

Abstract
Mitochondrial dysfunction and stem cell exhaustion are among the nine separate hallmarks of aging. Emerging evidence however suggests that mitochondrial activity can have a profound influence on the self-renewal and function of stem cells, thus mechanistically linking mitochondrial function and stem cell decline. In this review, we discuss how accumulation of mtDNA mutations or alterations in mitochondrial dynamics, turnover, and signaling can modulate age-dependent stem cell function. Finally, we also describe how mitochondrial substrate utilization influences stem and progenitor activity. Together, this growing body of evidence suggests that modulation of mitochondrial activity might provide a strategy to slow or reverse age-dependent stem cell decline, and potentially, slow or reverse human aging.
Introduction
The molecular basis of human aging remains obscure and no clear theoretical framework or agreed upon hypothesis currently exists. Perhaps the most well established construct is to define the molecular basis of aging through a series of distinct and seemingly universal molecular hallmarks of the process (Lopez-Otin et al., 2013). In the original formulation, this involved the delineation of nine separate hallmarks of aging that appeared to play a causal role in the process. These factors included molecular events such as telomere shortening and cellular senescence. Interestingly, two other delineated hallmarks were mitochondrial dysfunction and stem cell exhaustion. While initially portrayed as separate hallmarks, increasingly, it appears that the ability of the mitochondria to modulate aging might, in part, be driven by a particularly critical role of mitochondrial function in stem cell biology. Emerging evidence suggests that there are multiple aspects of mitochondrial biology that can impact the number and function of stem cells in various tissues. Here, we briefly review how mitochondria can modulate stem cells and how these two previously separate hallmarks of aging might in fact work in concert.
1. Mitochondria mutation and stem cell aging
Mitochondria possess their own genome (mtDNA), which is present in variable copies per cell. What regulates the mitochondrial copy number in a given cell is not well understood. Of note, while energetic demand likely determines that a beating cardiac myocyte will tend to have more mitochondria per cell than a quiescent endothelial cell, within similar cell types, such as a stem cell and its committed progenitors, mitochondrial mass and respiratory capacity are not particularly well correlated (de Almeida et al., 2017). mtDNA encodes 13 polypeptides of the respiratory chain, 2 ribosomal RNAs and 22 tRNAs that provide the machinery for mitochondria protein translation (Anderson et al., 1981; DiMauro and Schon, 2003). In comparison to nuclear DNA, mtDNA is believed to be more susceptible to DNA damage. The basis for this increased susceptibility has often been ascribed to the notion that the mitochondrial genome is subject to high levels of oxidative damage and lacks histone protection and the full range of DNA repair mechanisms (Alexeyev et al., 2013; Su et al., 2018). However, a significant role for oxidative damage driving mitochondrial mutations is not supported by more recent in vivo evidence. For instance, in Drosophila and in mice, deletion of mitochondrial superoxide dismutase (SOD2) or impairing the capacity to repair oxidative-damaged mitochondrial DNA (Itsara et al., 2014; Kauppila et al., 2018a) did not appear to significantly increase mitochondrial mutations. Similarly, while mitochondria lack histones, the transcription factor TFAM may play a similarly protective role (Kaufman et al., 2007; Kukat et al., 2015).
mtDNA mutations accumulate in aged tissues derived from human subjects (Corral-Debrinski et al., 1992; Kraytsberg et al., 2006; Reeve et al., 2009; Shin et al., 2004b; Wang et al., 2001) and animal models (Kujoth et al., 2005; Trifunovic et al., 2004). Initial estimates of the mutational rate for human mitochondrial DNA came from analyzing mtDNA in intestinal crypt cells in patients undergoing surgical resection for colonic tumors (Taylor et al., 2003). This study allowed for an estimated rate of somatic mtDNA mutation rate of approximately 5 × 10–5 mutations per genome per day in humans and suggested that the mutational rate per base pair was roughly 1000-fold higher for mtDNA than it was for nuclear DNA (Taylor et al., 2003). Subsequent analysis using more advanced and sensitive sequencing methods has suggested that the true mutational frequency in humans is likely 1–2 orders of magnitude lower than the initial estimate (Hoang et al., 2016)(Kennedy et al., 2013) (Fig. 1A). In contrast, whether somatic mtDNA mutations increase in aged mice is less clear, as at least some evidence suggests that the age-dependent increase in mtDNA mutations occur predominantly through a heteroplasmic rise of germline mtDNA variants in this species (Ma et al., 2018). It should be noted that mitochondrial mutations involve both point mutations (Greaves et al., 2012; Greaves et al., 2014; Taylor et al., 2003) and large deletions (Bender et al., 2006; Reeve et al., 2009) (Bua et al., 2006; Pallotti et al., 1996). The levels of both forms of mutations appear to increase with age (Kauppila et al., 2017). Again, while deletions are relatively easy to detect, advances in sequencing methodology, including the ability to sequence individual mitochondria, has allowed for a much clearer picture of the level of the harder-to-detect point mutations. These approaches have revealed significant single-nucleotide variant (SNV) heteroplasmy between mitochondria in the same cell (Morris et al., 2017). For mouse mitochondria, this appears to be roughly four SNVs per mitochondria. The physiological importance of this pervasive micro-heteroplasmy is unclear for either normal cell function or for stem and progenitor cell function. However, accumulation of mtDNA mutations are enriched in human induced pluripotent stem cells (iPSCs) derived from elderly patients when compared to similar stem cells derived from young adults (Kang et al., 2016). Moreover, human circulating hematopoietic stem cells (CD34+ clones) from adult bone marrow donors have significantly more mtDNA heterogeneity when compared to similar cells obtained from embryonic cord blood (Shin et al., 2004a). Interestingly, with the advent of single cell sequencing techniques, these natural mitochondrial mutations may provide a basis for barcoding cells thus providing a strategy to perform highly sensitive lineage tracing analysis in human samples (Ludwig et al., 2019).
Figure 1.
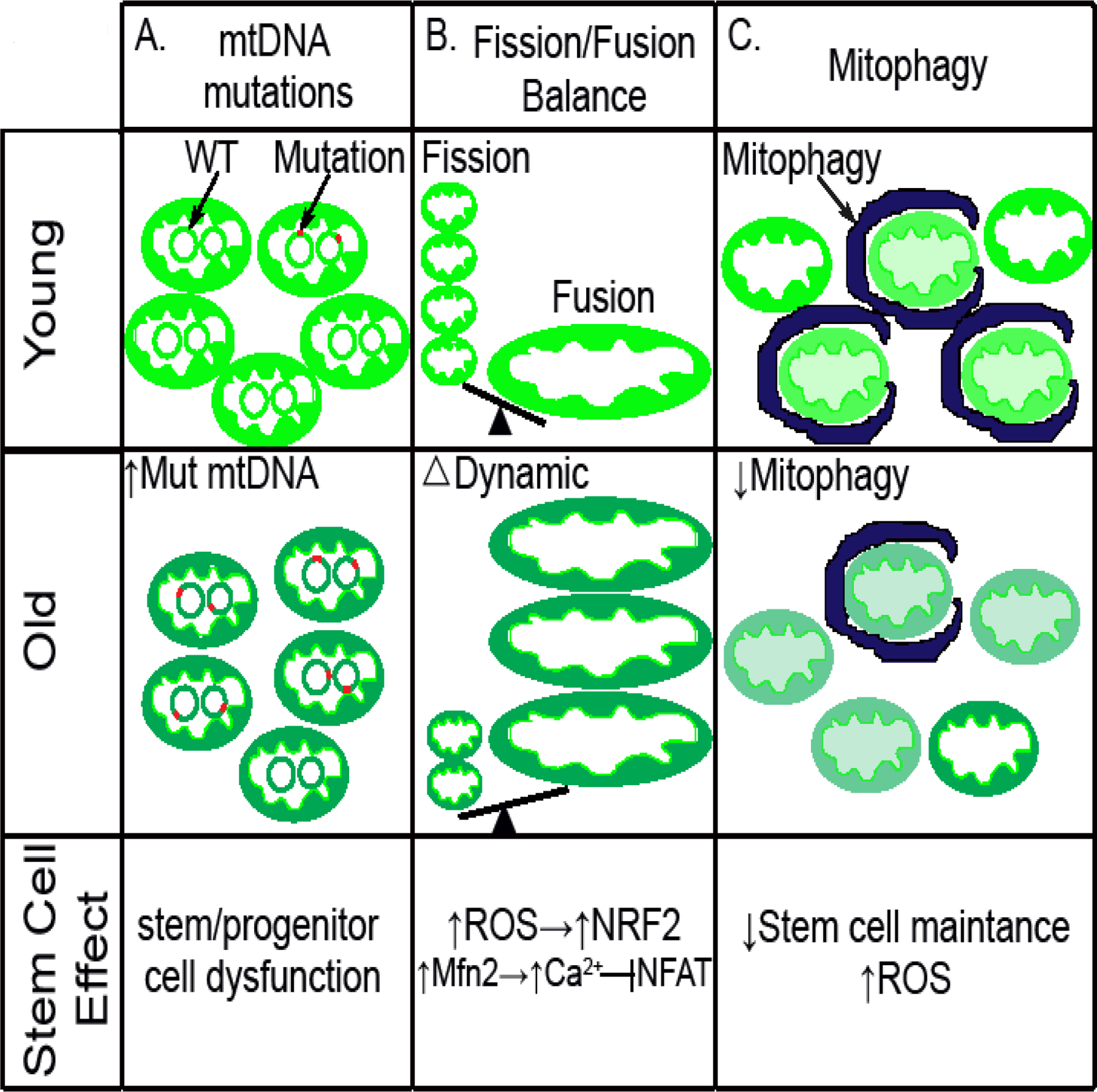
Mitochondrial DNA mutation and dynamics have a broad effect in stem cell aging. (A) mtDNA mutations that accumulate as stem cell age may partially contribute to dysfunctional stem/progenitor cells. (B) The mitochondrial fission/fusion balance go to the side of fusion as stem cell aged, leading to increased ROS mediated by NRF2 as well as altered Mfn2/Ca2+/Nfat signal cascade. (C) Mitochondrial quality is less controlled due to decreased mitophagy in old stem cells, leading to decreased stem cell maintenance, increased ROS levels and potential immune activation.
The studies described above clearly demonstrate that stem and progenitor cell aging is accompanied by an increase in mtDNA mutations, however, they do not per se casually link these mutations to any functional decline in stem and progenitor number or function. This mechanistic gap has been breached by a variety of mouse models. Perhaps the best characterized of these models is the so-called mitochondrial mutator mouse (Trifunovic et al., 2004) (Kujoth et al., 2005). These mice carry a knock-in mutation in the exonuclease domain of the mtDNA polymerase (Polg), leading to severe defects in the enzyme’s proofreading ability and the subsequent accumulation of widespread age-dependent mtDNA mutations. A subsequent genetic model involving double stranded mitochondrial breaks has also been described (Wang et al., 2013). The mutator mice were observed to undergo accelerated aging (Kujoth et al., 2005; Trifunovic et al., 2004) (Fig. 1A). These animals also exhibited various stem cell defects including impaired self-renewal ability of neural stem cells (NSCs) and abnormal hematopoiesis (Ahlqvist et al., 2012). Interestingly, while the accelerated aging phenotype of the mutator mice was viewed as ROS independent (Kujoth et al., 2005; Trifunovic et al., 2004), the progenitor cell defects were able to be rescued by N-acetylcysteine (NAC), a widely used antioxidant (Ahlqvist et al., 2012). It should be noted that the level of mitochondrial mutations in these animal models vastly exceeds what is observed in normal physiological aging. Moreover, while these models do develop age-dependent stem and progenitor dysfunction, these defects do not qualitatively mirror what is seen with physiological aging (Norddahl et al., 2011). Indeed, it would appear that mtDNA mutations need to accumulate to significant levels before there is any effect on either overall lifespan or stem cell function (Kauppila et al., 2018b). Except in rare instances, these levels of mitochondrial mutations do not occur with normal human aging and as such, the notion that mtDNA mutation drive stem cell aging is not clearly supported by the available data.
2. Mitochondria fission and fusion and stem cell aging
There is a growing appreciation that mitochondrial dynamics, the balance of mitochondrial fusion and fission, plays an important role in stem cell function (Chen and Chan, 2017). Most evidence points to fission predominating in self-renewing cells (Prieto et al., 2016; Wang et al., 2017), while more differentiated cells have increased levels of mitochondria fusion (Bahat et al., 2018; Forni et al., 2016; Kasahara et al., 2013). Nonetheless, examples exist where the opposite appears to hold (Khacho et al., 2016). Mitochondrial dynamics also plays an important role in maintaining cancer stem cell properties. Analyzing the stem-like cells (SLCs) derived from an immortalized mammary epithelial cell line, it was observed that mitochondrial fission was required to spatially restrict old mitochondria to the perinuclear region of the mother cell (Katajisto et al., 2015). Furthermore, it was noted that the distribution of mitochondria between daughter cells was asymmetric, with daughter cells that got young mitochondria able to maintain their stemness. In contrast, daughter cells that received old mitochondria went on to be more differentiated (Katajisto et al., 2015). Interestingly, these insights might lead to new mitochondrial-centric strategies to treat tumors (Civenni et al., 2019).
At present, much of the studies regarding mitochondrial dynamics and stem cell fate suffer from an incomplete understanding of the implications of a fused versus fragmented mitochondria. One potential explanation may be that mitochondrial dynamics alter mitochondrial-dependent signaling and can therefore exert lasting effects on cell fate. One example involving neural stem cells suggests that this might be through an ROS-dependent mechanism, where levels of fusion and fission determine ROS levels and the subsequent activation of the redox-dependent transcription factor NRF2 (Khacho et al., 2016) (Fig. 1B). Other examples in pluripotent stem cells suggest the fusion/fission balance alters calcium homeostasis, which in turn activates calcium-sensitive kinases and downstream transcription factors regulating stem cell fate (Zhong et al., 2019). Similarly, levels of the mitofusin 2 (mfn2), a gene required for mitochondrial fusion, appears to effect HSC lineage potential (i.e. the balance between lymphoid versus myeloid differentiation) by regulating calcium levels in stem cells and thereby influencing the subcellular localization and hence activity of nuclear factor of activated T cells (NFAT) (Luchsinger et al., 2016) (Fig. 1B). Interestingly, a requirement for mitofusin activity has also been recently established for the maintenance of Drosophila male germline stem cells (GSCs) (Senos Demarco et al., 2019). In this case, the absence of mitofusin impaired mitochondrial fatty acid oxidation, leading to depletion of GSCs. As such, it would appear that mitochondrial dynamics can modulate stem cell fate through multiple mechanisms including ROS levels, calcium signaling and substrate utilization.
3. Mitophagy and stem cell aging
Mitophagy is the selective degradation of damaged or dysfunctional mitochondria by the autophagy machinery. Reporter mice that allow for the detection of in vivo mitophagy have noted that areas in the brain enriched for stem and progenitor cells have significantly high basal rates of mitophagy (Sun et al., 2015). This is presumably because stem cells may require lower levels of ROS and therefore demand higher level of mitochondrial quality control. To what degree in vivo mitophagy acts as an important arm of quality control and surveillance (i.e. able to remove damaged but still functional mitochondria) versus acting solely to remove bioenergetically inert mitochondria is presently unclear. However, an intact mitophagy system is increasingly viewed as having important non-cell autonomous functions. This revolves around the ability of mitophagy to suppress the release of mitochondrial-derived damage-associated molecular patterns (DAMPs) that can stimulate the innate immune system (Sliter et al., 2018). Additional work is needed to understand whether this aspect of mitophagy represents it’s most important in vivo function. If so, this may help to explain the known age–dependent property termed ‘inflammaging’, since the key regulators of mitophagy appear to decrease in aged tissues (Rubinsztein et al., 2011).
In addition to its growing importance in innate immunity, autophagy and mitophagy appears to play important cell autonomous roles in stem and progenitor cells. In the muscle stem cell, autophagic activity is impaired during aging, leading to defective satellite cell maintenance (Garcia-Prat et al., 2016) (Fig. 1C). Much of this age-dependent muscle stem cell defect was attributed to a decline in mitophagy, leading to the persistence of damaged and defective mitochondria and hence a rise in ROS levels (Fig. 1C). Indeed, treatment with the antioxidant Trolox restored the cell-intrinsic proliferative and regenerative defect seen in these autophagy-compromised aged muscle stem cells. A similar critical requirement for mitophagy has been observed in the hematopoietic stem cell. Genetic disruption of autophagy in HSCs causes accelerated aging phenotypes, including loss of stem cell quiescence and accelerated myeloid differentiation of HSCs (Ho et al., 2017). In the case of HSCs, it would appear that disrupting global autophagy leads to a mitophagic defect and a subsequent alteration in mitochondrial metabolism. These metabolic alterations, in turn, alter metabolites that can modify the epigenetic landscape and modulate stem cell function (Ho et al., 2017). Other studies have implicated mitophagy in HSC self-renewal capacity (Ito et al., 2016).
In the previous example, mitophagy was important to restrain mitochondrial metabolism in HSCs. This fact has been exploited therapeutically with the observation that the NAD+-boosting agent nicotinamide riboside (NR) can also reduce mitochondrial activity within HSCs by increasing mitochondrial clearance (Vannini et al., 2019). Indeed, this connection between NAD levels and mitophagy may extend beyond the stem cell. In particular, there has been a number of recent studies attempting to boost NAD levels as a means of restoring mitophagy and treating age-dependent diseases. This includes rare accelerated aging syndromes such as Werner’s syndrome which appears to have reduced NAD levels and impaired mitophagy (Fang et al., 2019a), as well as more common age-related neurological diseases which are also characterized by insufficient mitophagic flux (Fang et al., 2019b). Finally, another parameter that might intersect with mitophagy is the mitochondrial membrane potential. Mitochondria with low membrane potential might be specifically targeted for mitophagic clearance. Indeed, the classic stimulus to induce mitophagy involves chemical uncouplers that abrogate the membrane potential across the inner mitochondrial membrane (Georgakopoulos et al., 2017). In this context, there are a number of studies that have linked mitochondrial membrane potential to stemness in populations ranging from embryonic stem cells (Schieke et al., 2008), to HSCs (Simsek et al., 2010) to stem-like immune cell populations (Sukumar et al., 2016).
4. UPRmt signaling
The mitochondrial unfolded protein response (UPRmt), trigged by accumulation of misfolded proteins within the mitochondrial matrix, is an evolutionarily protective mechanism to ensure cells survive under condition of stress (Yang et al., 2016; Zhang et al., 2018). While this pathway has been elegantly dissected in C. elegans, its overall importance and downstream effectors are considerably less clear in mammalian cells (Tran and Van Aken, 2020). For instance, evidence suggests that certain critical elements of the UPRmt identified in worms do not play a similar conserved function in mammals (Seiferling et al., 2016). With that caveat, it should be noted that several sets of observations suggest a role for UPRmt signaling in stem cell function. For instance, the transition from a largely quiescent stem cell to a dividing and activated progenitor cell is often accompanied by a burst of mitochondrial biogenesis (Folmes et al., 2012). This increase in mitochondrial number and hence mitochondrial protein synthesis represent a challenge to the mitochondrial quality control pathways, which appear to be specifically engaged when stem cells exit from quiescence (Mohrin et al., 2018). One important aspect of this quality control network involves SIRT7, a member of the sirtuin family of histone deacetylases. Evidence suggests that SIRT7 appears to mediate UPRmt signaling in HSCs (Mohrin et al., 2015). This occurs because of a direct interaction between SIRT7 and nuclear respiratory factor 1 (NRF1). NRF1 represents a master mitochondrial transcription factor (Scarpulla, 2008) and evidence suggests that SIRT7 inhibits aspects of NRF1-dependent transcriptional activation, thereby reducing mitochondrial stress. In particular, in the absence of SIRT7, HSCs had increased constitutive activation of the UPRmt, resulting in reduced quiescence and impaired functional activity of these stem cells (Mohrin et al., 2015). Interestingly, in the HSCs from older mice, levels of SIRT7 are reduced, and augmenting this expression, alleviates the age-dependent defects seen in these cells (Mohrin et al., 2015). Inappropriate or constitutive activation of the UPRmt can also alter stem cell function in other stem cell compartments. For instance, deletion of the mitochondrial chaperone protein HSP60 within intestinal epithelial cells leads to intestinal crypts with impaired stem cell function (Berger et al., 2016).
While the above studies highlight that constitutive or inappropriate UPRmt signaling can impair stem cell function, observations have also suggested that aging may result in a dampen UPRmt. This impaired UPRmt signaling appears to be a result of the natural age-dependent decline in NAD+ levels that occur in a wide range of organisms (Mouchiroud et al., 2013). Remarkably, increasing intracellular NAD+ levels, by treating animals with nicotinamide riboside, improved the functional properties of a wide range of mouse stem cells (Zhang et al., 2016). This effect was demonstrated to occur, at least in part, through the ability of NR to stimulate UPRmt signaling (Zhang et al., 2016). As such, either too little or too much UPRmt signaling appears to contribute to age-dependent stem cell defects.
5. Mitochondrial Fatty acid oxidation (FAO)
Mitochondrial substrate utilization is emerging as an important aspect through which stem cell fate and function can be modified. Perhaps the best-studied example involves the role that mitochondrial fatty acid oxidation (FAO) plays in modulating stem cell activity. Mitochondrial metabolism of long chain fatty acids requires the sequential activity of the mitochondrial outer membrane protein carnitine palmitoyltransferase-1 (CPT 1) and the activity of the mitochondrial inner membrane protein carnitine palmitoyltransferase-2 (CPT 2) to allow fatty acid transport across the double mitochondrial membrane and subsequent β-oxidation and ATP generation (Lundsgaard et al., 2018). It has been observed that maintenance of neural stem/progenitor cells (NSPCs) quiescence requires high levels of FAO with high expression of CPT1a. Treating NSPCs with the CPT1 inhibitor Etomoxir inhibits FAO leading NSPCs to exit the quiescence state and undergo apoptosis (Knobloch et al., 2017). Genetic inactivation of CPT1a in adult NSPCs also lead to decreased NSPCs expansion and enhanced NSPCs cell death. Moreover, mice administrated malonyl-CoA, an endogenous inhibitor of Cpt1a, promoted NSPCs to exit quiescence (Knobloch et al., 2017) (Fig. 2A). This combination of genetic and pharmacological approaches are important, as the use of etomoxir alone to disrupt FAO has led to potentially misleading conclusions (Divakaruni et al., 2018; Nomura et al., 2016; Raud et al., 2018).
Figure 2.
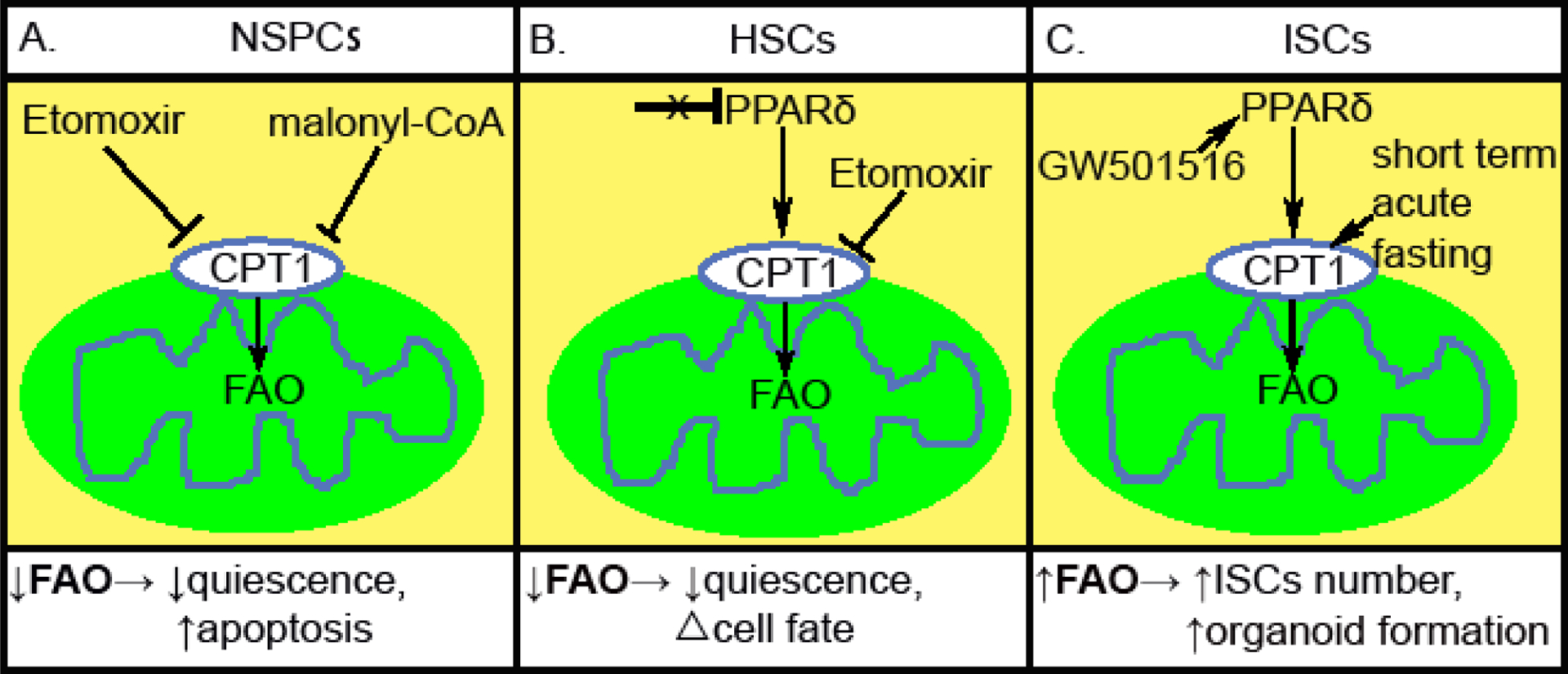
Modulating fatty acid oxidation (FAO) impacts tissue-specific stem cell function. (A) Inhibiting CPT1 by either Etomoxir or malonyl-CoA lead to decreased FAO in NSPCs, leading to reduced quiescence and increased apoptosis. (B) Inactivation of PPARδ or inhibiting CPT1 by Etomoxir lead to reduction of FAO in HSCs. This triggers an exit from quiescence and an alteration in symmetric versus asymmetric commitment. (C) Short term acute fasting or GW501516 treatment leads to activation of FAO in ISCs, and a subsequent increase in crypt Lgr5+ and Olfm4+ ISC/early progenitors, as well as crypt organoid-forming capacity.
In the context of the HSC, FAO appears to play an important role in stem cell maintenance as well as stem cell function. The ligand-activated transcription factor peroxisome proliferator-activated receptor δ (PPARδ) is the predominant mediator of FAO signaling in HSCs. Evidence suggests that genetic deletion of PPARδ, or treatment with etomoxir, leads to a reduction in FAO and a decrease in HSC repopulating ability (Ito et al., 2012). In contrast, activation of PPARδ with synthetic ligands that presumably mimic endogenous fatty acids, was shown to result in improved HSC function (Ito et al., 2012). Interesting, blocking FAO using etomoxir or PPARδ deletion also modulated HSC function, particularly the decision for an HSC to undergo symmetric (i.e. divide into two committed daughter cells) or asymmetric (give rise to one stem cell and one committed cell) division (Fig. 2B). Using single cell assays, it was demonstrated that FAO is necessary to maintain the requisite percentage of asymmetric divisions. These asymmetric division are, in turn, needed to regenerate and maintain the HSC pool and hence maintain bone marrow repopulation ability (Ito et al., 2012).
The role of FAO also extends to intestinal stem cells (ISCs). Recent evidence suggests that short-term acute fasting augments the organoid-forming capacity of ISCs in both young and aged mice, in part, by inducing a fatty acid oxidation (FAO) program. Interesting, in ISCs, fasting strongly induced expression of CPT1a, the rate-limiting enzyme in FAO. Inactivating CPT1a in the intestine led to reduced primary and secondary organoid formation of crypts, as well as decreased ISC number and long-term ISCs maintenance (Mihaylova et al., 2018). Moreover, the diminished regenerative capacity and number of aged ISCs appears to be responsive to manipulation of the FAO pathway. For instance, when aged mice were treated with GW501516, a potent transcriptional PPARδ activator, there was an induction in FAO and an increase in crypt Lgr5+ and Olfm4+ ISC/early progenitors (Mihaylova et al., 2018) (Fig. 2C).
Conclusion:
While initially viewed as separate hallmarks, the growing intersection between mitochondrial activity and stem cell function has clearly contributed to our overall understanding of aging. To what degree either a decline in mitochondrial function, or an impairment in stem cell function, actually contributes to human aging, awaits further validation. For many years, studies of mitochondrial dysfunction in human aging were confined largely to post-mitotic tissues such as skeletal muscle (Short et al., 2005). However, as highlighted here, the molecular impact of mitochondria on the aging process has focused increasingly on the stem and progenitor cell population. Our understanding of these processes remains woefully incomplete and questions remain as to which of the various properties of the mitochondria are the most critical for maintaining stem cell function in the aged organism. While we reviewed aspects of mtDNA mutations, dynamics, turnover, signaling and substrate utilization, other properties of mitochondria including mitochondrial biogenesis, calcium handling and ROS generation are also clearly relevant to stem cells. Nonetheless, an emerging picture has evolved in which the properties of stem cells, including their age-dependent maintenance and function appears to be actively modulated by the quality and function of intracellular mitochondria. Further refinement of these insights will undoubtedly lead to novel approaches to improve age-dependent stem cell decline and potentially, to slow or reverse human aging.
Highlights.
Mitochondrial function modulates stem cell function
Age-dependent decline in mitochondria might contribute to stem cell dysfunction
Mitochondrial dynamics, signaling, quality and substrate utilization effect stem cells
Acknowledgements:
The authors wish to thank members of the Finkel Lab for helpful comments. This work was supported by NIH grants 1R01 HL142663, 1R01HL142589 and P30 AG024827
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Ahlqvist KJ, Hamalainen RH, Yatsuga S, Uutela M, Terzioglu M, Gotz A, Forsstrom S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A, 2012. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab 15, 100–109. [DOI] [PubMed] [Google Scholar]
- Alexeyev M, Shokolenko I, Wilson G, LeDoux S, 2013. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harbor perspectives in biology 5, a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG, 1981. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. [DOI] [PubMed] [Google Scholar]
- Bahat A, Goldman A, Zaltsman Y, Khan DH, Halperin C, Amzallag E, Krupalnik V, Mullokandov M, Silberman A, Erez A, Schimmer AD, Hanna JH, Gross A, 2018. MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in embryonic stem cells. Nature communications 9, 5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM, 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38, 515–517. [DOI] [PubMed] [Google Scholar]
- Berger E, Rath E, Yuan D, Waldschmitt N, Khaloian S, Allgauer M, Staszewski O, Lobner EM, Schottl T, Giesbertz P, Coleman OI, Prinz M, Weber A, Gerhard M, Klingenspor M, Janssen KP, Heikenwalder M, Haller D, 2016. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat Commun 7, 13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM, 2006. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet 79, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2017. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab 26, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civenni G, Bosotti R, Timpanaro A, Vazquez R, Merulla J, Pandit S, Rossi S, Albino D, Allegrini S, Mitra A, Mapelli SN, Vierling L, Giurdanella M, Marchetti M, Paganoni A, Rinaldi A, Losa M, Mira-Cato E, D’Antuono R, Morone D, Rezai K, D’Ambrosio G, Ouafik L, Mackenzie S, Riveiro ME, Cvitkovic E, Carbone GM, Catapano CV, 2019. Epigenetic Control of Mitochondrial Fission Enables Self-Renewal of Stem-like Tumor Cells in Human Prostate Cancer. Cell Metab 30, 303–318 e306. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC, 1992. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature genetics 2, 324–329. [DOI] [PubMed] [Google Scholar]
- de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, Snoeck HW, 2017. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 21, 725–729 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA, 2003. Mitochondrial respiratory-chain diseases. The New England journal of medicine 348, 2656–2668. [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, Andreyev AY, Bowman CE, Caradonna K, Dranka BP, Ferrick DA, Liesa M, Stiles L, Rogers GW, Braas D, Ciaraldi TP, Wolfgang MJ, Sparwasser T, Berod L, Bensinger SJ, Murphy AN, 2018. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab 28, 490–503 e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Lautrup S, Jensen MB, Yang B, SenGupta T, Caponio D, Khezri R, Demarest TG, Aman Y, Figueroa D, Morevati M, Lee HJ, Kato H, Kassahun H, Lee JH, Filippelli D, Okur MN, Mangerich A, Croteau DL, Maezawa Y, Lyssiotis CA, Tao J, Yokote K, Rusten TE, Mattson MP, Jasper H, Nilsen H, Bohr VA, 2019a. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat Commun 10, 5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktaschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA, 2019b. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, Terzic A, 2012. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni MF, Peloggia J, Trudeau K, Shirihai O, Kowaltowski AJ, 2016. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem cells 34, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Munoz-Canoves P, 2016. Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos ND, Wells G, Campanella M, 2017. The pharmacological regulation of cellular mitophagy. Nat Chem Biol 13, 136–146. [DOI] [PubMed] [Google Scholar]
- Greaves LC, Elson JL, Nooteboom M, Grady JP, Taylor GA, Taylor RW, Mathers JC, Kirkwood TB, Turnbull DM, 2012. Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet 8, e1003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves LC, Nooteboom M, Elson JL, Tuppen HA, Taylor GA, Commane DM, Arasaradnam RP, Khrapko K, Taylor RW, Kirkwood TB, Mathers JC, Turnbull DM, 2014. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet 10, e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, Passegue E, 2017. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, Pandolfi PP, 2012. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 18, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, Mar JC, Singh R, Suda T, Lin CP, Frenette PS, Ito K, 2016. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, Cardozo-Pelaez F, Pallanck LJ, 2014. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet 10, e1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, Hayama T, Luo S, Harding CO, Amato P, Jensen J, Battaglia D, Lee D, Wu D, Terzic A, Wolf DP, Huang T, Mitalipov S, 2016. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell stem cell 18, 625–636. [DOI] [PubMed] [Google Scholar]
- Kasahara A, Cipolat S, Chen Y, Dorn GW 2nd, DScorranoorn L, 2013. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 342, 734–737. [DOI] [PubMed] [Google Scholar]
- Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM, 2015. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA, 2007. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell 18, 3225–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila JHK, Bonekamp NA, Mourier A, Isokallio MA, Just A, Kauppila TES, Stewart JB, Larsson NG, 2018a. Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res 46, 6642–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila TES, Bratic A, Jensen MB, Baggio F, Partridge L, Jasper H, Gronke S, Larsson NG, 2018b. Mutations of mitochondrial DNA are not major contributors to aging of fruit flies. Proceedings of the National Academy of Sciences of the United States of America 115, E9620–E9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila TES, Kauppila JHK, Larsson NG, 2017. Mammalian Mitochondria and Aging: An Update. Cell Metab 25, 57–71. [DOI] [PubMed] [Google Scholar]
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA, 2013. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 9, e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, Park DS, Slack RS, 2016. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 19, 232–247. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Pilz GA, Ghesquiere B, Kovacs WJ, Wegleiter T, Moore DL, Hruzova M, Zamboni N, Carmeliet P, Jessberger S, 2017. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep 20, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K, 2006. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature genetics 38, 518–520. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA, 2005. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. [DOI] [PubMed] [Google Scholar]
- Kukat C, Davies KM, Wurm CA, Spahr H, Bonekamp NA, Kuhl I, Joos F, Polosa PL, Park CB, Posse V, Falkenberg M, Jakobs S, Kuhlbrandt W, Larsson NG, 2015. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci U S A 112, 11288–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW, 2016. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, Pelka K, Ge W, Oren Y, Brack A, Law T, Rodman C, Chen JH, Boland GM, Hacohen N, Rozenblatt-Rosen O, Aryee MJ, Buenrostro JD, Regev A, Sankaran VG, 2019. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339 e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundsgaard AM, Fritzen AM, Kiens B, 2018. Molecular Regulation of Fatty Acid Oxidation in Skeletal Muscle during Aerobic Exercise. Trends Endocrinol Metab 29, 18–30. [DOI] [PubMed] [Google Scholar]
- Ma H, Lee Y, Hayama T, Van Dyken C, Marti-Gutierrez N, Li Y, Ahmed R, Koski A, Kang E, Darby H, Gonmanee T, Park Y, Wolf DP, Jai Kim C, Mitalipov S, 2018. Germline and somatic mtDNA mutations in mouse aging. PLoS One 13, e0201304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, Freinkman E, Dickey AS, La Spada AR, Huang Y, Bell GW, Deshpande V, Carmeliet P, Katajisto P, Sabatini DM, Yilmaz OH, 2018. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 22, 769–778 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D, 2015. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Widjaja A, Liu Y, Luo H, Chen D, 2018. The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell 17, e12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Na YJ, Zhu H, Lee JH, Giang H, Ulyanova AV, Baltuch GH, Brem S, Chen HI, Kung DK, Lucas TH, O’Rourke DM, Wolf JA, Grady MS, Sul JY, Kim J, Eberwine J, 2017. Pervasive within-Mitochondrion Single-Nucleotide Variant Heteroplasmy as Revealed by Single-Mitochondrion Sequencing. Cell Rep 21, 2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J, 2013. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T, 2016. Fatty acid oxidation in macrophage polarization. Nat Immunol 17, 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, Bryder D, 2011. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell stem cell 8, 499–510. [DOI] [PubMed] [Google Scholar]
- Pallotti F, Chen X, Bonilla E, Schon EA, 1996. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am J Hum Genet 59, 591–602. [PMC free article] [PubMed] [Google Scholar]
- Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, Lopez-Garcia C, Torres J, 2016. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nature communications 7, 11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stuve P, Arnold-Schrauf C, Guderian M, Lochner M, Rampertaap S, Romito K, Monsale J, Bronstrup M, Bensinger SJ, Murphy AN, McGuire PJ, Jones RG, Sparwasser T, Berod L, 2018. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 28, 504–515 e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Taylor G, Elson JL, Bender A, Taylor RW, Morris CM, Turnbull DM, 2009. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell 8, 496–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G, 2011. Autophagy and aging. Cell 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, 2008. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88, 611–638. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Ma M, Cao L, McCoy JP Jr., Liu C, Hensel NF, Barrett AJ, Boehm M, Finkel T, 2008. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem 283, 28506–28512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, Trifunovic A, 2016. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17, 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senos Demarco R, Uyemura BS, D’Alterio C, Jones DL, 2019. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat Cell Biol 21, 710–720. [DOI] [PubMed] [Google Scholar]
- Shin MG, Kajigaya S, McCoy JP Jr., Levin BC, Young NS, 2004a. Marked mitochondrial DNA sequence heterogeneity in single CD34+ cell clones from normal adult bone marrow. Blood 103, 553–561. [DOI] [PubMed] [Google Scholar]
- Shin MG, Kajigaya S, Tarnowka M, McCoy JP Jr., Levin BC, Young NS, 2004b. Mitochondrial DNA sequence heterogeneity in circulating normal human CD34 cells and granulocytes. Blood 103, 4466–4477. [DOI] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS, 2005. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 102, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA, 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ, 2018. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Su T, Turnbull DM, Greaves LC, 2018. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, Whitehill GD, Clever D, Eil RL, Palmer DC, Mitra S, Rao M, Keyvanfar K, Schrump DS, Wang E, Marincola FM, Gattinoni L, Leonard WJ, Muranski P, Finkel T, Restifo NP, 2016. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab 23, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA, Finkel T, 2015. Measuring In Vivo Mitophagy. Mol Cell 60, 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ, Greaves LC, Kirkwood TB, Turnbull DM, 2003. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest 112, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HC, Van Aken O, 2020. Mitochondrial unfolded protein-related responses across kingdoms: similar problems, different regulators. Mitochondrion 53, 166–177. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG, 2004. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. [DOI] [PubMed] [Google Scholar]
- Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, Ragusa S, Stefanidis E, Ryu D, Rainer PY, Nikitin G, Giger S, Li TY, Semilietof A, Oggier A, Yersin Y, Tauzin L, Pirinen E, Cheng WC, Ratajczak J, Canto C, Ehrbar M, Sizzano F, Petrova TV, Vanhecke D, Zhang L, Romero P, Nahimana A, Cherix S, Duchosal MA, Ho PC, Deplancke B, Coukos G, Auwerx J, Lutolf MP, Naveiras O, 2019. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 24, 405–418 e407. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X, Hu L, Tian S, Wu M, Hui L, Zhang H, Gao P, 2017. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. The EMBO journal 36, 1330–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pickrell AM, Rossi SG, Pinto M, Dillon LM, Hida A, Rotundo RL, Moraes CT, 2013. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum Mol Genet 22, 3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Michikawa Y, Mallidis C, Bai Y, Woodhouse L, Yarasheski KE, Miller CA, Askanas V, Engel WK, Bhasin S, Attardi G, 2001. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proceedings of the National Academy of Sciences of the United States of America 98, 4022–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cheung HH, Tu J, Miu KK, Chan WY, 2016. New insights into the unfolded protein response in stem cells. Oncotarget 7, 54010–54027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Menzies KJ, Auwerx J, 2018. The role of mitochondria in stem cell fate and aging. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J, 2016. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhong X, Cui P, Cai Y, Wang L, He X, Long P, Lu K, Yan R, Zhang Y, Pan X, Zhao X, Li W, Zhang H, Zhou Q, Gao P, 2019. Mitochondrial Dynamics Is Critical for the Full Pluripotency and Embryonic Developmental Potential of Pluripotent Stem Cells. Cell Metab 29, 979–992 e974. [DOI] [PubMed] [Google Scholar]