

Abstract
Endothelium-derived steroidogenic factor (EDSF) is an endothelial peptide that stimulates aldosterone release from bovine adrenal zona glomerulosa (ZG) cells. The regulation of aldosterone release by combinations of EDSF and angiotensin II (AII) or EDSF and ACTH was investigated. Endothelial cells (ECs) and EC-conditioned media (ECCM) increased aldosterone release from ZG cells, an activity attributed to EDSF. AII (10−12 to 10−8m) and ACTH (10−12 to 10−9m) also stimulated the release of aldosterone from ZG cells. The stimulation by AII, but not ACTH, was greatly enhanced when ZG cells were coincubated with ECs. AII was metabolized by ECs to peptides identified by mass spectrometry as angiotensin (1-7) and angiotensin IV. There was very little metabolism of AII by ZG cells. Neither of these two AII metabolites altered aldosterone release from ZG cells, so they could not account for the enhanced response with ECs. AII-induced aldosterone release from ZG cells was enhanced by ECCM but not cell-free conditioned medium. This enhanced response was not due to increased EDSF release from ECs by AII. The synergistic effect of EDSF and AII was apparent when AII was added during or after the generation of ECCM and not observed when the AII component of the enhancement was blocked by the AII antagonist, losartan. These studies indicate that EDSF enhances the steroidogenic effect of AII. In the adrenal gland, ECs are in close anatomical relationship with ZG cells and may sensitize ZG cells to the steroidogenic action of AII by releasing EDSF.
ENDOTHELIAL CELLS (ECS) release a factor that stimulates aldosterone release by zona glomerulosa (ZG) cells (1, 2). This factor has been named endothelium-derived steroidogenic factor (EDSF) and is a peptide discrete from endothelin-1 (ET-1), bradykinin (BK), angiotensin II (AII), and ACTH (2). The adrenal cortex is highly vascularized, with steroidogenic cells surrounding arterioles, capillaries, and sinusoids (3, 4). As a result, there is a close anatomical association of steroidogenic cells such as ZG cells with ECs (3, 4). This close relationship suggests a role for EDSF in the paracrine regulation of aldosterone release. Cultured ECs isolated from bovine coronary artery and adrenal cortex are capable of producing EDSF (1, 2). However, this activity was not produced by adrenal fibroblasts or pericytes. Thus, it was specific for ECs. The stimulation of aldosterone release occurs with direct exposure of ZG cells to ECs as well as with the transfer of EC-conditioned medium (ECCM) to ZG cells (1, 2). The factor is released within 30 min of EC incubation and stimulates aldosterone release to approximately 70% of a maximal stimulatory response to AII (1, 2).
The regulation of aldosterone release from ZG cells is a complex process involving interactions between a variety of stimulatory and inhibitory factors. The primary stimuli for aldosterone release are AII, ACTH, and potassium ion (5, 6). However, BK, ET-1, and various prostaglandins also stimulate aldosterone release (7–11). Previous studies indicate that some of the stimulators of aldosterone release interact synergistically and enhance each other’s effect. For example, potassium ion will enhance the steroidogenic effects of AII in vivo and in vitro (12–15). AII increases the plasma aldosterone concentration more in rats on a low-sodium diet than rats on a normal diet (16). ET-1 enhanced ACTH and 8-bromo-cyclic AMP-stimulated aldosterone release in cultured ZG cells but did not enhance the response to AII in the same study (17). In contrast, Cozza et al. (18) found that ET-1 enhanced AII-stimulated, but not potassium-stimulated aldosterone release. The mechanism for ET-1 potentiation of AII-stimulated aldosterone involved the activation of protein kinase C. Thus, the synergistic interactions of these peptides may involve increasing steroidogenic enzyme activity or alteration of signal transduction pathways. AII promotes extracellular calcium influx, and this increase in cellular calcium interacts synergistically with the ACTH-induced production of cAMP to increase the release of aldosterone in ZG cells (19). Finally, both AII-stimulated aldosterone release and increase in intracellular calcium were synergistically enhanced by PTH (20). Thus, the regulation of aldosterone release is modulated by the interactions between several secretagogues, many of which interact in synergistic fashion.
This study examined the interactions between EDSF and AII or ACTH in stimulating aldosterone release. The purpose of this study was to test the hypothesis that EDSF interacts with AII and ACTH to potentiate the release of aldosterone from ZG cells. These studies indicate that ECs and ECCM enhance the steroidogenesis by AII but not ACTH.
Materials and Methods
Cell culture
Fresh bovine adrenal glands were obtained from a local abattoir, placed in ice-cold buffer, and transported immediately to the laboratory. This procedure was reviewed and exempted by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Adrenal ZG cells were cultured from the fresh adrenals as described previously by Rosolowsky and Campbell (1, 2). ZG cells were plated at a density of 200,000 cells per well in 24-well tissue culture-treated plates and maintained in culture for 3 to 6 d. Bovine adrenal capillary ECs were cultured as previously described (2, 21). Adrenal cortical slices were prepared, and the ECs were removed by collagenase digestion. The purity of EC cultures was determined by their characteristic cobblestone morphology, as well as by uptake of diI-acetylated low-density lipoprotein, and lack of uptake of smooth muscle α-actin. The estimated purity was greater than 95%. ECs were fed every 2 d and were used after reaching 80–100% confluence.
EC/ZG cell coincubation experiments
ECs were grown to confluence on 225-cm2 tissue culture-treated flasks. Direct EC/ZG coincubations were performed by incubating subconfluent flasks of ECs with 100 mg of Cytodex microcarrier beads as previously described (1, 2). After 24 h, the ECs had coated the microcarrier beads and were used for experiments. The EC-coated beads were removed from the underlying monolayer by gentle washing and were then transferred to a sterile tube containing RPMI 1640 medium. The EC-coated beads were allowed to settle, and the medium was decanted. A second wash with RPMI 1640 medium was performed, and the beads were then suspended at 5–10 mg/ml in ZG steroidogenic medium consisting of modified Ham’s F-12 medium supplemented with 14 mm NaCl, 14 mm NaHCO3, 0.2% BSA, and 1.8 mm CaCl2. Microcarrier beads that had not been incubated with ECs were treated in an identical manner and used as a cell-free control. Suspensions of EC-coated beads or cell-free beads were transferred to multiwell plates of ZG cells, and varying concentrations of AII (10−12 to 10−8m) or ACTH (10−12 to 10−9m) were added. Coincubations of EC/ZG cells were maintained at 37 C in 5% CO2 in air for 2 h. The medium was then removed and stored at −40 C for subsequent aldosterone assay.
Conditioned-medium (CM) experiments
ECCM and cell-free conditioned medium (CFCM) were produced by incubating flasks or 12-well tissue culture-treated plates with Ham’s F12 medium supplemented with 14 mm NaCl, 14 mm NaHCO3, and 0.2% BSA. Confluent 225-cm2 flasks of ECs were washed once with 25 ml of CM and incubated with 20 ml of fresh medium. The cells were allowed to incubate for 1 or 2 h. The ECCM was then removed and stored in sterile aliquots at 4 C or directly transferred to ZG cells. CFCM was produced in cell-free culture flasks or plates and stored in an identical fashion. Before the start of a ZG cell experiment, ZG cells were washed twice with Ham’s F-12 medium supplemented with 14 mm NaCl, 14 mm NaHCO3, and 0.1% BSA. Cells were allowed to incubate in this wash buffer for 2 h to remove antioxidants present in the normal feed media. The wash buffer was then removed and replaced with ECCM or CFCM. ZG cells were stimulated with vehicle, AII, or ACTH and incubated for 2 h at 37 C. After the incubation period, the medium was removed and stored frozen at −40 C for subsequent aldosterone assay. In some experiments, ECCM was diluted with varying amounts of CFCM to achieve mixtures of 0–100% ECCM. The effect of AII on EC production of EDSF was examined by adding vehicle or AII to ECs on beads or cell-free beads during the conditioning period. After conditioning for 1 h, the CMs were removed and transferred to ZG cells that had been pretreated with the AII antagonist losartan (10−6m) or vehicle for 15 min. Losartan was added to the ZG cells to prevent the stimulation of aldosterone release by AII in the CM. The CM-treated ZG cells were incubated for 2 h and the media assayed for aldosterone.
Metabolism of 125I-AII by ECs
ECs were grown to confluence in 25-cm2 flasks. The flasks were washed twice with modified Ham’s F-12 medium supplemented with 14 mm NaCl, 14 mm NaHCO3, and 0.1% BSA. The medium was removed and replaced with 5 ml of Ham’s F12 medium supplemented with 14 mm NaCl, 14 mm NaHCO3, 0.2% BSA, and 130,000 cpm 125I-AII. The ECs were incubated at 37 C for 2 h, after which the medium was supplemented with 1000 ng of unlabeled AII and frozen in a dry ice/methanol bath. The medium was extracted over Baker C18 solid-phase extraction columns and washed successively with 10 ml each of water and 4% glacial acetic acid in water. The AII was eluted with 4% glacial acetic acid in ethanol and dried under nitrogen. AII metabolites were resolved by reverse phase HPLC using a Nucleosil C18 column and a linear elution gradient of 0–40% acetonitrile in 50 mm sodium phosphate buffer over 45 min. Metabolites of AII were identified by collecting 0.2-ml fractions of eluant and measuring radioactivity on a Packard γ-scintillation counter.
Identification of EC metabolites of AII by mass spectrometry (MS)
Analyses were carried out with a Waters-Micromass Quattro electrospray ionization-triple quadrupole MS/MS system coupled with a Waters 2695 high-performance liquid chromatograph (Waters Corporation, Milford, MA). The angiotensin peptides were resolved on a reversed-phase C18 column (Jupiter 2.0 × 250 mm, 5 μm; Phenomenex, Torrance, CA) using water-acetonitrile with 0.1% formic acid as a mobile phase at a flow rate of 0.2 ml/min. The mobile phase gradient started at 15% acetonitrile, linearly increased to 35% in 15 min, and then increased to 100%. All mass spectrometric experiments were performed in the positive ion mode using a capillary voltage of 3.5 kV, desolvation temperature of 350 C, desolvation gas flow of 600 liters/h, and source temperature of 130 C. The collision-induced dissociation was conducted using argon at 1.5 × 10−3 Pa. The angiotensin peptides show strong (M + 2H)2+ ions [m/z 450.0, 466.1, 523.5, 388.0, and 484.8 for angiotensin (1-7) or A (1-7), angiotensin III (AIII), AII, and angiotensin IV (AIV), and the Sar1 Ile8-AII internal standard, respectively]. Using these ions, the peptides were detected individually by the selected ion monitoring (SIM) mode using three SIM channels [channel 1, 0–4.6 min, for A (1-7) (m/z 450.0); channel 2, 4.6–6.5 min, for the internal standard Sar1 Ile8-AII (m/z 484.8); and channel 3, 6.5–12 min, for AIII, AII, and AIV (m/z 466.1, 523.5 and 388.0)]. The angiotensin peptides were separated by HPLC with elution times of 4.07, 7.37, 8.43, and 9.88 min for A (1-7), AIII, AII, and AIV, respectively. For MS/MS analysis, the (M + 2H)2+ ions underwent collision-induced dissociation to produce specific daughter ions of m/z 646.7, 484.8, 211.2, 110.0, and 262.9 for A (1-7), Sar1 Ile8-AII (internal standard), AIII, AII, and AIV, respectively. These ions were also used to detect the peptides individually and confirm their identity using the multireaction monitoring (MRM) mode.
Aldosterone assay
Aldosterone was measured by direct RIA or ELISA as previously described (1, 2, 22). Data represent averages of multiple incubations from at least two cell preparations or are a representative experiment from multiple cell preparations.
Materials
Reagents were obtained as follows: Ham’s F12 media and Cytodex 3 microcarrier beads, Sigma Chemical (St. Louis, MO); RPMI 1640 media, Life Technologies, Inc. (Grand Island, NY); fetal bovine serum, Hyclone (Logan, UT); AII, Beckman (Palo Alto, CA); 125I-AII, NEN Life Science Products (Boston, MA); losartan and angiotensin peptides, Bachem Biosciences Inc. (King of Prussia, PA); and Nucleosil C18 extraction columns, Varian Inc. (Palo Alto, CA). All other reagents were purchased from Sigma Chemical.
Results
Increasing concentrations of AII stimulated ZG cell aldosterone release in cell-free and EC coincubations (Fig. 1A). Experiments were performed with 1 mg of EC-coated beads to keep the EDSF activity low and prevent EC stimulation of aldosterone release from obscuring the stimulation by AII. EC treatment alone stimulated aldosterone release approximately 2-fold. No aldosterone was detected in ECs incubated in the absence of ZG cells as previously described (1, 2). The stimulation of aldosterone release by AII was enhanced by the presence of ECs on beads but not by cell-free beads. The AII concentration-response curve was shifted to the left by EC coincubation, and aldosterone release was maximal with 10−10m AII. The maximal response to AII was 4-fold greater in the presence of ECs on beads than with the cell-free bead control. At higher concentrations of AII, the enhanced effect was not apparent, and responses were not significantly different between EC-coated beads and cell-free beads.
Fig. 1.
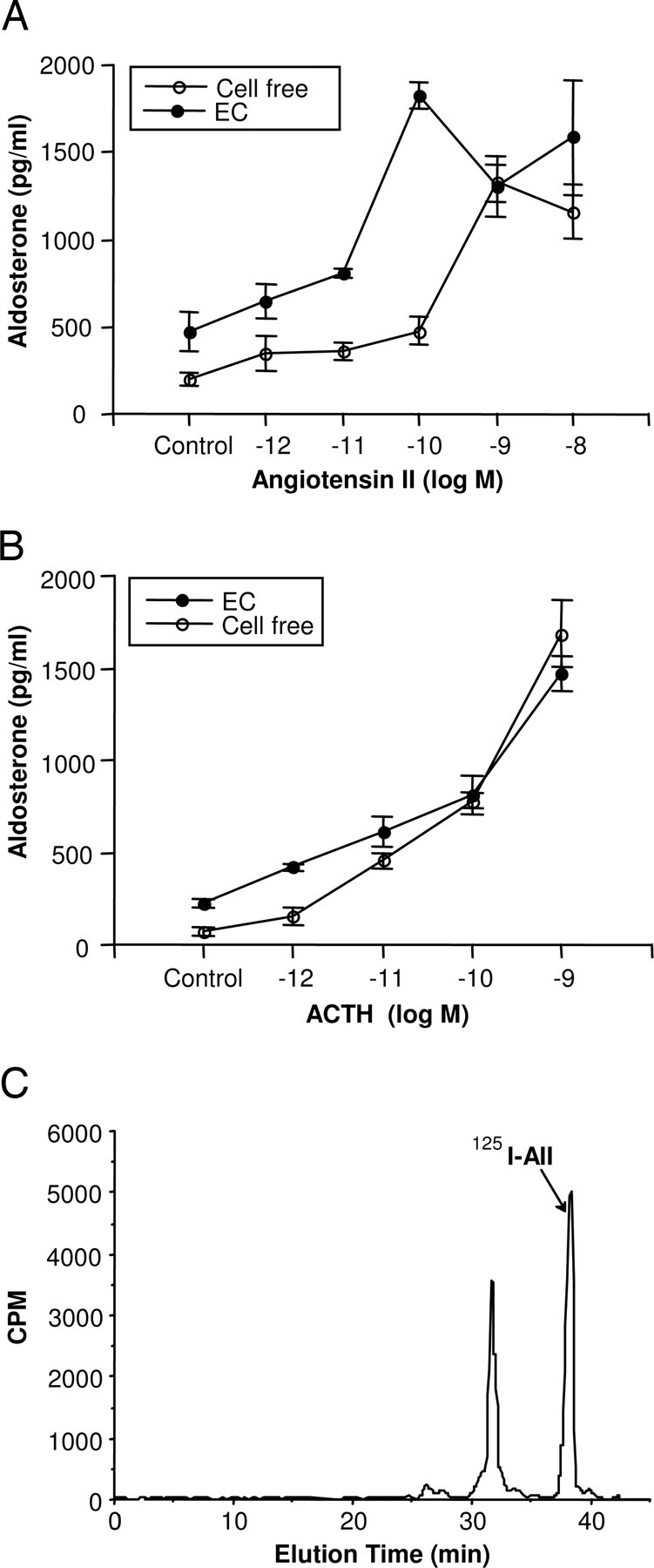
Effect of ECs on AII and ACTH stimulation of aldosterone release from ZG cells. Microcarrier beads coated with ECs or cell-free beads (1 mg) were transferred to wells of ZG cells permitting coincubation of the two cell types. AII or ACTH was added, and the incubation continued for 2 h at 37 C. Medium was then removed and assayed for aldosterone. Results are shown with AII (A) and ACTH (B) and expressed as the mean ± sem for n = 5–6. C, Metabolism of AII by ECs. ECs were incubated with 125I-AII for 2 h at 37 C. The medium was then removed, and the angiotensin peptides were separated by reverse phase HPLC.
ACTH stimulated aldosterone release in a concentration-related manner from ZG cells (Fig. 1B). This stimulation was only slightly increased at lower concentrations of ACTH by coincubation with ECs. EC coincubation itself increased aldosterone release by 3-fold, whereas 10−12m ACTH stimulated aldosterone release only slightly greater in ZG cells coincubated with ECs than in the cell-free control.
Because the AII metabolite AIII is more potent than AII in stimulating aldosterone release (23, 24), we wondered if the metabolism of AII to a more potent peptide might explain the enhanced response to AII in the presence of ECs. ECs were incubated with 125I-AII, and the AII metabolites were resolved by HPLC. 125I-AII had an elution time of 37 min in the cell-free control incubation (data not shown). When ECs were incubated with 125I-AII, the 37-min peak of 125I-AII was detected along with several unidentified, radiolabeled peaks, indicating EC-mediated degradation of 125I-AII (Fig. 1C). The major additional peak eluted at 32.5 min and accounted for 40% of the added 125I-AII. The earlier elution times of these peaks indicate that ECs metabolize 125I-AII to more polar products. The identity of these products could not be determined because the 125I-peptides did not coelute with the noniodionated angiotensins. As a result, we used liquid chromatography/tandem MS (LC/MS/MS) to identify the AII metabolites and confirm the metabolism of AII by ECs. ECs or ZGs were incubated with AII, and the incubation media analyzed by LC/MS/MS using SIM and MRM modes. ECs metabolized AII to two metabolites that comigrated with A (1-7) and AIV and had the same (M + 2H)2+ ions as these peptides (Fig. 2A). Further analysis of the peptides by LC/MS/MS using the MRM mode confirmed the identity of these peptides. The major endothelial metabolite of AII was A (1-7). When ZG cells were incubated with AII under the same conditions, there was very little metabolism of the peptide. A (1-7) and AIV were formed as minor metabolites, and AIII was not detected (Fig. 2B).
Fig. 2.

Identification of endothelial and ZG cell metabolites of AII by LC/MS/MS. ECs or ZG cells were incubated for 1 h at 37 C with AII (10−7m). The media were removed, extracted, and analyzed by LC/MS/MS. SIM ion chromatograms of AII metabolites of ECs (A) and ZG cells (B) monitoring specific (M + 2H)2+ ions for A (1-7), Sar1 Ile8-AII (internal standard), AIII, AII, and AIV. Elution times of known angiotensin standards are shown above the chromatograms. MRM ion monitoring of MS/MS daughter ions of A (1-7), AIII, AII, and AIV was used to confirm the identity of the angiotensin peptides (see Materials and Methods).
Both A (1-7) and AIV were tested for their ability to stimulate aldosterone release by ZG cells. Neither A (1-7) nor AIV (10−11 to 10−8m) changed aldosterone release (data not shown). Thus, the metabolism of AII to A (1-7) and AIV by ECs represents an inactivation pathway in the adrenal cortex and cannot explain the ability of ECs to enhance AII-stimulated aldosterone release.
The AII concentration-response experiment was repeated using ECCM in place of EC coincubation (Fig. 3). ECCM was diluted 1:4 with CFCM and caused a 2-fold increase in aldosterone release from ZG cells. Increasing concentrations of AII stimulated aldosterone release in ZG cells treated with both CFCM and ECCM (Fig. 3A). The AII stimulation was greatly enhanced by ECCM compared with CFCM as indicated by a leftward shift of the concentration-response curve with ECCM. The maximal effect occurred with 10−10m AII. Stimulation of ECCM-treated ZG cells with 10−10m AII resulted in aldosterone release that was 11-fold greater than the CFCM control. There was no detectable aldosterone release in the direct assay of ECCM.
Fig. 3.
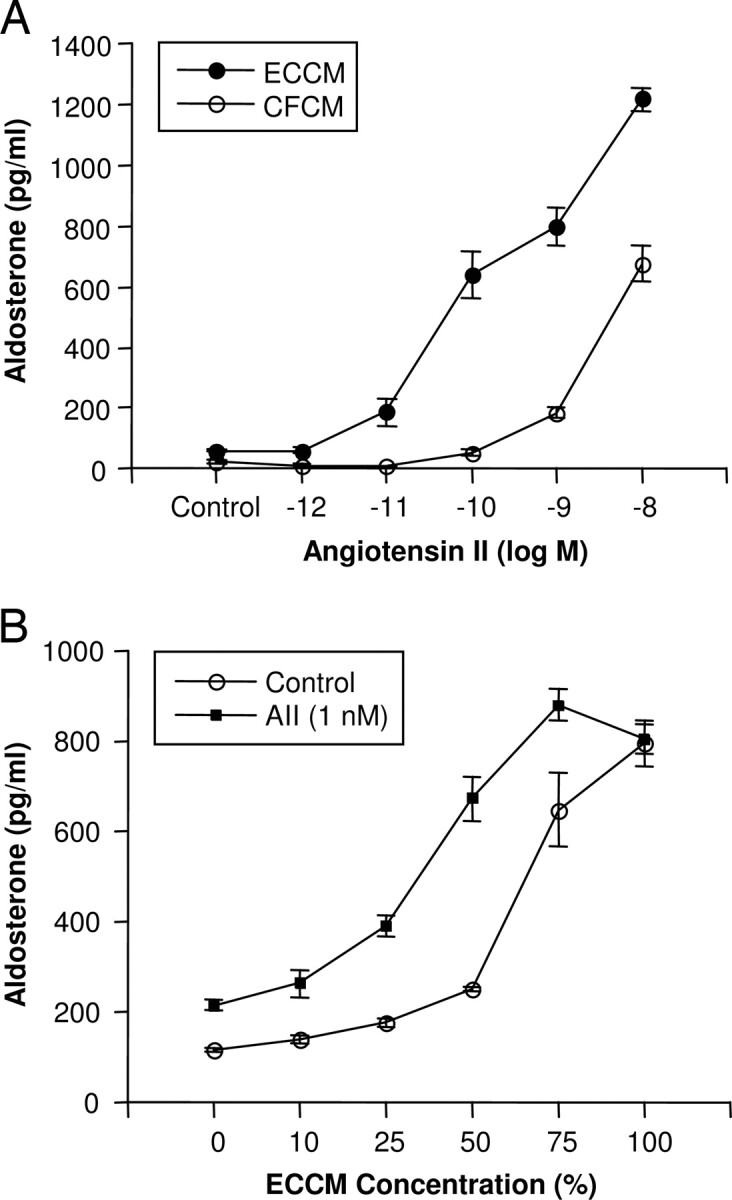
Effect of ECCM on AII stimulation of aldosterone release from ZG cells. ECCM or CFCM was produced by incubating ECs for 2 h at 37 C. A, ZG cells were incubated in 0.5 ml ECCM or CFCM, and AII was added. B, ECCM was diluted to the indicated concentrations with CFCM. ZG cells were incubated in 0.5 ml of the various dilutions and 1 nm AII or vehicle for 2 h. Medium was removed and assayed for aldosterone. Results are the mean ± sem for n = 6.
The effects of varying the ECCM concentration were determined by diluting ECCM with CFCM (Fig. 3B). ECCM stimulated aldosterone release in a concentration-related manner. Addition of 10−9m AII to 0% ECCM increased aldosterone release by 1.8-fold, whereas 100% ECCM could not be further stimulated by AII. Stimulation of ZG cells with 10−9m AII and 50% ECCM resulted in a 3-fold greater increase in aldosterone release than the vehicle control and 50% ECCM. These studies indicate that a soluble transferable factor, EDSF, mediates the ability of ECs to enhance AII-stimulated aldosterone release and further indicate that EC and ZG cell contact is not required for the activity.
ECs were treated with AII (10−10m) either during or after the conditioning period. Under both conditions, AII enhanced the aldosterone release from ZG cells by ECCM (Fig. 4A). However, the enhanced response was greater when AII was added to ECCM after the EC conditioning period. Addition of AII during EC conditioning resulted in a 4-fold increase in aldosterone release, whereas AII addition after EC conditioning resulted in a 12-fold increase in aldosterone release compared with cell-free controls. These findings further indicate that ECs metabolize AII to a less active peptide.
Fig. 4.
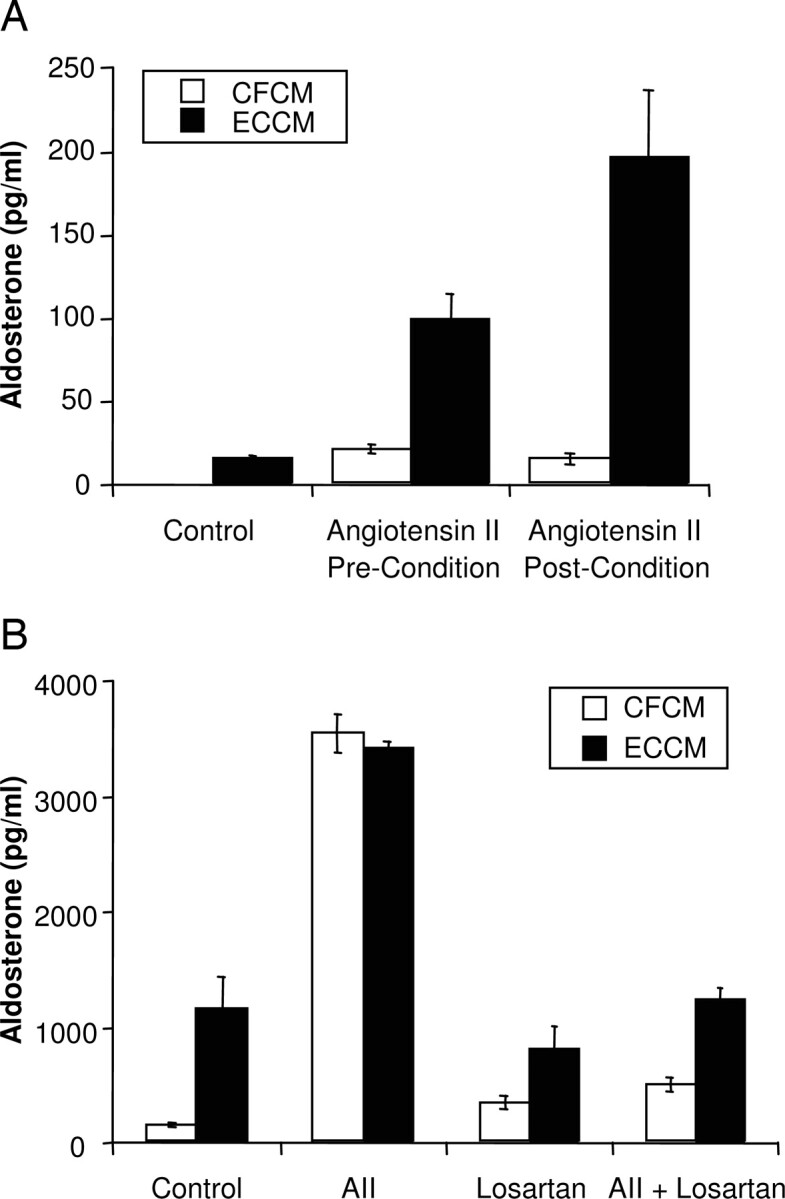
Effect of AII treatment on EDSF production by ECs. A, AII (10−10m) was added to the media before the conditioning period (pre-condition) or immediately after the conditioning period (post-condition). ZG cells were incubated in 1 ml of the CM for 2 h at 37 C. Medium was assayed for aldosterone. B, ECs or cell-free plates were conditioned as described with or without AII (10−10m). The CM were then transferred to wells of ZG cells that had been pretreated with losartan (10−6m) or vehicle for 15 min. ZG cells were incubated in the CM for 2 h at 37 C. Medium was assayed for aldosterone. Results are the mean ± sem for n = 6.
The possibility of AII directly stimulating the secretion of EDSF was examined by treating ECs with AII or its vehicle during the generation of CM. The ECCM was transferred to ZG cells pretreated for 15 min with vehicle or the AII antagonist losartan (10−6m). This concentration of losartan was previously determined to abolish AII-stimulated aldosterone release (1, 2). ECCM from vehicle-treated ECs increased aldosterone release in ZG cells pretreated with either vehicle or losartan (Fig. 4B). Stimulation with AII (10−10m) increased aldosterone release in ZG cells pretreated with vehicle but did not increase aldosterone release in ZG cells pretreated with losartan. AII (10−10m) was added to ECs during the conditioning period. Transfer of the AII-treated ECCM stimulated aldosterone release from ZG cells pretreated with vehicle to the same extent as AII in CFCM. In losartan pretreated ZG cells, the AII component was blocked so only the EDSF component remained. Under these conditions, AII-treated ECCM stimulated aldosterone release to the same extent as vehicle-treated ECCM. Thus, addition of AII to ECs during the conditioning period did not increase aldosterone release by stimulating more EDSF.
Discussion
Previous studies have demonstrated synergistic interactions between the various secretagogues of aldosterone release. For example, increasing the potassium concentration increases aldosterone release and enhances the ability of AII to stimulate aldosterone release (12–15). Similarly, ET-1 stimulates aldosterone release and enhances AII and ACTH-stimulated aldosterone release (7, 9, 17, 18). AII also enhances the aldosterone release by PTH and ACTH (19, 20). In this study, we characterized the effect of EDSF on AII and ACTH-stimulated aldosterone release. The study examined the EDSF response using EC/ZG coincubations as well as isolating the ZG from direct EC contact by adding ECCM to stimulate steroidogenesis.
For coincubation experiments, the number of ECs was selected that stimulated only moderate increases in aldosterone release. This allowed AII and ACTH to stimulate aldosterone release over a range of concentrations. The stimulation of aldosterone release by the combination of AII and EDSF was significantly greater than the sum of the stimulatory effect of AII and EDSF individually. This indicates a synergistic interaction. This synergism was particularly apparent at low concentrations of AII. These findings suggest that in the intact adrenal gland the close anatomical association between ECs and ZG cells may expose the ZG cells to EDSF and thereby enhance the sensitivity of ZG cells to AII stimulation of aldosterone release.
The enhanced steroidogenic action of AII in the presence of ECs led us to consider that ECs metabolize AII to another angiotensin peptide, and this peptide may have a greater steroidogenic effect than AII. For example, AIII is more potent than AII in stimulating steroidogenesis (23, 24). We investigated the metabolism of AII by ECs and ZG cells. When radiolabeled AII was incubated with ECs, several polar metabolites were detected indicating substantial metabolism of AII by ECs. Integration of the peak areas indicates that approximately 40% of the 125I -AII was metabolized during the course of a 2-h exposure to ECs. In additional studies, ECs were incubated with AII for 1 h, and the angiotensin peptide metabolites were identified by LC/MS/MS. These studies indicate that ECs metabolize AII predominantly to A (1-7) and to a lesser extent to AIV. Under identical conditions, there was little metabolism of AII by ZG cells. Neither of these angiotensins stimulated aldosterone release. Thus, the metabolism of AII to A (1-7) and AIV represents an inactivation pathway and cannot explain the ability of ECs to enhance AII-induced aldosterone release. Consistent with our findings, the conversion of AII to A (1-7) is catalyzed by angiotensin-converting enzyme 2, and this enzyme is present in ECs (25). ECs also release factors capable of inhibiting aldosterone release during conditioning. Endothelial nitric oxide release will inhibit ZG cell aldosterone release (22, 26). However, the short half-life of the nitric oxide in buffer makes it an unlikely inhibitory factor in CM (26).
When ECCM was transferred to ZG cells, it stimulated aldosterone release directly and enhanced the aldosterone release by AII. The ability of ECCM to enhance AII-stimulated aldosterone release in a similar manner to coincubations of ECs and ZG cells indicates that direct contact between ECs and ZG cells is not required and that the synergism is due to a soluble, transferable factor. This factor, EDSF, has been previously characterized as a peptide distinct from AII, ET-1, and BK (1, 2). The enhanced steroidogenic responses with ECCM were also apparent at low concentrations of AII and submaximal dilutions of ECCM.
Coincubations of ECs with ZG cells did not enhance the aldosterone stimulation by ACTH as they did with AII. A possible explanation for the selective effects of EDSF may be the interactions of the calcium signal transduction pathways. The mechanism of action of ACTH involves activation of adenylyl cyclase and increasing intracellular cAMP (27–29). In contrast, AII promotes aldosterone synthesis by increasing intracellular calcium (30–34). Thus, EDSF may amplify the calcium-mediated pathway but not the cAMP pathway. In ZG cells, EDSF caused a rapid increase in intracellular calcium that was maintained as a plateau (2). In contrast, AII caused a sharp, transient peak in intracellular calcium that was 7-fold greater than EDSF. This AII-stimulated calcium spike was followed by a plateau that approximated the plateau caused by EDSF. The stimulation of aldosterone release by EDSF was 70% of the increase caused by AII. These data, along with the synergistic nature of the EDSF and AII response indicate that EDSF must promote aldosterone biosynthesis by signaling mechanisms distinct from AII and may involve other transduction mechanisms in addition to an increase in intracellular calcium. It is possible that EDSF increases the expression of steroidogenic enzyme or AII receptors in ZG cells. However, ZG cells were only exposed to EDSF for 2 h. This short exposure time makes this mechanism unlikely. For example, cytokines increase the expression of AT1 receptors; however, an increase in receptor expression requires 6–12 h (35, 36). Similarly, expression of steroidogenic hydroxylase enzymes requires 6–10 h (37). Thus, the up-regulation of these proteins is too slow to explain EDSF enhancing the aldosterone stimulation by AII.
Our initial results suggested that exogenous AII may stimulate aldosterone release indirectly by acting on ECs to release EDSF as well as directly stimulating AII receptors on ZG cells. We considered the possibility that AII might stimulate the release of additional EDSF from ECs as a mechanism for the synergistic response. To examine this possibility, EDSF activity was monitored in ECCM generated in the presence and absence of AII. Any AII receptor isoform expressed on ECs would be activated during the generation of ECCM. Before transfer of ECCM to ZG cells, the AII antagonist, losartan, was added to the ZG cells to block their AT1 receptors and eliminate the direct contribution of AII in the ECCM. The aldosterone release due to the EDSF-component of ECCM could then be measured without the aldosterone release stimulated by AII present in ECCM. There was no difference in EDSF activity of ECCM generated in the presence or absence of AII. Thus, AII did not stimulate EDSF release from ECs.
The steroidogenic action of AII was enhanced by ECCM when AII was added either during or immediately after the conditioning period. These studies also indicate that contact between AII and ECs is not required for ECs to enhance the action of AII and support the conclusion that metabolism of AII by ECs is not involved in the synergistic response. However, the response to ECCM generated by ECs in the presence of AII was less than ECCM with AII added after conditioning. This difference can be explained, at least in part, by the endothelial metabolism of AII to inactive products.
Previous studies indicate that EDSF is rapidly released from ECs under basal conditions (2). The release of EDSF is detectable after 30 min and then quickly reaches a plateau. The cellular mechanism(s) and pathways that regulate the release and degradation of EDSF are not known. Thus, the physiological and pathological conditions that use EDSF for stimulating aldosterone release and amplifying the steroidogenic effects of AII require further study. It is possible that the synergistic interaction between AII and EDSF increases the sensitivity of the ZG cell to AII and emphasizes the importance of the endothelium in the control of aldosterone release. Factors that stimulate the production of EDSF could then cause an amplified response in the presence of an existing AII stimulus. These findings support the importance of intra-adrenal factors in the control of aldosterone release (38–40).
Acknowledgments
The authors thank Ms. Gretchen Barg for her secretarial assistance.
These studies were supported by National Institutes of Health Grants HL-52159, DK-58145, and HL-83297. The Waters-Micromass Quattro mass spectrometer was provided by a shared instrumentation grant from the National Institutes of Health (RR-17824).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations:
- A (1-7],
Angiotensin (1-7);
- AII,
angiotensin II;
- BK,
bradykinin;
- CFCM,
cell-free CM;
- CM,
conditioned medium;
- EC,
endothelial cell;
- ECCM,
EC-conditioned media;
- EDSF,
endothelium-derived steroidogenic factor;
- ET-1,
endothelin-1;
- LC/MS/MS,
liquid chromatography/tandem MS;
- MRM,
multireaction monitoring;
- MS,
mass spectrometry;
- SIM,
selected ion monitoring;
- ZG,
zona glomerulosa.
References
- 1. Rosolowsky LJ, Campbell WB. 1994. Endothelial cells stimulate aldosterone release from bovine adrenal zona glomerulosa cells. Am J Physiol 266:E107–E117 [DOI] [PubMed] [Google Scholar]
- 2. Rosolowsky LJ, Hanke CJ, Campbell WB. 1999. Adrenal capillary endothelial cells stimulate aldosterone release through a protein that is distinct from endothelin. Endocrinology 140:4411–4418 [DOI] [PubMed] [Google Scholar]
- 3. Vinson GP, Pudney JA, Whitehouse BJ. 1985. The mammalian adrenal circulation and the relationship between adrenal blood flow and steroidogenesis. Endocrinology 105:285–294 [DOI] [PubMed] [Google Scholar]
- 4. Dobbie JW, Symington T. 1991. The human adrenal gland with special reference to the vasculature. J Endocrinol 34:479–489 [DOI] [PubMed] [Google Scholar]
- 5. Quinn SJ, Williams GH. 1988. Regulation of aldosterone secretion. Ann Rev Physiol 50:409–426 [DOI] [PubMed] [Google Scholar]
- 6. Williams JS, Williams GH. 2003. 50th Anniversary of aldosterone. J Clin Endocrinol Metab 88:2364–2372 [DOI] [PubMed] [Google Scholar]
- 7. Morishita R, Higaki J, Ogihara T. 1989. Endothelin stimulates aldosterone biorelease by dispersed rabbit adreno-capsular cells. Biochem Biophys Res Commun 160:628–632 [DOI] [PubMed] [Google Scholar]
- 8. Hinson JP, Kapas S, Teja R, Vinson GP. 1991. Effect of the endothelins on aldosterone secretion by rat zona glomerulosa cells in vitro. J Steroid Biochem Mol Biol 40:437–439 [DOI] [PubMed] [Google Scholar]
- 9. Gomez-Sanchez CE, Cozza EN, Foecking MF, Chiou S, Ferris MW. 1990. Endothelin receptor subtypes and stimulation of aldosterone secretion. Hypertension 15:744–747 [DOI] [PubMed] [Google Scholar]
- 10. Rosolowsky LJ, Campbell WB. 1992. Bradykinin stimulates aldosterone release from cultured bovine adrenocortical cells through bradykinin B2 receptors. Endocrinology 130:2067–2075 [DOI] [PubMed] [Google Scholar]
- 11. Csukas S, Hanke CJ, Rewolinski D, Campbell WB. 1998. Prostaglandin E2-induced aldosterone release from bovine zona glomerulosa cells is mediated by an EP2 receptor. Hypertension 31:575–581 [DOI] [PubMed] [Google Scholar]
- 12. Fredlund P, Saltman S, Kondo T, Douglas J, Catt KJ. 1977. Aldosterone production by isolated glomerulosa cells: modulation of sensitivity to angiotensin II and ACTH by extracellular potassium concentration. Endocrinology 100:481–486 [DOI] [PubMed] [Google Scholar]
- 13. Young DB, Smith Jr MJ, Jackson TE, Scott RE. 1984. Multiplicative interaction between angiotensin II and K concentration in stimulation of aldosterone. Am J Physiol 247 (3 Pt 1): E328–E335 [DOI] [PubMed] [Google Scholar]
- 14. Pratt JH, Rothrock JK, Dominguez JH. 1989. Evidence that angiotensin-II and potassium collaborate to increase cytosolic calcium and stimulate the secretion of aldosterone. Endocrinology 125:2463–2469 [DOI] [PubMed] [Google Scholar]
- 15. Brickner RC, Raff H. 1991. Oxygen sensitivity of potassium- and angiotensin II-stimulated aldosterone release by bovine adrenal cells. J Endocrinol 129:43–48 [DOI] [PubMed] [Google Scholar]
- 16. Campbell WB, Schmitz JM, Itskovitz HD. 1977. The adrenal and vascular effects of angiotensin II and III in sodium depleted rats. Life Sci 20:803–809 [DOI] [PubMed] [Google Scholar]
- 17. Rosolowsky LJ, Campbell WB. 1990. Endothelin enhances adrenocorticotropin-stimulated aldosterone release from cultured bovine adrenal cells. Endocrinology 126:1860–1866 [DOI] [PubMed] [Google Scholar]
- 18. Cozza EN, Chiou S, Gomez-Sanchez CE. 1992. Endothelin-1 potentiation of angiotensin II stimulation of aldosterone production. Am J Physiol 262:R85–R89 [DOI] [PubMed] [Google Scholar]
- 19. Burnay MM, Vallotton MB, Capponi AM, Rossier MF. 1998. Angiotensin II potentiates adrenocorticotrophic hormone-induced cAMP formation in bovine adrenal glomerulosa cells through a capacitative calcium influx. Biochem J 330:21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isales CM, Barrett PQ, Brines M, Bollag W, Rasmussen H. 1991. Parathyroid hormone modulates angiotensin II-induced aldosterone secretion from the adrenal glomerulosa cell. Endocrinology 129:489–495 [DOI] [PubMed] [Google Scholar]
- 21. Folkman J, Haudenschild CC, Zetter BR. 1979. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci USA 76:5217–5221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanke CJ, Drewett JG, Myers CR, Campbell WB. 1998. Nitric oxide inhibits aldosterone release by a guanylyl cyclase-independent effect. Endocrinology 139:4053–4060 [DOI] [PubMed] [Google Scholar]
- 23. Campbell WB, Brooks SN, Pettinger WA. 1974. Angiotensin II- and angiotensin 3-induced aldosterone release in vivo in the rat. Science 184:994–996 [DOI] [PubMed] [Google Scholar]
- 24. Sarstedt CA, Vaughan Jr ED, Peach MJ. 1975. Selective inhibition by des-1-Asp-8-lle-angiotensin II of the steroidogenic response to restricted sodium intake in the rat. Circ Res 37:350–358 [DOI] [PubMed] [Google Scholar]
- 25. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanke CJ, Campbell WB. 2000. Endothelial cell nitric oxide inhibits aldosterone release in zona glomerulosa cells: modulation by oxygen. Am J Physiol 279:E846–E854 [DOI] [PubMed] [Google Scholar]
- 27. Sala GB, Hayashi K, Catt KJ, Dufau ML. 1979. Adrenocorticotropin action in isolated adrenal cells. The intermediate role of cyclic AMP in stimulation of corticosterone synthesis. J Biol Chem 254:3861–3865 [PubMed] [Google Scholar]
- 28. Fujita K, Aguilera G, Catt KJ. 1979. The role of cyclic AMP in aldosterone production by isolated zona glomerulosa cells. J Biol Chem 254:8567–8574 [PubMed] [Google Scholar]
- 29. Moyle WR, Kong YC, Ramachandran J. 1973. Steroidogenesis and cyclic adenosine 3′,5′-monophosphate accumulation in rat adrenal cells. Divergent effects of adrenocorticotropin and its o-nitrophenyl sulfenyl derivative. J Biol Chem 248:2409–2417 [PubMed] [Google Scholar]
- 30. Fakunding JL, Chow R, Catt KJ. 1979. The role of calcium in the stimulation of aldosterone production by adrenocorticotropin, angiotensin II, and potassium in isolated glomerulosa cells. Endocrinology 105:327–333 [DOI] [PubMed] [Google Scholar]
- 31. Capponi AM, Lew PD, Jornot L, Vallotton MB. 1984. Correlation between cytosolic free Ca2+ and aldosterone production in bovine adrenal glomerulosa cells. Evidence for a difference in the mode of action of angiotensin II and potassium. J Biol Chem 259:8863–8869 [PubMed] [Google Scholar]
- 32. Kramer RE. 1988. Angiotensin II causes sustained elevations in cytosolic calcium in glomerulosa cells. Am J Physiol 255 (3 Pt 1): E338–E346 [DOI] [PubMed] [Google Scholar]
- 33. Quinn SJ, Enyedi P, Tillotson DL, Williams GH. 1990. Cytosolic calcium and aldosterone response patterns of rat adrenal glomerulosa cells stimulated by vasopressin: comparison with angiotensin II. Endocrinology 127:541–548 [DOI] [PubMed] [Google Scholar]
- 34. Ganguly A, Davis JS. 1994. Role of calcium and other mediators in aldosterone secretion from the adrenal glomerulosa cells. Pharmacol Rev 46:417–447 [PubMed] [Google Scholar]
- 35. Kijima K, Matsubara H, Murasawa S, Maruyama K, Mori Y, Ohkubo N, Komuro I, Yazaki Y, Iwasaka T, Inada M. 1996. Mechanical stretch induces enhanced expression of angiotensin II receptor subtypes in neonatal rat cardiac myocytes. Circ Res 79:887–889 [DOI] [PubMed] [Google Scholar]
- 36. Ullian ME, Raymond JR, Willingham MC, Paul RV. 1997. Regulation of vascular angiotensin II receptors by EGF. Am J Physiol 273 (4 Pt 1): C1241–C1249 [DOI] [PubMed] [Google Scholar]
- 37. Waterman MR, Bischof LJ. 1997. Cytochromes P450 12: diversity of ACTH (cAMP)-dependent transcription of bovine steroid hydroxylase genes. FASEB J 11:419–427 [DOI] [PubMed] [Google Scholar]
- 38. Mulrow PJ. 1989. Adrenal renin: a possible local regulator of aldosterone production. Yale J Biol Med 62:503–510 [PMC free article] [PubMed] [Google Scholar]
- 39. Fallo F, Boscaro M. 1991. In vitro evidence for local generation of renin and angiotensin II/III immunoreactivity by the human adrenal gland. Acta Endocrinol 125 (3): 319–30 [DOI] [PubMed] [Google Scholar]
- 40. Sander M, Ganten D, Mellon SH. 1994. Role of adrenal renin in the regulation of adrenal steroidogenesis by corticotropin. Proc Natl Acad Sci USA 91:148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]