

Abstract
Understanding the physiologic mechanisms of wound healing has been the focus of ongoing research for many years. This research directly translates into changes in clinical standards used for treating wounds and decreasing morbidity and mortality for patients. Wound healing is a complex process that requires strategic cell and tissue interaction and function. One of the many critically important functions of wound healing is individual and collective cellular migration. Upon injury, various cells from the blood, surrounding connective, and epithelial tissues rapidly migrate to the wound site by way of chemical and/or physical stimuli. This migration response can largely dictate the outcomes and success of a healing wound. Understanding this specific cellular function is important to translational medicine that can lead to improved wound healing outcomes. Here, we describe a protocol used to better understand cellular migration as it pertains to wound healing, and how changes to the cellular environment can significantly alter this process. In this example study, dermal fibroblasts were grown in media supplemented with fetal bovine serum (FBS) as monolayer cultures in tissue culture flasks. Cells were aseptically transferred into tissue culture treated 12-well plates and grown to 100% confluence. Upon reaching confluence, the cells in monolayer were vertically scratched using a p200 pipet tip. Arsenic diluted in culture media supplemented with FBS was added to individual wells at environmentally relevant doses ranging from 0.1 – 10 μM. Images were captured every 4 hours over a 24-hour period using an inverted light microscope to observe cellular migration (wound closure). Images were individually analyzed using image analysis software, and percent wound closure calculated. Results demonstrate that arsenic slows wound healing. This technique provides a rapid, inexpensive first screen for evaluation of the effects of contaminants on wound healing.
Keywords: Scratch assay, cellular migration, wound healing, arsenic, environmental contaminants, human dermal fibroblasts
SUMMARY:
This study focuses on an in vitro model of wound healing (scratch assay) as a mechanism for determining how environmental contaminants such as arsenic influence cellular migration. The results demonstrate that this in vitro assay provides rapid and early indications of changes to cellular migration prior to in vivo experimentation.
INTRODUCTION:
Wound healing consists of a complex series of overlapping steps that involve various cell types, signaling molecules, and extracellular matrix components1. Together, these components work in concert to achieve wound resolution or repair, which ultimately leads to the formation of a protective fibrotic scar depending on the degree of trauma1. To capture the individual processes and functions of wound healing in one experiment is difficult; individual steps within the wound healing process need to be identified and tested separately. Among the many steps of wound healing, the process of cellular migration has been considered critical when evaluating healing rates2. Cellular migration can be observed in all phases of wound healing, and thus necessitates further understanding of how alterations to cell migration can impact wound closure. For example, cellular migration is seen in both early and late phase inflammation, with blood coagulation and platelet aggregation at the wound site3. These events prevent excessive blood loss by forming a temporary blockage to fill wounded areas devoid of tissue3. Platelets in circulation will synthesize and secrete vasoactive mediators along with chemotactic factors, which together signal for inflammatory leukocyte migration (white blood cells) to the wounded tissue for repair4. Re-epithelialization occurs within hours of tissue injury and involves covering the wound site through activity and migration of epithelial keratinocytes to prevent further contamination or invasion of foreign particles to the wound site2. These examples capture only a few of the many cell migration functions associated with wound healing.
The scratch assay has been described and used to better understand cell migration and its many roles in wound healing5,6. This assay is considered simplistic, provides high throughput, and enables the investigator to collect representative cell migration data. Migration assays have successfully demonstrated that certain compounds may accelerate or slow down the rate of cellular migration6. Furthermore, these assay results provide translational physiologic information that can be used to formulate target treatments, dosing concentrations, and genes of interest prior to in vivo experimentation. The assay requires basic cell culture techniques and supplies, a cell line of choice, and imaging technology to capture movement over time or movement in response to treatments.
The assay can be readily manipulated or tailored to suit the needs of the investigator. However, there are four basic questions to consider prior to experimentation. What general or specific question(s) is/are the investigator(s) trying to answer? This holds true for most hypotheses driven research and experiments; however, it is critical to this assay because it will determine many of the parameters needed to accurately and completely answer the question(s). What cell line or cell lines (if multiple) should be used to represent the physiologic system being studied? For example, in a cutaneous wound healing study, a dermal fibroblast,5,6,7 stem cell,8 or epidermal keratinocyte9 might be considered to represent cell populations that are normally found in abundance in skin tissue10. Alternatively, neutrophils, macrophages or other inflammatory cells could be evaluated in this system to represent cell migration of other components of wound healing within skin tissue10. Additionally, identifying the specific cell line being evaluated will help to identify which reagents are optimal to use to promote cell metabolic activity. How will cell migration be tracked/imaged? There are a number of techniques used to observe cell migration within a plate or flask. For example, dying or fluorescently tagging cells and using fluorescence or confocal imaging to track cells provides a strong contrast, which allows the investigator to clearly define cellular vs. non-cellular locations and cellular movement11. Another common technique, described in this study, is to use inverted light microscopy with phase contrast6,7. This alternative to cell dyes and fluorescence allows the user to live track cells without having to terminally fix cells prior to imaging and also allows for capturing images of the same individual wells containing wounded monolayers of cells across time points. What outcome measures will be used for analysis? Using the images gathered throughout the study, data can be generated in which changes to cellular migration rates can be understood. In our lab, microscopic images captured on the inverted light microscope are uploaded to image analysis software and the percent wound closure and summed area under the curve are (AUC) are calculated.
We will highlight the specific use of this assay to elucidate changes to dermal fibroblast migration in the presence of a known environmental contaminant, arsenic. Arsenic is a naturally occurring metalloid found within the earth’s crust. Various locations have been found with elevated levels of arsenic, which poses a risk to individuals who live near these locations. As a result, food and water sources have been shown to have increased levels of arsenic contamination, which increases the likelihood for individuals to become exposed to arsenic. Environmental arsenic exposure has been shown to have long-term health consequences in human populations above or even below the current United States Environmental Protection Agency (USEPA) Max Contaminant Level (MCL) of 10 ppb (10 μg/L)12,13. Although the USEPA has set this MCL and deemed it safe for drinking limits, arsenic concentrations that exceed this limit are not uncommon in water sources worldwide. In the Southwestern United States, numerous ground-water, well water, and springs have been documented to contain concerning levels of arsenic14. For example, in Verde Valley, AZ, Montezuma Well contains 210 μg/L of arsenic15. Groundwater around the Verde River, AZ contains 16 μg/L of arsenic on average, with peak values reaching 1.3 mg/L15,16. Notably, arsenic concentrations in many wells in the Verde River water shed exceed 50 μg/L15,16. With this knowledge, environmentally relevant arsenic concentrations were selected that range from below the MCL at 0.1 μM (7.5 ppb), to above the MCL at 10 μM (750 ppb) in the current study14–20.
The information gathered from these experiments will be used as an early indicator of potential changes to cellular migration rates in an in vivo wound contaminated with arsenic. Afterward, signaling molecules can be selected based on their role in facilitating wound healing and cellular migration, and future quantitative polymerase chain reaction (qPCR) based gene expression experiments may be set up upon completion of the current studies. If migration rates in this assay change in the presence of arsenic, specific gene targets involved in cellular migration may be the target for deleterious effects of arsenic, and thus may be the mechanism of disruption.
PROTOCOL:
1.). Preparation of biosafety cabinet II (BSC)
NOTE: The methods described in this protocol should be conducted with personal protective equipment, while using good laboratory practices to achieve aseptic technique.
1.1) Lift biosafety cabinet (BSC) protective glass to operating height and turn on laminar flow fans.
-
1.1.1)
Use 70% isopropanol aqueous solution to perform sanitation wipe of working area. Wipe BSC from back wall and side walls and work down to the base plate.
-
1.1.2)
Wipe all materials that will be used in assay with 70% alcohol (isopropanol or EtOH) and place inside the BSC.
2.). Human Dermal Fibroblasts Seeding in a T75 Flask
2.1) Obtain vials with the desired cryopreserved cell line (neonatal human dermal fibroblasts [hDFn], at cell concentrations of 500,000 cells/vial and at passages p3 – p5).
-
2.1.1)
Place a vial with cells in 1.0-cm-deep water bath at 37 °C to allow thawing. Thaw vial until approximately 70% of cell solution is liquid. Wipe down vial with 70% alcohol and place vial in vial rack inside BSC.
NOTE: Ensure water does not come in contact with threads or cap of vial to prevent potential contamination during cell seeding. Prevent complete thawing of cells in water bath, as failure to do so may cause unintentional cell death.
2.2) Draw out 10 mL of media supplemented with 10% Fetal Bovine Serum (FBS). Use an automatic pipettor and 10 mL pipette tip to aspirate into T75 tissue culture flask. Allow media to fully saturate the base of the flask by gently rocking the flask back and forth.
-
2.2.1)
Open vial with cell stock and aspirate solution from the vial, being mindful of pipetting speed, as individual cell membranes will be vulnerable from being in cryopreservation.
-
2.2.2)
Transfer cells to T75 tissue flask with media. Eject thawed cell into tissue flask, again being mindful of pipetting speed.
NOTE: Seeding density of cells in tissue flask is approximated to be 6,600 cells/cm2. Seeding density can be altered to accommodate the user and desired timing of experimentation.
-
2.2.3)
Measure out 1 mL of media from T75 tissue flask and transfer volume back into the vial. Transfer volume from the vial back into the T75 flask.
NOTE: Performing this step helps maximize cell yield from the vial.
-
2.2.4)
Tighten tissue culture flask and gently rock in efforts to uniformly disperse cells in suspension across entire surface. Check for even distribution of cells using an inverted microscope.
2.3) Label flask with cell type, date, initials, lineage, and purpose (e.g., hDFn, 07/11/2018, NDC, Passage 4, Arsenic Scratch Assay). Place tissue culture flask in a humidified incubator set to 37 °C and 5.0% CO2 for 72 h.
NOTE: Growth medium should be changed 12-24 h after initial seeding to remove trace amounts of dimethyl sulfoxide (DMSO) cryopreservative, then changed every 48 hours after that to ensure cells are properly nourished. The incubation and growth period will depend on the calculated number of cells needed for the scratch assay. The doubling time for hDFn cells is typically 16-24 h under normal conditions. However, doubling rate may change due to elevation, pH, temperature, humidity, medium type, etc., and must be accounted for when planning experiments.
3.). Cell Counting and Subculture into 12-Well Tissue Culture Plate
3.1) Retrieve T75 tissue culture flask from incubator. Observe adhered cells using inverted microscope at 10X objective magnification (100X total magnification) and determine approximate cell confluence.
-
3.1.1)
Scan the majority of the tissue culture flask when observing confluence to ensure the most representative percentage is approximated. Evaluate individual cell morphology for any abnormalities, or for bacterial and/or fungal contamination.
3.2) Assemble automatic pipettor with a 10 mL pipette tip. Use the pipettor to aspirate the full volume of media from the T75 and transfer solution to a waste container. Prevent the pipet tip from contacting the cell adhering surface. Discard pipette tip in a biohazard bin.
-
3.2.1)
Assemble automatic pipettor with a 5 mL pipette tip. Aspirate 5 mL of the balanced salt solution, dispense in the flask, and gently rock flask to rinse excess media, dead cells, and waste product.
-
3.2.2)
Draw the volume from the T75 and dispense into the waste container. Discard the pipette tip into the biohazard bin.
-
3.2.3)
Assemble automatic pipettor with 5 mL pipette adapter. Aspirate 2 mL of trypsin from stock, dispense in flask, and gently rock the flask to disperse the enzyme over the adhered cells. Ensure complete saturation of adherent surface. Place tissue flask in the incubator for 2-3 minutes (min).
NOTE: The maximum enzyme-substrate reaction should not exceed 10 minutes.
-
3.2.4)
Observe the tissue flask under inverted microscope at 10X objective lens magnification (100X total magnification). Apply gentle vibrations to facilitate cell detachment.
-
3.2.5)
Wipe the T75 flask with 70% alcohol and place inside the BSC.
-
3.2.6)
Assemble the automatic pipettor with 5 mL pipette tip. Aspirate 5 mL of media from the stock and add to the T75 tissue flask to stop the enzyme-substrate reaction. The media:typsin ratio should be 3:1. Pipet up and down the base of the flask 2-3 times to rinse and ensure that the reaction has stopped. Aspirate the complete volume of fluid from flask and transfer into a sterile 15 mL conical tube.
-
3.2.7)
Fill second 15 mL conical tube with an identical volume of media.
-
3.2.8)
Place both 15 mL conical tubes in an antipodal position to one another. Apply 200 g of centrifugal force for 5 minutes at 25° C by setting equipment’s running parameters.
-
3.2.9)
Wipe the 15 mL conical tube with pelleted cells with 70% alcohol and place inside the BSC.
-
3.2.10)
Using automatic pipettor with a 5 mL attachment pipet off media, leaving behind approximately 250 μL of fluid and cell pellet. Pay attention to the location of the pipet tip, as the cell pellet should remain undisturbed. Dispense volume collected into a waste container and discard the tip in a biohazard bin.
-
3.2.11)
Use a microcentrifuge tube rack to run the bottom of the conical tube across the vial slots, creating a swift vibration, to disturb and re-suspend the cell pellet (sometimes called “rack raking”).
NOTE: Avoid using a vortex mixer to perform this step. Gravity forces created by vortex mixers exceed cell g-force thresholds and can cause lysing. When completed correctly new cell solution will appear as a cloudy mixture with no remaining pellet. Diligence performing this step will oftentimes create a single cell suspension, leading to increased accuracy when counting cells.
-
3.2.12)
Add 2 mL of media to the cell stock or resuspension. Pipet the cells gently down the side of the tube 20 times to break up any clumps. Record the total cell suspension volume and set aside briefly.
3.3) Pipet 20 μL of Trypan Blue stock (0.4%) into an empty well of a 96-well plate using a sterile pipet.
-
3.3.1)
Mix the stock cell solution to generate a uniform cell suspension before transfer. Use a sterile pipet tip to remove 20 μL of cell stock (avoiding touching the tube walls). Add to the 20 μL Trypan Blue solution in the 96-well plate.
-
3.3.2)
Pipet gently to mix contents. Pipet 10 μL of the mixture between the coverslip and the counting chamber on the hemocytometer.
-
3.3.3)
Observe the cells and the grid under a 10X objective. Observe nine large squares within the field of view. Count cells only in the center and 4 corner squares (total of 5 squares).
NOTE: Look for a uniform distribution of cells across the entire grid to ensure an accurate count.
-
3.3.4)
Tally all cells (transparent and blue) in the 5 identified squares of the grid. Separately, tally only the nonviable (blue) cells in the same 5 squares.
-
3.3.5)
Calculate viable cell count using: x = a − b
where x = viable cell count, a = total cells, b = non-viable cells.
-
3.3.6)
Calculate the viable cell density using:
where x = viable cell density and y = total sum of viable cells.
-
3.3.7)
Calculate total viable cells: x = y × f
where x = total viable cells, y = viable cell density, f = cell suspension volume (mL).
-
3.3.8)
Calculate % cell viability:
where x = percent cell viability, y = viable cells counted on grid, f = total cells counted on grid.
3.4) Mark a horizontal line across the bottom of the 12-well plate to serve as reference marks during imaging.
-
3.4.1)
Use the viable cell density (calculated in 3.3.6) to calculate the volume of media to dilute cell stock in for seeding a 12-well tissue culture plate.
NOTE: The optimal cell seeding density for hDFn cells is 5,000 cells/cm2.
-
3.4.2)
Dilute cell stock to 19,000 cells/mL using media and seed each well with 1 mL of cell stock. Mix the cell stock periodically to ensure an even cell seeding across all 12 wells.
-
3.4.3)
Label each plate with the cell type, the lineage, the user initials, and the purpose. Place plates in incubator set to 37 °C and 5.0% CO2. Allow cells to grow until 100% confluence is achieved for scratch assay.
4.). Arsenic (As) stock solutions and calculations
4.1) Dilute sodium arsenite (NaAsO2) directly into cell culture media with 10% FBS at working concentrations of 10, 1.0, 0.1, and 0.01 μM As.
4.2) For this, make an initial 1 mM stock As solution by diluting 3.9 mg of sodium arsenite into 30 mL of media. Serially dilute the 1 mM stock for the remaining As solutions.
5.). Scratch assay technique and arsenic contamination
5.1) Obtain 12-well plate with cells from incubator. View cells at 10X objective magnification (100X total magnification). Determine cell confluence within each well.
NOTE: Each individual well must reach 100% confluence prior to scratching. This critical step ensures consistency within the assay and helps standardize assay protocols across experiments. If any wells have not reached 100% confluence, allow cells to incubate for additional time until all wells have reached 100% confluence. Additional growth time in wells that have already reached 100% confluence will not be affected. Confluent cells will typically become growth arrested and remain as a monolayer culture, while allowing sub-confluent wells to reach 100% confluence.
5.2) Assemble automatic pipettor with 10 mL pipette tip. Aspirate the growth media out of all wells. Dispose the volume in liquid waste and the pipette tip in the biohazardbin.
-
5.2.1)
Assemble a p200 pipettor with a 1-200 μL sterile tip. In each, well produce a scratch “mock wound” by gliding the tip across cell surface from a 12-o’clock to 6-o’clock location. NOTE: Three factors that will greatly affect consistency are speed, pressure, and tip angle. The scratch should be performed swiftly, with a constant pressure, while being mindful of the tip angle staying perpendicular to the base of the plate. Scratching too slowly will cause crooked lines, excess pressure can permanently score cell adhering surfaces, and a tip angle less than 90° will narrow the scratch width. Any of these common mistakes can result in altered migration rates and patterns.
-
5.2.2)
Clear away excess debris and cell clumps by rinsing with 1 mL of balanced salt solution per well. Rock the plate side-to-side to facilitate washing. Remove the balanced salt solution from wells and dispose in waste container. Dispose of the 5 mL pipette tip into the biohazards bin.
-
5.3)
Use a p1000 pipettor with a 1000 μL tip to pipet 1,000 μL from arsenic stocks into wells that have been randomly assigned with a treatment group. Use a new pipet tip for each well to prevent cross contamination. Record the time when treatments are added. Place 12-well plates in an incubator set at 37 °C and 5.0% CO2.
6.). Imaging
6.1) Capture images of each well at 10X objective magnification every 4 h over a 24 h period (0, 4, 8, 12, 16, 20, and 24 h). Reference the line previously drawn on the bottom of the plate to image the same location along the scratch within each well at subsequent time points.
NOTE: It is imperative that the same locations are photographed at each time point to allow for accurate image analysis and to obtain representative data.
7.). Image analysis
7.1) Upload images captured on the inverted light microscope to a computer and save as a JPEG image with a detailed file name.
7.2) Open ImageJ.
7.3) Upload an image of a stage micrometer with known distances to the software to convert pixels to millimeters (mm) when taking image measurements by dragging and dropping the file into the software window.
NOTE: The micrometer should have lines demarcating a known 1 mm length and the image must be taken at the same magnification used to capture images of cells. For example, in this study, images were taken at 100X total magnification, thus the image of the stage micrometer was taken at 100X total magnification.
-
7.3.1)
Click the Straight, Segmented, or Freehand Lines options.
-
7.3.2)
Draw a line of known distance on the micrometer image by using the tick-marks with a length associated to each tick marks. To draw a straight line: click mouse, hold, drag the cursor to desired location, then let go of mouse.
-
7.3.3)
Select Analyze then select Set Scale.
-
7.3.4)
Set Known Distance to the known length of the line drawn in previous step.
-
7.3.5)
Set Unit of Length to mm.
-
7.3.6)
Check the global box.
-
7.3.7)
Select OK to exit out of the menu.
-
7.3.8)
Close out of the stage micrometer image.
7.4) Upload one scratch assay image into ImageJ by dragging and dropping image file into the window.
-
7.4.1)
Draw a straight line across the width of the scratch (from left leading edge to right leading edge).
-
7.4.2)
Press the letter M on keyboard to record the measurement of the straight line drawn. A small Results window will immediately appear after typing the letter M. This is where the width measurement will be.
7.5) Repeat step 7.4 a total of 10 times to obtain 10 measurements along the length of the scratch
NOTE: It is important to generate the most representational width values down the entire length of the scratch. Make sure to space out the 10 width measurements equally along the length of the scratch from top to bottom.
7.6) Create a scratch assay spreadsheet file to copy and paste the measurements.
-
7.6.1)
Copy and paste the 10 width measurements from the Results window into appropriate column in a spreadsheet file (Table 2).
NOTE: When the measurements are copied from the Results window and pasted into the spreadsheet, there will be extraneous columns of values; these values should be deleted, preserving only the 10 width measurements.
Table 2. Raw width measurements table format.
This table shows the organizational structure for the raw measurements obtained.
| 10 random width measurements | 0 h | 4 h | 8 h | 12 h | 16 h | 20 h | 24 h |
|---|---|---|---|---|---|---|---|
| 1 | |||||||
| 2 | |||||||
| 3 | |||||||
| 4 | |||||||
| 5 | |||||||
| 6 | |||||||
| 7 | |||||||
| 8 | |||||||
| 9 | |||||||
| 10 | |||||||
| Average of 10 measurements |
7.7) Repeat the above step for the remainder of the scratch assay images.
7.8) Calculate the average of the 10 measurements at each time point (0-24 h) for every well.
7.9) Determine percent closure calculations using values from step 7.8.
-
7.9.1)
Create a table at the bottom of the spreadsheet titled “width measurement averages” (Table 3).
-
7.9.2)
Copy and paste the Average of 10 Measurements row calculated in step 7.8 into the “width measurement averages” table.
-
7.9.3)
Create another table next to the Width Measurement Averages table. Title this table Percent Wound Closure (Table 4).
NOTE: The value in the 0 h column for each well should be 0 because the scratch is 0% closed at the initial 0 h photo for all wells.
-
7.9.4)
Navigate to the next column over (4 h).
-
7.9.5)Calculate the percent closure for 4, 8, 12, 16, 20, and 24 h time points:
where, x = percent closure, i = initial scratch width (0 h width), f = final scratch width (4, 8, 12, 16, 20, or 24 h width).
Table 3. Width measurement averages table format.
This table shows the organizational structure for the width measurement averages calculated using the raw width measurements in Table 2.
| Well width averages | 0 h | 4 h | 8 h | 12 h | 16 h | 20 h | 24 h |
|---|---|---|---|---|---|---|---|
| 1a | |||||||
| 2a | |||||||
| 3a | |||||||
| 4a | |||||||
| 1b | |||||||
| 2b | |||||||
| 3b | |||||||
| 4b | |||||||
| 1c | |||||||
| 2c | |||||||
| 3c | |||||||
| 4c |
Table 4. Percent wound closure table format.
This table shows the organizational structure for the percent wound closure values calculated using the width measurement averages in Table 3.
| Well | 0 h | 4 h | 8 h | 12 h | 16 h | 20 h | 24 h |
|---|---|---|---|---|---|---|---|
| 1a | 0 | ||||||
| 2a | 0 | ||||||
| 3a | 0 | ||||||
| 4a | 0 | ||||||
| 1b | 0 | ||||||
| 2b | 0 | ||||||
| 3b | 0 | ||||||
| 4b | 0 | ||||||
| 1c | 0 | ||||||
| 2c | 0 | ||||||
| 3c | 0 | ||||||
| 4c | 0 |
7.10) Create another table next to the Percent Wound Closure table. Title this table AUC (Table 5).
-
7.10.1)Calculate the 0-4 h area under the curve (AUC) value in the 0-4 h column for each well:
where x = 0-4 h AUC value, a = % closure value for 0 h, b = % closure value for 4 h, h = the time between the two measures (4 h in this study). -
7.10.2)Calculate the 4-8 h area under the curve (AUC) value in the 4-8 h column for each well:
Where x = 4-8 h AUC value, a = % closure value for 4 h, b = % closure value for 8 h, h = the time between the two measures (4 h in this study).NOTE: One AUC value should be calculated for each 4 h time interval over the 24 h period for each well.
-
7.10.3)
Sum all of the AUC values from 0-24 h (calculated in steps 7.10.1-7.10.2) to get a summed AUC value for each well. Ensure these values are properly saved for statistical analysis.
Table 5. AUC table format.
This table shows the organizational structure for the AUC values calculated using the percent wound closure values in Table 4.
| Well | 0-4 h | 4-8 h | 8-12 h | 12-16 h | 16-20 h | 20-24 h | Summed AUC |
|---|---|---|---|---|---|---|---|
| 1a | |||||||
| 2a | |||||||
| 3a | |||||||
| 4a | |||||||
| 1b | |||||||
| 2b | |||||||
| 3b | |||||||
| 4b | |||||||
| 1c | |||||||
| 2c | |||||||
| 3c | |||||||
| 4c |
8.). Statistical analysis
8.1) Determine the statistical analysis method that best fits both the experimental design and summed AUC values obtained during the assay.
REPRESENTATIVE RESULTS:
Sample sizes (n) for all treatment groups was n = 28. Cells were successfully cultured following aseptic techniques and lab protocols (Figure 1). Uniform scratches were observed across all treatment groups with the mean scratch width measuring 0.8 mm ± 0.4 mm (Figure 2). Control groups had an average summed area under the curve value of 1,355.83; 0.01 μM As treatment groups had an average summed area under the curve value of 1,366.21; 0.1 μM As treatment groups had an average summed area under the curve value of 1,326.24, 1.0 μM As treatment group had an average summed area under the curve value of 1,295.10; and finally the 10 μM As treatment group had an average summed area under the curve value of 535.12, which was statistically lower compared to all other groups (p < 0.05) (Figure 3). R-program for statistical analysis was used to run a one-way ANOVA with a Tukey post hoc adjustment to determine statistical differences among treatment groups (p < 0.05).
Figure 1. Schematic representation of the scratch assay.
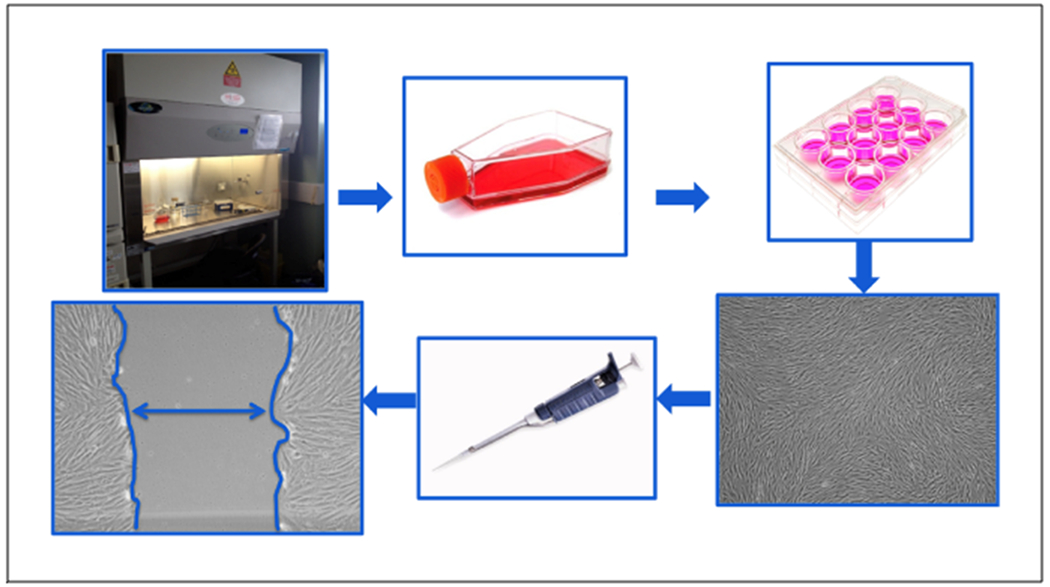
All work is done within an aseptic field to decrease the chance of contamination. Cells are seeded at a desired density in tissue culture flasks and grown in an incubator set to human physiologic conditions (5% CO2, 37° C). Upon reaching a desired confluence, cells are aseptically sub-cultured into 12-well tissue culture plates at a desired seeding density. Cells are grown to 100% confluence in each well of the 12-well plate and scratched using a sterile pipet tip. This “scratch” creates an in vitro mock wound (denoted by double headed arrow), in which cells naturally migrate to close the open area. Each well can be uniquely conditioned and cellular migration rates can be observed and quantified in response to different treatment conditions.
Figure 2. Representative scratch assay images over a 20-hour period.

Images 1A-4A represent cellular migration of control cells, images 1B-4B represent cellular migration of cells contaminated with 1.0 μM As, images 1C-4C represent cellular 424 migration of cells contaminated with 10 μM As. From these images, it is clear that cellular migration is slowed in the presence of arsenic over a 20-hour period. The control images show a completely “healed” scratch after 20 hours, while the other two As-treatment groups were not “healed”. The 10 μM As treatment inhibited complete closure altogether.
Figure 3. Arsenic slows cellular migration using the scratch assay.
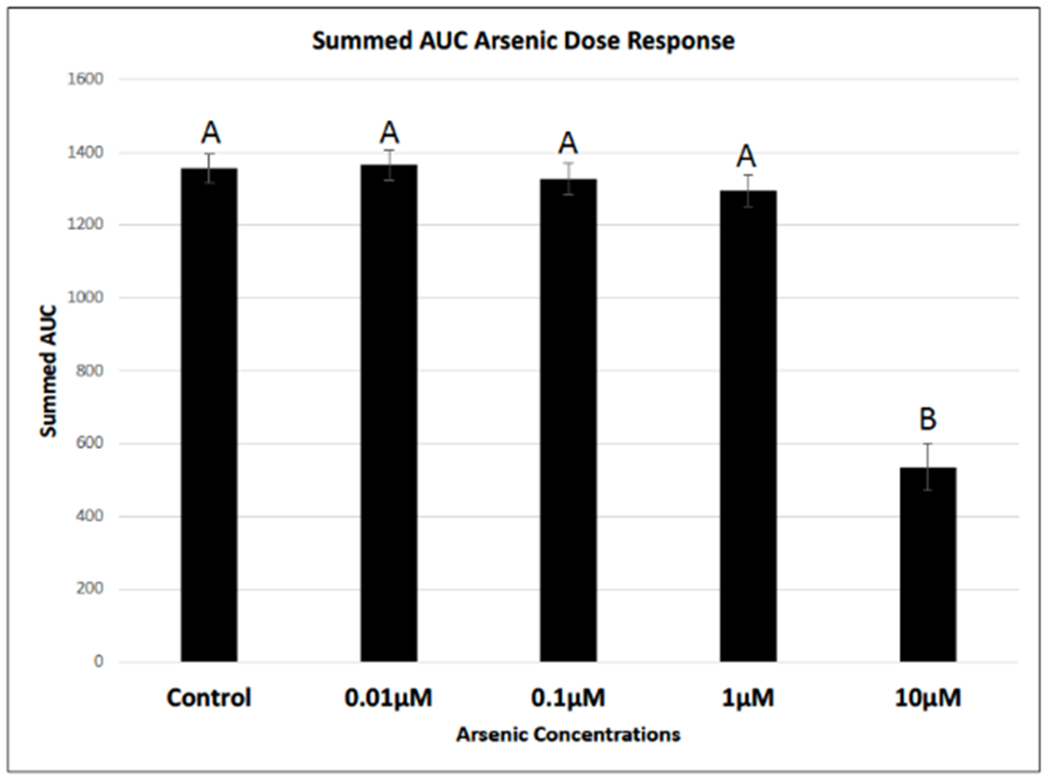
Images were captured over a 24-hour period to observe changes to cellular migration rates in the presence of varying concentrations of arsenic. Raw images were analyzed using an automated MatLab algorithm, which initially measured wound width, then calculated percent closure and finally area under the curve (AUC). 10 μM As treatment group statistically slowed cellular migration of human dermal fibroblasts neonatal (hDFn) compared to all other treatment groups using a one-way ANOVA with a Tukey post-hoc adjustment (p<0.05). Treatments that do not share a common letter are statistically significant from each other (p<0.05).
DISCUSSION:
Application of this assay provides a high-throughput and timely evaluation of cellular migration and may directly translate to complex physiological systems5,6,7. For this reason, the proposed bench-top model has been tuned and practiced to evaluate the effects of various therapeutic agents and environmental toxins on wound healing6. The concentrations of arsenic used in the current study were deemed environmentally relevant through extensive literature search of known exposure levels, and from retrospective review of various data bases14–21. Use of this in vitro scratch assay demonstrated that exposure to an arsenic concentration within the high, but environmentally relevant range, delayed wound closure (cellular migration).
This bench-top model was also employed to isolate and study an individual cell line (fibroblast) to further understand how fibroblast migration contributes to wound healing outcomes. Furthermore, it is difficult to study the functions of an individual cell line in complex in vivo systems with many other cells and tissues present that can interfere with the analysis. Additionally, the assay provides a means of achieving semi-quantitative percent wound closure data that can be used to target specific cellular mechanisms through which compounds may influence wound healing. For example, cells used in the scratch assay can be collected and stored in a desired reagent and used in gene expression (signal mRNA) or protein expression assays22. This information can be useful to the investigator if they seek to understand what signals or proteins may be largely contributing to changes in cellular migration in the presence of varying treatments (i.e., arsenic)22.
Human neonatal dermal fibroblasts were selected for this work based on their prominent role in wound healing. However, this scratch assay protocol has been implemented using various cell types and for a range of research purposes. For example, the scratch assay has been adopted into the field of oncology for studying migration inhibition of chemotherapy drugs23; for skeletal muscle cell migration, proliferation, and diffusion24; to study the effect of plant extracts on cellular migration25; and to understand how keratinocyte migration, proliferation, and to wound healing9. Additionally, a previous paper by Pinto et al. evaluated the effects of uranium (U) and platelet-rich plasma (PRP) on hDFn migration using the scratch assay6. The purpose of this work was to understand the extent to which this assay could test the effect of an agent thought to slow cellular migration (U), as well as an agent thought to speed up cellular migration (PRP). As can be seen in the aforementioned work and research by other investigators, application of the scratch assay has high potential for use in various experimental models26. In spite of the detail provided within the methods, variance should be expected between investigators and experiments. For example, cellular migration rates will likely fluctuate based on the cell line in use, in addition to the required micronutrients needed for unique cell types. Many of the important observations to aid in cell yield and viability are listed as NOTEs throughout this protocol. However, it is strongly recommended that investigators follow manufacturer’s recommendations for optimal growth conditions, and to use this method as supplementary instruction for general cell culture work.
Although the scratch assay is extremely versatile for study of cellular activity; unforeseen biases may arise in some measuring techniques. For example, when using Image J to manually trace cell migration fronts, variation may exist among users, which can limit accuracy and representative data collection. It should also be noted that migration fronts should be read blind to the treatment to minimize user bias. Furthermore, manually identifying migration fronts may result in miscalculation of average distance traveled due to pseudopod formation by cells and their ability to reach out to form focal adhesions, which can be visually misleading. In like manner, the use of fluorescent “cell tracker” dyes on cell surface or nuclear proteins to detect migration may unintentionally fix cells resulting in inaccuracies when measuring cellular migration over time. It is recommended that appropriate positive and/or negative controls be enrolled within the experiment to completely assess the effect of treatment on the cell lines. Practice of this assay must be completed in recognition of its limitations. This assay does not guarantee that cellular migration results obtained from in vitro experimentation will directly translate to in vivo outcomes. Would healing and cellular migration in a complex living system involves a multitude of cell types and molecular signals; each of which has the potential to influence healing responses.
In summary, the scratch assay has been found to provide high-throughput screening of cellular migration in response to changing environments6,25. We specifically utilized this protocol to successfully detect changes in cellular migration as a result of arsenic exposure at various environmentally relevant doses. These data have provided justification to use the scratch assay to further study and evaluate the mechanistic pathways of cellular migration in this assay and how these pathways are altered by arsenic exposure.
Movie 1.
Table 1. Arsenic stocks and serial dilutions.
This table explains the calculations used to create the As stock solutions and working stock solutions for treatment groups.
| 1000 μM As Stock Calculations | Arsenic Working Stock Concentrations | M1V1=M2V2 | Volume of Previous Arsenic Solution | Volume of Media | Total Volume of Arsenic Working Stock |
|---|---|---|---|---|---|
| NaAsO2 molecular weight: 129.9 g/mol | 10 μM As | (x mL)(1,000 μM) = (30 mL)(10 μM) x = 0.3 mL | 0.3 = 300 μL of 1mM As solution | 29.7 mL media | 30 |
| .03 L x .001 mol/L x 130 g/mol = 3.9 mg of Arsenic | 1.0 μM As | (x mL)(10 μM) = (30 mL)(1 μM) x = 3 mL | 3 mL of 10 μM As solution | 27 mL of media | 30 |
| Dilute 3.9 mg of sodium arsenite into 30 mL media | 0.1 μM As | (x mL)(1 μM) = (30 mL)(0.1 μM) x = 3 mL | 3 mL of 1 μM As | 27 mL of media | 30 |
| 0.01 μM As | (x mL)(0.1 μM) = (30 mL)(0.01 μM) x = 3 mL | 3 mL of 0.1 μM As | 27 mL of media | 30 |
Table of Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Class II A/B3 Biological Safety Cabinet | Forma Scientific | 15201-816 | |
| Nitrile Gloves | Kimberly-Clark | 55082 | |
| Water Bath | Precision | Model 182 | |
| Inverted Microscope | Leica | Q080318 | |
| CPR Centrifuge | Beckman | 349702 | |
| Analytical Scale | Ohaus | I093 1125062111P | |
| Weighting paper | VWR | H351125781212A | |
| Biohazard bags | Fisherbrand | 01-830A | |
| Water Jacketed CO2 Incubator | Thermo Scientific | 309739-31053 | |
| Hanks Balanced Salt Solution | Gibco | 14175-095 | |
| Dulbecco’s Modified Eagle Medium | Gibco | 10566-016 | |
| Fetal Bovine Serum | Gibco | 26140-079 | |
| Automatic Pipettor | Drummond | 140685 | |
| p200 Pipette | Gilson | ||
| p20 Pipette | Gilson | ||
| Denominator | Fisher Scientific | D59707 | |
| Trypan Blue Stain | Gibco | 15250-061 | |
| T75 tissue flask | Corning | 430725U | |
| 15 mL Conical Tube | Fisherbrand | 05-539-5 | |
| 50 mL Conical Tube | VWR | 21008-178 | |
| 5 mL Pipette | Fisherbrand | 13-678-11D | |
| 10 mL Pipette | Fisherbrand | 13-678-11E | |
| Tissue Marker | Securline | 92167 | |
| Wipes | Kintech | 34155 | |
| 70% Ethanol | Fisher | 63-67-0 | |
| Non-Filter Pipet Tips | Fisherbrand | 16160210 | |
| Bleach | Great Value | 220-04 | |
| 96-well | Falcon | 353915 | |
| Crovial Rach | Nalgene | 5030-0505 | |
| Hemacytometer | Gizmo Supply | B00SCOGY56 | |
| TrypLE Express | Gibco | 12605-028 | |
| Conical Tube Rack | n/a | n/a | |
| 12-well plate | Corning | 3598 | |
| Waste Beaker | n/a | n/a | |
| 1 mL Pipette | Fisherbrand | 13-678-11B | |
| Straight Edge Ruler | n/a | n/a | |
| Human neonatal dermal fibroblasts (hDFn) | lot: 1734, 2909 | 106-05n | |
| Rstudio Statistical Software | ver: 3.5.1 | ||
| ImageJ | NIH | ver: 1.8.0_112 |
ACKNOWLEDGMENTS:
Research reported in this publication was supported by the National Institute on Minority Health and Disparities of the National Institutes of Health under Award Number U54MD012388. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Diegelmann R, Evans M Wound healing: an overview of acute, fibrotic and delayed healing. Frontiers in BioScience. 9, 283–289 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Braiman-Wiksman L, Solomonik I, Spira R, et al. Novel insights into wound healing sequence of events. Toxicologic Pathology. 35, 767–779 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Michelson A Platelets 3rd edition Waltham, Massachusetts: (2013). [Google Scholar]
- 4.Aikawa M, Schoenbechler MJ, Barbaro JF, Sadun EH Interaction of rabbit platelets and leukocytes in the release of histamine. Electron microscopic observations. American Journal of Pathology. 63 (1), 85–98 (1971). [PMC free article] [PubMed] [Google Scholar]
- 5.Liang C, Park A, Guan J In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nature Protocols. 329–333 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Pinto B, et al. A bench-top in vitro wound assay to evaluate wound closure. Applied In Vitro Toxicology. 2 (3), 151–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinto B, et al. Estrogen mitigates the negative effects of arsenic contamination in an in vitro wound model. Applied In Vitro Toxicology. 4 (1), 24–29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roubelakis M, et al. Platelet-rich plasma (PRP) promotes fetal mesenchymal stem/stromal cell migration and wound healing process. Stem Cell Reviews and Reports. 10 (3), 417–428 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Shibata S, et al. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway. Journal of Immunology. 189 (6), 3231–3241 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Mendonca RJ, Coutinho-Netto J Cellular aspects of wound healing. Anais Brasileiros de Dermatologia. 84 (3), 257–262 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Beem E, Segal MS Evaluation of stability and sensitivity of cell fluorescent labels when used for cell migration. Journal of Fluorescence. 23 (5), 975–987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gentry PR, Clewell III HJ, Greene TB, et al. The impact of recent advances in research on arsenic cancer risk assessment. Regulatory Toxicology and Pharmacology. 69, 91–104 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Beamer PI, Klimecki WT, Loh M, et al. Association of children’s urinary CC16 levels with arsenic concentrations in multiple environmental media. International Journal of Environmental Research and Public Health.pii: E521.2016:13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson FN Arsenic in ground-water under oxidizing conditions, southwest United States. Environmental Geochemistry and Health. 11, 3–4 (1989). [DOI] [PubMed] [Google Scholar]
- 15.Foust RD, Mohapatra P, Compton-O’Brian AM, et al. Groundwater arsenic in the Verde Valley in central Arizona, USA. Applied Geochemistry. 19, 251–255 (2004). [Google Scholar]
- 16.Uhlman K Arsenic in Arizona Ground Water: Source and Transport Characteristics. Arizona Cooperative Extension, AZ 1453 The University of Arizona, Tucson, AZ: (2008). [Google Scholar]
- 17.Agusa T, et al. Human exposure to arsenic from drinking water in Vietnam. Science of the Total Environment. 488-489, 562–569 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Ayotte JD, et al. Factors affecting temporal variability of arsenic in groundwater used for drinking water supply in the United States. Science of the Total Environment.(2014). [DOI] [PubMed] [Google Scholar]
- 19.Dummer TJ, et al. Geostatistical modeling of arsenic in drinking water wells and related toenail arsenic concentrations across Nova Scotia, Canada. Science of the Total Environment. (2014). [DOI] [PubMed] [Google Scholar]
- 20.Sorg TJ, Chen AS, Wang L Arsenic species in drinking water wells in the USA with high arsenic concentrations. Water Research. 48, 156–169 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Beamer PI, et al. Association of Children’s Urinary CC16 Levels with Arsenic Concentrations in Multiple Environmental Media. International Journal of Environmental Research and Public Health. 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pomari E, Dalla Valle L, Pertile P, Colombo L, & Thornton M Intracrine sex steroid synthesis and signaling in human epidermal keratinocytes and dermal fibroblasts. The FASEB Journal. 29 (2), 508–524 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Gotsulyak Y, Kosach R, Cherednyk V, Tykhonkova O, Khoruzhenka I Optimization of cell motility evaluation in scratch assay. Biopolymers and Cell. 30, 223–228 (2014) [Google Scholar]
- 24.Goetsch KP, Niesler CU Optimization of the scratch assay for in vitro skeletal muscle wound healing analysis. Analytical Biochemistry. 411, 158–160 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Harishkumar M, et al. Revealing the mechanism of in vitro wound healing properties of citrus tamurana extract. BioMed Research International. 13, Article ID 963457, 8 pages (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnston ST, Simpson MJ, McElwain DLS How much information can be obtained from tracking the position of the leading edge in a scratch assay? Journal of the Royal Society Interface. 11, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]