

Abstract
Triple-negative breast cancers (TNBC) are aggressive and heterogeneous cancers that lack targeted therapies. We implemented a screening program to identify new leads for subgroups of TNBC using diverse cell lines with different molecular drivers. Through this program, we identified an extract from Calotropis gigantea that caused selective cytotoxicity in BT-549 cells as compared to four other TNBC cell lines. Bioassay-guided fractionation of the BT-549 selective extract yielded nine cardenolides responsible for the selective activity. These included eight known cardenolides and a new cardenolide glycoside. Structure activity relationships among the cardenolides demonstrated a correlation between their relative potencies toward BT-549 cells and Na+/K+ ATPase inhibition. Calotropin, the compound with the highest degree of selectivity for BT-549 cells, increased intracellular Ca2+ in sensitive cells to a greater extent than in the resistant MDA-MB-231 cells. Further studies identified a second TNBC cell line, Hs578T, that is also highly sensitive to the cardenolides and mechanistic studies were conducted to identify commonalities among the sensitive cell lines. Experiments showed that both cardenolide-sensitive cell lines expressed higher mRNA levels of the Na+/Ca2+ exchanger NCX1 than resistant TNBC cells. This suggests that NCX1 could be a biomarker to identify TNBC patients that might benefit from the clinical administration of a cardiac glycoside for anticancer indications.
Graphical Abstract
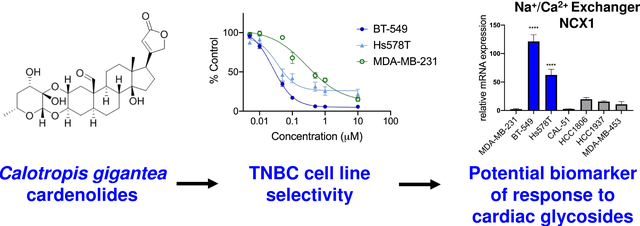
Triple-negative breast cancers (TNBC) are devoid of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (HER2) expression. TNBC represents 10–20% of all breast cancers and is noted for its aggressive phenotype, poor prognosis, and high mortality.1–3 The underlying challenge with TNBC is that they are highly heterogeneous and no single molecular driver has been identified which could be exploited using targeted therapies.4,5 Genomic profiling and gene expression studies show that TNBC can be sub-classified into distinct molecular subtypes. In 2011, Lehman and Bauer et al. identified six TNBC subtypes based on tumor gene expression analyses. A major outcome of this analysis was the identification of human TNBC cell lines representative of the clinical subtypes.6 More recently, these subtypes were reevaluated and refined into four subtypes.7 Other groups have identified distinct TNBC subgroups through evaluation of gene expression, DNA copy number alterations, and mutational signatures of TNBC tumors, and these subtypes have some commonalities with the Lehman and Bauer subtypes, but also some notable differences.8,9 These studies each highlight the heterogeneity of TNBC, but also demonstrated targetable commonalities among molecular subgroups of the disease. Despite these advances in identifying subgroups of TNBC, effective FDA-approved targeted therapies for TNBC are still out of reach, warranting continued efforts to identify subgroups of TNBC that respond to targeted therapies in the form of new drugs or repurposed existing therapeutics.
Natural products remain a rich source of anticancer agents, and more than half of all drugs used for the treatment of cancer are natural products or were inspired by natural products.10 We have a screening program aimed at identifying natural products that selectively target subgroups of TNBC as a mechanism to identify new therapeutic targets and drug leads for this disease. Our efforts have been successful in identifying multiple subtype-selective compounds from fungi and plants.11–14 Extracts from the National Cancer Institute (NCI) (USA) Natural Products Repository collection were screened for selective cytotoxicity in a panel of diverse TNBC cell lines that represent the heterogeneity found in TNBC patients. An extract from Calotropis gigantea (L.) W.T. Aiton (Apocynaceae) demonstrated selective cytotoxicity against BT-549 cells as compared to other TNBC cell lines. Calotropis gigantea, which is commonly known as the “giant milk weed” or “crown flower”, is found across South and Southeast Asia, as well as Africa, and is known for its many medicinal uses.15 Numerous metabolites that likely contribute to its reported medicinal properties have been isolated from Calotropis species, including sterols, flavonoids, oxypregnanes, terpenoids, and cardenolides, many of which have cytotoxic effects against cancer cells.16 Recently, 6 new cardenolides were isolated from C. gigantea, several of which displayed sub-micromolar potency against a pancreatic cancer cell line.17 Given this background, we speculated that cardenolides might be responsible for the selective activity of the C. gigantea crude extract. Indeed, bioassay-guided fractionation yielded nine cardenolides that exhibited selective cytotoxic activities against BT-549 cells. While eight of the metabolites were known, one was not previously reported.
Although the cytotoxic effects of this class of compounds are well known, selective cytotoxicity against a subgroup of TNBC cells at low concentrations is intriguing because it allows identification of a molecular susceptibility of these cells, which has not been investigated previously. Importantly, this selectivity provides the potential to identify a population of tumors that are exquisitely sensitive to this class of compounds at doses that are less toxic to patients, providing an improved therapeutic potential. Therefore, the mechanism of the selective cytotoxicity of calotropin, the cardenolide with the highest degree of selectivity and potency for BT-549 cells, was investigated. Calotropin caused a rapid increase in intracellular Ca2+ in the sensitive BT-549 cells that could be due in part to high intrinsic expression of the Na+/Ca2+ exchanger (NCX1) in these cells. A second calotropin sensitive cell line, Hs578T, also had high intrinsic levels of NCX1 mRNA. The significance of this is the possibility that NCX1 mRNA could be a biomarker for TNBC sensitive to FDA-approved cardiac glycosides, including digoxin.
RESULTS AND DISCUSSION
Calotropis gigantea Yields Cardenolides with Potent Cytotoxic Activities Against BT-549 Cells
To identify new compounds and targets for the treatment of TNBC subtypes, we evaluated plant extracts from the NCI collection in a mechanism-blind cytotoxicity screen using five TNBC cell lines representing molecularly distinct TNBC subtypes.6 The crude extract from C. gigantea demonstrated selective cytotoxic effects against BT-549 cells, which were 4–9 fold more sensitive as compared to cell lines representing other TNBC subtypes. Bioassay-guided fractionation using a combination of vacuum-liquid-chromatography (HP20ss), preparative HPLC (C18), and semi-preparative HPLC (C18, biphenyl, and pentafluorophenyl) led to the purification of nine cardenolides (1-9) (Chart). While carrying out dereplication, we discovered that some of the metabolites had been partially described in reports dating nearly 90 years prior. This raised several challenges for our team since dereplication strategies and spectroscopy tools have changed considerably during the intervening years, making definitive data comparisons nearly impossible to perform in several cases. Moreover, less rigid data reporting conventions for natural products during the period of discovery for 1-4 and 6-9 (1933–1966) resulted in several instances wherein insufficient data were available to independently confirm the accuracy of the structures proposed for these compounds. Furthermore, we found the rediscovery of some cardenolides had led to the fabrication of synonyms, which later had been misspelled in reports resulting in a convoluted scientific lineage for some of these metabolites. Thus, we performed a variety of analytical chemistry studies that are described in this report to authenticate the structures of these compounds, as well as to provide a current-day data set that will support the dereplication activities of other researchers probing this natural product family.

Chart 1
Uzarigenin (probable synonyms: uzarigenine, cerberigenin; echujetin; evonogenin; thevetigenin, odorigeni, urarigenin)18–20 (1) was purified as colorless crystals. Its molecular formula was determined to be C23H34O4 based on HRESIMS data (m/z 375.2540 [M + H]+, calcd 375.2530), which supported seven degrees of unsaturation. The 1H and 13C NMR data (Tables S1 and S2, Supporting Information), and 2D NMR spectra revealed the planar structure of this compound. The absolute configuration was determined based on a combination of data from ROESY and single-crystal X-ray diffraction experiments (Figure S1, Supporting Information).
Coroglaucigenin (no known synonyms)18,19 (2) was obtained as colorless crystals. The HRESIMS data (m/z 391.2491), together with the results from 1H and 13C NMR experiments (Tables S1 and S2, Supporting Information) led to its proposed molecular formula, C23H34O5. Compared to compound 1, the 19-CH3 was missing, and instead, it was replaced by a hydroxymethyl group, whose assignment was confirmed by 2D NMR experiments. A ROESY experiment helped determine the relative configuration of 2, while a single-crystal X-ray diffraction experiment was instrumental in finalizing its absolute configuration (Figure S1, Supporting Information).
Desglucouzarin (probable synonym: deglucouzarin)18–20 (3) was obtained as colorless crystals and its molecular formula was established to be C29H44O9 based on the HRESIMS data (m/z 537.3057[M +H]+,calcd 537.3058). Analysis of the 1H and 13C NMR datafornatural product 3 (Tables S1 and S2, Supporting Information) revealed that this compound was structurally similar to 1 with the addition of resonances characteristic for a glycosyl group. This conjecture was confirmed by an analysis of the HMBC data, which also showed the site of the glycosyl-ether linkage to be between C-3 and C-1’. Subsequent investigations of ROESY-derived data, 1H-1H coupling constant analysis (Table S1, Supporting Information), and results from a single-crystal X-ray diffraction experiment (Figure S1, Supporting Information) were instrumental in piecing together the working structure of 3; however, its absolute configuration could not be substantiated using these data. Instead, the absolute configuration of compound 3 was confirmed using a series of supporting experimental data. Those efforts focused on establishing the identity of the glycosyl group to be D-glucose based on acid hydrolysis coupled with LCMS comparison of its thiocarbamoyl-thiazolidine derivative with an authentic standard.21–25 Additionally, the C-1’ anomeric carbon was determined to be β-oriented based on analysisofits 1H NMR features ( δH 4.20, d, J= 7.8 Hz). Furthermore,the absoluteconfiguration of the molecule’s cardenolide core was confirmed as shown for compound 3 based on comparisons of ECD data for the aglycone to other compounds we purified in this analogue series (Figure S2, Supporting Information).
Another cardenolide glycoside, frugoside (probable synonyms: frugosid, cannogenol-3-O-beta-D-allomethyloside, coroglaucigenin-D-allomethylosid) 20,26,27 (4), was obtained that bore the same molecular formula as compound 3, but analyses of the 1D (1H and 13C) (Tables S1 and S2, Supporting Information) and 2D (HSQC and HMBC) NMR data pointed to two subtle structural changes that distinguished these metabolites. Specifically, compound 4 was found to be composed of the same cardenolide core as represented by 2, but the glyosidic portion of the compound was comprised of the monosaccharide 6-deoxy allopyranose. Placement of the C-3/C-1’ linkage between the cardenolide and glycoside portions of the compound was confirmed via an HMBC correlation from H-1’ to C-3. The relative configuration of the 6-deoxy allopyranose was revealed through ROESY correlations and the anomeric proton was established to be β-oriented based on analysis of its NMR data [(δH 4.73 (1H, d, J = 8.0 Hz) and δC 99.8 (CH)]. Whereas we were unable to generate crystals of 4 that were appropriate for X-ray diffraction analysis, we did find that a bromobenzoyl ester of 4 (Scheme 1) readily formed crystals that were suitable for X-ray diffraction studies. (Figure S1, Supporting Information). Thus, the structure of 4 was confirmed and its absolute configuration established as shown for the metabolite.
Scheme 1.

Linkage of bromobenzoyl to frugoside (4).
Compound 5 (no synonyms – new in this report) was obtained as a white amorphous powder and it was determined to have the molecular formula C29H42O10 based on HRESIMS analysis. A comparisonof 1H and 13C NMR data for compounds 5 and 4(Table 1, and Tables S1 and S2, Supporting Information) revealed that both compounds were structurally similar, but two significant differences stood out. First, the C19-CH2OH substituent in 4 (δH 3.86, d, J = 11.8 Hz; 3.74, d, J = 10.7 Hz and δC 60.0) had been replaced by an aldehyde group (δH 9.94, s and δC 209.6). Second, the C2-methylene in 4 (δH 1.89, overlap; 1.55, overlap, and δC 30.8) now bore an alcohol moiety (δH 3.20, m, and δC 69.9) in 5. These conclusions were confirmed by the 2D NMR correlations as illustrated in Figure 1 (e.g., COSY correlations between H-2 ↔ H-3 and H-3 ↔ H-4, together with HMBC correlations from H-1 to C-3, C-5, C-9, C-10, and C-19). The relative configuration of compound 5 was established via 1H-1H coupling constant analysis,as well as ROESY NMR correlations [for the cardenolide portion: H-22 and H-18, H-21 and H-18, H-18 and H-8, H-18 and 14-OH, H-8 and H-19, H-19 and H-2, H-19 and H-1eq (δH 2.37, dd, J = 12.9, 5.2 Hz), H-9 and H-15, H-1ax (δH 0.88, dd, J = 12.3, 12.1 Hz) and H-3,H-3 and H-5, and H-5 and H-1ax. Additionally, anexamination of the 1H and 13CNMR resonances and coupling constants associated with the glycoside in 5 (Table 1) revealed that it was identical to the 6-deoxy allopyranose observed in 4, (Tables S1–2, Supporting Information) and connected to the cardenolide moiety by means of a bond between C-1’ to C-3 (HMBC correlation from H-1’ to C-3) (Figure S40, Supporting Information). The anomeric proton was established to be β-oriented based on analysis of its NMR data [(δH 4.50 (1H, d, J = 8.0 Hz) and δC 99.4 (CH)]. To address the absolute configuration of 5, the natural product was subjected to hydrolysis and the glycosyl unit converted to a thiocarbamoyl-thiazolidine derivative. Analysis of the derivatized product by LCMS (retention time and mass) revealed a match to the product created when metabolite 4 was reacted using identical hydrolysis and derivatization steps.21–25 The ECD spectrum of 5 was obtained and found to be similar to the spectrum of compound 4 (Figure 2). Considering these data, along with their identical biogenic origins, the absolute configuration of 5 was established as shown and the compound was given the trivial name frugosidal.
Table 1.
1H (600 MHz) and 13C (150 MHz) NMR Data for Compound 5 in DMSO-d6
| NO. | δC, type | δH, mult. (J in Hz) |
|---|---|---|
| 1 | 38.9 CH2 | 2.37, dd (12.9, 5.2); 0.88, dd (12.3, 12.1) |
| 2 | 69.9 CH | 3.20, m |
| 3 | 83.0 CH | 3.30a |
| 4 | 34.7 CH2 | 1.75, overlap; 1.14, m |
| 5 | 42.3 CH | 1.39 overlap |
| 6 | 21.9 CH2 | 1.57, overlap; 1.05, m |
| 7 | 27.4 CH2 | 2.14, brd (12.6); 1.09, overlap |
| 8 | 42.1 CH | 1.42, ddd (13.0, 12.3, 3.1) |
| 9 | 47.8 CH | 1.27, overlap |
| 10 | 52.1 C | |
| 11 | 27.7 CH2 | 1.83, m; 1.52, overlap |
| 12 | 38.8 CH2 | 1.38, m; 1.29, overlap |
| 13 | 49.6 C | |
| 14 | 83.7 C | |
| 15 | 31.9 CH2 | 1.94, m; 1.55, overlap |
| 16 | 26.7 CH2 | 2.00, m; 1.76, overlap |
| 17 | 50.4 CH | 2.71, m |
| 18 | 15.9 CH3 | 0.69, s |
| 19 | 209.6 CH | 9.94, s |
| 20 | 176.6 C | |
| 21 | 73.6 CH2 | 4.93, d (18.1); 4.86, d (18.0) |
| 22 | 116.8 CH | 5.90, s |
| 23 | 174.3 C | |
| 1’ | 99.4 CH | 4.50, d (8.0) |
| 2’ | 70.7 CH | 3.13, dd (8.3, 2.9) |
| 3’ | 71.7 CH | 3.80, dd (3.0, 3.0) |
| 4’ | 73.0 CH | 3.00, dd (9.0, 2.4) |
| 5’ | 69.6 CH | 3.60, m |
| 6’ | 18.3 CH3 | 1.08, d (6.2) |
| 14-OH | 4.23, s |
overlapped with H2O peak and extracted from HSQC
Figure 1.

Selected 1H-1H COSY, HMBC, and ROESY correlations of compound 5
Figure 2.

ECD curves of compounds 4 and 5.
Glucofrugoside (no known synonyms)26(6) was found to have the molecular formula C35H54O14 based on HRESIMS that yielded a [M + Na]+ ion at m/z 721.3414 (calcd 721.3406). An examination of the 1H and 13C NMR data (Tables S2 and S3, Supporting Information) led to the conclusion that 6 contained a dissacharide linked to the same cardenolide as found in metabolite 4. This conclusion was supported by 2D NMR experiments, and the linkages between each portion of the molecule were substantiated by HMBC correlations (i.e., H-1’ to C-3 and H-1” to C-4’). The absolute configurations of the glycoside moieties were determined to match D-glucose and D-6-deoxy allopyranose based on LCMS analyses of their thiocarbamoyl-thiazolidine derivatives made from the hydrolysis products.21–25 The absolute configuration of the cardenolide portion of 6 was determined to be the same as the other metabolites in this series based on their similar ECD spectra and shared biogenic origins (Figure S2, Supporting Information).
Uscharin (probable synonym: uscharine)28,29 (7) and calotoxin (no known synonyms)30 (8) were purified from sequentially eluting fractions. Whereas 7 appeared as faintly yellow crystals and had the molecular formula C31H41NO8S, compound 8 was obtained as colorless crystals and had the molecular formula C29H40O10. The 1H and 13C NMR data for 7 and 8 (Tables S2 and S3, Supporting Information) revealed their high degree of structural similarity to one another, as well as to compound 5. A combination of 2D NMR experiments helped to secure the planar structures of 7 and 8 and their absolute configurations were obtained from their X-ray diffraction data (Figure S1, Supporting Information).
Calotropin (probable synonyms: pecilocerin A, pekilocerin A)28,30 (9) was obtained as a white, amorphous powder and its HRESIMS data (m/z 533.2748 [M + H]+, calcd 533.2745) supported a molecular formula of C29H40O9, which indicated the compound had an index of hydrogen deficiency equal to 10. The 1H and 13C NMR data (Tables S2 and S3, Supporting Information) for 9 indicated that it shared a high degree of structure similarity with 8, but it was missing the C-4” alcohol moiety, which instead was now a -CH2- group. The full bond-line structure of 9 was obtained from an analysis of the 2D NMR data and its relative configuration was secured based on interpretation of the ROESY data. Considering the high degree of similarity between the ECD data obtained for 8 and 9 (Figure S2, Supporting Information), as well as their shared biogenic sources, the absolute configuration of the metabolite was proposed as shown for compound 9.
Structure Activity Relationships
During the process of bioassay-guided fractionation to identify fractions that contained BT-549 selective activity, we identified a second TNBC cell line, Hs578T, that was also sensitive to the active constituents of this extract as compared to other TNBC cell lines, including MDA-MB-231, that were more resistant. The clinically approved cardiac glycoside, digoxin, was also evaluated in a larger panel of 10 TNBC cell lines, demonstrating that BT-549 and Hs578T cells are significantly more sensitive to digoxin than every other cell line tested (Table 2). These results confirm that BT-549 and Hs578T cells represent a distinct subgroup of TNBC that are selectively sensitive to cardenolides/cardiac glycosides. Concentration response curves of the cytotoxic effects of each of the purified compounds were determined in the sensitive BT-549 and Hs578T cells, as well as the resistant MDA-MB-231 cells (Figure S3, Supporting Information). While the potencies of the cardenolides were similar in BT-549 and Hs578T cells, the efficacies were greater in BT-549 cells for all compounds, causing essentially total cytotoxicity in BT-549 cells and for the most part only cytostatic effects in the Hs579T cells. The IC50 value, a concentration that caused a 50% decrease in cell number as compared to vehicle control, for each cardenolide was determined in the three cell lines (Table 3). Additionally, the selectivity index, defined as the IC50 value for each compound in MDA-MB-231 cells divided by the IC50 value in BT-549 cells, was calculated (Table 3). The IC50 values in BT-549 cells ranged from 14.6 nM for uscharin (7) to 3.9 μM for uzarigenin (1) and the selectivity index ranged from 6-fold for desglucouzarin (3) to 15-fold for calotropin (9). The potencies of the compounds correlated with their selectivity for BT-549 cells, except for uscharin (7), which was the most potent, but one of the least selective compounds in this series.
Table 2.
IC50 Values of Digoxin in TNBC Cell Lines
| cell Line | IC50 (μM) ± SEM |
|---|---|
| BT-549 | 0.051 ± 0.003 |
| Hs578T | 0.069 ± 0.008 |
| CAL-51 | 0.13 ± 0.03 |
| MDA-MB-468 | 0.23 ± 0.01 |
| MDA-MB-453 | 0.23 ± 0.06 |
| SUM185PE | 0.28 ± 0.02 |
| HCC1806 | 0.28 ± 0.01 |
| MDA-MB-231 | 0.48 ± 0.08 |
| HCC1937 | 0.55 ± 0.08 |
| HCC70 | 0.64 ± 0.02 |
Table 3.
IC50 Values and Selectivity of Cardenolides in TNBC Cell Lines
| IC50 (μM) ± SEM |
||||
|---|---|---|---|---|
| compound | BT-549 | Hs578T | MDA-MB-231 | SIa |
| uzarigenin (1) | 3.9 ± 0.4 | 3.6 ± 0.1 | > 30 | > 8 |
| coroglaucigenin (2) | 2.3 ± 0.3 | 1.8 ± 0.3 | 20 ± 4 | 9 |
| desglucouzarin (3) | 1.8 ± 0.2 | 1.57 ± 0.06 | 10 ± 1 | 6 |
| frugoside (4) | 0.121 ± 0.007 | 0.38 ± 0.06 | 1.25 ± 0.09 | 10 |
| frugosidal (5) | 1.4 ± 0.3 | 3.9 ± 0.9 | 10 ± 2 | 7 |
| glucofrugoside (6) | 0.070 ± 0.004 | 0.30 ± 0.05 | 0.98 ± 0.06 | 14 |
| uscharin (7) | 0.0146 ± 0.0001 | 0.034 ± 0.003 | 0.102 ± 0.004 | 7 |
| calotoxin (8) | 0.049 ± 0.002 | 0.12 ± 0.05 | 0.63 ± 0.04 | 13 |
| calotropin (9) | 0.030 ± 0.002 | 0.06 ± 0.01 | 0.44 ± 0.08 | 15 |
Selectivity Index = IC50 MDA-MB-231/ IC50 BT-549
The three most potent cardenolides of this series, uscharin (7), calotoxin (8), and calotropin (9), share the structural characteristic of a bridging ether bond between C-2 and C-2’, which is a feature unique to the cardenolides derived from plants in the subfamily Asclepiadoideae.31 While uscharin (7) was the most potent, calotoxin (8) and calotropin (9) were notably more selective for BT-549 cells. This finding suggested that the C-3’ thiazoline moiety in uscharin (7) is detrimental to selectivity, but advantageous for potency. Other structural features that appear to control potency include the glycosyl groups; for example, the aglycones uzarigenin (1) and coroglaucigenin (2) are less potent than desglucouzarin (3) and frugoside (4), respectively. Moreover, the aglycone portion of frugosidal (5) is identical to that found in the more potent compounds 7-9 that contain the bridging ether, further enforcing the importance of this unique structural feature.
Inhibition of Na+/K+ ATPase by Cardenolides Predicts Selectivity for BT-549 Cells
The correlation between the potent cytotoxic effects of cardenolides and the presence/absence of glycosyl groups is consistent with previous observations that the addition of certain carbohydrate moieties to digitalis-type cardiac glycosides increased potency for inhibition of Na+/K+ ATPase compared to their respective aglycones.32,33 To determine whether the selective activity of the cardenolides was related to inhibition of Na+/K+ ATPase, concentration response curves for inhibition of the purified enzyme were generated for each compound and the EC50 values, defined as the concentration causing 50% inhibition of enzyme activity compared to vehicle control were determined (Figure 3A, Table 4). The compounds displayed EC50 values ranging from 300 nM for calotoxin (8) to 2.7 μM for frugosidal (5). A linear regression analysis was performed to correlate potency for inhibition of Na+/K+ ATPase for each compound with cytotoxic potency in BT-549 cells, as well as the selectivity index for BT-549 as compared to MDA-MB-231 cells (Figure 3 B, C). In general, among this group of cardenolides, those that were most potent for inhibiting Na+/K+ ATPase were also the most potent and selective for causing cytotoxicity in BT-549 cells. A notable outlier in this analysis was uscharin (7), which was more potent in BT-549 cells than predicted from its relative potency for Na+/K+ ATPase inhibition (Figure 3B). However, the selectivity index for usharin was more highly correlated with its relative potency for inhibition of Na+/K+ ATPase, suggesting that potency for inhibition of Na+/K+ ATPase is a better predictor of BT-549 selectivity of these compounds than their cytotoxic potency.
Figure 3.

Effects of C. gigantea cardenolides on Na+/K+ ATPase activity and relationship to BT-549 selective cytotoxicity. A) Concentration response curves for inhibition of purified Na+/K+ ATPase represented as percent of control enzyme activity in the presence of vehicle. Each concentration was tested in duplicate in 2 independent experiments and the mean ± SEM is shown. In the lower panels the EC50 concentrations for inhibition of Na+/K+ ATPase were correlated with IC50 concentrations for BT-549 cytotoxicity (B) and selectivity index (C) and analyzed by simple linear regression.
Table 4.
EC50 Values of Cardenolides for Na+/K+ ATPase Inhibition
| compound | EC50 (μM) ± SEM |
|---|---|
| uzarigenin (1) | 2.1 ± 0.3 |
| coroglaucigenin (2) | 1.1 ± 0.3 |
| desglucouzarin (3) | 1.1 ± 0.3 |
| frugoside (4) | 0.50 ± 0.08 |
| frugosidal (5) | 2.5 ± 0.2 |
| glucofrugoside (6) | 0.6 ± 0.2 |
| uscharin (7) | 0.91 ± 0.03 |
| calotoxin (8) | 0.30 ± 0.06 |
| calotropin (9) | 0.36 ± 0.04 |
Differential Effects of Calotropin on Intracellular Ca2+ in TNBC Cells
The significant correlation between potency for inhibition of Na+/K+ ATPase and BT-549 selective cytotoxicity for the cardenolides suggested that the downstream effects of inhibiting Na+/K+ ATPase are central to their selective cytotoxic mechanisms. A major consequence of cardiac-glycoside-mediated inhibition of Na+/K+ ATPase is an increase in intracellular Na+, which inhibits the efflux of Ca2+ via the Na+/Ca2+ exchanger (NCX), ultimately leading to increased Ca2+ entry and elevated intracellular Ca2+ concentrations.34 The increase in intracellular Ca2+ improves cardiac muscle contractility, supporting the use of cardiac glycosides in congestive heart failure patients. Indeed, NCX activity was shown to be required for the ionotropic effects of cardiac glycosides in multiple models.34–36 However, increased intracellular Ca2+ can also trigger apoptosis, to which cancer cells with defects in Ca2+ signaling might be particularly vulnerable.37–39 We previously showed that BT-549 cells are selectively sensitive to englerin A, a TRPC1/4/5 cation channel agonist, which causes a rapid Ca2+ influx and mitochondrial depolarization in BT-549 cells, but not in resistant TNBC cells.14 These studies additionally demonstrated that digoxin caused an accumulation of Ca2+ in BT-549 cells, though with a slower onset than englerin A.14 Therefore, studies were undertaken with calotropin (9), the most selective and potent of the cardenolides, to evaluate its effects on intracellular calcium levels in the sensitive BT-549 cells as compared to resistant MDA-MB-231 cells.
First, the effects of 150 nM and 500 nM calotropin (9) on Ca2+ levels were determined in live BT-549 cells as measured by Cal520-AM fluorescence. Compared to vehicle-treated cells, calotropin (9) significantly increased the mean intracellular Ca2+ fluorescence after 3 h of treatment and this was sustained at 6 h (Figure 4A, B). Additionally, frequency distribution analysis of the cell population showed the much broader distribution of cellular Ca2+ fluorescence after a 3 h treatment with calotropin (9) compared to the population at the time of drug addition or in vehicle-treated cells (Figure 4C). Second, the early effects of calotropin (9) on Ca2+ fluorescence in BT-549 cells as compared to MDA-MB-231 cells were evaluated (Figure 4D, E). While 150 nM calotropin (9) increased Ca2+ fluorescence in BT-549 cells by almost 2-fold as early as 2 h, the change in Ca2+ was significantly less in MDA-MB-231 cells (Figure 4D, E). A similar pattern of results was also observed with 150 nM digoxin (Figure S4, Supporting Information). These results suggested that BT-549 cells are more sensitive to calotropin (9) and other cardiac glycosides due to increases in intracellular Ca2+, leading to cell death at lower concentrations than those required for most other TNBC cell lines.
Figure 4.

Effects of calotropin on intracellular Ca2+ in TNBC cells. A) Representative images at time of drug addition (T0) and after 3 h (T3) of BT-549 cells loaded with Cal520-AM and treated as indicated and B) quantification of Ca2+ fluorescence, significance was determined compared to DMSO at same time point by two-way ANOVA with Dunnett’s post hoc test n = 102–242 cells. C) Histograms of cellular Ca2+ fluorescence at T0 and T3, fit with Gaussian curves determined by non-linear regression. D) Images of the same fields of BT-549 and MDA-MB-231 cells loaded with Cal520-AM at T0 and at 2 and 3 h after drug addition and treated as indicated. E) Quantification of cellular Ca2+ fluorescence in BT-549 and MDA-MB-231 cells treated with 150 nM calotropin corresponding to images in D, significance was determined comparing BT-549 to MDA-MB-231 at each time point by two-way ANOVA with Bonferoni’s post hoc test, n = 291–531 cells, **** p < 0.0001.
Identifying a Biomarker for TNBCs Sensitive to Cardiac Glycosides
Due to the heterogeneity of cancers and their responses to treatment, major efforts are underway to identify biomarkers of response to traditional chemotherapy, targeted therapies, and immunotherapies. Preselection of patients for clinical trials based on biomarkers of response increases the likelihood of success. Others have sought to identify potential biomarkers of response to cardiac glycosides in preclinical models of both lung cancer and leukemia.40,41 In lung cancer models, STK11 mutations predict sensitivity and, in leukemia, MYC overexpression is correlated with sensitivity to cardiac glycosides. The selective cytotoxicity of cardenolides/cardiac glycosides in BT-549 and Hs578T cells suggested that these cells might share a biomarker that could predict TNBC patients that would benefit from treatment with a cardenolide/cardiac glycoside. Previously, we proposed expression of TRPC1/4/5 cation channels as a potential biomarker for cardiac glycoside sensitivity within TNBC.14 However, we sought to expand upon this work by investigating the expression of the known direct and downstream targets of cardiac glycosides in sensitive and resistant TNBC cell lines. Due to the significant correlation between BT-549 selective cytotoxicity and potency for inhibition of the Na+/K+ ATPase, we evaluated whether sensitive cell lines have differential expression of isoforms of the catalytic α subunit. Membrane-enriched cell lysates from a panel of untreated TNBC cells were evaluated for protein levels of the α1, α2 and α3 Na+/K+ ATPase isoforms (Figure 5A) and total protein (Figure S5, Supporting Information). Levels of the α3 isoform were the most similar among the cell lines. While there were some differences among the cell lines for baseline α2 expression, there was no correlation with sensitivity to cardiac glycosides. Interestingly, baseline α1 expression was somewhat inversely correlated to cardiac glycoside sensitivity, in that its expression was lowest in BT-549 cells. This was consistent with previous data suggesting that this isoform may be more resistant to inhibition by some cardiac glycosides.33 However, the other sensitive cells line, Hs578T, had an intermediate intrinsic level of α1 expression, thus not reflecting their sensitivity to cardiac glycosides.
Figure 5.

Expression of Na+/K+ ATPase isoforms and of Na+/Ca2+ exchanger in TNBC cells. Cell lines sensitive to cardenolides/cardiac glycosides in blue. A) Western blot of 30 ug of membrane enriched cell lysates were probed for Na+/K+ ATPase a subunit isoforms, α1, α2 and α3. B) mRNA expression of NCX1 and NCX3 determined by qRT-PCR and represented as fold difference compared to MDA-MB-231, n = 3–5 independent experiments. Significance determined by one-way ANOVA with Tukey’s post hoc test **** p < 0.0001, *** p < 0.001 C) Western blot of 75 μg of membrane enriched cell lysates for NCX1. Blots are representative of 3 independent experiments.
Another possible explanation for the higher levels of intracellular Ca2+ in calotropin-treated BT-549 cells and the sensitivity of BT-549 and Hs578T cells to cardiac glycosides could be due to differential baseline expression of NCX. As noted above, the NCX-mediated increase in Ca2+ is critical to the ionotropic effects of cardiac glycosides, making it a good candidate to explain the sensitivity of BT-549 and Hs578T cells to these agents. We previously reported that these cell lines have high expression of TRPC1/4/5 cation channels,14 and another study showed that they additionally have higher expression of the plasma membrane Ca2+ efflux pump, PMCA4, but lower levels of the endoplasmic reticulum Ca2+ transporter, SERCA2, as compared to other breast cancer cell lines.42 Therefore, we postulated that the altered ability of BT-549 and Hs578T cells to handle intracellular cations could be associated with alterations in other cation transporters, including NCX. Expression of the 3 NCX isoforms were evaluated in the panel of TNBC cell lines. Messenger RNA was only detected for NCX1 and NCX3 (Figure 5B). NCX1 mRNA expression was significantly higher in BT-549 and Hs578T cells than the other TNBC cell lines and was 120 and 60 times higher, respectively, in these cells as compared to MDA-MB-231 cells (Figure 5B). Interestingly, when evaluating protein levels of NCX1, expression was only detected by Western blots in membrane-enriched BT-549 cell lysates (Figure 5D, Figures S5, S6, Supporting Information). We further confirmed that the band was specific for NCX1 using siRNA knockdown (Figure S6, Supporting Information).
To test whether NCX1 is required to confer sensitivity of BT-549 cells to cardiac glycosides, CRISPR technology was used to knockout NCX1 in BT-549 cells with an inducible Cas9 system and three sgRNAs targeted to the NCX1 gene. Contrary to our hypothesis, removing NCX1 did not reduce the sensitivity of BT-549 cells to calotropin (Figure S7, Supporting Information). These results suggested that NCX1 protein overexpression alone is not required for BT-549 sensitively to cardiac glycosides, which is consistent with the fact that elevated levels of NCX1 protein were not detected in the other sensitive cell line, Hs578T. Therefore, we can postulate that elevated mRNA expression of NCX1 is an indicator of these cells attempting to compensate for broadly impaired cation handling due to multiple underlying defects that make them sensitive to cardiac glycosides. It is worth noting that inhibition of the Na+/K+ ATPase and the resulting increase and decrease in the normally tightly controlled intracellular Na+ and K+ concentrations, respectively, can affect a multitude of cellular functions. In addition to Na+/Ca2+ exchange, pathways and processes linked to cellular acidification, ROS production, Src pathway activation,43 and decreased intracellular K+, which is itself an apoptotic stimulus,44 are known to be affected. It is possible, given the many downstream consequences of cardiac glycosides, that other pathways compensate in BT-549 cells when NCX1 is not expressed and predominate in Hs578T cells. Regardless of the precise mechanism of cardiac glycoside-mediated cell death, high mRNA expression of NCX1 is a commonality of sensitive cell lines, and therefore is a potential biomarker for TNBCs that could benefit from treatment with a cardiac glycoside.
CONCLUSIONS
Here, we have demonstrated that mechanism-blind screening for natural products with selective cytotoxic activity against molecularly distinct TNBC cell lines can reveal novel molecular liabilities that, in this case, are targetable with existing drugs, namely cardiac glycosides. Cardiac glycosides have been recognized by many for their cytotoxic effects to cancer cells and their anticancer potential, although the specific population of patients who would benefit most from treatment has not been defined.43,45–47 Interestingly, a retrospective clinical study demonstrated a trend towards a decreased risk of breast cancer relapse during the first year for ER-negative patients on digoxin at the time of diagnosis, although the sample size was not large enough for statistical significance.48 This suggested that there may be a subset of patients within the ER-negative/TNBC subgroup that could derive benefit from a cardiac glycoside. Therefore, the identification of a biomarker is critical for determining the TNBC patients that could ultimately respond to a cardiac glycoside. Our findings established that among TNBC cell lines, high mRNA expression of NCX1 predicts sensitivity to cardenolides and might be an indicator of multiple defects in cation homeostasis. Additionally, mining the METABRIC dataset, we found that 4.8% (25/521) of ER-negative breast cancer patients had high mRNA expression of NCX1.49, 50 These data suggested that NCX1 expression could aid in the selection of TNBC patients for a clinical trial with a cardiac glycoside such as digoxin.
EXPERIMENTAL SECTION
Gaeneral Experimental Procedures
Optical rotation data were obtained on a Rudolph Research AUTOPOL® III automatic polarimeter. ECD data were obtained on a JASCO J-715 CD instrument. NMR data were collected on Varian 500 and 600 MHz NMR spectrometers. Intensity data were collected using a D8 Quest κ-geometry diffractometer with a Bruker Photon II cmos area detector and an Incoatec Iμs microfocus Mo Kα source (λ = 0.71073 Å). LC-MS data were obtained on a Shimadzu LC-MS 2020 system (ESI quadrupole) coupled to a photodiode array detector, with a Phenomenex Kintex 2.6 μm C18 column (100 Å, 75 × 3.0 mm, 0.4 mL/min). The preparative HPLC system utilized SCL-10A VP pumps and system controller with Phenomenex Gemini 5 μm C18 column (110 Å, 250 × 21.2 mm, 10 mL/min), the analytical and semi-preparative HPLC system utilized Waters 1525 binary pumps with Waters 2998 photodiode array detectors, and Phenomenex Gemini 5 μm Gemini C18, Phenomenex Kinetex 5 μm biphenyl, Phenomenex Kinetex 5 μm pentafluorophenyl (250 × 4.6 mm, 1 mL/min and 250 × 10 mm, 4 mL/min). Accurate mass data were collected on a Waters SYNAPT G2-Si mass spectrometer. All solvents were of ACS grade or better.
Extraction and Isolation of Natural Products
The plant Calotropis gigantea was collected in India in March 1993 by Dr Djaja D. Soejarto (University of Illinois at Chicago) under contract with the Natural Products Branch for the National Cancer Institute. The plant was identified by the taxonomist S. P. Birari and a voucher specimen (#U44Z-44009) was deposited at the Smithsonian Institution. An organic extract of this collection, N67393, was produced as reported previously51 and provided by NCI Natural Products Branch (Frederick, MD, USA). A portion of the crude extract (10.0 g) was fractionated over a vacuum-liquid-chromatography HP20ss column, eluted with a MeOH-H2O step-gradient (30:70, 50:50, 70:30, 90:10, and 100:0) and washed with CH2Cl2-MeOH (50:50). The third fraction (MeOH-H2O 70:30) and the fourth fraction (MeOH-H2O 90:10) were further fractionated by C18 preparative HPLC (MeCN-H2O 30:70 and MeCN-H2O 40:60, respectively) followed by semi-preparative HPLC using biphenyl and pentafluorophenyl columns, to obtain 1 (19.5 mg), 2 (22.0 mg), 3 (5.6 mg), 4 (36.0 mg), 5 (3.5 mg), 6 (3.1 mg), 7 (6.7 mg), 8 (5.5 mg), and 9 (1.0 mg).
Uzarigenin (1)
colorless crystal; [α]20 D+14(c 0.2, MeOH);1H and 13C NMR, see Tables S1 and S2, Supporting Information; HRESIMS m/z 375.2540 [M+H]+ (calcd for C23H35O4, 375.2530).
Coroglaucigenin (2)
colorless crystal; [α]20D +22(c 0.2, MeOH); 1H and 13C NMR, see Tables S1 and S2, Supporting Information; HRESIMS m/z 391.2491 [M+H]+ (calcd for C23H35O5, 391.2479).
Desglucouzarin (3)
colorless crystal; [α]20D −20 (c 0.2, MeOH); 1H and 13C NMR, see Tables S1 and S2, Supporting Information; HRESIMS m/z 537.3057 [M+H]+ (calcd for C29H45O9, 537.3058).
Frugoside (4)
white, amorphous powder; [α]C20D −20 (c 0.2, MeOH); 1H and 1C NMR, see Tables S1 and S2, Supporting Information; HRESIMS m/z 537.3068 [M+H]+ (calcd for C29H45O9, 537.3058).
Frugosidal (5)
white, amorphous powder; [α]20D −3 (c 0.014, MeOH); 1H and 13C NMR, see Table 1; HRESIMS m/z 551.2858 [M+H]+ (calcd for C29H43O10, 551.2851).
Glucofrugoside (6)
white, amorphous powder; α20 D-8(c 0.016, MeOH);1H and 13C NMR,see Tables S3 and S2, Supporting Information; HRESIMS m/z 721.3414 [M+Na]+ (calcd C35H54O14Na, 721.3406).
Uscharin (7)
light yellowish crystal; [α]20 D +34(c 0.2, MeOH); 1H and 13C NMR,se eTables S3 and S2, Supporting Information; HRESIMS m/z 588.2632 [M+H]+ (calcd for C31H42NO8S 588.2626).
Calotoxin (8)
colorless crystal; [α]20D +48 (c 0.2, MeOH); 1H and 13C NMR, see Tables S3 and S2, Supporting Information; HRESIMS m/z 549.2701 [M+H]+ (calcd C29H41O10, 549.2694).
Calotropin (9)
white, amorphous powder; [α]20D +40 (c 0.05, MeOH); 1H and 13C NMR, see Tables S3 and S2, Supporting Information; HRESIMS m/z 533.2748 [M+H]+ (calcd C29H41O9 533.2745).
X-ray Crystal Structure Analysis of 1–3, 7, 8, and Bromobenzoyl Derivative of 4
Intensity data for the compounds were collected using a D8 Quest κ-geometry diffractometer with a Bruker Photon II cmos area detector and an Incoatec Iμs microfocus Mo Kα source (κ = 0.71073 Å). The X-ray crystallographic data for these compounds have been deposited with the Cambridge Crystallographic Data Center under accession numbers CCDC1993992–1993997, respectively. These data can be accessed free of charge at http://www.ccdc.cam.ac.uk/. The cif documents of these compounds are also available free of charge on the ACS Publications website.
Acid Hydrolysis and Absolute Configuration Determination of Monosaccharide Moieties
Compounds 3–6 (0.5 mg for each) were hydrolyzed in the presence of 2 M HCl at 80 °C for 2 h. After extraction with EtOAc, the aqueous layers were neutralized with NaHCO3, dried under a vacuum evaporator, and dissolved in anhydrous pyridine (0.5 mL) with addition of L-cysteine methyl ester hydrochloride (2 mg). After the reaction mixtures were heated to 60 °C for 1.5 h, o-tolylisothiocyanate (50 μL) was added and the mixture was kept at 60 °C for 1 h. The reaction products were directly analyzed by LC-MS (MeCN-H2O, 1:3, with 0.1% formic acid; 0.4 mL/min). The monosaccharides D-glucose in 3 and 6, and D-6-deoxy allopyranose in 5–6 were identified based on comparison of the retention times with those of authentic samples [tR: D-glucose 4.53 min, L-glucose 4.23 min, and D-6-deoxy allopyranose 7.65 min (authentic D-6-deoxy allopyranose was from compound 4)].
Cell Lines and Reagents
BT-549 cells were acquired from the Lombardi Comprehensive Cancer Center of Georgetown University (Washington, DC, USA). CAL-51 cells were obtained from Creative Bioarray (Shirley, NY, USA), and SUM185PE cells were purchased from Asterand Bioscience (Detroit, MI, USA). All other cell lines were purchased from American Type Culture Collection (Manassas, VA, USA). Cell line identities were validated by STR profiling by Genetica DNA Laboratories (Burlington, NC, USA). MDA-MB-231, SUM185PE, and MDA-MB-453 cells were maintained in IMEM supplemented with 10% FBS and 25 μg/mL gentamicin. BT-549, CAL-51, HCC1806, HCC1937, MDA-MB-468, and HCC70 were maintained in RPMI-1640 supplemented with 10% FBS and 50 μg/mL gentamicin. Hs578T cells were maintained in DMEM supplemented with 10% FBS and 50 μg/mL gentamicin. All cell lines were grown at 37 °C in 5% CO2 humidified environment. Digoxin was purchased from Sigma (St. Louis, MO, USA).
Sulforhodamine B Assay
Cells were treated with indicated compounds for 48 h and antiproliferative and cytotoxic effects evaluated using the sulforhodamine B (SRB) assay as previously described.52,53 Concentration response curves were fit by four-parameter non-linear regression using GraphPad Prism 8. The IC50 concentrations were defined as the concentration that caused a 50% reduction in cell density as compared to vehicle (DMSO).
Na+/K+ ATPase Assay
Indicated concentrations of compounds were incubated with 6.25 mU of adenosine 5’-triphosphtase from porcine cerebral cortex (Sigma) and 2.5 mM ATP in 25 μL of 1X Na+/K+ ATPase reaction buffer (5X: 100 mM Tris (pH 7.8), 2.8 mM EDTA, 100 mM MgCl2, 15 mM KCl, 665 mM NaCl) at 37 °C for 15 min. An aliquot (25 μL) of ADP-Glo™ Reagent (Promega, Madison, WI, USA) was added to each reaction for 40 min to deplete the remaining ATP, followed by 50 μL of ADP-Glo™ Max Detection Reagent (Promega) for 1 h to convert ADP to ATP and generate a luciferase reaction. Luminescence was measured using the Cytation 5 plate reader and Gen5™ software (BioTek, Winooski, VT, USA). Concentration response curves were fit by four-parameter non-linear regression using GraphPad Prism 8. EC50 concentrations were defined as the concentration that caused a 50% reduction in luminescence compared to vehicle (DMSO) control.
Intracellular Ca2+ Imaging
Cells were seeded either on glass coverslips (Figure 4A–C) or in a 96 well optical bottom black-sided plate (Thermo Fisher Scientific, Waltham, MA, USA) (Figure 4D–E) and allowed to reach 60–80% confluency. Cells were loaded with 2.5 μM Cal-520 AM (ATT Bioquest, Sunnyvale, CA, USA) in complete RPMI media supplemented with 0.2% Pluronic F-127 (Biotium, Fremont, CA, USA) for 30 min at 37 °C. Cells were washed with PBS and kept in the dark in complete RPMI media for 15 min at room temperature prior to imaging. Images were acquired at baseline and the time of compound addition (T0) and at indicated time points. Cells were returned to 37 °C in between acquisitions. For Figure 4A–C, images were acquired using a Nikon Eclipse TE2000-U inverted microscope with a 20× objective and an Andor iXon camera using Metamorph software. Multiple fields of view were imaged for each time point. For Figure 4D–E, images were acquired on the Cytation 5 with the 4× objective and Gen5™ software (BioTek). The same field of view was imaged at each time point. Cell-by-cell analysis of fluorescence intensity was performed using ImageJ software, and data are represented as corrected total cell fluorescence (CTCF) = integrated density − (cell area × mean background fluorescence) normalized to the T0 values. Data were analyzed by two-way ANOVA with either Dunnett’s or Bonferroni’s multiple comparisons post hoc test using GraphPad Prism 8. For Figure 4C, frequency distribution of the cell-by-cell data was generated using GraphPad Prism 8 and the histograms were analyzed by non-linear regression to generate a Gaussian curve.
Membrane Enrichment
Cells were lysed in a digitonin buffer (10 mM PIPES, pH 6.8, 0.015% (w/v) digitonin, 100 mM NaCl, 300 mM sucrose, 5 mM MgCl2, 5 mM EDTA) supplemented with 1 mM PMSF (Sigma) and Halt™ protease inhibitor cocktail (Thermo Fisher Scientific) to release cytoplasmic contents. The membrane fraction was separated from the cytosol by centrifugation at 400 ×g for 15 min at 4 °C and then solubilized in Triton X-100 buffer (10 mM PIPES, pH 7.4, 0.5% (v/v) Triton X-100, 100 mM NaCl, 300 mM sucrose, 5 mM MgCl2, 5 mM EDTA) supplemented with 1 mM PMSF (Sigma) and Halt™ protease inhibitor cocktail (Thermo Fisher Scientific). Insoluble proteins and cellular components were separated from the solubilized membranes by centrifugation at 5,000 ×g for 10 min at 4 °C.
Western Blotting
Equal amounts of membrane and cytosolic protein were resolved by SDS PAGE and transferred to Immobilon-FL PVDF membranes (EMD Millipore, Burlington, MA, USA) for immunodetection of proteins. Non-reducing conditions were used for NCX1 detection. A detailed list of primary antibodies is provided in Supplementary Table S4. IRDye® secondary antibodies were used for all western blots (LI-COR Biosciences, Lincoln, NE, USA) and imaged on the Odyssey® FC (LI-COR). The REVERT™ total protein stain (LI-COR) and Image Studio software (LI-COR) was used to quantify total protein levels.
qRT-PCR
RNA was isolated using the TRIzol method and purity and concentration levels were assessed using a Cytation 5 plate reader, Take3™ micro-volume plate, and Gen5™ software (BioTek). The iScript™ Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA) was used to make cDNA in the T100™ thermal cycler (Bio-Rad). Transcript abundances were evaluated using iTaq™ Universal SYBR Green Supermix (Bio-Rad) on the CFX Connect™ (Bio-Rad) real-time PCR instrument. mRNA fold differences were calculated by the 2-ΔΔCt method54 with GAPDH designated the control gene. Primer oligos were purchased from Sigma: GAPDH F: GCAAATTCCATGGCACCGT, R: TCGCCCCACTTGATTTTGG, NCX1/SLC8A1 F: AGTGCTGGGGAAGATGATGACGACG, R: AGGATGGAGACAATGAAACACGCCC,55 NCX3/SLC8A3 F: GCATTGCCAGGGTCATTGTCT R: CCATAAGGGTCAGGTTGGAGA56
Supplementary Material
ACKNOWLEDGMENTS
Support for this project was provided by NIH grant U01CA182740 to R.H.C. and S.L.M and the Greehey Endowment (S.L.M.). Training support for P.J.P by the South Texas Medical Scientist Training Program (NIH T32GM113896) is acknowledged. The LC-MS instrument used for this project was provided in part by a Challenge Grant from the Office of the Vice President for Research, University of Oklahoma, Norman Campus and an award through the Shimadzu Equipment Grant Program (R.H.C.). The authors thank the National Science Foundation (grant CHE-1726630) and the University of Oklahoma for funds to purchase of the X-ray instrument and computers. The authors thank Dr. Mark Shapiro (University of Texas Health Science Center at San Antonio) for providing instruments for conducting intracellular Ca2+ imaging experiments. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. NMR data tables of compounds 1–4 and 6–9, ECD curves of compounds 1–3 and 6–9, X-ray structures of compounds 1, 2, 3, 7, 8, and bromobenzoyl derivative of compound 4, and 1D, 2D NMR and HRESIMS spectra for compounds 1–9, 1D NMR spectra for bromobenzoyl derivative of compound 4 are provided, antibody information, the effects of digoxin on intracellular Ca2+, membrane enrichment validation, total protein stains for western blots, data for NCX1 siRNA and CRISPR experiments, and supplementary methods for siRNA transfection and CRISPR gene knockout are provided.
REFERENCES
- (1).Li X; Yang J; Peng L; Sahin AA; Huo L; Ward KC; O’Regan R; Torres MA; Meisel JL Breast Cancer Res. and Treat 2017, 161, 279–287. [DOI] [PubMed] [Google Scholar]
- (2).Malorni L; Shetty PB; De Angelis C; Hilsenbeck S; Rimawi MF; Elledge R; Osborne CK; De Placido S; Arpino G Breast Cancer Res. Treat. 2012, 136, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dent R; Trudeau M; Pritchard KI; Hanna WM; Kahn HK; Sawka CA; Lickley LA; Rawlinson E; Sun P; Narod SA Clin. Cancer Res. 2007, 13, 4429–34. [DOI] [PubMed] [Google Scholar]
- (4).Garrido-Castro AC; Lin NU; Polyak K Cancer Discov. 2019, 9, 176–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bianchini G; Balko JM; Mayer IA; Sanders ME; Gianni L Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lehmann BD; Bauer JA; Chen X; Sanders ME; Chakravarthy AB; Shyr Y; Pietenpol JA J. Clin. Invest. 2011, 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lehmann BD; Jovanovic B; Chen X; Estrada MV; Johnson KN; Shyr Y; Moses HL; Sanders ME; Pietenpol JA PLoS ONE 2016, 11, e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Burstein MD; Tsimelzon A; Poage GM; Covington KR; Contreras A; Fuqua SA; Savage MI; Osborne CK; Hilsenbeck SG; Chang JC; Mills GB; Lau CC; Brown PH Clin. Cancer Res. 2015, 21, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jiang Y-Z; Ma D; Suo C; Shi J; Xue M; Hu X; Xiao Y; Yu K-D; Liu Y-R; Yu Y; Zheng Y; Li X; Zhang C; Hu P; Zhang J; Hua Q; Zhang J; Hou W; Ren L; Bao D; Li B; Yang J; Yao L; Zuo W-J; Zhao S; Gong Y; Ren Y-X; Zhao Y-X; Yang Y-S; Niu Z; Cao Z-G; Stover DG; Verschraegen C; Kaklamani V; Daemen A; Benson JR; Takabe K; Bai F; Li D-Q; Wang P; Shi L; Huang W; Shao Z-M Cancer Cell 2019, 35, 428–440. [DOI] [PubMed] [Google Scholar]
- (10).Newman DJ; Cragg GM J. Nat. Prod. 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- (11).Robles AJ; Du L; Cichewicz RH; Mooberry SL J. Nat. Prod. 2016, 79, 1822–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Robles AJ; Cai S; Cichewicz RH; Mooberry SL Breast Cancer Res. Treat. 2016, 157, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Robles AJ; McCowen S; Cai S; Glassman M; Ruiz F, 2nd; Cichewicz RH; McHardy SF; Mooberry SL J Med. Chem. 2017, 60, 9275–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grant CV; Carver CM; Hastings SD; Ramachandran K; Muniswamy M; Risinger AL; Beutler JA; Mooberry SL Breast Cancer Res. Treat. 2019, 177, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kadiyala M; Ponnusankar S; Elango KJ Ethnopharmacol. 2013, 150, 32–50. [DOI] [PubMed] [Google Scholar]
- (16).Chan EWC; Sweidan NI; Wong SK; Chan HT Rec. Nat. Prod. 2017, 11, 334. [Google Scholar]
- (17).Nguyen MTT; Nguyen KDH; Dang PH; Nguyen HX; Awale S; Nguyen NT J. Nat. Prod. 2020, 83, 385–391. [DOI] [PubMed] [Google Scholar]
- (18).Gohar AA; El-Olemy M; Abdel-Sattar E Nat. Prod. Sci, 2000, 6, 142–146. [Google Scholar]
- (19).Hosseini SH; Masullo M; Cerulli A; Martucciello S; Ayyari M; Pizza C; Piacente SJ Nat. Prod. 2019, 82, 74–79. [DOI] [PubMed] [Google Scholar]
- (20).Elgamal MHA; Hanna AG; Morsy NAM; Duddeck H; Simon A; Gáti T; Tóth GJ Mol. Struct. 1999, 477, 201–208. [Google Scholar]
- (21).Tanaka T; Nakashima T; Ueda T; Tomii K; Kouno I Chem. Pharm. Bull. (Tokyo) 2007, 55, 899–901. [DOI] [PubMed] [Google Scholar]
- (22).Muhit MA; Umehara K; Mori-Yasumoto K; Noguchi HJ Nat. Prod. 2016, 79, 1298–1307. [DOI] [PubMed] [Google Scholar]
- (23).Ge Y-W; Tohda C; Zhu S; He Y-M; Yoshimatsu K; Komatsu KJ Nat. Prod. 2016, 79, 1834–1841. [DOI] [PubMed] [Google Scholar]
- (24).Odonbayar B; Murata T; Batkhuu J; Yasunaga K; Goto R; Sasaki KJ Nat. Prod. 2016, 79, 3065–3071. [DOI] [PubMed] [Google Scholar]
- (25).Kil Y-S; Kim SM; Kang U; Chung HY; Seo EK J. Nat. Prod. 2017, 80, 2240–2251. [DOI] [PubMed] [Google Scholar]
- (26).Clarkson C; Hansen SH; Jaroszewski JW Anal. Chem. 2005, 77, 3547–3553. [DOI] [PubMed] [Google Scholar]
- (27).Sawlewicz L; Weiss E; Reichstein T Helv. Chim. Acta 1967, 50, 504–530. [DOI] [PubMed] [Google Scholar]
- (28).Cheung HTA; Chiu FCK; Watson TR; Wells RJ J. Chem. Soc. Perkin. Trans. 1, 1983, 0, 2827–2835. [Google Scholar]
- (29).Van Quaquebeke E; Simon G; André A; Dewelle J; Yazidi ME; Bruyneel F; Tuti J; Nacoulma O; Guissou P; Decaestecker C; Braekman J-C; Kiss R; Darro FJ Med. Chem 2005, 48, 849–856. [DOI] [PubMed] [Google Scholar]
- (30).Cheung HTA; Nelson CJ; Watson TR J. Chem. Soc. Perkin. Trans. 1, 1988, 7, 1851–1857. [Google Scholar]
- (31).Agrawal AA; Petschenka G; Bingham RA; Weber MG; Rasmann S New Phytol. 2012, 194, 28–45. [DOI] [PubMed] [Google Scholar]
- (32).Schonfeld W; Weiland J; Lindig C; Masnyk M; Kabat MM; Kurek A; Wicha J; Repke KR N.-S. Arch. Pharmacol. 1985, 329, 414–426. [DOI] [PubMed] [Google Scholar]
- (33).Katz A; Lifshitz Y; Bab-Dinitz E; Kapri-Pardes E; Goldshleger R; Tal DM; Karlish SJD J. Biol. Chem 2010, 285, 19582–19592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Altamirano J; Li Y; Desantiago J; Piacentino V; Houser SR; Bers DM J. Physiol. 2006, 575, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Reuter H; Henderson SA; Han T; Ross RS; Goldhaber JI; Philipson KD Circ. Res. 2002, 90, 305–308. [DOI] [PubMed] [Google Scholar]
- (36).Bai Y; Morgan EE; Giovannucci DR; Pierre SV; Philipson KD; Askari A; Liu L Am. J. Physiol. Heart Circ 2013, 304, H427–H435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).So CL; Saunus JM; Roberts-Thomson SJ; Monteith GR Semin. Cell Dev. Biol. 2019, 94, 74–83. [DOI] [PubMed] [Google Scholar]
- (38).Frandsen SK; Krüger MB; Mangalanathan UM; Tramm T; Mahmood F; Novak I; Gehl J Cancer Res. 2017, 77, 4389–4401. [DOI] [PubMed] [Google Scholar]
- (39).Bong AHL; Monteith GR Biochim. Biophys. Acta Mol. Cell Res 2018, 1865, 1786–1794. [DOI] [PubMed] [Google Scholar]
- (40).Kim N; Yim HY; He N; Lee C-J; Kim JH; Choi J-S; Lee HS; Kim S; Jeong E; Song M; Jeon S-M; Kim W-Y; Mills GB; Cho Y-Y; Yoon S Sci. Rep. 2016, 6, 29721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Da Costa EM; Armaos G; McInnes G; Beaudry A; Moquin-Beaudry G; Bertrand-Lehouillier V; Caron M; Richer C; St-Onge P; Johnson JR; Krogan N; Sai Y; Downey M; Rafei M; Boileau M; Eppert K; Flores-Díaz E; Haman A; Hoang T; Sinnett D; Beauséjour C; McGraw S; Raynal NJMJ Exp. Clin. Cancer Res 2019, 38, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Varga K; Hollósi A; Pászty K; Hegedüs L; Szakács G; Tímár J; Papp B; Enyedi Á; Padányi R BMC Cancer 2018, 18, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Slingerland M; Cerella C; Guchelaar HJ; Diederich M; Gelderblom H Invest. New Drugs 2013, 31, 1087–1094. [DOI] [PubMed] [Google Scholar]
- (44).Yu SP Prog. Neurobiol. 2003, 70, 363–386. [DOI] [PubMed] [Google Scholar]
- (45).Prassas I; Diamandis EP Nat. Rev. Drug Discov. 2008, 7, 926–935. [DOI] [PubMed] [Google Scholar]
- (46).Schneider NFZ; Cerella C; Simões CMO; Diederich M; Schneider NFZ; Cerella C; Simões CMO; Diederich M Molecules 2017, 22, 1932. [Google Scholar]
- (47).Menger L; Vacchelli E; Kepp O; Eggermont A; Tartour E; Zitvogel L; Kroemer G; Galluzzi L Oncoimmunology 2013, 2, e23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Biggar RJ; Andersen EW; Kroman N; Wohlfahrt J; Melbye M Breast Cancer Res. 2013, 15, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cerami E; Gao J; Dogrusoz U; Gross BE; Sumer SO; Aksoy BA; Jacobsen A; Byrne CJ; Heuer ML; Larsson E; Antipin Y; Reva B; Goldberg AP; Sander C; Schultz N Cancer Discov. 2012, 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gao J; Aksoy BA; Dogrusoz U; Dresdner G; Gross B; Sumer SO; Sun Y; Jacobsen A; Sinha R; Larsson E; Cerami E; Sander C; Schultz N Sci Signal 2013, 6, pI1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).McCloud TG Molecules 2010, 15, 4526–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Skehan P; Storeng R; Scudiero D; Monks A; McMahon J; Vistica D; Warren JT; Bokesch H; Kenney S; Boyd MR J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- (53).Shaffer CV; Cai S; Peng J; Robles AJ; Hartley RM; Powell DR; Du L; Cichewicz RH; Mooberry SL J. Nat. Prod. 2016, 79, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Livak KJ; Schmittgen TD Methods 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- (55).Muñoz JJ; Drigo SA; Barros-Filho MC; Marchi FA; Scapulatempo-Neto C; Pessoa GS; Guimarães GC; Trindade Filho JCS; Lopes A; Arruda MAZ; Rogatto SR J. Urol. 2015, 194, 245–251. [DOI] [PubMed] [Google Scholar]
- (56).Pelzl L; Hosseinzadeh Z; Al-Maghout T; Singh Y; Sahu I; Bissinger R; Schmidt S; Alkahtani S; Stournaras C; Toulany M; Lang F Cell Physiol. Biochem. 2017, 42, 1240–1251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.