybridized to the street, and subsequent barcode readout begins by binding of the first barcode digit by a labeled 8-mer, which is then ligated and imaged. The 8-mer is then cleaved between nucleotides (nts) 5 and 6, leaving the first 5-nts and removing the label, allowing for the next digit to be read. Excluding the primer binding site, barcodes were 23 nts long and sufficient to accommodate 4 rounds of sequencing ([4 rounds of sequencing * 5-nts per digit] + the 3 nts left uncleaved after the 4th round of sequencing); when fully utilized, 4 or 8-digit barcodes have the potential to distinguish 256 (44) or 65,536 targets (48), respectively. Using O-LIT on Chr19–20K, we recovered 4-digit barcodes from 92.1% ± 5.7% of PGP1f cells (n = 85 from 4 replicates; Fig. 1f).
In the case of O-SIT, (Fig. 1d and Extended Data Fig. 1b), barcodes are sequenced using Illumina NextSeq chemistry24 via the extension of primers one base at a time and using only two fluorophores; one fluorophore is assigned to deoxycytidine (C), the other is assigned to deoxythymidine (T), both are assigned simultaneously to deoxyadenosine (A), and deoxyguanosine (G) is left unlabeled (Fig. 1d,f.). With each digit of the barcode being only a single nucleotide (nt), SIT barcodes are compact, with an 8-nt long barcode theoretically able to identify 65,536 targets (48). Applying O-SIT to Chr19–20K, we recovered 4-digit barcodes from 90.8 ± 5.6% of PGP1f cells (n = 66 from 4 replicates; Fig. 1f).
Chr19–20K can also be co-opted for hybridization-based interrogation of targets (HIT) through SBH (Fig. 1a), reminiscent of strategies that have enabled Oligopaints to facilitate transcriptome profiling6,8,12,18. Here, we introduce SBH for 3D spatial mapping of chromosomal DNA. In particular, we implemented OligoFISSEQ-HIT (O-HIT) by appending SBH barcodes via two bridge oligos14,19,20,26, one hybridizing to the junction of the LIT barcode and its primer sequence on Mainstreet and the other hybridizing to the junction of the SIT barcode and its primer sequence on Backstreet; SBH barcodes can also be embedded directly into the streets. As each bridge carries two 20-nt barcode positions, each position encoding one of six possible barcodes, the resulting 24 (4*6) barcodes have the potential to identify 1,296 (64) targets (Fig. 1e). Each barcode is identified via complementary labeled secondary oligos and, thus, using three fluorophore species, 8 rounds of hybridization (8*3) are sufficient to identify all 24 barcodes in this iteration of O-HIT, with the option to increase target capacity through additional barcode positions, barcode sequences, and/or fluorophore species. Using O-HIT on Chr19–20K, we successfully recovered 4-digit barcodes from 91.6% ± 3.8% of PGP1f cells (n = 79 from 4 replicates; Fig. 1f and Extended Data Fig. 1b).
Mapping 66 genomic targets with O-LIT
We next assessed the potential of OligoFISSEQ to address multiple regions on multiple chromosomes, choosing to work with O-LIT because we anticipated it to scale well. For example, O-LIT would be expected to scale without the increased costs predicted to accompany the scaling of purely hybridization-based technologies, such as O-HIT, for which the number of species of labeled oligos, and thus their cost, would increase as the number of targets increases. In contrast, O-LIT reagents would remain the same regardless of whether they target 1, hundreds, or thousands of regions. Furthermore, because the 5-nt O-LIT barcode digits are relatively compact, they decrease the requisite length of Oligopaint oligos, further reducing cost. In addition, because O-LIT delivers a positive signal at each round of sequencing, in contrast to O-SIT and O-HIT, which contain “blank” readouts, its barcoding is more robust.
To assess the scalability of O-LIT, we designed an Oligopaint library (36plex-5K; Fig. 2a) targeting six regions along each of six chromosomes: Chromosome 2 (Chr2; 242 Mb), Chr3 (198 Mb), Chr5 (181 Mb), Chr16 (90 Mb), Chr19 (58 Mb), and ChrX (156 Mb), with a unique barcode for each of the thirty-six targets. Thus, 36plex-5K targets a total of 66 regions in PGP1f cells (6 targets for each of 2 homologs of the 5 autosomes + 6 targets on the single X), each tiled by 5,000 Oligopaint oligos and, together, encompassing 31.6 Mb, with targeted regions ranging in size between 642 kb and 1.22 Mb (876 kb average). We chose gene poor (5.4 – 6.1 genes/Mb; Chr 2, 3, 5, X) and gene rich (10.8 and 23 genes/Mb; Chr 16 and 19, respectively) as well as large (Chr2: 242 Mb) and small chromosomes (Chr 19: 58 Mb) and positioned three targets along each chromosome arm – one as close as possible to the telomere, one in the center of the arm, and one as close as possible to the centromere, with inter-target distances ranging from 7 to 74.9 Mb (28.8 Mb average). The number of Oligopaint oligos per target (5,000) was kept constant to assess the robustness of LIT with respect to target size and different densities of oligo binding sites (4 – 7.7 binding sites/kb, 5.8 average). In addition, because all 36plex-5K Oligopaint oligos targeting the same chromosome share the same reverse primer sequence, it was possible to use indirect labeling to produce a 6-banded pattern along all targeted chromosomes in metaphase and distinctly colored territories in interphase cells (Fig. 2b). This outcome confirmed the accuracy of the library.
Fig. 2. OligoFISSEQ-LIT on 36plex-5K.
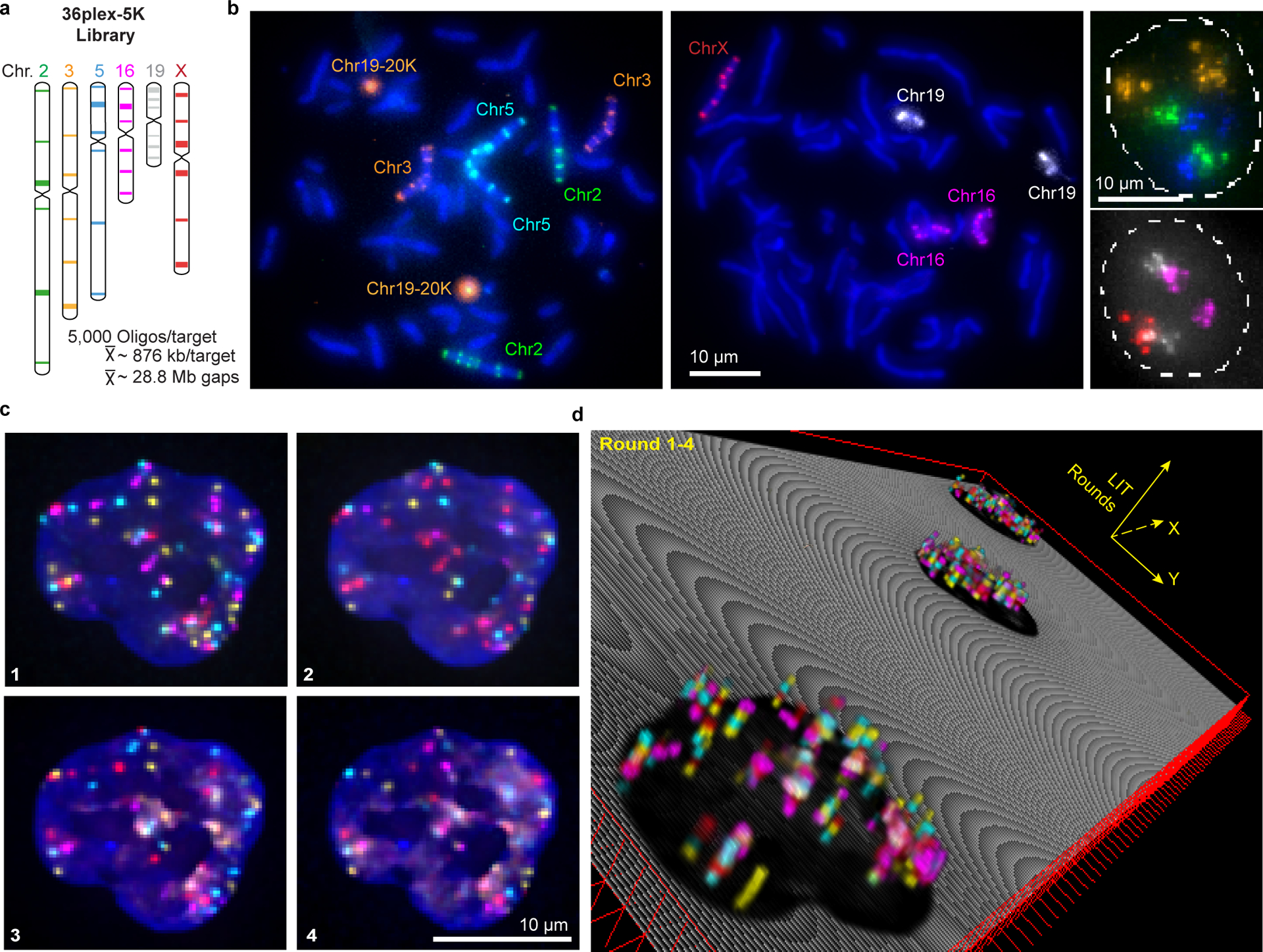
a) Targets of the library. Chromosome number color-coded to correspond to images in panel b. Each target corresponds to a unique barcode.
b) Metaphase chromosome spreads of male lymphoblast cells (left; cells from Applied Genetics; Methods) and interphase nuclei from PGP1f cells (right) representative of four replicates. All six targets on any single chromosome were labeled with secondary oligos carrying the same species (color) of fluorophore. Chr19–20K was used as a positive control in metaphase chromosome spreads. Maximum z-projections.
c) Four rounds of O-LIT off of both streets of 36plex-5K. Images are from deconvolved, five-color merged maximum z-projections. n = 1.
d) 3D representation of a field of view (FOV) containing three cells sequenced with four rounds of O-LIT. Sequencing rounds are represented on the z-axis, with the first being closest to the DAPI-determined nuclear outline (black). Maximum z-projection of sequencing signal from each round was taken, duplicated (2-images total for better visualization), and then stacked on top of each other. The lower left cell corresponds to the cell in panel c.
An every-pixel automated analysis pipeline
To improve target detection, we sequenced simultaneously off of Mainstreet and Backstreet (Fig. 2c–d and Extended Data Fig. 1c–f), which, in the case of 36plex-5K, carried the same barcode. Indeed, this strategy identified 100% of the 66 targets in PGP1f cells via manual decoding (n = 2 from 2 replicates; Extended Data Fig. 1f). However, as manual decoding does not scale well, we developed an automated pipeline to address a range of signal intensities and sizes by interrogating every pixel individually (Fig. 3a); a centroid-based pipeline did not perform as well (29.93 ± 4.9% vs. 62.8% ± 4.8%, n = 111 cells from 3 replicates; Extended Data Fig. 1g).
Fig. 3. Every-pixel analysis pipeline on 36plex-5K.
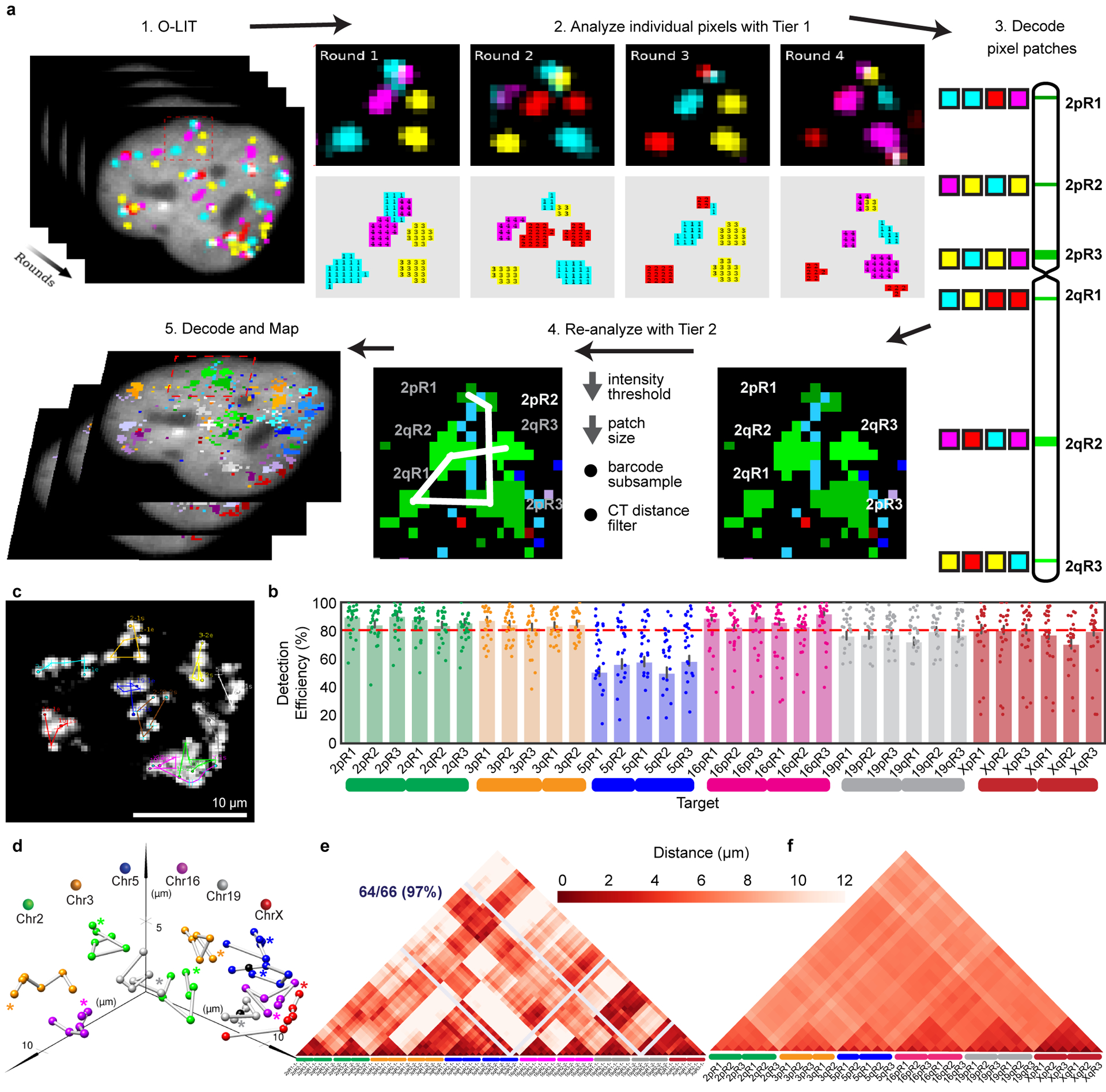
a) Analysis pipeline. Sequencing rounds (Step 1) are analyzed at the level of individual pixels using Tier 1 parameters with thresholds for signal intensity and pixel patch size (Step 2) and then decoded (Step 3). Missing targets and false positives are filtered by re-analyzing images with Tier 2 parameters (Step 4) to produce traces (Step 5). Tier 2 decreases thresholds for signal intensity and pixel patch size, subsamples barcodes, and applies filters for chromosome territories. 1 = FITC, 2 = Cy3, 3 = TxRed, 4 = Cy5.
b) 36plex-5K Tier 2 detection efficiency after sequencing off of both streets. 80.2 ± 7.3% of targets are detected in 611 cells from 15 replicates. X-axis, chromosomal targets. Detection efficiency from individual replicates are plotted. Dashed red line marks the mean over all chromosomal targets. 3qR3 and 5pR3 share a barcode and are not included (see text). For unknown reasons, Chr5 target detection was less robust. Error bars represent 95% bootstrap confidence interval of the mean.
c) Chromosome traces of Figure 2c nucleus after Tier 2. 64/66 (97%) of targets were detected. N = 1.
d) Ball and stick traces of panel b nucleus. Colored spheres represent targets; black spheres represent undetected targets and, thus, positioned by calculating the median proportionate distance between flanking detected spheres. Grey lines between signals, extrapolations. Asterisks, beginning of chromosomes.
e) Single-cell pairwise spatial distance matrix after Tier 2 detection of panel b nucleus. Homologs separately displayed. Centroids of targets were used for this and all subsequent spatial distance matrices. Grey lines, undetected targets.
f) 36plex-5K population pairwise spatial distance measurements after Tier 1 detection (n = 611 from 15 replicates). Homologous target measurements were combined. Overall greater distances of Chr2, Chr3, Chr5, and X may reflect more peripheral positioning, while lesser distances of Chr16 and Chr19 may reflect more central positioning.
The every-pixel pipeline detected 95 ± 5.15% of 36plex-5K targets, but with many false positives (FPs) (574.86 ± 325.38 FPs/nucleus; n = 611 from 15 replicates; Extended Data Fig. 2a–b). Thus, we developed a two-tier system (Fig. 3a), wherein Tier 1 filtered out pixels below a minimum signal intensity and/or patch size, reducing FPs 165-fold (3.49 ± 1.36 FPs/nucleus; 5.29 ± 2.06%;) while detecting 62.2 ± 6.68% of the targets (~41/66) in each nucleus (n = 611 from 15 replicates; Extended Data Fig. 2c–d). In Tier 2, requirements for pixel intensity and patch size were lowered, after which barcode subsampling was applied, with all newly detected signals from the same chromosome required to be within 4.5 μm of Tier 1 targets. This proximity-based filtering reflects the propensity of chromosomes to occupy distinct territories2 as well as measurements of distances between consecutive Tier 1 regions along a chromosome (Methods; Supplementary Figure 1) although, in the context of chromosome rearrangements, it would need to be modified. Tier 2 eliminated all FPs while detecting 80.2 ± 7.3% (~52/66) of targets in each nucleus with at least 70% (~46/66) of targets recovered in ~70% of cells (Fig. 3b and Extended Data Fig. 2e–g). The centroids of all detected targets were then conceptually connected to produce ball and stick renditions of chromosomes, with undetected targets positioned by calculating the median distance between flanking centroids (Fig. 3c–d); ball and stick strategies have been used elsewhere to trace chromosome paths and are useful when assessing chromosome structure and positioning9,13,17,20,21. Note that two targets, 3qR3 and 5pR3, that were designed to share barcodes were both detected at 69% efficiency, boding well for the consistency and robustness of barcode recovery. Similarly, 15 replicates using PGP1f cells produced similar ranges of barcode recovery, with no significant differences after principal component analysis (Extended Data Fig. 2h).
Development of eLIT to interrogate fine scale genome organization
O-LIT mapping of 36plex-5K revealed the paths of all 6 chromosomes (Fig. 3c–d and Extended Data Fig. 3a–b), producing single-cell spatial genomics data (Fig. 3e–f and Extended Data Fig. 3c–e) that align with previous studies and thus argue the potential of OligoFISSEQ to be informative. First, the chromosomes fell into different territories3, with the smaller (Chr16, Chr19) and larger (Chr2, Chr3, Chr5) chromosomes positioned towards the center and periphery of the nucleus, respectively (Extended Data Fig. 3f), in line with observations of a radial positioning of chromosomes that places smaller chromosomes more centrally2,27. Consistent with this, median inter-homolog distances for the smaller chromosomes were less than that for the larger chromosomes across hundreds of cells (Extended Data Fig. 4a; p = 4.3 × 10−37). These robust sample sizes also enabled consideration of suggestions that diploid genomes can, under some circumstances, separate into two spatially distinct haploid sets28–30. Here, cluster analyses of 36plex-5K maps revealed that the 5 targeted PGP1f autosomes spatially separated into two haploid sets in 6.9% (18/258) of cells (Extended Data Fig. 4b–e), which, however, was statistically similar to proportions expected from randomized controls (5% and 5.4% for Directed random and Completely random). While definitive descriptions await the analysis of complete genomes, this observation, compounded with studies of homolog pairing and anti-pairing31 highlight the possibility that it is in cell types that do not segregate the genome into haploid sets that inter-homolog interactions will prevail.
We also aggregated single-cell 36plex-5K data from 611 cells to produce an average distance matrix, this time combining data for homologous chromosomes (Fig. 3e–f). Comparison of this matrix to a Hi-C map of PGP1f cells14 revealed a strong correlation (r = 0.705, p = 1.77 × 10−174; Extended Data Fig. 3e), once more indicating the robustness of O-LIT. Nevertheless, the matrices also differ, with O-LIT producing sub-chromosomal stripes of greater or lesser distance, and the Hi-C matrix being more mottled. While stripes may reflect discontinuities along a chromosome, they may also suggest chromosome-specific9,21 and interchromosome-specific signatures. For example, chromosomal regions that are overall further from other regions may be relatively more buried within a chromosome territory or nearer the nuclear membrane, while those that are closer may be nearer to the surface of the territories or less constrained to the nuclear membrane. As for the mottled appearance of the Hi-C matrix, it suggests that, at the scale of whole chromosomes, distances on the order of microns may not always correlate with interaction frequencies and distances amenable to Hi-C; indeed, absence of correlation may indicate that proximity and interaction are distinct features. Thus, O-LIT matrices of distance and Hi-C matrices of interaction frequency may, when considered together, provide layers of information that neither alone can provide.
Confirmations of the usefulness of OligoFISSEQ in hand, we next refined O-LIT so that it can target smaller genomic regions as well as trace chromosomes at higher genomic resolution. As fate would have it, it was at this time that the commercial production of SOLiD reagents was discontinued, leading us to develop a method that, ultimately, improved signal detection. We begin with a description of SOLiD chemistry. SOLiD chemistry reads sequences as dinucleotides using labeled 8-nt oligos (TGNNNIII, where the first two positions are used to represent all 16 dinucleotide combinations, positions 3–5 are degenerate, and positions 6–8 are universal), thus entailing 1,024 (16*43) oligo species24. Because this level of complexity is excessive for O-LIT, where barcodes are defined by the user, we aimed to reduce the complexity of the oligo pool to the minimum necessary for decoding O-LIT barcodes, reasoning further that a minimally complex oligo pool might increase signal over background. Thus, taking advantage of the universal base, deoxyinosine32, we reduced the complexity of the oligo pool from 1,024 to 4, referring to this strategy as just enough barcodes (JEB) and the LIT chemistry using it as eLIT (Fig. 4a–b). Application of OligoFISSEQ using eLIT (O-eLIT) to a library targeting 9,267 Oligopaint oligos to Chr19 (Chr19–9K) proved successful, yielding a 3.3-fold brighter signal-to-nuclear background ratio as compared to the application of LIT to the same library using SOLiD oligos (n > 55 from 2 replicates; Extended Data Fig. 5a–b).
Fig. 4. OligoFISSEQ-eLIT.
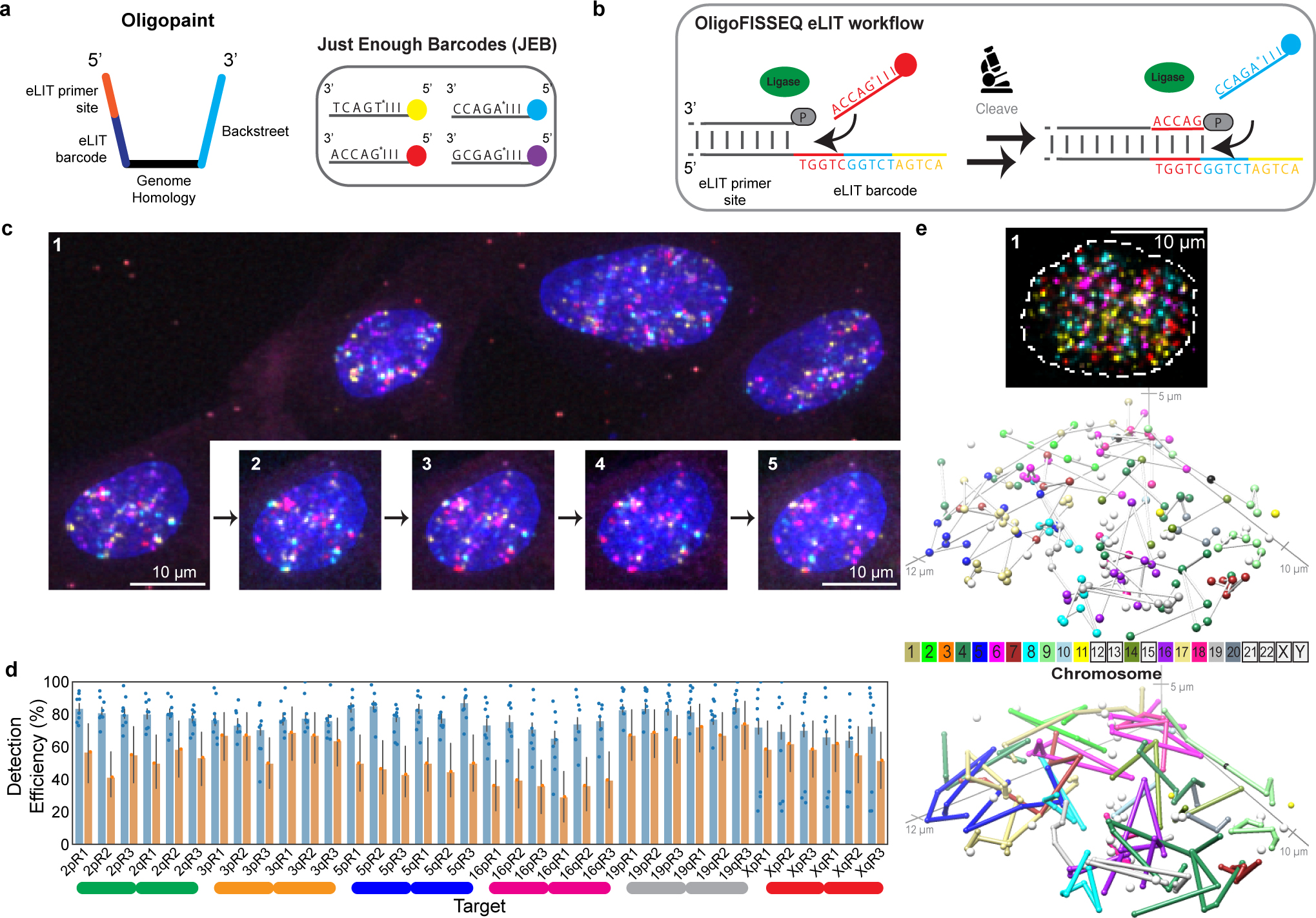
a) JEB technology reduces the pool of labeled 8-mers to four. Left, design of Oligopaint oligos in libraries, such as 36plex-1K, that use eLIT. Right, JEB labeled 8-mers complementary to the 5-nt eLIT barcode digit. I, universal base, deoxyinosine. While eLIT is compatible with a variety of barcode configurations, our current application uses barcodes consisting of 5 digits each, wherein each digit is one of only four distinct 5-nt sequences. To further reduce the complexity of the pool of 8-nt oligos, we also use the universal base, deoxyinosine32 in positions 6, 7, and 8. In short, JEB reduces the pool of labeled oligos from 1,024 to 4 (Extended Data Fig. 5a–b).
b) eLIT workflow with JEB. See legend to Figure 1c for details.
c) Five rounds of sequencing with O-eLIT. Left, PGP1f cells after first round of sequencing. Right, images from five rounds of sequencing (1–5) of one nucleus from panel c (yellow square); T, totality of targets labeled simultaneously with a secondary oligo complementary to a barcode present on each oligo. Extranuclear puncta are fiducial tetraspeck beads (Thermo Fisher). Images are deconvolved maximum z-projections. n = 1.
d) Tier 2 target detection efficiency of 36plex-1K after five rounds of O-LIT with SOLiD reagents (orange; average of 54.6%; n = 41) or O-eLIT with JEB (blue; average of 74 ± 11.2%; n = 440 from 9 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
e) First O-eLIT round of 129-plex (top; deconvolved maximum-intensity z projection; n = 1). Tier 2 tracings (middle; white spheres are tier 1 duplicated barcodes that did not move to tier 2, with untraced chromosomes boxed in color key). Sticks color-coded to facilitate visualization (bottom). Oligonucleotide target density was 5.8 to 11.9 per kb.
Anticipating that the improved signal-to-nuclear background ratio would improve genomic resolution, we generated a library identifying smaller genomic regions (173 kb average) by directing Oligopaint oligos to only the first 1,000 of the 5,000 oligo targets defined by 36plex-5K for each designated genomic region (Extended Fig. 5c caption). Then, to benchmark this library, called 36plex-1K, against 36plex-5K, we adopted the same barcodes for 35 of the 36 targets, the exception being 5pR3, which was given a new barcode; it had previously shared a barcode with 3qR3 to enable assessment of barcode detection across different regions. Five rounds of O-eLIT using only Mainstreet of 36plex-1K yielded a Tier 2 barcode recovery efficiency of 74 ± 11.2% (48/66) (n = 440 from 9 replicates) that was higher than that obtained with 5 rounds of O-LIT (54.6%, n = 41; Fig. 4c–d and Extended Data Fig. 5c–d). Not surprisingly, O-eLIT of 36plex-1K was also able to generate homolog-resolved, single-cell spatial data across all the targeted chromosomes with comparable efficiency (Extended Data Fig. 5e–ii). These findings argued that O-eLIT would be useful genome wide. We recently imaged 249 regions with a genome-wide library (129plex) corresponding to 129 100-kb targets spanning all the autosomes (120 targets), ChrX (6 targets) and ChrY (3 targets). Five rounds of sequencing confirmed genome-wide capacity (Methods); although inadvertent barcode duplications complicated analyses, tier 2 can nevertheless detect 95% (165 of 174) of unique barcodes, while tier 1 can detect 44% (33 of 75) of duplicated barcodes (Fig. 4e and Supplementary Table 12).
Fine ChrX tracing and suggestions of chromosome signatures
To test the potential of O-eLIT to achieve finer genomic resolution, we applied an Oligopaint library (ChrX-46plex-2K) targeting 2,000 oligos to each of 46 regions along human ChrX, the number of targets aligning with a previous study using a hybridization-based Oligopaint strategy to image 40 regions of this chromosome9. The targets ranged in size from 253 kb to 1.22 Mb (445 kb average), with the average distance between targets being 2.75 Mb and total coverage being 20.4 Mb or 13.3% of the chromosome (Fig. 5a). As such, ChrX-46plex-2K served as an informative proxy for assessing the capacity of OligoFISSEQ to accommodate all other chromosomes. Here, we applied O-eLIT to both streets and achieved a Tier 2 barcode recovery efficiency of 74.3 ± 2.5% (~34/46 targets, n = 177 from 7 replicates; Supplementary Figure 2) in PGP1f cells (Fig. 5b–c and Extended Figure 6a–b), interpolating the positions of any target that had escaped detection (Methods). Although 3 targets were difficult to recover (X15, X19, X31), the quality of the data nevertheless permitted 176 traces spanning the entirety of the X, single-cell spatial distance matrices, and a population-based spatial distance matrix that was strongly correlated with a corresponding Hi-C map (r = 0.641, p = 7.074e-245) and inversely correlated with Hi-C interaction frequencies (r = −0.84, p = 5.08 × 10−275), the latter producing a scaling factor of 0.18 (Fig. 5d–g and Extended Data Fig. 6c–j), similar to that observed previously9. Furthermore, the chromosome traces revealed two major clusters (Extended Data Fig. 7a–c; Calinski-Harabasz index of 213.71) that differed in their radii of gyration (t = −10.1; p = 3.9 × 10−19; Extended Data Fig. 7d), one consisting of 156 (89%) and the other of 20 (11%) chromosomes. While the basis for this heterogeneity will require additional study, be it the cell cycle, chromatin accessibility, and/or overall chromosome activity, these findings emphasize the potential of O-eLIT to advance our understanding of the manner in which chromosomal material can be packaged and whether that packaging correlates with function.
Fig. 5. Tracing 46 regions along Chromosome X.
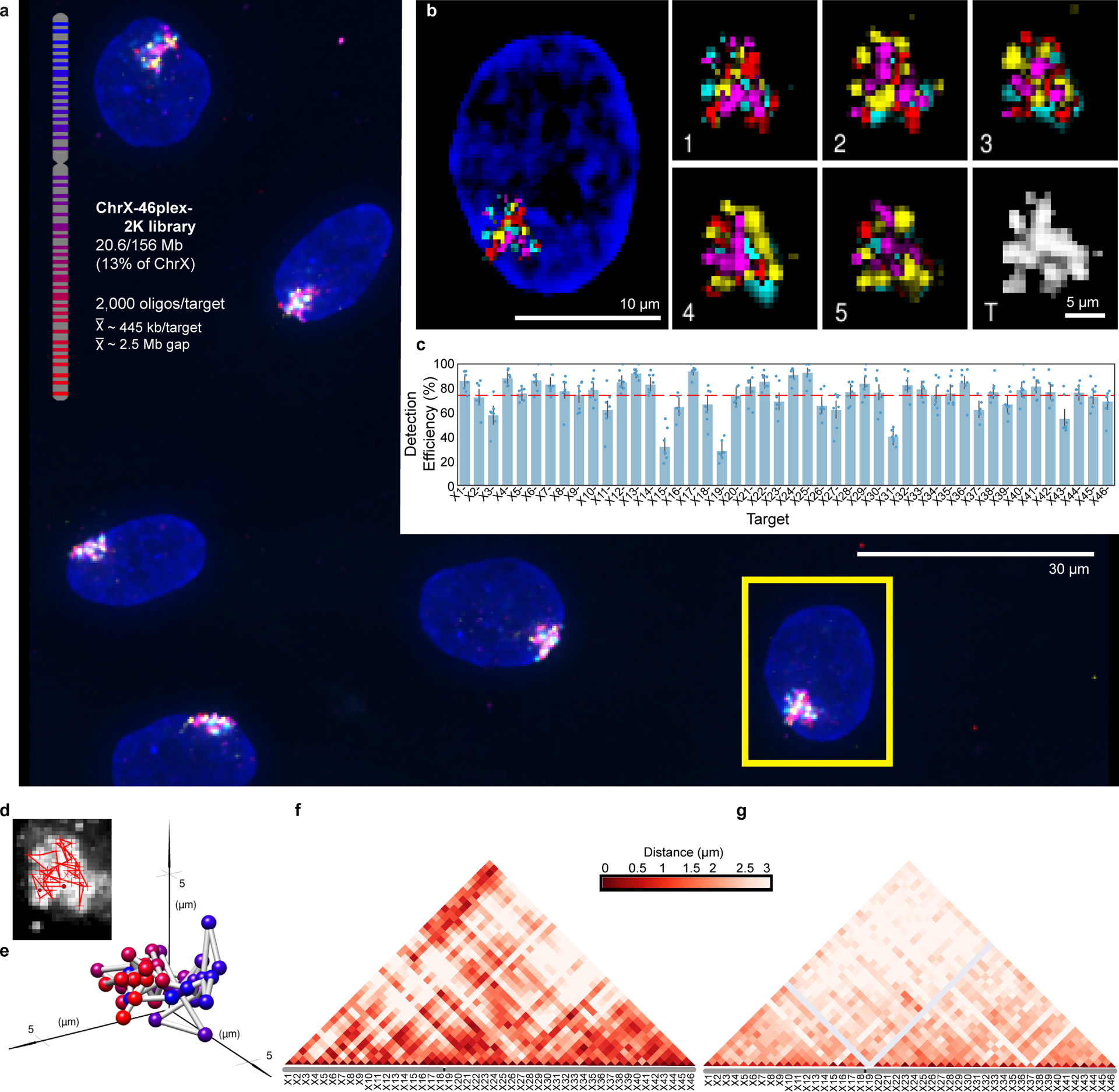
a) Targets of ChrX-46plex-2K and nuclei after a first round of O-eLIT sequencing off of both streets in PGP1f. Images are from deconvolved maximum intensity z-projection. n = 1.
b) Five rounds of sequencing with O-eLIT off of both streets. Nucleus from panel a (yellow square). Left, view of DAPI stained nucleus after first round of sequencing. White numbers, round of sequencing. T, totality of targets labeled simultaneously with a secondary oligo complementary to a barcode present on all oligos. Images are from deconvolved maximum z-projection. n = 1.
c) Tier 2 target detection efficiency after five rounds of O-eLIT off of both streets in PGP1f cells. The mean detection efficiency marked by the red dashed line was 74.29 ± 2.5% (n = 177 from 7 replicates), averaging detection efficiencies off of one street (73.7 ± 2.97%, n = 122 from 5 replicates) and off of both streets (75.3 ± 1.97%, n = 55 from 2 replicates). Particularly difficult was X19, being detectable only 0% and 28% of the time after Tier 1 (Extended Data Fig. 6a) and Tier 2, respectively Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
d-e) Chromosome traces (d) and 3D visualization (e) of the nucleus in Figure 5b after Tier 2 analysis and interpolation of missing targets. Sphere color corresponds to chromosome cartoon in panel a. n = 1.
f) Single-cell pairwise spatial distances after interpolation of missing targets of nucleus in panel b.
g) Population pairwise spatial distances (n = 177 from 7 replicates) after Tier 1 detection (combining reads off of Mainstreet with reads off of both streets).
36plex-5K and 36plex-1K have also enabled analyses of chromosome folding. Combining the two datasets (for 36plex-5K, n = 611 from 15 replicates; for 36plex-1K, n = 440 from 9 replicates) we evaluated the angles formed by the chromosomal segments flanking the centromeres (Extended Data Fig. 8a), observing that only a minority, if any, of the chromosomes extend their p and q arms in polar opposite directions or are folded into a hairpins; median values for the angles ranged from 74° to 94° (Extended Data Fig. 8b–c). Furthermore, assessment of the angles formed by the two contiguous chromosomal segments lying within each arm (Extended Data Fig. 8a) showed that the p and q arm angles were significantly different for Chr2, 3, 16, and 19 (n= 686, 668, 586, and 760, respectively; p = 4.15 × 10−16, 0.004, 1.36 × 10−14, and 3.33 × 10−11, respectively; Extended Data Fig. 8c). As the larger angle was associated with the p (shorter) arm of Chr2 and Chr19 and with the q (longer) arm of Chr3 and Chr16, these findings cannot be explained solely by relative arm lengths. Consistent with this, arm angle and arm length were not significantly correlated (r = 0.26, p = 0.42, Extended Data Fig. 8d), leaving open the possibility that arm angles reflect the impact of centromere structure on flanking genomic regions and/or interdependence of the p and q arms, the constraints of chromosomal territories or other intrinsic organizational principles, Rabl configurations resulting from the last cell division, and/or the state of gene activity, such as accessibilities underlying allelic skewing. Regardless, these observations of ChrX conformations (Extended Data Fig. 7a–d) and arm angles (Extended Data Fig. 8a–d) demonstrate the potential of chromosome-wide imaging to address whether there are chromosome-level structural signatures, such as may be indicative of cell type, cell state, and/or cellular health or age, with a recent study of two chromosomes in C. elegans aligning with these possibilities21. Chromosome organization may also reflect the evolutionary history of a chromosome33,34. The capacity of OligoFISSEQ to generate large datasets will facilitate the study of these potential paradigms of genome organization.
Single-gene identification, immunofluorescence, and acceleration of super-resolution imaging
OligoFISSEQ has proven versatile in our hands, capable of imaging single regions in the size range of tens of kilobases, accommodating immunofluorescence, as well as accelerating super-resolution imaging (Fig. 6a–e, Extended Data Fig. 9a–b, and Extended Data Fig. 10a–b). With respect to single regions, we applied O-eLIT to six genes ranging in size from 11 kb to 136 kb (Fig. 6a–b): HES5 (11 kb, Chr1), MMP2 (27 kb, Chr16), FL11 (39 kb, Chr11), ABL (45 kb, Chr9), BCR (100 kb, Chr22), and DXZ4 (136 kb; ChrX). Detection of the larger targets hovered between 43% and 80%, reaching as high as 83.7 ± 4.38% for ABL (n = 61 cells from 2 replicates; Fig. 6b), and, although detection of the smallest target, HES5, was low (9.82 ± 3.79%), we expect that, with the incorporation of amplification strategies35,36, detection of targets as small as, or even smaller than, HES5 should become robust.. Regarding immunofluorescence, we conducted 4 rounds of O-LIT using 36plex-5K and sequencing off of both streets, followed by immunocytochemical detection of antibodies directed against α-tubulin, GAPDH, and TOMM20 and were able to trace all 6 chromosomes as well as obtain strong signals for all three proteins (Fig. 6c; Extended Data Fig. 10a). We have also applied ChrX-46plex-2K to IMR-90 human fibroblast cells (XX) and then distinguished the active X (Xa) from the inactive X (Xi) through immunofluorescence to macroH2A.1, which preferentially binds the latter (Extended Data Fig. 9a–l). Xi displayed a lower radius of gyration (p = 9.07 × 10−5; Extended Data Fig. 9h) as well as megadomain structures (Extended Data Fig. 9k,l), consistent with Hi-C and FISH studies9,37–43, further validating the use of O-eLIT for high-resolution chromosome tracing. Taken together, these findings confirm the potential of OligoFISSEQ to enable discoveries regarding the genome-wide spatial relationship between genes and their epigenetic partners.
Fig. 6. OligoFISSEQ extensions and applications.
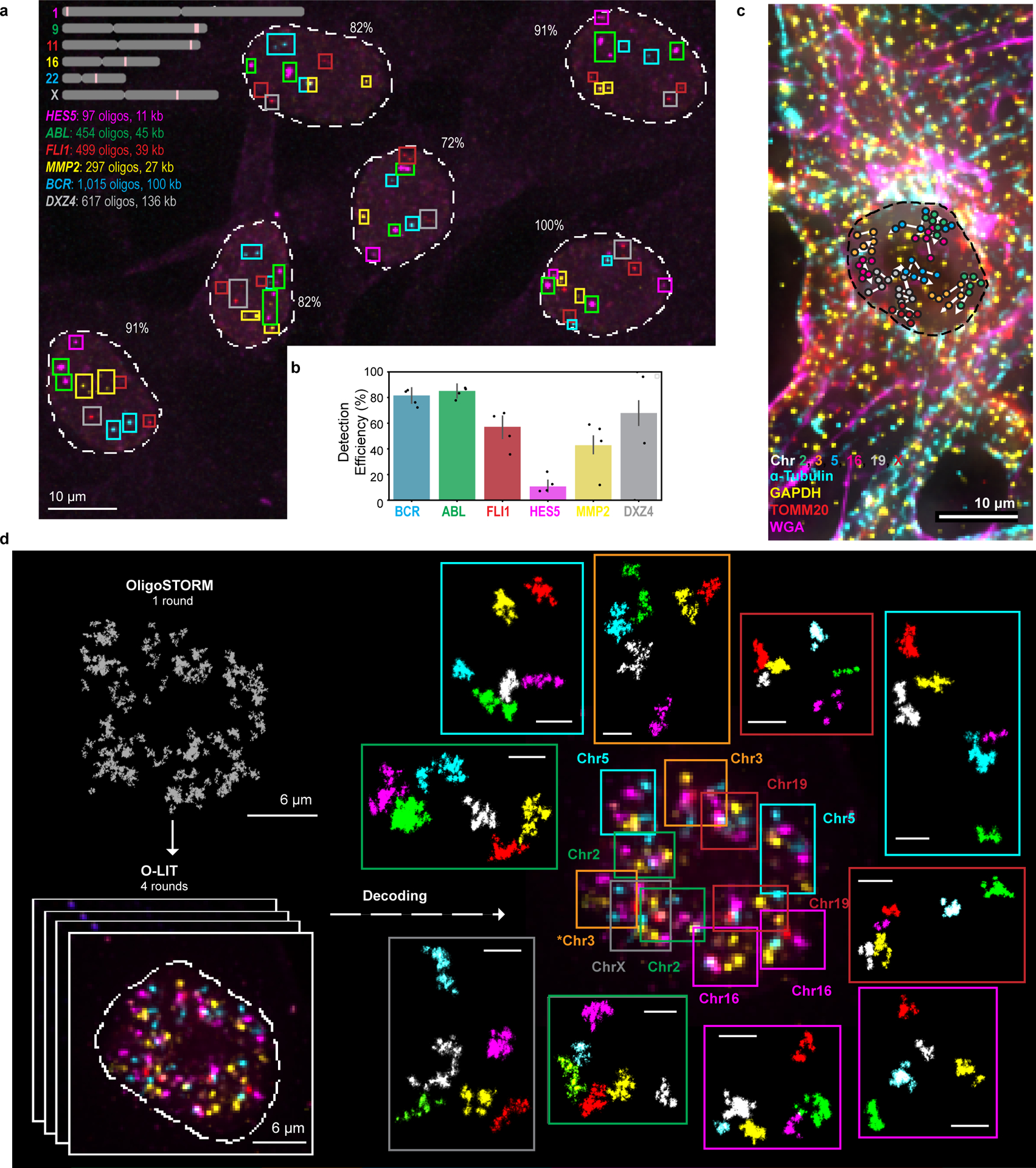
a) O-eLIT detection of single gene targets after sequencing off of both streets. Colored squares mark gene targets identified after 5 rounds of sequencing. Number reflects percentage of targets detected out of 11 (5 autosomal genes x 2 in addition to DXZ4 on the X). Image from deconvolved maximum intensity z-projection and representative of 2 replicates.
b) Tier 1 target detection efficiency from experiment in panel a (n = 61 from 2 replicates). Tier 2 inapplicable due to lack of targets from the same chromosome. Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
c) Combining O-LIT and immunofluorescence. 36plex-5K was sequenced for four rounds with O-LIT off of both streets, followed by immunofluorescence and staining with wheat germ agglutinin (WGA). Image from deconvolved maximum intensity z projection with chromosome traces overlaid. n = 1.
d) 36plex-5K was hybridized to PGP1f cells and imaged with 1 round of OligoSTORM (2 hours) to visualize all 66 targets simultaneously, followed by 4 rounds of O-LIT (2 – 3 hours per round) to decode targets. Top left, OligoSTORM image showing entire field of view with all unidentified targets. Bottom left, micrograph from first round of O-LIT; image from deconvolved maximum z-projection. Right, all 6 chromosomes identified (central image decorated with colored squares, color coded by chromosome as shown) and then arrayed, in super-resolution, around the central nucleus. All 66 targets excepting one region on Chr16, were detected and identified by O-LIT, with one homolog of Chr3 (*) not captured by OligoSTORM because it happened to fall outside the field of view. All scale bars for OligoSTORM images, 1 μm.
e) Each chromosomal region imaged with OligoSTORM from panel d is displayed separately; orientation may differ from that in panel e. n = 1.
Lastly, we demonstrated the capacity of OligoFISSEQ to improve the speed with which genomic regions can be imaged using single-molecule localization microscopy. Here, we focused on OligoSTORM5,10, which combines Oligopaints4 with Stochastic Optical Reconstruction Microscopy44 to provide super-resolution images of genomic regions in a space-filling fashion and thus reveal detailed volumetric structures5,7,13,14,16. The throughput of OligoSTORM, however, hovers at tens to a few hundred cells per experiment, and its imaging times can run in the range of 2 hours. In contrast, because OligoFISSEQ can be carried out with diffraction-limited microscopy, it has the capacity to image hundreds to thousands of cells per experiment, with relatively negligible imaging times. Thus, we explored the possibility of accelerating super-resolution genome imaging by combining O-LIT with OligoSTORM (Fig. 6d–e and Extended Data Fig. 10b).
First, using 36plex-5K and bridge oligos containing binding sites for secondary oligos labeled with a fluorophore suitable for OligoSTORM (Alexa Fluor 647), we captured all 66 targets simultaneously in a single 2-hour round of OligoSTORM (Fig. 6d; also Chr2–6plex, as shown in Extended Data Fig. 10b). Then, with only four rounds of O-LIT, we identified all 66 targets. Thus, by combining OligoSTORM with OligoFISSEQ, we enabled a 36-fold reduction in imaging time and data storage demands (from ~2.73 Terabytes to ~76 Gigabytes; Fig. 6d–e), while achieving 17 ± 5 nm lateral and 50 ± 10 nm axial precision and 40 ± 5 nm lateral and 60 ± 5 nm axial resolution. Extrapolating to all 46 chromosomes of a diploid human nucleus and anticipating many more than 6 targets per chromosome, this study demonstrates the feasibility of simultaneously OligoSTORMing hundreds of regions genome-wide. O-LIT should also permit OligoSTORM walking along the genome, with many walks per nucleus. Previously, we accomplished multi-walk imaging through temporal barcoding14. Here, multiple rounds of OligoSTORM would produce super-resolved walks in multiple regions of the genome, simultaneously, after which all regions would be identified with O-LIT. In sum, given the potential of O-LIT to identify hundreds to perhaps thousands of regions, OligoSTORM should scale similarly.
Discussion
There is a growing need for methods that will enable the imaging of entire genomes at high genomic and optical resolution while also supporting the levels of throughput and reproducibility that are becoming increasingly essential for understanding biological entities as dynamic as the genome. To this end, we have described proof-of-principle for OligoFISSEQ, a set of three methods for in situ genome mapping, demonstrating the potential of these methods to scale towards whole-genome imaging. OligoFISSEQ also has the capacity to meld with other technologies and thus extend its usefulness yet further. For example, combined with homolog-specific Oligopaints (HOPs)5, it should enable genome-wide studies in the context of parent-of-origin and, with adjustments to the barcodes, OligoFISSEQ could also enable multiplexed and/or multi-color visualization of chromosome folding in combination with other technologies, such as OligoDNA-PAINT5, Hi-M17, and optical reconstruction of chromatin architecture (ORCA)20. In terms of scaling, our capacity to map 46 regions on the X at ~1 genomic target per 2.75 Mb predicts that OligoFISSEQ should be able to accommodate a thousand or more targets in human nuclei, with the potential to increase that number through reduction in target size, temporal barcoding to better resolve targets, additional rounds of sequencing, and incorporation of expansion microscopy45; preliminary studies show that Chr19–9K can support 8 rounds of O-LIT (Extended Data Fig. 10c) and OligoFISSEQ to be feasible in the context of hydrogels (Extended Data Fig. 10d–e). Scaling could also be enhanced via microfluidics, which would significantly reduce the time required for each round of sequencing by 15–20%. Indeed, with the advent of improved enzymatics, methods for amplifying signal (e.g. SABER35 and ClampFISH36) and superior imaging, OligoFISSEQ should become applicable to the study of smaller targets, such as enhancers and promoters. As important will be improvements in image analysis. For example, implementation of point spread function fitting algorithms should improve spatial resolution and thus, scalability46, while a reduction in the dependence on proximity of signals to affirm true signal would permit better detection of chromosome rearrangements, where targets that are expected to be near each other are, instead, widely separated. Finally, OligoFISSEQ should interface beautifully with other FISSEQ-based technologies to achieve multi-omic views of the genome, with each round of sequencing visualizing DNA, RNA22,23, and protein47 simultaneously.
Finally, we note that, as OligoFISSEQ has the capacity for significant genome coverage as well as the potential to consistently identify the same targets across thousands of cells, it is well-suited for studying variability at a handful of regions as well as for addressing this challenging topic at the level of the entire genome. Structural variability of specific genomic features has now been widely observed7,9,11,13,14,16,17,20,21,48 and (recently reviewed by49) and, while it is often thought of locally, its impact may reach globally14. Even a minor, seemingly inconsequential change in one part of the nucleus may have a profound “butterfly effect”50 on the global scale, its impact potentially contributing to and/or propagating gene regulatory states and phase separations, perhaps even constituting essential, potentially heritable, signatures of the genome. Thus, although variability may appear random at the local level, a genome-wide perspective may reveal that apparent randomness actually reflects global responsiveness, an exquisitely controlled regulatory program that directs structural conformations across the entire nucleus, as much the outcome of evolution as any other honed genetic function.
Online Methods
Materials and Methods
List of reagents (including catalog numbers) can be found in Supplementary Table 1. List of oligo sequences can be found in Supplementary Tables 2–8. Information regarding each library (coordinates, barcodes, density, etc.) can be found in Supplementary Table 12.
Oligopaint library design
All Oligopaint oligo sequences and coordinates for libraries used in this study can be found in Supplementary Tables 2–6. Oligopaints4 leverages the ability to computationally design and synthesize sequence specific oligonucleotide probes for FISH4, also see Supplementary Note 1 for additional examples. Oligopaint FISH probes were computationally designed for optimal hybridization as well as high specificity. Oligopaint genome binding sequences were obtained from the Oligopaints website (https://oligopaints.hms.harvard.edu/)51, using hg19 genome and balanced settings. 129plex sequences were obtained using OligoMiner on soft-masked hg38 sequence using a Tm window of 42–47 °C and a length range of 30–37 nucleotides51. Genome homology sequences ranged from 35–41 nt. Universal forward and reverse priming sequences were appended to each Oligopaint oligo using OligoLEGO (https://github.com/gnir/OligoLego), allowing the libraries to be PCR amplified and renewable. The universal priming sequences also served as various OligoFISSEQ primer/bridge sites. Each library used in this study was designed with specific features and is described in detail in the supplemental file specific for each set.
Ligation based Interrogation of Targets
For Chr19–20K library, a portion of the universal forward priming sequence was used as the LIT primer binding site, followed by the LIT barcode. Barcode and color-code designation is as follows: 4 = Cy5/Alexa-647, 3 = TxRd, 2 = Cy3, 1 = FITC/Alexa-488.
36plex-5K library shared the same universal forward priming sequence among all oligos and contained chromosome specific universal reverse priming sequences. Individual chromosome targets could be amplified, hybridized, and detected by using the universal reverse priming sequence. Universal forward priming sequences were used as LIT primer binding site. LIT primers used are 18-nt. In cases where O-LIT was performed off of both Mainstreet and Backstreet, a LIT primer binding site was hybridized to the Backstreet. Barcodes were specified using sequences from OligoLego (https://github.com/gnir/OligoLego). Candidate barcodes sequences were decoded to reveal color-code using a MATLAB (https://www.mathworks.com/) script. To maintain color-code diversity between neighboring targets, barcodes were manually assigned to targets (e.g. barcodes were specified so that neighboring targets would have different colors in the first round). Each LIT barcode digit requires a 5-nt sequence, while the last barcode digit requires 8-nt to allow adequate space for 8mer binding. Thus, a 4-digit barcode requires 23-nt in total. For 36plex-5K, targets 3qR3 and 5pR2 contained the same barcode sequences to assess barcode recovery from separate genomic targets.
JEB/O-eLIT barcodes
For 36plex-1K, ChrX-23plex-Odd, ChrX-23plex-Even (ChrX-46plex-2K is a combination of ChX-23plex-Odd and ChrX-23plex-Even). 36plex-1K library targets a subregion of 36plex-5K targets, with 1,000 Oligopaint oligos per target instead of 5,000. Additionally, 36plex-1K targets contained JEB compatible barcode digits. 36plex-1K targets contained the same barcode digit color code as 36plex-5K, with target 5pR3 as the exception. 36plex-1K can only be sequenced using Mainstreet and not both streets.
ChrX-46plex library was designed to span the entire human X chromosome with 2,000 Oligopaint oligos per target. The library was divided into two sub-libraries (ChrX-23plex-odd and ChrX-23plex-even), with each sub-library targeting the odd (X1, X3, X5, etc.) or even targets (X2, X4, X6, etc.). Each sub-library contained the same universal forward priming sequences and different universal reverse priming sequences. ChrX-46plex barcodes contained JEB digits and were also manually assigned to maintain color-code diversity between neighboring targets. ChrX-46plex is compatible with sequencing off of both streets.
6 gene library shared the same universal forward priming sequence and different universal reverse priming sequences. Barcodes were manually specified using JEB digits. 6 gene library is compatible with sequencing off of both streets.
The 129plex genome-wide library aims at imaging each chromosome arm of the human genome using OligoFISSEQ. We selected the regions based on the density of Oligopaint oligonucleotides that could be targeted (average, 8.6 oligonucleotide targets per kb) and position on the chromosome arm. First, using a custom-curated R script, we used a sliding window of 100 kb along all chromosomes to calculate oligonucleotide target densities. Then, wherever possible, we selected three regions for each chromosome arm: one near the telomere, another near the centromere, and a third more centrally located, selecting regions where the density of oligonucleotide targets would be above 6 per kb. For some chromosome arms, we selected fewer than three regions owing to the constraints of oligonucleotide target density. Each region corresponded to a 5-digit barcode. The 129plex was sequenced off both streets. Due to 21 inadvertently duplicated barcodes, 42 of the targets could not be assigned (Supplementary Table 12).
Synthesis based Interrogation of Targets barcode
For Chr19–20K library, universal reverse priming sequence was used as SIT primer binding site, followed by SIT barcode sequence. Barcode and color-code designation is as follows: 4 = Cy5, 3 = Cy5 + Cy3, 2 = Cy3, 1 = blank.
For 36plex-1K, universal reverse priming sequence was used as SIT primer binding site, followed by SIT barcodes. Target color-code was designed to be the same as 36plex-5K but with SIT reagents.
Hybridization based Interrogation of Targets barcode
For Chr19–20K library, bridging oligos (HIT bridge) were designed to hybridize to Mainstreet and Backstreet. HIT bridges contained binding sites for HIT readout oligos. HIT readout oligo sequences were derived from OligoLego. Barcode and color-code designation is as follows: 0 = blank, 1 = Alexa 647/Cy5, 2 = Cy3B/Cy3, 3 = FAM/Alexa 488.
For 36plex-5K library, HIT bridges were designed to hybridize to street specific sequences for each target. This was done by designing bridges flanking universal priming sites (forward and reverse) as well as 5’ or 3’ end of LIT barcodes, due to similar LIT barcodes being present on both streets. HIT bridges contained binding sites for HIT readout oligos derived from OligoLego.
Oligopaint probe synthesis
Oligopaint oligos were purchased as single stranded Oligopools from CustomArray (http://www.customarrayinc.com/oligos_main.htm) or Twist Biosciences (https://www.twistbioscience.com/) in 12K and 92K chip formats. Oligopools were amplified as previously described10,14 with minor modifications. Step by step protocol can be found in Supplementary Protocol 1. Briefly, PCR conditions for each library and sub-library were optimized using real-time PCR to obtain optimal template concentration, primer concentration, and annealing temperature. Next, libraries were linearly amplified with low-cycle PCR using Kapa Taq reagents. dsDNA PCR products were purified using Zymo columns and eluted with ultra-pure water (UPW). T7 RNA promoter sequence was then appended to Oligopaints using REV primers containing the T7RNAP on the 5’ end. Note that some users may opt to add the T7RNAP straight from the raw library. dsDNA PCR products were purified using Zymo columns and eluted with UPW. PCR products were then in-vitro transcribed using NEB HiScribe (NEB E2040S) overnight at 37oC to make RNA.
RNA products were reverse transcribed with Thermo Maxima H Minus RT (Thermo Fisher, EP0753) to make cDNA. RNA was then digested to leave ssDNA. This product was purified using Zymo columns. Final ssDNA Oligopaint oligos were resuspended at 100 μM in UPW and stored at −20oC until use. Linear PCR, touched-up PCR, and ssDNA Oligopaint oligos were quality checked by running on 2% Agarose DNA gels to confirm single bands migrating at expected sizes during synthesis.
Other oligonucleotides
Sequences for all other oligos can be found in Supplementary Tables 7–8. Primers, secondary fluorophore labeled oligos, LIT sequencing primers, SIT sequencing primers, JEB oligos, and MIPs were purchased from IDT (https://www.idtdna.com/). HIT secondary oligos were purchased from Biosynthesis (https://www.biosyn.com/). Alexa405 activator fluorophore was purchased from Thermo Fisher (https://www.thermofisher.com/).
Cell culture
Our study used two human cell lines: PGP1f and IMR-90. PGP1f are primary human fibroblast from male donor PGP1 (Coriell; GM23248)52. They were previously found to be of normal karyotype14,53. PGP1f were cultured in DMEM (Gibco) supplemented with 10% Fetal Bovine Serum (Thermo Fisher; A3160401), 1X Penicillin-Streptomycin (Thermo Fisher, 15140122), and 1X Non-essential amino acids (Thermo Fisher, 11140050). PGP1f cells were cultured for no more than 5 passages before thawing new cultures. IMR-90 were cultured in DMEM supplemented with 10% Fetal Bovine Serum, 1X Penicillin-Streptomycin, Cells were cultured in 37oC incubator at 5% CO2.
Sample preparation for OligoFISSEQ
Ibidi Sticky Slide VI (https://ibidi.com/, 80608) were used for all experiments except for metaphase spreads (Fig. 2b) and hydrogel (Extended Data Fig. 10d–e). Ibidi slides were assembled and allowed to cure overnight at 37oC prior to use. Each well requires 100–200 μL of reagent and we generally designated one hole as the inlet and the other as the outlet. PGP1f cells from ~70% confluent 10cm dishes were detached from dishes using 1 mL trypsin (Thermo Fisher, 25–200-056), neutralized with 2–3 mL fresh media. 100 μL of cells in suspension were added to each Ibidi well and allowed to adhere and recover overnight at 37oC incubator. The following day, media was aspirated, and wells were washed with 1X PBS and fixed for 10 min with 4% formaldehyde (Electron Microscopy Sciences, 15710) in final concentration of 1X PBS (Thermo Fisher, 10010–023). Fixative was removed and cells were rinsed with 1XPBS. Cells were then permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, T8787–250ML) in 1X PBS final for 15 min on a rotator. Permeabilization reagent was aspirated and cells were rinsed in 0.1% triton/ 1X PBS and stored in this or PBS at 4oC until use. Samples were used within 2–3 weeks after fixation.
Cell samples for MIP/hydrogel experiments were grown on rectangular glass microscope slides. Cells were plated similarly to Ibidi, except 150 μL of cells in suspension were plated onto discrete areas on rectangular slides (previously etched with glass etching pen (to note the region) and incubated overnight at 37oC incubator in 10 cm petri dish. The following day, the same steps as with Ibidi above were performed but in 50 mL coplin jars. Cells were stored in 1XPBT in coplin jars until use.
Metaphase spreads were purchased from Applied Genetics (Product: HMM).
DNA FISH
Step by step protocols can be found in Supplementary Protocols 2–3 and is adapted from4 and based on54,55. All OligoFISSEQ methods begin with hybridization of primary Oligopaint libraries overnight and then deviate. Common to LIT, SIT, and HIT with Ibidi slides (all steps done on rotator unless specified): Ibidi wells washed with 0.1% PBT at RT for 5 minutes and incubated with 0.1 N HCl for 8 minutes. 2XSSCT washes were performed. Cellular RNA was digested with 50 μL of 2 ug/mL RNAseA (Thermo Fisher, EN0531) in 2XSSCT for each well. Slide was incubated in 37oC humid chamber for 1 hour. RNAseA was washed out by adding 2XSSCT. Pre-hybridization began by adding 50% formamide/ 2XSSCT for 10 min at RT. Pre-hybridization continued with prewarmed (60oC) 50% formamide/ 2XSSCT being added and by placing the slide on top of heat block set in 60oC water bath for 20 min. Next, primary Oligopaint library was added, the samples were aspirated and 50 μL total of primary Oligopaint oligo library (2 uM final) were added in hybridization mix (50% formamide, 2XSSCT, 10% Dextran Sulfate). Samples with primary Oligopaint oligo libraries were then denatured, wells were sealed with parafilm to prevent evaporation and slide was placed on pre-heated hot block in 80oC water bath for 3 minutes under the weight of a rubber plug. Oligopaint oligo library hybridization to samples was performed by placing samples in humid chamber at 42oC incubator for >16 hours. The next day, probes that did not hybridize were washed out by adding prewarmed (60oC) 2XSSCT directly to each well containing primary hybe mix and aspirated. New prewarmed 2XSSCT was added and samples were incubated on hot block for 15 min. This was repeated one time and then another time at RT. After this wash is where the protocol deviates for the techniques (see below). Note that cellular DNA was stained after every 2 rounds of sequencing to maintain adequate DAPI signal.
For detection of Oligopaints via secondary hybridization, samples were then prepared for secondary oligo hybridization to primary oligo streets for detection. Samples were washed with 30% formamide/2XSSCT for 8 min and 50 μL total of secondary oligos and/or bridge oligos were added at 1.2 uM in 30% formamide/2XSSCT to each well. Samples were incubated in humid chamber for 45 min at RT dark. Non hybridized secondary oligos were washed out with 30% formamide/2XSSCT being added directly in, aspirated, and incubated 2× 15min on rotator. Samples were washed with 2XSSCT two times 5 min. In some experiments, DNA was counterstained with DAPI (Thermo Fisher, D1306) in PBS for 10 min. Samples were then washed with 1XPBS x2 5 min and imaged in 1XPBS or imaging buffer containing.
For cells on rectangular slides, same overall protocol as above was performed but in coplin jars, scaling wash volumes accordingly (25 μL volumes for primary and secondary hybridizations). The protocol is modified as follows: RNAse was added directly to cells on rectangular slide and covered with 22×22mm coverslip. Post RNAse washes were performed by transferring slide and coverslip to coplin jar and “sliding” the cover slip off. Same approach was performed for secondary hybridization. Primary Oligopaint hybridization was performed by adding primary Oligopaint mix directly to cells on rectangular slide, covering with 22×22mm coverslip, and sealing edges with rubber cement (Elmer’s). Rubber cement was allowed to dry for 3 min and sample was denatured on heat block, similar to Ibidi.
Ligation based Interrogation of Targets (LIT)
LIT is built upon Oligopaint4, SBL56, and FISSEQ technologies22,23,57 and also see Supplementary Note 2 for recent iterations. Step by step protocol can be found in Supplementary Protocol 3. After hybridization of primary Oligopaint library, for O-LIT, samples required treatment with phosphatase to deplete endogenous phosphates that could prime ligation, contributing to background and poor signal. The samples were washed with 50 μL of 1X NEB CutSmart buffer for 8 min. Next, 50μL of shrimp alkaline phosphatase (rSAP; NEB, M0371L) (7.5 μL rSAP in 1X CutSmart) was added to each well and incubated at 37oC humid for 1 hour. To inactivate phosphatase, sample was then transferred to pre-heated heat block in 65oC water batch for 5 min, and washed x2 with preheated (65oC) 2XSSCT on heat block for 5 min each. RT 2XSSCT was added for 5 min. Samples were then prepared for LIT primer binding by washing with 30% formamide/2XSSCT for 8 min and 50 μL total of LIT sequencing primer was added at 1.2 uM in 30% formamide/2XSSCT to each well. Samples were incubated in humid chambers for 45 min. Non-hybridized LIT primers were washed out with 30% formamide/2XSSCT being washed directly in, aspirated, and incubated 2× 15min on rotator. Samples were washed with 2XSSCT two times 5 min. Next, samples were prepared for first round of LIT by adding 100 μL of 1X Quick Ligation buffer (NEB, B6058S) for 8 min and aspirated. LIT reaction mix (recipe in Supplementary Protocol 3) was prepared on ice. Before adding ligases (added last), vigorous vortexing was performed on the LIT reaction mix. After vortexing, ligases were added and mixed thoroughly by pipetting. O-eLIT reagent was performed similarly but instead of SOLiD purple reagent mix, 40 pmol of each JEB oligo was added and UPW was adjusted accordingly. 100 μL of this mix was added to each well and samples were incubated in humid chamber at 25oC for 55 min. LIT reaction mix was then aspirated and samples were rinsed with 1M Guanadine Hydrochloride (Sigma-Aldrich, G3273), and washed x2 15 min on nutator at RT. 1XPBS wash 5 min was performed. Cellular background fluorescence was reduced by treating the samples with 100 μL True Black (Biotum, 23007) in 70% EtOH for 2 min. 3× 1XPBS quick rinses, and then 10 min wash was performed. Samples were then imaged in 1XPBS or imaging buffer (see Doc_S3 for recipe). Before proceeding to the next LIT round, non-ligated phosphates are treated with phosphatase (Quick CIP; NEB, M0508L) for 30 min at 37oC. Quick CIP is then washed out with 3x GHCL washes 5 min. Previous LIT round is cleaved to release fluorophore and regenerate 5’ PO4 by rinsing and then 15 min incubation at RT rotator with Cleave 1, and then the same for Cleave 2. Samples are then rinsed x3 with GHCL and washed x2 5 min. The next round of LIT can proceed with the pre-ligation step. After the last barcode digit is read, the fluorophore can be cleaved, and all targets can be detected by hybridizing specific bridges and fluorophores as in DNA FISH method.
Synthesis based Interrogation of Targets (SIT)
SIT is based upon Oligopaint4 and SBS58 technologies using Illumina NextSeq 500/550 TG Kit (Illumina, TG-160–2002). After hybridization of primary Oligopaint library, samples were then prepared for SIT primer binding by washing with 30% formamide/2XSSCT for 8 min and 50 μL total of LIT sequencing primer was added at 1.2 uM in 30% formamide/2XSSCT to each well. Samples were incubated in humid chambers for 45 min. Non-hybridized SIT primers were washed out with 30% formamide/2XSSCT being washed directly in, aspirated, and incubated 2× 15min on rotator. Samples were washed with 2XSSCT two times 5 min. First round of SIT proceeds by rinse with 100 μL of pre-warmed (60oC) NextSeq polymerase solution (from reservoir 31), and then incubation on 60oC heat block in water bath for 5 min. Sample is aspirated and washed with 2XSSCT x3 10 min. 1XPBS wash was performed and samples were imaged in 1XPBS or imaging buffer. Before proceeding onto the next SIT round, sample is treated with NextSeq cleave solution (from reservoir 29) with a rinse, then 5 min incubation on 60oC heat block in water bath. Sample is then washed 3× 10min in 2XSSCT. The next round of SIT can now proceed. For all target identification, SIT primers containing an Alexa488 can be used or secondary oligos with bridges can be added.
Hybridization based Interrogation of Targets (HIT)
HIT is based on Oligopaint4 and SBH technologies6,12,59. After hybridization of primary Oligopaint library, samples were then prepared for HIT bridge oligo hybridization to primary oligo streets for detection. HIT bridges for 36plex-5K were designed to span the universal priming region and part of either the Mainstreet barcode or Backstreet barcode. Samples were washed with 30% formamide/2XSSCT for 8 min and 50 μL total of bridge oligos were added at 1.2 uM in 30% formamide/2XSSCT to each well. Samples were incubated in humid chamber for 45 min at RT dark. Non hybridized bridge oligos were washed out with 30% formamide/2XSSCT being added directly in, aspirated, and incubated 2× 15min on rotator. The first round of HIT proceeds by addition of 50 μL to each well with round specific HIT secondary oligos at 1.2 uM of each in 30% formamide/2XSSCT for 45 min at RT dark humid chamber. Non hybridized HIT secondary oligos were washed out with 30% formamide/2XSSCT being added directly in, aspirated, and incubated 2× 15min on rotator. Samples were washed with 2XSSCT x2 5min and then 1XPBS for 5 min. Samples were imaged in 1XPBS or imaging buffer. Before proceeding to the next round, previous HIT round secondary oligo fluorophores are cleaved via rinse and incubation for 15 min with 1 mM TCEP (Sigma-Aldrich, 646547–10X1ML). 3x PBS rinse was performed and the next HIT round can proceed.
Immunofluorescence
To visualize proteins, samples were subjected to immunofluorescence. After OligoFISSEQ, Oligopaint oligos were removed by washing with 80% formamide/2XSSCT 2× 7 min. Next, samples were washed with 2XSSCT for 3 min, rinsed with 1X PBS and fixed in 4% Formaldehyde/PBS for 10 min. After PBS rinses and permeabilization in 0.5% Triton/PBS for 10 min, samples were blocked in 3% BSA/PBT for 1 hr. Primary antibodies diluted in 1% BSA/PBT were then added to each well, sealed with parafilm, and incubated O/N at 4oC for > 12 hrs. The next day, primary antibody was removed and 3x PBT washes were performed. Secondary antibodies (Supplementary Table 1) diluted in 1% BSA/PBT were then added at 1:500 dilution for each for 1 hr at R/T shaker. Wheat Germ Agglutinin (WGA, Thermo-Fisher, W11261) (1:20) could also be added during the 2o incubation step. 3x PBT washes for 5 min each were performed, and samples were restained with DAPI (1:1000) for 10 min and imaged in imaging buffer.
Hydrogel
Hydrogel embedding was based on60. Step by step protocol can be found in Supplementary Protocol 4. Cells for hydrogel embedding were grown on rectangular glass slides. FISH was performed on these slides as described in “DNA FISH” section. After primary Oligopaint library hybridization, samples were washed in 60oC 2XSSCT for 20 min, followed by a 10 min wash at RT, then 1XPBS for 5 min. In preparation for hydrogel embedding, slides were air dried for 5 min and area around cells was wiped dry with Kim wipe. Hydrogel reagents were combined in Eppendorf tubes on ice and degassed on ice in vacuum chamber (Thermo Fisher, 08–642-7) during incubations. Cells were then washed for 10 min at 4oC with hydrogel mix without APS/TEMED. Hydrogel mix was then removed from sample and ~20 uL of hydrogel solution (recipe in Supplementary Protocol 4) was spotted onto parafilm on gelation chamber slide (rectangular slide wrapped in parafilm, using 2 22×22mm coverslips as spacers on each end of the slide), slide sample was then flipped onto hydrogel solution/gelation chamber, being careful to spread the hydrogel solution without forming bubbles. Sample was then incubated at 37°C for 1 hr in vacuum chamber. After incubation, gelation chamber was carefully removed. Edges of hydrogel disc were trimmed, and diamond etching pen was used to break rectangular slide, preserving the gel/glass slide portion. Gel/glass slide portion was then transferred to 35 mm petri dish and digested in 2 mL digestion buffer (recipe Supplementary Protocol 4 from60) O/N at 37oC. After O/N digestion, cell/hydrogel dissociates from the glass slide so extra care should be taken to avoid hydrogel damage. Digestion buffer and glass slide is removed, and hydrogel is washed in 2XSSC for 3× 20 min. The hydrogel can be divided into smaller pieces for downstream applications. To note orientation, hydrogel pieces can be cut into distinct shapes, which will make imaging and alignment easier downstream. After cutting, the hydrogel sample can be transferred to 1.5 mL Eppendorf tubes for easier handling.
Metaphase FISH
Steps were performed using coplin jars except for where noted. RNAseA treatment was performed by adding 25 μL and sandwiching under 22×22mm coverslip and incubated in humid chamber. Primary Oligopaint hybridization was performed the same way.
Diffraction-Limited Microscopy
OligoFISSEQ and diffraction limited microscopy was carried out using a widefield epifluorescence setup. A Nikon Eclipse Ti body was equipped with a Nikon 60× 1.4NA Plan Apo lambda (Nikon MRD01605) objective lens, Andor iXon Ultra EMCCD camera (DU-897U: 512 × 512 pixel FOV, 16 μm pixel size), X-Cite 120 LED Boost light source, motorized stage, and off the shelf filter sets from Chroma (~488nm 49308 C191880, ~532 nm 49309 C191881, ~594nm 49310 C191882, ~647nm 49009 C177216). Images were obtained with ND4 and ND8 filters in place. Microscope operation was handled by Nikon NIS elements software. In general, z-stacks were obtained with 0.3 μm slices with 2–300 ms exposure time and 20–60% LED intensity, depending on library being imaged. XYZ stage position was maintained within .nd2 metadata and was essential for returning to the same FOV. Orientation of sample into the stage and sample holder was carefully maintained as to enable returning to the same FOV. This was important, as the sample was removed after imaging and between sequencing rounds.
OligoSTORM imaging
In order to combine OligoFISSEQ with OligoSTORM, we first performed one round of OligoSTORM imaging on all the targets (Chr2–6plex or 36plex-5K) inside a PGP1f male fibroblast cells by hybridizing Alexa 647 labeled secondary oligos that binds to the bridges (present in the backstreet of individual Oligopaint oligos, each chromosome containing specific barcodes) containing a binding site for secondary oligos. OligoSTORM samples were imaged on a Vutara 352 biplane system with an Olympus 60× 1.3NA Silicone objective (UPLSAPO60XS2). For single molecule blinking, we used a switching buffer containing 2-Mercaptoethanol and GLOXY14. The excitation laser power was set at 60% on the software (6.3 kW/cm2 at the objective) for the 640 nm laser and 0.5% on the software (0.08 kW/cm2 at the objective) for the photoactivation laser of 405 nm. We have used 30–40 Z slices of 0.1 μm thickness for each Z slice. 10–12 photoswitching cycles of 250 frames per cycle was used for each Z slice.
The OligoSTORM images were analyzed using Vutara SRX software14. DBSCAN clustering algorithm was used to identify the clusters from the raw image. 50 particles within a 0.1 μm distance was used for clustering. The mean axial precision was 50 +/− 10 nm in Z and mean radial precision was 17+\- 5 nm in XY. The resolution of the super-resolved structures were calculated by Fourier ring correlation analysis (a built-up feature in SRX software). Resolution in XY was 40 +/−5 nm and resolution in Z was 60 +/−5 nm.
Data visualization
Images were processed using either Nikon Elements or ImageJ/FIJI61. .nd2 images were imported using Bio-formats plugin62. Figure 2d was generated using ImageJ (Plugins > 3D Viewer)63. Chromosome schematics were generated using ChromoMap64. Figures were assembled in Adobe Illustrator. Micrograph images for publication figures were post-processed using Brightness and contrast enhancement (ImageJ > Image > Adjust > Brightness/Contrast). Prism by GraphPad was also used for graphs. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM10331165.
Tier 1 detection
Preprocessing.
Each round of OligoFISSEQ is imaged using 5 channels: Alexa 647, Texas Red, Cy3, Alexa 488 and DAPI and a series of z-slices. The z-stacks are deconvolved and background corrected using 20 iterations of the Richardson-Lucy algorithm using a theoretically calculated point spread function with Nikon software66.
Rounds are compiled into hyperstacks composed by the 5 channels, a series of z-slices and one frame per round. If an image where all the puncta are labelled, like in toto image, is available it is included as a new additional frame. The hyperstacks are aligned using Fiji plugin “Correct 3D Drift”67. Images of DAPI stained nuclei are used to perform threshold segmentation and extract each individual cell from the initial image as a separate region of interest (ROI). The segmentation provides information about location and envelope of individual nuclei that compose each hyperstack. Nuclei with areas below 25 μm2 are discarded.
Detection of barcodes.
To compare intensities from different channels images are normalized by dividing its intensities by the maximum intensity among the values of all the z-slices in the same round and channel.
For the detection of barcodes and for each round the intensities of every-pixel position is compared across different channels. A centroid based pipeline using TrackMate68, did not perform as well in our hands, thus we moved forward with this every-pixel approach. The channel with the highest value is kept as the prevalent. At every-pixel position the transition between channels along the different rounds is compared with the list of expected barcodes. A barcode is assigned to a pixel position if the set of transitions coincides with the one associated to the barcode. A maximum intensity projection (MiP) image is built by averaging the intensities of the prevalent channels of every round. Connected pixels having the same barcode are grouped to form 3D patches. The following information is collected and saved for each patch:
Barcode
Center position
Number of pixels forming part of the patch (size)
Maximum intensity of the pixels of the patch
Pixel position having the maximum intensity of the pixels of the patch
If we have an image with all puncta labelled the information of the intensity of each pixel position is stored in an additional file.
Tier 2 detection
Chromosome tracing.
Patches composed by a single pixel location are discarded. The rest of the patches are used in the tracing disregarding its intensity or size.
Patches with high intensity values are selected as the most confident and used to find the chromosome centers. We use an implementation of the Constrained K-means algorithm69 to find the center of the set of barcodes belonging to the same chromosome. To separate the homologs, we use a cannot-link constraint in the two copies of the same regions to avoid having them in the same cluster. We use a sphere of radius 4.5 μm with origin in the centers to delimit the chromosome territory and filter out patches located outside.
The Domino sampler of the Integrative Modelling Platform70 is the core element of the chromosome tracing. In Domino each locus is represented by a particle with a finite set of different possible locations in the image. The locations are extracted from the list of patches having the same barcode as the one assigned to the locus. The remaining factors of the proposed problem are encoded in the system as restraints to the list of possible solutions. The following restraints are imposed to the system to filter compatible solutions:
Two particles cannot share the same location/patch
Two consecutive particles of the same chromosome should be closer than a distance of 4 μm for 36plex dataset and 1 μm for ChrX-46plex.
Chromosomes must be confined in territories modelled as spheres of radius 4.5 μm
Chromosome territory and distance between consecutive regions are inferred as explained in section Inferring chromosome territory and maximum distance between consecutive regions. By applying these additional constraints to the barcodes, we are able to use patches having intensities that are below but not far from the detection thresholds (Supplementary Table 14) and are likely to be true positives. Patches with higher intensities and sizes are most likely to be true positive regions. Therefore, a score based on intensity and size is assigned to each patch as a measure of the likelihood of the patch to be a true positive detection. The list of patches is sorted by score and used as input data to an iterative process to find the most probable path of each chromosome (Supplementary Figure 3).
The iterative process of tracing the chromosomes starts by assigning patches with high score to the corresponding regions. The process is executed one time per chromosome considering all homologs at the same time because barcodes are not designed to distinguish them. Domino is used to list all possible solutions that are compatible with the imposed restraints. Each solution has a total score obtained by the addition of the scores of the individual patches selected in that particular solution. We select the conformation having the highest total score. In case two or more solutions yield an identical total score, we select the solution which conforms the shortest chromosome spatial length. Regions assignment is done in an iterative process by lowering the threshold to use more patches as input and use the previous approach to select the remaining unassigned regions. This iterative process finishes when all regions have been identified or there is no more input data to feed Domino.
Detection efficiency and False Positives ratios
To calculate the detection efficiency per barcode the datasets are filtered using intensity thresholds (Supplementary Table 14) that are optimized for every experimental condition. Patches formed by one single pixel are also discarded regardless of its intensity.
For 36plex datasets we calculated the mean of barcodes detected per nuclei excluding the ones assigned to the X chromosome. In the ideal case and due to the ploidy, we expect two barcodes per nucleus. In reality the datasets may eventually include false positives or duplicates of patches that are probably belonging to the same oligo which will rise the ratio. Nucleus with a mean of more than 2.5 barcodes are discarded because they are most likely in a mitotic process. For the ChrX-46plex we followed a similar procedure and discarded nuclei which mean of detected barcodes was higher than 1.5.
For each of the remaining nuclei we compute the ratio of detected versus expected barcodes. We expect two barcodes per cell except for the barcodes belonging to the chromosome X. The ratios per barcode and per cell are cap to 1.0 and averaged over all the cells to produce the detection efficiency. For the False Positive ratio of the barcode, instead, we calculate the excess of detections as the detected minus the expected value in the cases where detected is over expected, and then compute the ratio excess versus expected.
Distance heat-maps and Hi-C maps
For every traced nucleus, we calculated all pairwise distances between the detected regions and averaged the result among all cells. For the average heatmap of 36plex-5K LIT dataset regions 3qR3 and 5pR3 were not taken into consideration because they shared the same barcode and were therefore indistinguishable. Hi-C maps of PGP1f cells were obtained from previous in situ Hi-C experiments14. The values of the interaction frequencies in the included Hi-C maps were extracted from observed values of interaction matrices produced at 5Kbp resolution. The submatrices formed by the genomic regions of each pair of probes were aggregated to obtain the inter-regions observed interaction. Single cell heatmaps were built with the identification of homologous chromosomes. The list of barcodes are traced according to the procedure described in the methodology. Then all pairwise distances of the traced regions are calculated. Not identified regions appear as grayed columns/rows.
Inferring chromosome territory and maximum distance between consecutive regions
To infer the maximum distance between consecutive regions used in the chromosome tracing the list of detected barcodes for all 36plex datasets was filtered to discard mitotic cells as explained in the Detection efficiency section. Patches formed by one single pixel were also filtered out. After the filtering process 36plex dataset was composed of 1,171 nuclei and 48,352 barcodes. Then we calculated the distances between consecutive regions for each chromosome in each nucleus (Supplementary Figure 1). The plots of the histograms of those distances shows the expected bimodal distributions for the chromosomes except for the chromosome X as foreseen from male cells. Bimodality is more evident in bigger chromosomes because those tend to be in the periphery of the nucleus while smaller chromosomes prefer the interior.
After the inspection of the histograms we selected 4 μm as a general maximum distance between consecutive regions and a slightly higher value of 4.5 μm for the chromosome territory.
In the case of ChrX-46plex we followed a similar approach. After the filtering process the ChrX-46plex dataset contained 189 nuclei and 7752 barcodes. Based on the histograms of distances between consecutive regions we selected 2.5 μm as a general maximum distance (Supplementary Figure 2).
Clustering of 3D structures for ChrX-46plex
After tier 2 detection we had 177 cells for the chrX-46plex library with an average of 34 detected regions. We discarded one of the cells having less than 23 identified barcodes as to require at least 50% detection efficiency per cell in all the 3D structures. Then we calculated for each chromosome the pairwise distances between all of their detected targets and use those as a measure of similarity to built distance matrix. We use the coincident distances between structures to cluster them hierarchically using the Ward method.
The Calinski-Harabasz criterion for clustering evaluation was used to evaluate the optimal number of clusters.
ChrX-46plex-2K tracing in IMR-90 cells
O-eLIT with ChrX-46plex-2K library in IMR-90 cells was performed as in PGP1f. Five rounds of sequencing was performed off both streets followed by immunostaining for MacroH2A.1 (Abcam: ab183041) at 1:250 dilution to mark the inactive X chromosome. For every-pixel analysis, Chromosome traces with less than 13 identified regions were filtered out.
MacroH2A.1 IF images were aligned and segmented with the DAPI channel of their OligoFISSEQ correspondence. For each nucleus the position of maximum intensity of the IF image was compared with the geometric center of the traced X chromosomes. In order to filter out images without a clear IF signal we only consider nuclei where their maximum IF intensity was greater than two times the average intensity inside. If such center is closer than 3.5 um the X chromosome is considered IF positive and annotated as inactive (Xi). The other X chromosome in the nucleus is annotated as active. (Xa) In the cases where both homologs are closer
to 3.5 um to the IF signal, the closest is annotated as Xi and the farthest as Xa. The nuclei were manually checked to discard errors mainly due to overlapping cells ending up with 40 Xi chromosomes for which we could trace 31 homologs that were identified as Xa.
Generation of random nuclei for haploid separation
For the Directed random nuclei, we first calculated the average and standard deviation of the distance to the nuclear envelope of every chromosome (Supplementary Table 15). We used the information to generate a set of random nuclei where the chromosomes are randomly placed following a normal distribution which mean and standard deviation are equal to the calculated in the observed data. The positions of the large chromosomes in the synthetic nuclei will be biased towards the periphery while the positions of small chromosomes to the interior.
No spatial bias was used for the completely random nuclei.
Histogram of split homologs by K-means
For the analysis we selected 258 nuclei for which all centers of the 11 chromosomes are known. We use the conventional K-means algorithm to cluster the positions of the chromosomes in two groups and report how many autosomes are split by the clustering, that is how many autosomes have one copy in one of the group and the homolog in the other group.
Method for the alignment of the nuclei
For the analysis we selected 258 nuclei for which all centers of the 11 chromosomes are known. We use an implementation of the Constrained K-means algorithm69 to cluster the chromosomes in two groups: one will contain one copy of each autosome and the other group will contain the homolog. The X chromosome will be assigned to the closest group. The geometric centers of the clusters are joined and the resulting segment together with all the positions of the chromosomes rotated as to be parallel to the x axis and moved to leave the middle point to the origin x=0, y=0. In the rotation of the nuclei we leave the group containing the X chromosome at the left of the y axis.
Density plots
The density plots are built using the Kernel density estimation (KDE) of the projection to the x-y plane of the position of the chromosomes.
Number of split homologs
We checked each aligned nucleus and report how many autosomes can be split by a virtual line along the y axis, that is how many autosomes present one of the copies in left of the y axis and the other in the right.
Number of split homologs left to right
We checked each aligned nucleus and report how many autosomes can be split by a virtual line parallel to the y axis at different distances from the origin, that is how many autosomes present one of the copies in left of the line and the other in the right.
Datasets
Information regarding all datasets (cells, replicates, filters, etc.) can be found in Supplementary Table 9.
Data and materials availability:
All data is available in the main text or the supplementary materials, all other data are available in supplementary materials, and other materials are available upon request.
Code availability:
All code is available at https://github.com/3DGenomes/OligoFISSEQ
Supplementary Material
Extended Data
Extended Data Fig. 1. Chr19–20K and 36plex-5K-O-LIT optimization.
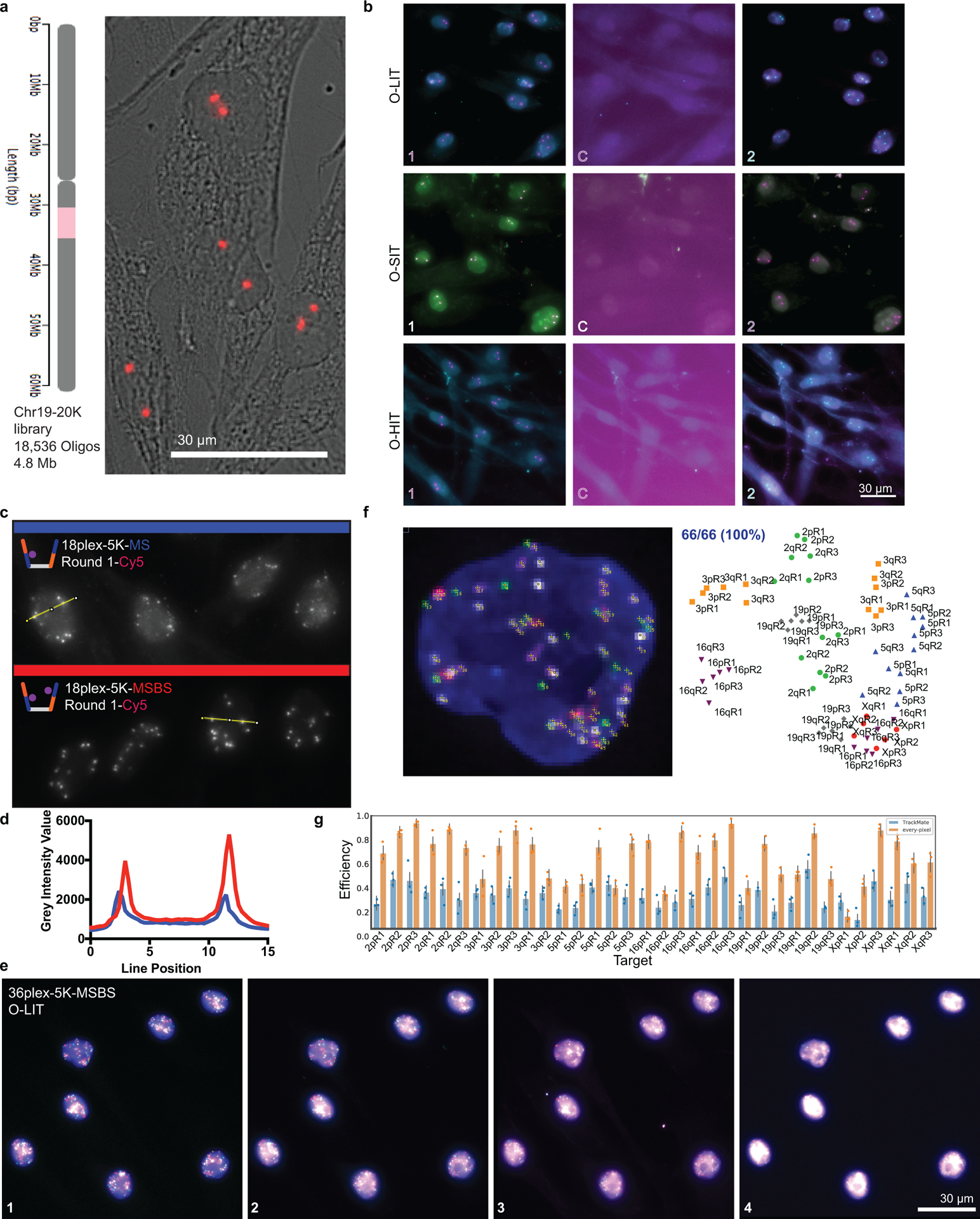
a) Chr19–20K targets 18,536 Oligopaint oligos to human chromosome 19. Right, Chr19–20K detection with secondary oligo (red) in PGP1f cells representative of 5 replicates.
b) Signal is completely removed in each OligoFISSEQ method after cleavage. Images showing two rounds of sequencing with a cleavage step (C) and representative of 4 replicates.
c) 36plex-5K O-LIT off of both Mainstreet and Backstreet (MSBS; bottom, red) produces stronger signal than off of Mainstreet (MS; top, blue). Cy5 channel from first round of O-LIT. n = 1.
d) O-LIT off of both streets produces stronger signal than off of MS. Grey intensity value measurements from yellow lines in panel c. n = 1.
e) Raw, non-deconvolved field of view of cell from Figures 2c–d and 3a–c. Maximum z-projection. n = 1.
f) Manual decoding of cell from panel c and Figures 2c–d and 3a–c yields 100% target recovery. n = 1.
g) Tier 1 detection efficiency after 36plex-5K O-LIT off of both streets and detected with TrackMate (blue, 29.93 ± 4.9%) or Every-pixel (orange, 62.8% ± 4.8%). n = 111 cells from 3 replicates. Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
Extended Data Fig. 2. Detection efficiency after 36plex-5K O-LIT.
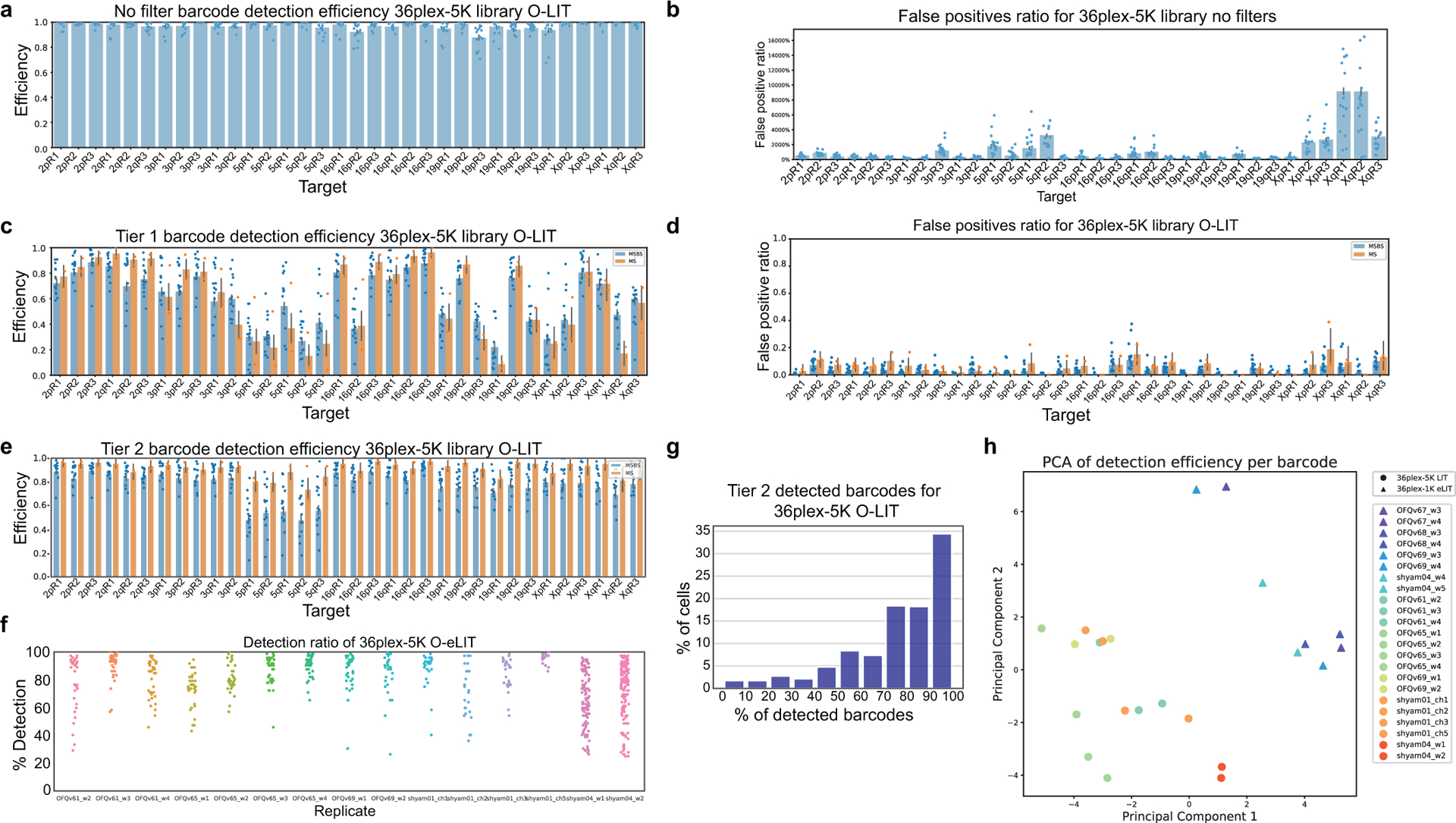
a) Detection efficiency without filtering after 36plex-5K O-LIT off of both streets. 95 ± 5.15% of targets are detected (n = 611 from 15 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
b) False positive (FP) discovery rate from panel a. FP discovery rate from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
c) Tier 1 detection efficiency after 36plex-5K O-LIT off of Mainstreet (orange, 61.93 ± 12%, n = 53 from 2 replicates) versus off of both streets (blue, 62.17% ± 6.68%, n = 611 cells from 15 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
d) FP discovery rate from panel c. Using Mainstreet = 8.64% and using both streets = 5.29%. FP discovery rate from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
e) Tier 2 detection efficiency after 36plex-5K off of Mainstreet (orange, 92.3% ± 3.42% from 53 cells from 2 replicates) versus off of both streets (blue, 80.19 ± 7.29%, n = 611 cells from 15 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
f) Detection efficiency after 36plex-5K O-LIT off of both streets for individual cells from 15 replicates in panel e.
g) Percentage of cells displaying a range of efficiencies of barcode detection after 36plex-5K O-LIT off of both streets. Data taken from panel e.
h) Principal component analysis showing lack of batch effect in 36plex datasets (n = 1171 cells from 15 36plex-5K O-LIT replicates and 8 36plex-1K O-eLIT replicates).
Extended Data Fig. 3. O-LIT with 36plex-5K to interrogate genome organization.
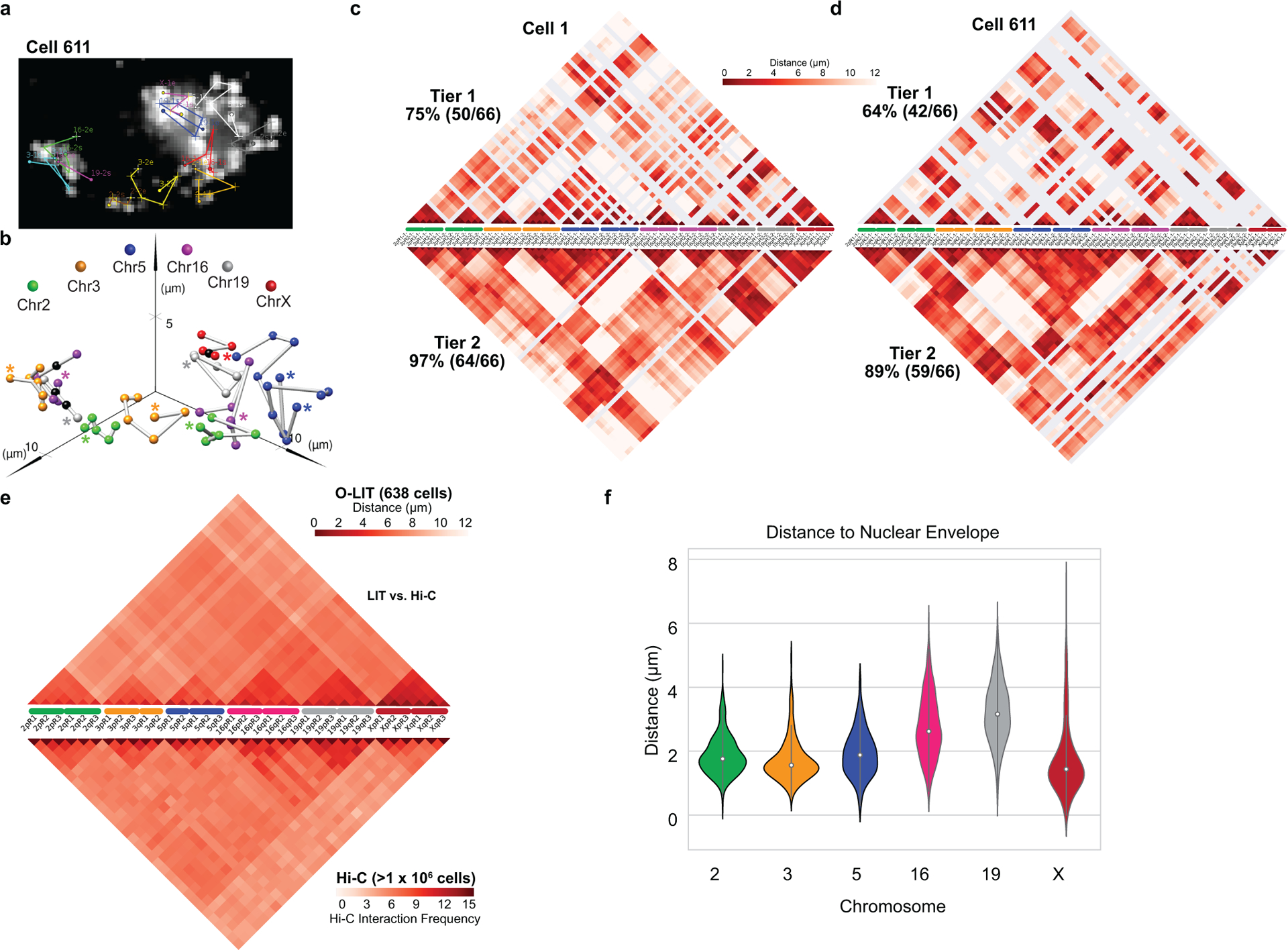
a) Chromosome traces of Cell 611 after Tier 2 detection of cell 611 after four rounds of O-LIT 36plex-5K off of both streets. 59/66 (89%) of 36plex-5K targets were detected. Image is from the first round of O-LIT with target identities. n = 1.
b) Ball and stick of Cell 611. Colored spheres represent chromosomal targets, while black spheres represent targets that were not detected and, thus, were placed by calculating the median proportionate distance between flanking detected targets. Beginning of chromosome (e.g. 2pR1) marked by an asterisk.
c) Single-cell pairwise spatial distance matrix after Tier 1 (top) and Tier 2 (bottom) detection of the nucleus in Figure 3. Targets are represented on the x-axis with homologs separately displayed. Undetected targets are represented by grey lines.
d) Single-cell pairwise spatial distance matrix after Tier 1 (top) and Tier 2 (bottom) detection of Cell 611. Targets are represented on the x-axis with homologs separately displayed. Undetected targets are represented by grey lines.
e) 36plex-5K population pairwise spatial distances (top, from Fig. 3f). Average pairwise spatial distances from cell population after Tier 1 detection (n = 611 from 15 replicates). (Spearman’s rank correlation 0.705, two-sided p-value for a hypothesis test whose null hypothesis is that two sets of data are uncorrelated = 1.77e-174). Measurements from homologous targets were combined. Bottom, Hi-C data of 36plex-5K targets obtained from (Nir et al. 2018).
f) Average distances between the nuclear membrane and the closest of the six targets imaged for each chromosome. (n = 686, 668, 364, 586, 760, and 494 for Chr2, 3, 5, 16, 19, and X, respectively.) The thick line in each violin plot represents the Interquartile range (IQR), the white dot marks the median and the thin lines extend 1.5 times the IQR.
Extended Data Fig. 4. O-LIT with 36plex-5K to interrogate homolog organization.
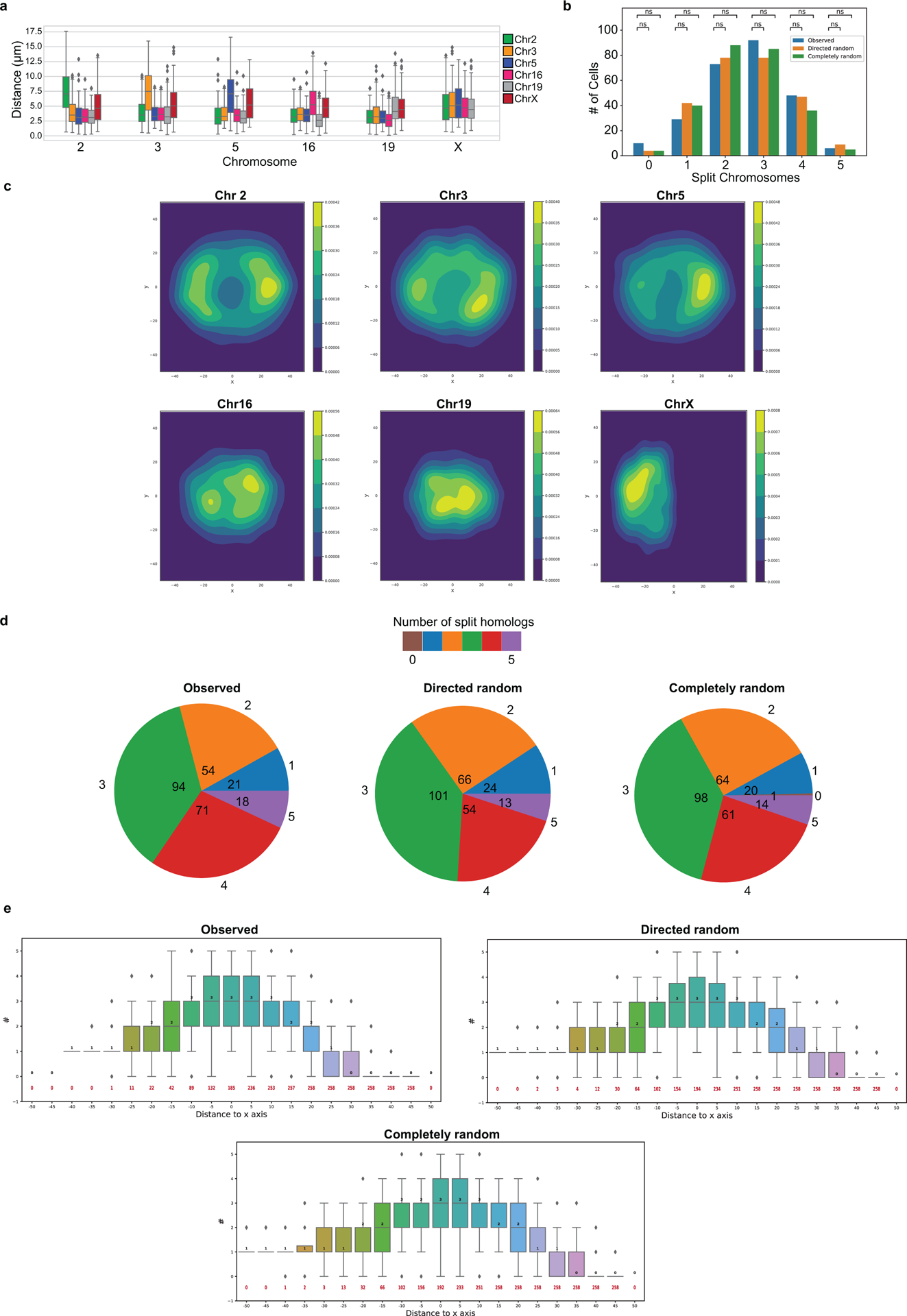
a) Minimum distances between heterologous and homologous chromosomes. All measurements represent distances between the geometric centers of chromosomes for which all six targets were imaged. Distances between a chromosome and a heterologous chromosome is the shorter of the two distances between that chromosome and the two homologous copies of the heterologous chromosome (n = 686, 668, 364, 586, 760, and 494 for Chr2, 3, 5, 16, 19, and X, respectively). Inter-homolog distances for Chr16 and 19 are less than those for Chr2, 3, and 5 (independent-samples t-test p = 4.28 × 10–37). Boxes represent the IQR (25th, 50th and 75th percentiles) and whiskers extend 1.5 times the IQR
b) Number of cells with varying numbers of homologs split by K-means clustering. The K-means algorithm was applied to 258 nuclei, individually, to cluster chromosomes into two groups based on proximity and then report the number of homolog pairs that were split by the clustering. A value of “5” indicates that the homologs from each five pairs of imaged autosomes in a single nucleus clustered into two spatially separate groups. Observed, PGP1f cells. Directed random, raw positions in Observed but with the chromosome identities of all positions randomized, with the larger chromosomes (2, 3, 5) biased towards the nuclear periphery and smaller chromosomes (16 and 19) biased towards the nuclear interior. Completely random category, randomization of the chromosome identities carried out with no spatial bias. The significance of each pair was evaluated from a two proportion z-test with n=258 for each category with a null hypothesis of equal proportion and a significance level of 0.05.
c) Density plots of homolog positions. Built by using Kernel density estimation (KDE) of nuclei projected and aligned along the x-y plane of the position of the chromosomes.
d) Pie charts of total number of cells with homologs split by a virtual line along the y-axis.
e) Number of aligned cells with homologs split by a virtual line parallel to the y-axis at different distances from the origin, that is, number of autosomes with one of their homologs on the left of the line and the other on the right (n = 258 for each category). Boxes represent the IQR (25th, 50th and 75th percentiles) and whiskers extend 1.5 times the IQR.
Extended Data Fig. 5. O-eLIT with JEB.
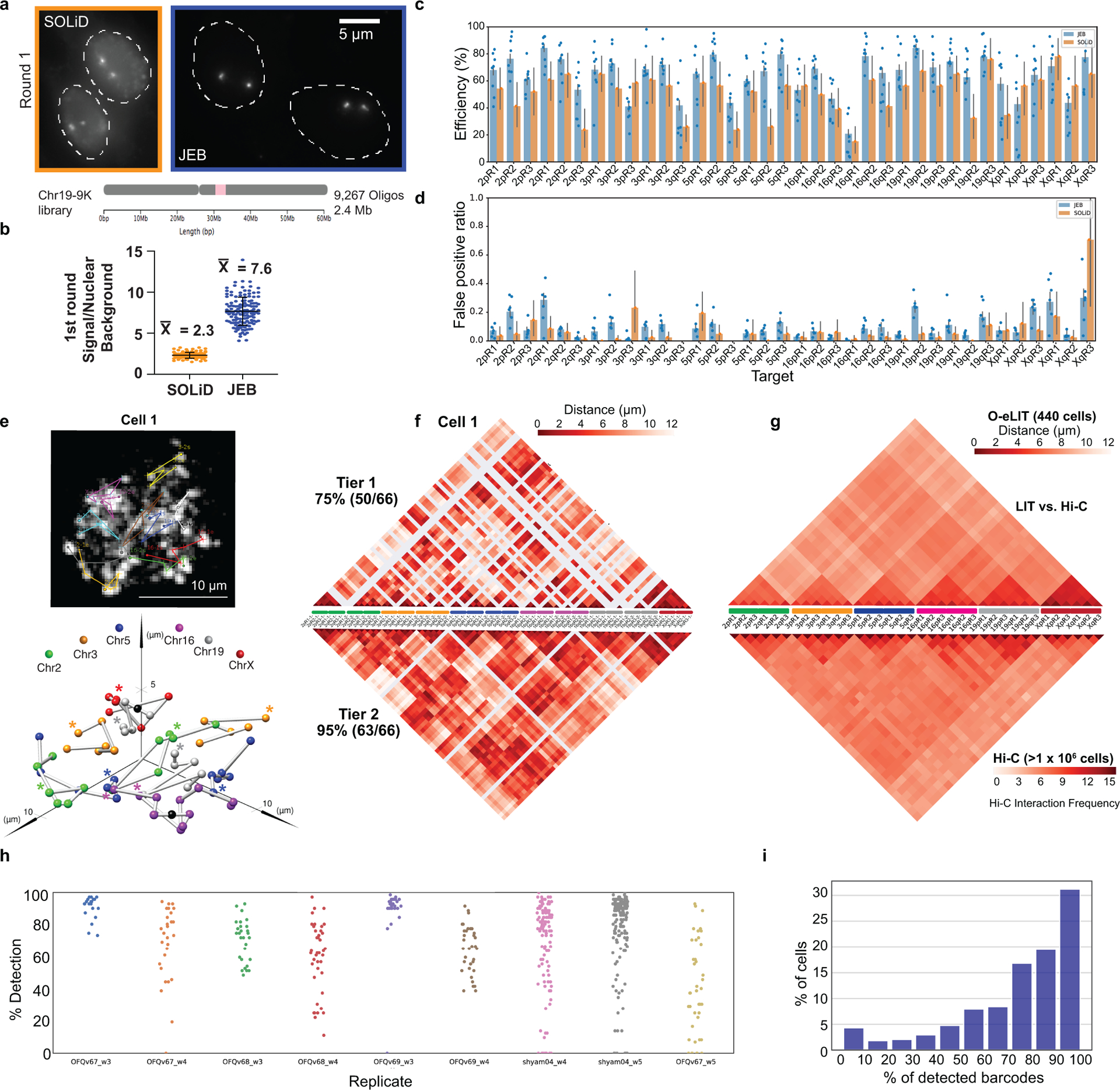
a) Chr19–9K. One round of O-LIT (SOLiD) or O-eLIT (JEB) off of Mainstreet. Maximum z-projections representative of 2 replicates.
b) Chr19–9K signal over nuclear background measurements after one round of O-LIT (orange; n = 113 puncta from 55 cells from 2 replicates) or O-eLIT (blue; n = 136 puncta from 57 cells from 2 replicates). Bar is the mean and SD.
c) Tier 1 detection of 36plex-1K after five rounds of O-LIT with SOLiD reagents (orange; average of 51.75%, n = 41) or O-eLIT with JEB (blue; average of 61.2 ± 10.2%, n = 440 from 9 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean. 36plex-1K library shares first 1,000 Oligopaint oligos of each target in 36plex-5K. For example, for target 2pR1, 36plex-5K spans the chromosomal region from nt position 1,002,895 to 1,660,898 (~658 kb), whereas 36plex-1K spans the region from nt 1,002,895 to 1,147,495 (~144 kb).
d) FP discovery rate from panel c. SOLiD = 7.49% and JEB = 8.95%. FP discovery rate from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
e) Chromosome traces and ball and stick of Fig. 4c cell after Tier 2 detection and five rounds of O-eLIT 36plex-1K. 63/66 (95%) targets were detected. Asterisks, beginning of chromosomes. n = 1.
f) Single-cell pairwise spatial distance matrices of panel C cell.
g) 36plex-1K population pairwise spatial distance measurements (top, from Fig. 3f). Average pairwise spatial distance from cell population after Tier 1 detection (n = 440 from 9 replicates). Measurements from homologous targets were combined. Bottom, Hi-C data of 36plex-5K targets obtained from (Nir et al. 2018).
h) 36plex-1K detection rate for individual cells from 9 replicates.
i) Percentage of cells displaying a range of efficiencies of barcode detection after 36plex-1K O-eLIT off of Mainstreet.
Extended Data Fig. 6. O-eLIT with ChrX-46plex-2K.
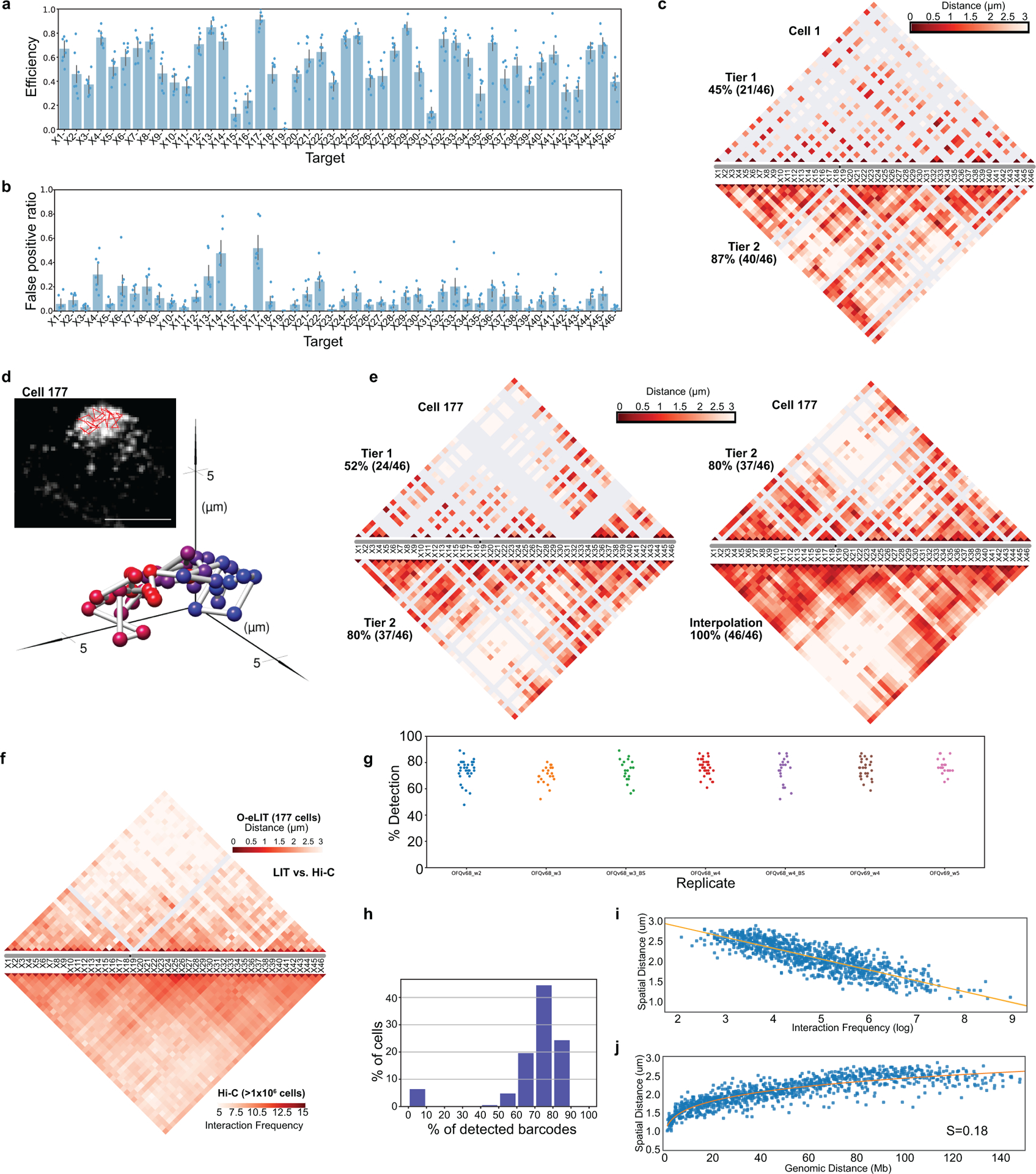
a) ChrX-46plex-2K O-eLIT Tier 1 detection off of one street and off of both streets combined (52.86 ± 5.78% from 177 cells from 7 replicates). Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
b) FP discovery rate from panel a. Error bars represent 95% bootstrap confidence interval of the mean.
c) Single-cell pairwise spatial distance matrix after Tier 1 (top) and Tier 2 (bottom) detection of Cell 1 from Figure 5b. Undetected targets are represented by grey lines.
d) Chromosome traces (top) and ball and stick representation (bottom) of Cell 177 after Tier 2 detection and interpolation and five rounds of O-eLIT on ChrX-46plex-2K off of both streets. Image is from the first round of O-eLIT with target identities. n = 1.
e) Single-cell pairwise spatial distance matrix after Tier 1 (top), Tier 2 (bottom) of Cell 177 (left), and Tier 2 (top) and interpolation (bottom) of same cell (right). Undetected targets are represented by grey lines.
f) ChrX-46plex-2K population pairwise spatial distances (top). Average pairwise spatial distances from cell population after Tier 1 detection (n = 177 from 7 replicates). Bottom, Hi-C (Nir et al. 2018) data of ChrX-46plex-2K targets. (Spearman’s rank correlation 0.641, two-sided p-value for a hypothesis test whose null hypothesis is that two sets of data are uncorrelated = 7.074e-245).
g) ChrX-46plex-2K detection rate for individual cells from 7 replicates.
h) Percentage of cells displaying a range of efficiencies of barcode detection after ChrX-46plex-2K O-eLIT.
i) Mean spatial distance versus Interaction frequency of Hi-C (Nir et al. 2018) of ChrX-46plex-2K targets. Pearson correlation coefficient (r = −0.84) and p-value = 5.08E-275 (two-sided, using slope = 0 for null hypothesis and Wald Test with t-distribution as test statistic) of the linear least-squares regression.
j) Mean spatial distance versus genomic distance for all pairwise ChrX-46plex-2K targets (n = 177 from 7 replicates).
Extended Data Fig. 7. O-eLIT identifies clusters after ChrX-46plex O-eLIT.
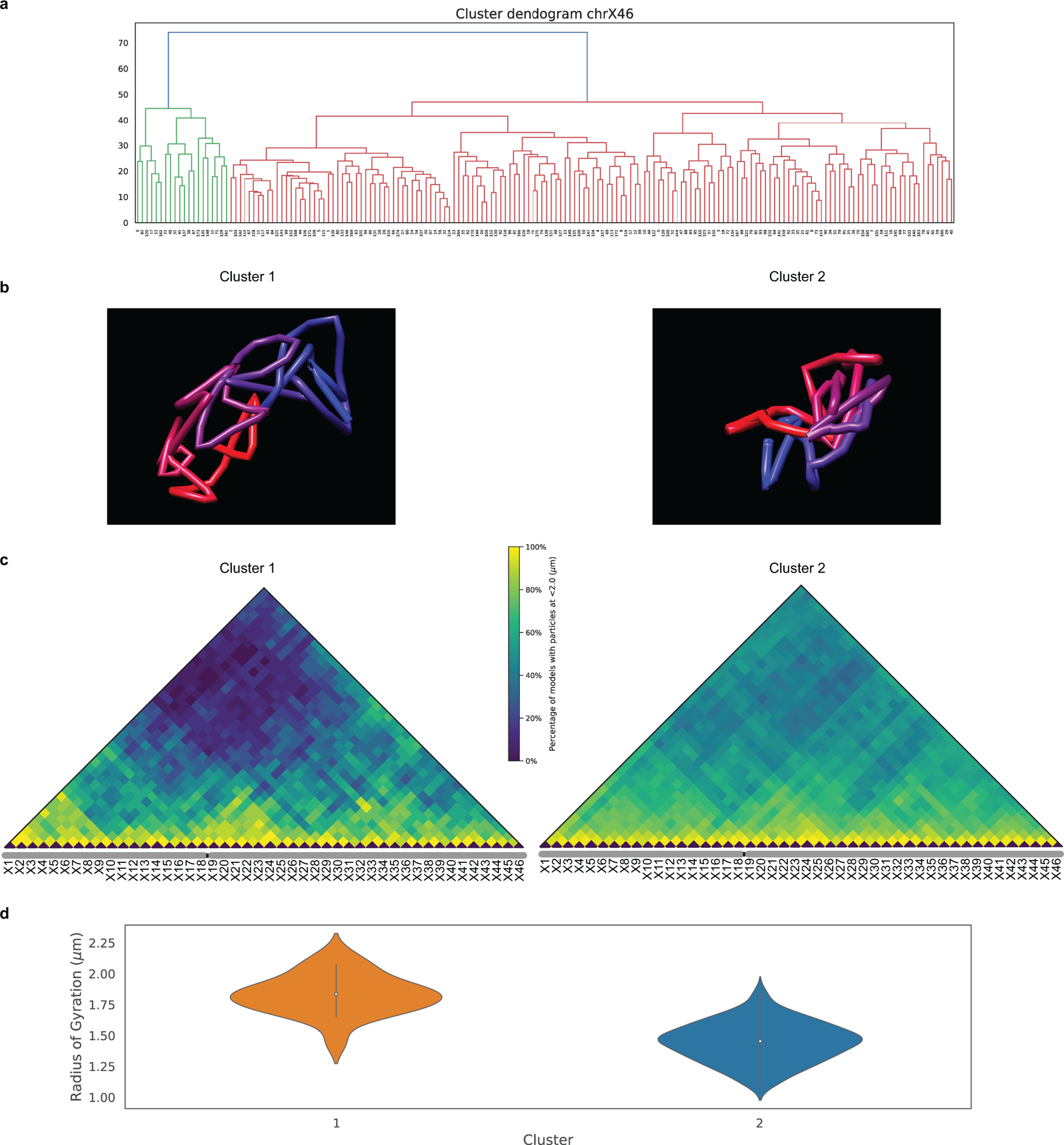
a) Hierarchical clustering based on structure of ChrX traces from ChrX-46plex after 5 rounds of O-eLIT and Tier 2 detection yielded two clusters (Cluster 1 = 20; Cluster 2 = 156). See Methods for more details.
b) ChrX representative models (existing traces that are closer to the virtual centroid) of the two clusters obtained after Hierarchical clustering in panel a.
c) ChrX-46plex-2K population contact matrix of two clusters derived after Hierarchical clustering in panel a where pairwise spatial distances are considered to be in contact if less than 2 μm apart.
d) Radius of gyration for the two clusters (Cluster 1 = 20; Cluster 2 = 156) derived after the hierarchical clustering shown in panel a. The thick line in each violin plot represents the Interquartile range (IQR), the white dot marks the median and the thin lines extend 1.5 times the IQR.
Extended Data Fig. 8. Angles from 36plex.
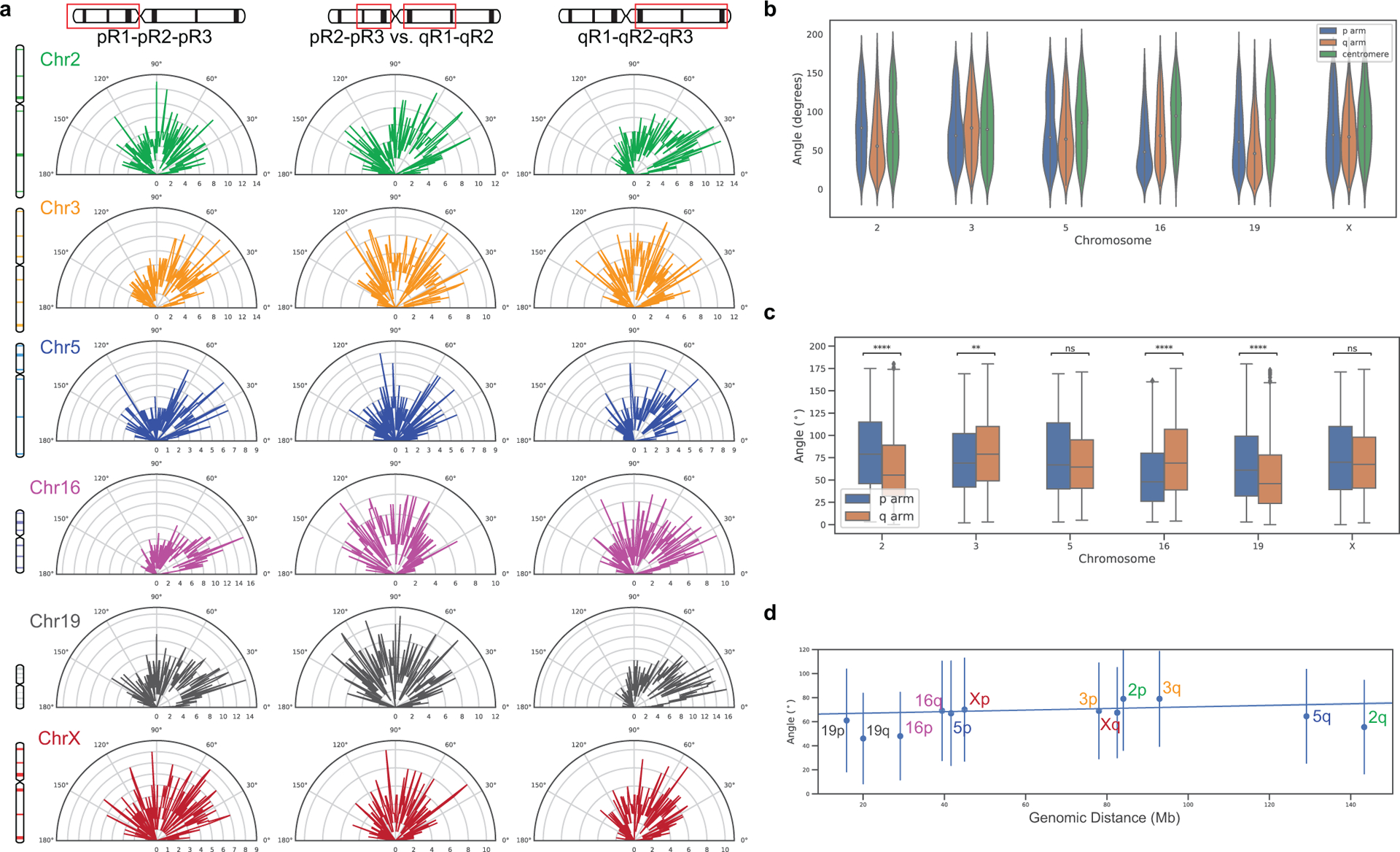
a) Measurements of angles formed by three points along the p arm (left), q arm (right), and intersection of vectors formed by pR2-pR3 and qR1-qR2 (middle) for each chromosome. Measurements were obtained by combining data from 36plex-5K and 36plex-1K analyses and selecting chromosomes that had all six targets identified. Chr2: n = 686, Chr3: n = 668, Chr5: n = 363, Chr16: n = 586, Chr19: n = 760, ChrX: n = 493 (n = 1,051 cells from 24 replicates; for 36plex-5K, n = 611 from 15 replicates; for 36plex-1K, n = 440 from 9 replicates).
b) Distribution of angles formed by segments in panel a. The thick line in each violin plot represents the Interquartile range (IQR), the white dot marks the median and the thin lines extend 1.5 times the IQR.
c) Box plots comparing p and q arm angles. Two-sided student’s t-test with null hypothesis of equal mean was performed to compare arms, ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Boxes represent the IQR (25th, 50th and 75th percentiles) and whiskers extend 1.5 times the IQR. Sample size information in a). Exact p-values for each chromosome: Chr.2 = 4.149e-16, Chr.3 = 0.004, Chr.5 = 0.093, Chr.16 = 1.357e-14, Chr.19 = 3.325e-11, Chr.X = 0.101.
d) Linear least-squares regression between arm angle and arm length with Pearson correlation coefficient r = 0.26 and p-value = 0.42 (two-sided, using slope = 0 for null hypothesis and Wald Test with t-distribution as test statistic).
Extended Data Fig. 9. O-eLIT comparison of X chromosomes in female IMR-90 cells after ChrX-46plex-2K O-eLIT off of both streets.
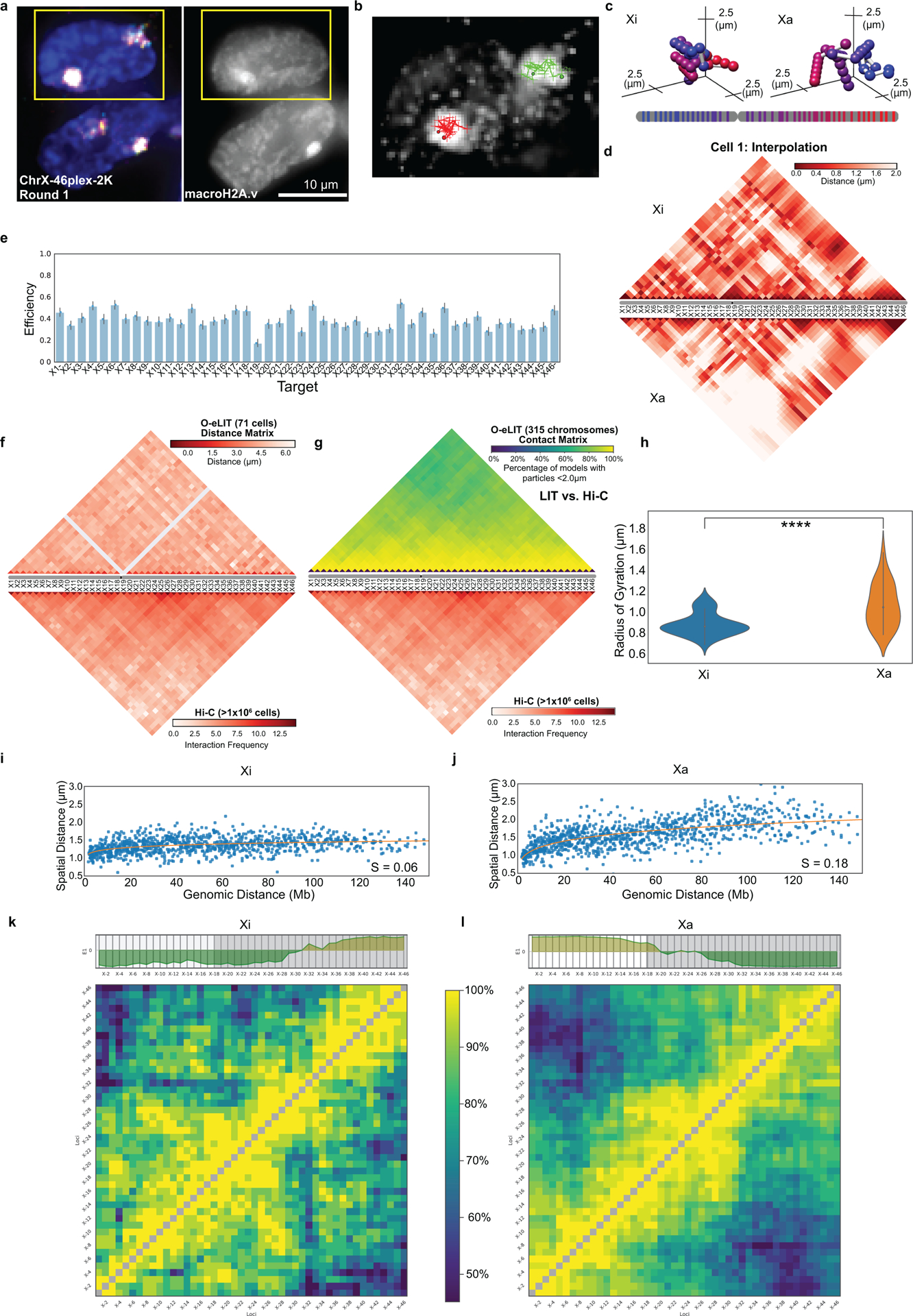
a) First round of O-eLIT sequencing. MacroH2A.1 immunostaining after five rounds of O-eLIT marks the Xi. n = 1.
b-c) Xi and Xa traces (b) and ball and stick (c) of panel a nucleus after Tier 2 analysis and interpolation of missing targets. Sphere color corresponds to chromosome cartoon. n = 1.
d) Single-cell pairwise spatial distances after interpolation of missing targets in panel a.
e) Tier 2 target detection efficiency after five rounds of O-eLIT. 38.57% of targets are detected in 71 cells. Detection efficiency from individual replicates are plotted. Error bars represent 95% bootstrap confidence interval of the mean.
f) Population pairwise spatial distances after Tier 1 detection (n = 71 cells) and Hi-C data of IMR-90 cells (Rao et al. 2014).
g) Population contact maps (top) where two targets are considered to be in contact if less than 2 μm apart (n = 315 chromosomes). Bottom, Hi-C data as in panel f. (Spearman’s rank correlation with the Hi-C matrix is r =0.733, two-sided p-value for a hypothesis test whose null hypothesis is that two sets of data are uncorrelated = 2.564 × 10e-175).
h) Radius of gyration for the Xi (n = 40 chromosomes) and Xa (n = 31 chromosomes). The thick line in each violin plot represents the Interquartile range (IQR), the white dot marks the median and the thin lines extend 1.5 times the IQR. P-value = 7.08 × 10e-6 (two-sided t-test whose null hypothesis is equal means).
i-j) Linear plot of the mean spatial distance versus the genomic distance for all pairwise targets for Xi (n = 40 chromosomes) and Xa (n = 31 chromosomes).
k-l) Population contact maps for Xi (n = 40 chromosomes) and Xa (n = 31 chromosomes) with eigenvector analysis used to identify different domains. X1-X18 (white) and X19-X46 (grey) targets p and q arms, respectively.
Extended Data Fig. 10. OligoFISSEQ applications.
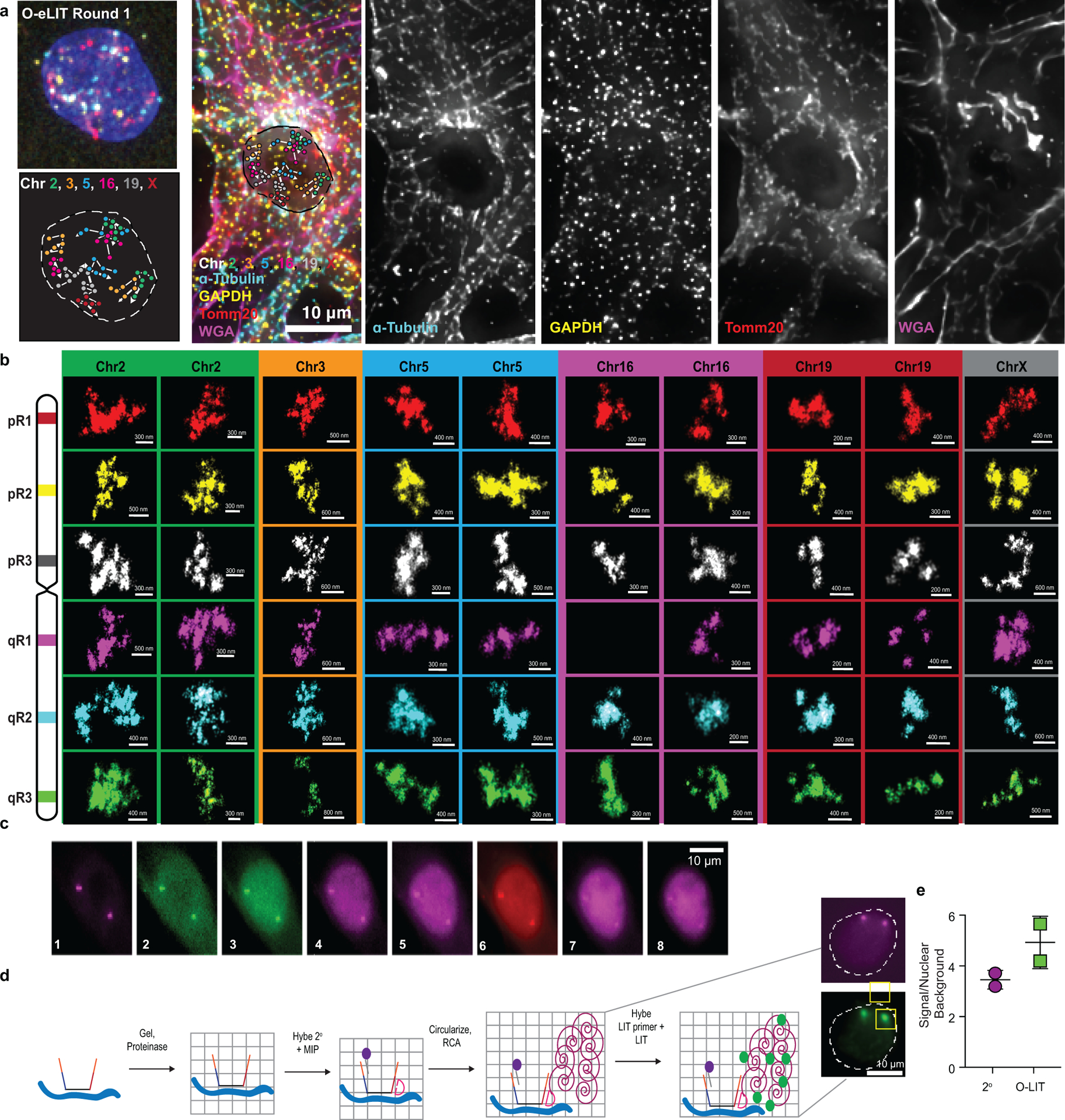
a) O-eLIT and immunofluorescence (IF). 36plex-1K was sequenced 5 rounds with O-eLIT off Mainstreet. Then, the same sample was prepared for IF and stained with antibodies. Samples were counterstained with wheat germ agglutinin (WGA) to stain membranes. Images are from deconvolved, maximum z-projections representative of 2 replicates.
b) Chromosomal regions imaged with OligoSTORM from Fig. 6d enlarged and displayed separately. Orientation may differ from Fig. 6d. n = 1.
c) 8 rounds of O-LIT sequencing of Chr19–9K off of Mainstreet. Images are maximum z-projections. Signal is detectable in all rounds even though the imaging was conducted without the advantage of eLIT, suggesting that 8 rounds of O-eLIT will produce even stronger signals. Images are representative of 2 replicates.
d) O-LIT is compatible with gel embedding and target amplification via rolling circle amplification (RCA). Chr19–9K was hybridized to PGP1f cells, after which the sample was embedded in a hydrogel and then cleared of cellular background with proteinase. Next, a molecular inversion probe (MIP) was hybridized to a Chr19–9K specific barcode on Backstreet as well as a fluorophore labeled (purple) secondary oligo to Mainstreet to visualize Chr19–9K Oligopaint oligos. MIPs were circularized via ligation and RCA, after which the first digit of the barcode was sequenced using O-LIT (green). Images representative of 2 replicates.
e) Comparison of secondary fluorophore signal (2°) versus first round sequencing signal (LIT) from puncta in panel b images. Center values are mean values (3.4 for 2° and 4.9 for O-LIT) with SD.
Acknowledgments:
We apologize to the authors of publications we were unable to cite due to reference constraints. We also gratefully acknowledge members of the Marti-Renom and Wu laboratories for technical and conceptual support, especially T. Ryu, A. Lioutas, and S. Aufmkolk as well as J. AlHaj Abed, S. D. Lee, J. Erceg, and T. Hatkevich; B. Beliveau, H. Sasaki, J. Horrell, L. Cai, J. Kishi, and P. Soler-Vila for discussion; D. Barclay, R. Kohman, E. Iyer, K. Rodgers, A. Skrynnyk, J. Tam, and R. Terry for discussion about FISSEQ and sequencing reagents; S. Alon, F. Chen, Z. Chiang, D. Goodwin, A. Payne, A. Sinha, and O. Wassie for discussion about FISSEQ; C. Ebeling, J. Rosenberg, and J. Stuckey for discussion and technical assistance; F. Pan and A. Hutchinson for assistance in procuring SOLiD reagents; P. Montero-Llopis and the MicRoN imaging core at Harvard Medical School; the ImageJ discussion forum; as well as StackOverflow.
Funding:
This work was supported by awards from the NSERC of Canada (PGS D) to PR, the NIH (HG005550 and HG008525) and NSF (DGE1144152) to ERD, the European Research Council under the 7th Framework Program (FP7/2007-2013 609989), the European Union’s Horizon 2020 Research and Innovation Program (676556), and the Spanish Ministerio de Ciencia, Innovación y Universidades (BFU2017-85926-P) to MAM-R, the Centro de Excelencia Severo Ochoa 2013-2017 (SEV-2012-0208) and the CERCA Programme/Generalitat de Catalunya to the CRG, from NIH to GMC (RM1HG008525-03), and the NIH (DP1GM106412, R01HD091797, R01GM123289) to C-tW.
Footnotes
Competing Interests:
Harvard University has filed patent applications on behalf of C-tW, HQN, and SC, pertaining to Oligopaints and related oligo-based methods for genome imaging. ERD is currently an employee of ReadCoor and has an equity interest in ReadCoor. Potential conflicts of interests for GMC are listed on http://arep.med.harvard.edu/gmc/tech.html. C-tW has an equity interest in ReadCoor and an active research collaboration with Bruker Nano, Inc. in her laboratory at Harvard Medical School.
References:
- 1.Hu Q, Maurais EG & Ly P Cellular and genomic approaches for exploring structural chromosomal rearrangements. Chromosome Res 28, 19–30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolzer A et al. Three-Dimensional Maps of All Chromosomes in Human Male Fibroblast Nuclei and Prometaphase Rosettes. PLoS Biology 3, e157 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cremer T & Cremer M Chromosome territories. vol. 2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proceedings of the National Academy of Sciences 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beliveau BJ et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nature Communications 6, 7147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boettiger AN et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah S, Lubeck E, Zhou W & Cai L In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron (2016) doi: 10.1016/j.neuron.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang S et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beliveau BJ et al. In situ super-resolution imaging of genomic DNA with OligoSTORM and OligoDNA-PAINT. Methods Mol Biol 1663, 231–252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cattoni DI et al. Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nature Communications 8, 1753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eng C-HL, Shah S, Thomassie J & Cai L Profiling the transcriptome with RNA SPOTs. Nature Methods 14, 1153–1155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science (New York, N.Y.) 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nir G et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLOS Genetics 14, e1007872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosin LF, Nguyen SC & Joyce EF Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLOS Genetics 14, e1007393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabo Q et al. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci Adv 4, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardozo Gizzi AM et al. Microscopy-Based Chromosome Conformation Capture Enables Simultaneous Visualization of Genome Organization and Transcription in Intact Organisms. Molecular Cell 74, 212–222.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019 1 (2019) doi: 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fields BD, Nguyen SC, Nir G & Kennedy S A multiplexed DNA FISH strategy for assessing genome architecture in Caenorhabditis elegans. eLife 8, e42823 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mateo LJ et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 1 (2019) doi: 10.1038/s41586-019-1035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawh AN et al. Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. elegans Embryos. Molecular Cell S1097276520300769 (2020) doi: 10.1016/j.molcel.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke R et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nature Methods 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metzker ML Sequencing technologies — the next generation. Nature Reviews Genetics 11, 31–46 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Shendure J et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Player AN, Shen L-P, Kenny D, Antao VP & Kolberg JA Single-copy Gene Detection Using Branched DNA (bDNA) In Situ Hybridization. J Histochem Cytochem. 49, 603–611 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Heride C et al. Distance between homologous chromosomes results from chromosome positioning constraints. J Cell Sci 123, 4063–4075 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Mayer W, Smith A, Fundele R & Haaf T Spatial Separation of Parental Genomes in Preimplantation Mouse Embryos. Journal of Cell Biology 148, 629–634 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hua LL & Mikawa T Mitotic antipairing of homologous and sex chromosomes via spatial restriction of two haploid sets. PNAS 115, E12235–E12244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichmann J et al. Dual-spindle formation in zygotes keeps parental genomes apart in early mammalian embryos. Science 361, 189–193 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Joyce EF, Erceg J & Wu C. -ting. Pairing and anti-pairing: a balancing act in the diploid genome. Curr Opin Genet Dev 37, 119–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watkins NE, SantaLucia J, SantaLucia J & Jr. Nearest-neighbor thermodynamics of deoxyinosine pairs in DNA duplexes. Nucleic acids research 33, 6258–67 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yunis JJ & Prakash O The origin of man: a chromosomal pictorial legacy. Science 215, 1525–1530 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Tjong H et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. PNAS 113, E1663–E1672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishi JY et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nature Methods 16, 533–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouhanifard SH et al. ClampFISH detects individual nucleic acid molecules using click chemistry–based amplification. Nat Biotechnol 37, 84–89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao SSP et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng X et al. Bipartite structure of the inactive mouse X chromosome. Genome Biol 16, 152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minajigi A et al. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darrow EM et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. PNAS 113, E4504–E4512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giorgetti L et al. Structural organization of the inactive X chromosome in the mouse. Nature 535, 575–579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C-Y, Jégu T, Chu H-P, Oh HJ & Lee JT SMCHD1 Merges Chromosome Compartments and Assists Formation of Super-Structures on the Inactive X. Cell 174, 406–421.e25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rust MJ, Bates M & Zhuang XW Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen F, Tillberg PW & Boyden ES Expansion microscopy. Science 347, 543 – 548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sage D et al. Super-resolution fight club: assessment of 2D and 3D single-molecule localization microscopy software. Nature Methods 16, 387–395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohman RE & Church GM Fluorescent in situ sequencing of DNA barcoded antibodies. Preprint at http://biorxiv.org/lookup/doi/10.1101/2020.04.27.060624 (2020) doi: 10.1101/2020.04.27.060624. [DOI]
- 48.Finn EH et al. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 176, 1502–1515.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finn EH & Misteli T Molecular basis and biological function of variability in spatial genome organization. Science 365, eaaw9498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorenz EN Deterministic Nonperiodic Flow. J. Atmos. Sci 20, 130–141 (1963). [Google Scholar]
- 51.Beliveau BJ et al. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proceedings of the National Academy of Sciences of the United States of America 115, E2183–E2192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ball MP et al. A public resource facilitating clinical use of genomes. Proc Natl Acad Sci USA 109, 11920–11927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang K et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods 6, 613–618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pardue ML et al. Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proceedings of the National Academy of Sciences of the United States of America 64, 600–4 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauman JGJ, Wiegant J, Borst P & van Duijn P A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochrome-labelled RNA. Experimental Cell Research 128, 485–490 (1980). [DOI] [PubMed] [Google Scholar]
- 56.Shendure J et al. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 309, 1728–1732 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Valouev A et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Research (2008) doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo J et al. Four-color DNA sequencing with 3’-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proceedings of the National Academy of Sciences (2008) doi: 10.1073/pnas.0804023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single cell in situ RNA profiling by sequential hybridization. Nat Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moffitt JR et al. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. PNAS 113, 14456–14461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Linkert M et al. Metadata matters: access to image data in the real world. The Journal of Cell Biology 189, 777–782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmid B, Schindelin J, Cardona A, Longair M & Heisenberg M A high-level 3D visualization API for Java and ImageJ. BMC Bioinformatics 11, 274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anand L chromoMap: An R package for Interactive Visualization and Annotation of Chromosomes. bioRxiv 605600 (2019) doi: 10.1101/605600. Preprint at https://www.biorxiv.org/content/10.1101/605600v2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pettersen EF et al. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Richardson WH Bayesian-Based Iterative Method of Image Restoration*. J. Opt. Soc. Am 62, 55 (1972). [Google Scholar]
- 67.Parslow A, Cardona A & Bryson-Richardson RJ Sample Drift Correction Following 4D Confocal Time-lapse Imaging. J Vis Exp (2014) doi: 10.3791/51086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tinevez J-Y et al. TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Wagstaff K, Cardie C, Rogers S & Schroedl S Constrained K-means Clustering with Background Knowledge. 8. [Google Scholar]
- 70.Russel D et al. Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLoS Biol 10, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.