

Abstract
Lessons Learned
Despite strong preclinical rationale, combined cobimetinib‐mediated MEK inhibition and GDC‐0994‐mediated ERK inhibition was not tolerable on two 28‐day dosing schedules in which GDC‐0994 was given for 21 days continuously and cobimetinib administered over 21 days either continuously or intermittently.
Adverse events were as expected for mitogen‐activated protein kinase pathway inhibition, but overlapping and cumulative toxicities could not be managed on either dosing schedule.
Pharmacokinetic parameters of cobimetinib and GDC‐0994 given in combination were similar to those previously observed in monotherapy studies, so that there was no evidence of drug–drug interaction.
Cycle 1 metabolic responses were observed by 18F‐fluorodeoxyglucose‐positron emission tomography but were not predictive of outcome measured by RECIST 1.1.
Background
Simultaneous targeting of multiple nodes in the mitogen‐activated protein kinase (MAPK) pathway offers the prospect of enhanced activity in RAS‐RAF‐mutant tumors. This phase Ib trial evaluated the combination of cobimetinib (MEK inhibitor) and GDC‐0994 (ERK inhibitor) in patients with locally advanced or metastatic solid tumors.
Methods
Cobimetinib and GDC‐0994 were administered orally on two separate dosing schedules. Arm A consisted of concurrent cobimetinib and GDC‐0994 once daily for 21 days of a 28‐day cycle; Arm B consisted of intermittent dosing of cobimetinib on a 28‐day cycle concurrent with GDC‐0994 daily for 21 days of a 28‐day cycle.
Results
In total, 24 patients were enrolled. For Arm A, owing to cumulative grade 1–2 toxicity, the dose of cobimetinib was decreased. For Arm B, dose increases of GDC‐0994 and cobimetinib were intolerable with grade 3 dose‐limiting toxicities of myocardial infarction and rash. Pharmacokinetic data did not show evidence of a drug–drug interaction. Overall, seven patients had a best overall response of stable disease (SD) and one patient with pancreatic adenocarcinoma had an unconfirmed partial response.
Conclusion
The safety profile of MEK and ERK inhibition demonstrated classic MAPK inhibitor–related adverse events (AEs). However, overlapping AEs and cumulative toxicity could not be adequately managed on either dosing schedule, restricting the ability to further develop this combination.
Discussion
Dysregulation of the MAPK pathway, initiated by mutations in the RAS or BRAF oncogenes or through upstream growth factor signaling, leads to tumorigenesis 1 and has been implicated in ∼30% of all human cancers 2. Although targeting RAF and/or MEK has proved clinically effective, single‐agent kinase inhibitor regimens have demonstrated limited clinical benefit outside of BRAF‐mutant metastatic melanoma 3, 4, 5, 6. Combinations of inhibitors that target multiple nodes within the MAPK pathway, such as MEK and ERK, may therefore be necessary in order to effectively suppress pathway signaling to enhance activity in RAS‐ and RAF‐mutant tumors 7, 8.
GDC‐0994 is a novel orally bioavailable small molecule inhibitor selective for extracellular signal responsive kinase 1/2 (ERK1/2) kinase activity 9, 10. Cobimetinib is a potent and highly selective inhibitor of MAP kinase/ERK kinase 1/2 (MEK1/2). In KRAS‐mutant cancer cell lines, inhibition of MEK or ERK alone, by cobimetinib or GDC‐0994, respectively, only transiently suppresses the MAPK pathway 11, 12. In contrast, cotargeting MEK and ERK with these agents in MAPK pathway–dysregulated cancer cells and tumor models demonstrates strong antitumor activity. The combination of cobimetinib and GDC‐0994 may therefore be an effective therapeutic strategy in patients with MAPK pathway–dysregulated tumors, such as KRAS‐mutant non‐small cell lung cancer, colorectal cancer, and pancreatic cancer.
The primary objective of this study was to evaluate the safety and tolerability of cobimetinib and GDC‐0994 in patients with locally advanced or metastatic solid tumors. Secondary objectives were to assess pharmacokinetic, pharmacodynamic, and antitumor activity. The safety profile of this combination of MEK and ERK inhibition demonstrated classic MAPK inhibitor–related AEs. No new safety signals were identified, and there was no evidence of interaction between the study drugs. However, overlapping grade 1–2 AEs and cumulative on‐target toxicity could not be adequately managed with supportive care on either dosing schedule. Of 15 patients who underwent at least one tumor assessment, best responses included stable disease in 7 patients (29%) and an unconfirmed partial response (SLD reduction −69%) in 1 patient with pancreatic adenocarcinoma (Fig. 1).
Figure 1.
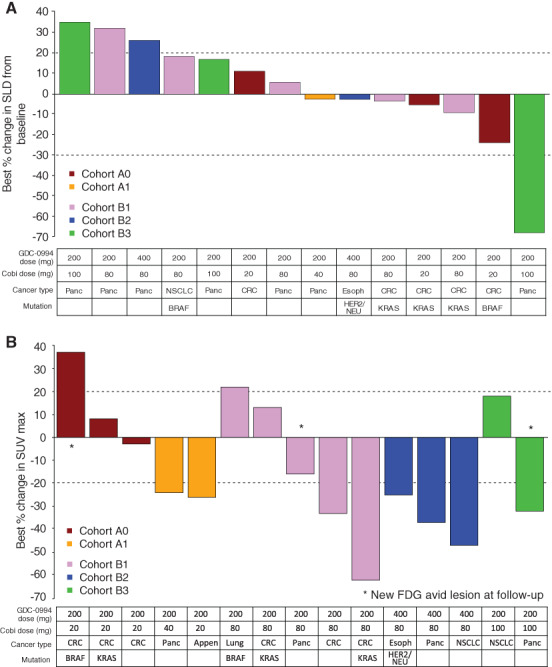
Cobimetinib + GDC‐0994 antitumor activity. (A): Patients with on‐study tumor assessments and (B) metabolic responses by FDG‐positron emission tomography (PET). GDC‐0994 and cobimetinib dose and cohort assignment, cancer type, and mutation status are shown below the chart and correspond to each patient. Note: Not all patients were evaluable/comparable because FDG‐PET scans were obtained earlier in the course of treatment than computed tomography scans.Abbreviations: Appen, appendiceal; Cobi, cobimetinib; CRC, colorectal cancer, Esoph, esophageal cancer, FDG, fluorodeoxyglucose; HER2, human epidermal growth receptor 2; NSCLC, non‐small cell lung cancer; Panc, pancreatic adenocarcinoma; SLD, sum of the longest diameters; SUVmax, maximum standardized uptake value.
Although nonclinical data with MEK‐ERK inhibition were promising in RAS‐ and RAF‐mutant tumor models, the intolerability of combined MEK‐ERK inhibition would not allow for sustained dosing at potentially therapeutic levels.
Trial Information
| Disease | Advanced cancer |
| Stage of Disease/Treatment | Metastatic/advanced |
| Prior Therapy | None |
| Type of Study | Phase I; 3 + 3 |
| Primary Endpoints | Safety, tolerability, maximum tolerated dose |
| Secondary Endpoints | Pharmacokinetics, pharmacodynamic, efficacy |
| Additional Details of Endpoints or Study Design | |
| Patients: Patients with histologically or cytologically documented, locally advanced or metastatic solid tumors for which standard therapy either does not exist or has proved ineffective or intolerable were eligible for participation in this study. Other inclusion criteria included an age of at least 18 years; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; adequate hematologic, hepatic, and renal function; and evaluable disease or disease measurable per RECIST 1.1. Patients had fluorodeoxyglucose (FDG)‐positron emission tomography (PET) avid disease on baseline scan. Exclusion criteria included history of prior significant toxicity from another MEK inhibitor or ERK inhibitor requiring discontinuation of treatment; brain metastases that were untreated, symptomatic, or require therapy to control symptoms; evidence of visible retinal pathology as assessed by ophthalmologic examination that was considered a risk factor for retinal vein thrombosis (RVO), history of glaucoma, intraocular pressure >21 mmHg as measured by tonometry, or predisposing factors to RVO; recent radiotherapy, surgery, anticancer therapy, uncontrolled systemic disease, or cardiac or liver dysfunction. | |
| Study Design: This was an open‐label, multicenter, global phase Ib dose escalation study designed to assess the safety, tolerability, and pharmacokinetics of oral dosing of cobimetinib and GDC‐0994 administered in combination in patients with histologically confirmed, locally advanced, or metastatic solid tumors for which standard therapies either do not exist or have proved ineffective or intolerable. Patients were enrolled in a standard 3 + 3 dose escalation study designed to establish the combination maximum tolerated dose (MTD) for cobimetinib and GDC‐0994 on two dosing schedules. Arm A consisted of concurrent cobimetinib (20 or 40 mg) and GDC‐0994 (200 mg) given once daily (QD) on a 21‐day‐on/7‐day‐off schedule. Arm B consisted of intermittent dosing of cobimetinib (100 or 120 mg) on Days 1, 4, 8, 11, 15, and 18 concurrent with GDC‐0994 (200 or 400 mg) QD for 21 consecutive days of a 28‐day cycle (Fig. 2). Treatment continued until disease progression, unacceptable toxicity, or any other discontinuation criterion was met. | |
| Assessments: Safety was assessed through incidence, nature, and severity of AEs, dose‐limiting toxicities (DLTs), changes in select laboratory test results, changes in vital signs, electrocardiograms, and cobimetinib and GDC‐0994 exposures. AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. DLTs were defined as grade ≥3 nonhematologic or grade ≥4 hematologic toxicities attributed to treatment occurring in the first cycle. The MTD was defined as the highest dose at which ≤1 out of 6 patients experienced a DLT. All AEs were recorded until 30 days after the last dose of study treatment or until initiation of another anticancer therapy. After this period, only treatment‐related serious AEs were recorded. Protocol‐defined adverse events of special interest included grade ≥1 retinal vein occlusion, grade ≥2 visual disturbances, grade ≥2 decrease of left ventricular ejection fraction, grade ≥3 rash for >7 days, grade ≥3 diarrhea for >3 days, any DLT, grade 3 hepatotoxicity, and cases of potential drug‐induced liver injury that include an elevated alanine aminotransferase or aspartate aminotransferase in combination with either an elevated bilirubin or clinical jaundice, as defined by Hy's law, and suspected transmission of an infectious agent by the study drug. | |
| Blood samples were collected for pharmacokinetic characterization of GDC‐0994 and cobimetinib (alone or in combination) and to explore the relationship between cobimetinib and GDC‐0994 concentrations and antitumor activity. The pharmacokinetic parameters for cobimetinib and GDC‐0994 were determined in all patients who received study treatment using noncompartmental analysis. | |
| Tumor assessments were performed at baseline and every two cycles after initiation of study treatment. Tumor response was assessed by the investigator according to RECIST version 1.1 and occurred during Days 21–28 in Cycle 2 and on Day 25 in Cycle 4, and then every 8 weeks or as clinically indicated throughout the duration of the study. | |
| To assess pharmacodynamic effects of cobimetinib and GDC‐0994 administered in combination, FDG‐PET scans were mandatory at screening and at Cycle 1, Day 15, a time point at which plasma concentrations of GDC‐0994 were expected to be at steady state. | |
| Statistical Analysis: The final analysis was based on patient data collected through study discontinuation. The analyses were based on the safety‐evaluable population, defined as patients receiving ≥1 dose of study drug. Design considerations were not made with regard to explicit power and type I error considerations but to obtain preliminary safety, pharmacokinetic, and pharmacodynamic information in this patient population. Overall, approximately 72 patients were planned to be enrolled in the study according to the study stages that included dose escalation and indication‐specific dose expansions. As the study was terminated early by the sponsor, a total of 24 patients were enrolled in the dose escalation phase of the study. The indication‐specific dose expansion stage was not initiated. | |
| Changes in Study Conduct or Planned Analyses: The sponsor decided to terminate the study given the lack of tolerability of the combination of cobimetinib and GDC‐0994. Consequentially, a planned indication‐specific dose‐expansion stage designed to investigate the potential recommended phase II dose in five indication‐specific expansion cohorts was not initiated. The exploratory and pharmacodynamic analyses were not performed owing to limited number of patients. | |
| Investigator's Analysis | Poorly tolerated/not feasible |
Figure 2.
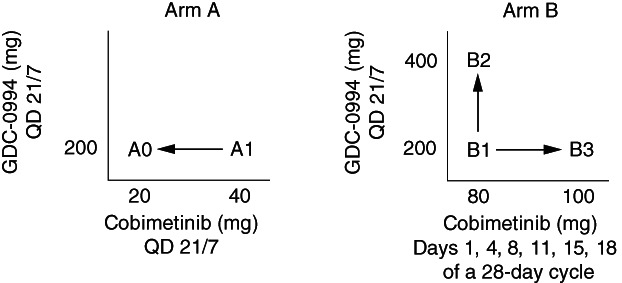
Study schema and dose schedules.Abbreviation: QD, once daily.
Drug Information
| Drug 1 | |
| Generic/Working Name | Cobimetinib |
| Company Name | Genentech, Inc. |
| Drug Type | Small molecule |
| Drug Class | MEK |
| Dose | Arm A: 20 or 40 mg; Arm B: 100 or 120 mg per flat dose |
| Route | p.o. |
| Schedule of Administration |
Arm A: Once daily on a 21‐day‐on/7‐day‐off schedule. Arm B: Days 1, 4, 8, 11, 15, and 18 of a 28‐day cycle. |
| Drug 2 | |
| Generic/Working Name | GDC‐0994 |
| Company Name | Genentech, Inc. |
| Drug Type | Small molecule |
| Drug Class | Other |
| Dose | Arm A: 200 mg; Arm B: 200 or 400 mg per flat dose |
| Route | p.o. |
| Schedule of Administration |
Arm A: Once daily on a 21‐day‐on/7‐day‐off schedule. Arm B: QD for 21 consecutive days of a 28‐day cycle. |
Dose Escalation Table
| Dose level | Dose of drug: cobimetinib | Dose of drug: GDC‐0994 | No. enrolled | No. evaluable for toxicity |
|---|---|---|---|---|
| Arm A | 20 | 200 | 5 | 5 |
| Arm A | 40 | 200 | 3 | 3 |
| Arm B | 80 | 200 | 6 | 6 |
| Arm B | 80 | 400 | 4 | 4 |
| Arm B | 100 | 200 | 6 | 6 |
Patient Characteristics
| Number of Patients, Male | 7 |
| Number of Patients, Female | 17 |
| Age | Median (range): 55 (28–75) years |
| Number of Prior Systemic Therapies | Median (range): 9 (1–27) |
| Performance Status: ECOG |
0 — 7 1 — 17 2 — 0 3 — 0 Unknown — 0 |
| Other |
Mutations (Local and/or central of archive tissue): BRAF, 3 (25%); KRAS, 9 (75%); NRAS, 0 (0) Mutations, Other (includes HER2/NEU, or not available): 7 (100%) |
| Cancer Types or Histologic Subtypes | Colon, 7; Pancreas, 7; Lung, 5; Esophagus, 1; Rectum, 1; Other, 3 |
Primary Assessment Method
| Title | Primary assessments |
| Number of Patients Screened | 24 |
| Number of Patients Enrolled | 24 |
| Number of Patients Evaluable for Toxicity | 24 |
| Number of Patients Evaluated for Efficacy | 24 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 1 (4%) |
| Response Assessment SD | n = 7 (29%) |
| Response Assessment PD | n = 0 (0%) |
| Response Assessment OTHER | n = 16 (67%) |
| Outcome Notes | PR is unconfirmed. |
Adverse Events
See Table 1
Table 1.
Adverse events related to cobimetinib and GDC‐0994 occurring in ≥4 patients (safety‐evaluable)
| MedDRA preferred term | Arm A (n = 8) | Arm B (n = 16) | All patients (n = 24) |
|---|---|---|---|
| Diarrhea | 7 (88%) | 13 (81%) | 20 (83%) |
| Vomiting | 2 (25%) | 10 (63%) | 12 (50%) |
| Nausea | 5 (63%) | 9 (56%) | 14 (58%) |
| Acneiform dermatitis | 4 (50%) | 7 (44%) | 11 (46%) |
| Fatigue | 3 (38%) | 7 (44%) | 10 (42%) |
| Decreased appetite | 3 (38%) | 5 (31%) | 8 (33%) |
| Vision blurred | 2 (25%) | 4 (25%) | 6 (25%) |
Serious Adverse Events
| Serious AE | Arm A (n = 8) | Arm B (n = 16) | All patients (n = 24) |
|---|---|---|---|
| Diarrhea | 1 (13%) | 1 (13%) | 2 (8%) |
| Serious Adverse Events Legend | Serious adverse events regardless of relationship to study drug in >2 safety‐evaluable patients overall. | ||
Dose‐Limiting Toxicities
| Cobimetinib dose | GDC‐0994 dose | No. enrolled | No. evaluable for toxicity | No. with a dose‐limiting toxicity | Dose‐limiting toxicity information |
|---|---|---|---|---|---|
| 20 mg | 200 mg | 5 | 5 | 0 | |
| 40 mg | 200 mg | 3 | 3 | 1 | Grade 3 diarrhea |
| 80 mg | 200 mg | 6 | 6 | 2 | Grade 3 myocardial infarction; nonserious grade 3 events of rash and dermatitis acneiform |
| 80 mg | 400 mg | 4 | 4 | 0 | |
| 100 mg | 200 mg | 6 | 6 | 0 |
Pharmacokinetics/Pharmacodynamics for Phase I New Arm
| Dose level | Dose of drug: cobimetinib | Dose of drug: GDC‐0994 | Cmax (range), ng/L | Tmax (%), hours | AUC0–12 (%), ng × hours/mL) |
|---|---|---|---|---|---|
| Arm A | 20 | 200 | 3.0 (2.0–4.0) | 1.8 (74%) | 23.5 (73%) |
| Arm A | 40 | 200 | 24.0 | 2.01 | 24.1 |
| Arm A | 80 | 200 | 2.0 (2.0–3.0) | 2.26 (23%) | 33.3 (66%) |
| Arm A | 80 | 300a | 3.0 | 2.56 | 48.3 |
| Arm A | 80 | 400 | 3.0 | 2.09 | 37.2 |
| Arm A | 100 | 200 | 2.0 (2.0–2.0) | 2.31 (28%) | 34.0b |
| Arm B | 200 | 20 | 1.5 (1.0–2.0) | 48.5 (121%) | 508 (255%) |
| Arm B | 200 | 40 | 24.0 | 204 | 2,460 |
| Arm B | 200 | 80 | 1.0 (0.5–2.0) | 399 (46%) | 4,320 (37%) |
| Arm B | 200 | 100 | 2.0 (2.0–2.0) | 431 (63%) | 4,070c |
| Arm B | 300 | 80 | 1 | 73.0 | 908 |
| Arm B | 400 | 80 | 2.0 | 284 | 3,410 |
Dose reduced from 400 mg to 300 mg.
n = 1.
n = 1.
Assessment, Analysis, and Discussion
| Completion | Study terminated before completion |
| Terminated Reason | Toxicity |
| Investigator's Assessment | Poorly tolerated/not feasible |
This trial aimed to test the combination of two inhibitors, cobimetinib and GDC‐0994, designed to target multiple nodes within the mitogen‐activated protein kinase (MAPK) pathway to achieve vertical inhibition in MAPK‐dysregulated tumors. The safety profile of this combination of MEK and ERK inhibition demonstrated classic MAPK inhibitor–related adverse events (AEs), indicating on‐target effects as predicted by nonclinical data; however, overlapping and cumulative toxicity could not be adequately managed on either dosing schedule. The maximum tolerated dose (MTD) was not formally determined in schedule A and exceeded in cohort B1, and the trial was stopped by the sponsor.
The efficacy of the cobimetinib and GDC‐0994 combination was limited in this study. Figure 1 shows patients with on‐study tumor assessments and metabolic responses. Of 15 patients who underwent at least one tumor assessment, best responses included stable disease in 7 patients (29%) and an unconfirmed partial response (SLD reduction −69%) in 1 patient with pancreatic adenocarcinoma. A metabolic response by fluorodeoxyglucose (FDG)‐positron emission tomography (PET) was achieved in 8 of 15 (53%) patients who underwent a pre‐ and on‐treatment FDG‐PET scan, although metabolic responses did not predict tumor responses by RECIST 1.1. Not all patients were evaluable/comparable because FDG‐PET scans were obtained earlier in the course of treatment than computed tomography scans. Furthermore, limitations in sample size and times on study preclude confident interpretations of pharmacodynamic or antitumor activity.
To identify an MTD, this study was designed to allow dose reductions in both cobimetinib and GDC‐0994 in the dose escalation stage in response to toxicities (Fig. 1). In Arm A, a single dose‐limiting toxicity (DLT) of grade 3 diarrhea occurred in cohort A1 (40 mg cobimetinib +200 mg GDC‐0994). Because of this DLT and additional cumulative grade 1–2 toxicities, the dose of cobimetinib was decreased to 20 mg in cohort A0. However, four of five patients in cohort A0 withdrew consent for the study, indicating poor tolerability due to cumulative on‐target toxicity. For Arm B, cohort B1 enrolled patients at a dose of 80 mg cobimetinib +200 mg GDC‐0994. Dose escalation to cohort B2 (GDC‐0994 increased to 400 mg) and cohort B3 (cobimetinib increased to 100 mg) resulted in intolerability owing to cumulative grade 1–2 AEs in both cohorts. As a result, two additional patients were enrolled in cohort B1. One of these patients experienced a DLT of grade 3 myocardial infarction. A third patient who was enrolled in cohort B1 experienced a DLT of grade 3 rash and dermatitis acneiform, indicating that the MTD had been exceeded.
Sixteen patients experienced a total of 24 AEs of special interest: 5 patients (21%) experienced grade ≥3 diarrhea, 3 patients (13%) experienced grade ≥2 left ventricular ejection fraction decrease, 4 patients (17%) experienced grade ≥2 visual disturbances, 3 patients (13%) experienced grade ≥3 hepatotoxicity (ascites [n = 2] and grade 3 aspartate aminotransferase [AST] increased [n = 1]), 2 (8%) experienced grade ≥3 rash, and 1 patient (4%) experienced elevated AST >3× baseline and an elevated bilirubin >2× upper limit of normal. These AEs were commensurate with the safety profile expected for MAP kinase pathway inhibition.
Pharmacokinetic evaluations showed that at a single dose, GDC‐0994 tmax ranged from 2.00 to 24.0 hours at doses ranging from 200 to 400 mg, and that there were overlapping steady‐state exposures between the doses. GDC‐0994 exposure in combination with cobimetinib was similar to monotherapy GDC‐0994 exposure 13. Similarly, cobimetinib exposures were within the range observed after cobimetinib monotherapy in published data, and cobimetinib exposure in combination with GDC‐0994 was similar to historic cobimetinib exposure 14. These data indicate that there is no evidence of a drug–drug interaction between these two compounds, even though both are metabolized by CYP3A.
In conclusion, despite promising preclinical biology supporting the evaluation of cobimetinib and GDC‐0994 to enhance inhibition of the MAPK pathway 8, the combination was associated with intolerable toxicity and its development was discontinued. Although combining BRAF and MEK inhibitors for MAPK pathway inhibition has resulted in approved, effective, and tolerable treatment combinations, unfortunately, this has not been shown with combined MEK and ERK inhibition owing to intolerability of the combination at the doses tested. Indeed, the approved dosing schedule for cobimetinib is 60 mg, 21 days on/7 days off (in combination with vemurafenib) 6, 15, which was not reached in this combination. ERK inhibition may have value after development of acquired resistance to MEK or BRAF/MEK inhibition. Additionally, these data do not preclude evaluation of combined BRAF and ERK inhibition as another strategy to achieve vertical MAPK pathway suppression.
Disclosures
Colin Weekes: Genentech, Inc. (RF, uncompensated); Patricia LoRusso: Abbvie, Agios, Five Prime, GenMab, Halozyme, Roche‐Genentech, Genentech, CytomX, Takeda, Sotio, Cybrexa, Agenus, Tyme, IQVIA, TRIGR, Pfizer, I‐Mab, ImmunoMet, Black Diamond, Gloxo‐Smith Kline, QED Therapeutics, AstraZeneca, EMD Serono, Shattuck, Astellas, Salarius, Silverback, MacroGenics (C/A); Elaine Murray: Roche, Genentech (E), Roche Stock Options (OI); Erica Park: Genentech (E), Roche (OI); Mike Tagen: Genentech, Inc. (E), Roche (OI); Jatinder Singh: Genentech, Inc. (E), Roche (OI); Indrani Sarkar: Genentech, Inc. (E), Roche (OI); Lars Mueller: Genentech (E), Roche (OI); Hatem Dokainish: Genentech, Inc. (E), Roche (E, OI); Geoffrey Shapiro: Eli Lilly, Merck KGaA/EMD‐Serono, Merck, Sierra Oncology (RF), Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD‐Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, and Daiichi Sankyo, Boehringer Ingelheim, Agios, ImmunoMet, Asana (SAB); Howard Burris: Daiichi Sankyo, Pfizer (C/A), HCA/Sarah Cannon (E), Celgene, AstraZeneca, FORMA Therapeutics, Incyte (C/A, institutional), Jounce Therapeutics, Janssen, BIND Therapeutics, Pfizer, Vertex, Gilead Sciences, Bayer, Bristol‐Myers Squibb, Macrogenics, CytomX Therapeutics, Arch, Revolution Medicines, MedImmune, BI, Merck, Moderna Therapeutics, Verastem, Harpoon Therapeutics, Lilly, Tesaro, Millennium, miRNA Therapeutics, Incyte, AZ, Novartis, Seattle Genetics, GSK, BioAtla, Agios, BioMed Valley Discoveries, TG Therapeutics, eFFECTOR Therapeutics, Ciclo Med, Array BioPharma, Kymab, Roche/Genentech, Arvinas (RF, institutional), HCA/Sarah Cannon (OI), Novartis (ET, institutional). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Figure and Table
Acknowledgments
This study was funded by Genentech, Inc., South San Francisco, CA. We thank the patients and their families as well as the study investigators and staff who participated in this study. Editorial and writing support was provided by Genentech, Inc.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Footnotes
- ClinicalTrials.gov Identifier: NCT02457793
- Sponsor: Genentech, Inc.
- Principal Investigator: Colin Weekes
- IRB Approved: Yes
References
- 1. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003;3:11–22. [DOI] [PubMed] [Google Scholar]
- 2. Hoshino R, Chatani Y, Yamori T et al. Constitutive activation of the 41‐/43‐kDa mitogen‐activated protein kinase signaling pathway in human tumors. Oncogene 1999;18:813–822. [DOI] [PubMed] [Google Scholar]
- 3. Flaherty KT, Robert C, Hersey P et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 2012;367:107–114. [DOI] [PubMed] [Google Scholar]
- 4. Long GV, Stroyakovskiy D, Gogas H et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF‐mutant melanoma: A multicentre, double‐blind, phase 3 randomised controlled trial. Lancet 2015;386:444–451. [DOI] [PubMed] [Google Scholar]
- 5. Moriceau G, Hugo W, Hong A et al. Tunable‐combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015;27:240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ascierto PA, McArthur GA, Dréno B et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)‐mutant melanoma (coBRIM): Updated efficacy results from a randomised, double‐blind, phase 3 trial. Lancet Oncol 2016;17:1248–1260. [DOI] [PubMed] [Google Scholar]
- 7. Yap JL, Worlikar S, MacKerell AD et al. Small‐molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem 2011;6:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merchant M, Moffat J, Schaefer G et al. Combined MEK and ERK inhibition overcomes therapy‐mediated pathway reactivation in RAS mutant tumors. PLoS One 2017;12:e0185862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blake JF, Burkard M, Chan J et al. Discovery of (S)‐1‐(1‐(4‐Chloro‐3‐fluorophenyl)‐2‐hydroxyethyl)‐4‐(2‐((1‐methyl‐1H‐pyrazol‐5‐yl)amino)pyrimidin‐4‐yl)pyridin‐2(1H)‐one (GDC‐0994), an extracellular signal‐regulated kinase 1/2 (ERK1/2) inhibitor in early clinical development. J Med Chem 2016;59:5650–5660. [DOI] [PubMed] [Google Scholar]
- 10. Varga A, Soria J‐C, Hollebecque A, et al. A first‐in‐human phase I study to evaluate the ERK1/2 inhibitor GDC‐0994 in patients with advanced solid tumors. Clin Cancer Res 2020. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 11. Hatzivassiliou G, Haling JR, Chen H et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS‐ versus BRAF‐driven cancers. Nature 2013;501:232–236. [DOI] [PubMed] [Google Scholar]
- 12. Lito P, Saborowski A, Yue J et al. Disruption of CRAF‐mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014;25:697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varga A, Soria JC, Hollebecque A et al. A first‐in‐human phase I study to evaluate the ERK1/2 inhibitor GDC‐0994 in patients with advanced solid tumors. Eur J Cancer 2016;69(suppl 1):S11. [DOI] [PubMed] [Google Scholar]
- 14. Rosen LS, LoRusso P, Ma WW et al. A first‐in‐human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Invest New Drugs 2016;34:604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larkin J, Ascierto PA, Dréno B et al. Combined vemurafenib and cobimetinib in BRAF‐mutated melanoma. N Engl J Med 2014;371:1867–1876. [DOI] [PubMed] [Google Scholar]