

SUMMARY
P. falciparum infection and malaria remain a risk for millions of children and pregnant women. Here, we seek to integrate knowledge of mouse and human T helper cell (Th) responses to blood-stage Plasmodium infection to understand their contribution to protection and pathology. Although there is no complete Th subset differentiation, the adaptive response occurs in two phases in non-lethal rodent Plasmodium infection, coordinated by Th cells. In short, cellular immune responses limit the peak of parasitemia during the first phase; in the second phase, humoral immunity from T cell dependent germinal centers is critical for complete clearance of rapidly changing parasite. A strong IFN-γ response kills parasite, but an excess of TNF compared to regulatory cytokines (IL-10, TGF-β) can cause immunopathology. This common pathway for pathology is associated with anemia, cerebral malaria and placental malaria. These two phases can be used to both understand how the host responds to rapidly growing parasite, and how it attempts to control immunopathology and variation. This dual nature of T cell immunity to Plasmodium is discussed, with particular reference to the protective nature of the continuous generation of effector T cells, and the unique contribution of effector memory T cells.
Keywords: CD4 T cells, T follicular, Plasmodium, Cytokines
1. INTRODUCTION
CD4 T helper (Th) cells are required to control both parasitemia and pathology in Plasmodium chabaudi, yoelii, and berghei infection of mice, and CD4 T cell expansion correlates with protection in human P. falciparum infection. Several mouse studies have shown the effect of CD4 T cells in combination with B cells and phagocytes in protection from malaria in mouse models 1–3. Upon infection with Plasmodium spp., many of the known innate immune sensors are triggered, and multiple adaptive immune mechanisms are activated. Many of these mechanisms are coordinated by CD4 Th cells, a regulatory cell type. Their predominant effector mechanism is production of secreted cytokines, which have functions both locally and systemically. Therefore, understanding the mechanisms by which CD4 Th direct the immune response can suggest points of potential intervention upstream of many other cell types. Th cytokines are important in response to Plasmodium for both helping B cells to make antibody, and activating innate defense mechanisms of parasite killing. Systemic cytokine production, such as that seen when parasitemia reaches high levels, is responsible for the induction of host defense mechanisms that can also cause disease 4. Despite this tremendous response, without treatment, infection with Plasmodium spp. becomes persistent. P. falciparum can be persistent for at least a year in humans 5,6. Persistence is also evident in other settings of co-evolution, such as in the thicket rats (Thamnomys) where the parasites now used in mouse models were originally discovered 7. In response to the urgent need to control the parasite to prevent lethal parasite-driven damage, the mammalian host has evolved a two-phase immune response to Plasmodium infection, which we will describe in detail in this review. The first phase following innate responses that fail to eliminate parasite is a strong cellular response, followed by a prolonged humoral response that can completely clear the parasite.
In response to infection with Plasmodium spp., acute phase protein production includes complement 8, platelet activation 9, and coagulation cascade proteins induced in the liver 10,11. In addition, endothelia are activated to make cytokines and coagulation regulators by local immune cell and parasite adhesion, as well as circulating cytokines. Inflammatory cytokines increase tissue surveillance by upregulation of adhesion molecules and chemokines. The early CD4 T cell response to blood-stage Plasmodium infection promotes increased phagocytosis in the spleen and liver 12. This increase is due to the combination of recruitment of innate cells, such as CD8 DCs, to sites of parasite killing, and activation of phagocytosis and parasiticidal effector molecules within immune and structural cells 13. Unfortunately, while this coordinated effort activates a diverse set of effector mechanisms, these early T cell responses also result in significant damage to host tissues. The spectrum of malaria disease ranges from mild to severe, where multiple organs can be affected at once, however, Th cells also regulate inflammation. In this review, we will describe studies performed in mice and humans to understand the role of CD4 T cells in coordinating this balanced response to infection with Plasmodium spp., and how these cells contribute to pathology and/or protection from infection and disease.
Most findings in P. chabaudi infection of mice regarding adaptive immunity and pathology have been verified in P. falciparum infection 14. P. yoelii has increasingly also been used to study basic immunology of Plasmodium infection, and while many features of the immune responses to these two rodent pathogens are similar, we also highlight responses that appear to be different. In this review, we will progress from what is known about the earliest activation of T cells, through interactions with B cell biology, to generation of memory T cells. Through the lens of cytokines produced by CD4 T cells, we will attempt to integrate the known mechanisms of pathology caused by T cells with what is known about how malaria disease pathology occurs and is regulated. Along the way, we will identify some of the many unknowns and the prognosis for this basic knowledge to be applied to successful vaccination.
2. SPECIFICITY OF THE RESPONSE TO PLASMODIUM
2.1. T cell detection in Plasmodium-immune subjects
Since the early 1990’s it has been possible to detect T cell responses to P. falciparum in humans. Peptides have been identified from parasite antigens to which there are antibodies in the serum of exposed people, and various assays used to detect them. While proliferation in response to a peptide indicates the presence of specific T cells, many studies relied instead on cytokine production. Different cytokines in response to these peptides were found to correlate with different aspects of the response and clinical symptoms of malaria, and not necessarily to in vitro proliferation 15. For example, in 1990, IL-4 in response to peptides from surface antigen RESA, was found to be correlated to the serum antibody 16, in agreement with the Th1/Th2 paradigm, first described in 1986. Plasmodium-specific CD4 T-cell responses was further demonstrated later by several studies both in human and murine model using intracellular staining by multi-parameter flow cytometry 17–19. Studies in mouse models have made strides towards understanding the role of T cell specificity in the response to Plasmodia since generation of the first T cell receptor transgenic mice for CD4 and CD8 T cells specific for malaria 2,20–22, and some mouse MHC tetramer reagents 23. However, the protective CD4 T cell epitopes, if there are specific protective ones, have not been identified in either mouse or human.
Overall, 596 epitopes have been identified for CD4+ T cells and 147 CD8+ epitopes from the Plasmodium-specific response in humans and mice 24. However, the nature of specificity of the natural T cell response in terms of contribution of each epitope response to protection is less well understood. It would be useful to identify potentially protective T cell epitopes that could stimulate IFN-γ, and also help B cells make protective antibodies. This would have to be done by determining all peptides generated by eluting peptides from human antigen presenting cells MHCII from immune individuals and testing them for protection. However, thus far, it has not been feasible to test overlapping peptides from all 5,369 predicted proteins of P. falciparum for stimulation and protection. Due to the requirement for both B and T cell responses, it is challenging to prove that a given CD4 T cell specificity is particularly protective. Due to the mechanisms of phagocytosis and antigen presentation, T cells can recognize peptides from proteins even inside the parasite, leading to a likelihood of their recognizing a broader repertoire of proteins than B cells from the first infection. In addition, internal epitopes are likely to have less variation due to the reduction in selection by antibody-mediated neutralization and other clearance mechanisms. It should be noted that more conserved T cell epitopes could be used to enhance protective B cell epitopes. In addition, epitopes that are conserved are generally selected by an essential function, and immune responses that can target such a critical function are much more likely to be successful.
There are two primary mechanism employed by the parasite in addition to the sheer number of proteins. As an example, merozoite surface protein-1 (MSP-1) has been very well studied. Very few T cell epitopes come from the protective B cell conformational epitope in the C-terminus of MSP-1. While tight conserved essential structures is are challenging for antigen presenting cells to digest, the looser external structure is easier 25,26. Similar camouflaged invasion mechanisms in other parasites and viruses suggest that hiding essential epitopes is a great evasion strategy. In contrast, the exposed and, hence, more variant regions are more likely to induce antibodies. Importantly, there is more latitude than commonly considered for finding T cell helper epitopes. Even though the B5 transgenic T-cell receptor (TCR) recognizes MSP-1, B5 CD4 T cell epitope is on an exposed fragment of MSP1 that is shed into the serum each time merozoites invade, while the protective 19-kDa C-terminal region of MSP-1 (p19) remains attached within the infected red blood cell, B5-specific T cells are able to help MSP-1 p19-specific B cells 2. Unfortunately, this looser conformation also imparts more latitude for variation in the sequence of the T cell epitopes. Variation can affect the T cell response, with some alternate versions of epitopes even inhibiting other variants 27. It would be interesting to understand the mechanisms involved in B cell help from T cells when the epitopes are not physically connected. While soluble antigen is presented to B cells by follicular dendritic cells and subcapsular macrophages, the mechanism of large antigen capture by B cells remains obscure 28. A recent paper has shed some light on B cells capturing multiple antigens from infected cells that are nearby in space on the membrane, rather than part of the same amino acid chain 29. This suggests that a B cell with a B cell receptor (BCR) that recognizes merozoite surface proteins can be helped by T cells recognizing any other merozoite surface proteins. In addition, DCs may be able to present antigen in immune complexes to B cells in the marginal zone by a newly described pathway using FcγRIIB that seems to bypass T cell tolerance mechanisms 30. As T cell epitopes are less subject to selection than exposed B cell epitopes, adding T cell epitopes from less exposed sites could be a powerful method of improving vaccination for broadly neutralizing antibodies. Further study using functional assays to determine the protective contribution of particular epitopes will be essential in building up an armory of protective and immunogenic epitopes for the next generation of multi-stage, multi-antigen malaria vaccines.
2.2. Polyclonal adaptive immune activation in Plasmodium infection
The co-evolution of P. falciparum with H. sapiens is evident in the modern immune response to infection 31. This process has resulted in some helpful alleles, including germline T cell receptors and natural IgM responsive to infected red blood cells 32–35. Upon infection with Plasmodium chabaudi or yoelii, about half of the immune repertoire gets activated 36,37. This has been observed for both CD4 T and B cells. This massive activation may be due to the huge number of proteins expressed by Plasmodium (from an estimated 5,369 genes 38,39), compared to a bacterial or viral pathogen. In addition, the immunodominant repeat regions, and the switching of surface proteins with variant domains add a huge variety of unhelpful specificities. There are also polyclonal B and T cell stimulatory molecules in P. falciparum, which could account for this large non-specific response 40–43. Antibodies that recognize non-Plasmodium antigens have even been measured in serum from Plasmodium-infected animals 44. The B cell polyclonal stimulator has not been specifically identified yet. However, PfEMP1 can induce IL-12 and IL-18 from antigen presenting cells, which stimulate IFN-γ, even in the absence of MHCII stimulation 42.
2.3. The three signals of T cell activation in response to infection with Plasmodium spp.
Activation of CD4 T cells during Plasmodium infection promotes parasite-specific B cell activation 1,45, and phagocytosis of the parasite by secretion of Th1 cytokines by cognate interaction of the T cell receptor (TCR) with the major histocompatibility complex class II (MHCII) 3. However, these interactions are likely to be obscured by the large polyclonal response described above. Interestingly in this regard, it was found that a vast number of CD8 T cells would be required to be generated to find all infected hepatocytes in P. yoelii infection and affect parasitemia 46. Given the less direct action of CD4 T cells compared to CD8, through cytokines rather than cytotoxicity, it is not known if the same barrier exists for CD4 T cell numbers, which generally proliferate less 47.
Three signals are required for Th cell stimulation in general: MHCII/TCR, co-stimulation, and cytokines. However, the sequence of events leading to T cell expansion and generation of effector and memory T cells in response to Plasmodium infection are under investigation. CD28 is the primary co-stimulatory molecule in the dendritic cell T cell interaction, binding to CD80 and CD86. CD28 is known to be responsible for recruitment of many of the transcription factors required for IL-2 production 48,49. IL-2 promotes T cell expansion; however, though CD28 is a costimulatory signal thought to be required for IL-2 production, CD28−/− mice infected with P. chabaudi have similar numbers of CD4 T cells on days 7 or 11-13, suggesting that costimulatory pathways other than CD28 may contribute to T cell activation 50,51. In infected CD28−/− mice, IFN-γ is significantly reduced compared to wildtype, as is switching to IgG2a/c, a critical immunoglobulin isotype in parasite clearance 52. In addition to CD28, T cells in mouse Plasmodium infections upregulate several co-stimulatory and inhibitory surface molecules 22,51. Expression of co-stimulatory molecule OX40 is beneficial for co-stimulation of T cells, regulating the size of the effector T cells (Teff) response to P. yoelii 53,54. ICOS expression, while known for its enhancement of Tfh function, inhibits control of peak P. chabaudi parasitemia, while anti-ICOS prolongs P. yoelii infection suggesting an important immunoregulatory role for this molecule that differs in the two infections 55.
As in other infections, the co-inhibitory molecules programmed cell death-1 (PD-1), B and T lymphocyte attenuator (BTLA), and cytotoxic T lymphocyte-associated antigen 4 (CTLA4) are upregulated on T cells upon Plasmodium infection. Inhibition of these molecules demonstrates the extensive network and successive waves of regulation involved in regulating the balance of parasiticidal and immuno-pathological outcomes of Plasmodium infections. While there is PD-1 expression on T cells in all Plasmodium infections studied to date, it may represent a baseline level that is present on CD4 Teff 56,57. The most striking finding about T cell co-receptors in infection is that while blocking the inhibitory co-receptors PD-1 and LAG3 inhibited T cell mediated control of the peak parasitemia in P. yoelii 58. PD-1 signaling on the other hand allows persistence later on in P. chabaudi, and this is modulated by PD-L2 59. CTLA4 on regulatory T cells (Treg) cells during priming was shown to play a critical role in regulation of the T cell response in the second week of P. yoelii infection 60. BTLA, highly expressed on B and T cells, also has a regulatory effect that should continue to be studied, as it is similar to PD-1 in that it directly affects TCR signaling 61 and parasite clearance. BTLA deficient mice controlled P. yoelii strikingly well due to intrinsic effects on both B and T cells affecting both antibody and IFN-γ, suggesting a strong inhibitory effect by BTLA throughout infection 62. In summary, co-stimulatory and inhibitory receptors are acting in concert and competition to regulate the very strong multi-modal activation initiated by Plasmodium infection. However, there may or may not be enough overlap between mouse models to inform human malaria therapeutics.
The third required signal in T cell stimulation is cytokines. The innate cytokine response is critical for regulation of the size and effectiveness of the T cell response. Early host factors like IFN-γ from NK cells and Type I IFN-γ from plasmacytoid cells are critical mediators of adaptive immunity 63–66. Each parasite strain and species appears to induce a variety of early responses from different cell types 67–69, and research urgently needs to be done about the parasite alleles that can regulate innate responses changing the adaptive response. IL-2 production by CD4 T cells regulates the Treg/Teff ratio, and is required for optimal T cell expansion. However, IL-2 may be produced at low levels in Plasmodium infection, likely reducing levels of IFN-γ produced 70,71. IL-12 made early on by innate cells like macrophages and dendritic cells is the predominant determinant of Th1 differentiation, and also promotes IL-10 production in Th1 cells, as does IL-27 72,73. Unexpectedly, IL-27 is made by a subset of T cells in Plasmodium infection and inhibits T cell expansion. It has been suggested that IL-27 expression reduces IL-2 which also affects later IFN-γ production, thereby inhibiting the ability of the immune response to control the peak of parasitemia 70,71. IL-27 also regulates IL-10 production by T cells later in infection 74. Many aspects of the T cell cytokine response have been recently reviewed 58. Both host and parasite factors regulate the size of the T cell response. For example, parasite-encoded Macrophage Migration Inhibitory Factor (pMIF) interferes with T cell activation dramatically, including driving upregulation of inhibitory co-receptor PD-1 75. There should be a concerted effort to understand other mechanisms of parasite virulence using immune-evasion, for example, screening a library of parasites mutated one gene at a time to discover parasite genes that drive the compromise with the host and thereby determine host morbidity and mortality.
2.4. The cellular activation pathway of effector CD4+ T cells in response to Plasmodium infection
In order to begin to understand the process of CD4 T cell activation in P. chabaudi infection, we studied surface phenotypes and identified a progression of T cell phenotypes, from more to less pluripotent, and from less to more terminally differentiated 76. We hope that this cellular activation pathway will promote further study of both CD4 T cell effector and memory development in the context of Plasmodium and other infections. Using this new set of markers, one can separate terminally differentiated effector T cells, from those that are still expressing higher levels of anti-apoptotic genes. More work is needed to understand mechanisms driving the transitions from quiescent Early Teff (CD127loCD127−CD62Lhi) to either proliferating Intermediate and Late Teff (CD62Llo), or pre-memory T cells (CD127+). It will be interesting to use this progression of distinct subsets to investigate integration of signaling for proliferation, metabolic shifts, and survival involved in T cell expansion and contraction. For example, we have recently identified the fatty acid synthesis pathway intrinsic to T cells as specifically important in generation of pre-memory T cells in prolonged P. chabaudi infection 77. Research into early T cell decisions have converged on IL-2 as a critical cytokine driving memory CD4 T cell differentiation 78–81. Another interesting application of this system of markers is to understand maintenance of T cell responsiveness or quiescence. For example, we and others have found that while CD27 is reported to be required for memory T cell survival, CD4+CD27− Teff and effector memory T cells (Tem) can re-express CD27 in some conditions, presumably prolonging their survival 76,82,83. As CD27+ Teff produce the most cytokines, it will be important to understand additional factors involved in their survival. Understanding the basic mechanisms of T cell activation in Plasmodium infection will help us find pathways that could be manipulated to improve the response in vaccination.
3. TWO PHASES OF THE IMMUNE RESPONSE TO PLASMODIUM SPP.
To understand immunity to P. falciparum, which has a variety of outcomes due the immense variation and number of strains, it would seem instructive to compare the two most common mouse models used to understand anti-parasite immunity. While P. yoelii XNL infection of wildtype mice lasts for about twenty days 84, P. chabaudi AS infected mice can have relapsing remitting parasitemia for between two and three months 85. In fact, there are reports of sub-patent P. yoelii infections up to 7 weeks, though that may be the result of uncloned lines being used 86. Furthermore, the peak of P. yoelii does not occur until around day 15 post-infection, while P. chabaudi peaks between days 8-10. Two differences between these two infections that affect the immune response are the cell types they infect, and the timing of their emergence from red blood cells. Firstly, P. yoelii parasites prefer immature erythrocytes, called reticulocytes, while P. chabaudi (Figure 1A), like P. falciparum, are less particular. The low initial frequency of reticulocytes probably explains the slower growth of the parasite (Figure 1B). Secondly, P. chabaudi, like P. falciparum, emerges synchronously from infected erythrocytes while P. yoelii does not, a disease-causing process that controls release of cytokines into the serum. While kinetics of parasitemia differ, both infections require both B and T cells to fully clear infection 87. We will discuss below the role of T cells in the early control of parasite, and the role of antibody in full clearance starting with these two rodent infections.
Figure 1.

Two phase adaptive immune response to Plasmodium spp.. Alignment of parasitemia of A) P. chabaudi chabaudi (AS) with B) P. yoelii (17XNL) and C) kinetics of T cell phenotypes in mice each week of infection. Th1-like response peaks preceeding strong germinal center response, which does not coincide with increase in GC-Tfh cells. Based on data from 36
As originally proposed by Jean Langhorne in 1998 88, fast-growing P. chabaudi elicits a notable IFN-γ response in the first week of infection. This response is followed by a change. T cells recovered after two weeks, when the parasite is well controlled, produce cytokines that help B cells make antibody (Figure 1C). This intriguing shift in the immune response over time is also seen in infection with M. tuberculosis, suggesting it is driven by persistent infection 89. The mechanisms of this switch have been elucidated in that there is a change in the available antigen presenting cells from CD8a+ to CD8α− dendritic cells after day 10, which induce activation of less inflammatory T cells 90. It was recently shown that B cells are also critical antigen presenting cells for generating GC Tfh cells in the first three days of P. chabaudi 91. The peak of the Th1 response in both P. chabaudi and P. yoelii (which is mixed with Tfh features) occurs during the first week of infection, after which levels of the Th1 cytokine IFN-γ and T cell expression of the Th1-master regulatory transcription factor T-bet decrease 36,92. The contraction of IFN-γ+ effector T cells corresponds with the increase in GC B cells seen over the course of infection (Figure 1C)
Control of early parasitemia is due to T cells in both P. chabaudi and P. yoelii. For example, when CD4 T cells were depleted in P. yoelii or P. chabaudi infection, the parasitemia increased dramatically in the first week to ten days 93,94. CD4 T cells responding to P. yoelii generate a larger IFN-γ response than that made by T cells responding to P. chabaudi. These highly activated T cells become exhausted by the time P. yoelii parasite growth peaks at the time of reticulocytosis. Inhibition of nitric oxide affects parasitemia in the second week of P. chabaudi infection, but not P. yoelii 12,95. The strong Th1 response to fast growing parasite leads to pathology downstream of the cytokine response in P. chabaudi. However, P. yoelii is initially well controlled by a stronger Th1 response that induces anemia, but is not associated with fever or hypothermia 96–98
Strikingly, control of T cell exhaustion by anti-PD-L1 plus anti LAG3 antibodies induced excellent control of the peak of parasitemia in P. yoelii, but not P. chabaudi 58. It is possible that this early T cell exhaustion in P. yoelii is caused by the continuous presence of parasite due to the unsynchronized release of P. yoelii, which is atypical in malaria-causing parasites. While PD-1 has been detected on T cells in all infections 56, that is likely to represent T cell activation rather than exhaustion 57, since anti-PD-1 does not improve T cell responsiveness in P. chabaudi. However a variety of co-inhibitory pathways may be involved in regulating the potentially pathogenic response. In P. vivax, simultaneous blockade of CLTA-4, PD-1, and T-cell immunoglobulin and mucin-3 (Tim3) ex vivo increases cytokine production by antigen-specific CD4 T cells 99.
T cells may be helped in their control of the initial parasitemia in P. yoelii due to this parasite’s preference for the smaller population of immature erythrocytes, compared to the large population of normocytes targeted by P. chabaudi, leading to almost unhindered parasite growth. Or, perhaps the stronger immune response is due to the preference for immature (MHCI+) reticulocytes, leading to better control, perhaps with CD8 participation. Regardless, it has been known for many years that the peak of P. chabaudi infection can be reduced best, and even delayed, by a decent IFN-γ or IL-12 biased response, while the peak of P. yoelii is best reduced by antibody 2,12,100. Although parasitemia of some strains of P. yoelii do not seem to be affected at all by inhibition of IFN-γ 68, other T cell cytokines such as LT or TNF may be at play because deleting CD4 T cells does change parasitemia from P. yoelii XNL both early and late 101.
While we acknowledge differences in the success of the adaptive response against the two parasite species P. yoelii and P. chabaudi, we hypothesize that the apparent difference between required immune responses in control of the peak of parasitemia is primarily due to the timing of the appearance of susceptible erythrocytes. This idea is based on the similar kinetics of the adaptive immune response with an increase in hybrid Th1/Tfh cells in both, as in P. falciparum, and a similar gradual increase in germinal center B cells 36,102. This is a hypothesis that can be tested, for example, using various parasite strains to induce different levels of innate responses by NK cells and pDCs and, thereby, contributing more or less to early control. Some published work does test the hypothesis. For example, while ICOS deficient mice have very reduced peak P. chabaudi parasitemia, anti-ICOS actually prolongs P. yoelii infection. ICOS deficient T cells become more Th1-like to promote control of early parasitemia, but inhibits the clearance of parasite by GC-dependent antibody 55. Therefore, ICOS deficiency may have differential effects in each infection due to its effect on IFN-γ, which may control peak P. chabaudi, but inhibit the germinal center for longer supporting the hypothesis we are proposing here. The differential effect of acute gamma herpesvirus reduction of humoral responses on P. chabaudi and P. yoelii also supports this idea 103.
The nature of the adaptive response is regulated very early in infection, as shown in studies cited above, where the innate response regulates the degree of the adaptive response 104. We also show that allowing prolonged infection beyond day 3 post-infection leads to a complete change in the Th phenotypes generated compared to extended infection 36,76. Therefore, it is critical to note that there are no known pathogenic Plasmodium species that are controlled completely in the first week of infection by the innate response, as are many viruses. One interpretation is that many of the features of the T cell response, including the hybrid Th1/Tfh phenotype, and the predominance of Tem compared to central memory T cell (Tcm), are due to successful immune evasion by the parasite using unknown early mechanisms.
3.1. Early roles of B cells and T cells
Transfer of serum from immune individuals can protect less immune individuals 105,106, and antibody levels can correspond to protection after vaccination 107. Another clue pertaining to the importance of antibody is that hyperimmune serum, antiserum generated by immunization, or purified IgG or IgM significantly reduces parasitemia of a first infection in mice better than serum from a single infection 108. Repeated infection with P. chabaudi or P. yoelii makes the immune serum more effective in parasite control, though CD4 T cells are required for this effect. These historic data support the interpretation that affinity maturation of antibody does eventually occur and that both T cells and antibody are crucial for continued immunity 109,110. Serum from protected mice opsonizes parasite for phagocytosis 111, and B cell deficient mice never clear parasitemia 1. Although we have not yet been able to deplete plasma cells at specific timepoints, overall there is clearly a role for antibody in protection from malaria and parasitemia. However, there is significant debate about the timing that specific antibody begins to play an effective role in elimination of parasite.
The early response includes significant serum antibody, and there is a large activation of CD19+ B cells in the first week of infection. Various studies have shown a significant population of Syndecan-1 (CD138+) B cells, likely to be plasmablasts, and measurable P. chabaudi MSP1-specific serum IgM, though little IgG 85,112,113. The early CD138+ B cells are CD4 and ICOS dependent 55. These B cells are sometimes challenging to measure in ELISpot as antibody secreting cells either ex vivo, or stimulated, likely because this response is transient, with a peak of short-lived CD138+ B220int cells lasting only one day 114–116. However, they were detected beautifully as MSP-1 p19 tetramer-binding T cells in a new flow cytometry assay 113. Strikingly, the T cell co-stimulatory molecule CD28 is not required for the generation of a large polyclonal and potentially non-specific IgM that is seen early in the response and provides some protection from fast growing parasitemia 50,117.
The localization and kinetics of B cell activation leading to early IgM and their dependence on T cells are just starting to become clear. However, the B cell and T cell interactions leading to the later antibody response dominated by IgG2a-c are more likely to be canonical. Generation of the antibodies that clear infection of mice with P. chabaudi is clearly T cell dependent, and likely to require the germinal center (GC) 1,45. IgM is produced throughout the infection and IgM deficient mice show significantly increased early parasitemia 112,117. In support of an early antibody production, there is early activation of AID, an enzyme critical for both isotype switching and affinity maturation 118, though the cell types represented and effects of this are not yet clear. Further investigation is needed into potential early isotype switching or affinity maturation, as GC are not required for isotype switching 119,120. Intriguingly, unswitched IgM memory T cells have been recently shown to be important in reinfection, despite polyclonal antigen recognition 50,117,121. However, while ICOS is clearly required for early CD138+ B cells, unswitched antibodies are often T cell-independent, and these IgM+ memory B cells (Bmem) have been shown to require Bcl6 intrinsically, while Bcl6 and CXCR5 expression are not required in T cells for generation of IgM+ Bmem 122. The mechanisms and roles for antibody in this early response are being investigated in multiple laboratories.
IL-21, made by Teff, promotes B cell responses, including isotype switching. IL-21 is a critical actor in both early and late immune responses. This is evident from the parasitemia curve of IL-21 and IL-21R deficient mice infected with P. chabaudi 123. The peak of parasitemia is significantly higher in IL-21 KO compared to wildtype controls, despite equivalent amounts of IFN-γ. High levels of IL-21 from T cells produced in this infection may also have other functions on T cells including, intriguingly, an ability to cooperate with transforming growth factor beta (TGF-β) to inhibit Treg expansion 124,125. This is particularly relevant given that an excess of Tregs in P. chabaudi was shown to lead to death of the mice with greatly reduced IFN-γ and IL-10 126. IL-21 has also been reported to inhibit IL-2 production by Teff 127,128, which could also affect the Treg response. However, anti-IL-2 did not affect parasitemia 129. The potential roles of Tregs in human parasitemia are under intense study; however, more work needs to be done on TGF-β and IL-21. Next, we will discuss what is known about regulation of T cell phenotype, T cells in the antibody response, and return to the role of the early cytokines in pathology and in subsequent sections.
3.2. Regulation of the Th phenotype in Plasmodium infection
A significant amount is known about Th differentiation in vitro and in acute infection. However, using these definitions we have established that IFN-γ+ T cells in Plasmodium infections do not match any of the classical Th subsets (Figure 2). This has been reported for chronic LCMV as well, with the state of CD4 T cells in that infection being called dysregulated 130. In vitro and in acute Plasmodium infections, fully differentiated Th1 cells express high levels of T-bet transcription factor, which determines long-term commitment of a T cell to production of IFN-γ 131. CXCR3 is the primary Th1-specific chemokine receptor, and is induced by IFN-γ 132. Bcl6 and cMaf are the primary determining transcription factors in Tfh differentiation, though this is still an area of active research 133,134. CXCR5 is the defining chemokine receptor of Tfh cells because it allows them to traffic to the B cell area. cMaf has recently been shown to regulate IL-10 production, and also contributes to IL-21 production 135. In P. chabaudi infection, all of these markers and transcription factors are expressed, but not at high levels as in terminally differentiated Th1 cells 92. Most of the effector T cells activated by Plasmodium express many markers of both Th1 (specifically, IFN-γ, T-bet, CXCR3, and IL-12Rβ2) and Tfh (specifically, CXCR5, IL-21, Bcl6, ICOS, and BTLA) studied to date 45,92. The first demonstration of this was in humans. Teff in P. falciparum infected people express the chemokine receptors of both Th1 and Tfh, CXCR5+CXCR3+, as well as T-bet 102. In mice, as described above, the early response has classically been described as IFN-γ+ Th1. Many IFN-γ+ T cells generated in response to Plasmodium turned out to be the now well accepted T-bet+ IFN-γ+IL-10+ Th1, also known as Tr1 cells 18,136, in both mouse and human Plasmodium infection, and in other infections that are not immediately controlled by the innate immune system 18,130,137,138. As in some other infections, the IL-10 produced by these cells is under the control of IL-27, cMaf, and Blimp-1 74,135,139. More recently, the IFN-γ+IL-10+ T cells were also shown to express IL-21 123. This IL-21 and the IL-10 also appear to be regulated by cMaf 135, which also represses IL-2, corresponding with the interpretation that autocrine or paracrine IL-2 regulates the Th1/Tfh phenotype gradient 140. More specifically, in the first 7-10 days of infection in mouse malaria (P. chabaudi and P. yoelii) almost all of the IFN-γ producers also make IL-21, and about half of these make IL-10, complicating our understanding of Th subset differentiation. While IFN-γ+ T cells are commonly assumed to be Th1 cells and promote phagocytosis and parasite killing, both IL-21 and IL-10 promote B cell responses 123,141, even though they have been shown to be able to be produced by either classical Th1 or Tfh cells 136,142,143. The localization and full functionality of these cells in protection from malaria is not yet known.
Figure 2.
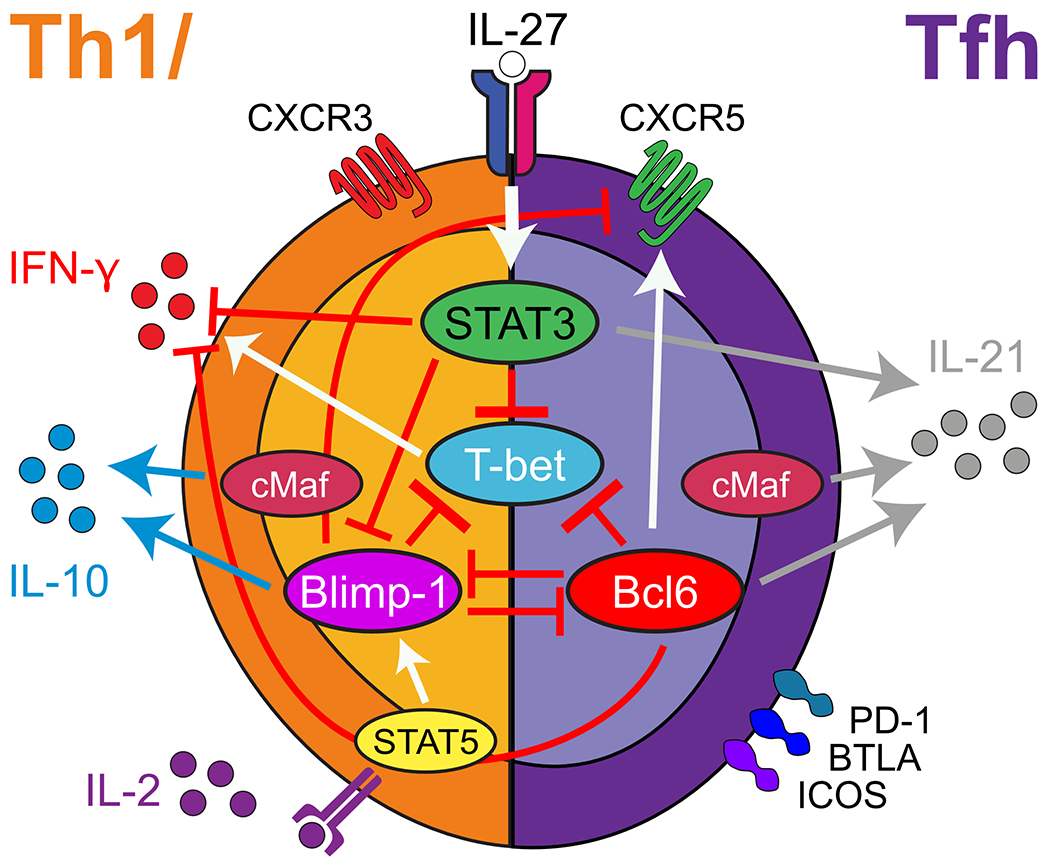
Several transcription factors regulate T-bet in Plasmodium-induced hybrid Th1/Tfh cells. Each marker and cytokine is regulated by a unique set of overlapping signals. Based on data from 36
While Teff in P. chabaudi infection express T-bet, they also express cMaf, Bcl6, and Blimp-1. We have shown that the T-bet/Bcl6 protein ratio in IFN-γ+ Teff correlates positively with IFN-γ transcription 92, and deficiency in T-bet significantly reduces, but does not completely abolish IFN-γ. Deficiency in Bcl6 also does not dramatically increase IFN-γ. In addition, Bcl6 is not the only transcription factor regulating T-bet. Blimp-1 also inhibits T-bet as does STAT3 36, as illustrated in Figure 2. While mechanisms of generation of these cells is under intense study in Plasmodium infection and other persistent infections, some driving cytokines have been identified. IL-27 and Type I Interferon (IFN-I) are definitely involved in inducing the hybrid T cells 66,74, while IL-12, cMaf and Blimp-1 have also been implicated in induction of IL-10 in IFN-γ+ T cells 144,145.
There is an appreciable literature about cytokines and transcription factors involved in regulating the spectrum from Th1 to Tfh 140. However, the combination of cytokines in each infection and timepoint is likely to generate a large variety of possible conformations. We found that deficiency of the Tfh defining transcription factor Bcl6 affects CXCR5 expression much more than the Tfh effector molecule, IL-21. The CXCR5int T cell phenotype in response to P. chabaudi was additionally shown to be determined by ICOS and/or CD40L ligation in the first three days of infection 91. On the other hand, deficiency of the Th1 master regulator, T-bet, significantly affects the generation of CXCR5+ IFN-γ+IL-21+ T cells. Classical Th1 culture also generates transient IFN-γ+IL-21+ Th1 cells in vitro 142. This phenotype includes transient expression of Bcl6, which can bind and inhibit T-bet. 36 The early Th1-like response seen in these studies, which is highly related to mixed but transient phenotypes seen in classical Th1 culture, led us to propose the nomenclature “Th1/Tfh hybrid” for cells expressing markers of both Th1 and Tfh cells 92, rather than the previously used “Th1-like Tfh” 102. T-bet regulates IFN-γ expression, though possibly to varying levels when using different parasites 146,147. Therefore, we propose a Th1 lineage for these hybrid cells despite their expression of intermediate levels of CXCR5 36. In addition to this dominant Th1/Tfh hybrid population, there is a small, steady, and important Bcl6hi CXCR5hi PD-1hi population of GC-T follicular helper (Tfh) cells (in both P. chabaudi and P. yoelii), as shown in Figure 1 36. This population of genuine GC Tfh cannot be generated without B cell interaction in the first three days of P. chabaudi infection 91. T-bet expression in B cells has been demonstrated to have strong effects on GC formation as well 141,148.
Despite the seeming overlap of functional markers by flow cytometry in Th1/Tfh, single cell transcriptional analysis of individual effector T cells suggests that there are likely to be populations discernable at each end of the Th1-Tfh spectrum 149. Indeed, these are evident in one of the antibody combinations we have used to evaluate immune phenotype by flow cytometry (CXCR5, T-bet) 36 and using CXCR6, which is not expressed on CXCR5+ cells.150. We were able to explore the functional importance of shifts along this spectrum. Upon infection of STAT3fl/fl-CD4-Cre mice, which have a specific deletion of STAT3 in cells that have expressed Cd4, the Th1/Tfh hybrid cell type was shifted towards Th1, with more IFN-γ+IL-21− cells being generated, and fewer CXCR5intIFN-γ+IL-21+ cells 36.
One interpretation is that the clear separation of Th1 and Tfh lineages seen in unbiased bioinformatics analysis represents two separable terminally differentiated Teff 149, though it is not clear if Tfh can be a terminally differentiated phenotype 143. It is also possible that the separation of two lineages within the single cell transcriptomics data is due to the high level of analogy between Tfh and Tcm expression patterns 151, which is not yet fully understood. In this reading, the early divergence of Tcm from Teff could explain the separation of Tcm/Tfh from Th1/Teff starting n day 4 post-infection in the single cell transcriptomics data. In the future, these two interpretations might be discernible using downregulation of CD62L or CCR7 on Tfh effector cells, but not on Tcm 152–154, if that is apparent in the transcriptome. The authors considered this possibility and studied memory differentiation in a separate work, which suggests that quiescence is acquired slowly over time in the effector population. While they consider a third phenotype during peak parasitemia unlikely, they allow for the possibility that the Tfh phenotype defined within the effector phase overlaps significantly with a mitochondrially fueled memory precursor that is protected from death in the contraction phase, which is challenging to detect with this method.150 In culture, IFN-γ+IL-21+ Th1 cells can be driven towards a Tfh-like phenotype by varying levels of IL-2 in the culture 78,140. This is interesting, as IL-21 seems to counter-regulate responsiveness to IL-2 135,155. The balance is at least partially regulated by IL-2 driven STAT5 activation, which increases transcription of Blimp-1, that can inhibit both Bcl6 and T-bet transcription. However, STAT3 has not been previously studied in this regard 140,156,157.
Regulation of Th1 and Tfh cytokines via this complex system of regulation throughout a long battle with the parasite clearly has evolutionary and pathological implications. The increase in IFN-γ+IL-21− T cells in infected STAT3fl/fl-CD4-Cre mice did lead to some increase in observed pathology, though no mice died. However, in a second infection of these mice, there was an immediate control of parasitemia, suggesting the importance of studying the cost/benefit ratio to plasticity in Th cells in responses to Plasmodium spp. It will be important to test if parameters of Th1 and Tfh hybrid versus IFN-γ+IL-10−IL-21− or classical GC-Tfh cells correlate with various tests of protection in humans or not.
3.3. T cell dependent antibody in the two stages of Plasmodium infection
The second half of the infection, where parasite is completely cleared, absolutely depends on the generation of antibody 1,2,45. Antibody definitely reduces peak P. yoelii parasitemia, while this effect is harder to measure in P. chabaudi. The earliest role of antibody is easiest to measure in P. yoelii, as the peak of parasitemia happens in the second week when germinal center B cells can be detected 87. The effect of T cell produced IL-21 on the later P. chabaudi parasitemia is much more pronounced than that of the early effect described above. IL-21 deficient mice do not clear infection at all, and over weeks 2-4 post-infection, the parasitemia climbs back to peak levels 123. This model would predict that both peak and subsequent parasitemia would be affected in P. yoelii infection of IL-21 deficient mice, which might even die of hyper-parasitemia, while P. chabaudi infected IL-21 deficient mice do not die. However, it is less likely that parasitemia would be affected days 8-10 in P. yoelii, as seen in P. chabaudi, since T cells are quite effective at this timepoint in this infection. The late action of IL-21 on P. chabaudi persistence was shown to be due to the presence of IL-21R on B cells and not T cells 123. Therefore, the role of IL-21 on the persistent responsiveness of CD8 T cells shown in chronic LCMV does not seem to play a role in CD4 T cell responsiveness here 123. Protective MSP-1 p19-specific IgG antibody is not detectable until the third week of infection in P. chabaudi infection 85,112. IgG responses to the other fragments of MSP-1 have similar kinetics 2. Therefore, it is striking that generation of species-specific isotype-switched IgG2 antibodies in both infections is around day 11p.i. 158. IgG2 antibody has been shown to be able to inhibit parasite growth better than IgM 52,111,159.
The interactions between B and T cells leading to clearance of parasitemia in the later phase are gradually becoming clearer. In the first two weeks of a P. chabaudi infection, parasitemia in mice with Bcl6-deficient T cells is equivalent to wildtype 45,92. However, Bcl6 in T cells is essential in controlling full clearance of the infection in the second two weeks 85. As there are no GC present in mice with either T cells or B cells that are missing Bcl6, and no GC Tfh, this data suggest that GC are required for full clearance of parasite. It is important to note that most pathology occurs during the first two weeks, suggesting that in the first infection, it may not be antibody that allows survival. This is a critical point in determining what germinal centers means for patients with Plasmodium infection. Persistent parasitemia, particularly in immune people, does not result in clinical malaria. However, germinal centers must be important for generating high affinity neutralizing antibodies, and possibly for control of variation in parasite strains.
All subsets of cytokine-producing CD4 T cells are known to be capable of promoting B cell antibody production in vitro 160. However, a highly-specialized subset of activated T cells is now known to specifically inhabit the GC and promote the affinity and diversification of the antibody response 161. GC Tfh are defined by their location, and in flow cytometry usually as PD-1hiCXCR5hi, with both markers having critical functions for these cells. The critical cytokines for GC function are IL-10, IL-4, and IL-21162–164. All three of these cytokines have overlapping and unique functions in promoting B cell antibody secretion and plasma cell generation. IL-21 and IL-4 are B cell growth factors. While IL-4 also promotes IgG1 specifically, IL-21 promotes isotype switching in general. While Tfh expressing CXCR5 can be generated in cell culture with IL-21 165, IL-21 is not sufficient for the formation of GC Tfh in vivo 166. It is important to note that even though Bcl6 regulates the master regulators of other Th subsets, Tfh and other programs co-exist 160.
The potential of Th1/Tfh hybrid Teff T cells to contribute to antibody production has been shown in both human and mouse (CXCR3+CXCR5+, Ly6c+T-bet+, or NK1.1+ICOS+) Plasmodium infection 102,141,167. However, they do not help as much as fully differentiated GC Tfh 102. Part of the difficulty in determining the function and relevance of CXCR5-expressing Teff in Plasmodium infection lies in the emerging details of their differentiation pathway. For example, some CXCR5intICOS+ T cells are pre-Tfh and are known to require further interaction with B cells (CD40L), and further upregulation of Bcl6 and CXCR5 to enter the GC 168,169. Indeed, the large population of CXCR5int effector T cells generated in P. berghei ANKA infection had the potential to transition into GC-Tfh in IRF4 deficient animals 170, indicating their potential as pre-Tfh. From our perspective, the important question about hybrid Th1/Tfh cells in malaria is if they are desirable for either phase of the infection, or if they primarily contribute to prolonging the infection and/or worsening the pathology. If they are unprotective, then understanding their generation could help to avoid their promotion by vaccine strategies.
3.4. Using basic biology of B and T cells to understand weapons available against persistent and repeated Plasmodium infection
Variation of parasite surface proteins is a serious challenge for control of any infection by the immune response. Antigenic variation within Plasmodium strains can lead to low-grade chronic infection 110,171–174, and is likely a major contributor to the slow acquisition of immunity to malaria disease 175. The most striking evidence for this is that patients whose parasites can be recognized by their own serum antibody are less likely to be severely ill 176–178. While GCs generate high affinity antibodies, and are therefore the center for improving neutralizing specificity, the contribution of GCs to the control of persistent and repeated infection in the field is unknown, and should be studied in connection with parasite variation. However, reagents are just being developed to allow detection of variant antigens and variant-specific B cells in parallel 179–181.
On the other hand, the large repertoire of parasite antigens, the highly immunogenic but rarely protective surface antigen specificities contribute to a smokescreen of non-protective epitopes that probably protect the parasite from destruction. Not many protective B or T cell epitopes are well-defined for Plasmodium spp that are relevant to naturally acquired immunity 182–184, however, making the smokescreen hypothesis challenging to test. Here, we will attempt to use existing evidence to build a model of how germinal center biology could help the host recognize conserved protective antigens and compete with antigenic variation. While natural immunity may not be the best guide to possible vaccine targets, understanding the potential of the germinal center reaction will also be necessary to design synthetic vaccine antigens. We will also review the evidence that antibody specificity improves over time. Hyperimmune mice are refractory to further challenge with homologous P. chabaudi parasites after six or seven infections, but remain susceptible to heterologous challenge 110, similar to that seen in protective immunity in humans. Supporting a role for the evolution of antibodies in Plasmodium infection, hyperimmune serum is significantly better at controlling P. yoelii infection than serum collected after one infection 111. Indeed, we observed continuous increases in antibody affinity to parasite lysate over 6 weeks post P. chabaudi infection 85. The dramatic reduction of parasitemia from 30% infected erythrocytes in the first P. chabaudi infection at the peak, to less than 1% in the second infection is also likely due to plasma cells in the bone marrow which will continue to generate antibody over the long-term. GC have also been shown to expand much faster in the second infection than in the first 2,116,151.
GC have been studied for over 40 years, and significant detail about mechanisms of affinity maturation have emerged 185. The GC contains a limited number of helper T cells, and also follicular dendritic cells holding antigen on Fc receptors, that serve to select B cells with ever higher affinity antibody on their surface. Higher affinity B cells take up more antigen via their surface antibody, leading to more MHC-peptide to stimulate T cells, and therefore, more B cell growth factors from T cells and stronger clonal expansion. Since selection in the GC depends on antigens captured by follicular dendritic cells, the GC would seem to be the ideal mechanism for keeping up with evolving parasite. However, protease expression is limited in the GC, and antigen can remain on FcR of follicular dendritic cells for up to three months 186, potentially allowing variants to escape.
It seems plausible that the evolution of the immune response to one P. chabaudi infection, which varies expression of variant antigens weekly, may be similar to repeated exposure, even within one day to infection with multiple strains and/or even species of parasite (in endemic areas). On the other hand, competing parasite strains, and even species, may result in significantly more variation in humans than in the mouse model. The kinetics of B cell division (~12 hours per division 187, parasite division (~24-72 hours per 8-32 new parasites depending on parasite species, and immune success), and the mechanisms and kinetics of variation (e.g. pre-programmed) will determine the success of one against the other. Some modelling has been done based on current knowledge of P. falciparum var switching and antibody responses, supporting the role of variant antigen switching in the slow acquisition of immunity, and the selection of variants by the antibody response 188–190. While we do not know how the kinetics of antibody generation compares to that of the parasite variation, based on the evidence that variation leads to persistence, the kinetics of the germinal center reaction in Plasmodium infection may represent a weakness that the parasite is able to circumvent to persist in the host. GC B cells can be detected as early as day 8 post infection, and GCs are detectable by immunohistochemistry staining with peanut agglutinin on day 10 post infection with P. chabaudi 113,114,191. While IgG takes some time to appear, IgM concentration stays constant throughout, and affinity also increases 113. The kinetics of the GC, as also seen for T cell expansion, are slightly slower in the response to Plasmodium than after immunization with simple antigens. In a response to model antigen, GC are detectable within the first week and higher affinity antibody is evident in the second week 192, suggesting there are factors at play to delay their formation in response to Plasmodium. In agreement with this interpretation, immunohistochemistry of GCs (GL7+) in P. berghei indicates a paucity in the time before death on day 8. Early simultaneous inhibition of IFN-γ and TNF increased the early appearance of the GC in P. berghei infection, and T cell intrinsic T-bet was critical for the delay of splenic GC 170. In Salmonella infection, it is IL-12Rβ2 expression on T cells that inhbits accumulation of GC-Tfh 193. It is possible that this is a broad mechanism delaying T cell dependent antibody, however, there are a few caveats worth considering. Firstly, P. berghei may inhibit GCs more than observed for other infections 194. In addition, in contrast to this data suggesting that the early Th1-like response to Plasmodium inhibits GC formation, recent data indicates that STAT4, IFN-γR, and T-bet are all required in both B and T cells for generation of the protective isotypes IgG2a-c in other systems 87,195,196. Indeed, we found a strong reduction in GC-Tfh cells in STAT4 deficient mice infected with P. chabaudi 36. As GC-Tfh numbers did not vary significantly over the time course of P. chabaudi or P. yoelii infections, it may be that variation in the expression of T-bet during infection, such as the decrease seen from day 7 to 9 post-infection with P. chabaudi, allows the emergence of the GC structure. It is likely that the degree of the cytokine response determines the result of counter-regulation, allowing for a balanced and regulated Th1-biased GC response.
In addition to the evidence that GC size may be regulated early in the response, there is some evidence that GC structure is changed in the response to Plasmodium. It has been reported that the Plasmodium GC is unusually heavy in centroblasts with a loss of definition between the dark and light zones in P. berghei ANKA, and in P. falciparum-infected Saimiri sciureus monkeys 197,198. This lack of definition in the GC structure in the first 7-10 days of infection was confirmed in P. chabaudi, and affinity maturation to the model antigen, chicken gamma globulin, during infection was shown to be reduced 191. This result could indicate that proliferating centroblasts are not being properly selected in the light zone where follicular dendritic cells are located. It is not possible to predict if the parasite switches faster than the GC antibody can mature for effective neutralization. However, the evidence suggests that variation is a successful evasion mechanism in mice and NHP to prolong the infection 199, supporting the interpretation that the parasite surface probably changes too quickly for the B cell response to keep up, or that the parasite population as a whole becomes too diverse. More research is required to understand how the process of affinity maturation is used against variants, heterologous infection, and persistent infection in order to identify a potential for intervention.
3.5. Broadly neutralizing antibodies take several years to develop
While Th1-isotypes are important, neutralization, or invasion-blocking specificities are also proven to be able to reduce parasitemia in Plasmodium infection. Neutralizing antibody can inhibit invasion of erythrocytes by blocking surface binding, or inhibiting proteolytic reactions that expose invasion ligands 200. However, it is clear that of all the antibodies made to MSP-1 by exposed people, only a small fraction is neutralizing. HIV only has two antigens on its surface for the immune system to focus on, and yet, neutralizing antibodies are still rare. Mechanisms of generation of broadly neutralizing antibodies (bnAbs), as opposed to highly strain-specific antibodies, to HIV gp120 have started to become clear. However, the process of evolving bnAbs can take several years in vivo 201, and the role of T cell helper specificity is very poorly understood. Models of B cell activation show multiple critical variables, namely precursor frequency, antigen affinity, and avidity, to inform B cell vaccination strategies to promote bnAb selection in GCs 202. Importantly, this work also suggests that if, perhaps stochastically, a suitable precursor arrives to the GC reaction, it can survive selection and the reaction is complete within a few weeks. Therefore, it is likely that the rarity of these precursor B cells is the rate-limiting step, and that their rarity may also be a result of the evolution of a successful pathogen’s antigen structure and variety. Therefore, it is likely that vaccination with synthetic antigens that bypass the rarity of natural protective epitopes and promote rapid refinement in the Germinal center will be necessary to increase immunity more quickly than natural infection.
Much work is currently going into defining protective antibodies by cloning human BCRs from immune patients. For example, cloning P. falciparum-specific B cells led to the discovery of a host mechanism of resistance where a germline sequence, leukocyte-associated immunoglobulin-like receptor-1 (LAIR) is inserted to Immunoglobulin and adds specificity to variant antigen family epitopes 203. This work is leading to the identification of new and promising epitopes 204,205 and will improve vaccine constructs aimed at increasing their representation in the repertoire 206. However, work on T cell epitopes is very far behind this effort. The most likely way to identify T cell epitopes is to elute peptides from immune patients’ antigen presenting cells 207–211. In order to determine their role in protection, most likely this would require testing T cells specific to that peptide by adoptive transfer in mice. Since T cells tend to use more conserved epitopes, epitope discovery for suitable Tfh vaccine antigens could lead to more neutralizing B cell repertoires. Understanding the role of T cells in promoting the evolution of neutralizing antibody to conserved and variable protective epitopes will allow us to design ever more effective vaccine strategies.
4. THE BALANCE OF INFLAMMATORY CYTOKINES AND REGULATORY CYTOKINES PLAYS A MAJOR ROLE IN ALL MALARIA PATHOLOGY
The protective role of CD4 T cell cytokines in malaria infection has been demonstrated in murine and human Plasmodium infection. Most research on protection and pathology thus far has focused on the role of several highly-expressed cytokines: interferon (IFN-γ), tumor necrosis factor (TNF), and interleukin (IL)-10. Most studies analysing T cell cytokines in human malaria have generally used either serum ELISA cytokine measurements, or ELISA of culture supernatants after stimulation of PBMC with malaria peptide antigens. However, several studies lately have been studying cytokine production by intracellular cytokine staining, and experimental infections 17,18,212. Below we discuss the roles of these cytokines in both control of parasitemia and regulation of pathology.
4.1. CD4 T cell cytokine responses to Plasmodium infection: Distinguishing the roles of IFN-γ and TNF in malaria immunity
The primary role of IFN-γ in cellular immunity is to activate phagocytosis and parasite killing mechanisms within macrophages and other phagocytic cells, which promotes parasite killing. Indeed, IFN-γ enhances phagocytosis of P. chabaudi iRBCs and frees merozoites in vitro by primary mouse macrophages 213. Importantly, this critical parasiticidal mechanism can also be inhibited by IL-10 214. In P. chabaudi infection of mice, the IFN-γ-induced antibody isotypes, IgG2a-c, have also been shown to play an important role in parasite clearance 52. This suggests that antibody-mediated phagocytosis via the Fc receptors, bound best by (mouse) IgG2a-c (or human IgG1, 3) rather than other isotypes, is a critical contributor to parasite control in Plasmodium infection. However, this is challenging to test in humans where only correlation is possible, and specificity is confounding. IgG2a-c, In mice, antibody-coated parasites are phagocytosed better than un-opsonized parasite in vitro and in vivo 159,215, and transfer of IgG2a-c definitively promotes parasite clearance in mice 40. It would be very important to screen various parasite lines for mechanisms used to evade killing by macrophages and neutrophils 215.
Antigen-specific IFN-γ in response to both erythrocyte and liver stage malaria antigens in exposed humans has been associated with protection 216,217. For example, over the course of multiple experimental human challenges with blood-stage P. falciparum, parasitemia after a challenge infection negatively correlated with numbers of circulating IFN-γ-producing CD4+ T cells 218, and IFN-γ+CD62Llo T cells responding to MSP-1 show a similar trend in naturally exposed people 219. Inhibition of major histocompatibility complex class II in these experiments leads to loss of IFN-γ production in vitro, identifying CD4 T cells as the main source of IFN-γ production upon Plasmodium infected red blood stimulation in humans 214,217. The superior resistance inherited by members of the Fulani ethnic group to Plasmodium infection in Mali has also been positively correlated with serum IFN-γ 220. In addition, there are vaccine strategies where IFN-γ producing T cells correlate best with protection. In infection-and-drug-cure immunization of people and mice, the IFN-γ-secreting CD4 T cell response correlates better with malaria protection than serum concentration of antibody 218,221. The response to the recombinant circumsporozoite protein-viral particle-based malaria vaccine RTS,S that correlates best is the combination of anti-circumsporozoite protein antibodies and IFN-γ production 222,223.
Mouse studies primarily support the importance of IFN-γ in reducing parasitemia, in that deficiency in IFN-γ and IL-12, a strong inducer of IFN-γ-producing T cells, controls the peak of P. chabaudi parasitemia, representing the first phase of parasite control 52. Early production of IFN-γ by NK and γ/δ T cells can also play a critical role in promoting very early control of P. yoelii parasitemia 69,224. The early response by early sensors of infection, STING, cGAS, IL-18, and IFN-I, also critically regulate the adaptive immune response 65,104,225–228. However, as phagocytosis and oxidative radicals do not uniformly eliminate parasites in vivo or in vitro, the precise mechanism of parasite killing is less clear 229. Data suggests, for example, that although nitric oxide, a primary macrophage lysosome product, can kill parasites in macrophages, it is not a major effector mechanism in all models. Therefore, more work needs to be done to understand the mechanisms at work in order to determine if interventions could be developed.
An important role of TNF in the immune response to malaria has also been demonstrated in many studies, though it is even more challenging to define. Serum TNF can correlate with the synchronized rupture of infected red blood cells, and also with the severity of malaria disease in some studies on P. falciparum 230,231. TNF has several measurable effects during Plasmodium infection, for example, on parasite killing by neutrophils 232. However, the roles of TNF in vivo, either for parasite control or pathology in malaria, are not yet defined in a fully mechanistic manner 233. In malaria endemic areas, higher frequencies of P. falciparum–specific CD4 T cells co-producing IFN-γ and TNF were observed in immune adults 17. However, comparing the immune response in the first week of infection in mice using two sets of more and less lethal parasites revealed that differences in serum IFN-γ correlate better than TNF with parasite control 199. While TNF may drive a fraction of total IFN-γ production in P. chabaudi infection 234, but not in P. vinkei 235, IFN-γ seems to be more beneficial in parasite control. Based on evidence from both mice and humans, the balance between TNF (but not IFN-γ) and IL-10 regulates some modes of pathology associated with malaria disease, as discussed further below 236.
Mouse data suggest a key role of TNF family members in malaria associated pathology such as cerebral malaria and anemia, rather than in immune-mediated parasite control, however TNF family members clearly can induce parasiticidal mechanisms. TNF does not appear to be primarily made by T cells, at least early in mouse Plasmodium infections 69; however, TNF and T cell-produced IFN-γ promote each other’s production. We do not know which cell types in addition to T cells make TNF during the later stages, when excess production of a TNF family member can cause pathology 234. One study showed a lethal early increase in parasitemia in TNF deficient mice using P. chabaudi adami 237. This interpretation is supported by a study showing that recombinant human TNF reduced parasitemia from P. berghei K173 enough to diminish death from cerebral complications 238. In addition, the results of many studies of TNF itself are in question due to the use of antibodies that cross-react with lymphotoxin alpha (LTα), and TNF deficient mice that also express lower levels of LTα 234,239. Critically, LTα kills parasites in neutrophils ten times better than TNF, and also causes hypoglycemia 232. TNF family members have a critical role in driving the microstructure of the spleen, including the marginal zone, and separation of B and T cell areas, further clouding these results. Finally, TNF family members LTα2β1 and lymphotoxin-related inducible ligand that competes for glycoprotein D binding to herpesvirus entry mediator on T cells (LIGHT), also known as TNF superfamily (TNFSF)14/CD258, also play roles in Plasmodium infection of mice 240. Studies of P. berghei ANKA infection-induced experimental cerebral malaria demonstrate that lymphotoxin beta receptor (LTα2β1), rather than the TNFRI, contributes most to pathology in the brain 241. Infected LTβR deficient animals show similar parasitemia, but no death from P. berghei ANKA infection. In addition, they show strong reductions in ICAM expression and parasite binding in the cerebral vascular endothelium. However, the effects of the absence of both of these receptors on splenic microarchitecture makes it very challenging to interpret these results 242. LIGHT is made by activated T cells, constitutively expressed on DCs, and can co-stimulate or inhibit immune responses depending on the collective activity of TNF family receptors 139. Deficiency of LIGHT leads to increased parasite killing in P. berghei ANKA infection, suggesting that LIGHT plays an inhibitory role of T cells early in infection 243. This effect is so strong that it inhibits development of experimental CM. Therefore, the role of TNF family members in lethal malaria is critical, and also very much open for further study, particularly for development of therapeutics.
Tregs are also sometimes associated with inhibition of the response 60,244,245. The role of Treg in immunity to mouse or human malaria is unclear, as conflicting results have been observed 244–247. The strongest evidence so far is that upon experimental P. falciparum infection, increased levels of the Treg determining transcription factor, FoxP3, and one Treg mechanism, TGF-β, were increased upon infection and their levels correlated with higher rates of parasite growth and lower antigen-specific immune responses 247. However, subsequently conflicting findings make it unclear if Tregs continue to correlate with parasite load over subsequent infections in P. falciparum or P. vivax endemic areas 245. Interestingly, Treg activity during acute infection in children in Gambia was found to be inversely correlated with malaria-specific memory responses, suggesting a mechanism for generating quiescent memory T cells despite a prolonged inflammatory response 245. There are also recently reported CXCR3+T-bet+FoxP3+ Treg cells found in P. chabaudi 126. In mouse models, even though there is a relatively stable number of Tregs throughout infection, increasing Tregs using a FoxP3 transgenic mouse, or IL-2 immune complex, does inhibit control of the peak of parasitemia 248,249. Overall data support the conclusion that Tregs are highly regulated in Plasmodium infection and have the potential to inhibit parasite control, but are not otherwise central regulators of malaria pathology. Tregs may be more important in infants, as discussed below. In summary, the IFN-γ and TNF family member responses are the cytokines most implicated in increasing phagocytic uptake of parasites, and mechanisms of macrophage killing of these parasites. However, as we describe below, these same inflammatory cytokines are involved in pathology and in regulation of the humoral immune response.
4.2. Role of IL-10 and TGF-β and Tregs in regulation of malaria disease
Much of the evidence of the role of TNF in malarial pathology comes from studies that also demonstrate how IL-10 regulates this damaging potential. Without regulation, an excessive pro-inflammatory response controlling the parasitemia peak or parasite growth can cause tissue damage. Therefore, a balance of cytokines with the potential to regulate the effects of hyper-inflammation is important in Plasmodium infection to control the outcomes of the infection. IL-10 was first described in Plasmodium infected people in 1994 250. The earliest study describing the impact of the balance of anti- and pro-inflammatory response and malaria complication showed an association between a significantly higher TNF:IL-10 ratio and severe anemia in children in Ghana 251. Further study in western Kenya confirmed this observation, showing a higher TNF:IL-10 ratio in children with severe malarial anemia compared to children with uncomplicated malaria 252. Following this, May et al. observed that children with severe malarial anemia in Gabon had higher TNF:IL-10 ratios compared to non-anemic children 253. In fact, a higher IL-10:TNF ratio in the plasma was shown to be predictive of severe anemia in this study. Those studies confirm that the balance between anti- and pro-inflammatory responses is critical for protection from complications and severe disease in malaria. Elevated levels of TNF have also been documented in cerebral malaria 254,255. While anti-TNF antibodies, which have revolutionized treatment for some inflammatory conditions, did not ameliorate cerebral malaria 230,231,255–257, it will be important to try next generation, more specific TNF family reagents, some of which can enter the brain 258.
TGF-β is the other major regulatory cytokine known to play a role in Plasmodium immunity and pathology . Severe P. falciparum infection in children was associated with elevated levels of TGF-β in plasma whereas IL-2 or IL-12 were lower in uncomplicated malaria 259. Some mechanisms of IL-10 and TGF-β in regulation of the pro-inflammatory cytokine response in Plasmodium infection have been investigated in mouse studies. IL-10 knock-out mice infected with P. chabaudi revealed worse pathology (hypothermia, hyperglycemia, weight loss, and anemia) and 60% mortality compared t wildtype 234,260. Some signs of cerebral malaria were detected downstream of hyper-inflammation including cerebral edema, hemorrhages, behavioral changes, and vascular congestion including leukocytes 261,262. Neutralization of TGF-β in IL-10 KO mice leads to uncontrolled pathology and mortality increased to 100% of the mice 234. Although many cell types can secrete IL-10 (Breg, T cells subsets, NK cells and γδ T cells), it has been shown that the IL-10 that protects animals from P. chabaudi-induced mortality does come from T cells, as IL-10fl/flCD4Cre mice do not die 74. IL-10 induced by P. chabaudi and P. yoelii is downstream of IL-27 73,74. The potential effect of IL-10 driven by Plasmodium infection on clinical illness has been highlighted by the recent finding that the fraction of P. falciparum-specific IL-10-positive T cells (IFN-γ−TNF−) correlates well with decreased odds of symptoms in children from an area of high exposure 18. It will be interesting to determine if these serve a more regulatory or Tfh function (e.g. IL-21 or CXCR5+), as IL-10 also promotes antibody secreting B cells. In human Plasmodium infection, it is not yet known if the majority of the specific IL-10 is made by CD4 T cells.
4.3. Autoregulation of T cell responses affects development of malaria immunity
In addition to the roles of IL-10 in regulating pathology, IL-10 and TGF-β can also regulate the innate response by antigen presenting cells and phagocytes. It is well known that IL-10 inhibits IL-12 production and antigen presentation, thereby reducing the proliferation and Th1 differentiation of T cells at the time of priming 263. In the field, detectable plasma IL-10 and T cells that can make IL-10 in the intracellular cytokine assay were correlated with recent P. falciparum positive smears, or high parasite burden during a recent infection 18,264–266, suggesting that a possible reduction in immunity to parasite growth correlated with IL-10 production. In mouse studies, IL-10+IFN-γ+ T cells also directly correlate with the fraction of infected erythrocytes, suggesting a potential cofounding factor in that the same T cells make both paraciticial and anti-inflammatory cytokines 36. Indeed, a high IL-10:TNF ratio in the plasma was predictive of hyperparasitemia (>250,000 parasites/µL) in exposed Gabonese children 253.
IL-10 and Tregs have also been found to correlate with permissive parasite growth in neonates. Lower proportions of Teff and higher IL-10 response in cord blood were associated with an increased risk of P. falciparum infection during the first year of life 267,268. In utero exposure to Plasmodium also increased of Treg cells in cord blood 268–270. Though much more needs to be done to understand the mechanisms involved in susceptibility to infection of newborns, this data supports the interpretation that IL-10 and Tregs may play an inhibitory role on the immune response most conducive to parasite elimination in some settings.
These opposing functions of IL-10 can also be detected in the mouse Plasmodium literature. For example, IL-10 deficient animals show decreasing parasitemia in some laboratories, but primarily increasing malarial pathology in others 139,145,234,262. Similarly, although infection of Blimp-1 deficient mice results in quite a substantial reduction in IL-10 production, this results in the death of animals from P. chabaudi AS in some laboratories, but not others 36,145. Perhaps a difference in the strain of parasite or mouse strains, or microbiomes evolving over time in different laboratories causes these differential outcomes. In fact, both TNF and IL-10 are differentially induced by different clones of P. chabaudi and different species, supporting the interpretation that IL-10 plays a double-edged role in immunity to Plasmodium infection 139,271.
TGF-β has similar dual functions 272. TGF-β can suppress macrophage activation and inhibit antigen presentation, antagonizing upregulation of IL-12 in synergy with IL-10. In mice, increasing TGF-β led to reduced inflammatory cytokine production and decreased severity of malaria disease, while neutralization of TGF-β also led to more rapid parasite growth 97,234. In human infection, upregulation of TGF-β, FoxP3, and CD25+ CD4+ T cells was also shown to correlate with more rapid parasite growth in P. falciparum sporozoite challenge 247. However, though TGF-β levels correlated with Treg numbers, about half of the TGF-β detectable as intracellular in PBMCs was found in monocytes. Interestingly, although TGF-β needs to be cleaved to be active, several Plasmodium species have a metalloprotease that actives TGF-β 273. The critical variables shifting the importance of IL-10 and TGF-β in inhibiting pathogenic outcomes versus inhibiting control of parasitemia for the two major outcomes of P. falciparum infection are not yet known.
An interesting insight, however, is emerging from immunological studies in a highly endemic area of Uganda. Frequency of exposure is an important variable for determining the amount of IL-10, with more IL-10 being found co-produced by effector T cells from children in a higher transmission setting compared to lower transmission 17. The authors report that IL-10 decayed over time since last exposure, but also correlated inversely with the ability of T cells to proliferate ex vivo to parasite antigens. Jagannathan et al., therefore, tested the potential of anti-IL-10Rα to enhance T cell proliferation and found a small but significant role for IL-10 in inhibition of T cell proliferation to parasite lysate that was confirmed in 8/9 subjects 265. Therefore, heavily exposed children maintain a high enough level of IL-10 to actually inhibit T cell proliferation. This has also been seen in cord blood, as IL-10 inhibited IFN-γ production 274. The mechanism is suspected to be either by a T cell autocrine loop, or via inhibition of MHCII and antigen presentation 275. This work suggests that the T cell specificities are still present despite frequent parasitemia, which could suggest an absence of Plasmodium-specific T cells. Overall the data indicates that the downside of inhibition of the immune response by IL-10 is balanced by the upside of reduction of pathology only in a highly exposed host.
4.4. Role of CD4 cytokines in regulating malaria pathologies in newborns and children
While acknowledging that T cell regulation can have a cost in parasite growth, an excessive inflammatory T cell response to P. falciparum can have a devastating effect in newborns or toddlers, and therefore does require regulation. CD4 T cells clearly play a critical role in both the protective immune response to malaria in the first years of life, and the immunoregulation of the dangerous side-effects. Several studies have shown the important role of CD4 T cells in the regulation and protective immune response to malaria in the first years of life with increasing Treg or Teff frequencies 268,270,276,277. Inflammatory mediators such as IL-1β, IL-6, and IL-8, which have been implicated in fever, are made in response to P. falciparum-infected erythrocytes, even by uninfected children’s immune cells. However, during fever-inducing infection, these children’s PBMCs, produced lower levels of pro-inflammatory cytokines and higher levels of anti-inflammatory cytokines (IL-10, TGF-β) 277. This regulatory effect has been attributed to specific CD4+ Teff that co-produce IL-10, IFN-γ, and TNF. Therefore, T cells regulate the exposure-dependent P. falciparum–specific reduction in pathologic fever response 277. Additional mechanisms are likely to be at play in regulation of the fever response including changes in antigen presenting cell responsiveness known as TLR tolerance 278,279. Similar tolerance mechanisms may be at play in anemia, which is transient, even in the presence of prolonged infection 234,280. As reviewed in detail elsewhere some kind of tolerance also regulates the newborn immune response after exposure of the fetus to malaria during pregnancy 281. Essentially, the tolerance to fever induced by repeated Plasmodium infections malaria may lead to the side-effect of increased susceptibility to infections in general.
Some mechanistic insights into disease tolerance suggest roles for TLR tolerance and IL-10. P. falciparum infection during pregnancy is associated with increased frequencies of Tregs and alterations in the response to TLR ligands in newborns exposed to P. falciparum during first months of life 267,268. We also saw evidence of higher IL-10 upon stimulation of whole blood with TLR stimulation in newborns born to infected mothers. Therefore, elevated IL-10 activity in exposed newborns may inhibit T cell responsiveness via inadequately activated antigen presenting cells. Supporting this idea, increased proportions of Tregs, have been associated with high concentrations of IL-10 in cord blood in children exposed in utero to Plasmodium infection 269,270,282,283 .
4.5. Role of CD4 T cells in tissue pathology of malaria infection
Cerebral malaria is the most common and most lethal of the outcomes of Plasmodium infection, and still kills more than half a million people worldwide every year. Severe malarial anemia is also very common, as are malaria-associated poor outcomes in pregnancy and birth. The role of cytokines is appreciated as a cause of placental malaria, and there is growing appreciation for the role of inflammatory cytokines in human cerebral malaria. As CD4 T cells regulate cytokine production by innate and adaptive cell types thus controlling both parasite killing and inflammatory pathology, they are at the center of these lethal pathologies.
There is an ongoing debate about the causes of cerebral malaria, which can be resolved by allowing that it is a multi-factorial syndrome. The most striking histopathological observation ever made in autopsy studies from cerebral malaria patients is the heavy parasitization of the blood vessels of the brain. Historically, the prevalence of parasites found adherent to vascular endothelium and within vessels, and the relative paucity of leukocytes found in the brain upon autopsy, led to the common interpretation that sequestration, not inflammation (interpreted as accumulation of leukocytes rather than cytokine production), drives cerebral malaria 284. However, the concept of inflammation includes systemic and local cytokine and chemokine levels, and not just infiltration of a target organ by leukocytes. In addition, there is a growing appreciation for the effect of leukocytes within the vasculature, including T cells on the integrity of the endothelium and tissue function 285–288. The role of T cell IL-10 in P. chabaudi infection is the perfect example. IL-10 produced by T cells is required to protect the host from dying of a syndrome that includes severe cerebral symptoms, and yet, T cells do not leave the vasculature in the brain 74,262. T cell cytokines also have the potential to cause endothelial activation, an important pathological feature of cerebral malaria, including upregulation of the adhesion molecules used by the parasite to adhere for sequestration 240. T cells can also recognize and kill endothelial cells with captured Plasmodium antigens 289–295. Therefore, there is strong significance to the study of inflammation in mouse models that recapitulate some of the features of cerebral malaria, such as P. berghei ANKA infection of C57/BL6 mice. In this infection, a deficiency in either CD4 or CD8 T cells, as well as several T cell cytokines, effector molecules, chemokines, and several adhesion molecules can eliminate the behavioural symptoms associated with cerebral dysfunction 13. CD8 T cells were recently identified in the cerebral vasculature of people who died from cerebral malaria 296. As the immune response to P. berghei ANKA, and its relation to cerebral malaria has been extensively reviewed elsewhere 295,297, we will not dwell on it here, except where mechanisms seem to overlap with anemia and placental malaria.
Severe malarial anemia is also at least partially driven by and regulated by T cells. Anemia is not only the result of the death of parasite-infected erythrocytes. While this contributes to the reduction of functional erythrocytes, additional mechanisms are being discovered. In addition to inappropriate killing of uninfected erythrocytes, dyserythropoesis is evident in mouse and human. The serum cytokine levels in patients with severe malarial anemia have a measurably different ratio of IL-10 to IL-12 than serum from malaria patients with less severe disease, suggesting a role for T cell regulation, as shown for fever 298,299. TNF inhibits erythroid expansion in vitro, as does addition of infected red blood cells or infected cell lysates 300,301. In this review, we have chosen to use placental malaria as an example of what is known about how regulation of inflammatory cytokines by CD4 T cells contribute to the pathological consequences of this blood-borne infection.
4.6. Role of CD4 T cells in placental malaria immune cell infiltration and pathology
Plasmodium-infected erythrocytes adhere to endothelial cells and placental trophoblasts using host cell receptors contributing to severe complications, including cerebral or placental malaria 302. Among the parasite surface proteins that contribute to sequestration, PfEMP1 (P. falciparum-Erythrocyte Membrane Protein-1) locus encodes about 60 members of the VAR gene family. The VAR2CSA-type has unique affinity to placental-expressed chondroitin sulfate A (CSA) 302,303 leading to binding of infected erythrocytes in the placenta 304–306. Essentially, binding to VAR2CSA selects parasite variants that can expand in immune adults by infecting the new tissue formed only during pregnancy. In addition to maternal malaria, there are adverse effects of systemic inflammation induced by infection on outcomes of pregnancy, including pre-eclampsia 307. High levels of infected red blood cells at the placental:fetal interface leads to a pro-inflammatory response in the placenta to kill the parasite, which also leads to pathological damage to the placenta. This localized pathology limits the function of the intervillious interface, which is to nourish the fetus and provide oxygen and therefore can induce intrauterine fetal death, maternal morbidity, premature birth, and low birth-weight infants, all of which is also seen in mouse models 308–311. Additional effects on newborns as they grow up are just now becoming clear.
Placental pathology showed syncytiotrophoblast disruption in Gambian women 312. Chronic intervillositis, or cellular infiltration of the open circulation in the placental intervilious space, has been reported, that ultimately affects maternal-fetal exchange 313. Several studies have shown the expansion of circulating CD4+ Teff during malaria infection during pregnancy 266,314. Furthermore, the presence of immune T cells and monocyte/macrophages has been observed in Plasmodium-infected placentas. Increased frequencies of CD4+ T cells secreting-IFN-γ and TNF were demonstrated in P. falciparum infected placenta compared to uninfected 315. This study also found IL-12+ monocyte/macrophages within placental tissue by flow cytometry. Elevated cytokine-secreting T cells and pro-inflammatory cytokine levels in placenta indicate that T cells actively participate to inflammatory responses to Plasmodium infection in the intervillous space. Several studies have demonstrated the impact of placental cytokine levels due to malaria infection with poor pregnancy outcomes. Specifically, increased concentrations of TNF, IFN-γ, IL-2, and IL-10 have been measured 316,317. IFN-γ and IL-2 indicates the presence of CD4 T cells and suggests local proliferation, which can also regulate the other cell types identified in the tissue 318. Investigators showed that higher TNF and IFN-γ in infected placentas from Kenyan women with P. falciparum were associated with both low birth weight and maternal anemia 319.
Mouse and human placentas share the hemotrichorial type and some, but not all features; therefore, we include relevant lessons learned from mouse models here 320. Placentas of P. berghei-infected mice and rats show necrosis as well as hyperplasia of trophoblasts, peri-vascular edema, and sinusoid constriction 311,321–323. Fibrin-type fibrinoid deposition was also seen associated with placental necrosis 312. Pathology is also evident in the placenta of P. chabaudi-infected dams including placental disruption and fibrin thrombi 324. Fibrin accumulation is also seen in mouse and human cerebral malaria and anti-coagulants have shown promise as adjuvant therapy in both pathologies in mice 324,325. While sequestration contributes to congestion in vessels in the brain, lungs, kidney, and placenta, there are other important factors such as coagulation, leukocyte binding and activation, and endothelial and immune cell activation contributing to dysfunction of parenchymal tissue. It is also important to note that while there is definitely vascular congestion, there is little to no evidence that brain or placental pathology is downstream of hypoxia or necrosis caused by vessel blockage. Therefore, it is critical to continue searching for all of the contributing mechanisms of malaria pathology.
In P. chabaudi infected pregnant mice, high systemic production of the proinflammatory cytokines IFN-γ and IL-1β was seen while sustained TNF was measured in the placenta itself more than systemically 326. Systemic TNF has also been shown to be able to induce the loss of the fetus, with infected dams more sensitive to its effects 327. Monocyte/macrophages have been identified in the placentas of P. falciparum, P. chabaudi, and P. berghei 328,329. Regulatory IL-10 and inflammatory chemokines (IP-10, MIP-1, MCP and I-309), and cytokines (IFN-γ and TNF) 118,319,330,331, are produced in response to infected erythrocytes by placental mononuclear cells and lymphocytes from patients infected during pregnancy. Systemic neutralization of TNF (and LT) preserved placental architecture, which was severely affected in infected pregnant animals.
The data so far suggest that as in experimental cerebral malaria, T cells both make inflammatory cytokines in placental circulation that promote macrophage activation, and that must be regulated by T cell cytokines. In many immunopathologies, T cell cytokines amplify and regulate inflammatory monocyte function, which do not perform prolonged inflammatory functions in tissues without this positive and negative regulation. Both cytokine producing T cells and monocytes have been increased in infected placentas compared to uninfected placenta 315, and are likely to be the leukocytes seen in damaged areas of the placenta 332. Damaged areas can also be marked by parasite hemozoin indicating prior parasite binding in these same locations. Potentially as a result of damage to the maternal trophoblast barrier to direct blood exchange, parasite antigens are thought to gain access to fetal blood, leading to P. falciparum-specific T and B cell responses by fetal umbilical cord mononuclear cells 282.
Overall these studies suggest a cellular inflammatory etiology to the damage in the placenta that may be done by hyper-activated inflammatory monocytes that leads to poor maternal fetal exchange that is both exacerbated and regulated by T cells. While it is not obvious that this same cellular mechanism can occur in the brain without significant cellular infiltration, these interactions may occur in the vasculature. Another possibility is that the brain is exquisitely sensitive to systemic cytokines, and that local amplification by tissue sentinel cells such as microglia drives neurological dysfunction. Better understanding of the interactions in the brain vasculature and intervillous space could also apply in the open circulation of the bone marrow where cytokines can affect erythropoiesis. Cytoadhesion of the parasite in various organs is also common to tissue pathologies, suggesting another potential contributor to induction of local inflammation. A new understanding of the specific pathologic mechanisms downstream of the contributions of TNF family members and coagulation to various pathways of pathology could lead to new clinical trials.
5. CD4 T cell memory to Plasmodium spp.
As CD4 T cells are so critical both in regulating effector functions important to parasite control and elimination, as well as effector functions that contribute to pathology, we must endeavour to understand both the quantity and quality of durable T cells that remain after an initial infection. Detecting Plasmodium-specific T cells in humans has been challenging, as described above. However in the last 20 years, development of mice with defined T cell receptors has allowed study of Plasmodium-specific T cells as they develop into effector and memory T cells 2,20,22. Both in humans and mice, the majority of CD4 memory phenotype T cells specific to MSP-1 have an effector memory phenotype 219,333. However, the CD62L or CCR7lo phenotype generally used includes both short-lived Teff and longer-lived Tem. We have recently taken advantage of the complete but transient downregulation of the IL-7Rα (CD127) on effector T cells to distinguish them from the effector memory phenotype T cells generated by P. chabaudi infection. The mixture of cells present two months after infection includes a mixture of effector, effector memory, and central memory T cells (in a ratio of about 10:80:10, 333). Persistent infection, which maintains protection in humans and mice, also maintains Ifng+ T cells 333, which are a mixture of effector and effector memory T cells 334. While the mixture of T cells divide in response to re-infection, they do not accumulate in number, or “boost,” and their production of cytokines may be their most significant response 335,336. When we tested all of the various T cell phenotypes found in the memory phase for protection, we found that the most differentiated, late effector memory phenotype T cells (Tem Late) contributed more to protection of immunocompromised mice than memory T cells with a central memory phenotype 337. In addition, the Tem Late subset maintains a proportion of IFN-γ+ T cells while they are alive 334. We have recently shown that depletion of IFN-γ prior to re-infection inhibits the improved immunity to reinfection imparted by persistent infection 334. This is in agreement with the finding that long-term immunity is boosted by recombinant IFN-γ 338. While IFN-γ is likely to boost phagocytosis and maintain a state of heightened basal innate immunity, the mechanisms at play in persistent or recurrent Plasmodium infection have not yet been defined. Decay of immunity in the absence of exposure has been shown to correlate better with decay of T cells rather than antibody 221, and we recently showed that effector T cells actually protect better than any subset of memory T cells 337. Intriguingly, this suggests that long-lived memory T cells generated to natural infection are not effective. That does not mean that there is not a potentially protective type of memory T cell that could be generated by vaccination. Further studies of the function 335 and metabolism 77 of these cells may provide some insight into differences with Tmem that protect in other infections. For example, there is a significantly different mitochondrial response to inhibition of fatty acid synthesis in persistently Plasmodium exposed T cells compared to Tmem generated in a shortened infection 77. This could suggest that central memory T cells generated by acute stimulii 339, which can be promoted with drugs that stimulate fatty acid oxidation, rather than fatty acid synthesis alone, would be a better match for the parasite.
As suggested above, the surface phenotype can be one indication of protectiveness, however balanced cytokine production and effector function, including help for B cells, must be a critical component. Our work and that of others suggests that improving the production of IFN-γ in the memory phase, or continuous generation of this powerful cytokine may be as critical as maintaining protective levels of neutralizing antibodies in maintaining protection. In addition, stimulation of regulatory cytokines that promote less pathogenic responses to infection are critical. IL-10 increases over the course of one infection in P. chabaudi 74, and is made earlier in a second infection than a first 335,336. While regulation of IL-10 transcription involves Blimp-1, and cMaf, it is not well understood, and mouse T cells in vitro do not show maintenance of IL-10 over time, like a fully differentiated subset. Similarly, most IL-10 producers in P. yoelii were short-lived 72. Nevertheless, patients last exposed to P. vivax up to six years in the past still had T cells that could make IL-10 (in a six-day assay), but not IFN-γ (24-hour assay). While children make more IL-10 in highly exposed settings, adults do not, supporting the apparent self-limiting production of this cytokine 265. This is particularly important now that IL-10 has been correlated not only with immunity to severe disease, but also to tolerance to fever downstream of the inhibition of multiple inflammatory responses from innate cells 277. In this context, it is important to understand which cytokine responses are the best for reinfection, particularly with heterologous parasite, as in the field.
Both P. falciparum and P. vivax can be quite persistent, with P. vivax persisting in the liver and P. falciparum being able to evade immunity for up to two years, even in the absence of re-exposure 5,6. As in other parasite infections, and other persistent pathogens, persistent infection can be the best prevention of recurrent pathology. For example, the Leishmanization strategy with active parasites still employed in Iran to avoid Leishmania 340. In P. falciparum, IFN-γ+ MSP-1-specific T cells with a CD62Llo Teff/Tem phenotype accumulate over time with exposure 219. While Tem with a CD27− phenotype are somewhat efficacious in mice, the efficacy of central versus effector memory T cells in humans against Plasmodium spp. is unknown 337. Recent studies in highly exposed people in Uganda suggest that cytokines associated with the central memory T cell phenotype (TNF, IL-2) correlate with better T cell proliferation and lower parasitemia 341. Re-expansion is often used as a ruler to measure efficacy of memory T cell responses; however, only central memory T cells appear to have this capacity. Much more needs to be understood about effector memory T cells before we can conclude whether they are useful for protection. In our transcriptome analysis comparing Tcm to Tem, we have found much more similarity between all memory T cell subsets than difference. For example, despite the expression of several “activation markers,” Tem do not show a significantly different metabolic transcriptome, or a broadly different balance of pro- and anti-survival or cell cycle genes than Teff (our unpublished observations). This phenotypic information can be used to reverse engineer vaccine strategies that will promote long-lived Plasmodium-specific T cell responses for long-term protection. Therefore, the take home message is likely to be that maintenance of effector T cells rather than memory T cells is a dominant mechanism protecting most adults from clinical malaria. This suggests that new mechanisms of chronic vaccination may be successful at prolonged protection, whereas all current methods of protection by vaccination are short-lived.
As we have seen above, information gleaned from the surface phenotype needs to be combined with information about protective functionality, including cytokine expression for antibody help, phagocyte activation, and also regulatory capacity. The last is likely the most critical aspect for understanding human immunity to disease. This is highlighted by the clear absence of sterilizing immunity in many people, especially if they leave an endemic area 175. Cytokine studies are often performed separately from surface phenotype analysis due to technical limitations. However, we can understand cytokine production in immune people by the relationship with the age of the persistently exposed person. As this field is developing, variation in the frequency of exposure and parasite species still confound integration of the data collected on this point so far.
6. CONCLUDING REMARKS AND FUTURE DIRECTIONS
In summary, the two primary functions of T cells in protection from Plasmodium infection itself are help for phagocytosis and antibody. While these two facets of the response are largely separated in time, they are carried out by the same T cells interfering with the exquisite immune architechture that separates these functions and thereby drives affinity maturation. As we have already seen, increased understanding of the molecular mechanisms of Th phenotype regulation will contribute to better therapeutics for many immune-mediated and chronic diseases. The symptoms caused by the inflammatory response are self-regulated by T cell cytokines as well. The overlapping functions within effector T cells are likely to lead to vulnerabilities that the parasite is able to use to evade immunity using its large genome, and capacity for antigenic variation. While regulatory cytokines are necessary to ensure the survival of the host, they also inhibit the parasiticidal immune response enough to allow persistent infection. Although much is understood about this response, there are still many outstanding questions, and we highlight some below. A major limitation to studying the Plasmodium-specific CD4 T cell response in humans is the lack of known protective T cell epitopes or MHCII tetramers. Without these reagents, we will not be able to compare mechanistic observations from mouse infection with Plasmodium-specific human T cell responses, or understand the role of the polyclonal T cell response in control of parasitemia or pathology in humans. However, the search to define proteins and epitopes that can drive protective responses by vaccination is well under way. To prevent disease caused by blood-stage infection, it is likely to be important to add blood-stage epitopes to the current liver-stage targeted vaccines, unless their effectiveness can be increased to 100%.
Another critical area of study is to determine the major determinants of protection provided by CD4 T cells. While effector T cells can protect in the presence of continuous or repeated infection, central memory T cells cannot be effectively generated under these conditions. Further work on new methods to generate memory and effector T cells with new vaccine platforms, particularly to drive particularly protective effector function combinations will be essential. It will be important to determine if strain variation is the primary reason that we continue to be susceptible to malaria disease long after the first infection, unlike in chicken pox. We now have the tools to detect PfEMP-1 variant gene expression and it will be important to understand the correlation of switching, which seems to be programmed, with selection by antibodies to exposed parts of the molecule. T cell epitopes, which can be derived from unexposed portions of proteins, and thus may be less variable, could provide a reliable vaccine component. While it is known that chronic infection is possible in part due to expression of variant antigens by the parasite, it is harder to determine the role of the immune response in the strain variation present in each population center. However, tools are being developed to answer these questions, though they will require close collaboration between immunologists and those studying genetic diversity of the parasite.
Another urgent need is to understand the role of TNF family members in pathology caused mostly, but not solely, by P. falciparum. As the balance of TNF and IL-10 has been clearly linked to each of the most severe pathologies, we should search for vulnerabilities in this system to utilize in therapeutics. Further understanding of the cell types involved in production and the various actions of TNF on the diverse tissue types targeted by immunopathology in malaria are required to identify druggable targets among the broad and cross-reactive TNF and TNFR families. In addition to these pressing, clinically relevant questions, there are many ways that understanding of the response to Plasmodium infection can inform our understanding of inflammation, immunopathology, and basic immunology.
Recent work from many new and established laboratories are using Plasmodium spp. as a tool to define basic mechanisms of B and T cell biology, and we have highlighted a few here. A major conundrum will be deciphering mechanisms of affinity maturation that contribute to the clearance of the persistent parasite, and determining the role of this process in repeated and heterologous infection in humans. Some interesting areas are how the persistent infection or the host metabolic response to infection affect the T cell response, or how the parasite or T cell response regulates exhaustion. Now that parasite genetics are tractable, there should be a concerted effort to understand mechanisms of parasite virulence and immune-evasion, for example, screening a library of parasites mutated one gene at a time to discover parasite genes that drive the compromise with the host. This complex and important pathogen provides an amazing opportunity to delve into the weapons of war used by the host and pathogen and better understand their strengths and limitations.
Supplementary Material
Acknowledgements:
We would like to thank past and present members of the Stephens Lab for their effort in this research and valuable discussion, particularly Drs. Samad Ibitokou, Michael Opata, Victor Carpio, and Kyle Wilson. We thank Dr. Linsey Yeager for editorial assistance. The authors are grateful to NIH NIAID for support from R01AI135061 (RS, KG), R01NS106597 (RS).
Footnotes
Conflict of Interest Statement: The authors have no conflicts of interest related to this manuscript
REFERENCES
- 1.Meding SJ, Langhorne J. CD4+ T cells and B cells are necessary for the transfer of protective immunity to Plasmodium chabaudi chabaudi. Eur J Immunol. 1991;21(6):1433–1438. [DOI] [PubMed] [Google Scholar]
- 2.Stephens R, Albano FR, Quin S, et al. Malaria-specific transgenic CD4(+) T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood. 2005;106(5):1676–1684. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson PG, Doherty PC. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J Virol. 1999;73(2):1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirunpetcharat C, Finkelman F, Clark IA, Good MF. Malaria parasite-specific Th1-like T cells simultaneously reduce parasitemia and promote disease. Parasite Immunol. 1999;21(6):319–329. [DOI] [PubMed] [Google Scholar]
- 5.Collins WE, Jeffery GM. A retrospective examination of the patterns of recrudescence in patients infected with Plasmodium falciparum. Am J Trop Med Hyg. 1999;61(1 Suppl):44–48. [DOI] [PubMed] [Google Scholar]
- 6.Hamad AA, El Hassan IM, El Khalifa AA, et al. Chronic Plasmodium falciparum infections in an area of low intensity malaria transmission in the Sudan. Parasitology. 2000;120 ( Pt 5):447–456. [DOI] [PubMed] [Google Scholar]
- 7.Landau I, Killick-Kendrick R. Rodent plasmodia of the Republique Centrafricaine: the sporogony and tissue stages of Plasmodium chabaudi and P. berghei yoelii. Trans R Soc Trop Med Hyg. 1966;60(5):633–649. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt CQ, Kennedy AT, Tham WH. More than just immune evasion: Hijacking complement by Plasmodium falciparum. Mol Immunol. 2015;67(1):71–84. [DOI] [PubMed] [Google Scholar]
- 9.Biryukov S, Angov E, Landmesser ME, Spring MD, Ockenhouse CF, Stoute JA. Complement and Antibody-mediated Enhancement of Red Blood Cell Invasion and Growth of Malaria Parasites. EBioMedicine. 2016;9:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francischetti IM. Does activation of the blood coagulation cascade have a role in malaria pathogenesis? Trends Parasitol. 2008;24(6):258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waisberg M, Molina-Cruz A, Mizurini DM, et al. Plasmodium falciparum infection induces expression of a mosquito salivary protein (Agaphelin) that targets neutrophil function and inhibits thrombosis without impairing hemostasis. PLoS Pathog. 2014;10(9):e1004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevenson MM, Tam MF, Wolf SF, Sher A. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J Immunol. 1995;155(5):2545–2556. [PubMed] [Google Scholar]
- 13.Howland SW, Claser C, Poh CM, Gun SY, Renia L. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015;37(3):221–231. [DOI] [PubMed] [Google Scholar]
- 14.Stephens R, Culleton RL, Lamb TJ. The contribution of Plasmodium chabaudi to our understanding of malaria. Trends Parasitol. 2012;28(2):73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flanagan KL, Lee EA, Gravenor MB, et al. Unique T cell effector functions elicited by Plasmodium falciparum epitopes in malaria-exposed Africans tested by three T cell assays. J Immunol. 2001;167(8):4729–4737. [DOI] [PubMed] [Google Scholar]
- 16.Troye-Blomberg M, Riley EM, Kabilan L, et al. Production by activated human T cells of interleukin 4 but not interferon-gamma is associated with elevated levels of serum antibodies to activating malaria antigens. Proc Natl Acad Sci U S A. 1990;87(14):5484–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyle MJ, Jagannathan P, Bowen K, et al. Effector Phenotype of Plasmodium falciparum-Specific CD4+ T Cells Is Influenced by Both Age and Transmission Intensity in Naturally Exposed Populations. J Infect Dis. 2015;212(3):416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle MJ, Jagannathan P, Bowen K, et al. The Development of Plasmodium falciparum-Specific IL10 CD4 T Cells and Protection from Malaria in Children in an Area of High Malaria Transmission. Front Immunol. 2017;8:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roetynck S, Olotu A, Simam J, et al. Phenotypic and functional profiling of CD4 T cell compartment in distinct populations of healthy adults with different antigenic exposure. PLoS One. 2013;8(1):e55195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sano G, Hafalla JC, Morrot A, Abe R, Lafaille JJ, Zavala F. Swift development of protective effector functions in naive CD8(+) T cells against malaria liver stages. J Exp Med. 2001;194(2):173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau LS, Fernandez-Ruiz D, Mollard V, et al. CD8+ T cells from a novel T cell receptor transgenic mouse induce liver-stage immunity that can be boosted by blood-stage infection in rodent malaria. PLoS Pathog. 2014;10(5):e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez-Ruiz D, Lau LS, Ghazanfari N, et al. Development of a Novel CD4(+) TCR Transgenic Line That Reveals a Dominant Role for CD8(+) Dendritic Cells and CD40 Signaling in the Generation of Helper and CTL Responses to Blood-Stage Malaria. J Immunol. 2017;199(12):4165–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poh CM, Howland SW, Grotenbreg GM, Renia L. Damage to the blood-brain barrier during experimental cerebral malaria results from synergistic effects of CD8+ T cells with different specificities. Infect Immun. 2014;82(11):4854–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heide J, Vaughan KC, Sette A, Jacobs T, Schulze Zur Wiesch J. Comprehensive Review of Human Plasmodium falciparum-Specific CD8+ T Cell Epitopes. Front Immunol. 2019;10:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hensmann M, Li C, Moss C, et al. Disulfide bonds in merozoite surface protein 1 of the malaria parasite impede efficient antigen processing and affect the in vivo antibody response. Eur J Immunol. 2004;34(3):908. [DOI] [PubMed] [Google Scholar]
- 26.Quin SJ, Seixas EM, Cross CA, et al. Low CD4(+) T cell responses to the C-terminal region of the malaria merozoite surface protein-1 may be attributed to processing within distinct MHC class II pathways. Eur J Immunol. 2001;31(1):72–81. [DOI] [PubMed] [Google Scholar]
- 27.Flanagan KL, Wilson KL, Plebanski M. Polymorphism in liver-stage malaria vaccine candidate proteins: immune evasion and implications for vaccine design. Expert Rev Vaccines. 2016;15(3):389–399. [DOI] [PubMed] [Google Scholar]
- 28.Cyster JG. B cell follicles and antigen encounters of the third kind. Nat Immunol. 2010;11(11):989–996. [DOI] [PubMed] [Google Scholar]
- 29.Sanderson NS, Zimmermann M, Eilinger L, et al. Cocapture of cognate and bystander antigens can activate autoreactive B cells. Proc Natl Acad Sci U S A. 2017;114(4):734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23(5):503–514. [DOI] [PubMed] [Google Scholar]
- 31.Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77(2):171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Currier J, Beck HP, Currie B, Good MF. Antigens released at schizont burst stimulate Plasmodium falciparum-specific CD4+ T cells from non-exposed donors: potential for cross-reactive memory T cells to cause disease. Int Immunol. 1995;7(5):821–833. [DOI] [PubMed] [Google Scholar]
- 33.Good MF, Zevering Y. Malaria-specific memory T cells: putative roles of different types of memory responses in immunity and disease. Res Immunol. 1994;145(6):455–460. [DOI] [PubMed] [Google Scholar]
- 34.Montes de Oca M, Kumar R, Rivera FL, et al. Type I Interferons Regulate Immune Responses in Humans with Blood-Stage Plasmodium falciparum Infection. Cell Rep. 2016;17(2):399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zenklusen I, Jongo S, Abdulla S, et al. Immunization of Malaria-Preexposed Volunteers With PfSPZ Vaccine Elicits Long-Lived IgM Invasion-Inhibitory and Complement-Fixing Antibodies. J Infect Dis. 2018;217(10):1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carpio VH, Aussenac F, Wilson KD, Villarino AV, Dent AL, Stephens R. Increased Th1 bias in memory T cells corresponds with protection from reinfection in Plasmodium infection, and is regulated by T cell-intrinsic STAT3. bioRxiv. August 05, 2019. Accessed November 08, 2019 [Pre-Print] DOI: 10.1101/724963 [DOI] [Google Scholar]
- 37.Stephens R, Seddon B, Langhorne J. Homeostatic proliferation and IL-7R alpha expression do not correlate with enhanced T cell proliferation and protection in chronic mouse malaria. PLoS One. 2011;6(10):e26686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanger Institute. Plasmodium falciparum. https://www.sanger.ac.uk/resources/downloads/protozoa/plasmodium-falciparum.html. Accessed November 7, 2019.
- 40.Donati D, Mok B, Chene A, et al. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J Immunol. 2006;177(5):3035–3044. [DOI] [PubMed] [Google Scholar]
- 41.Lucas B, Engels A, Camus D, Haque A. T-cell recognition of a cross-reactive antigen(s) in erythrocyte stages of Plasmodium falciparum and Plasmodium yoelii: inhibition of parasitemia by this antigen(s). Infect Immun. 1993;61(11):4863–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ndungu FM, Sanni L, Urban B, et al. CD4 T cells from malaria-nonexposed individuals respond to the CD36-Binding Domain of Plasmodium falciparum erythrocyte membrane protein-1 via an MHC class II-TCR-independent pathway. J Immunol. 2006;176(9):5504–5512. [DOI] [PubMed] [Google Scholar]
- 43.Simone O, Bejarano MT, Pierce SK, et al. TLRs innate immunereceptors and Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) CIDR1alpha-driven human polyclonal B-cell activation. Acta Trop. 2011;119(2–3):144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falanga PB, D’Imperio Lima MR, Coutinho A, Pereira da Silva L. Isotypic pattern of the polyclonal B cell response during primary infection by Plasmodium chabaudi and in immune-protected mice. Eur J Immunol. 1987;17(5):599–603. [DOI] [PubMed] [Google Scholar]
- 45.Perez-Mazliah D, Nguyen MP, Hosking C, et al. Follicular Helper T Cells are Essential for the Elimination of Plasmodium Infection. EBioMedicine. 2017;24:216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt NW, Butler NS, Harty JT. Plasmodium-host interactions directly influence the threshold of memory CD8 T cells required for protective immunity. J Immunol. 2011;186(10):5873–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Boer RJ, Homann D, Perelson AS. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol. 2003;171(8):3928–3935. [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Llordella M, Esensten JH, Bailey-Bucktrout SL, et al. CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J Exp Med. 2013;210(8):1603–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh YM, Park HB, Shin JH, et al. Ndrg1 is a T-cell clonal anergy factor negatively regulated by CD28 costimulation and interleukin-2. Nat Commun. 2015;6:8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borges da Silva H, Machado de Salles E, Lima-Mauro EF, Sardinha LR, Alvarez JM, D’Imperio Lima MR. CD28 deficiency leads to accumulation of germinal-center independent IgM+ experienced B cells and to production of protective IgM during experimental malaria. PLoS One. 2018;13(8):e0202522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elias RM, Sardinha LR, Bastos KR, et al. Role of CD28 in polyclonal and specific T and B cell responses required for protection against blood stage malaria. J Immunol. 2005;174(2):790–799. [DOI] [PubMed] [Google Scholar]
- 52.Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J Immunol. 2002;168(3):1348–1355. [DOI] [PubMed] [Google Scholar]
- 53.Zander RA, Vijay R, Pack AD, et al. Th1-like Plasmodium-Specific Memory CD4(+) T Cells Support Humoral Immunity. Cell Rep. 2017;21(7):1839–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zander RA, Obeng-Adjei N, Guthmiller JJ, et al. PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe. 2015;17(5):628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The Costimulatory Molecule ICOS Regulates Host Th1 and Follicular Th Cell Differentiation in Response to Plasmodium chabaudi chabaudi AS Infection. J Immunol. 2016;196(2):778–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Illingworth J, Butler NS, Roetynck S, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. 2013;190(3):1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reiley WW, Shafiani S, Wittmer ST, et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A. 2010;107(45):19408–19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butler NS, Moebius J, Pewe LL, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2011;13(2):188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karunarathne DS, Horne-Debets JM, Huang JX, et al. Programmed Death-1 Ligand 2-Mediated Regulation of the PD-L1 to PD-1 Axis Is Essential for Establishing CD4(+) T Cell Immunity. Immunity. 2016;45(2):333–345. [DOI] [PubMed] [Google Scholar]
- 60.Kurup SP, Obeng-Adjei N, Anthony SM, et al. Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nat Med. 2017;23(10):1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lepenies B, Pfeffer K, Hurchla MA, et al. Ligation of B and T lymphocyte attenuator prevents the genesis of experimental cerebral malaria. J Immunol. 2007;179(6):4093–4100. [DOI] [PubMed] [Google Scholar]
- 62.Adler G, Steeg C, Pfeffer K, et al. B and T lymphocyte attenuator restricts the protective immune response against experimental malaria. J Immunol. 2011;187(10):5310–5319. [DOI] [PubMed] [Google Scholar]
- 63.Artavanis-Tsakonas K, Riley EM. Innate immune response to malaria: rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. J Immunol. 2002;169(6):2956–2963. [DOI] [PubMed] [Google Scholar]
- 64.Haque A, Best SE, Ammerdorffer A, et al. Type I interferons suppress CD4(+) T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol. 2011;41(9):2688–2698. [DOI] [PubMed] [Google Scholar]
- 65.Yu X, Cai B, Wang M, et al. Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity. 2016;45(5):1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zander RA, Guthmiller JJ, Graham AC, et al. Type I Interferons Induce T Regulatory 1 Responses and Restrict Humoral Immunity during Experimental Malaria. PLoS Pathog. 2016;12(10):e1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choudhury HR, Sheikh NA, Bancroft GJ, Katz DR, De Souza JB. Early nonspecific immune responses and immunity to blood-stage nonlethal Plasmodium yoelii malaria. Infect Immun. 2000;68(11):6127–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Couper KN, Blount DG, Hafalla JC, van Rooijen N, de Souza JB, Riley EM. Macrophage-mediated but gamma interferon-independent innate immune responses control the primary wave of Plasmodium yoelii parasitemia. Infect Immun. 2007;75(12):5806–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Souza JB, Williamson KH, Otani T, Playfair JH. Early gamma interferon responses in lethal and nonlethal murine blood-stage malaria. Infect Immun. 1997;65(5):1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho M, Webster HK, Green B, Looareesuwan S, Kongchareon S, White NJ. Defective production of and response to IL-2 in acute human falciparum malaria. J Immunol. 1988;141(8):2755–2759. [PubMed] [Google Scholar]
- 71.Kimura D, Miyakoda M, Honma K, et al. Production of IFN-gamma by CD4(+) T cells in response to malaria antigens is IL-2 dependent. Int Immunol. 2010;22(12):941–952. [DOI] [PubMed] [Google Scholar]
- 72.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. [DOI] [PubMed] [Google Scholar]
- 73.Findlay EG, Greig R, Stumhofer JS, et al. Essential role for IL-27 receptor signaling in prevention of Th1-mediated immunopathology during malaria infection. J Immunol. 2010;185(4):2482–2492. [DOI] [PubMed] [Google Scholar]
- 74.Freitas do Rosario AP, Lamb T, Spence P, et al. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J Immunol. 2012;188(3):1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baeza Garcia A, Siu E, Sun T, et al. Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nat Commun. 2018;9(1):2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Opata MM, Carpio VH, Ibitokou SA, Dillon BE, Obiero JM, Stephens R. Early effector cells survive the contraction phase in malaria infection and generate both central and effector memory T cells. J Immunol. 2015;194(11):5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ibitokou SA, Dillon BE, Sinha M, et al. Early Inhibition of Fatty Acid Synthesis Reduces Generation of Memory Precursor Effector T Cells in Chronic Infection. J Immunol. 2018;200(2):643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DiToro D, Winstead CJ, Pham D, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science. 2018;361(6407). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKinstry KK, Alam F, Flores-Malavet V, et al. Memory CD4 T cell-derived IL-2 synergizes with viral infection to exacerbate lung inflammation. PLoS Pathog. 2019;15(8):e1007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat Immunol. 2011;12(6):467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saparov A, Wagner FH, Zheng R, et al. Interleukin-2 expression by a subpopulation of primary T cells is linked to enhanced memory/effector function. Immunity. 1999;11(3):271–280. [DOI] [PubMed] [Google Scholar]
- 82.Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol. 2005;17(3):275–281. [DOI] [PubMed] [Google Scholar]
- 83.Nolz JC, Rai D, Badovinac VP, Harty JT. Division-linked generation of death-intermediates regulates the numerical stability of memory CD8 T cells. Proc Natl Acad Sci U S A. 2012;109(16):6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy JR, Lefford MJ. Host defenses in murine malaria: evaluation of the mechanisms of immunity to Plasmodium yoelii infection. Infect Immun. 1979;23(2):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Achtman AH, Stephens R, Cadman ET, Harrison V, Langhorne J. Malaria-specific antibody responses and parasite persistence after infection of mice with Plasmodium chabaudi chabaudi. Parasite Immunol. 2007;29(9):435–444. [DOI] [PubMed] [Google Scholar]
- 86.Freeman RR, Parish CR. Plasmodium yoelii: antibody and the maintenance of immunity in BALB/c mice. Exp Parasitol. 1981;52(1):18–24. [DOI] [PubMed] [Google Scholar]
- 87.Weinbaum FI, Evans CB, Tigelaar RE. Immunity to Plasmodium Berghei yoelii in mice. I. The course of infection in T cell and B cell deficient mice. J Immunol. 1976;117(5 Pt.2):1999–2005. [PubMed] [Google Scholar]
- 88.Langhorne J, Cross C, Seixas E, Li C, von der Weid T. A role for B cells in the development of T cell helper function in a malaria infection in mice. Proc Natl Acad Sci U S A. 1998;95(4):1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sander B, Skansen-Saphir U, Damm O, Hakansson L, Andersson J, Andersson U. Sequential production of Th1 and Th2 cytokines in response to live bacillus Calmette-Guerin. Immunology. 1995;86(4):512–518. [PMC free article] [PubMed] [Google Scholar]
- 90.Sponaas AM, Belyaev N, Falck-Hansen M, Potocnik A, Langhorne J. Transient deficiency of dendritic cells results in lack of a merozoite surface protein 1-specific CD4 T cell response during peak Plasmodium chabaudi blood-stage infection. Infect Immun. 2012;80(12):4248–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arroyo EN, Pepper M. B cells are sufficient to prime the dominant CD4(+) Tfh response to Plasmodium infection. J Exp Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carpio VH, Opata MM, Montanez ME, Banerjee PP, Dent AL, Stephens R. IFN-gamma and IL-21 Double Producing T Cells Are Bcl6-Independent and Survive into the Memory Phase in Plasmodium chabaudi Infection. PLoS One. 2015;10(12):e0144654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sebina I, James KR, Soon MS, et al. IFNAR1-Signalling Obstructs ICOS-mediated Humoral Immunity during Non-lethal Blood-Stage Plasmodium Infection. PLoS Pathog. 2016;12(11):e1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suss G, Eichmann K, Kury E, Linke A, Langhorne J. Roles of CD4- and CD8-bearing T lymphocytes in the immune response to the erythrocytic stages of Plasmodium chabaudi. Infect Immun. 1988;56(12):3081–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kobayashi F, Ishida H, Matsui T, Tsuji M. Effects of in vivo administration of anti-IL-10 or anti-IFN-gamma monoclonal antibody on the host defense mechanism against Plasmodium yoelii yoelii infection. J Vet Med Sci. 2000;62(6):583–587. [DOI] [PubMed] [Google Scholar]
- 96.Day JF, Edman JD. The importance of disease induced changes in mammalian body temperature to mosquito blood feeding. Comp Biochem Physiol A Comp Physiol. 1984;77(3):447–452. [DOI] [PubMed] [Google Scholar]
- 97.Omer FM, Riley EM. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J Exp Med. 1998;188(1):39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roberts DW, Weidanz WP. Splenomegaly, enhanced phagocytosis, and anemia are thymus-dependent responses to malaria. Infect Immun. 1978;20(3):728–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Costa PA, Leoratti FM, Figueiredo MM, et al. Induction of Inhibitory Receptors on T Cells During Plasmodium vivax Malaria Impairs Cytokine Production. J Infect Dis. 2015;212(12):1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vukovic P, Chen K, Qin Liu X, et al. Single-chain antibodies produced by phage display against the C-terminal 19 kDa region of merozoite surface protein-1 of Plasmodium yoelii reduce parasite growth following challenge. Vaccine. 2002;20(21–22):2826–2835. [DOI] [PubMed] [Google Scholar]
- 101.Jayawardena AN, Targett GA, Carter RL, Leuchars E, Davies AJ. The immunological response of CBA mice to P. yoelii. I. General characteristics, the effects of T-cell deprivation and reconstitution with thymus grafts. Immunology. 1977;32(6):849–859. [PMC free article] [PubMed] [Google Scholar]
- 102.Obeng-Adjei N, Portugal S, Tran TM, et al. Circulating Th1-Cell-type Tfh Cells that Exhibit Impaired B Cell Help Are Preferentially Activated during Acute Malaria in Children. Cell Rep. 2015;13(2):425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matar CG, Anthony NR, O’Flaherty BM, et al. Gammaherpesvirus Co-infection with Malaria Suppresses Anti-parasitic Humoral Immunity. PLoS Pathog. 2015;11(5):e1004858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hahn WO, Butler NS, Lindner SE, et al. cGAS-mediated control of blood-stage malaria promotes Plasmodium-specific germinal center responses. JCI Insight. 2018;3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cohen S, Mc GI, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. 1961;192:733–737. [DOI] [PubMed] [Google Scholar]
- 106.McGregor IA CSP, S. Cohen S. . Treatment of East African P. falciparum malaria with West African human γ-globulin. Transactions of The Royal Society of Tropical Medicine and Hygiene. 1963; 57(3):170–175. [Google Scholar]
- 107.Foquet L, Hermsen CC, van Gemert GJ, et al. Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection. J Clin Invest. 2014;124(1):140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Daly TM, Long CA. Humoral response to a carboxyl-terminal region of the merozoite surface protein-1 plays a predominant role in controlling blood-stage infection in rodent malaria. J Immunol. 1995;155(1):236–243. [PubMed] [Google Scholar]
- 109.Jayawardena AN, Targett GA, Leuchars E, Davies AJ. The immunological response of CBA mice to P. yoelii. II. The passive transfer of immunity with serum and cells. Immunology. 1978;34(1):157–165. [PMC free article] [PubMed] [Google Scholar]
- 110.Jarra W, Hills LA, March JC, Brown KN. Protective immunity to malaria. Studies with cloned lines of Plasmodium chabaudi chabaudi and P. berghei in CBA/Ca mice. II. The effectiveness and inter- or intra-species specificity of the passive transfer of immunity with serum. Parasite Immunol. 1986;8(3):239–254. [DOI] [PubMed] [Google Scholar]
- 111.Mota MM, Brown KN, Do Rosario VE, Holder AA, Jarra W. Antibody recognition of rodent malaria parasite antigens exposed at the infected erythrocyte surface: specificity of immunity generated in hyperimmune mice. Infect Immun. 2001;69(4):2535–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Quin SJ, Langhorne J. Different regions of the malaria merozoite surface protein 1 of Plasmodium chabaudi elicit distinct T-cell and antibody isotype responses. Infect Immun. 2001;69(4):2245–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krishnamurty AT, Thouvenel CD, Portugal S, et al. Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity. 2016;45(2):402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Achtman AH, Khan M, MacLennan IC, Langhorne J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J Immunol. 2003;171(1):317–324. [DOI] [PubMed] [Google Scholar]
- 115.Nduati EW, Ng DH, Ndungu FM, Gardner P, Urban BC, Langhorne J. Distinct kinetics of memory B-cell and plasma-cell responses in peripheral blood following a blood-stage Plasmodium chabaudi infection in mice. PLoS One. 2010;5(11):e15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ndungu FM, Cadman ET, Coulcher J, et al. Functional memory B cells and long-lived plasma cells are generated after a single Plasmodium chabaudi infection in mice. PLoS Pathog. 2009;5(12):e1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Couper KN, Phillips RS, Brombacher F, Alexander J. Parasite-specific IgM plays a significant role in the protective immune response to asexual erythrocytic stage Plasmodium chabaudi AS infection. Parasite Immunol. 2005;27(5):171–180. [DOI] [PubMed] [Google Scholar]
- 118.Abrams ET, Brown H, Chensue SW, et al. Host response to malaria during pregnancy: placental monocyte recruitment is associated with elevated beta chemokine expression. J Immunol. 2003;170(5):2759–2764. [DOI] [PubMed] [Google Scholar]
- 119.Roco JA, Mesin L, Binder SC, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity. 2019;51(2):337–350 e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Toellner KM, Gulbranson-Judge A, Taylor DR, Sze DM, MacLennan IC. Immunoglobulin switch transcript production in vivo related to the site and time of antigen-specific B cell activation. J Exp Med. 1996;183(5):2303–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arama C, Skinner J, Doumtabe D, et al. Genetic Resistance to Malaria Is Associated With Greater Enhancement of Immunoglobulin (Ig)M Than IgG Responses to a Broad Array of Plasmodium falciparum Antigens. Open Forum Infect Dis. 2015;2(3):ofv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pritchard GH, Krishnamurty AT, Netland J, Arroyo EN, Takehara KK, Pepper M. The Development of Optimally Responsive Plasmodium-specific CD73+CD80+ IgM+ Memory B cells Requires Intrinsic BCL6 expression but not CD4+ Tfh cells. BioRxiv. March 01, 2019. Accessed November 08, 2019 [Pre-Print] DOI: 10.1101/564351 [DOI] [Google Scholar]
- 123.Perez-Mazliah D, Ng DH, Freitas do Rosario AP, et al. Disruption of IL-21 signaling affects T cell-B cell interactions and abrogates protective humoral immunity to malaria. PLoS Pathog. 2015;11(3):e1004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. [DOI] [PubMed] [Google Scholar]
- 125.Tian Y, Zajac AJ. IL-21 and T Cell Differentiation: Consider the Context. Trends Immunol. 2016;37(8):557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Berretta F, Piccirillo CA, Stevenson MM. Plasmodium chabaudi AS Infection Induces CD4(+) Th1 Cells and Foxp3(+)T-bet(+) Regulatory T Cells That Express CXCR3 and Migrate to CXCR3 Ligands. Front Immunol. 2019;10:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Attridge K, Wang CJ, Wardzinski L, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood. 2012;119(20):4656–4664. [DOI] [PubMed] [Google Scholar]
- 128.Schmitz I, Schneider C, Frohlich A, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog. 2013;9(5):e1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zago CA, Bortoluci KR, Sardinha LR, et al. Anti-IL-2 treatment impairs the expansion of T(reg) cell population during acute malaria and enhances the Th1 cell response at the chronic disease. PLoS One. 2012;7(1):e29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. 2014;40(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. [DOI] [PubMed] [Google Scholar]
- 132.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187(6):875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu X, Nurieva RI, Dong C. Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol Rev. 2013;252(1):139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mathew R, Mao AP, Chiang AH, et al. A negative feedback loop mediated by the Bcl6-cullin 3 complex limits Tfh cell differentiation. J Exp Med. 2014;211(6):1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gabrysova L, Alvarez-Martinez M, Luisier R, et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4(+) T cells. Nat Immunol. 2018;19(5):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jankovic D, Kullberg MC, Feng CG, et al. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204(2):273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. 2005;79(16):10514–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105(51):20428–20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kumar R, Ng S, Engwerda C. The Role of IL-10 in Malaria: A Double Edged Sword. Front Immunol. 2019;10:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oestreich KJ, Weinmann AS. Transcriptional mechanisms that regulate T helper 1 cell differentiation. Curr Opin Immunol. 2012;24(2):191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guthmiller JJ, Graham AC, Zander RA, Pope RL, Butler NS. Cutting Edge: IL-10 Is Essential for the Generation of Germinal Center B Cell Responses and Anti-Plasmodium Humoral Immunity. J Immunol. 2017;198(2):617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakayamada S, Kanno Y, Takahashi H, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. 2011;35(6):919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wali S, Sahoo A, Puri S, Alekseev A, Nurieva R. Insights into the development and regulation of T follicular helper cells. Cytokine. 2016;87:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O’Garra A. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31(2):209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Montes de Oca M, Kumar R, de Labastida Rivera F, et al. Blimp-1-Dependent IL-10 Production by Tr1 Cells Regulates TNF-Mediated Tissue Pathology. PLoS Pathog. 2016;12(1):e1005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oakley MS, Sahu BR, Lotspeich-Cole L, et al. T-bet modulates the antibody response and immune protection during murine malaria. Eur J Immunol. 2014;44(9):2680–2691. [DOI] [PubMed] [Google Scholar]
- 147.Oakley MS, Sahu BR, Lotspeich-Cole L, et al. The transcription factor T-bet regulates parasitemia and promotes pathogenesis during Plasmodium berghei ANKA murine malaria. J Immunol. 2013;191(9):4699–4708. [DOI] [PubMed] [Google Scholar]
- 148.Ly A, Liao Y, Pietrzak H, et al. Transcription Factor T-bet in B Cells Modulates Germinal Center Polarization and Antibody Affinity Maturation in Response to Malaria. Cell Rep. 2019;29(8):2257–2269 e2256. [DOI] [PubMed] [Google Scholar]
- 149.Lonnberg T, Svensson V, James KR, et al. Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol. 2017;2(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Soon MSF, Lee HJ, Engel JA, et al. Transcriptome dynamics reveals progressive transition from effector to memory in CD4+ T cells. bioRxiv. 2019. [Pre-Print] 10.1101/675967 [DOI] [Google Scholar]
- 151.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35(4):583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Choi YS, Yang JA, Crotty S. Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Curr Opin Immunol. 2013;25(3):366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stone EL, Pepper M, Katayama CD, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity. 2015;42(2):239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weber JP, Fuhrmann F, Feist RK, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med. 2015;212(2):217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jandl C, Liu SM, Canete PF, et al. IL-21 restricts T follicular regulatory T cell proliferation through Bcl-6 mediated inhibition of responsiveness to IL-2. Nat Commun. 2017;8:14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209(2):243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee JY, Skon CN, Lee YJ, et al. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity. 2015;42(2):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Langhorne J, Evans CB, Asofsky R, Taylor DW. Immunoglobulin isotype distribution of malaria-specific antibodies produced during infection with Plasmodium chabaudi adami and Plasmodium yoelii. Cell Immunol. 1984;87(2):452–461. [DOI] [PubMed] [Google Scholar]
- 159.Mota MM, Brown KN, Holder AA, Jarra W. Acute Plasmodium chabaudi chabaudi malaria infection induces antibodies which bind to the surfaces of parasitized erythrocytes and promote their phagocytosis by macrophages in vitro. Infect Immun. 1998;66(9):4080–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Crotty S Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. [DOI] [PubMed] [Google Scholar]
- 161.Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol. 2016;34:335–368. [DOI] [PubMed] [Google Scholar]
- 162.Linterman MA, Beaton L, Yu D, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010;207(2):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17(10):1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yusuf I, Kageyama R, Monticelli L, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol. 2010;185(1):190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nurieva RI, Chung Y, Hwang D, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29(1):138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eto D, Lao C, DiToro D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6(3):e17739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wikenheiser DJ, Brown SL, Lee J, Stumhofer JS. NK1.1 Expression Defines a Population of CD4(+) Effector T Cells Displaying Th1 and Tfh Cell Properties That Support Early Antibody Production During Plasmodium yoelii Infection. Front Immunol. 2018;9:2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Choi YS, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34(6):932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kroenke MA, Eto D, Locci M, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol. 2012;188(8):3734–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ryg-Cornejo V, Ioannidis LJ, Ly A, et al. Severe Malaria Infections Impair Germinal Center Responses by Inhibiting T Follicular Helper Cell Differentiation. Cell Rep. 2016;14(1):68–81. [DOI] [PubMed] [Google Scholar]
- 171.Brown IN, Brown KN, Hills LA. Immunity to malaria: the antibody response to antigenic variation by Plasmodium knowlesi. Immunology. 1968;14(1):127–138. [PMC free article] [PubMed] [Google Scholar]
- 172.Handunnetti SM, Mendis KN, David PH. Antigenic variation of cloned Plasmodium fragile in its natural host Macaca sinica. Sequential appearance of successive variant antigenic types. J Exp Med. 1987;165(5):1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jemmely NY, Niang M, Preiser PR. Small variant surface antigens and Plasmodium evasion of immunity. Future Microbiol. 2010;5(4):663–682. [DOI] [PubMed] [Google Scholar]
- 174.McLean SA, Pearson CD, Phillips RS. Plasmodium chabaudi: antigenic variation during recrudescent parasitaemias in mice. Exp Parasitol. 1982;54(3):296–302. [DOI] [PubMed] [Google Scholar]
- 175.Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. 2008;9(7):725–732. [DOI] [PubMed] [Google Scholar]
- 176.Mackintosh CL, Mwangi T, Kinyanjui SM, et al. Failure to respond to the surface of Plasmodium falciparum infected erythrocytes predicts susceptibility to clinical malaria amongst African children. Int J Parasitol. 2008;38(12):1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cunningham DA, Jarra W, Koernig S, et al. Host immunity modulates transcriptional changes in a multigene family (yir) of rodent malaria. Mol Microbiol. 2005;58(3):636–647. [DOI] [PubMed] [Google Scholar]
- 178.Spence PJ, Jarra W, Levy P, et al. Vector transmission regulates immune control of Plasmodium virulence. Nature. 2013;498(7453):228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gitau EN, Tuju J, Karanja H, et al. CD4+ T cell responses to the Plasmodium falciparum erythrocyte membrane protein 1 in children with mild malaria. J Immunol. 2014;192(4):1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Langhorne J, Duffy PE. Expanding the antimalarial toolkit: Targeting host-parasite interactions. J Exp Med. 2016;213(2):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Spence PJ, Brugat T, Langhorne J. Mosquitoes Reset Malaria Parasites. PLoS Pathog. 2015;11(7):e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dent AE, Nakajima R, Liang L, et al. Plasmodium falciparum Protein Microarray Antibody Profiles Correlate With Protection From Symptomatic Malaria in Kenya. J Infect Dis. 2015;212(9):1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Osier FH, Mackinnon MJ, Crosnier C, et al. New antigens for a multicomponent blood-stage malaria vaccine. Sci Transl Med. 2014;6(247):247ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Richards JS, Arumugam TU, Reiling L, et al. Identification and prioritization of merozoite antigens as targets of protective human immunity to Plasmodium falciparum malaria for vaccine and biomarker development. J Immunol. 2013;191(2):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev. 2010;237(1):72–89. [DOI] [PubMed] [Google Scholar]
- 186.Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. 2014;14(7):495–504. [DOI] [PubMed] [Google Scholar]
- 187.Firl DJ, Degn SE, Padera T, Carroll MC. Capturing change in clonal composition amongst single mouse germinal centers. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nielsen MA, Staalsoe T, Kurtzhals JA, et al. Plasmodium falciparum variant surface antigen expression varies between isolates causing severe and nonsevere malaria and is modified by acquired immunity. J Immunol. 2002;168(7):3444–3450. [DOI] [PubMed] [Google Scholar]
- 189.Bull PC, Pain A, Ndungu FM, et al. Plasmodium falciparum antigenic variation: relationships between in vivo selection, acquired antibody response, and disease severity. J Infect Dis. 2005;192(6):1119–1126. [DOI] [PubMed] [Google Scholar]
- 190.Eckhoff PP falciparum infection durations and infectiousness are shaped by antigenic variation and innate and adaptive host immunity in a mathematical model. PLoS One. 2012;7(9):e44950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cadman ET, Abdallah AY, Voisine C, et al. Alterations of splenic architecture in malaria are induced independently of Toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect Immun. 2008;76(9):3924–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16(11):2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Elsner RA, Shlomchik MJ. IL-12 Blocks Tfh Cell Differentiation during Salmonella Infection, thereby Contributing to Germinal Center Suppression. Cell Rep. 2019;29(9):2796–2809 e2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wedderburn N, Turk JL, Davies DR, Hutt MS. Chronic malarial infection of mice: A comparison of single and multiple infections with Plasmodium berghei following P. yoelii. Trans R Soc Trop Med Hyg. 1978;72(6):610–614. [DOI] [PubMed] [Google Scholar]
- 195.Ma CS, Suryani S, Avery DT, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. 2009;87(8):590–600. [DOI] [PubMed] [Google Scholar]
- 196.Stone SL, Peel JN, Scharer CD, et al. T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-gamma on B Cells. Immunity. 2019;50(5):1172–1187 e1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Alves FA, Pelajo-Machado M, Totino PR, et al. Splenic architecture disruption and parasite-induced splenocyte activation and anergy in Plasmodium falciparum-infected Saimiri sciureus monkeys. Malar J. 2015;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Carvalho LJ, Ferreira-da-Cruz MF, Daniel-Ribeiro CT, Pelajo-Machado M, Lenzi HL. Germinal center architecture disturbance during Plasmodium berghei ANKA infection in CBA mice. Malar J. 2007;6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jarra W, Brown KN. Protective immunity to malaria: studies with cloned lines of rodent malaria in CBA/Ca mice. IV. The specificity of mechanisms resulting in crisis and resolution of the primary acute phase parasitaemia of Plasmodium chabaudi chabaudi and P. yoelii yoelii. Parasite Immunol. 1989;11(1):1–13. [DOI] [PubMed] [Google Scholar]
- 200.Pleass RJ, Holder AA. Opinion: antibody-based therapies for malaria. Nat Rev Microbiol. 2005;3(11):893–899. [DOI] [PubMed] [Google Scholar]
- 201.Liao HX, Lynch R, Zhou T, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496(7446):469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Abbott RK, Lee JH, Menis S, et al. Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity. 2018;48(1):133–146 e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pieper K, Tan J, Piccoli L, et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature. 2017;548(7669):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kisalu NK, Idris AH, Weidle C, et al. A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat Med. 2018;24(4):408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tan J, Sack BK, Oyen D, et al. A public antibody lineage that potently inhibits malaria infection through dual binding to the circumsporozoite protein. Nat Med. 2018;24(4):401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Alanine DGW, Quinkert D, Kumarasingha R, et al. Human Antibodies that Slow Erythrocyte Invasion Potentiate Malaria-Neutralizing Antibodies. Cell. 2019;178(1):216–228 e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Barra C, Alvarez B, Paul S, et al. Footprints of antigen processing boost MHC class II natural ligand predictions. Genome Med. 2018;10(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Clement CC, Becerra A, Yin L, et al. The Dendritic Cell Major Histocompatibility Complex II (MHC II) Peptidome Derives from a Variety of Processing Pathways and Includes Peptides with a Broad Spectrum of HLA-DM Sensitivity. J Biol Chem. 2016;291(11):5576–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Croft NP, Purcell AW, Tscharke DC. Quantifying epitope presentation using mass spectrometry. Mol Immunol. 2015;68(2 Pt A):77–80. [DOI] [PubMed] [Google Scholar]
- 210.Paul S, Karosiene E, Dhanda SK, et al. Determination of a Predictive Cleavage Motif for Eluted Major Histocompatibility Complex Class II Ligands. Front Immunol. 2018;9:1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tang Q, Nie F, Kang J, Ding H, Zhou P, Huang J. NIEluter: Predicting peptides eluted from HLA class I molecules. J Immunol Methods. 2015;422:22–27. [DOI] [PubMed] [Google Scholar]
- 212.Teirlinck AC, McCall MB, Roestenberg M, et al. Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog. 2011;7(12):e1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Amani V, Vigario AM, Belnoue E, et al. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol. 2000;30(6):1646–1655. [DOI] [PubMed] [Google Scholar]
- 214.King T, Lamb T. Interferon-gamma: The Jekyll and Hyde of Malaria. PLoS Pathog. 2015;11(10):e1005118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ferrante A, Kumaratilake L, Rzepczyk CM, Dayer JM. Killing of Plasmodium falciparum by cytokine activated effector cells (neutrophils and macrophages). Immunol Lett. 1990;25(1–3):179–187. [DOI] [PubMed] [Google Scholar]
- 216.Luty AJ, Lell B, Schmidt-Ott R, et al. Interferon-gamma responses are associated with resistance to reinfection with Plasmodium falciparum in young African children. J Infect Dis. 1999;179(4):980–988. [DOI] [PubMed] [Google Scholar]
- 217.Moormann AM, Sumba PO, Chelimo K, et al. Humoral and cellular immunity to Plasmodium falciparum merozoite surface protein 1 and protection from infection with blood-stage parasites. J Infect Dis. 2013;208(1):149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pombo DJ, Lawrence G, Hirunpetcharat C, et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360(9333):610–617. [DOI] [PubMed] [Google Scholar]
- 219.Chelimo K, Embury PB, Sumba PO, et al. Age-related differences in naturally acquired T cell memory to Plasmodium falciparum merozoite surface protein 1. PLoS One. 2011;6(9):e24852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.McCall MB, Hopman J, Daou M, et al. Early interferon-gamma response against Plasmodium falciparum correlates with interethnic differences in susceptibility to parasitemia between sympatric Fulani and Dogon in Mali. J Infect Dis. 2010;201(1):142–152. [DOI] [PubMed] [Google Scholar]
- 221.Freitas do Rosario AP, Muxel SM, Rodriguez-Malaga SM, et al. Gradual decline in malaria-specific memory T cell responses leads to failure to maintain long-term protective immunity to Plasmodium chabaudi AS despite persistence of B cell memory and circulating antibody. J Immunol. 2008;181(12):8344–8355. [DOI] [PubMed] [Google Scholar]
- 222.Dobano C, Sanz H, Sorgho H, et al. Concentration and avidity of antibodies to different circumsporozoite epitopes correlate with RTS,S/AS01E malaria vaccine efficacy. Nat Commun. 2019;10(1):2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sun P, Schwenk R, White K, et al. Protective immunity induced with malaria vaccine, RTS,S, is linked to Plasmodium falciparum circumsporozoite protein-specific CD4+ and CD8+ T cells producing IFN-gamma. J Immunol. 2003;171(12):6961–6967. [DOI] [PubMed] [Google Scholar]
- 224.Ho M, Tongtawe P, Kriangkum J, et al. Polyclonal expansion of peripheral gamma delta T cells in human Plasmodium falciparum malaria. Infect Immun. 1994;62(3):855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wykes MN, Liu XQ, Beattie L, et al. Plasmodium strain determines dendritic cell function essential for survival from malaria. PLoS Pathog. 2007;3(7):e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sebina I, Haque A. Effects of type I interferons in malaria. Immunology. 2018;155(2):176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Spaulding E, Fooksman D, Moore JM, et al. STING-Licensed Macrophages Prime Type I IFN Production by Plasmacytoid Dendritic Cells in the Bone Marrow during Severe Plasmodium yoelii Malaria. PLoS Pathog. 2016;12(10):e1005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cramer JP, Lepenies B, Kamena F, et al. MyD88/IL-18-dependent pathways rather than TLRs control early parasitaemia in non-lethal Plasmodium yoelii infection. Microbes Infect. 2008;10(12–13):1259–1265. [DOI] [PubMed] [Google Scholar]
- 229.Mercer F, Ng SH, Brown TM, Boatman G, Johnson PJ. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol. 2018;16(2):e2003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci U S A. 1992;89(8):3200–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kern P, Hemmer CJ, Van Damme J, Gruss HJ, Dietrich M. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med. 1989;87(2):139–143. [DOI] [PubMed] [Google Scholar]
- 232.Kumaratilake LM, Ferrante A, Rzepczyk CM. Tumor necrosis factor enhances neutrophil-mediated killing of Plasmodium falciparum. Infect Immun. 1990;58(3):788–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Korner H, McMorran B, Schluter D, Fromm P. The role of TNF in parasitic diseases: still more questions than answers. Int J Parasitol. 2010;40(8):879–888. [DOI] [PubMed] [Google Scholar]
- 234.Li C, Sanni LA, Omer F, Riley E, Langhorne J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor alpha and exacerbated by anti-transforming growth factor beta antibodies. Infect Immun. 2003;71(9):4850–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Clark K, Kulk N, Amante F, Haque A, Engwerda C. Lymphotoxin alpha and tumour necrosis factor are not required for control of parasite growth, but differentially regulate cytokine production during Plasmodium chabaudi chabaudi AS infection. Parasite Immunol. 2007;29(3):153–158. [DOI] [PubMed] [Google Scholar]
- 236.Luty AJ, Perkins DJ, Lell B, et al. Low interleukin-12 activity in severe Plasmodium falciparum malaria. Infect Immun. 2000;68(7):3909–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hernandez-Valladares M, Naessens J, Musoke AJ, et al. Pathology of Tnf-deficient mice infected with Plasmodium chabaudi adami 408XZ. Exp Parasitol. 2006;114(4):271–278. [DOI] [PubMed] [Google Scholar]
- 238.Postma NS, Hermsen CC, Crommelin DJ, Zuidema J, Eling WM. Treatment with recombinant human tumour necrosis factor-alpha reduces parasitaemia and prevents Plasmodium berghei K173-induced experimental cerebral malaria in mice. Parasitology. 1999;118 ( Pt 1):7–15. [DOI] [PubMed] [Google Scholar]
- 239.Clark IA, Gray KM, Rockett EJ, et al. Increased lymphotoxin in human malarial serum, and the ability of this cytokine to increase plasma interleukin-6 and cause hypoglycaemia in mice: implications for malarial pathology. Trans R Soc Trop Med Hyg. 1992;86(6):602–607. [DOI] [PubMed] [Google Scholar]
- 240.Engwerda CR, Mynott TL, Sawhney S, De Souza JB, Bickle QD, Kaye PM. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med. 2002;195(10):1371–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Togbe D, de Sousa PL, Fauconnier M, et al. Both functional LTbeta receptor and TNF receptor 2 are required for the development of experimental cerebral malaria. PLoS One. 2008;3(7):e2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Randall LM, Engwerda CR. TNF family members and malaria: old observations, new insights and future directions. Exp Parasitol. 2010;126(3):326–331. [DOI] [PubMed] [Google Scholar]
- 243.Randall LM, Amante FH, Zhou Y, et al. Cutting edge: selective blockade of LIGHT-lymphotoxin beta receptor signaling protects mice from experimental cerebral malaria caused by Plasmodium berghei ANKA. J Immunol. 2008;181(11):7458–7462. [DOI] [PubMed] [Google Scholar]
- 244.Torcia MG, Santarlasci V, Cosmi L, et al. Functional deficit of T regulatory cells in Fulani, an ethnic group with low susceptibility to Plasmodium falciparum malaria. Proc Natl Acad Sci U S A. 2008;105(2):646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Walther M, Jeffries D, Finney OC, et al. Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog. 2009;5(4):e1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Finney OC, Riley EM, Walther M. Regulatory T cells in malaria--friend or foe? Trends Immunol. 2010;31(2):63–70. [DOI] [PubMed] [Google Scholar]
- 247.Walther M, Tongren JE, Andrews L, et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity. 2005;23(3):287–296. [DOI] [PubMed] [Google Scholar]
- 248.Berretta F, St-Pierre J, Piccirillo CA, Stevenson MM. IL-2 contributes to maintaining a balance between CD4+Foxp3+ regulatory T cells and effector CD4+ T cells required for immune control of blood-stage malaria infection. J Immunol. 2011;186(8):4862–4871. [DOI] [PubMed] [Google Scholar]
- 249.Haque A, Best SE, Amante FH, et al. CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo. PLoS Pathog. 2010;6(12):e1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schmitt N, Morita R, Bourdery L, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity. 2009;31(1):158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kurtzhals JA, Adabayeri V, Goka BQ, et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet. 1998;351(9118):1768–1772. [DOI] [PubMed] [Google Scholar]
- 252.Othoro C, Lal AA, Nahlen B, Koech D, Orago AS, Udhayakumar V. A low interleukin-10 tumor necrosis factor-alpha ratio is associated with malaria anemia in children residing in a holoendemic malaria region in western Kenya. J Infect Dis. 1999;179(1):279–282. [DOI] [PubMed] [Google Scholar]
- 253.May J, Lell B, Luty AJ, Meyer CG, Kremsner PG. Plasma interleukin-10:Tumor necrosis factor (TNF)-alpha ratio is associated with TNF promoter variants and predicts malarial complications. J Infect Dis. 2000;182(5):1570–1573. [DOI] [PubMed] [Google Scholar]
- 254.Grau GE, Piguet PF, Vassalli P, Lambert PH. Tumor-necrosis factor and other cytokines in cerebral malaria: experimental and clinical data. Immunol Rev. 1989;112:49–70. [DOI] [PubMed] [Google Scholar]
- 255.van Hensbroek MB, Palmer A, Onyiorah E, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis. 1996;174(5):1091–1097. [DOI] [PubMed] [Google Scholar]
- 256.Kwiatkowski D, Hill AV, Sambou I, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336(8725):1201–1204. [DOI] [PubMed] [Google Scholar]
- 257.Kwiatkowski D, Molyneux ME, Stephens S, et al. Anti-TNF therapy inhibits fever in cerebral malaria. Q J Med. 1993;86(2):91–98. [PubMed] [Google Scholar]
- 258.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lourembam SD, Sawian CE, Baruah S. Dysregulation of cytokines expression in complicated falciparum malaria with increased TGF-beta and IFN-gamma and decreased IL-2 and IL-12. Cytokine. 2013;64(2):503–508. [DOI] [PubMed] [Google Scholar]
- 260.Li C, Corraliza I, Langhorne J. A defect in interleukin-10 leads to enhanced malarial disease in Plasmodium chabaudi chabaudi infection in mice. Infect Immun. 1999;67(9):4435–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sanni LA, Jarra W, Li C, Langhorne J. Cerebral edema and cerebral hemorrhages in interleukin-10-deficient mice infected with Plasmodium chabaudi. Infect Immun. 2004;72(5):3054–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wilson KD, Stutz SJ, Ochoa LF, et al. Behavioural and neurological symptoms accompanied by cellular neuroinflammation in IL-10-deficient mice infected with Plasmodium chabaudi. Malar J. 2016;15(1):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fiorentino DF, Zlotnik A, Vieira P, et al. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146(10):3444–3451. [PubMed] [Google Scholar]
- 264.Ibitokou SA, Bostrom S, Brutus L, et al. Submicroscopic infections with Plasmodium falciparum during pregnancy and their association with circulating cytokine, chemokine, and cellular profiles. Clin Vaccine Immunol. 2014;21(6):859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Jagannathan P, Eccles-James I, Bowen K, et al. IFNgamma/IL-10 co-producing cells dominate the CD4 response to malaria in highly exposed children. PLoS Pathog. 2014;10(1):e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Requena P, Barrios D, Robinson LJ, et al. Proinflammatory responses and higher IL-10 production by T cells correlate with protection against malaria during pregnancy and delivery outcomes. J Immunol. 2015;194(7):3275–3285. [DOI] [PubMed] [Google Scholar]
- 267.Gbedande K, Varani S, Ibitokou S, et al. Malaria modifies neonatal and early-life toll-like receptor cytokine responses. Infect Immun. 2013;81(8):2686–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Nouatin O, Gbedande K, Ibitokou S, et al. Infants’ Peripheral Blood Lymphocyte Composition Reflects Both Maternal and Post-Natal Infection with Plasmodium falciparum. PLoS One. 2015;10(11):e0139606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Flanagan KL, Halliday A, Burl S, et al. The effect of placental malaria infection on cord blood and maternal immunoregulatory responses at birth. Eur J Immunol. 2010;40(4):1062–1072. [DOI] [PubMed] [Google Scholar]
- 270.Prahl M, Jagannathan P, McIntyre TI, et al. Timing of in utero malaria exposure influences fetal CD4 T cell regulatory versus effector differentiation. Malar J. 2016;15(1):497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Long GH, Chan BH, Allen JE, Read AF, Graham AL. Parasite genetic diversity does not influence TNF-mediated effects on the virulence of primary rodent malaria infections. Parasitology. 2006;133(Pt 6):673–684. [DOI] [PubMed] [Google Scholar]
- 272.Hansen DS, Schofield L. Natural regulatory T cells in malaria: host or parasite allies? PLoS Pathog. 2010;6(4):e1000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Omer FM, de Souza JB, Corran PH, Sultan AA, Riley EM. Activation of transforming growth factor beta by malaria parasite-derived metalloproteinases and a thrombospondin-like molecule. J Exp Med. 2003;198(12):1817–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Brustoski K, Moller U, Kramer M, et al. IFN-gamma and IL-10 mediate parasite-specific immune responses of cord blood cells induced by pregnancy-associated Plasmodium falciparum malaria. J Immunol. 2005;174(3):1738–1745. [DOI] [PubMed] [Google Scholar]
- 275.de Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174(4):915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Odorizzi PM, Jagannathan P, McIntyre TI, et al. In utero priming of highly functional effector T cell responses to human malaria. Sci Transl Med. 2018;10(463). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Portugal S, Moebius J, Skinner J, et al. Exposure-dependent control of malaria-induced inflammation in children. PLoS Pathog. 2014;10(4):e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Franklin BS, Parroche P, Ataide MA, et al. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci U S A. 2009;106(14):5789–5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol. 2005;174(10):5921–5925. [DOI] [PubMed] [Google Scholar]
- 280.White NJ. Anaemia and malaria. Malar J. 2018;17(1):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Harrington WE, Kakuru A, Jagannathan P. Malaria in pregnancy shapes the development of foetal and infant immunity. Parasite Immunol. 2019;41(3):e12573. [DOI] [PubMed] [Google Scholar]
- 282.Brustoski K, Moller U, Kramer M, et al. Reduced cord blood immune effector-cell responsiveness mediated by CD4+ cells induced in utero as a consequence of placental Plasmodium falciparum infection. J Infect Dis. 2006;193(1):146–154. [DOI] [PubMed] [Google Scholar]
- 283.Malhotra I, Dent A, Mungai P, et al. Can prenatal malaria exposure produce an immune tolerant phenotype? A prospective birth cohort study in Kenya. PLoS Med. 2009;6(7):e1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Clark IA. Along a TNF-paved road from dead parasites in red cells to cerebral malaria, and beyond. Parasitology. 2009;136(12):1457–1468. [DOI] [PubMed] [Google Scholar]
- 285.Anderson KG, Mayer-Barber K, Sung H, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9(1):209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Sakai S, Kauffman KD, Schenkel JM, et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. 2014;192(7):2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Suidan GL, McDole JR, Chen Y, Pirko I, Johnson AJ. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS One. 2008;3(8):e3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.van de Berg PJ, Yong SL, Remmerswaal EB, van Lier RA, ten Berge IJ. Cytomegalovirus-induced effector T cells cause endothelial cell damage. Clin Vaccine Immunol. 2012;19(5):772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Howland SW, Poh CM, Renia L. Activated Brain Endothelial Cells Cross-Present Malaria Antigen. PLoS Pathog. 2015;11(6):e1004963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Razakandrainibe R, Pelleau S, Grau GE, Jambou R. Antigen presentation by endothelial cells: what role in the pathophysiology of malaria? Trends Parasitol. 2012;28(4):151–160. [DOI] [PubMed] [Google Scholar]
- 291.Carvalho LJ. Murine cerebral malaria: how far from human cerebral malaria? Trends Parasitol. 2010;26(6):271–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hunt NH, Grau GE, Engwerda C, et al. Murine cerebral malaria: the whole story. Trends Parasitol. 2010;26(6):272–274. [DOI] [PubMed] [Google Scholar]
- 293.Riley EM, Couper KN, Helmby H, et al. Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010;26(6):277–278. [DOI] [PubMed] [Google Scholar]
- 294.Stevenson MM, Gros P, Olivier M, Fortin A, Serghides L. Cerebral malaria: human versus mouse studies. Trends Parasitol. 2010;26(6):274–275. [DOI] [PubMed] [Google Scholar]
- 295.White NJ, Turner GD, Medana IM, Dondorp AM, Day NP. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26(1):11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Barrera V, Haley MJ, Strangward P, et al. Comparison of CD8(+) T Cell Accumulation in the Brain During Human and Murine Cerebral Malaria. Front Immunol. 2019;10:1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.de Souza JB, Hafalla JC, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology. 2010;137(5):755–772. [DOI] [PubMed] [Google Scholar]
- 298.Lamikanra AA, Brown D, Potocnik A, Casals-Pascual C, Langhorne J, Roberts DJ. Malarial anemia: of mice and men. Blood. 2007;110(1):18–28. [DOI] [PubMed] [Google Scholar]
- 299.Wroczynska A, Nahorski W, Bakowska A, Pietkiewicz H. Cytokines and clinical manifestations of malaria in adults with severe and uncomplicated disease. Int Marit Health. 2005;56(1–4):103–114. [PubMed] [Google Scholar]
- 300.Casals-Pascual C, Kai O, Cheung JO, et al. Suppression of erythropoiesis in malarial anemia is associated with hemozoin in vitro and in vivo. Blood. 2006;108(8):2569–2577. [DOI] [PubMed] [Google Scholar]
- 301.Felli N, Pedini F, Zeuner A, et al. Multiple members of the TNF superfamily contribute to IFN-gamma-mediated inhibition of erythropoiesis. J Immunol. 2005;175(3):1464–1472. [DOI] [PubMed] [Google Scholar]
- 302.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415(6872):673–679. [DOI] [PubMed] [Google Scholar]
- 303.Jensen AR, Adams Y, Hviid L. Cerebral Plasmodium falciparum malaria: The role of PfEMP1 in its pathogenesis and immunity, and PfEMP1-based vaccines to prevent it. Immunol Rev. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Fried M, Domingo GJ, Gowda CD, Mutabingwa TK, Duffy PE. Plasmodium falciparum: chondroitin sulfate A is the major receptor for adhesion of parasitized erythrocytes in the placenta. Exp Parasitol. 2006;113(1):36–42. [DOI] [PubMed] [Google Scholar]
- 305.Maubert B, Fievet N, Tami G, Boudin C, Deloron P. Cytoadherence of Plasmodium falciparum-infected erythrocytes in the human placenta. Parasite Immunol. 2000;22(4):191–199. [DOI] [PubMed] [Google Scholar]
- 306.Muthusamy A, Achur RN, Valiyaveettil M, et al. Chondroitin sulfate proteoglycan but not hyaluronic acid is the receptor for the adherence of Plasmodium falciparum-infected erythrocytes in human placenta, and infected red blood cell adherence up-regulates the receptor expression. Am J Pathol. 2007;170(6):1989–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Zarate A, Saucedo R, Valencia J, Manuel L, Hernandez M. Early disturbed placental ischemia and hypoxia creates immune alteration and vascular disorder causing preeclampsia. Arch Med Res. 2014;45(7):519–524. [DOI] [PubMed] [Google Scholar]
- 308.Desai M, ter Kuile FO, Nosten F, et al. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis. 2007;7(2):93–104. [DOI] [PubMed] [Google Scholar]
- 309.Feeney ME. The immune response to malaria in utero. Immunol Rev. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kwak-Kim JYH, Gilman-Sachs A, Kim CE. T helper 1 and 2 immune responses in relationship to pregnancy, nonpregnancy, recurrent spontaneous abortions and infertility of repeated implantation failures. Chem Immunol Allergy. 2005;88:64–79. [DOI] [PubMed] [Google Scholar]
- 311.Neres R, Marinho CR, Goncalves LA, Catarino MB, Penha-Goncalves C. Pregnancy outcome and placenta pathology in Plasmodium berghei ANKA infected mice reproduce the pathogenesis of severe malaria in pregnant women. PLoS One. 2008;3(2):e1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Crocker IP, Tanner OM, Myers JE, Bulmer JN, Walraven G, Baker PN. Syncytiotrophoblast degradation and the pathophysiology of the malaria-infected placenta. Placenta. 2004;25(4):273–282. [DOI] [PubMed] [Google Scholar]
- 313.Matteelli A, Caligaris S, Castelli F, Carosi G. The placenta and malaria. Ann Trop Med Parasitol. 1997;91(7):803–810. [DOI] [PubMed] [Google Scholar]
- 314.Ibitokou S, Oesterholt M, Brutus L, et al. Peripheral blood cell signatures of Plasmodium falciparum infection during pregnancy. PLoS One. 2012;7(12):e49621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Diouf I, Fievet N, Doucoure S, et al. IL-12 producing monocytes and IFN-gamma and TNF-alpha producing T-lymphocytes are increased in placentas infected by Plasmodium falciparum. J Reprod Immunol. 2007;74(1–2):152–162. [DOI] [PubMed] [Google Scholar]
- 316.Fievet N, Moussa M, Tami G, et al. Plasmodium falciparum induces a Th1/Th2 disequilibrium, favoring the Th1-type pathway, in the human placenta. J Infect Dis. 2001;183(10):1530–1534. [DOI] [PubMed] [Google Scholar]
- 317.Rogerson SJ, Hviid L, Duffy PE, Leke RF, Taylor DW. Malaria in pregnancy: pathogenesis and immunity. Lancet Infect Dis. 2007;7(2):105–117. [DOI] [PubMed] [Google Scholar]
- 318.Taylor-Robinson AW, Smith EC. Modulation of experimental blood stage malaria through blockade of the B7/CD28 T-cell costimulatory pathway. Immunology. 1999;96(3):498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Fried M, Muga RO, Misore AO, Duffy PE. Malaria elicits type 1 cytokines in the human placenta: IFN-gamma and TNF-alpha associated with pregnancy outcomes. J Immunol. 1998;160(5):2523–2530. [PubMed] [Google Scholar]
- 320.Furukawa S, Kuroda Y, Sugiyama A. A comparison of the histological structure of the placenta in experimental animals. J Toxicol Pathol. 2014;27(1):11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Oduola AM, Holbrook TW, Galbraith RM, Bank H, Spicer SS. Effects of malaria (Plasmodium berghei) on the maternal-fetal relationship in mice. J Protozool. 1982;29(1):77–81. [DOI] [PubMed] [Google Scholar]
- 322.Oduola AM, Phillips JH, Spicer SS, Galbraith RM. Plasmodium berghei: histology, immunocytochemistry, and ultrastructure of the placenta in rodent malaria. Exp Parasitol. 1986;62(2):181–193. [DOI] [PubMed] [Google Scholar]
- 323.Tegoshi T, Desowitz RS, Pirl KG, Maeno Y, Aikawa M. Placental pathology in Plasmodium berghei-infected rats. Am J Trop Med Hyg. 1992;47(5):643–651. [DOI] [PubMed] [Google Scholar]
- 324.Avery JW, Smith GM, Owino SO, et al. Maternal malaria induces a procoagulant and antifibrinolytic state that is embryotoxic but responsive to anticoagulant therapy. PLoS One. 2012;7(2):e31090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Wilson KD, Ochoa LF, Solomon OD, et al. Elimination of intravascular thrombi prevents early mortality and reduces gliosis in hyper-inflammatory experimental cerebral malaria. J Neuroinflammation. 2018;15(1):173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Poovassery JS, Sarr D, Smith G, Nagy T, Moore JM. Malaria-induced murine pregnancy failure: distinct roles for IFN-gamma and TNF. J Immunol. 2009;183(8):5342–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Clark IA, Chaudhri G. Tumor necrosis factor in malaria-induced abortion. Am J Trop Med Hyg. 1988;39(3):246–249. [DOI] [PubMed] [Google Scholar]
- 328.Hviid L, Marinho CR, Staalsoe T, Penha-Goncalves C. Of mice and women: rodent models of placental malaria. Trends Parasitol. 2010;26(8):412–419. [DOI] [PubMed] [Google Scholar]
- 329.Poovassery J, Moore JM. Murine malaria infection induces fetal loss associated with accumulation of Plasmodium chabaudi AS-infected erythrocytes in the placenta. Infect Immun. 2006;74(5):2839–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Chene A, Briand V, Ibitokou S, et al. Placental cytokine and chemokine profiles reflect pregnancy outcomes in women exposed to Plasmodium falciparum infection. Infect Immun. 2014;82(9):3783–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Suguitan AL Jr., Leke RG, Fouda G, et al. Changes in the levels of chemokines and cytokines in the placentas of women with Plasmodium falciparum malaria. J Infect Dis. 2003;188(7):1074–1082. [DOI] [PubMed] [Google Scholar]
- 332.Mens PF, Bojtor EC, Schallig HD. Molecular interactions in the placenta during malaria infection. Eur J Obstet Gynecol Reprod Biol. 2010;152(2):126–132. [DOI] [PubMed] [Google Scholar]
- 333.Stephens R, Langhorne J. Effector memory Th1 CD4 T cells are maintained in a mouse model of chronic malaria. PLoS Pathog. 2010;6(11):e1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Ibitokou S, Gbedande K, Opata M, Carpio V, Marshall K, Stephens R. Decay of IFN-g+ Effector T cells after Infection Leads to Loss of the Protection Provided by Persistent Plasmodium Infection. In preparation. [Google Scholar]
- 335.Opata MM, Stephens R. Chronic Plasmodium chabaudi Infection Generates CD4 Memory T Cells with Increased T Cell Receptor Sensitivity but Poor Secondary Expansion and Increased Apoptosis. Infect Immun. 2017;85(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Villegas-Mendez A, Inkson CA, Shaw TN, Strangward P, Couper KN. Long-Lived CD4+IFN-gamma+ T Cells rather than Short-Lived CD4+IFN-gamma+IL-10+ T Cells Initiate Rapid IL-10 Production To Suppress Anamnestic T Cell Responses during Secondary Malaria Infection. J Immunol. 2016;197(8):3152–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Opata MM, Ibitokou SA, Carpio VH, et al. Protection by and maintenance of CD4 effector memory and effector T cell subsets in persistent malaria infection. PLoS Pathog. 2018;14(4):e1006960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.da Silva HB, de Salles EM, Panatieri RH, et al. IFN-gamma-induced priming maintains long-term strain-transcending immunity against blood-stage Plasmodium chabaudi malaria. J Immunol. 2013;191(10):5160–5169. [DOI] [PubMed] [Google Scholar]
- 339.Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Okwor I, Mou Z, Liu D, Uzonna J. Protective immunity and vaccination against cutaneous leishmaniasis. Front Immunol. 2012;3:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Jagannathan P, Bowen K, Nankya F, et al. Effective Antimalarial Chemoprevention in Childhood Enhances the Quality of CD4+ T Cells and Limits Their Production of Immunoregulatory Interleukin 10. J Infect Dis. 2016;214(2):329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.