

Summary
Leukaemic stem cells (LSC) have been experimentally defined as the leukaemia‐propagating population and are thought to be the cellular reservoir of relapse in acute myeloid leukaemia (AML). Therefore, LSC measurements are warranted to facilitate accurate risk stratification. Previously, we published the composition of a one‐tube flow cytometric assay, characterised by the presence of 13 important membrane markers for LSC detection. Here we present the validation experiments of the assay in several large AML research centres, both in Europe and the United States. Variability within instruments and sample processing showed high correlations between different instruments (R pearson > 0·91, P < 0·001). Multi‐centre testing introduced variation in reported LSC percentages but was found to be below the clinical relevant threshold. Clear gating protocols resulted in all laboratories being able to perform LSC assessment of the validation set. Participating centres were nearly unanimously able to distinguish LSChigh (>0·03% LSC) from LSClow (<0·03% LSC) despite inter‐laboratory variation in reported LSC percentages. This study proves that the LSC assay is highly reproducible. These results together with the high prognostic impact of LSC load at diagnosis in AML patients render the one‐tube LSC assessment a good marker for future risk classification.
Acute myeloid leukaemia (AML) is a heterogeneous group of diseases, with the shared feature of proliferation of immature myeloid blasts in the bone marrow and blood. The classification of AML has changed dramatically over the last decades, and is mainly based on the chromosomal abnormalities and gene mutations underlying each individual’s disease. 1 Despite this advancement, risk classification remains suboptimal as a proportion of patients will relapse regardless of the absence of poor‐risk factors at diagnosis. Improved detection of measurable residual disease (MRD) during therapy by immunophenotypic and molecular methods has shown low levels of persisting disease in patients in morphologic remission, 2 essential for further therapy choices. This MRD compartment presumably encompasses leukaemic stem cells (LSCs). LSCs are pivotal for underlying leukaemia propagation, therapy resistance and as a cellular reservoir of relapse. 3 , 4 , 5 Recent studies have correlated high LSC frequencies at the time of diagnosis with the presence of MRD and subsequent poor prognosis. 6 , 7 The implementation of LSC measurements in the clinic is therefore instrumental for risk stratification and facilitating the selection of appropriate treatment protocols. 8 , 9
Several studies identified LSCs by (cyto)genetic and functional characteristics. 4 , 9 , 10 Apart from these assays, LSCs can be immunophenotypically identified based on the principle that LSC can aberrantly express antigens. These flow cytometric assays can easily be implemented in most AML diagnostic workups. Although different cellular compartments are shown to possibly contain leukaemia‐initiating cells, 9 , 11 the CD34+CD3‒ compartment is the most established. 6 , 12 , 13 The use of an antibody panel with CD34 and CD38 has therefore been the basis of many studies, discriminating the CD34+CD38‒cell fraction, which contains both LSCs and normal haematopoietic stem cells (HSCs), from other cells.
For optimal discrimination between LSCs and HSCs, multiple markers were identified, highlighting the heterogeneity of AML LSCs. 6 , 7 We previously tested many of the proposed LSC markers in a large cohort of AML patients and selected those which showed the best distinction between HSC and LSC and, moreover, identified the highest LSC burden. 14 After omitting redundant markers, 13 markers (i.e. CLEC12A, TIM‐3, CD7, CD11b, CD22, CD56, CD33, CD45RA, CD123, CD44 and backbone markers CD34, CD38, CD45) remained necessary for correct identification. This panel of markers was arranged in a single eight‐colour flow cytometry antibody panel, combining the first six markers together in one fluorescence channel, hereinafter referred to as the ‘Combi’ channel, with the potential to be easily implemented in other laboratories. 14
Medical laboratory assays are essential to support clinicians to provide optimal treatment choices for patients; hence their results and reports should be of the best achievable quality. Research in improving these assays is ongoing, but is also directed towards standardisation. 15 , 16 to facilitate multi‐institutional collaborations.
Here we evaluated the technical and analytical feasibility of the eight‐colour LSC single tube assay, as well as standardisation of the process. The study is conducted in several large research centres both in Europe and the United States, with extensive flow cytometry experience, but not with the assessed LSC assay. We show that limited training leads to highly concordant results, allowing other centres to independently validate the clinical utility of LSC testing in AML. These results, together with the high prognostic impact of the LSC load at AML diagnosis, render the one‐tube LSC assessment a good marker for future risk classification.
1. Materials and methods
1.1. Instruments, setups and samples
The instruments used are listed in Table SI. We previously described the setup of flow cytometers, as based on EuroFlow instructions. 15 , 17 Sample information and details regarding harmonisation of all machines are described in supplemental materials and methods.
1.2. Study setup
A schematic overview of the study setup is shown in Fig 1 and described in detail in the supplementary text. In short, three cryopreserved diagnosis samples were used to evaluate inter‐instrument variance and four cryopreserved diagnosis samples were used for inter‐laboratory processing. Gating was trained on these latter four samples, and validated in 10 FCS files of representative diagnosis AML samples as listed in Table I.
Fig. 1.

Schematic overview of study setup. The study can be divided into four parts: (A) pre‐analysis by the central site, (B) analysis of four initial samples by six researchers of the central site and participating centres (C) identification of critical gating steps and (D) the validation of both the coordinating centre and the participating centres.
Table I.
Conclusive results training and validation set: percentages and events.
| Sample | T1 | T2 | T3 | T4 | V1 | V2 | V3 | V4 | V5 | V6 | V7 | V8 | V9 | V10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra‐laboratory | |||||||||||||||
| Technician 1 | % | 0.0012 | 0.0024 | 0.0000 | 0.0010 | 0.0000 | 0.0003 | 0.0020 | 0.0006 | 0.0505 | 0.0183 | 0.1206 | 2.3590 | 0.0007 | 0.2224 |
| Events | 24 | 91 | 0 | 42 | 0 | 11 | 19 | 15 | 612 | 364 | 2509 | 35 834 | 8 | 3669 | |
| Technician 2 | % | 0.0011 | 0.0008 | 0.0000 | 0.0027 | 0.0000 | 0.0002 | 0.0473 | 0.0003 | 0.0514 | 0.0178 | 0.1245 | 2.3689 | 0.0007 | 0.2046 |
| Events | 32 | 32 | 0 | 92 | 0 | 7 | 457 | 7 | 627 | 352 | 2528 | 36 939 | 9 | 3453 | |
| Technician 3 | % | 0.0022 | 0.0008 | 0.0000 | 0.0011 | 0.0000 | 0.0002 | 0.0018 | 0.0001 | 0.0521 | 0.0171 | 0.1249 | 2.3881 | 0.0001 | 0.2166 |
| Events | 48 | 28 | 1 | 42 | 0 | 8 | 18 | 3 | 634 | 334 | 2513 | 36 086 | 1 | 3492 | |
| Technician 4 | % | 0.0003 | 0.0007 | 0.0000 | 0.0007 | 0.0001 | 0.0002 | 0.0031 | 0.0003 | 0.0373 | 0.0139 | 0.1115 | 2.0514 | 0.0002 | 0.1765 |
| Events | 16 | 40 | 0 | 42 | 3 | 7 | 31 | 6 | 460 | 280 | 2275 | 31 804 | 105 | 6339 | |
| Technician 5 | % | 0.0026 | 0.0026 | 0.0003 | 0.0020 | 0.0000 | 0.0021 | 0.0085 | 0.0003 | 0.0509 | 0.0192 | 0.1009 | 2.4384 | 0.0005 | 0.2303 |
| Events | 113 | 181 | 1 | 75 | 1 | 82 | 85 | 7 | 620 | 397 | 2127 | 37 642 | 6 | 3938 | |
| Technician 6 | % | 0.0008 | 0.0016 | 0.0000 | 0.0016 | 0.0000 | 0.0003 | 0.0012 | 0.0001 | 0.0481 | 0.0200 | 0.1167 | 2.2837 | 0.0003 | 0.2041 |
| Events | 48 | 86 | 0 | 92 | 0 | 10 | 12 | 3 | 580 | 397 | 2451 | 34 002 | 4 | 3451 | |
| Inter‐laboratory | |||||||||||||||
| Laboratory 1 | % | 0.0001 | 0.0005 | 0.0067 | 0.0002 | 0.0514 | 0.0181 | 0.1405 | 2.2475 | 0.0012 | 0.2657 | ||||
| Events | 3 | 17 | 64 | 5 | 627 | 351 | 2829 | 33 456 | 14 | 4388 | |||||
| Laboratory 2 | % | 0.0001 | 0.0001 | 0.0011 | 0.0000 | 0.0466 | 0.0109 | 0.0855 | 1.9329 | 0.0002 | 0.0983 | ||||
| Events | 2 | 5 | 11 | 1 | 573 | 223 | 1803 | 30 155 | 2 | 1668 | |||||
| Laboratory 3 | % | 0.0001 | 0.0001 | 0.0007 | 0.0001 | 0.0290 | 0.0105 | 0.0703 | 1.5800 | 0.0005 | 0.1100 | ||||
| Events | 2 | 4 | 7 | 3 | 446 | 223 | 1534 | 28 601 | 6 | 2422 | |||||
| Laboratory 4 | % | 0.0003 | 0.0005 | 0.0275 | 0.0002 | 0.0358 | 0.0135 | 0.0854 | 2.0172 | 0.0000 | 0.1850 | ||||
| Events | 8 | 16 | 285 | 4 | 435 | 277 | 1859 | 30 494 | 0 | 2995 | |||||
| Laboratory 5 | % | 0.0000 | 0.0002 | 0.0021 | 0.0000 | 0.0362 | 0.0139 | 0.1061 | 2.3287 | 0.0003 | 0.2444 | ||||
| Events | 0 | 6 | 21 | 0 | 444 | 250 | 2302 | 32 983 | 4 | 3884 | |||||
| Laboratory 6 | % | 0.0000 | 0.0003 | 0.0034 | 0.0002 | 0.0485 | 0.0134 | 0.0875 | 3.0156 | 0.0007 | 0.0698 | ||||
| Events | 0 | 9 | 30 | 5 | 519 | 241 | 1698 | 32 568 | 8 | 1082 | |||||
| Laboratory 7 | % | 0.0001 | 0.0006 | 0.0077 | 0.0003 | 0.0441 | 0.0135 | 0.0772 | 2.3639 | 0.0016 | 0.2992 | ||||
| Events | 2 | 22 | 83 | 8 | 574 | 268 | 610 | 37 017 | 19 | 5180 | |||||
| Intra‐laboratory | Mean | 0.0014 | 0.0015 | 0.0001 | 0.0015 | 0.0000 | 0.0006 | 0.0107 | 0.0003 | 0.0484 | 0.0177 | 0.1165 | 2.3149 | 0.0004 | 0.2091 |
| Minimum | 0.0003 | 0.0007 | 0.0000 | 0.0007 | 0.0000 | 0.0002 | 0.0012 | 0.0001 | 0.0373 | 0.0139 | 0.1009 | 2.0514 | 0.0001 | 0.1765 | |
| Maximum | 0.0026 | 0.0026 | 0.0003 | 0.0027 | 0.0001 | 0.0021 | 0.0473 | 0.0006 | 0.0521 | 0.0200 | 0.1249 | 2.4384 | 0.0007 | 0.2303 | |
| Variance | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0003 | 0.0000 | 0.0000 | 0.0000 | 0.0001 | 0.0160 | 0.0000 | 0.0003 | |
| Inter‐laboratory | Mean | 0.0001 | 0.0003 | 0.0070 | 0.0001 | 0.0417 | 0.0134 | 0.0932 | 2.2123 | 0.0006 | 0.1818 | ||||
| Minimum | 0.0000 | 0.0001 | 0.0007 | 0.0000 | 0.0290 | 0.0105 | 0.0703 | 1.5800 | 0.0000 | 0.0698 | |||||
| Maximum | 0.0003 | 0.0006 | 0.0275 | 0.0003 | 0.0514 | 0.0181 | 0.1405 | 3.0156 | 0.0016 | 0.2992 | |||||
| Variance | 0.0000 | 0.0000 | 0.0001 | 0.0000 | 0.0001 | 0.0000 | 0.0005 | 0.1713 | 0.0000 | 0.0071 | |||||
1.3. Statistics
All results showed complete gating strategy, listing the number of events for all relevant populations (lymphocytes, (CD34+) blasts, CD34+CD38‒, LSC and HSC). LSC percentages ≥0·03% was classified as LSChigh or LSClow <0·03%. 6 , 16 Since percentages found in LSClow patients are low, variances calculated as a coefficient of variation are high. ±0·5 log was considered as acceptable error with limited effect on prognostic value (Figure S1). Variation was calculated using the Excel function VAR.P. Pearson correlation coefficients were calculated when reported percentages were compared between laboratories or machines.
2. Results
2.1. Central site inter‐instrument processing
To evaluate the influence of different flow cytometers, the assay was first assessed on different platforms at the central site. Expression plots of the total blast population and CD34+ blast subpopulation of two representative samples measured on BD LSRFortessa and BC Gallios EX were compared to BD FACSCanto II. See Fig 2. Lymphocyte and LSC percentages were analysed in all samples and showed a high correlation, with results on BD FACSCanto II Fig 2C.
Fig. 2.
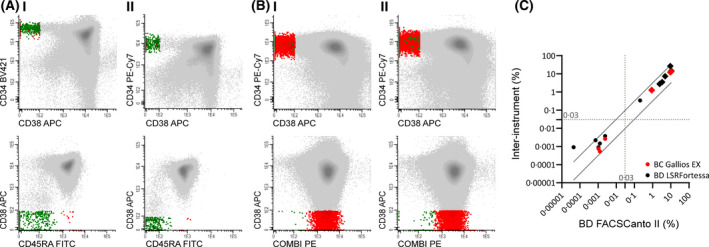
Central site inter‐instrument processing. Multiple samples with similar processing were measured across multiple flow cytometric instruments (A) or platforms (B). (A) Training sample 1 was measured on BD FACSCanto II (I) and BD LSRFortessa (II). FACS plots of complete blasts (top row) and CD34+ blasts (bottom row). CD33 and CD34 were exchanged in the channel (see Table S3) for standardisation within the coordinating institute. (B) A diagnostic AML sample was measured on BD FACSCanto II (I) and BC Gallios EX (II). FACS plots for complete blast population (top row) and CD34+ blast population (bottom row). (C) LSC percentages (dots) and lymphocyte percentages (diamonds) analysed in samples measured on BD LSRFortessa (black) and BC Gallios EX (red), compared to BD FACSCanto II. 0·5 log error is depicted as diagonal lines. 0·03% clinical stem cell cut‐off depicted as dotted lines. Pearson correlation coefficients between BC Gallios EX and BD FACSCanto II, and BD LSRFortessa and BD FACSCanto II were r = 1·00, P =< 0·001 and r = 0·91, P =< 0·0001 respectively. Grey: (CD34+) population; Red: LSC; Green: HSC.
2.2. Training
2.2.1. FCS files generated at central site with analysis by six researchers from central site
Gating reproducibility within the trained team at the central site was evaluated by having six researchers perform LSC assessments on FCS files, generated at central site (n = 4). See Fig 3 and Figure S2. Since LSCs are heterogeneous in marker expression, the gating of all individual markers was analysed (Table SIV). The largest variance was found for LSCs gated positive for Combi (mean 2·7*10−6, range 0·0–9·7*10−6). Without prior knowledge of inclusion of a CD34‒ patient, 18 all researchers from the central site were able to identify this CD34‒ sample (T3).
Fig. 3.
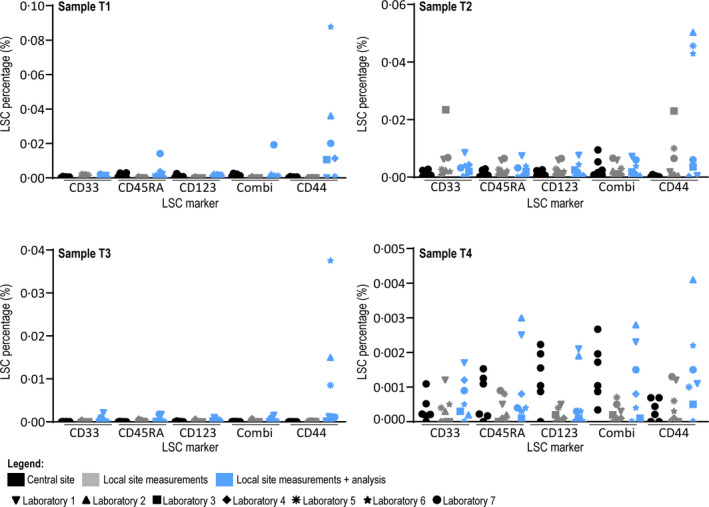
Results of central site and local sites on initial samples. Results of analysis of FCS files generated at central site by six researchers from central site (black), results of FCS files generated at local sites with analysis by one researcher from central site (grey) and results of FCS files generated at local sites with analysis from local site (blue) on samples T1–T4. All individual results are shown as the percentage of leukaemic stem cells of the complete white blood cell compartment. Laboratories are specified using different symbols, showing that differences in LSC percentages is not consistently explained by one laboratory. Axes run from 0·000% to 0·030% (clinical relevant cut‐off) for sample T1 and T2, and 0·000–0·003% for samples T3 and T4. CD44 is not depicted due to high variance (shown in Figure S3).
2.2.2. FCS files generated at local sites with analysis by one researcher from central site
The same set of cryopreserved samples were sent to all participating laboratories, accompanied by a protocol describing flow cytometer setup (see supplemental text). Generated FCS files were uploaded by the local sites to a designated repository and checked by one researcher from the central site. Percentages of LSC were analysed in all samples Fig 3, Figure S3 and Table SV). While there was variation in the number of WBCs measured (T1 mean 1 551 352 events, range 460 484–4 538 067; T2 mean 2 137 499 events, range 475 915–3 153 669; T3 mean 1 807 224 events, range 548 304–4 218 299; T4 mean 1 318 815 events, range 478 873–3 808 045), measurements were overall highly comparable. Representative expression plots of the total blast population and expression plots of the CD34+ blast subpopulation of sample T2 show a high resemblance between different local sites (Figure S2). Analysis performed by one researcher from the central site resulted in LSC percentages resembling those found in the analysis of files generated within the central site Fig 3. Sample T1 from Laboratory 4 contained 295 370 WBC and could therefore not be analysed.
2.2.3. FCS files generated at local sites with analysis from local site
The participating centres were asked for analysis of the files using the advised gating strategy (see supplemental data 1). Analyses were uploaded to the repository and reviewed by the central site. When gating could evidently be optimised (i.e. WBC gate included debris), feedback was sent to the local site and analysis could be revised. An average of 1·9 (range 1–3) analysis rounds were needed to come to final gating results (two laboratories did not need feedback, four laboratories needed feedback once, while one laboratory needed feedback twice). Results reported by the local sites showed more variability (especially in sample T3 and T4), compared to analyses within the central site (Fig 3, Figure S3 and corresponding Table SVI). In all samples, the largest variation was found in percentages CD44+ LSC (mean 0·0003621%, range 0·0000016–0·0008108%). In conclusion, variance mostly results from data analysis, not data collection.
2.3. Critical steps in gating strategy
As a consequence of training the participating centres, a number of critical steps in the gating strategy were elucidated.
Exact gating of WBC compartment. Since the burden of LSC is presented as a percentage of the complete WBC load, correct gating of this population is critical. However, gating for WBCs was not described. Furthermore, as no live‐dead marker is included in the stem cell tube, gating for viable WBC versus debris is solely based on scatter properties. Therefore, it is recommended to start with gating the lymphocytes in a CD45/SSC plot. Since lymphocytes are low in side‐ and forward‐scatter properties, they can act as a starting point for the WBC gate (Figure S4A).
Discriminating CD34+ blasts from CD34‒ blasts. Leukaemic blasts can differ in CD34 expression, and gating of CD34+ blasts can therefore be challenged by CD34dim blasts ‒ the CD34/CD38 plot can be illustrative (Figure S4B). After gating LSC and HSC, the gating strategy supports so‐called back‐gating. Both LSC and HSC often present as clusters in SSC/FSC, CD45/SSC and CD34/SSC plots. Scattered LSC (or HSC) events low in the CD34+ gate therefore lead to narrowing of the CD34gate (Figure S4C).
All tips for optimal gating were reported to all laboratories before the validation step was initiated.
2.4. Validation
2.4.1. Additional FCS files generated at central site with analysis by researchers from central site
To evaluate the reproducibility of LSC analysis in clinical practice, ten additional representative diagnostic FSC files were selected for analysis. Of these samples, four were LSChigh (V5, V7, V8 and V10), five were LSClow (V1, V2, V3, V4, V6, V9), of which one was CD34‒ (V1). Two samples were around the cut‐off (V5 LSChigh, V6 LSClow). Results similar to routine practice were reported: which includes gating of all LSC markers (i.e. CD33, CD44, CD123, CD45RA and Combi) separately and selecting the best marker to ultimately report as LSC load (Figure S5A, detailed in Table I. As LSCs are frequently covered by more than one LSC marker, the selection of a different marker did not always result in identification of a distinct different population (see supplemental gating strategy for examples). Discrimination between high (≥0·03%) and low (<0·03%) LSC load was concurrent among all researchers in 13/14 samples (93%). Sample V3 showed discordance between the researchers, as one researcher included CD45high cells as leukaemic blasts and CD34+CD38‒ cells with higher scatter properties as LSC, in absence of LSC markers. The selected ‘best’ marker (Table SVII) showed high resemblance (i.e. 5/6 or 6/6 researchers chose the same marker) in some of the samples (6/14; 43%), and lower resemblance (i.e. 3–4 of the six researchers chose the same marker) in the other samples (8/14; 57%). CD45RA was selected as best LSC marker in 63%, followed by Combi (18·3%), CD123 (13·3%) and CD33 (5·0%). Marker CD44 was never selected.
Repeat analyses by individual operators are evaluated in a select set of samples and is shown in Figure S6 and corresponding Table SVIII. The variance introduced by repeated analyses is minor, and had no effect on the outcome (i.e. LSClow remained LSClow, LSChigh remained LSChigh).
2.4.2. Additional FCS files generated at central site with analysis by local sites
LSC analyses of the local sites were evaluated in the same set of ten FSC files (Figure S5B). Participating centres were unanimously able to distinguish LSChigh samples V7, V8 and V10, but LSC percentages Table I and selected markers differed (Table SVII). Sample V5 was identified in 6/7 laboratories as LSChigh, but identified as LSClow in one participating laboratory with 0·029% (nevertheless very close to the cut‐off of 0·03%). All results in LSChigh patients were within ±0·5 log error Fig 4, which was identified as acceptable error (see Materials and methods above). There was a high correlation in detected LSC burden (mean r = 0·999, range 0·998–1·000, P < 0·001) between participating centres and the central site. In analyses of the local sites, CD45RA was selected as best marker in most samples (38·6%), followed by Combi (27·1%). CD44 was selected as best marker in 17·1%, while never being selected by researchers from the central site. While none of the researchers from the central site selected the same marker for all samples, three participating institutes did.
Fig. 4.
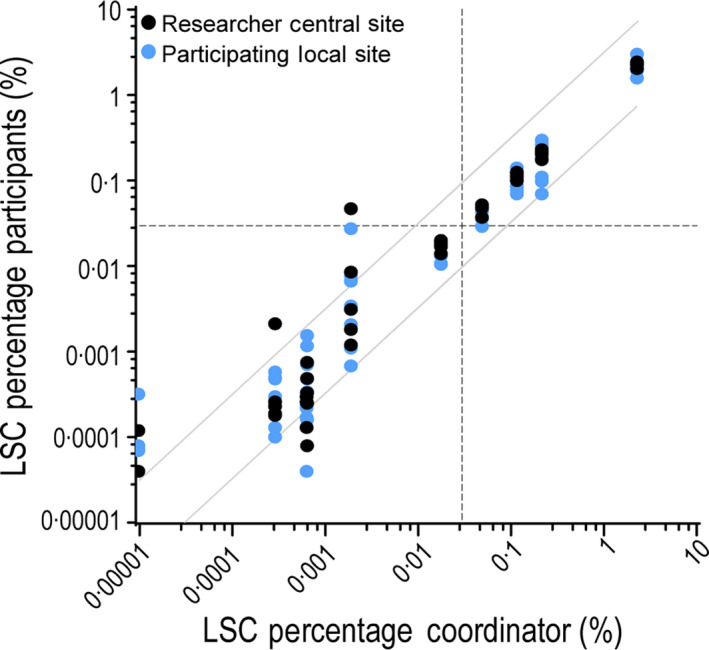
Results of central site and local sites on validation FCS files. Ten representative diagnostic AML samples were selected and corresponding flow cytometry files were sent to six researchers from the central site and seven participating centres for analysis. Results were reported back as the leukaemic stem cell percentage analysed by the most reliable stem cell marker (or markers). Results reported by one researcher from the central site compared to LSC percentages reported by all other participants (researchers from the central site in black, participating laboratories in blue). Previously determined (and validated) cut‐off of 0·03% is shown as a dotted line. Results above the clinical validated cut‐off fall within ±0·5 log error, shown as grey diagonal lines. Lower percentages fall outside ±0·5 log error but are clinically irrelevant.
2.5. Clinical implications
Implementation of the assay has important clinical implications as it allows the identification of patients who have a distinctly different prognosis, and could therefore be added to future risk classification. Correct characterisation of patients with a significant poor outcome (LSChigh) and patients with a significant better outcome (i.e. CD34‒ 18 ) was evaluated. The 14 samples analysed within this study included four patients with high LSC load and two CD34‒ patients. Three patients were correctly identified as LSChigh in all analyses. The remaining sample were correctly categorised in 12/13 analyses. The two CD34‒ patients were recognised as LSClow in all analyses, but only researchers from the coordinating institute noted that these patients were CD34‒. It is important to note that the participating centres were not specifically asked for this conclusion. Fig 4C shows a higher variation in samples below the clinical 0·03% cut‐off, as is to be expected. However, the clinical value of the exact frequency of LSC at time of the diagnosis is not thoroughly established and is possibly obscured due to higher intra‐ and inter‐laboratory variability. In summary, the data reveal that the current dichotomisation between LSChigh and LSClow is more robust, and should therefore be adopted.
3. Discussion
Identification of patients with a high LSC load at time of diagnosis, allows identification of patients with poor disease outcome very early in the disease course. 6 , 16 Similarly, identification of patients who lack aberrant leukaemic CD34+ (stem) cells allows identification of patients with a distinct and better prognosis before the response of therapy can be perceived. 18 While the contribution of CD34+CD38‒ LSC to poor disease outcome is demonstrated in several studies, 19 , 20 , 21 LSC measurements are not clinically implemented because of the seemingly complex process which requires specific experience and standardisation in the laboratories involved. Among eight institutes, this study shows that the one‐tube LSC assay is highly reproducible between several large‐flow cytometry AML centres in both Europe and the United States after a relatively simple training.
Sophisticated 8–10 colour flow cytometry is at the basis of the diagnosis, characterisation and monitoring of haematological malignancies. Correct implementation of the technique and standardisation in its applications is of high importance and several guidelines to achieve this have recently been published. 22 , 23 In this study, we demonstrate that harmonisation between flow‐cytometers is required for comparable results. Here, the use of BD's FC beads or setup according to Euroflow protocol was adequate to result in comparable measurements as percentages of lymphocytes, blasts, CD34+ blasts, CD34+ CD38dim and CD34+CD38‒ fractions were decidedly comparable among all institutes (data not shown).
A defined gating strategy is essential for laboratories aspiring to incorporate any flow cytometric assessment. To highlight the effectiveness of our gating strategy, our training focused on samples low in LSC frequency (three LSClow, one CD34‒), since analyses of low‐frequent cell populations is sensitive to errors. Nonetheless, limited feedback from experienced researchers was sufficient to train new researchers to gate according to protocol and achieve a high degree of comparison with those results found by experienced researchers. During the training phase, a number of critical steps in the gating strategy were elucidated and are emphasised in this article.
The development of a standardised antibody panel for LSC detection 14 helps to aim at standardisation. This tube simplifies LSC assessment in routine AML‐flow cytometry work‐up, as well as limiting the costs and the number of cells needed. While the LSC tube consists of the best (most discriminating, high negative predictive value and most sensitive) markers for identification of LSC, some were preferred over others. Markers CD45RA and Combi often show distinct separation between HSC and LSC as two separate ‘tails’ within the CD34+CD38‒ fraction. CD44 is used as a marker for LSC 14 based on overexpression of CD44 compared to expression on normal haematopoietic (stem) cells, 24 but correct identification of normal‐high expression versus overexpression is difficult when only one ‘tail’ is present. For specific purposes, exclusion of CD44 could be suggested to allow incorporation of additional antibodies.
The training in analysis was confirmed by ten representative diagnostic AML samples, ranging from high levels of LSC to absence of LSC, mimicking the clinical setting. As expected, a high degree of correlation is found in samples with higher frequencies of LSCs, in contrast to samples with low frequencies. Previously, a cut‐off of 0·03% was identified as clinically and prognostically relevant. 6 , 16 In our validation, five samples with percentages proximal to this cut‐off were correctly classified in 4/5 cases, with the remaining sample being misclassified as LSClow by one institute (with 0·029% just below cut‐off). Analysis of the exact percentage of LSC below the 0·03% cut‐off is prone to higher variation, but critical analysis of LSClow patients is crucial for the identification of CD34‒ patients, associated with an overall good prognosis. 16 , 18 Further research should be undertaken to evaluate whether CD34‒ patients are correctly identified and discriminated from LSClow patients.
Although addition of LSC measurements at diagnosis may lead to further improvement of risk group stratification, post‐induction MRD measurements are valuable for guiding post‐remission strategies. 25 MRD in AML is a rapidly evolving area with fast developments in designs and approaches. While the introduction of next‐generation sequencing MRD detection certainly holds promise for the future, the combination with flow cytometry showed that both techniques contributed independently to the prognostic value of the patient cohort. 2 Combining flow cytometry MRD measurements with post‐induction LSC measurements improved the prognostic classification further. 16 Since LSC in MRD situations are rare events, correct gating of LSCs, but also measurement of sufficient WBCs is critical. The possibility and practicability of LSC MRD measurements are not yet described in this manuscript and should be explored as part of the proceedings of this multicentre international group.
Current limitations of this study include the selection of a restricted set of patient samples which conceivably does not cover the complete cellular heterogeneity seen within the AML population. It could therefore be argued that the implementation of the assay in centres needs to be evaluated in prospective multicentre studies. Furthermore, all samples measured were cryopreserved mononuclear cells. Ideally, fresh samples would be first measured at the coordinating institute, deemed suited for the training and then immediately sent to the participating centres and measured. This was considered impractical due to introducing more variability because of poorer viability. As the effect of different sample processing could therefore not be analysed extensively, the use of a standardised protocol is therefore warranted. 15 , 26
In summary, we show that the one‐tube LSC assay is highly reproducible for many different FC experienced laboratories after a relatively simple training. Since the tube is useful for finding almost all CD34+CD38‒ stem cells and requires limited samples, it can be implemented in clinical studies. The high concordance between different laboratories is particularly valuable for use in multicentre studies. These results, together with the high prognostic impact of the LSC load at diagnosis in AML patients render the one‐tube LSC assessment a good marker for future risk classification.
Author contributions
DH, GJS and JC initiated and designed the study. DH, AK, WJS, NK, MM, MIC, MS, AdJ, SOA and MW analysed the flow cytometry data. DH coordinated the study, collected all data, conducted and analysed all integrated analyses. DH performed the statistical analysis. DH wrote the manuscript which was further revised by CB and JC and reviewed by all coauthors.
Conflict of interest
Financial support for part of this research has been received by BD Biosciences. A lyophilised version of the LSC tube is currently in production and will become available, with royalty payments for intellectual property rights to the VUmc.
Supporting information
Data S1. Supplemental information and figures
Data S2. Gating strategy.
Fig S1. 0.5 log error on LSC data.
Fig S2. Representative example of FCS files generated at local sites with analysis by one researcher from central site.
Fig S3. Results of central site and local sites on initial samples.
Fig S4. Critical steps in gating strategy.
Fig S5. Results of central site and local sites on validation FCS files.
Fig S6. Individual operator variability.
Table SI. Instruments and setup.
Table SII. Reference target values FC beads.
Table SIII. LSC antibody panel.
Table SIV. Results of analysis by six researchers from the central site on FCS files generated at the central site.
Table SV. Results of FCS files generated at local sites with analysis by one researcher from central site.
Table SVI. Results of FCS files generated at local sites with analysis from local site.
Table SVII. Selected markers.
Table SVIII. Repeat analyses from individual operators.
Acknowledgements
The authors thank all MRD team members of the Amsterdam UMC for participation in the intra‐laboratory analyses. We thank Erik Huys and Frank Preijers from the Department of Laboratory Medicine/Laboratory for Hematology, Radboud UMC for their measurements on BC Navios and discussions on harmonisation of flow cytometry platforms. We thank Hetty J. Bontkes, Martine Reijm and Jolien C. Hollander from the Department of Clinical Chemistry, Medical Immunology (Amsterdam UMC, location VUmc) for help and access to BC Gallios. Antibodies used for these validation studies were provided by BD.
References
- 1. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27895058 [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jongen‐Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, al Hinai A, Zeilemaker A, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med. 2018;378(13):1189–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29601269 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 3. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2. Available from: http://papers3://publication/doi/10.1038/nm0909‐1010 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 4. Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34(+)CD38(−) cells in acute myeloid leukemia. Blood. 2012;119:3571–7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3325044&tool=pmcentrez&rendertype=abstract [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Rhenen A, Feller N, Kelder A, Westra A, Rombouts E, Zweegman S, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–7. Available from: http://clincancerres.aacrjournals.org/cgi/doi/10.1158/1078‐0432.CCR‐05‐0468 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 6. Terwijn M, Zeijlemaker W, Kelder A, Rutten AP, Snel AN, Scholten WJ, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE. 2014;9:e107587 Available from: http://dx.plos.org/10.1371/journal.pone.0107587 [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. Int J Hematol. 2017;105:549–57. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28357569 [cited 2017 August 4]. [DOI] [PubMed] [Google Scholar]
- 8. Hourigan CS, Gale RP, Walter RB. Refining AML outcome prediction. Leukemia. 2019;33:283–4. [DOI] [PubMed] [Google Scholar]
- 9. Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17‐gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7. Available from: http://www.nature.com/doifinder/10.1038/nature20598 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 10. Won E, Kim H, Park R, Choi S‐Y, Shin JH, Suh S‐P, et al. Direct confirmation of quiescence of CD34+CD38− leukemia stem cell populations using single cell culture, their molecular signature and clinicopathological implications. BMC Cancer. 2015;15:217 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4391681&tool=pmcentrez&rendertype=abstract [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarry J, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc‐deficient mice. J Clin Invest. 2011;121:384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Rhenen A, van Dongen GAMS, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell associated antigen CLL‐1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–66. Available from: http://www.bloodjournal.org/cgi/doi/10.1182/blood‐2007‐03‐083048 [cited 2018 June 14]. [DOI] [PubMed] [Google Scholar]
- 13. Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut J, et al. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas‐induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Can Res. 2000;60:4403–11. [PubMed] [Google Scholar]
- 14. Zeijlemaker W, Kelder A, Oussoren‐Brockhoff Y, Scholten Wj, Snel An, Veldhuizen D, et al. A simple one‐tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia. 2016;30(2):439–46. 10.1038/leu.2015.252 [DOI] [PubMed] [Google Scholar]
- 15. Cloos J, Harris JR, Janssen JJWM, Kelder A, Huang F, Sijm G, et al. Comprehensive protocol to sample and process bone marrow for measuring measurable residual disease and leukemic stem cells in acute myeloid leukemia. J Vis Exp. 2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29553571 [cited 2018 June 14]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeijlemaker W, Grob T, Meijer R, Hanekamp D, Kelder A, Carbaat‐Ham JC, et al. CD34+CD38− leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia. 2019;33(5):1102–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30542144 [cited 2019 March 5]. [DOI] [PubMed] [Google Scholar]
- 17. Kalina T, Flores‐Montero J, van der Velden VHJ, Martin‐Ayuso M, Böttcher S, Ritgen M, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26:1986–2010. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3437409&tool=pmcentrez&rendertype=abstract [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeijlemaker W, Kelder A, Wouters R, Valk PJM, Witte BI, Cloos J, et al. Absence of leukaemic CD34+ cells in acute myeloid leukaemia is of high prognostic value: a longstanding controversy deciphered. Br J Haematol. 2015;171:227–38. [DOI] [PubMed] [Google Scholar]
- 19. Plesa A, Dumontet C, Mattei E, Tagoug I, Hayette S, Sujobert P, et al. High frequency of CD34+CD38‐/low immature leukemia cells is correlated with unfavorable prognosis in acute myeloid leukemia. World J Stem Cells. 2017;9(12):227–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29321824 [cited 2019 July 4]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jentzsch M, Bill M, Nicolet D, Leiblein S, Schubert K, Pless M, et al. Prognostic impact of the CD34+/CD38− cell burden in patients with acute myeloid leukemia receiving allogeneic stem cell transplantation. Am J Hematol. 2017;92(4):388–96. Available from: http://doi.wiley.com/10.1002/ajh.24663 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 21. Hwang K, Park C‐J, Jang S, Chi H‐S, Kim D‐Y, Lee J‐H, et al. Flow cytometric quantification and immunophenotyping of leukemic stem cells in acute myeloid leukemia. Ann Hematol. 2012;91(10):1541–6. Available from: http://link.springer.com/10.1007/s00277‐012‐1501‐7 [cited 2019 July 4]. [DOI] [PubMed] [Google Scholar]
- 22. Lacombe F, Bernal E, Bloxham D, Couzens S, Porta MGD, Johansson U, et al. Harmonemia: a universal strategy for flow cytometry immunophenotyping—A European LeukemiaNet WP10 study. Leukemia. 2016;30:1769–72. Available from: http://www.nature.com/articles/leu201644 [cited 2018 November 29]. [DOI] [PubMed] [Google Scholar]
- 23. Solly F, Angelot‐Delettre F, Ticchioni M, Geneviève F, Rambaud H, Baseggio L, et al. (2019) Standardization of flow cytometric immunophenotyping for hematological malignancies: the FranceFlow Group Experience . Cytom Part A, cyto.a.23844. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cyto.a.23844 [cited 2019 August 23]. [DOI] [PubMed] [Google Scholar]
- 24. Cao H, Heazlewood SY, Williams B, Cardozo D, Nigro J, Oteiza A, et al. The role of CD44 in fetal and adult hematopoietic stem cell regulation. Haematologica. 2016;101(1):26–37. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26546504 [cited 2019 April 10]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buccisano F, Maurillo L, Schuurhuis GJ, Del Principe MI, Di Veroli A, Gurnari C, et al. The emerging role of measurable residual disease detection in AML in morphologic remission. Semin Hematol. 2019;56(2):125–30. Available from: https://www.sciencedirect.com/science/article/abs/pii/S003719631830101X?via%3Dihub [cited 2019 August 23]. [DOI] [PubMed] [Google Scholar]
- 26. Schuurhuis GJ, Heuser M, Freeman S, Béné M‐C, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29330221 [cited 2018 January 31]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental information and figures
Data S2. Gating strategy.
Fig S1. 0.5 log error on LSC data.
Fig S2. Representative example of FCS files generated at local sites with analysis by one researcher from central site.
Fig S3. Results of central site and local sites on initial samples.
Fig S4. Critical steps in gating strategy.
Fig S5. Results of central site and local sites on validation FCS files.
Fig S6. Individual operator variability.
Table SI. Instruments and setup.
Table SII. Reference target values FC beads.
Table SIII. LSC antibody panel.
Table SIV. Results of analysis by six researchers from the central site on FCS files generated at the central site.
Table SV. Results of FCS files generated at local sites with analysis by one researcher from central site.
Table SVI. Results of FCS files generated at local sites with analysis from local site.
Table SVII. Selected markers.
Table SVIII. Repeat analyses from individual operators.