

Abstract
In all orders of life, cell cycle progression in proliferating cells is dependent on cell growth, and the extent of growth required for cell cycle progression is proportional to growth rate. Thus, cells growing rapidly in rich nutrients are substantially larger than slow-growing cells. In budding yeast, a conserved signaling network surrounding Tor complex 2 (target of rapamycin complex 2; TORC2) controls growth rate and cell size in response to nutrient availability. Here, a search for new components of the TORC2 network identified a pair of redundant kinase paralogues called Ark1 and Prk1. Previous studies found that Ark/Prk play roles in endocytosis. Here, we show that Ark/Prk are embedded in the TORC2 network, where they appear to influence TORC2 signaling independently of their roles in endocytosis. We also show that reduced endocytosis leads to increased cell size, which suggests that cell size homeostasis requires coordinated control of plasma membrane growth and endocytosis. The discovery that Ark/Prk are embedded in the TORC2 network suggests a model in which TORC2-dependent signals control both plasma membrane growth and endocytosis, which would ensure that the rates of each process are matched to each other and to the availability of nutrients so that cells achieve and maintain an appropriate size.
INTRODUCTION
Growth and inheritance are defining features of life. Much is known about inheritance, yet surprisingly little is known about growth. Growth requires myriad biosynthetic processes, all of which must be precisely coordinated with one another and matched to the availability of nutrients. Moreover, the extent of growth must be tightly controlled to ensure that cells maintain an appropriate size.
Not surprisingly, growth is overseen by master regulators that integrate nutrient-dependent signals as well as information from feedback loops to ensure coordination of biosynthetic events. Among the most important master regulators of growth in eukaryotic cells are the Tor kinases, which are assembled into two large multiprotein complexes called TORC1 (target of rapamycin complex 1) and TORC2 (Loewith et al., 2002; Wedaman et al., 2003; González and Hall, 2017). TORC1 is inhibited by rapamycin, a macrolide with potent immunosuppressant activity (Heitman et al., 1991). TORC1 has therefore been extensively studied and has well-established roles in control of ribosome biogenesis, translation, nutrient import, and autophagy (Wullschleger et al., 2006).
Much less is known about TORC2. In budding yeast, TORC2 directly phosphorylates and activates a pair of redundant kinase paralogues called Ypk1 and Ypk2, which are homologues of vertebrate SGK kinases (Casamayor et al., 1999; Kamada et al., 2005; Niles et al., 2012). An important function of Ypk1/2 is to control production of ceramide, a low-abundance lipid that plays roles in signaling (Aronova et al., 2008; Breslow et al., 2010; Roelants et al., 2011; Muir et al., 2014). Ceramide is also a precursor for production of complex ceramides that serve as components of the plasma membrane. Ypk1/2 control production of ceramide in two ways: they stimulate production of sphingolipids that serve as precursors for ceramide synthesis, and they directly activate ceramide synthase, which builds ceramide from the sphingolipid precursors. The TORC2 signaling network includes a poorly understood negative feedback loop in which ceramide-dependent signals appear to inhibit TORC2 signaling (Roelants et al., 2011; Berchtold et al., 2012; Alcaide-Gavilan et al., 2018; Lucena et al., 2018). The TORC2 network may also respond to changes in membrane tension (Riggi et al., 2018).
Recent work has shown that the TORC2 network relays nutrient-dependent signals that influence cell size and growth rate (Alcaide-Gavilan et al., 2018; Lucena et al., 2018). In cells that span all orders of life, the growth rate of proliferating cells is proportional to nutrient availability, and cell size is proportional to growth rate (Schaechter et al., 1958; Hirsch and Han, 1969; Johnston et al., 1977; Ferrezuelo et al., 2012; Leitao and Kellogg, 2017). In budding yeast, the TORC2 network is required for these proportional relationships (Lucena et al., 2018; Leitao et al., 2019). TORC2 signaling is high in rich carbon and low in poor carbon. Moreover, reduced activity of Ypk1/2 leads to a large decrease in cell size, and inhibitors of sphingolipid or ceramide synthesis cause dose-dependent decreases in cell size and growth rate. Mutant cells that cannot make ceramide show a complete failure in nutrient modulation of growth rate and cell size, as well as a failure in nutrient modulation of TORC2 signaling.
A better understanding of the TORC2 network will require identification and characterization of additional components of the network. Here, we searched for additional proteins that play roles in the TORC2 network. A key component of the TORC2 network is Rts1, a conserved regulatory subunit for protein phosphatase 2A. Loss of Rts1 causes a failure in nutrient modulation of cell size, as well as defects in nutrient modulation of TORC2 signaling (Artiles et al., 2009; Lucena et al., 2018; Leitao et al., 2019). Therefore, to identify new components of the TORC2 network, we searched for targets of PP2ARts1. In previous work, we compared the phospho-proteomes of wild-type and rts1∆ cells to identify proteins that undergo large changes in phosphorylation in rts1∆ cells (Zapata et al., 2014). Of the many thousands of phosphorylation events detected by mass spectrometry, a subset of 241 sites on 156 proteins were identified as high-confidence phosphorylation sites that undergo substantial changes in phosphorylation in rts1∆ cells. We queried this data set to identify candidate signaling proteins that could work in the TORC2 network. This led to the identification of a kinase called Ark1 as a candidate component of the TORC2 network. Previous studies found that Ark1 and its redundant paralogue Prk1 play roles in controlling late endocytic events (Cope et al., 1999; Zeng et al., 2001; Toshima et al., 2005). Here, we discovered that Ark1 and Prk1 are also required for normal control of TORC2 signaling.
RESULTS
The Ark1 and Prk1 kinases are required for nutrient modulation of TORC2 signaling
We first tested whether Ark1 and Prk1 are required for normal signaling in the TORC2 network. TORC2 directly phosphorylates Ypk1 and Ypk2 to stimulate their activity (Kamada et al., 2005). A phosphospecific antibody that recognizes a TORC2 site present on both Ypk1 and Ypk2 therefore provides a readout of TORC2 signaling (Niles et al., 2012; Lucena et al., 2018). We found that loss of Ark1 or Prk1 alone caused no effect, but loss of both caused a reduction in TORC2-dependent signaling to Ypk1/2 ( A). Loss of Ark/Prk also caused an increase in the amount of Ypk1 protein, as well as an increase in the electrophoretic mobility of Ypk1. Previous work found that large changes in Ypk1 electrophoretic mobility are due to the activity of Fpk1 and Fpk1, a pair of redundant kinase paralogues that play poorly understood roles in regulation of Ypk1/2 (Roelants et al., 2010).
FIGURE 1:

Ark1 and Prk1 are required for nutrient modulation of TORC2 signaling. (A) Cells of the indicated genotypes were grown to log phase in YPD medium at 30°C, and levels of Ypk-pT662 and Ypk1 protein were assayed by Western blot. An asterisk indicates a background band that overlaps with the Ypk1 signal. Quantification of Ypk-pT662 and total Ypk1 protein are shown. Error bars represent the SD of the mean of three biological replicates. *** indicates a p value smaller than 0.005 compared with the wild type. (B) Cells of the indicated phenotype were grown in YPG/E medium until early log phase, and 2% galactose was added to overexpress Ark1. Cells were collected at the indicated time intervals and levels of Ark1, Ypk-pT662 and Ypk1 protein were assayed by Western blot. An asterisk indicates a background band. (C) Wild-type and ark1∆ prk1∆ cells were grown in YPD medium to early log phase and were then rapidly washed into YPG/E medium at 30°C. Cells were collected at the indicated time intervals, and Western blotting was used to assay levels of Ypk-pT662 and Ypk1 protein. (D) A series of 10-fold dilutions of the indicated strains were grown at 30°C for 2 d on rich (YPD) or poor (YPG/E) nutrient conditions. A small number of colonies that have suppressor mutations appear when ark1∆ prk1∆ cells are grown on YPG/E medium.
We next tested the effects of inhibiting an analogue-sensitive allele of PRK1 in an ark1∆ background (prk1-as ark1∆) (Sekiya-Kawasaki et al., 2003). Inhibition of prk1-as caused a reduction in TORC2 signaling within 60 min, as well as an increase in Ypk1 protein (Supplemental Figure S1A). Inhibition of prk1-as did not cause substantial effects before 60 min. Since analogue-sensitive kinases are rapidly inhibited by PP1 analogues in vivo (Harvey et al., 2005), the data suggest that Ark/Prk are not capable of rapid modulation of TORC2 activity.
We also tested whether overexpression of Ark/Prk influences TORC2 activity. Overexpression of ARK1 from the GAL1 promoter caused a decrease in TORC2-dependent signaling to Ypk1/2, as well as an increase in Ypk1 protein levels (Figure 1B). Thus, overexpression of ARK1 appears to cause dominant negative effects that are similar to the effects caused by loss of Ark/Prk. A previous study found that overexpression of PRK1 is lethal, which suggests that PRK1 overexpression also causes dominant negative effects (Zeng and Cai, 1999).
A protein kinase named Akl1 is a third member of the Ark/Prk kinase family. Analysis of akl1∆ ark1∆ prk1∆ cells showed that akl1∆ did not cause additive effects on TORC2 signaling in ark1∆ prk1∆ cells (Supplemental Figure S1B).
We next tested whether Ark/Prk are required for modulation of TORC2 signaling in response to changes in carbon source. In wild-type cells, a shift from rich carbon (2% glucose) to poor carbon (2% glycerol, 2% ethanol) causes a rapid reduction in TORC2-dependent signaling to Ypk1/2 (Lucena et al., 2018). Over longer times, TORC2 signaling in poor carbon partially recovers, but remains reduced relative to TORC2 signaling in rich carbon. Modulation of TORC2 signaling in response to carbon source failed to occur in ark1∆ prk1∆ cells (Figure 1C). In addition, ark1∆ prk1∆ cells failed to proliferate on poor carbon media, consistent with a role for Ark/Prk in the TORC2 signaling network that influences the response to changes in carbon source (Figure 1D).
Together, these observations suggest that Ark/Prk execute functions that are required for normal functioning of the TORC2 network.
Ark1 and Prk1 respond to nutrient-dependent signals that modulate TORC2 activity
The TORC2 network responds rapidly to changes in carbon source (Lucena et al., 2018). Therefore, to further test whether Ark1 and Prk1 play roles in TORC2 signaling, we tested whether their phosphorylation state or abundance responds rapidly to changes in carbon source. We first tested whether Ark1 and Prk1 are phosphoproteins. Multiple forms of both Ark1-3XHA and Prk1-6XHA were detected by Western blot. Treatment with phosphatase caused the forms to collapse into one band, confirming that Ark1 and Prk1 are phosphorylated (Figure 2A).
FIGURE 2:

Ark1 and Prk1 respond to nutrient-dependent signals. (A) Extracts from Ark1:3xHA cells and Prk1:6xHA cells were treated with lambda-phosphatase and analyzed by Western blot. (B) Ark1:3xHA cells and Prk1:6xHA cells were grown at room temperature in YPD medium to early log phase and were then rapidly washed into YPG/E medium. The behavior of Ark1:3xHA and Prk1:6xHA was assayed by Western blot with anti-HA antibody. (C) Ark1:3xHA cells and Prk1:6xHA cells were grown overnight at room temperature in YPD or YPG/E medium and the behavior of Ark1:3xHA and Prk1:6xHA was assayed by Western blot. The asterisk indicates a nonspecific band.
We next shifted wild-type cells from rich to poor carbon and assayed the behavior of Ark1 and Prk1 by Western blot (Figure 2B). A fraction of Ark1 underwent rapid phosphorylation in response to poor carbon, which was followed by a gradual decrease in protein levels over 2 h. Upon prolonged incubation in poor carbon, a fraction of Ark1 persisted in a hyperphosphorylated state and the amount of protein remained reduced (Figure 2C). Prk1 also underwent rapid hyperphosphorylation in response to poor carbon but showed no change in protein levels. Phosphorylation of Prk1 appeared to return to the starting baseline by the end of the time course. The rapid changes in the phosphorylation state of Ark1 and Prk1 occurred with a timing that was similar to the timing of changes in TORC2 signaling (Figure 1B and Lucena et al., 2018).
Ark1 and Prk1 carry out functions in the TORC2 signaling network
To further investigate roles of Ark1 and Prk1 in the TORC2 signaling network, we tested for functional interactions with components of the network. A simplified model that summarizes proposed functional relationships in the TORC2 network is shown in Figure 3A. Signaling in the network is strongly influenced by feedback loops. Thus, ceramides produced in response to signals from Ypk1/2 relay poorly understood signals that inhibit TORC2 signaling. The simplified model does not capture all of the data from previous studies. Nevertheless, it provides a framework for interpreting data regarding functional interactions in the network.
FIGURE 3:

Ark1 and Prk1 carry out functions in the TORC2 signaling network. (A) Simplified model of the TORC2 signaling network. (B) Series of 10-fold dilutions of the indicated strains was grown at 30°C in the presence or absence of 0.1 µg/ml myriocin. (C) The indicated strains were grown at room temperature in YPD medium to early log phase, and cells were collected before and after 1 h after addition of 50 µM 3MOB-PP1. Ark1:3xHA, Prk1:6xHA, Ypk-pT662, and Ypk1 proteins were analyzed by Western blot. (D) Ark1:3xHA and Prk1:6xHA cells were grown at room temperature in YPD medium to early log phase. Myriocin (0.5 µg/ml) or DMSO (control) was added, and cells were collected at the indicated times to analyze Ark1:3xHA, Prk1:6xHA, Ypk-pT662, and Ypk1 proteins by Western blot. (E) Ark1:3xHa and Prk1:6xHA cells were grown at room temperature in YPD medium to early log phase. Palmitoylcarnitine (PalmC) (20 µM) was added at the indicated time points, and Ark1:3xHA, Prk1:6xHA, Ypk-pT662, and Ypk1 proteins were analyzed by Western blot.
We first tested for genetic interactions with Ypk1 and Ypk2. Loss of Ypk2 causes little or no phenotype, while loss of Ypk1 causes a reduced rate of proliferation and a large reduction in cell size (Lucena et al., 2018). We were unable to recover ypk1∆ ark1∆ prk1∆ triple mutants. We also found that ark1∆ prk1∆ cells are hypersensitive to myriocin, an inhibitor of serine palmitoyltransferase, which is stimulated by Ypk1/2 and catalyzes the first step in production of sphingolipids that are required for normal signaling in the TORC2 network (Figure 3B).
We next tested whether inhibition of Ypk1/2 causes effects on Ark1 or Prk1. To do this, we used an analogue-sensitive allele of Ypk1 in a ypk2∆ background (ypk1-as ypk2Δ) (Sun et al., 2012). Addition of analogue inhibitor to ypk1-as ypk2∆ cells caused an increase in Ark1 protein levels and a change in Prk1 phosphorylation within 1 h (Figure 3C). Changes were not observed at shorter time points. Phos-tag gels that provide better resolution of phosphorylated forms revealed that phosphorylation of Ark1 was reduced in ypk1-as ypk2∆ cells (Supplemental Figure S2A). The reduced phosphorylation of Ark1 in ypk1-as ypk2∆ cells was observed even in the absence of analogue inhibitor. Previous studies have shown that analogue-sensitive kinases can have reduced activity in the absence of inhibitor (Bishop et al., 2000). Addition of analogue inhibitor to ypk1-as ypk2∆ cells caused further loss of phosphorylated forms of Ypk1. A dephosphorylated form of Ark1 was observed in the ypk1-as ypk2∆ cells that was not observed in wild-type cells (marked with an asterisk in Supplemental Figure S2A). We were not able to obtain clear results for Prk1 on Phos-tag gels.
Ypk1/2 promotes synthesis of ceramide from sphingolipid precursors, and ceramide is required for feedback signals that repress TORC2 signaling (Roelants et al., 2011; Muir et al., 2014; Lucena et al., 2018). Therefore, we next tested how inhibiting production of sphingolipids and ceramide influences the Ark/Prk proteins. Addition of myriocin to cells caused an increase in Ark1 protein levels (Figure 3D) and a decrease in Ark1 phosphorylation (Supplemental Figure S2B), similar to the effects caused by ypk1-as ypk2∆. However, there were two differences between the effects of myriocin and the effects of ypk1-as ypk2∆. First, the effects of myriocin took longer to appear. Second, ypk1-as ypk2∆ caused the appearance of a dephosphorylated form of Ark1 that was not observed in cells treated with myriocin (Supplemental Figure S2B). These differences suggest that myriocin and inhibition of Ypk1/2 may influence different or overlapping sets of phosphorylation sites. For example, Ypk1/2 could influence Ark1 phosphorylation via fast signaling that acts on one set of sites, and via slow signaling that influences different or overlapping sites and works more indirectly through the sphingolipid-dependent feedback loop that is inhibited by myriocin.
To further test whether components of the TORC2 network influence signaling to Ark/Prk, we used palmitoylcarnitine, a long-chain fatty acid derivative of carnitine that was previously shown to cause a rapid decrease in TORC2 activity (Riggi et al., 2018). We found that palmitoylcarnitine caused rapid loss of TORC2 activity, as well as a decrease in Ark1 protein levels and increased phosphorylation of Prk1 (Figure 3E). Thus, the effects of palmitoylcarnitine were similar to the effects of a shift to poor nutrients, consistent with a role for the TORC2 network in modulating the functions of Ark/Prk. However, palmitoylcarnitine did not cause the hyperphosphorylation of Ark1 that was observed upon a shift from rich to poor carbon (Figure 2B).
We also tested for functional relationships with PP2ARts1. Loss of Rts1 causes increased signaling in the TORC2 network (Lucena et al., 2018). Here, we found that increased TORC2 signaling in rts1∆ cells is dependent on Ark1 and Prk1 (Figure 4A). We further found that rts1∆ enhanced the slow proliferation phenotype caused by ark1∆ prk1∆ (Figure 4B).
FIGURE 4:

Ark1 and Prk1 show genetic interactions with Rts1. (A) Cells of the indicated genotypes were grown at room temperature to log phase in YPD medium. Ypk-pT662 and total Ypk1 were assayed by Western blot. An asterisk indicates a background band that overlaps with the Ypk1 signal. Quantifications of Ypk-T662 phosphorylation and total Ypk1 protein are shown. Error bars represent the SD of the mean of three biological replicates. *** indicates a p value smaller than 0.005; ** indicates a p value between 0.01 and 0.005 when the indicated strain is compared with the wild type. (B) A series of 10-fold dilutions of the indicated strains were grown at 25°C on YPD medium. (C) Wild-type and rts1∆ cells were grown to log phase in YPD medium, and phosphorylation of Ark1:3xHA and Prk1:6xHA was assayed by Phos-tag Western blot. (D) Wild-type and ark1∆ prk1∆ kinase mutants were grown at 30°C in YPD medium and were then shifted to YPG/E medium. Rts1 was detected by Western blotting.
Since our previous mass spectrometry analysis suggested that Ark1 could be a target of PP2ARts1, we also analyzed the phosphorylation state of Ark1 and Prk1 in rts1∆ cells. No change in phosphorylation was observed by normal Western blot in log-phase cells or in nutrient-shifted cells (Supplemental Figure S3, A and B). However, analysis of phosphorylation using Phos-tag gels in rapidly growing cells and nutrient-shifted cells showed a loss of Ark1 phosphorylation in rts1∆ cells, consistent with the mass spectrometry data (Figure 4C and Supplemental Figure S2C). No changes in phosphorylation of Prk1 protein that could be detected by standard SDS–PAGE were observed.
In previous work, we found that Rts1 undergoes TORC2-dependent hyperphosphorylation in response to a shift to poor carbon (Alcaide-Gavilan et al., 2018). Here, we found that hyperphosphorylation of Rts1 in response to poor carbon was strongly reduced in ark1∆ prk1∆ cells (Figure 4D). The effects of ark1∆ prk1∆ on Rts1 hyperphosphorylation are similar to the effects caused by inactivation of Ypk1/2 or other components of the TORC2 network (Alcaide-Gavilan et al., 2018).
Elm1, a budding yeast homologue of vertebrate LKB1 kinase, is required for normal levels of TORC2 signaling and for nutrient modulation of TORC2 signaling (Alcaide-Gavilan et al., 2018). We therefore tested whether Elm1 is required for nutrient-dependent signaling to Ark1 and Prk1. We found that phosphorylation of Ark1 and Prk1 in response to poor carbon was not dependent on Elm1 (Supplemental Figure S3, D and E). We also found that elm1∆ is synthetically lethal with ark1∆ prk1∆, which suggests that Elm1 could promote TORC2 signaling via a pathway that works in parallel with Ark/Prk.
Together, the data establish that Ark/Prk show complex functional interactions with multiple components of the TORC2 signaling network. To summarize, poor nutrients and palmitoylcarnitine, which both cause decreased TORC2 signaling, also cause a decrease in Ark1 protein levels. Conversely, myriocin and inhibition of Ypk1/2 both cause increased levels of the Ark1 protein, and both of these conditions are thought to cause increased TORC2 signaling due to loss of negative feedback (Roelants et al., 2011; Lucena et al., 2018). Thus, Ark1 protein levels are correlated with TORC2 signaling. Similarly, Ark1 phosphorylation increases in poor nutrients (low TORC2 activity) and decreases in myriocin-treated cells, rts1∆ cells, and ypk1-as ypk2∆ cells (high TORC2 activity). However, although palmitoylcarnitine caused a decrease in TORC2 signaling, it did not cause an increase in Ark1 phosphorylation. Thus, Ark1 phosphorylation is less correlated with TORC2 signaling. Prk1 phosphorylation changed in response to conditions that alter TORC2 signaling, but it was difficult to define a clear correlation with the level of TORC2 signaling. The complexity of TORC2 signaling, as well as our limited understanding of signaling mechanisms within the TORC2 network, make it difficult to fit these observations into a simple and consistent model (see Discussion).
Cell size defects in ark1∆ prk1∆ cells are likely caused by defects in endocytosis
Ark/Prk kinases are known to regulate endocytic vesicle uncoating via phosphorylation of multiple coat proteins, including Pan1, Ent1, and Sla1 (Watson et al., 2001; Zeng et al., 2001). Previous work has shown that endocytic proteins Ent1 and Pan1 are TORC2/Ypk1 effectors (Rispal et al., 2015). Since ark1∆ prk1∆ cells are abnormally large, we reasoned that their increased size could be due to defects in TORC2 signaling, defects in endocytosis, or both. In previous work, we showed that defects in TORC2 signaling cause defects in nutrient modulation of cell size, as well as defects in cell size modulation in response to sublethal doses of myriocin (Lucena et al., 2018). However, the discovery that ark1∆ prk1∆ cells fail to proliferate in poor carbon sources or in the presence of low doses of myriocin made it impossible to test whether they show normal cell size responses to these conditions.
To determine whether the large size of ark1∆ prk1∆ cells could be due to defects in endocytosis, we tested whether reduced endocytosis causes increased cell size. Ent1 and Ent2 are redundant paralogues that drive early steps in endocytosis (Wendland et al., 1999). To inactivate Ent1/2, we generated an auxin-inducible (AID) degron version of ENT2 in an ent1∆ background (ent2-AID ent1∆). The ent2-AID ent1∆ cells were viable in the absence of auxin and failed to proliferate in the presence of 15 µM auxin (Supplemental Figure S4A). The ent2-AID protein disappeared within 10 min of adding auxin (Supplemental Figure S4B).
In the absence of auxin, ent2-AID ent1∆ cells were abnormally large, similar to the increase caused by ark1∆ prk1∆ (Figure 5A). Sublethal doses of auxin caused ent2-AID ent1∆ cells to become even larger, suggesting that reduced endocytosis strongly influences cell size. Thus, the large size of ark1∆ prk1∆ cells may be due largely to defects in endocytosis. Previous studies have shown that increased cell size can be a consequence of excessive cell growth during prolonged cell cycle delays. However, analysis of cell growth and cell cycle progression in synchronized ent2-AID ent1∆ cells showed that reduced activity of Ent1/2 caused prolonged cell cycle delays, yet also caused reduced growth rate (Figure 5B). Increased cell size caused by decreased endocytosis may therefore be the outcome of defects that accumulate over multiple cell cycles. One explanation could be that the cell size defects arise because exocytic events that drive plasma membrane growth are no longer properly balanced with endocytosis.
FIGURE 5:
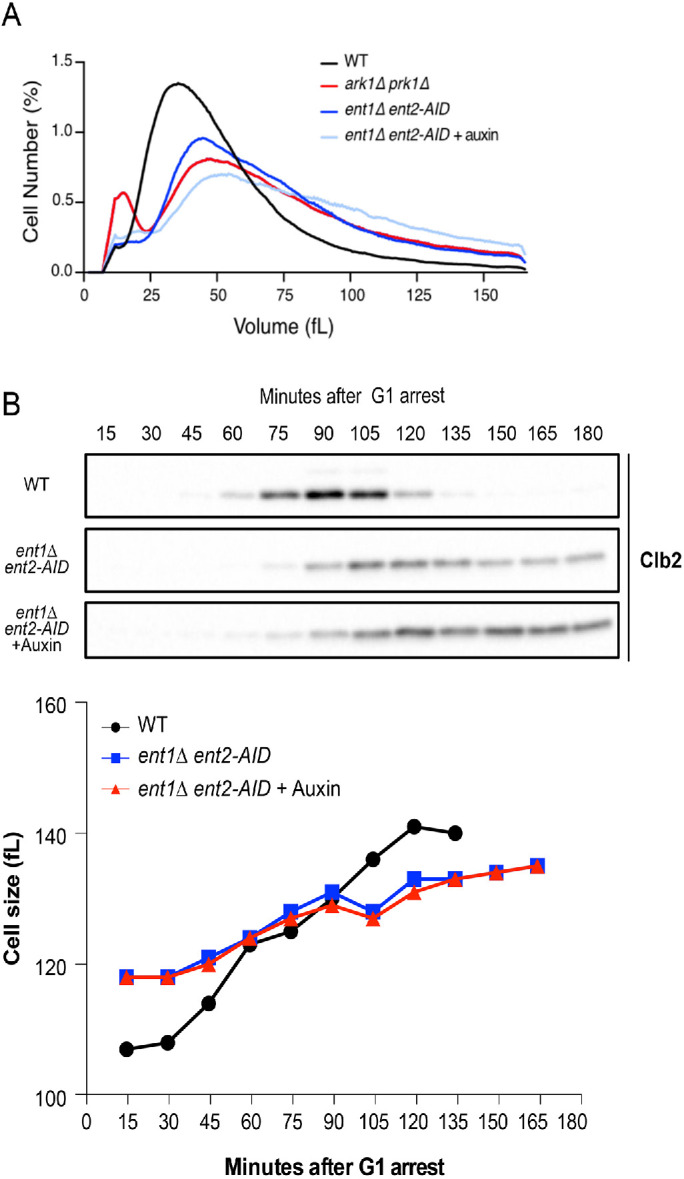
Cell size defects in ark1∆ prk1∆ cells are likely caused by defects in endocytosis. (A) Cells of the indicated genotypes were grown to log phase at 25°C and cell size distributions were determined using a Coulter counter. (B) ent2-AID ent1∆ cells were grown in the absence or presence of 5 µM auxin. Each plot is the average of three biological replicates. For each biological replicate, three technical replicates were analyzed and averaged.
The effects of Ark/Prk on TORC2 signaling are unlikely to be a consequence of reduced endocytosis
Since Ark/Prk are required for normal endocytosis, we tested whether defects in TORC2 signaling caused by ark1∆ prk1∆ could be a consequence of defects in endocytosis. To do this, we examined how mutants in several endocytic proteins that are thought to be targets of Ark/Prk influence TORC2 signaling. We first tested the effects of loss of function of Ede1, which is required for efficient execution or early endocytic steps. Previous work found that ede1∆ causes a 35% reduction in endocytosis (Gagny et al., 2000). We found that ede1∆ had no effect on the level of TORC2 signaling in unsynchronized cells growing in rich carbon but caused an increase in the amount of Ypk1 protein (Figure 6A). We also found that ede1∆ had no effect on the response of the TORC2 network to a shift from rich to poor carbon (Figure 6B).
FIGURE 6:

Early and intermediate endocytic mutants have no effects on TORC2 signaling. (A) Wild-type and ede1∆ cells were grown at 30°C to log phase in YPD medium, and Ypk-pT662 and Ypk1 protein were assayed by Western blot. Quantification of Ypk-T662 phosphorylation and total Ypk1 protein are shown. (B) Wild-type and ede1∆ cells were grown in YPD medium to early log phase and were then rapidly washed into YPG/E medium at 30°C. Cells were collected at the indicated time intervals, and levels of Ypk-pT662 and Ypk1 protein were assayed by Western blot. An asterisk indicates a background band that overlaps with the Ypk1 signal. (C) Cells of the indicated genotypes were grown at 30°C to log phase in YPD medium. Auxin (1 mM) was added, and cells were collected and analyzed by Western blot at the indicated time points. Quantification of Ypk-T662 phosphorylation and total Ypk1 protein are shown. An asterisk indicates a background band that overlaps with the Ypk1 signal. (D) Cells of the indicated genotypes were grown in YPD medium to early log phase. Auxin (1 mM) was added, and after 30 min cells were washed into YPG/E medium at 30°C containing 1 mM auxin. Cells were collected at the indicated time intervals, and Ypk-pT662 and Ypk1 were assayed by Western blot. An asterisk indicates a background band that overlaps with the Ypk1 signal. Error bars represent the SD of the mean of three biological replicates. *** indicates a p value smaller than 0.005 and * indicates a p value between 0.05 and 0.01 when the indicated strain is compared with the wild type .
We next tested essential components of the endocytic machinery. Inactivation of Ent1/2 in rapidly growing cells had no effect on the level of TORC2-dependent phosphorylation of Ypk1/2 but did cause an increase in levels of the Ypk1 protein (Figure 6C). Growing ent2-AID ent1∆ cells overnight in the presence of a low nonlethal dose of auxin that caused an increase in cell size (Figure 5) had no effect on TORC2 signaling (Supplemental Figure S4A). Moreover, inactivation of Ent1/2 30 min before a shift to poor carbon had no effect on modulation of TORC2 signaling in response to a shift to poor carbon (Figure 6D). Finally, we observed that ede1∆ and ent2-AID ent1∆ mutants are resistant to myriocin (Supplemental Figure S5B). In contrast, ark1∆ prk1∆ cells are highly sensitive to myriocin (Figure 3B).
A key target of Ark/Prk in the endocytic pathway is Pan1, an epsin-like protein that promotes polymerization of actin to help drive formation and internalization of endocytic vesicles. There are 19 potential Ark/Prk phosphorylation sites in Pan1, and it has been demonstrated that Ark/Prk directly phosphorylate and inhibit Pan1 (Toshima et al., 2005, 2016). Since Pan1 is an essential player in the last steps of endocytosis, we tested whether loss of Pan1 could have an effect on TORC2 signaling. We used an aAID version of Pan1 to analyze how inactivation of Pan1 influences TORC2 signaling (Bradford et al., 2015). Inactivation of pan1-AID did not have a significant effect on TORC2 signaling in cells growing in rich carbon or in cells shifted from rich to poor carbon (Figure 7, A and B). Inactivation of pan1-AID caused increased levels of Ypk1 protein (Figure 7A).
FIGURE 7:

Pan1 mutants do not show significant defects in TORC2 signaling. (A) Wild-type and pan1-AID cells were grown at 30°C to log phase in YPD medium. Auxin (1 mM) was added, cells were collected at the indicated intervals, and Ypk-pT662 and Ypk1 protein levels were analyzed by Western blot. Quantification of Ypk-T662 phosphorylation and total Ypk1 protein are shown. (B) Wild-type and pan1-AID cells were grown in YPD medium to early log phase. Auxin (1 mM) was added, and after 30 min cells were washed into YPG/E medium at 30°C containing 1 mM auxin. Cells were collected at the indicated intervals, and Ypk-pT662 and Ypk1 protein levels were assayed by Western blot. (C) Cells of the indicated genotypes were grown in YPD medium to early log phase at 30°C, and Ypk-pT662 and Ypk1 protein were assayed by Western blot. An asterisk indicates a background band in the Ypk1 blot that overlaps with the Ypk1 signal. Error bars represent the SD of the mean of three biological replicates. *** indicates a p value smaller than 0.005, and ** indicates a p value between 0.01 and 0.005 when the indicated strain is compared with the wild type. * indicates a p value between 0.05 and 0.01.
We next considered the possibility that the effects of ark1∆ prk1∆ on TORC2 signaling are due to hyperactivity of Pan1. Previous work found that a phospho-site mutant of Pan1 that cannot be phosphorylated by Ark/Prk (pan1-18TA) phenocopies the effects of ark1∆ prk1∆ on actin organization and endocytosis (Toshima et al., 2016). We found that the pan1-18TA mutant had no effect on TORC2 signaling or total Ypk1 protein (Figure 7C).
Since ark1∆ prk1∆ do not proliferate on poor carbon (Figure 1C), we tested whether endocytic mutants influence growth on poor carbon. However, we found that ede1∆ and pan1-18TA had no effect on proliferation on poor carbon (Figure S5, C and E). Growth of ent2-AID ent1∆ or pan1-AID in the presence of sublethal doses of auxin also had no effect on proliferation in poor carbon (Supplemental Figure S5, B and D).
Together, the data suggest that Ark/Prk influence the level of TORC2 signaling independently of their role in endocytosis. Furthermore, defects in endocytosis, caused by loss of Ark/Prk or endocytic proteins, cause increased levels of Ypk1 protein.
DISCUSSION
Previous studies suggested that Ark1 and Prk1 function at a late stage of endocytosis to inhibit actin polymerization and promote disassembly of endocytic coat proteins (Cope et al., 1999; Watson et al., 2001; Zeng et al., 2001; Henry et al., 2003; Toshima et al., 2005, 2016). Here, we show that Ark/Prk are also required for normal control of TORC2 signaling. Loss of Ark/Prk causes reduced TORC2 signaling, as well as a failure in modulation of TORC2 signaling in response to carbon source. Ark/Prk also show strong genetic interactions with multiple components of the TORC2 signaling network. Thus, ark1∆ prk1∆ are synthetically lethal with loss of YPK1 or ELM1, both of which are required for normal levels of TORC2 signaling. In addition, ark1∆ prk1∆ cells are exquisitely hypersensitive to myriocin, which inhibits production of sphingolipids that relay signals in the TORC2 network. Ark/Prk show functional interactions with Rts1, a regulatory subunit of PP2A that controls TORC2 signaling. Loss of Rts1 causes increased signaling in the TORC2 network that is dependent on Ark/Prk, and normal control of Rts1 phosphorylation is also dependent on Ark/Prk.
Analysis of changes in Ark/Prk phosphorylation and abundance provided further evidence that Ark1 and Prk1 play roles in the TORC2 network. Ark1 undergoes rapid and sustained changes in both abundance and phosphorylation in response to a switch from rich to poor carbon that drives a rapid decrease in TORC2 signaling. Prk1 also shows changes in phosphorylation in response to a shift to poor carbon. Multiple components of the TORC2 network are required for normal control of the phosphorylation state and abundance of Ark/Prk.
A model that summarizes functional relationships that we observed between the TORC2 network and Ark/Prk is shown in Figure 8. However, it is not yet possible to fit the data into a simple model that is consistent with all the observations. Interpretation of the data is complicated by the presence of feedback loops and by our limited understanding of signaling mechanisms within the TORC2 network. In addition, proteins like Ypk1/2 could have multiple distinct functions at different locations with the cell, which could lead to complex genetic interactions. Furthermore, myriocin, Ypk1/2, and Rts1 all influence phosphorylation of Ark1, yet they appear to work on different time scales and cause substantially different effects on the pattern of Ark1 phosphorylation, which suggests that they influence different or partially overlapping sets of phosphorylation sites. Finally, data from previous studies have not yet yielded a clear picture of signaling within the TORC2 network that would provide a strong foundation for building models. For example, there are data that suggest that TORC2-dependent signals activate PP2ARts1 (Lucena et al., 2018), as well as data that suggest that TORC2-dependent signals inhibit PP2ARts1 (Alcaide-Gavilan et al., 2018). Distinguishing models will require a more detailed mechanistic understanding of how signals are relayed in the TORC2 network.
FIGURE 8:
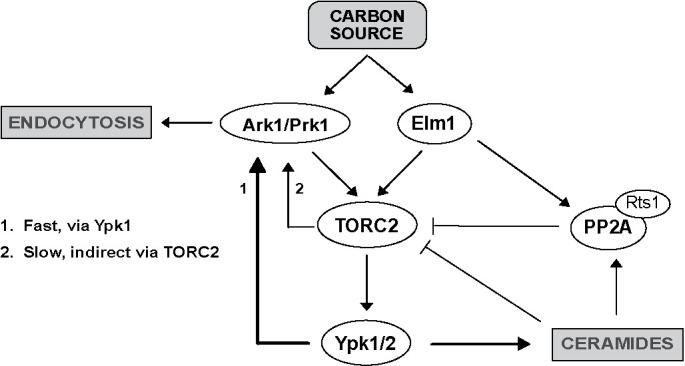
Model summarizing hypothesized functional interactions between Ark-family kinases and the TORC2 signaling network.
A number of observations suggest that Ark/Prk do not influence TORC2 signaling solely via their roles in endocytosis. For example, loss of Ark/Prk causes decreased TORC2 signaling and increased Ypk1 levels, whereas inhibition of endocytosisonly causes increased Ypk1 levels. In addition, a Pan1 phosphorylation site mutant that lacks all of the sites targeted by Ark/Prk phenocopies the effects of ark1∆ prk1∆ on endocytosis yet causes no effects on TORC2 signaling. Finally, the inviability of ark1∆ prk1∆ cells on poor carbon is difficult to explain if one assumes that Ark/Prk function solely in endocytosis, since there is no evidence that efficient endocytosis is required for survival on poor carbon.
An interesting possibility is that Ark/Prk are embedded in the TORC2 network, where they influence TORC2 signaling while also relaying TORC2-dependent signals that influence endocytosis. Previous studies found that TORC2-dependent signals are required for endocytosis. For example, inhibition of TORC2 activity causes a rapid block to endocytosis (Rispal et al., 2015). Similarly, inhibiting the synthesis of sphingolipids that are required for TORC2-dependent signals causes a rapid block to endocytosis (Zanolari et al., 2000). Our discovery that reduced endocytosis leads to increased cell size suggests that plasma membrane growth and endocytosis must be precisely coordinated to ensure that the two processes are appropriately balanced to achieve and maintain an appropriate cell size. Furthermore, when cells are shifted from rich to poor carbon, the rates of biosynthetic processes that drive plasma membrane growth decrease, so it is likely that the rate of endocytosis must also be decreased to ensure that cells reach an appropriate size. Having common TORC2-dependent signals control both membrane growth and endocytosis could ensure that the rates of each process are appropriately matched to each other and to the availability of nutrients so that cells maintain an appropriate size.
MATERIALS AND METHODS
Yeast strains and media
All strains are in the W303 background (leu2-3,112 ura3-1 can1-100 ade2-1 his3-11,15 trp1-1 GAL+ ssd1-d2), with the exception of strains DDY903, DDY2544, and JJTY0888 used in Figure 7C and Supplemental Figure S5E, which is in the S288C background (his3-∆200, ura3-52, lys2-801, leu2-3, 112). Table 1 shows additional genetic features. One-step PCR-based gene replacement was used for making deletions and adding epitope tags at the endogenous locus (Longtine, 1998; Janke et al., 2004). Cells were grown in YP medium (1% yeast extract, 2% peptone, 40 mg/l adenine) supplemented with 2% dextrose (YPD), 2% glycerol/ethanol (YPG/E), or 2% galactose (YPGal). For nutrient shifts, cells were grown in YPD medium overnight to log phase. Cells were then washed three times with YPG/E medium and resuspended in YPG/E medium.
Table 1.
Strains used in this study.
| Strain | MAT | Genotype | Source |
|---|---|---|---|
| DK186 | a | bar1 | Altman and Kellogg, 1997 |
| DDY903 | a | S288C background | Gift of David Drubin |
| DK3416 | a | bar1 ark1Δ::HIS | This study |
| DK3414 | a | bar1 prk1Δ::kanMX6 | This study |
| DK3460 | a | bar1 ark1Δ::HIS prk1Δ::kanMX6 | This study |
| DDY2544 | a | his3 -Δ200 leu2-3 , 112 ura3-52 ark1Δ ::HIS3 prk1Δ ::URA3 | Drubin Lab |
| DK3769 | a | bar1 ark1-3xHA::HIS | This study |
| DK3771 | a | bar1 prk1-6xHA::HIS | This study |
| DK3862 | a | bar1 ark1-3xHA::HIS ypk1-as (L424A) ypk2Δ::HIS3 | This study |
| DK3863 | a | bar1 prk1-6xHA::KanMX ypk1-as (L424A) ypk2Δ::HIS3 | This study |
| DK647 | a | bar1 rts1Δ::kanMX6 | Artiles et al., 2009 |
| DK3495 | α | bar1 ark1Δ::HIS prk1Δ::kanMX6 rts1Δ::NatNT2 | This study |
| DK3770 | a | bar1 ark1-3xHA::HIS rts1Δ::kanMX6 | This study |
| DK3772 | a | bar1 prk1-6xHA::HIS rts1Δ::kanMX6 | This study |
| DK1853 | a | bar1 ede1Δ::kanMX6 | This study |
| DK3699 | a | bar1 ent1Δ::NatNT2 ent2-AID::KanMX pTIR4::LEU | This study |
| DK3815 | α | bar1 ADH1-OsTIR1-9myc::URA3 ade2::ADE2 HisΔ::URA Pan1-AID::G418 | Bradford et al., 2015 |
| JJTY0888 | a | his3-Δ200 leu2-3, 112 ura3-52 lys2-801 pan1Δ::pan1-18TA::LEU2 | Toshima et al., 2016 |
| DK3935 | a | bar1 ark1Δ::KanMX6 prk1-as::URA | This study |
| DK3400 | α | bar1 akl1Δ::HYG | This study |
| DK3421 | a | bar1 ark1Δ::HIS akl1Δ::Hyg | This study |
| DK3464 | a | bar1 prk1Δ::KanMX6 akl1Δ:: Hyg | This study |
| DK3462 | a | bar1 ark1Δ::HIS akl1Δ::Hyg prk1Δ::kanMX6 | This study |
| DK3832 | a | bar1 elm1Δ::TRP ark1-3xHA::HIS | This study |
| DK3116 | a | bar1 ypk1Δ::HIS | Lucena et al., 2018 |
| DK2546 | a | bar1 ypk2Δ::HIS | Lucena et al., 2018 |
Myriocin (Sigma) was dissolved in 100% methanol to make a 500 µg/ml stock solution. The same volume of methanol was added in control experiments. We observed significant differences in the effective concentration of myriocin in different batches from the same supplier. Palmitoylcarnitine (Sigma) was dissolved in 100% dimethyl sulfoxide (DMSO) to make a 5 mM stock solution.
Production of polyclonal Ypk1 antibody
An antibody that recognizes Ypk1 was generated by immunizing rabbits with a fusion protein expressed in bacteria. Briefly, a PCR product that includes the full-length open reading frame for Ypk1 was cloned into the entry vector pHis::parallel. The resulting plasmid expresses Ypk1 fused at its N-terminus to 6XHis-TEV. The 6XHis-TEV-Ypk1 fusion was expressed in Rosetta cells and purified using standard procedures, yielding 80 mg of protein from 3 l of bacterial culture. A milligram of the purified protein was used to immunize a rabbit. The 6XHis-TEV-Ypk1 fusion protein was coupled to Affigel 10 (Bio-Rad Laboratories) to create an affinity column for purification of the antibody. The specificity of the antibody was tested by Western blot using samples from wild-type, ypk1∆, and ypk2∆ strains (Supplemental Figure S6).
Western blotting
For Western blots using cells growing in early log phase, cultures were grown overnight at the specified temperature to an OD600 of less than 0.8. After adjusting optical densities to normalize protein loading, 1.6 ml samples were collected and centrifuged at 13,000 rpm for 30 s. The supernatant was removed and 150 µl of glass beads was added before freezing in liquid nitrogen.
To analyze cells shifted from rich to poor nutrients, cultures were grown in YPD medium overnight at 25°C to an OD600 of less than 0.8. After adjusting optical densities to normalize protein loading, cells were washed three times with the same volume of YPG/E medium and then incubated at 30°C in YPG/E medium for the time course. Samples (1.6 ml) were collected at each time point.
Cells were lysed into 140 μl of sample buffer (65 mM TrisHCl, pH 6.8, 3% SDS, 10% glycerol, 50 mM NaF, 100 mM β-glycerophosphate, 5% 2mercaptoethanol, and bromophenol blue). Phenylmethylsulfonyl fluoride (PMSF) was added to the sample buffer to 2 mM immediately before use. Cells were lysed in a Mini-bead-beater 16 (BioSpec) at top speed for 2 min. The samples were removed and centrifuged for 15 s at 14,000 rpm in a microfuge and placed in boiling water for 5 min. After being boiled, the samples were centrifuged for 5 min at 15,000 rpm and loaded on an SDS–polyacrylamide gel.
Samples were analyzed by Western blot as previously described (Harvey et al., 2011). SDS–PAGE gels were run at a constant current of 20 mA, and electrophoresis was performed on gels containing 10% polyacrylamide and 0.13% bis-acrylamide. Proteins were transferred to nitrocellulose using a Trans-Blot Turbo system (Bio-Rad Laboratories). Blots were probed with primary antibody overnight at 4°C. Proteins tagged with the human influenza hemagglutinin (HA) epitope were detected with the 12CA5 anti-HA monoclonal antibody (gift of David Toczyski, University of California, San Francisco) or an anti-HA polyclonal antibody developed in our lab. Rabbit anti–phospho-T662 antibody (gift of Ted Powers, University of California, Davis) was used to detect TORC2-dependent phosphorylation of YPK1/2 at a dilution of 1:20,000 in TBST (10 mM Tris-Cl, pH 7.5, 100 mM NaCl, and 0.1% Tween 20) containing 3% milk. Total Ypk1 was detected using anti-Ypk1 antibody (Santa Cruz Biotechnology; catalogue number sc-12051, not produced anymore) at a dilution of 1:2000 or the Ypk1 polyclonal antibody previously described. V5 Tag monoclonal antibody (E10/V4RR; Thermo-Fisher MA5-15253) was used to detect ent2-AID at 1:2500 in phosphate-buffered saline containing 3% milk.
For Phos-tag Western blots, cells were lysed by bead beating into sample buffer without phosphatase inhibitors. After cell lysis, samples were centrifuged for 1 min at 13,000 rpm at 4°C and quickly placed in a boiling water bath for 5 min. Samples were loaded into 10% SDS–PAGE gels supplemented with 60 mM Phos-t and 120 mM MnCl2. To prepare Phos-tag gels, the gel mixture was degassed for 1 min prior to the addition of N,N,N′,N′-tetramethylethylenediamine (TEMED), and polymerization was allowed to occur for 1–2 h at room temperature followed by overnight at 4°C. Gels were run at 10 mA for 6 h until a 29-kDa marker was at the bottom of the gel. The gel was incubated for 10 min in transfer buffer supplemented with 2 mM EDTA, followed by a second incubation without EDTA. Gels were transferred onto nitrocellulose via the Trans-Blot Turbo Transfer System (Bio-Rad).
All blots were probed with a horseradish peroxidase (HRP)-conjugated donkey anti-rabbit secondary antibody (GE Healthcare; catalogue number NA934V), HRP-conjugated donkey anti-mouse antibody (GE Healthcare; catalogue number NXA931), or HRP-conjugated donkey anti-goat (Santa Cruz Biotechnology; catalogue number sc-2020) for 45–90 min at room temperature. Secondary antibodies were detected via chemiluminescence with Advansta ECL reagents.
Densitometric quantification of Western blot signals was performed using ImageJ (Schneider et al., 2012). Quantification of Ypk-pT662 phosphorylation was calculated as the ratio of the phosphospecific signal over the total Ypk1 protein signal, with wild-type signal normalized to a value 1. At least three biological replicates were analyzed and averaged to obtain quantitative information.
Lambda phosphatase treatment
Lambda phosphatase treatment of Ark1 and Prk1 in cell extracts was carried out as previously described (Lucena et al., 2017).
Statistical analysis
A minimum of three independent biological replicates were performed for each experimental condition. For the statistical analyses, a one-tailed unpaired t test was performed using Prism 5 (GrahPad). p values are described in the figure legends.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplementary Material
Acknowledgments
We thank members of the laboratory for advice and support. We also thank Ted Powers (University of California, Davis) for the Ypk-pT662 phosphospecific antibody, David Toczyski (University of California, San Francisco) for 12CA5 antibody, and Kevan Shokat (University of California, San Francisco) for providing 1NA-PP1 and 3MOB-PP1. We are grateful to Yidi Sun (University of California, Berkeley) and Beverly Wendland (Johns Hopkins University) for strains and Carrie Partch (University of California, Santa Cruz) for pHis::parallel plasmid. This work was supported by National Institutes of Health grant GM053959.
Abbreviations used:
- AID
auxin-inducible degron
- HA
human influenza hemagglutinin
- TORC2
target of rapamycin complex 2.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-12-0685) on July 2, 2020.
REFERENCES
- Alcaide-Gavilan M, Lucena R, Schubert KA, Artiles KL, Zapata J, Kellogg DR. (2018). Modulation of TORC2 signaling by a conserved Lkb1 signaling axis in budding yeast. Genetics , 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronova S, Wedaman K, Aronov PA, Fontes K, Ramos K, Hammock BD, Powers T. (2008). Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab , 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artiles K, Anastasia S, McCusker D, Kellogg DR. (2009). The Rts1 regulatory subunit of protein phosphatase 2A is required for control of G1 cyclin transcription and nutrient modulation of cell size. PLoS Genet , e1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. (2012). Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol , 542–547. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al (2000). A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature , 395–401. [DOI] [PubMed] [Google Scholar]
- Bradford MK, Whitwoth K, Wendland B. (2015). Pan1 regulates transitions between stages of clathrin-mediated endocytosis. Mol Biol Cell , 1371–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Weissman JS. (2010). Membranes in balance: mechanisms of sphingolipid homeostasis. Mol Cell , 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. (1999). Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr Biol , 186–197. [DOI] [PubMed] [Google Scholar]
- Cope MJ, Yang S, Shang C, Drubin DG. (1999). Novel protein kinases Ark1p and Prk1p associate with and regulate the cortical actin cytoskeleton in budding yeast. J Cell Biol , 1203–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrezuelo F, Colomina N, Palmisano A, Garí E, Gallego C, Csikász-Nagy A, Aldea M. (2012). The critical size is set at a single-cell level by growth rate to attain homeostasis and adaptation. Nat Commun , 1012. [DOI] [PubMed] [Google Scholar]
- Gagny B, Wiederkehr A, Dumoulin P, Winsor B, Riezman H, Haguenauer-Tsapis R. (2000). A novel EH domain protein of Saccharomyces cerevisiae, Ede1p, involved in endocytosis. J Cell Sci (Pt 18), 3309–3319. [DOI] [PubMed] [Google Scholar]
- González A, Hall MN. (2017). Nutrient sensing and TOR signaling in yeast and mammals. EMBO J , 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SL, Charlet A, Haas W, Gygi SP, Kellogg DR. (2005). Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell , 407–420. [DOI] [PubMed] [Google Scholar]
- Harvey SL, Enciso G, Dephoure N, Gygi SP, Gunawardena J, Kellogg DR. (2011). A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol Biol Cell , 3595–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. (1991). Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science , 905–909. [DOI] [PubMed] [Google Scholar]
- Henry KR, D’Hondt K, Chang JS, Nix DA, Cope MJTV, Chan CSM, Drubin DG, Lemmon SK. (2003). The actin-regulating kinase Prk1p negatively regulates Scd5p, a suppressor of clathrin deficiency, in actin organization and endocytosis. Curr Biol , 1564–1569. [DOI] [PubMed] [Google Scholar]
- Hirsch J, Han PW. (1969). Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J Lipid Res , 77–82. [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast , 947–962. [DOI] [PubMed] [Google Scholar]
- Johnston GC, Pringle JR, Hartwell LH. (1977). Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp Cell Res , 79–98. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Fujioka Y, Suzuki NN, Inagaki F, Wullschleger S, Loewith R, Hall MN, Ohsumi Y. (2005). Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol , 7239–7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitao RM, Jasani A, Talavera RA, Pham A, Okobi QJ, Kellogg DR. (2019). A conserved PP2A regulatory subunit enforces proportional relationships between cell size and growth rate. Genetics , 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitao RM, Kellogg DR. (2017). The duration of mitosis and daughter cell size are modulated by nutrients in budding yeast. J Cell Biol , 3463–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell , 457–468. [DOI] [PubMed] [Google Scholar]
- Lucena R, Alcaide-Gavilan M, Anastasia SD, Kellogg DR. (2017). Wee1 and Cdc25 are controlled by conserved PP2A-dependent mechanisms in fission yeast. Cell Cycle , 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena R, Alcaide-Gavilan M, Schubert K, He M, Domnauer MG, Marquer C, Klose C, Surma MA, Kellogg DR. (2018). Cell size and growth rate are modulated by TORC2-dependent signals. Curr Biol , 196–210.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J. (2014). TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife , e03779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niles BJ, Mogri H, Hill A, Vlahakis A, Powers T. (2012). Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci USA , 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi M, Niewola-Staszkowska K, Chiaruttini N, Colom A, Kusmider B, Mercier V, Soleimanpour S, Stahl M, Matile S, Roux A, Loewith R. (2018). Decrease in plasma membrane tension triggers PtdIns(4,5)P2 phase separation to inactivate TORC2. Nat Cell Biol 20, 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rispal D, Eltschinger S, Stahl M, Vaga S, Bodenmiller B, Abraham Y, Filipuzzi I, Movva NR, Aebersold R, Helliwell SB, Loewith R. (2015). Target of rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J Biol Chem , 14963–14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants FM, Baltz AG, Trott AE, Fereres S, Thorner J. (2010). A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci USA , 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. (2011). Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci USA , 19222–19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaechter M, Maaloe O, Kjeldgaard NO. (1958). Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol , 592–606. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods , 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya-Kawasaki M, Groen AC, Cope MJ, Kaksonen M, Watson HA, Zhang C, Shokat KM, Wendland B, McDonald KL, McCaffery JM, Drubin DG. (2003). Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis. J Cell Biol , 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Miao Y, Yamane Y, Zhang C, Shokat KM, Takematsu H, Kozutsumi Y, Drubin DG. (2012). Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol Biol Cell , 2388–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Martin AC, Drubin DG. (2005). Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat Cell Biol , 246–254. [DOI] [PubMed] [Google Scholar]
- Toshima JY, Furuya E, Nagano M, Kanno C, Sakamoto Y, Ebihara M, Siekhaus DE, Toshima J. (2016). Yeast Eps15-like endocytic protein Pan1p regulates the interaction between endocytic vesicles, endosomes and the actin cytoskeleton. eLife , 3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson HA, Cope MJ, Groen AC, Drubin DG, Wendland B. (2001). In vivo role for actin-regulating kinases in endocytosis and yeast epsin phosphorylation. Mol Biol Cell , 3668–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedaman KP, Reinke A, Anderson S, Yates J 3rd, McCaffery JM, Powers T. (2003). Tor kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Mol Biol Cell , 1204–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B, Steece KE, Emr SD. (1999). Yeast epsins contain an essential N-terminal ENTH domain, bind clathrin and are required for endocytosis. EMBO J , 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. (2006). TOR signaling in growth and metabolism. Cell , 471–484. [DOI] [PubMed] [Google Scholar]
- Zanolari B, Friant S, Funato K, Sütterlin C, Stevenson BJ, Riezman H. (2000). Sphingoid base synthesis requirement for endocytosis in Saccharomyces cerevisiae. EMBO J , 2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata J, Dephoure N, Macdonough T, Yu Y, Parnell EJ, Mooring M, Gygi SP, Stillman DJ, Kellogg DR. (2014). PP2ARts1 is a master regulator of pathways that control cell size. J Cell Biol , 359–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Cai M. (1999). Regulation of the actin cytoskeleton organization in yeast by a novel serine/threonine kinase Prk1p. J Cell Biol , 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Yu X, Cai M. (2001). Regulation of yeast actin cytoskeleton-regulatory complex Pan1p/Sla1p/End3p by serine/threonine kinase Prk1p. Mol Biol Cell , 3759–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.