

SUMMARY
Nonalcoholic fatty liver disease is strongly associated with hepatic insulin resistance (HIR); however, the key lipid species and molecular mechanisms linking these conditions are widely debated. We developed a subcellular fractionation method to quantify diacylglycerol (DAG) stereoisomers and ceramides in the endoplasmic reticulum (ER), mitochondria, plasma membrane (PM), lipid droplets and cytosol. Acute knockdown (KD) of diacylglycerol acyltransferase-2 in liver induced HIR in rats. This was due to PM sn-1,2-DAG accumulation, which promoted PKCε activation, and insulin receptor kinase (IRK)-T1160 phosphorylation resulting in decreased IRK-T1162 phosphorylation. Liver PM sn-1,2-DAG content and IRK-T1160 phosphorylation were also higher in humans with HIR. In rats, liver-specific PKCε KD ameliorated high-fat diet-induced HIR by lowering IRK-T1160 phosphorylation, while liver-specific overexpression of constitutively active PKCε-induced HIR by promoting IRK-T1160 phosphorylation. These data identify PM sn-1,2-DAGs as the key pool of lipids that activate PKCε and that hepatic PKCε is both necessary and sufficient in mediating HIR.
Graphical Abstract
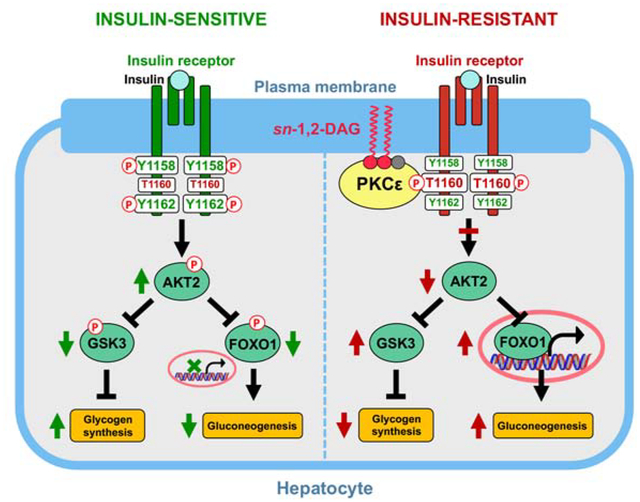
eTOC
Lipid-induced hepatic insulin resistance is mediated by plasma membrane bound sn-1,2-diacylglycerols, which promote PKCε translocation to the plasma membrane. PKCε is both necessary and sufficient for mediating lipid-induced hepatic insulin resistance through phosphorylation of insulin receptor at Thr1160.
INTRODUCTION
The postprandial rise in plasma insulin concentration activates hepatic insulin signaling pathways, which promote the storage of ingested carbohydrates into hepatic glycogen and suppress endogenous glucose production (EGP) to maintain normoglycemia within a relatively narrow range. Decreased hepatic insulin action disrupts these processes and contributes to fasting and postprandial hyperglycemia in type 2 diabetes (T2D) (Shulman, 2000). While the cellular and molecular mechanisms responsible for HIR are unresolved, ectopic lipid accumulation in the liver, leading to nonalcoholic fatty liver disease (NAFLD), is strongly associated with HIR and often precedes the development of T2D. However, the key lipid species and molecular mechanisms by which ectopic lipids mediate HIR are widely debated, with pathways mediated by DAGs and ceramides positioned at the center of this controversy (Chavez and Summers, 2012; Kusminski and Scherer, 2019; Petersen and Shulman, 2017).
DAGs, the penultimate intermediate in triglyceride synthesis, have been shown to be highly associated with hepatic steatosis and HIR in both rodents and humans (Abulizi et al., 2017; Kumashiro et al., 2011; Luukkonen et al., 2016; Magkos et al., 2012; Perry et al., 2015b; Samuel and Shulman, 2018; Ter Horst et al., 2017). In liver, DAGs have been proposed to induce HIR by activating PKCε (Kumashiro et al., 2011; Raddatz et al., 2011; Samuel et al., 2004; Samuel et al., 2007). When activated, PKCε phosphorylates T1160, a key threonine residue on IRK, which is situated between two critical IRK activation sites, Y1158 and Y1162, to destabilize IRK’s activation loop (Petersen et al., 2016). However, there are still gaps in this mechanistic model and experimental support establishing the direct causal relationship between DAG accumulation and the development of HIR is lacking. Further, the role of hepatic PKCε and IRK-T1160 phosphorylation in regulating hepatic glucose metabolism has recently been challenged (Brandon et al., 2019).
Among three DAG stereoisomers (sn-1,2-DAG, sn-2,3-DAG and sn-1,3-DAG), sn-1,2-DAG was shown to be the only stereoisomer that activates novel PKCs (Boni and Rando, 1985; Nomura et al., 1986; Rando and Young, 1984) and therefore is potentially the key DAG stereoisomer that is responsible for impeding insulin signaling. In addition, the subcellular distribution of DAGs is likely important in this regard. For example, in mouse models of hepatic CGI-58 KD and Hdac3 depletion, accumulation of lipid droplet DAGs are not associated with HIR (Cantley et al., 2013; Sun et al., 2012). Therefore, it is essential to determine the associations between subcellular DAG stereoisomer content, liver-specific PKCε activity, IRK-T1160 phosphorylation and hepatic insulin action in vivo.
To address these questions, we developed a liquid-chromatography tandem mass spectrometry (LC-MS/MS) method to quantify DAG stereoisomers, as well as ceramides in five hepatocellular compartments: ER, mitochondria, PM, lipid droplet and cytosol. We then applied these methods in combination with in vivo assessment of hepatic insulin signaling (pIRK-T1160, pIRK-Y1162, pAkt-S473, pGSK3β-S9 and pFOXO1-S256 and EGP, utilizing the hyperinsulinemic-glucose clamp technique, in awake rats with acute liver-specific diacylglycerol acyltransferase-2 (DGAT2) KD to acutely raise hepatic DAG content. Next, in order to determine if the results translate to humans, we quantified liver DAG stereoisomers and ceramides in the five subcellular compartments as well as pIRK-T1160 in humans with and without NAFLD and HIR. Finally, in order to determine the role of PKCε in regulating hepatic insulin signaling, we assessed hepatic glycogen synthesis, EGP and hepatic insulin signaling in liver-specific PKCε KD rats and rats with liver-specific overexpression (OE) of constitutively activated PKCε. Using this comprehensive approach, we show that an accumulation of PM sn-1,2-DAG content is sufficient to cause HIR at the level of IRK, which in turn could be attributed to activation of PKCε and phosphorylation of IRK-T1160 in vivo. We further establish that liver PKCε is both necessary and sufficient in the development of lipid-induced HIR.
RESULTS
Acute Hepatic DGAT2 KD Induces HIR
DGAT2 is the critical ER-located enzyme that catalyzes the esterification of DAGs with acyl-CoAs to produce triglyceride. We previously reported that chronic hepatic DGAT2 KD paradoxically reverses high-fat diet (HFD)-induced DAG accumulation and HIR, at least partially due to suppression of SREBP-1c-mediated lipogenesis (Choi et al., 2007). However, we hypothesized that an acute DGAT2 KD might provide a narrow window in which DAGs transiently accumulate, allowing us to further delineate their association with PKCε activation and HIR. To test this hypothesis, we dosed regular chow-fed male Sprague Dawley rats with an antisense oligonucleotide (ASO) targeting DGAT2. Two days after the dosing, liver DGAT2 protein content was ~50% lower in acute hepatic DGAT2 KD rats compared to the control group and no KD effect was observed in white adipose tissue (WAT) (Figure 1A). During a hyperinsulinemic-euglycemic clamp (Figures S1A–S1D), the acute hepatic DGAT2 KD impaired insulin’s ability to suppress EGP (Figure 1B). Acute hepatic DGAT2 KD impaired hepatic insulin signaling at the level of IRK. Insulin-stimulated IRK-Y1162 phosphorylation was >50% lower and was accompanied by impaired downstream insulin-stimulated Akt-S473, GSK3β-S9 and FOXO1-S256 phosphorylation in acute hepatic DGAT2 KD rats compared to the control group (Figure 1C). This defect in hepatic insulin signaling was associated with ~2-fold higher PKCε activation (Figure 1D). PKCε has been previously shown to phosphorylate IRK at T1160 to impair hepatic insulin signaling (Petersen et al., 2016). We used a phospho-specific monoclonal antibody to assay the phosphorylation of this residue in liver and observed ~75% higher pIRK-T1160 in acute hepatic DGAT2 KD rats compared to the control group (Figure 1E).
Figure 1. Acute Hepatic DGAT2 KD Induces HIR.
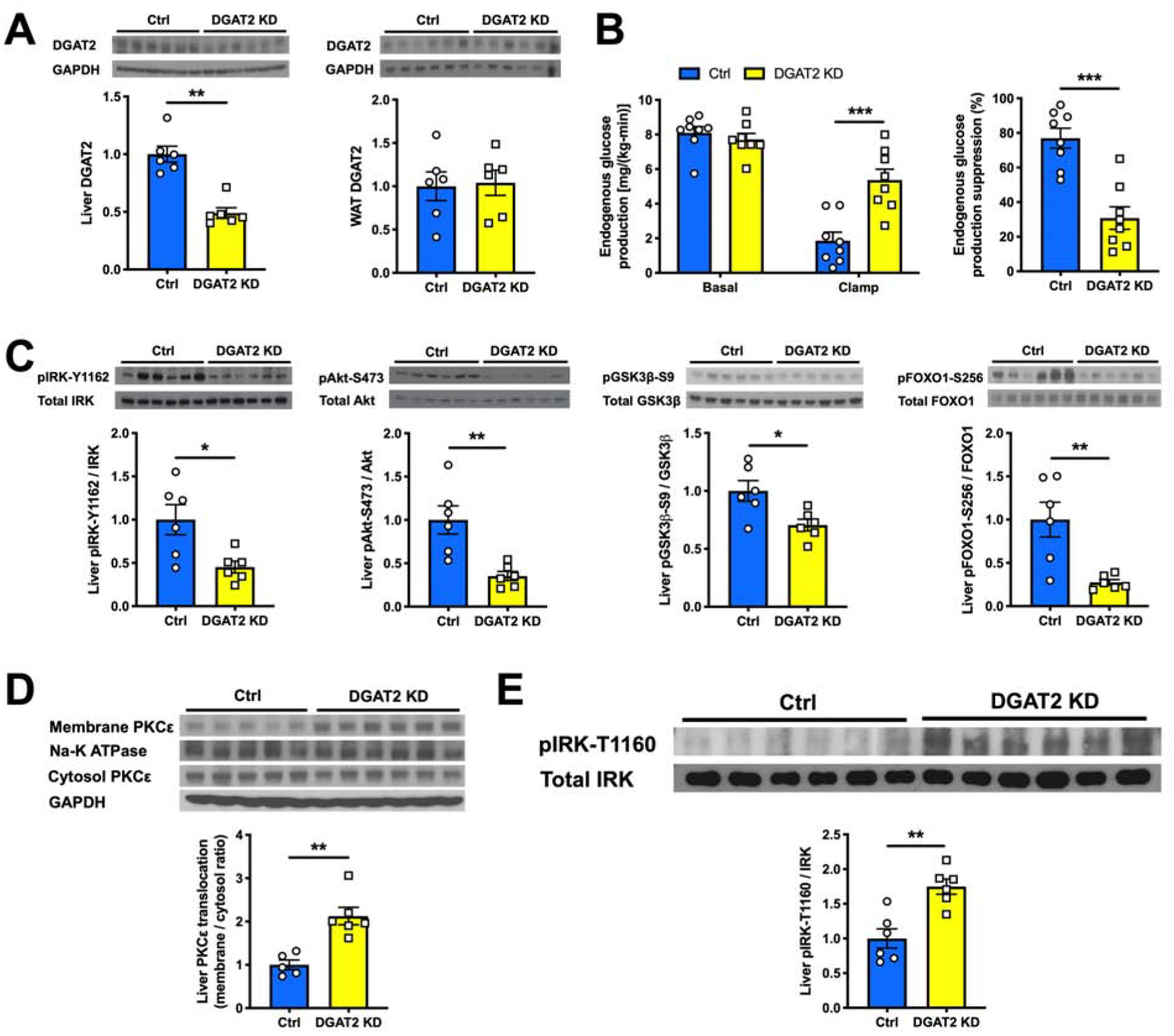
(A) Hepatic and WAT DGAT2 protein content measured by western blot (top) and its quantification (bottom).
(B) EGP and its suppression by insulin during a hyperinsulinemic-euglycemic clamp in Ctrl vs acute hepatic DGAT2 KD rats.
(C) Levels of insulin-stimulated liver pIRK-Y1162, pAkt-S473, pGSK3β-S9 and pFOXO1-S256 as measured by western blot (top) and with its quantification (bottom).
(D) Liver PKCε translocation from cytosol to membrane as measured by western blot (top) and with its quantification (bottom).
(E) Levels of liver pIRK-T1160 in Ctrl vs acute hepatic DGAT2 KD rats as measured by western blot (top) and with its quantification (bottom).
In all panels, data are the mean±S.E.M. In (A) and (C), n = 6 per group. In (B), n = 8 per group. In (D), n = 5 or 6 per group. In (E), n = 5 per group. *P < 0.05, **P < 0.01 and ***P < 0.001.
Liver PM sn-1,2-DAG Content Tracks with HIR in Rats and Humans
Next to examine the associations of DAG stereoisomer content in different subcellular compartments with HIR, we separated liver tissues into ER, mitochondria, PM, lipid droplet and cytosol using differential centrifugation (Figures 2A and S2A) and measured their DAG stereoisomer content utilizing a novel LC/MS-MS method (Figure 2B). Acute hepatic DGAT2 KD caused the sn-1,2-DAG content to be ~50% higher in the ER and ~80% higher in the PM (Figure 2C). In contrast, there were no discernible differences observed for sn-2,3- and sn-1,3-DAGs (Figures 2D and 2E). We also observed slightly higher hepatic cytosolic C16:0, C22:0 and C24:0 ceramide content following acute DGAT2 KD, potentially due to substrate (acyl-CoA) accumulation (Figure S2B). In order to determine if these results would translate into humans, we also examined liver tissues from a cohort of human individuals who were either hepatic insulin-sensitive or hepatic insulin-resistant as previously characterized using hyperinsulinemic clamp methodology (Ter Horst et al., 2017) (Table S1). Consistent with the results in rodents, liver PM sn-1,2-DAG content was ~5-fold higher in individuals with HIR and NAFLD compared to individuals without HIR (Figure 2F), without discernible differences in ceramide content (Figure S2C). Furthermore, the higher PM sn-1,2-DAG content was associated with an ~3-fold higher level of pIRK-T1160 in individuals with HIR compared to individuals without HIR (Figure S2D).
Figure 2. Liver PM sn-1,2-DAG Content Tracks with HIR in Rats and Humans.
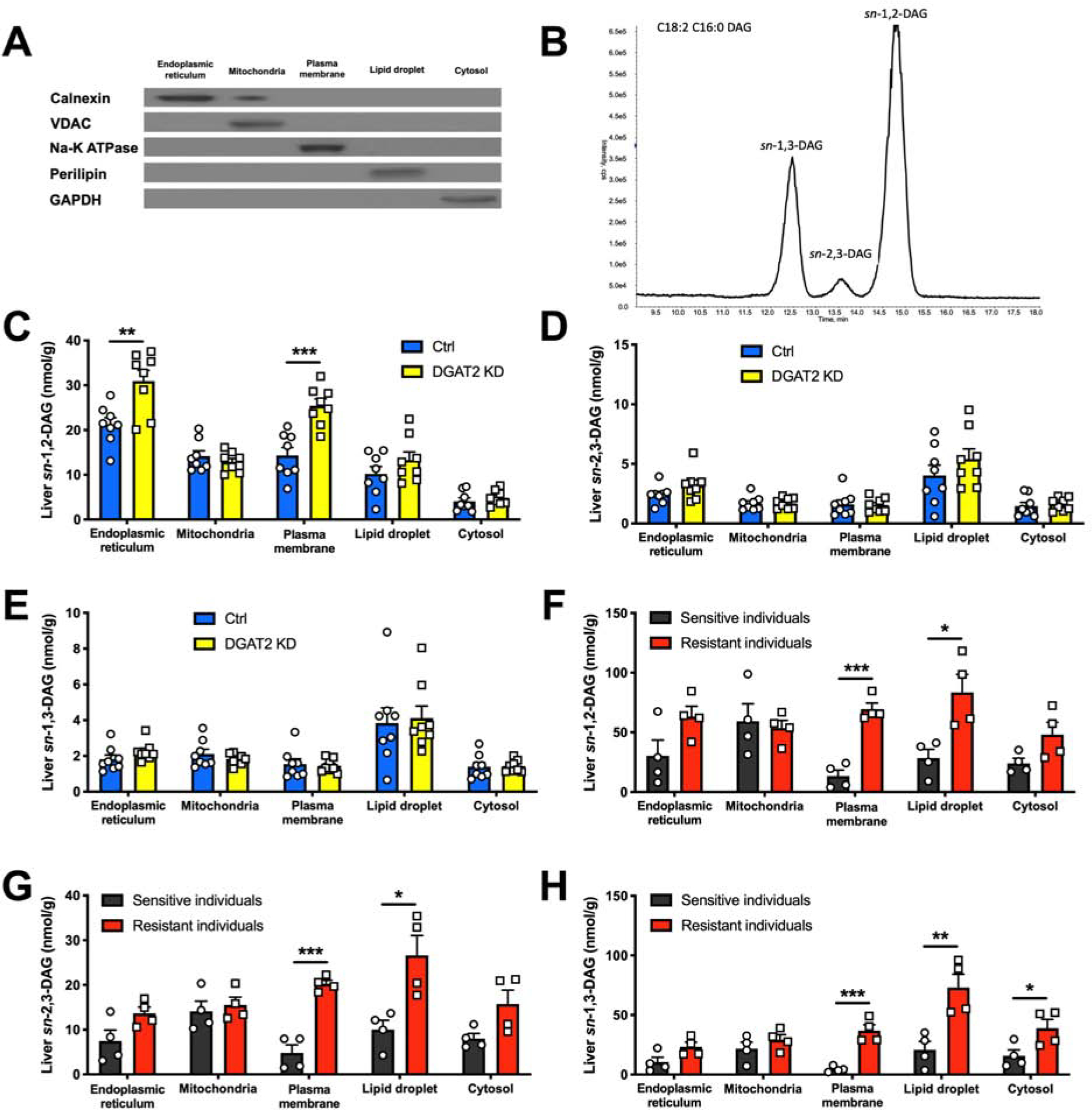
(A) Separation of five subcellular compartments in liver measured by western blot.
(B) Representative chromatogram (n = 13) of DAG stereoisomer separation on LC/MS-MS.
(C), (D) and (E) Liver DAG stereoisomer content in five subcellular compartments in Ctrl vs DGAT2 KD rats.
(F), (G) and (H) Liver DAG stereoisomer content in five subcellular compartments in human individuals who were insulin-sensitive (black) or insulin-resistant (red).
In all panels, data are the mean±S.E.M. In (C), (D) and (E), n = 8 per group. In (F), (G) and (H), n = 4 per group. *P < 0.05, **P < 0.01 and ***P < 0.001.
Liver-Specific PKCε KD Ameliorates HFD- and Acute DGAT2 KD-Induced HIR
A recent study in liver-specific PKCε knockout mice challenged the notion that hepatic PKCε activation is required for the development of lipid-induced HIR (Brandon et al., 2019), seemingly refuting a prior study using 2’-O-(2-methoxy)-O-ethyl (2’-MOE) ASO against PKCε in which the KD of both hepatic and WAT PKCε were associated with improvements in hepatic and WAT insulin action in awake rats (Samuel et al., 2007). To further examine the role of PKCε in regulating insulin action in the liver, we used a next-generation 2’-MOE ASO additionally modified with N-acetyl galactosamine (GalNAc). GalNAc modification facilitates ASO’s delivery into hepatocytes (Prakash et al., 2014), permitting liver-specific PKCɛ KD in adult rats over the course of 3-week ASO dosing. MOE-GalNAc-modified PKCε ASO lowered liver PKCε protein content by ~70% without altering WAT PKCε protein content (Figure 3A). Insulin action was then assessed in these rats after a 4-day HFD feeding which is a common model to induce acute hepatic steatosis and HIR. During a hyperinsulinemic-hyperglycemic clamp (Figures S3A–S3F), liver-specific PKCε KD improved insulin’s ability to suppress EGP (Figure 3B). We chose to use hyperinsulinemic-hyperglycemic clamp since both hyperinsulinemia and hyperglycemia are required for maximumly promoting hepatic glycogen synthesis (Nozaki et al., 2020), and therefore provides an optimal condition to detect potential differences in the rates of glycogen synthesis during the hyperinsulinemic clamp. Hepatic glycogen synthesis (specifically the direct pathway: glucose → glucose-6-phosphate → UDP glucose → glycogen) is the most robust in vivo flux indicator of direct insulin action on hepatic glucose metabolism, while the suppression of EGP is also influenced indirectly (i.e. independent of insulin signaling in liver) by insulin’s regulation of WAT lipolysis (Perry et al., 2015a; Petersen and Shulman, 2018). Liver-specific PKCε KD led to an ~2-fold higher insulin-stimulated hepatic glycogen synthesis rate compared to the control group, resulting in higher post-clamp hepatic glycogen content (Figure 3C).
Figure 3. Liver-Specific PKCε KD Ameliorates HFD- and Acute DGAT2 KD-Induced HIR.
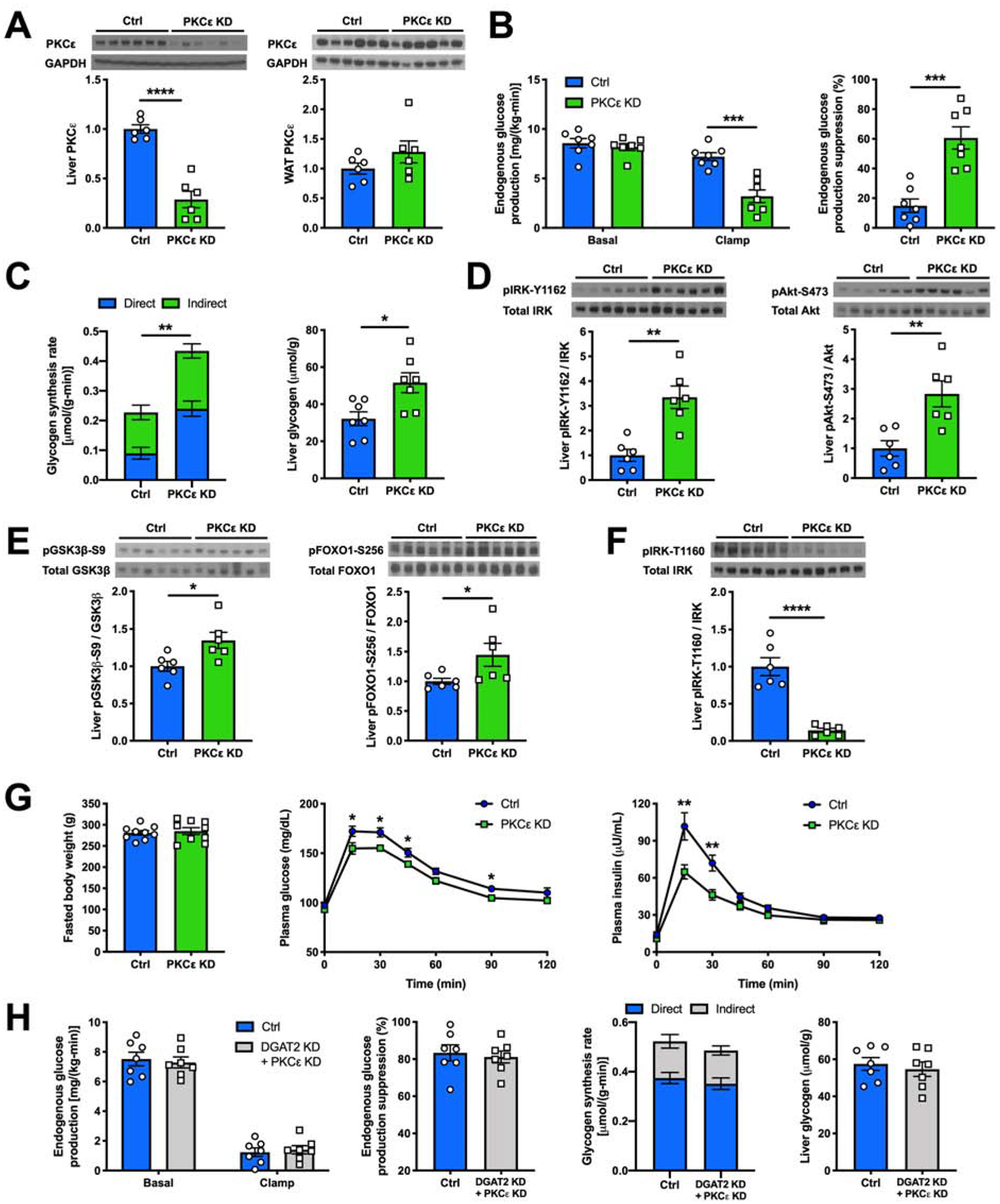
(A) Hepatic and WAT PKCε protein content measured by western blot (top) and its quantification (bottom).
(B) EGP and its suppression by insulin during a hyperinsulinemic-hyperglycemic clamp in Ctrl vs hepatic PKCε KD rats.
(C) Hepatic glycogen synthesis rate during a hyperinsulinemic-hyperglycemic clamp and post-clamp hepatic glycogen content.
(D) and (E) Levels of insulin-stimulated liver pIRK-Y1162, pAkt-S473, pGSK3β-S9 and pFOXO1-S256 as measured by western blot (top) and with its quantification (bottom).
(F) Levels of liver pIRK-T1160 as measured by western blot (top) and with its quantification (bottom).
(G) Fasted body weight, plasma glucose and insulin levels during an oGTT in Ctrl vs hepatic PKCε KD rats.
(H) EGP, EGP’s suppression by insulin and hepatic glycogen synthesis rate during a hyperinsulinemic-hyperglycemic clamp and post-clamp hepatic glycogen content in Ctrl vs hepatic PKCε KD rats.
In all panels, data are the mean±S.E.M. In (A), (D), (E) and (F), n = 6 per group. In (B), (C) and (H), n = 7 per group. In (G), n = 9 per group. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
We then assayed key components of the insulin signaling pathway and found that in rats treated with the liver-specific PKCε ASO, the improved hepatic insulin sensitivity was associated with ~3-fold higher insulin-stimulated pIRK-Y1162 (Figure 3D). This in turn was accompanied by significant improvements in downstream insulin-stimulated Akt-S473, GSK3β-S9 and FOXO1-S256 phosphorylation (Figures 3D and 3E). We also observed that there was markedly lower pIRK-T1160 levels with liver-specific PKCε KD compared to the control group (Figure 3F).
Furthermore, with the same 4-day HFD feeding, rats with liver-specific PKCε KD had lower plasma glucose and insulin concentrations during an oral glucose tolerance test (oGTT) despite similar body weights (Figure 3G), demonstrating that hepatic PKCε activation contributes to HFD-induced hyperglycemia and more prominently hyperinsulinemia during a physiological feeding behavior by directly mediating HIR. Additionally, in regular chow-fed rats, liver-specific PKCε KD abrogated acute DGAT2 KD’s induction of HIR during a hyperinsulinemic-hyperglycemic clamp (Figures 3H and S3G), indicating that hepatic PKCε is the necessary mediator of PM sn-1,2-DAG accumulation-induced HIR in the acute DGAT2 KD model. Liver-specific PKCε KD in regular chow-fed rats, however, did not further enhance hepatic insulin sensitivity (Figure S3H).
Thus, liver-specific KD of PKCε with an MOE-GalNAc-modified liver-targeted ASO for PKCε specifically ameliorated lipid-induced HIR in HFD-fed rats.
Liver-Specific OE of Constitutively Active PKCε Induces HIR
Taken together, these data so far demonstrate that PKCε is necessary for the development of sn-1,2-DAG-mediated HIR. To determine whether PKCε activation per se is sufficient to cause HIR, we used an AAV vector to overexpress a constitutively active isoform of PKCε (PKCε A159E) (Takeishi et al., 2000) driven by the liver-specific promoter, thyroxine binding globulin (TBG), in regular chow-fed rats. The A159E mutation prevents the binding of the pseudo-substrate domain to the catalytic domain, rendering the kinase constitutively active (Takeishi et al., 2000). The AAV injection doubled the amount of total PKCε content in the liver with ~6-fold higher translocation without altering WAT PKCε content (Figure 4A). The higher hepatic constitutively active PKCε content impaired insulin-stimulated hepatic glycogen synthesis by ~40% during a hyperinsulinemic-hyperglycemic clamp, resulting in lower post-clamp hepatic glycogen content (Figures 4B and S4A–S4G). HIR was observed at the level of IRK, reflected by >50% lower insulin-stimulated pIRK-Y1162 levels, accompanied by impaired downstream insulin-stimulated Akt-S473, GSK3β-S9 and FOXO1-S256 phosphorylation in hepatic PKCε OE rats compared to the control group (Figures 4C and 4D). Furthermore, we also observed that there was ~2-fold higher pIRK-T1160 levels with liver-specific OE of constitutively active PKCε (Figure 4E).
Figure 4. Liver-Specific OE of Constitutively Active PKCε Induces HIR.
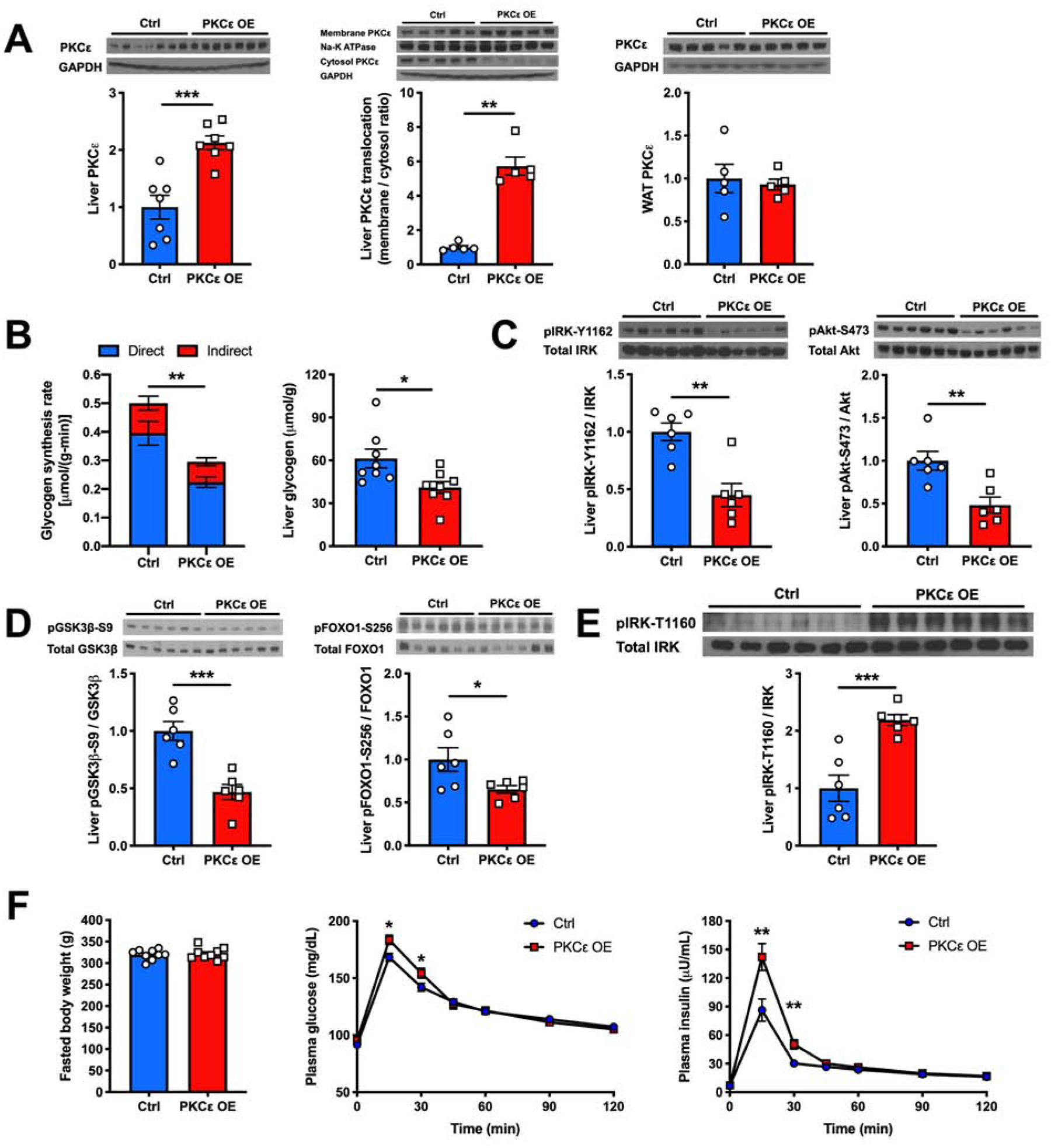
(A) Hepatic PKCε protein content, hepatic PKCε translocation from cytosol to membrane and WAT PKCε content measured by western blot (top) and its quantification (bottom).
(B) Hepatic glycogen synthesis rate during a hyperinsulinemic-hyperglycemic clamp and post-clamp liver glycogen content in Ctrl vs hepatic PKCε OE rats.
(C) and (D) Levels of insulin-stimulated liver pIRK-Y1162, pAkt-S473, pGSK3β-S9 and pFOXO1-S256 as measured by western blot (top) and with its quantification (bottom).
(E) Levels of liver pIRK-T1160 as measured by western blot (top) and with its quantification (bottom).
(F) Fasted body weight, plasma glucose and insulin levels during an oGTT in Ctrl vs hepatic PKCε OE rats.
In all panels, data are the mean±S.E.M. In (A), n = 5 or 7 per group. In (B), n = 8 per group. In (C), (D) and (E), n = 6 per group. In (F), n = 9 per group. *P < 0.05, **P < 0.01 and ***P < 0.001.
Despite marked reductions in hepatic insulin signaling and hepatic glycogen synthesis, hepatic OE of PKCε-A159E did not affect EGP during the clamp (Figure S4E). This result is consistent with prior studies demonstrating a relatively minor role for direct hepatic insulin signaling in regulating hepatic glucose production in mice and rats without hepatic steatosis and the critical role of insulin to suppress WAT lipolysis to regulate hepatic glucose production by the indirect pathway (Perry et al., 2015a; Petersen and Shulman, 2018; Samuel and Shulman, 2016, 2018). Consistent with this model, plasma non-esterified fatty acid (NEFA) concentrations were equally suppressed by >90% in both groups resulting in near complete suppression of EGP in both groups during the clamp due to the indirect pathway regulation (Figure S4B).
Taken together, these data support the sufficiency of hepatic PKCε in mediating HIR. Additionally, regular chow-fed rats with liver-specific OE of constitutively active PKCε had higher plasma glucose and insulin concentrations during an oGTT despite similar body weights (Figure 4F), demonstrating that hepatic PKCε activation per se is sufficient to drive hyperglycemia and more prominently hyperinsulinemia during a physiological feeding behavior by directly mediating HIR.
DISCUSSION
NAFLD and HIR are both highly prevalent conditions that are strongly associated with each other, with the underlying molecular links between them being widely debated. Multiple studies have reported the dissociation between hepatic triglyceride accumulation and HIR (Brown et al., 2010; Cantley et al., 2013; Farese et al., 2012; Luukkonen et al., 2016; Minehira et al., 2008; Monetti et al., 2007; Sun et al., 2012), suggesting that triglycerides are likely not the mediating factor in causing HIR. Instead, among all the lipid species associated with NAFLD, DAGs and ceramides are the leading candidates thought to be involved (Chavez and Summers, 2012; Kusminski and Scherer, 2019; Petersen and Shulman, 2017; Samuel and Shulman, 2016). Since the mechanisms currently proposed for ceramides to induce hepatic insulin resistance target the distal insulin signaling steps (e.g. at the level of Akt) (Chavez and Summers, 2012), they cannot explain the occurrence of proximal defects in hepatic insulin signaling at the level of the insulin receptor (Caro et al., 1986; Samuel et al., 2007).
We established the importance of PM sn-1,2-DAGs in the development of HIR using a model of acute DGAT2 KD. Since DGAT2 has been shown to be an ER-resident protein (McFie et al., 2011; Stone et al., 2009), the accumulation of sn-1,2-DAGs in the ER following the acute hepatic DGAT2 KD is likely a direct result of transient substrate backup, while the extra DAGs are rapidly shuttled to other membrane compartments such as the PM, where they promote translocation of PKCε to the PM to induce insulin resistance.
Consistent with the rodent studies, we found that liver PM sn-1,2-DAG content and pIRK-T1160 levels also tracked with HIR in humans. The concomitant accumulation of PM sn-2,3-DAGs and sn-1,3-DAGs in the livers of individuals with HIR may simply reflect the condition of chronic excessive fatty acid supply, lipogenesis and/or lipolysis in the livers with NAFLD and likely does not contribute to the defects in hepatic insulin signaling. We also observed an accumulation of lipid droplet DAGs in liver samples from humans with HIR, consistent with our prior report where we saw a similar accumulation in lipid droplet DAGs in liver samples of humans with HIR using a relatively crude three-compartment subcellular fractionation method (Kumashiro et al., 2011). Though we also saw an association between plasma membrane DAG content and insulin resistance, the association between the lipid droplet DAGs and insulin resistance was stronger. However, this likely reflects the concurrent expansion of the lipid droplet pool in fatty liver in subjects with obesity under chronic conditions of increased substrate flux to the liver. Consistent with the hypothesis, we have previously demonstrated that this pool of lipid droplet DAGs does not contribute directly to HIR as demonstrated in a mouse model where CGI-58 ASO treated mice, with a marked accumulation of hepatic lipid droplet DAG content, were protected from lipid-induced HIR and manifested reduced hepatic PKCɛ translocation (Cantley et al., 2013). In addition, in the study of Ter Horst et al. (Ter Horst et al., 2017), higher cytosolic DAGs were detected in human livers with HIR. However, the high cytosolic DAG content reported likely reflects contamination from lipid droplet and membrane fractions, while our new fractionation methodology utilized in the present study has been our best attempt to resolve content of DAGs and ceramides in different subcellular compartments.
Our finding on the importance of PM sn-1,2-DAGs in mediating PKCε translocation, IRK-T1160 phosphorylation and HIR is consistent with studies demonstrating that only sn-1,2-DAGs activate novel PKCs (Boni and Rando, 1985; Nomura et al., 1986; Rando and Young, 1984), as well as that the PM is the primary site of action for PKCε to induce HIR (Petersen et al., 2016; Samuel et al., 2007). Since lipid droplet lipolysis has been shown to produce mostly sn-2,3- and sn-1,3-DAGs, which do not activate novel PKCs (Eichmann et al., 2012), our data could also provide an explanation for a few experimental models where CGI-58 and HSL (enzymes in lipolysis pathway) inhibition caused accumulation of DAGs (likely sn-2,3- and sn-1,3-DAGs) in the absence of PKCε activation and HIR (Brown et al., 2010; Haemmerle et al., 2002; Osuga et al., 2000).
In contrast to the consistent association between PM sn-1,2-DAGs and HIR, we observed no consistent association of any ceramide species in any subcellular compartment with HIR. This is consistent with prior studies that have dissociated hepatic ceramide content from HIR (Magkos et al., 2012; Perry et al., 2018; Perry et al., 2015b).
Multiple studies have suggested the involvement of PKCε in mediating lipid-induced HIR (Kumashiro et al., 2011; Raddatz et al., 2011; Samuel et al., 2004; Samuel et al., 2007), though many of the interventions used in these studies affected PKCε globally or in multiple tissues. In this regard, a recent study challenged the role of hepatic PKCε in regulating hepatic glucose metabolism (Brandon et al., 2019). Their findings argue that PKCε in liver is not important in regulating glucose metabolism, and that only PKCε in WAT is important in this matter. Consistent with our prior study (Samuel et al., 2007) Brandon et al. found that WAT PKCε has a role in the regulation of glucose metabolism. However, in contrast to the conclusions of Brandon et al., we find that hepatic PKCε also has an important role in lipid-mediated HIR and hypothesize that the discrepancies between our study and their study may be due to multiple technical issues. Firstly, as hepatic glucose production receives regulatory input from multiple sources in addition to the liver (e.g. WAT and skeletal muscle), and that insulin’s acute effects to suppress hepatic glucose production reflects insulin’s ability to directly stimulate hepatic glycogen synthesis and indirectly inhibit hepatic gluconeogenesis through inhibition of WAT lipolysis (Perry et al., 2015a; Petersen and Shulman, 2018; Samuel and Shulman, 2016, 2018), it is essential to assess insulin’s direct activation of hepatic insulin signaling parameters (e.g. IRK-Y1162, Akt-S473, GSK3β-S9 and FOXO1-S269 phosphorylation), as well as hepatic glycogen synthesis, to reliably assess the direct effects of insulin on hepatic glucose metabolism and HIR. Unfortunately, none of these parameters were assessed in the liver-specific PKCε knockout mice in the previous study (Brandon et al., 2019). Secondly, there may have been technical differences between the studies that preclude direct comparison. For example, the plasma glucose concentrations in some of the experiments of the previous study (Brandon et al., 2019) were reported to be 12–13 mM for the basal state and increased to 30–35 mM during the GTTs. Despite this severe hyperglycemia, plasma insulin response curves during the GTTs were relatively flat without a physiological response to the extremely high plasma glucose concentrations, suggesting that these mice may have been stressed during the procedures, which in turn will mask any effects that PKCε may have on hepatic insulin action. Thirdly, the Cre-lox genetic knockout mouse model used in the previous study (Brandon et al., 2019) may naturally produce different results compared to the short-term gene KD and OE rat models utilized in our studies due to potential developmental effects or strong compensatory effects in long-term genetic manipulation conditions. Finally, while it is possible that species differences might also explain the different observations, we do not think this is likely given that we and others have established a strong relationship between hepatic DAG content, PKCε translocation and HIR in mice (Abulizi et al., 2017; Camporez et al., 2019; Tillander et al., 2018) and most recently demonstrated that IRK-T1160A mice are protected from lipid-induced HIR (Petersen et al., 2016).
In order to address these limitations, we performed a series of studies in short-term genetically modified awake rats. There are multiple advantages and strengths of the awake rat model compared to the awake mouse model including the fact that rates of EGP and insulin-stimulated whole-body glucose turnover in the awake rat are much closer to rates of EGP and turnover in humans than mice (Perry et al., 2015a; Petersen and Shulman, 2018), and compared to a life-long genetic knockout model, short-term genetic modulation eliminates potential confounding developmental compensatory effects on hepatic insulin action. Using this approach, our results support both the necessity and sufficiency of hepatic PKCε activation in mediating HIR.
Taken together with the fact that IRK-T1160 is highly conserved throughout evolution down to Drosophila melanogaster, we can hypothesize that the PM sn-1,2-DAG-PKCε-IRK-T1160 axis may be evolutionarily preserved to limit insulin’s effects on promoting hepatic glycogen synthesis and reducing EGP during re-feeding following fasting and starvation with the intention to preserve blood glucose in favor of supporting central nervous system function (Petersen and Shulman, 2018). Furthermore, given the presence of PKCε in other insulin responsive tissues, such as skeletal muscle and WAT, it is likely that PM sn-1,2-DAG activation of PKCε may also be playing a similar role in causing lipid-induced insulin resistance in these tissues. Finally, given that PKCε has multiple targets besides IRK-T1160, it is likely that PKCε activation may also impact additional downstream targets to alter insulin action in these tissues (Gassaway et al., 2018).
Limitations of Study
The number of human liver samples from subjects with and without NAFLD and HIR were relatively small, and we were limited by the amount of had tissue available from each biopsy. We therefore to prioritize our analyses to yield the most novel information. We therefore prioritized these samples to assess DAG stereoisomers and ceramides in the five compartments in subjects with adequate liver tissues and subjected the rest of the tissue samples to an IRK-T1160 phosphorylation assay. Unfortunately, we did not have enough remaining tissues to assess PKCε translocation in these liver biopsies and future studies will be required to further address this relationship. However, we have previously established that increased PKCε translocation correlates with HIR in humans with NAFLD in the same cohort of human subjects (Ter Horst et al., 2017).
Additionally, our study mostly investigated the importance of PM sn-1,2-DAGs and hepatic PKCε in relatively acute models of HIR. However, our data from human subjects suggest that the proposed model may also play an important role in long-term conditions of lipid-induced HIR associated with NAFLD.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Gerald I. Shulman (gerald.shulman@yale.edu).
Materials Availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rat Models
All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards. 150–250 g male Sprague-Dawley rats at ~6–8 weeks of age were purchased from Charles River Laboratories (Wilmington, MA) and were housed in the Yale Animal Resources Center at 23°C under 12-hour light (7:00am–7:00pm)/12-hour dark cycles (7:00pm–7:00am). The rats were fed ad libitum with free access to water and were in good health condition. Upon arrival from the vendor, the rats remained on their regular living condition without any experimental perturbations for at least 3–4 days for acclimation and stress-minimization. The rats were group housed (2–3 per cage) until they underwent surgery under general isoflurane-induced anesthesia for insertion of polyethylene catheters in the common carotid artery (PE50 tubing, Instech Solomon, Plymouth Meeting, PA), the jugular vein (PE90 tubing, Instech) and the stomach (PE90 tubing, Instech) or injection of AAV solution into the portal vein, after which they were singly housed until time of killing. The rats were either fed with a regular chow diet (Harlan Teklad #2018, Indianapolis, IN) or a safflower oil-based HFD with 60 kcal% from fat (Dyets #112245, Bethlehem, PA) for the amount of time as indicated in the main text. The rats were randomly allocated into experimental groups with matched body weight.
Human Subjects
Human subjects were recruited from the outpatient clinics of two obesity centers in the Amsterdam metropolitan area. Inclusion and exclusion criteria have been described (Ter Horst et al., 2017). Liver fat content was measured using proton magnetic resonance spectroscopy as described (Ter Horst et al., 2017). Insulin sensitivity was measured during a two-step hyperinsulinemic-euglycemic clamp using a stable isotope-labeled glucose tracer as described (Ter Horst et al., 2017). Liver biopsies were collected from the right liver lobe by an experienced surgeon during bariatric surgeries scheduled <2 weeks after clinical assessments. All human studies were approved by the Amsterdam University Medical Center medical ethics committee. The study was registered in the Netherlands Trial Register (NTR4666). All subjects provided written informed consent prior to inclusion. Please refer to Table S1 for the baseline characteristics of the human subjects included in this study.
METHOD DETAILS
Animal Studies
At the end of all animal studies, rats were killed by intravenous pentobarbital sodium injection (150 mg/kg) with tissues and plasma collected for further analysis. Tissues were rapidly harvested and snap-frozen with clamps precooled in liquid nitrogen and then stored at −80°C for further analyses. For clamps, all tracers, insulin and glucose were infused via a catheter placed in the carotid artery at least one week before the study, and all blood collections were performed via a catheter placed in the jugular vein together with the arterial catheter.
For the DGAT2 ASO dosing, control and DGAT2 ASO (dissolved in PBS) were dosed once at the dose of 100 mg/kg via intraperitoneal (IP) injection two days before the clamp/tissue collection studies. For the PKCε ASO dosing, control and PKCε ASO (dissolved in PBS) were dosed twice per week at the dose of 0.75 mg/kg via IP injection for three weeks prior to the clamp/oGTT/tissue collection studies. For the PKCε A159E AAV dosing, control (AAV8-TBG-eGFP, dissolved in PBS with 5% glycerol) and PKCε A159E AAV (AAV8-TBG-rPKCe A159E, dissolved in PBS with 5% glycerol) were dosed once at the dose of 2e13 GC/kg via portal vein injection four weeks prior to the clamp/oGTT/tissue collection studies.
Hyperinsulinemic-Glucose Clamps
For the hyperinsulinemic-euglycemic clamp, after 14–16 hours of overnight fasting, [1,2,3,4,5,6,6-2H7] glucose was infused at the rate of 0.1 mg/[kg-min] for 2 hours during basal infusion (−120 min to 0 min) to measure basal glucose turnover rates. Blood samples were collected at −20 min, −10 min and 0 min to measure plasma glucose, insulin, NEFA concentrations and tracer APE. Immediately after the basal infusion, insulin was infused at the rate of 4 mU/[kg-min] to achieve physiological hyperinsulinemia during clamp (0 min to 120 min). At the same time, 20% dextrose was infused at variable rates to maintain plasma glucose level at ~100–110 mg/dL. Infusion of [1,2,3,4,5,6,6-2H7] glucose continued at the rate of 0.1 mg/[kg-min] during the clamp to measure insulin-stimulated glucose turnover rates. Blood samples were collected at 100 min, 110 min and 120 min to measure plasma glucose, insulin, NEFA concentrations and tracer APE. At the end of the clamp, rats were euthanized with tissues collected as described above.
For the hyperinsulinemic-hyperglycemic clamp, after 14–16 hours of overnight fasting, [6,6-2H2] glucose was infused at the rate of 0.1 mg/[kg-min] for 2 hours during basal infusion (−120 min to 0 min) to measure basal glucose turnover rates. Blood samples were collected at −20 min, −10 min and 0 min to measure plasma glucose, insulin, NEFA concentrations and tracer atom percent excess (APE). Immediately after the basal infusion, insulin was infused at the rate of 4 mU/[kg-min] achieve physiological hyperinsulinemia during clamp (0 min to 90 min). A 1:1 mixture of somatostatin-14 and somatostatin-28 (w/w) was also infused at the rate of 4 μg/[kgmin] to suppress endogenous insulin secretion. At the same time, 20% dextrose labeled with 50% [13C6] glucose was infused at variable rates to maintain plasma glucose level at ~150–160 mg/dL, measure rates of insulin-stimulated hepatic glycogen synthesis and glucose turnover. Blood samples were collected at 10 min, 20 min, 30 min, 40 min, 50 min, 60 min, 75 min and 90 min to measure plasma glucose, insulin, NEFA concentrations and tracer APE. At the end of the clamp, rats were euthanized with tissues collected as described above.
Oral Glucose Tolerance Tests
After 14–16 hours of overnight fasting, a single dose of 1 g/kg dextrose was injected via the gastric catheter placed at least one week before the study, and blood samples were collected at 0 min, 15 min, 30 min, 45 min, 60 min, 90 min and 120 min via the jugular vein catheter placed together with the gastric catheter to measure plasma glucose and insulin levels. At the end of the oral glucose tolerance tests, rats were euthanized with tissues collected as described above.
Flux Measurements
All turnover rates were calculated based on the following equation:
APE designates the atom percent enrichment measured by gas chromatography-mass spectrometry (GC-MS). Briefly, glucose samples were deproteinized with 1:1 ZnSO4:Ba(OH)2, derivatized with 1:1 acetic anhydride:pyridine (v/v) at 65°C with methanol added afterwards to quench the reaction. Derivatized samples were then analyzed on GC-MS with CI mode to measure enrichment.
For the calculation of glycogen synthesis rates, liver glycogen m+6 APE () and plasma glucose m+6 APE () were determined by GC-MS. Liver UDP-glucose m+6 APE () was determined by liquid chromatography-MS/MS (LC-MS/MS). Rates of liver glycogen synthesis direct pathway Vdirect were calculated based on the following equation:
The fractional direct pathway contribution D was calculated as:
Total glycogen synthesis rates Vtotal were calculated as:
To measure liver total glycogen, liver samples were first homogenized in 0.6 N perchloric acid (PCA). Part of the homogenate was then neutralized with 1M KHCO3 and digested with amyloglucosidase (2 mg/mL) dissolved in acetate buffer (0.4 M, pH 4.8) for 2 hours at 37–40°C. Glucose levels were determined in samples before (free glycose) and after the amyloglucosidase digestion (total glucose) to calculate liver total glycogen content.
To measure liver glycogen m+6 APE, liver samples were ethanol-precipitated from PCA homogenates, dialyzed extensively against deionized water to remove free glucose before digested with amyloglucosidase. Digested glucose samples were then derivatized as described above prior to GC-MS analysis.
Biochemical Analyses
Plasma glucose was measured enzymatically using the YSI Glucose Analyzer (Yellow Springs, OH). Plasma insulin was measured by the Yale Diabetes Research Center Radioimmunoassay Core using radioimmunoassay. Plasma NEFA was measured using the enzymatic colorimetric method by Wako reagents (Wako Diagnostics, Mountain View, CA). Liver triglyceride was first extracted with 2:1 chloroform:methanol (v/v). Samples were then mixed with sulfuric acid (1 M) and centrifugated to achieve phase separation. The organic phase was used to measure triglyceride content with the Sekisui triglyceride-SL reagent spectrophotometrically. Liver glycogen content measurement was described above.
Tissue Analyses
DAGs and ceramides were extracted from tissues with 2:1 chloroform:methanol (v/v) with 0.01% butylated hydroxytoluene. Known amounts of internal standards (1,2-dinonadecanoin for DAGs, N-heptadecanoyl-D-erythro-sphingosine for ceramides) were added during the extraction process. Afterwards, samples were dried down and re-dissolved in 95:5:0.5 hexane/methylene chloride/ethyl ether (v/v/v) before analyzed on LC/MS-MS as described below.
For western blots, tissues were homogenized in ice-cold homogenization buffer (20 mM Tris·HCl, pH 7.4, 5 mM EDTA, 0.25 mM EGTA, 10 mM Na4P2O7, 1% Nonidet P-40, and protease and phosphatase inhibitor mixtures; Roche Diagnostics) and centrifuged at 13,000 g at 4 °C for 10 min to collect supernatant. Protein concentration was then determined by the Bradford method (Thermo Scientific). Protein samples were then diluted to 2 μg/μL with the addition of SDS buffer and β-mercaptoethanol and boiled for 5 min at 95°C. Afterwards, 20 μg of protein was loaded and resolved by SDS/PAGE using 4–12% gradient gels (Life Technologies) and transferred to polyvinylidene difluoride membranes (DuPont) using semi-dry transfer. Membranes were then blocked for 60 min at room temperature in 5% bovine serum albumin (BSA) and incubated overnight with primary antibody. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 1 hour. Detection was performed with enhanced chemiluminescence.
For assaying the IRK-T1160 phosphorylation, after protein concentration quantitation, protein samples were first immunoprecipitated by Dynabeads M-270 Epoxy (Invitrogen) conjugated with D2 anti-IR alpha-subunit antibody. Primary antibody solution was diluted 1:100 – 1:200 for pIRK-T1160 detection.
PKCε Translocation Assay
Tissue samples were homogenized in ice-cold buffer A (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.25 mM EGTA, 250 mM sucrose, and freshly added protease and phosphatase inhibitors, Roche Diagnostics). Debris was removed by centrifugation (5 min, 280 g, 4°C). Lysate was centrifuged (60 min, 100,000 g, 4°C), and an aliquot of the supernatant was saved as the cytosolic fraction. The pellet was washed once in ice-cold buffer B (250 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.25 mM EGTA, 2% Triton X-100, and freshly added protease and phosphatase inhibitors). The pellet was resuspended in buffer B by sonication, incubated at 4°C for 45 minutes to solubilize membrane proteins, and centrifuged (60 min, 100,000 g, 4°C). An aliquot of the supernatant was saved as the membrane fraction. The resulting protein samples were subjected to western blot analysis as described above. The ratio between membrane PKCε (normalized to Na-K ATPase intensity) to cytosolic PKCε (normalized to GAPDH intensity) was calculated as an index of PKCε translocation.
Five-compartment Ultracentrifugation Fractionation
The protocol was adapted and modified based on previous studies (Bogan et al., 2001; Castle, 2003). Fresh tissues (60–150 mg) were first homogenized in cold (4°C) TES buffer (250 mM sucrose, 10 mM Tris - pH 7.4, 0.5 mM EDTA) with a Dounce homogenizer. The homogenate was then centrifuged (at 12,000 rpm with SS-34 rotor or 17,000 g, 15 min, 4°C) to separate pellet A and supernatant A. The top lipid layer was collected as lipid droplet fraction. Pellet A was washed by being resuspended in TES buffer and centrifuged (at 12,000 rpm with SS-34 rotor or 17,000 g, 20 min, 4°C). Pellet A was then collected and resuspended in TES buffer and gently layered on top of 1.12 M sucrose buffer cushion in ultracentrifuge tubes. The pellet A samples were then centrifuged (at 35,000 rpm with TLS-55 rotor or 105,000 g, 20 min, 4°C) to separate pellet B, interface B and supernatant B. The interface B was collected, resuspended in TES buffer and centrifuged (at 37,000 rpm with TLA-100.2 rotor or 60,000 g, 9 min, 4°C) to obtain pellet C. Pellet C was then washed by being resuspended in TES buffer and centrifuged (at 37,000 rpm with TLA-100.2 rotor or 60,000 g, 9 min, 4°C) and was designated as plasma membrane fraction. The pellet B was washed by being resuspended in TES buffer and centrifuged (at 12,000 rpm with SS-34 rotor or 17,000 g, 15 min, 4°C) and was collected as mitochondria fraction. The supernatant A was centrifuged (at 65,000 rpm with Ti-70.1 rotor or 390,000 g, 75 min, 4°C) to separate pellet D and supernatant D. Pellet D was washed by being resuspended in TES buffer and centrifuged (at 65,000 rpm with Ti-70.1 rotor or 390,000 g, 60 min, 4°C) and was collected as the endoplasmic reticulum fraction. Supernatant D was collected as the cytosol fraction.
Diacylglycerol Stereoisomer Separation and Quantitation
Chiral analysis of DAGs were performed by LC-MS/MS using electrospray ionization on an AB Sciex Qtrap 6500 interfaced to Shimadzu UFLC with 2 LC-20AD pumps (and degassers), SIL-20AC xR autosampler. Chromatographic separation was performed using Luna 5u Silica (100A, 250×2.0mm) and LUX 5u Cellulose-1 (250×4.6mm) columns connected in series with an isocratic solvent of hexane:isopropanol (300:7) with a flow rate of 0.6 ml/min. TAGs elute between ~4 to 8 minutes, and DAGs elute between ~11 and 17 minutes. For DAGs with the same FA composition, the order of elution was sn-1,3-DAG → sn-2,3-DAG → sn-1,2-DAG. For any specific stereoisomer (i.e., sn-1,2-DAG, sn-2,3-DAG, or sn-1,3-DAG) the order of elution generally increases with the degree of unsaturation. For example, C18:1 C18:1 → C18:1 C18:2 → C18:2 C18:2. Standards were used to establish retention times, matrix effects, and response relative to the internal standard (C17, C17-DAG). No matrix effects were detected in the five subcellular compartments. However, the position of the FAs on the DAGs have not been established. For example, for the sn-1,2 DAG assigned as C18:0 C16:0, C16:0 could be on either the 1 or the 2 positions. In the process of optimization, it was found that the use of cartridges (e.g., DiOH) or silica TLC plates to pre-separate the DAGs from TAGs results leads to intramolecular trans-esterification (e.g. the pre-separation of a sn-1,2 DAG will produce a mixture of sn-1,2, sn-2,3, and sn-1,3 DAGs). Thus, no additional separation steps following centrifugation to obtain cellular fraction were done prior to LC-MS/MS analysis.
Generation of Rabbit-Mouse Chimeric Anti-pIRK-T1160 Monoclonal Antibody
All experimental procedures involving animals were performed under a license granted by the national Danish authority, The Animal Experiment Inspectorate. Rabbits were immunized four times with a 1:1 mixture of CYEpTDY and YEpTDYC conjugated to keyhole limpet hemocyanin via the terminal cysteines. All immunizations were performed subcutaneously using 50 μg of antigen in Ribi adjuvant. Blood was drawn ten days after the last immunization and used for sera tests. Splenic B cells were harvested from rabbits with best titers and single-cell sorted on a Sony SH800 cell sorter using FITC-labelled anti-rabbit IgG (Jackson ImmunoResearch) and Biotin-GGSGGSDIYEpTDYYRKG labelled with Streptavidin-PE (Jackson ImmunoResearch). Sytox red (Invitrogen) was included as a dead live stain. Sorted cells were expanded in vitro for 10 days on a layer of irradiated EL4-B5 cells, and the culture supernatant was harvested and used for ELISA screening. ELISA-positive clones were defined as binding the peptide used for immunization, but not binding the non-phosphorylated version of the same peptide. Hits were cloned as described in Lightwood et al. 2006 (Lightwood et al., 2006). In short, the heavy/light chain variable regions were cloned using RT-PCR with gene specific primers and the amplicons inserted into an expression vector containing mouse IgG1/kappa constant regions. The construct was expressed in HEK cells, and the specificity of the chimeric rabbit/mouse antibody was validated by ELISA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Most of our data follow normal distribution, and comparisons were performed using the unpaired 2-tailed Student’s t-test, with significance defined as a P-value < 0.05. For data that do not follow normal distribution, non-parametric test was used to calculate the P-value. GraphPad Prism 8.0 (San Diego, CA) was used for all statistical analysis. In most cases, n = 6–8 per group, unless otherwise indicated in the figure legends. Data are presented as the mean±S.E.M.
All the tracer APE analyses (measured by GC-MS), DAG and ceramide content analyses (measured by LC-MS/MS) and insulin level assays were performed in a blinded fashion. For the animal studies, sample sizes were preselected using power calculation. Exclusion was applied only when the technical procedure failed (e.g. catheter failure causing inability to complete the clamp). Approximately 1 or 2 out of 10 samples in the animal studies were exclude based on this criteria.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DGAT2 | Santa Cruz Biotechnology | Cat#sc-293211 |
| GAPDH | Cell Signaling Technology | Cat#5174 |
| IRK pY1162 | Cell Signaling Technology | Cat#3918 |
| IRK | Cell Signaling Technology | Cat#3025 |
| Akt pS473 | Cell Signaling Technology | Cat#4060 |
| Akt | Cell Signaling Technology | Cat#2920 |
| GSK3β pS9 | Cell Signaling Technology | Cat#5558 |
| GSK3β | Cell Signaling Technology | Cat#12456 |
| FOXO1 pS256 | Cell Signaling Technology | Cat#84192 |
| FOXO1 | Cell Signaling Technology | Cat#2880 |
| PKCε | BD Biosciences | Cat#610086 |
| Na-K ATPase | Abcam | Cat#ab7671 |
| IRK pT1160 | This paper | N/A |
| Calnexin | Abcam | Cat#ab22595 |
| VDAC | Abcam | Cat#ab14734 |
| Perilipin | Cell Signaling Technology | Cat#9349 |
| Bacterial and Virus Strains | ||
| AAV8-TBG-eGFP | Vector Biolabs | SKU#VB1743 |
| AAV8-TBG-rPKCe(A159E) | Vector Biolabs | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human insulin | Novo Nordisk | NDC 0169-1833-11 |
| [1,2,3,4,5,6,6-2H7] glucose | Cambridge Isotopes | Cat#DLM-2062 |
| [6,6-2H2] glucose | Cambridge Isotopes | Cat#DLM-349 |
| [1,2,3,4,5,6-13C6] glucose | Cambridge Isotopes | Cat#CLM-1396 |
| Somatostatin-14 | Bachem | Cat#H-1490 |
| Somatostatin-28 | Bachem | Cat#H-4955 |
| Amyloglucosidase | Millipore-Sigma | Cat#A7420 |
| Diacylglycerol concentration standard | Indofine Chemical | Cat#32–1902 |
| Ceramide concentration standard | Avanti Polar Lipids | Cat#860517P |
| Critical Commercial Assays | ||
| HR Series NEFA-HR (2) Color Reagent A | Wako | Cat#999–34691 |
| HR Series NEFA-HR (2) Solvent A | Wako | Cat#995–34791 |
| HR Series NEFA-HR (2) Color Reagent B | Wako | Cat#991–34891 |
| HR Series NEFA-HR (2) Solvent B | Wako | Cat#993–35191 |
| NEFA Standard Solution | Wako | Cat#276–76491 |
| Triglyceride-SL reagent | Sekisui | Cat#236–99 |
| Experimental Models: Organisms/Strains | ||
| Sprague-Dawley rats | Charles River | Strain code:400 |
| Oligonucleotides | ||
| MOE control ASO: CCTTCCCTGAAGGTTCCTCC | Ionis Pharmaceuticals | ISIS-141923 |
| DGAT2 ASO: GCATTACCACTCCCATTCTT | Ionis Pharmaceuticals | ISIS-369235 |
| MOE GalNAc control ASO: CCTTCCCTGAAGGTTCCTCC | Ionis Pharmaceuticals | ISIS-716837 |
| PKCε ASO: GCCAGCTCGATCTTGCGCCC | Ionis Pharmaceuticals | ISIS-1289956 |
| Software and Algorithms | ||
| GraphPad Prism 8.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| Regular chow diet | Envigo | Cat#2018 |
| High-fat diet | Dyets | Cat#112245 |
Highlights.
Plasma membrane bound sn-1,2-diacylglycerols cause hepatic insulin resistance
PKCε is necessary and sufficient for mediating lipid-induced hepatic insulin resistance
PKCε promotes hepatic insulin resistance via phosphorylating insulin receptor Thr1160
Ceramides do not consistently track with hepatic insulin resistance
Context and Significance.
Nonalcoholic fatty liver disease (NAFLD) affects 1 in 3 Americans and is strongly associated with liver insulin resistance and type 2 diabetes. However, the specific lipid molecules, their intracellular location, and the cellular mechanisms for lipid-induced insulin resistance are widely debated. Here Lyu et al. show that sn-1,2-diacylglycerols, bound to the plasma membrane, are the key lipid species responsible for activation of PKCε, an enzyme that is key to mediating liver insulin resistance. They further show that PKCε is both necessary and sufficient for the development of lipid-induced liver insulin resistance. These findings provide important new insights into the pathogenesis of liver insulin resistance associated with NAFLD and type 2 diabetes.
ACKNOWLEDGMENTS
The authors thank Jianying Dong, John Stack and Xianman Zhang for their excellent technical assistance. This study was supported by grants from the United States Public Health Service (R01 DK116774, R01 DK119968, R01 DK113984, P30 DK045735, R01 DK092661) and a VA Merit Award (I01 BX000901).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that no relevant conflicts of interest exist.
REFERENCES
- Abulizi A, Perry RJ, Camporez JPG, Jurczak MJ, Petersen KF, Aspichueta P, and Shulman GI (2017). A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J 31, 2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS, McKee AE, and Lodish HF (2001). Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol Cell Biol 21, 4785–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni LT, and Rando RR (1985). The nature of protein kinase C activation by physically defined phospholipid vesicles and diacylglycerols. J Biol Chem 260, 10819–10825. [PubMed] [Google Scholar]
- Brandon AE, Liao BM, Diakanastasis B, Parker BL, Raddatz K, McManus SA, O’Reilly L, Kimber E, van der Kraan AG, Hancock D, et al. (2019). Protein Kinase C Epsilon Deletion in Adipose Tissue, but Not in Liver, Improves Glucose Tolerance. Cell Metab 29, 183–191 e187. [DOI] [PubMed] [Google Scholar]
- Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer J, Shah R, et al. (2010). CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res 51, 3306–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camporez JP, Lyu K, Goldberg EL, Zhang D, Cline GW, Jurczak MJ, Dixit VD, Petersen KF, and Shulman GI (2019). Anti-inflammatory effects of oestrogen mediate the sexual dimorphic response to lipid-induced insulin resistance. J Physiol 597, 3885–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, et al. (2013). CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A 110, 1869–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro JF, Ittoop O, Pories WJ, Meelheim D, Flickinger EG, Thomas F, Jenquin M, Silverman JF, Khazanie PG, and Sinha MK (1986). Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J Clin Invest 78, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle JD (2003). Purification of organelles from mammalian cells. Curr Protoc Immunol Chapter 8, Unit 8 1B. [DOI] [PubMed] [Google Scholar]
- Chavez JA, and Summers SA (2012). A ceramide-centric view of insulin resistance. Cell Metab 15, 585–594. [DOI] [PubMed] [Google Scholar]
- Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, et al. (2007). Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 282, 22678–22688. [DOI] [PubMed] [Google Scholar]
- Eichmann TO, Kumari M, Haas JT, Farese RV Jr., Zimmermann R, Lass A, and Zechner R (2012). Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem 287, 41446–41457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV Jr., Zechner R, Newgard CB, and Walther TC (2012). The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15, 570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassaway BM, Petersen MC, Surovtseva YV, Barber KW, Sheetz JB, Aerni HR, Merkel JS, Samuel VT, Shulman GI, and Rinehart J (2018). PKCepsilon contributes to lipid-induced insulin resistance through cross talk with p70S6K and through previously unknown regulators of insulin signaling. Proc Natl Acad Sci U S A 115, E8996–E9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, and Zechner R (2002). Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 277, 4806–4815. [DOI] [PubMed] [Google Scholar]
- Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, et al. (2011). Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 108, 16381–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, and Scherer PE (2019). Lowering ceramides to overcome diabetes. Science 365, 319–320. [DOI] [PubMed] [Google Scholar]
- Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, and Lawson AD (2006). Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods 316, 133–143. [DOI] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, Hyotylainen T, and Yki-Jarvinen H (2016). Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 64, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, Varela JE, Brunt EM, Patterson BW, and Klein S (2012). Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142, 1444–1446 e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFie PJ, Banman SL, Kary S, and Stone SJ (2011). Murine diacylglycerol acyltransferase-2 (DGAT2) can catalyze triacylglycerol synthesis and promote lipid droplet formation independent of its localization to the endoplasmic reticulum. J Biol Chem 286, 28235–28246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minehira K, Young SG, Villanueva CJ, Yetukuri L, Oresic M, Hellerstein MK, Farese RV Jr., Horton JD, Preitner F, Thorens B, et al. (2008). Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res 49, 2038–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV Sr., et al. (2007). Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6, 69–78. [DOI] [PubMed] [Google Scholar]
- Nomura H, Ase K, Sekiguchi K, Kikkawa U, Nishizuka Y, Nakano Y, and Satoh T (1986). Stereospecificity of diacylglycerol for stimulus-response coupling in platelets. Biochem Biophys Res Commun 140, 1143–1151. [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Petersen MC, Zhang D, Vatner DF, Perry RJ, Abulizi A, Haedersdal S, Zhang XM, Butrico GM, Samuel VT, et al. (2020). Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc Natl Acad Sci U S A 117, 8166–8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, et al. (2000). Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci U S A 97, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. (2015a). Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, Song JD, Zhang D, Zhang XM, Nozaki Y, Dufour S, et al. (2018). Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. Cell Metab 27, 210–217 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang D, Zhang XM, Boyer JL, and Shulman GI (2015b). Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang XM, et al. (2016). Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 126, 4361–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2017). Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci 38, 649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 98, 2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, et al. (2014). Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res 42, 8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, Wilks D, Preston E, Hegarty BD, Leitges M, et al. (2011). Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 54, 1447–1456. [DOI] [PubMed] [Google Scholar]
- Rando RR, and Young N (1984). The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 122, 818–823. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, and Shulman GI (2004). Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279, 32345–32353. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, and Shulman GI (2007). Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2018). Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab 27, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI (2000). Cellular mechanisms of insulin resistance. J Clin Invest 106, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Zhou P, Han J, Walther TC, and Farese RV Jr. (2009). The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem 284, 5352–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, et al. (2012). Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med 18, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, and Walsh RA (2000). Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circ Res 86, 1218–1223. [DOI] [PubMed] [Google Scholar]
- Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, Romijn JA, Nieuwdorp M, Zhang D, Samuel VT, et al. (2017). Hepatic Diacylglycerol-Associated Protein Kinase Cepsilon Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep 19, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillander V, Minami A, Alves-Bezerra M, Coleman RA, and Cohen DE (2018). Thioesterase superfamily member 2 promotes hepatic insulin resistance in the setting of Glycerol-3-phosphate acyltransferase 1-induced steatosis. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not generate any unique datasets or code.