

Abstract
Excessive bradykinin (BK) stimulation is responsible for the exaggerated permeabilization of the endothelium in angioedema. However, the molecular mechanisms underlying these responses have not been investigated. BK receptors are Gq-protein-coupled receptors phosphorylated by G protein-coupled receptor kinase 2 (GRK2) with a hitherto unknown biological and pathophysiological significance. In the present study, we sought to identify the functional role of GRK2 in angioedema through the regulation of BK signaling. We found that the accumulation of cytosolic Ca2+ in endothelial cells induced by BK was sensitive to GRK2 activity, as it was significantly augmented by inhibiting the kinase. Accordingly, permeabilization and NO production induced by BK were enhanced, as well. In vivo, mice with reduced GRK2 levels in the endothelium (Tie2-CRE/GRK2fl+/fl−) exhibited an increased response to BK in terms of vascular permeability and extravasation. Finally, patients with reduced GRK2 levels displayed a severe phenotype of angioedema. Taken together, these findings establish GRK2 as a novel pivotal regulator of BK signaling with an essential role in the pathophysiology of vascular permeability and angioedema.
Keywords: Angioedema, Bradykinin, Calcium, CaMKII, Endothelium, GPCR, GRK2
Graphical Abstract
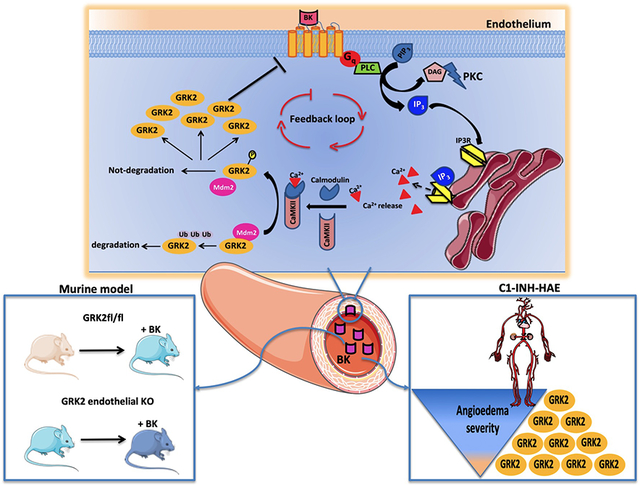
Introduction
Angioedema is a clinical condition characterized by blood vessel dilation and increased vascular permeability, which can be life-threatening when it involves the upper airways or gastrointestinal tract1-6. The functional role of vasoactive and proinflammatory mediators released, mainly bradykinin (BK), in determining vascular responses during angioedema attacks, has been extensively studied7-10. On the other hand, much less is known on the molecular and biochemical features of the vascular tree in patients with angioedema, which in turn could explain the hefty variability of the clinical outcome. Accordingly, the emerging association with atherosclerosis and endothelial dysfunction strongly supports the view of angioedema as an endothelial disorder9, 11, 12. Therefore, while BK accumulation is seen as a causative mechanism, especially in C1 inhibitor (C1-INH) deficiency and angiotensin converting enzyme (ACE) inhibitor-related forms of angioedema1, 13, 14, the contribution of additional mechanisms linked to endothelial properties has to be considered.
BK is a vasoactive nonapeptide derived from enzymatic digestion of high molecular weight kininogen by kallikreins, and is an important regulator of endothelial homeostasis15-17. Indeed, BK induces endothelial-dependent vasodilation18, 19 and permeability20 by activating specific cell surface receptors (BK Receptors, BRs), belonging to the broad family of G-protein coupled receptors (GPCRs)21-27. BRs are coupled to Gαq28 and their stimulation elicits calcium (Ca2+) mobilization from the endoplasmic reticulum via inositol 1,4,5-trisphosphate receptors29, 30. BRs are regulated by desensitization through phosphorylation events on serine and threonine residues on their C-terminus31, 32. Despite the potential pathophysiological relevance of this phenomenon in ensuring the attenuation of BK signaling upon activation, the underlying molecular mechanisms have been poorly investigated. Phosphorylation of GPCRs is mediated by the members of G protein-coupled receptor kinases (GRKs) family, including GRK233-36. Receptor activation triggers the subcellular accumulation of GRK2, which in turn phosphorylates the active conformation of GPCRs37-39. GRK2 overexpression has been shown to modify the phosphorylation patterns of BRs and to reduce BR-mediated signaling in human embryonic kidney 293 (HEK-293) cells32, 40.
In this study, we assess the involvement of GRK2 in the regulation of functional endothelial responses to BK, the underlying molecular mechanisms, and the implication in human angioedema.
Methods
Data from this study are available from the corresponding author upon reasonable request. The extended version of Materials and Methods section – which includes a detailed description of our in vitro, in vivo, and human studies34, 41-52 – is available in the supplementary material.
In vitro studies were performed in bovine aortic endothelial cells (BAECs), cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS) at standard conditions of 37°C in 95% air and 5% CO2. On this cell line we studied BK signaling, assessing Ca2+ mobilization (Fluo-4 based assay), NO production (DAF-FM Diacetate based assay), and cell-permeabilization in response to GRK2 modulation45, 51.
In vivo studies were conducetd in Tie2CRE-GRK2fl/− (heterozygous endothelial-specific GRK2 knock-out) mice. In these rodents, the Miles assay was performed to measure the vascular permeabilization and edema formation in response to BK.
For human studies, we identified a population of 15 patients with a confirmed diagnosis of C1-INH hereditary angioedema (C1-INH-HAE) and 10 healthy controls. From patients and controls, we collected peripheral blood mononuclear cells (PBMCs) in which we assessed protein levels of GRK2, as we previously described34.
Study approval
Experimental protocols on mice were carried out in accordance with NIH and were approved by the Ethical Committee for Animal Studies of the University of Salerno (#713/2015-PR). For human studies, the Ethical Committee of “Federico II” University of Naples approved that blood obtained during routine diagnostics could be used to investigate the physiopathology of human ereditary angioedema (#216/16); written informed consent was obtained from patients according to the principles expressed in the Declaration of Helsinki.
Statistical Analysis
All values are presented as mean ± SD. All experiments were performed at least in triplicate by blinded investigators. Shapiro-Wilk test was applied to verify the normal distribution of values; unpaired t-test or ANOVA followed by Bonferroni post hoc testing were performed as appropriate, were applicable. A correlation analysis was performed to evaluate the relationship between GRK2 levels and severity score of angioedema phenotype. A significance level of p<0.05 was assumed for all statistical evaluations. Statistics were computed with GraphPad Prism (v. 8.4.0) software (San Diego, CA).
Results
Inhibition of GRK2 enhances endothelial responsiveness to BK
To establish the physiological role of GRK2 in the regulation of BK signaling, we evaluated the effect of its inhibition on the endothelial responses to BK using the GRK2 inhibitor KRX-C7 (1μM, 1h). In vitro, the stimulation of BK receptors in BAEC rapidly induced Ca2+ mobilization producing a peak of Ca2+ released in the cytosol, followed by a phase of progressive reduction (Fig. 1A-B). Instead, when the same cells were pre-treated with KRX-C7, Ca2+ mobilization response to BK was significantly enhanced (Fig. 1A-B).
Figure 1. Effects of GRK2 inhibition on functional endothelial responses to BK.
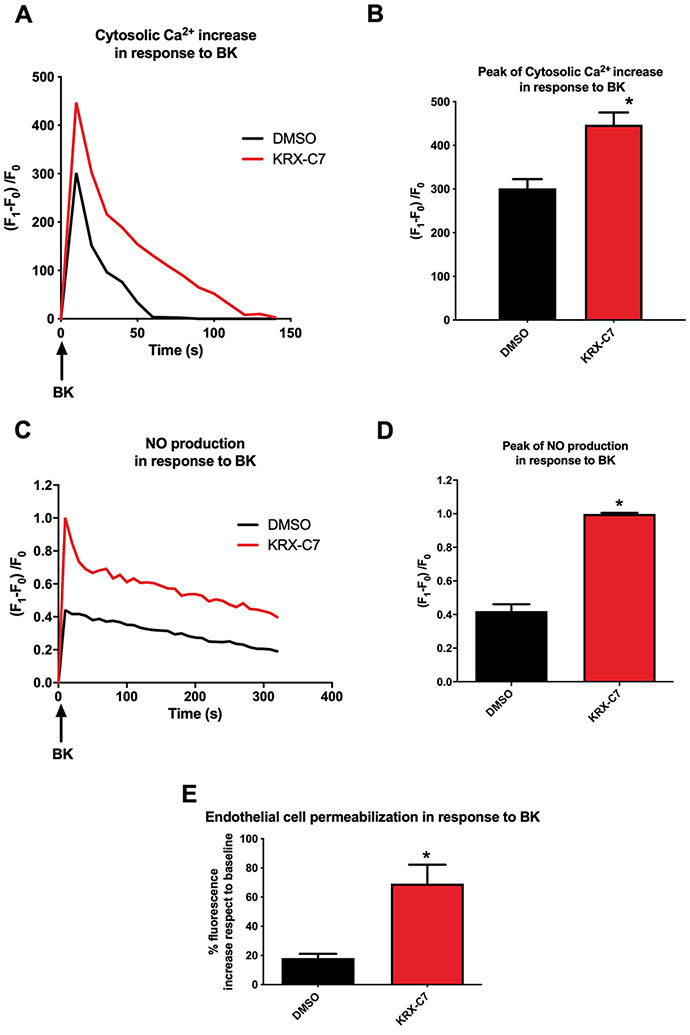
The kinetic of Ca2+ release in response to BK (30 nM), was evaluated in BAEC pre-treated with GRK2 inhibitor KRX-C7 (1 μM, 30 min) vs vehicle-treated cells (DMSO), showing a significant increase in BK-dependent Ca2+ release when GRK2 is inhibited. The image is representative of at least three independent experiments (A). The maximum increase of cytosolic Ca2+ concentration is represented as peak of fluorescence increase (DMSO 301.5 ± 21.21 vs KRX-C7 447.0 ± 28.28, t-test: p<0.001, n≥3) (B). NO production in response to BK (30 nM) was evaluated in BAEC pre-treated with KRX-C7 (1 μM, 30 min) or with vehicle (DMSO). BK-dependent NO production is significantly higher in presence of GRK2 inhibitor KRX-C7. The image is representative of at least three independent experiments (C). The maximum production of NO was represented as peak of fluorescence increase in response to BK (DMSO 0.420 ± 0.07 vs KRX-C7 0.998 ± 0.01, t-test: p<0.05, n≥3) (D). BK induced cell permeabilization was evaluated in BAEC pre-treated with KRX-C7 (1 μM, 30 min) compared to veichle treated cells, performing an in vitro permeability assay. Fluorescent Dextran was added to endothelial monolayer plated on the upper side of a double chamber system. The entity of endothelial permeabilization was determined measuring in the bottom chamber the fluorescence of Dextran that passed through the cell monolayer. The cell monolayer treated with GRK2 inhibitor displayed higher permeability in response to BK. The fluorescence is expressed as percent of increase in response to BK respect to baseline (DMSO 18.17 % ± 2.98 vs KRX-C7 69.15 % ± 12.97, t-test: p<0.05, n≥3) (E).
BK is known to increase NO production by activating eNOS through a calmodulin-dependent mechanism53. Consistent with this phenomenon, in our experimental setting BK-induced a significant increase in NO production by reflecting Ca2+ mobilization kinetics (Fig. 1C-D). Again, GRK2 inhibition enhanced BK dependent NO production (Fig. 1C-D). The effect of GRK2 inhibition on BK induced Ca2+ mobilization and NO production does not depend on extracellular Ca2+, as we recorded a similar trend in cells exposed to Ca2+-free medium (Fig. S1 A-C).
To evaluate the effects of GRK2 inhibition on BK dependent endothelial permeabilization, we performed an in-vitro vascular permeability assay. BK stimulation increased cell permeabilization, as expected (Fig. 1E). However, the pre-treatment with KRX-C7 determined a significantly higher response in terms of BK-induced permeabilization of the endothelial monolayer (Fig. 1E).
Overall, these data demonstrate that GRK2 inhibition enhances the effects of BK on the EC, suggesting that this kinase can act as an endogenous inhibitor of BK signaling.
BK induces rapid accumulation of GRK2 in endothelial cells
The relevant effects of GRK2 inhibition on acute responses induced by BK suggest a rapid recruitment of the kinase in the BK signaling pathway. To verify this hypothesis, we evaluated whether changes in the expression levels or trafficking of GRK2 occurred in response to BK stimulation. BAEC were acutely exposed to BK (30 nM) for 5 and 15 min and GRK2 protein levels were evaluated in whole cell lysate, in the membrane, mitochondrial, and cytosolic extracts (Fig. 2A-D). At 5 min, BK stimulation significantly increased GRK2 total levels. After 15 min, this phenomenon was attenuated and GRK2 returned to baseline levels (Fig. 2A). The analysis of different cellular compartments revealed that at 5 min from BK stimulation the accumulation of GRK2 occurred in membranes (Fig. 2B), mitochondria (Fig. 2C), and cytosol (Fig. 2D). 15 min after stimulation, levels of GRK2 returned to baseline in mitochondria and cytosol, remaining prevalently localized at the plasma membranes (Fig. 2B-D). These data indicate that BK causes GRK2 accumulation and polarization mainly to the plasma membrane.
Figure 2. Evaluation of GRK2 expression and subcellular localization in response to BK.
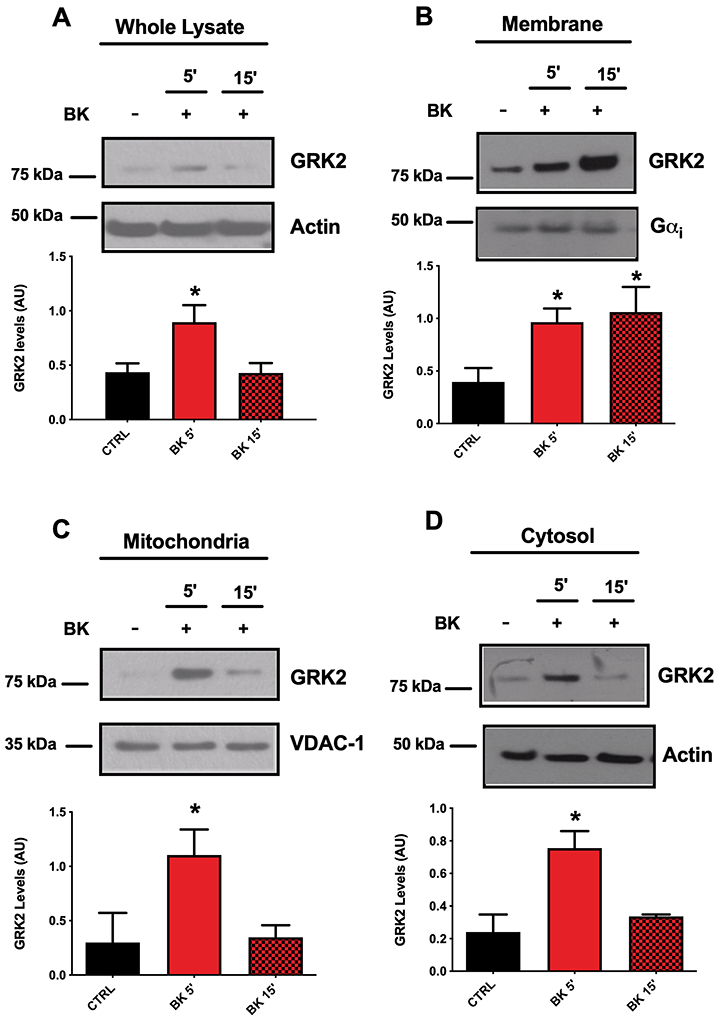
After stimulation with BK (30 nM) for 5 and 15 min, BAEC were collected and processed to isolate cellular compartments or to obtain whole cell lysate where GRK2 level was evaluated by western blot. In whole cell lysate, GRK2 increased at 5 min from BK stimulation and returned to baseline levels at 15 min (A). In membrane extract, GRK2 accumulates at 5 min from BK stimulation and continued to be higher at 15 min (B). Mitochondrial and cytosolic levels of GRK2 increased at 5 min from BK stimulation, reflecting the levels of GRK2 in whole cell lysate (C-D). Actin was used as a loading control for whole cell lysate and for cytosolic extracts, while Gαi and VDAC were used as loading control for membrane and mitochondrial extracts, respectively. Images are representative of at least three independent experiments (One-way ANOVA: *p<0.05, n≥3).
BK blocks the proteasome-dependent degradation of GRK2
The rapid GRK2 accumulation occurring after BK stimulation cannot result from GRK2 de-novo synthesis, as a timeframe of 5 min is not enough for protein production. Therefore, we hypothesized that the observed increase in GRK2 level relates to the ubiquitin-dependent proteasome system (UPS), which is known to rapidly modify the intracellular levels of specific proteins54. Indeed, UPS regulates GRK2 levels through the E3 ubiquitin ligase Mdm2 that binds and promotes GRK2 poli-ubiquitination and degradation55. As shown in Fig. 3A, a significant decrease in GRK2 ubiquitination levels occurred after BK exposure.
Figure 3. Evaluation of GRK2 degradation pattern in response to BK.
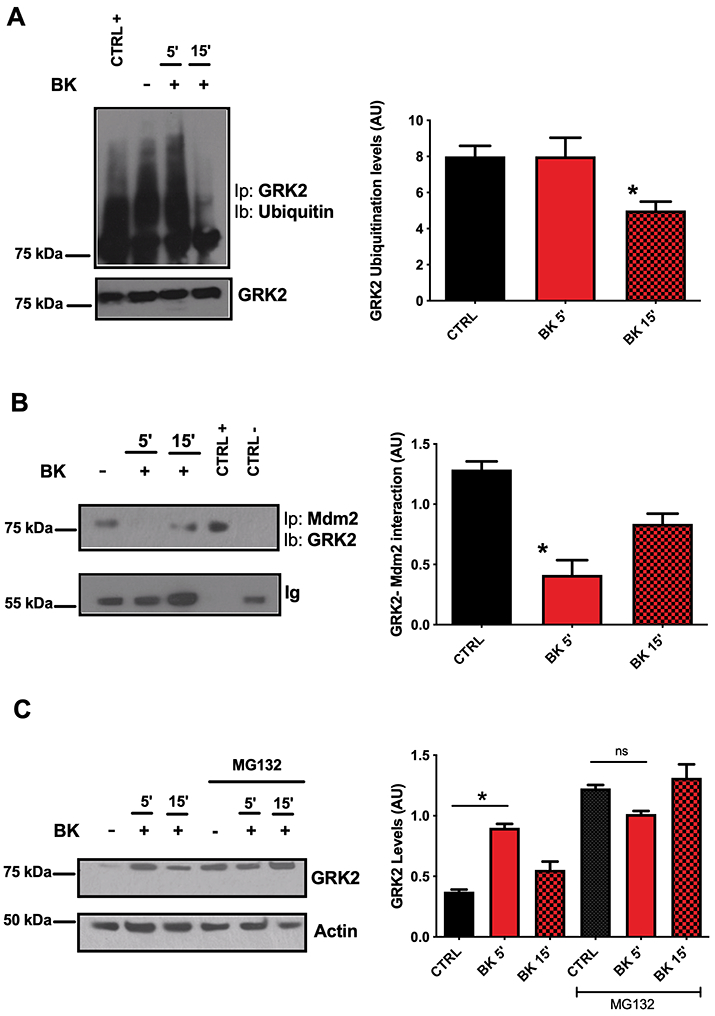
GRK2 ubiquitination levels were assessed in response to BK acute stimulation (30 nM). GRK2 was immune-precipitated and the ubiquitination levels of the kinase were determined by western blot using a specific antibody for ubiquitin. In response to BK acute stimulation, the ubiquitination levels of GRK2 were significantly decreased. Whole cell lysate was used as positive control. The levels of immunoprecipitated GRK2 were used as loading control (A). The interaction between GRK2 and its E3 ubiquitin ligase Mdm2 was evaluated by immunoprecipitation. BK stimulation at 5 min interrupts GRK2 and Mdm2 interaction, which was recovered at 15 min from stimulation. The immunoprecipitated sample obtained using the secondary antibody was used as negative control, while whole cell lysate as positive control. The levels of immunoglobulins (Ig) were used as loading control (B). To prove the key role of proteasome in the regulation of GRK2 availability in response to BK, the kinase levels were determined in cells pre-treated with MG132 (5 μM, 30 min) a specific inhibitor of proteasome activity, compared to no treated cells. Western blot analysis confirmed that the accumulation of GRK2 in response to BK did not occur when the proteasome was inhibited. Actin was used as loading control (C). Images are representative of at least three independent experiments (One-way ANOVA: *p<0.05, n≥3).
Accordingly, after BK stimulation, the interaction between GRK2 and Mdm2 was significantly decreased at 5 min and recovered at 15 min (Fig. 3B). The treatment with proteasome inhibitor MG132 led to GRK2 accumulation in response to BK (Fig. 3C). Overall, these data indicate that BK reduces GRK2/Mdm2 interaction, thereby resulting in a reduced degradation and accumulation of the kinase.
CaMKII regulates the activation of GRK2 in response to BK
To explore the mechanism of BK regulation of GRK2 ubiquitination, we evaluated the role of Ca2+-calmodulin-dependent kinase type II (CaMKII), since this kinase mediates most of the cellular effects of BK in endothelial cells56. Indeed, GRK2 and CaMKII significantly interact within 5 min from BK stimulation (Fig. 4A). Moreover, when cells were pre-treated with the specific CaMKII inhibitor peptide C17β, the BK dependent accumulation of GRK2 did not occur (Fig. 4B). Thus, we evaluated the effect of C17β also on GRK2 degradation pattern. The reduction of GRK2 ubiquitination levels occurring in response to BK was prevented in BAEC pre-treated with CaMKII inhibitor (Fig. 4C). Similarly, CaMKII inhibition preserved GRK2/Mdm2 interaction after BK stimulation (Fig. 4D), confirming the involvement of CaMKII in GRK2 accumulation induced by BK. Collectively, these results unveil a molecular mechanism by which BK intrinsically activates the pathway for the attenuation of its own signaling, according to a pattern of GPCR desensitization, mediated by CaMKII and proteasome. To further prove the functional role of CaMKII in mediating the downregulation of BK signaling by GRK2 recruitment, we measured two well-established BK-dependent phenotypes (i.e. NO production and endothelial permeabilization) in presence of CaMKII inhibition. We observed that the inhibition of CaMKII significantly increases BK-induced NO production and permeabilization (Fig. S2 A-B), producing an effect similar to GRK2 inhibition. The combination of the two inhibitors (KRX-C7 and C-17β) further increase BK-induced responses as compared to the two compound admistrated alone (Fig. S2 A-B). Of note, these responses were not affected when the experiments were performed in absence of extracellular Ca2+ (Fig. S2 C-D).
Figure 4. Involvement of CaMKII in BK-mediated GRK2 accumulation.
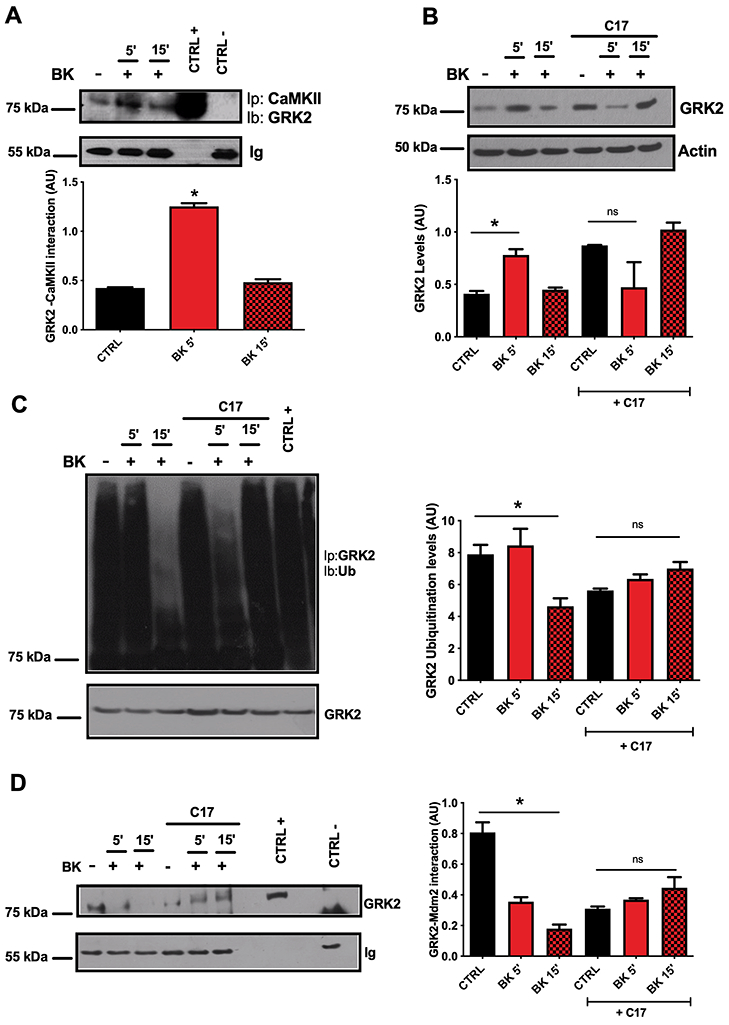
The interaction between CaMKII and GRK2 was evaluated at 5 and 15 min from BK stimulation (30 nM) by co-immunoprecipitation assay. The interaction between the two molecules was significantly increased at 5 min from BK exposure, and returned to baseline levels at 15 min from stimulation. The immunoprecipitated sample obtained using the secondary antibody was used as negative control while, whole cell lysate as positive control. The levels of immunoglobulins (Ig) were used as loading control (A). To evaluate the role of CaMKII in proteasome dependent accumulation of GRK2, the effects of BK on GRK2 ubiquitination and interaction with Mdm2 were evaluated in cells pre-treated with CaMKII inhibitor C-17β (5μM, 30 min) compared to control cells (DMSO). The ubiquitination test showed that in cells pre-treated with CaMKII inhibitor BK exposure does not significantly affect the levels of GRK2 ubiquitiation. GRK2 immuneprecipitated was used as loading control (B). The interaction between GRK2 and mdm2 was assessed through co-immunoprecipitation assay. In the cells where CaMKII was inhibited, the reduction of GRK2 and Mdm2 interaction does not occur. The immunoprecipitated sample obtained using the secondary antibody was used as negative control, while the whole cell lysate was used as positive control. The levels of immunoglobulins (Ig) were used as loading control (C). Images are representative of at least three independent experiments (One-way ANOVA: *p<0.05, n≥3).
GRK2 selective endothelial knock-out increases vascular permeability in response to BK in vivo
To translate our findings in an in vivo setting, we assessed vascular permeability in response to acute administration of BK in endothelial GRK2 hemizygous mice (Tie2-CRE/GRK2fl+/fl−) vs control (GRK2fl+/fl+) littermates. Interestingly, Tie2-CRE/GRK2fl+/fl− mice showed a wide blue coloration already in basal condition, particularly evident around the nose, feet, and internal organs such as the intestine; BK administration (30 μg/Kg for 30 min) further increased the difference between endothelial GRK2 hemizygous and control mice (Fig. 5A). The quantitative analysis of Evans Blue extravasation provided statistical confirmation of the phenomenon; indeed, in basal conditions, extravasation was significantly higher in Tie2-CRE/GRK2fl+/fl− mice compared to controls. BK administration enhanced Evans Blue extravasation in such organs for both animal groups; however, this phenomenon was significantly more evident in Tie2-CRE/GRK2fl+/fl− mice compared to control littermates (Fig. 5B). The data collected from in vivo experiments demonstrate the physiological role of GRK2 in the regulation of endothelial responsiveness to BK.
Figure 5. GRK2 regulates BK dependent vascular permeability in vivo.
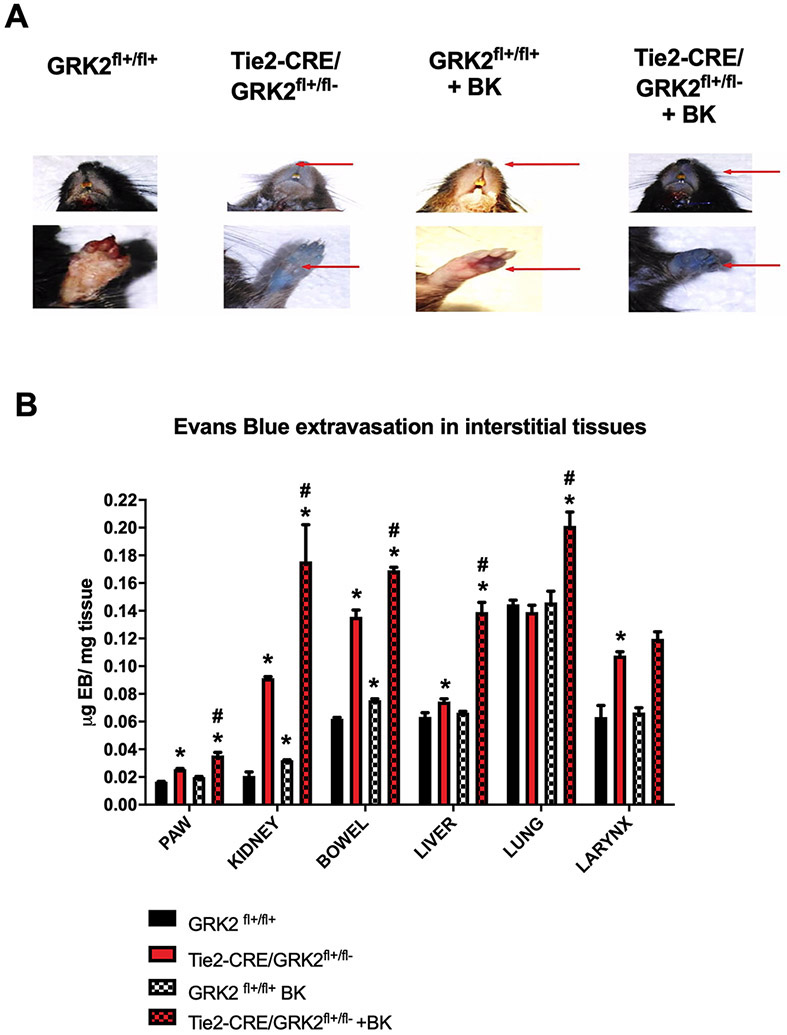
To evaluate the effect of BK on vascular permeability in vivo, Myles assay was performed in control (GRK2fl/fl) and endothelial selective heterozygous GRK2 KO mice (Tie2-CRE/GRK2fl+/fl−). Evans Blue (EB, 200 μl 0,5% in PBS) was intravenously injected in basal condition and after BK stimulation (30 μg/Kg). Mice were euthanized and macroscopic evaluation of EB distribution in tissues was performed. The images are representative of at least three independent experiments (A). The organs were collected and exposed to a formamide solution. Spectrophotometric analysis (610 nm) of several organs was performed, and the quantity of EB incorporated by tissue (μg EB/mg tissue) was calculated. The quantitative analysis showed that already in basal condition, the amount of EB extravasated was higher in mice with endothelial GRK2 KO for several districts (kidney, bowel, larynxs). BK administration exacerbated the differences between groups (Two-way ANOVA: *p<0.05 vs GRK2fl/fl, #p<0.05 vs Tie2-CRE/GRK2fl/fl) (B).
GRK2 levels inversely correlate with severity of C1-INH deficiency hereditary angioedema: a human model of altered response to BK
The above data indicate GRK2 as a key regulator of endothelial responsiveness to BK with potential implications in the pathophysiological conditions characterized by increased vascular permeability. Then, we hypothesized that different GRK2 levels in patients with C1-INH-HAE could correlate with the severity of the disease.
We isolated peripheral blood mononuclear cells (PBMCs) from patients with C1-INH-HAE and age-matched control subjects (Table S1) and evaluated the levels of GRK2 by immunoblot analysis. No significant differences were found in the average levels of GRK2 between C1-INH-HAE patients and controls (Fig. 6A). However, our C1-INH-HAE population displayed a larger variability in the GRK2 levels and a significant inverse correlation between GRK2 levels and the severity score related to clinical phenotype42 (Fig. 6B). These data indicate that the reduced levels of GRK2 contribute to increasing endothelial sensitivity, enhancing the BK dependent pathological responses in angioedema.
Figure 6. Evaluation of GRK2 levels in C1-INH-HAE patients and correlation with severity of the phenotype.
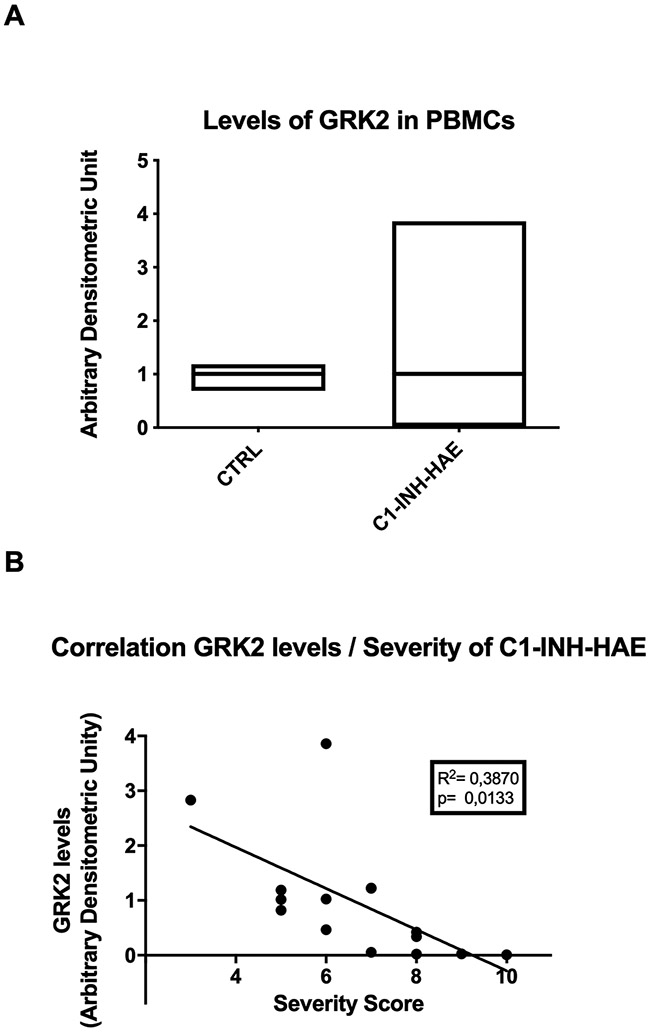
PBMCs were collected from C1-INH-HAE patients and healthy controls, and levels of GRK2 were evaluated. No significant differences were observed in the average level of GRK2 between patients and controls, despite the patient group displayed higher variability of GRK2 quantity (A). Levels of GRK2 measured in patients were correlated to severity score of angioedema in order to evaluate whether the wide distribution of GRK2 could reflect different clinical phenotypes. The correlation analysis confirmed a significant inverse correlation between GRK2 and severity score (linear regression, p<0.05) (B).
Discussion
This work is among the first studies approaching angioedema focusing on the contribution of endothelial alterations. Indeed, while the alterations linked to BK synthesis or catabolism occurring in angioedema patients have been extensively investigated, the molecular events occurring downstream of BK release and BK signaling regulation in endothelial cells, have been poorly assessed.
This is the first comprehensive investigation of BK signaling regulation by GRK2, from molecular mechanisms in cells to human condition. Indeed, we show that GRK2 regulates endothelial responses to BK, through a feedback loop that involves CaMKII and Mdm2. This mechanism is physiologically relevant since transgenic mice with reduced endothelial levels of GRK2 display an increased vascular permeability in response to BK.
These observations have an important translational impact in the clinical scenario. Indeed, C1-INH-HAE is a hereditary disease in which BK accumulation leads to transient and localized massive vasodilatation and permeabilization, producing local self-limiting edema57. However, patients with the same genetic mutation show a large variability in clinical phenotypes, spanning from mild to severe manifestations without a well-established explanation58. Our data show that in C1-INH-HAE, GRK2 levels inversely correlate with the severity score related to clinical phenotype. The reciprocal regulation between GRK2 and BK signaling through a proteasome-Ca2+ interface is a completely unknown molecular mechanism with relevant consequences for vascular physiopathology.
GRK2 regulates several intracellular signaling pathways, by turning off the downstream transduction of both GPCR and non-GPCR activated pathways38, 59. BK receptors belong to GPCR and previous studies have shown the ability of GRK2 to phosphorylate BK receptor32, 40; however, the biological significance of such event has never been addressed hitherto. The present study demonstrates for the first time that GRK2 regulates BK signaling in the endothelium. First, BK stimulation of endothelial cells induced a rapid GRK2 accumulation by reducing its interaction with Mdm2 and blocking its proteasome-dependent degradation. This phenomenon has been also observed in response to other acute stimuli60; in fact, the block of degradation is a strategy known to quickly increase the bioavailability of proteins with a fundamental role in acute responses61. In this regard, it is imperative to discern between the molecular effects of acute and chronic GPCRs stimulation since they involve distinct signal pathways leading to different effects. Indeed, whereas an acute stimulation induces the recruitment and the activation of early signaling protein like GRK2 to attenuate the cellular response (thus avoiding the cell hyperexcitation), a chronic stimulation of GPCRs can induce cell remodeling to support the adaptation, often involving changes in the gene transcription program with the synthesis of new molecular partners and new signaling. In this work we evaluated the acute response to BK based on the evidence that this peptide acutely induces phenotypes like angioedema when its circulating levels rapidly increase. Besides, BK has a short half-life, so it is rapidly removed, further supporting our theory that the acute phase of BK exposure is the most interesting and useful window to study in human disease. Our findings follow this groove and provide further evidence that GRK2 belongs to a conserved biological mechanism that cellular systems employ for the fine-tuning of several intracellular pathways62, essential to avoid the deleterious consequences of a highly activated signaling.
The mechanism through which BK induces GRK2 accumulation relies on Ca2+ and CaMKII. Indeed, in presence of CaMKII inhibition, BK stimulation does not significantly affect GRK2/Mdm2 interaction nor GRK2 accumulation (Fig 4) or CaMKII inhibition. These findings indicate that in response to BK, CaMKII meddles on the GRK2/Mdm2 interaction, reducing ubiquitination and promoting the intracellular increase of GRK2. These signalling events are independent of extracellular Ca2+, further proving that GRK2 specifically acts on BK receptor signalling, which indeed induces Ca2+ mobilization exclusively from intracellular stores.
Collectively, our results demonstrate that BK exposure of endothelial cells prompts the activation of a previously unknown functional axis among BK receptors, CaMKII, and GRK2 that finely regulate GRK2 levels, promoting the switch-off of BK receptor signaling (see graphical abstract).
Furthermore, GRK2 inhibition enhances the main cellular outcomes induced by BK (i.e. Ca2+ mobilization, NO production, cell permeability), proving that GRK2 negatively regulates BK signaling. These results confirm the functional role for GRK2 in mediating the physiological attenuation of BK signaling through a feedback loop which likely terminates with the desensitization of BK receptors. Accordingly, CaMKII inhibition also enhanced BK dependent endothelial responses (e.g. NO production and permeabilization63) by preventing GRK2 accumulation, whereas the combination of CaMKII and GRK2 inhibition sinergistically exacerbates these phenomena (Fig. 4 and S2).
BK is an important determinant of vascular function by inducing endothelial vasodilatation and permeabilization22. In the last years, several studies explored the possibility to target BK receptors to treat different pathological conditions; the antagonism of BK receptor, for example, has been proposed as anti-inflammatory therapy, while the sustained activation of BK receptor has been tested as a potential strategy for cardio-protection64, 65. However, no investigation focused on the intracellular regulation of BK signaling transduction. Previous studies from our and other groups proved the physiological role of GRK2 in the maintenance of vascular homeostasis. Mice with endothelial homozygous GRK2 gene ablation display defective angiogenesis, increased expression of chemokine, and adhesion molecules with the development of early atherosclerosis66, 67. These mice present severe structural abnormalities at the vascular level66. Therefore, in the present study, we used hemizygous mice, which on the contrary have preserved vasculature (Fig. S3). Our results show that reduced endothelial levels of GRK2 trigger a significant increase in vascular permeability both in basal conditions and upon BK administration. The wide and intense response to BK in Tie2-CRE/GRK2fl+/fl− mice respect to controls highlights the increased endothelial sensitivity to BK when GRK2 is genetically down-regulated. This phenomenon occurs in several tissues, including districts with high clinical relevance like bowel and larynx. The different response rate recorded among tissues could be linked to the physiological heterogeneity of the endothelium between organs also attributable to the variable physiological expression of endothelial permeability-related molecules68. The ability of GRK2 to regulate endothelial responsiveness to BK can have important pathological implications, especially for BK-related disorders. In C1-INH-HAE, several genetic defects in C1-INH production/activity cause BK overproduction with a resultant increased vascular permeability and associated edema. However, C1-INH-HAE patients show high heterogeneity in terms of severity of clinical manifestations, which does not correlate with the specific genetic mutation58. This evidence suggests that the heterogeneity in the effects of increased BK levels in the bloodstream could be imputable to different responsiveness of the endothelium to BK. Our data support this hypothesis, since the inverse correlation between GRK2 levels and severity score could imply that low levels of GRK2 are associated with a higher amplitude of endothelial responses and of the clinical manifestations observed in C1-INH-HAE patients.
The absence of significant differences in the average of GRK2 levels between C1-INH-HAE patients and healthy controls demonstrates that GRK2 is not the cause of the disease; rather, the kinase can act as “phenotype modifier”, as its levels reflect different sensitivity of the endothelium to BK, which accumulates due to C1-INH mutations.
Our study does have some limitations. First and foremost, we only measured GRK2 levels in PBMCs, whereas to translate our preclinical findings in the clinical scenario an assessment of GRK2 level/activity in human endothelium from angioedema patients is warranted, along with signaling studies. Therefore, our human data should be considered as a starting point for future studies aimed at the characterization of the functional role of GRK2 in human angioedema. Nevertheless, the data obtained from PBMCs respond to the need of a biomarker of disease severity measurable in a non-invasive manner (peripheral blood), and GRK2 level and activity in PBMCs have been shown to mirror the ones in other organs or tissues, including heart and blood vessels27, 30, 51-55.
Our data do not allow to define the exact mechanisms responsible for the different GRK2 levels observed in C1-INH-HAE patients. Genetic and/or epigenetic factors may be involved, resulting in an altered GRK2 degradation/expression. For instance, a recent report demonstrated that the C825T genetic variant in exon 10 of GNB3 gene affects GRK2 ubiquitination, eventually resulting in reduced GRK2 degradation56; this variant has been also associated with the hypertensive phenotype56. Hence, these findings strongly suggest that genetic variants can be accountable for the different GRK2 availability and then affect vascular responses. Therefore, we cannot exclude that in patients with angioedema genetic variants reducing GRK2 levels/activity can promote the devolpment of a severe phenotype.
In conclusion, our report indentifies a new regulatory mechanism of BK-signalling mediated by GRK2, able to switch-off BK receptor, thereby attenuating the downstream endothelial responses, as shown both in vitro and in vivo. Despite the need for further dedicated clinical investigations, these findings have interesting translational implications, as GRK2 can be exploited as a novel reliable biomarker of severity as well as a novel molecular target for BK-mediated angioedema.
Perspectives
The results of the present study prompt intruiguing fields of investigation, such as the search of GRK2 genetic variants or related genes potentially associated with the development of human angioedema. Besides C1-INH deficiency, alterations of the GRK2 level/activity can influence other models of BK-mediated angioedema, like ACE-inhibitors-related angioedema. It is known that the administration of an ACE-inhibitor determines BK accumulation, however, only a small portion of subjects develop detrimental vascular reactions, and the reasons behind these phenomena are mostly unknown4, 13. We can speculate that the different amplitude of endothelial sensitivity to BK could be a key determinant for the occurrence of angioedema in response to ACE-inhibitors, and endothelial GRK2 could be among the factors influencing the amplitude of vascular responses. However, due to the high variability in ACE-inhibitors-related angioedema – compared to the genetically determined forms of angioedema – and the frequent presence of comorbidities that can independently affect GRK2 levels37, a large-study population will be required to specifically investigate these aspects.
In conclusion, GRK2 is known to be involved in cardiovascular and metabolic disorders, as well as in inflammation mechanisms48, 69-71; similarly, BK has been shown to mediate many other phenomena beyond angioedema, including blood pressure regulation, nociception, inflammation, metabolism, and oxidative stress17, 72-80. Therefore, this unprecedent GRK2-mediated BK-signaling regulation could be functionally involved also in other vascular and non-vascular disorders, paving the way for new investigations.
Supplementary Material
Novelty and Significance.
What Is New?
GRK2 regulates the endothelial responsiveness to BK enhancing BK-induced NO production and permeabilization, in vitro as well as in vivo.
This new insight in vascular biology advances the pathophysiological role of GRK2 in human angioedema.
What Is Relevant?
GRK2 is a predicitor of angioedema severity, as its levels inversely correlate with angioedema severity score.
Summary.
Physiologically, GRK2 mediates the attenuation of BK signaling through a feedback loop that involves CaMKII. Reduced availability of GRK2 in endothelial cells triggers enhanced NO production and permeabilization in response to BK. Herein, we demonstrate for the first time that this mechanism has crucial clinical implications in angioedema, a pathological condition characterised by the excessive BK stimulation of the endothelium. Indeed, we show that in patients with angioedema, the reduced levels of GRK2 are associated with a severe phenotype of the disease, since lower GRK2 availability compromises the attenuation of the hyperactivated BK signaling. The variability of GRK2 levels in angioedema patients can explain the differences in the clinical phenotype of this disease, prompting GRK2 as a novel biomarker of severity in angioedema.
Acknowledgments
Sources of Funding
The study was supported by FARB-2018 Salerno (to M.C.), by the American Heart Association (POST35211151 to J.G.), and by the National Institutes of Health (NIH: R01-DK123259, R00-DK107895, R01-HL146691, R56-AG066431, and R01-DK033823 to G.S.).
Footnotes
Disclosures
None.
References
- 1.Tarbox JA, Bansal A and Peiris AN. Angioedema. JAMA. 2018;319:2054. [DOI] [PubMed] [Google Scholar]
- 2.Bova M, De Feo G, Parente R, De Pasquale T, Gravante C, Pucci S, Nettis E and Triggiani M. Hereditary and Acquired Angioedema: Heterogeneity of Pathogenesis and Clinical Phenotypes. Int Arch Allergy Immunol. 2018;175:126–135. [DOI] [PubMed] [Google Scholar]
- 3.Aygoren-Pursun E, Magerl M, Maetzel A and Maurer M. Epidemiology of Bradykinin-mediated angioedema: a systematic investigation of epidemiological studies. Orphanet journal of rare diseases. 2018;13:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carucci L, Bova M, Petraroli A, Ferrara AL, Sutic A, de Crescenzo G, Cordisco G, Margaglione M, Gambardella J, Spadaro G, Genovese A and Loffredo S. Angiotensin-Converting Enzyme Inhibitor-Associated Angioedema: From Bed to Bench. J Investig Allergol Clin Immunol. 2020;30:272–280. [DOI] [PubMed] [Google Scholar]
- 5.Riedl MA, Grivcheva-Panovska V, Moldovan D, Baker J, Yang WH, Giannetti BM, Reshef A, Andrejevic S, Lockey RF, Hakl R, Kivity S, Harper JR, Relan A and Cicardi M. Recombinant human C1 esterase inhibitor for prophylaxis of hereditary angio-oedema: a phase 2, multicentre, randomised, double-blind, placebo-controlled crossover trial. Lancet. 2017;390:1595–1602. [DOI] [PubMed] [Google Scholar]
- 6.Mormile I, Cocchiaro A, Bova M, Loffredo S, de Paulis A, Spadaro G and Petraroli A. Gastrointestinal manifestations of angioedema: a potential area of misdiagnosis. Eur J Gastroenterol Hepatol. 2020: -DOI: 10.1097/MEG.0000000000001848. [DOI] [PubMed] [Google Scholar]
- 7.Cicardi M and Zuraw BL. Angioedema Due to Bradykinin Dysregulation. J Allergy Clin Immunol Pract. 2018;6:1132–1141. [DOI] [PubMed] [Google Scholar]
- 8.Cesoni Marcelli A, Loffredo S, Petraroli A, Carucci L, Mormile I, Ferrara AL, Spadaro G, Genovese A and Bova M. Nailfold Videocapillaroscopic Findings in Bradykinin-Mediated Angioedema. J Investig Allergol Clin Immunol. 2020: -DOI: 10.18176/jiaci.0524. [DOI] [PubMed] [Google Scholar]
- 9.Riedl MA. Angioedema: Challenges and Insights. Immunol Allergy Clin North Am. 2017;37:xv–xvi. [DOI] [PubMed] [Google Scholar]
- 10.Bossi F, Fischetti F, Regoli D, Durigutto P, Frossi B, Gobeil F Jr., Ghebrehiwet B, Peerschke EI, Cicardi M and Tedesco F. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J Allergy Clin Immunol. 2009;124:1303–10 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demirturk M, Polat N, Guz G, Gurdal A, Altun I, Gelincik A, Toz B, Oflaz H, Colakoglu B, Dal M and Buyukozturk S. There is an increased risk of atherosclerosis in hereditary angioedema. International immunopharmacology. 2012;12:212–6. [DOI] [PubMed] [Google Scholar]
- 12.Firinu D, Bassareo PP, Zedda AM, Barca MP, Crisafulli A, Mercuro G and Del Giacco S. Impaired Endothelial Function in Hereditary Angioedema During the Symptom-Free Period. Frontiers in physiology. 2018;9:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller DR, Oliveria SA, Berlowitz DR, Fincke BG, Stang P and Lillienfeld DE. Angioedema incidence in US veterans initiating angiotensin-converting enzyme inhibitors. Hypertension. 2008;51:1624–30. [DOI] [PubMed] [Google Scholar]
- 14.Bas M, Adams V, Suvorava T, Niehues T, Hoffmann TK and Kojda G. Nonallergic angioedema: role of bradykinin. Allergy. 2007;62:842–56. [DOI] [PubMed] [Google Scholar]
- 15.Gryglewski RJ, Uracz W, Chlopicki S and Marcinkiewicz E. Bradykinin as a major endogenous regulator of endothelial function. Pediatr Pathol Mol Med. 2002;21:279–90. [DOI] [PubMed] [Google Scholar]
- 16.Marceau F, Bachelard H, Bouthillier J, Fortin JP, Morissette G, Bawolak MT, Charest-Morin X and Gera L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. International immunopharmacology. 2020;82:106305. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan AP. Bradykinin-mediated diseases. Chem Immunol Allergy. 2014;100:140–7. [DOI] [PubMed] [Google Scholar]
- 18.Cameron AC, Welsh P, Neves KB, Newby DE, Touyz RM and Lang NN. Acute vascular effects of vascular endothelial growth factor inhibition in the forearm arterial circulation. J Hypertens. 2020;38:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su JB. Role of Bradykinin in the Regulation of Endothelial Nitric Oxide Synthase Expression by Cardiovascular Drugs. Curr Pharm Des. 2017;23:6215–6222. [DOI] [PubMed] [Google Scholar]
- 20.Han ED, MacFarlane RC, Mulligan AN, Scafidi J and Davis AE 3rd. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scharfstein J, Ramos PIP and Barral-Netto M. G Protein-Coupled Kinin Receptors and Immunity Against Pathogens. Adv Immunol. 2017;136:29–84. [DOI] [PubMed] [Google Scholar]
- 22.Howl J and Payne SJ. Bradykinin receptors as a therapeutic target. Expert Opin Ther Targets. 2003;7:277–85. [DOI] [PubMed] [Google Scholar]
- 23.Wilson PC, Lee MH, Appleton KM, El-Shewy HM, Morinelli TA, Peterson YK, Luttrell LM and Jaffa AA. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J Biol Chem. 2013;288:18872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santulli G and Iaccarino G. Pinpointing beta adrenergic receptor in ageing pathophysiology: victim or executioner? Evidence from crime scenes. Immun Ageing. 2013;10:10 DOI: 10.1186/1742-4933-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joedicke L, Mao J, Kuenze G, Reinhart C, Kalavacherla T, Jonker HRA, Richter C, Schwalbe H, Meiler J, Preu J, Michel H and Glaubitz C. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat Chem Biol. 2018;14:284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matarese A, Gambardella J, Lombardi A, Wang X and Santulli G. miR-7 Regulates GLP-1-Mediated Insulin Release by Targeting beta-Arrestin 1. Cells. 2020;9:1621 -DOI: 10.3390/cells9071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M and Lefkowitz RJ. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Weerd WF and Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–66. [DOI] [PubMed] [Google Scholar]
- 29.Billups D, Billups B, Challiss RA and Nahorski SR. Modulation of Gq-protein-coupled inositol trisphosphate and Ca2+ signaling by the membrane potential. J Neurosci. 2006;26:9983–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gambardella J, Lombardi A, Morelli MB, Ferrara J and Santulli G. Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. J Clin Med. 2020;9: 1096 -DOI: 10.3390/jcm9041096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munoz CM, Cotecchia S and Leeb-Lundberg LM. B2 kinin receptor-mediated internalization of bradykinin in DDT1 MF-2 smooth muscle cells is paralleled by sequestration of the occupied receptors. Arch Biochem Biophys. 1993;301:336–44. [DOI] [PubMed] [Google Scholar]
- 32.Blaukat A, Pizard A, Breit A, Wernstedt C, Alhenc-Gelas F, Muller-Esterl W and Dikic I. Determination of bradykinin B2 receptor in vivo phosphorylation sites and their role in receptor function. J Biol Chem. 2001;276:40431–40. [DOI] [PubMed] [Google Scholar]
- 33.Sorriento D, Ciccarelli M, Cipolletta E, Trimarco B and Iaccarino G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure - A Review on Available GRK2 Inhibitors. Frontiers in cardiovascular medicine. 2016;3:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santulli G, Campanile A, Spinelli L, Assante di Panzillo E, Ciccarelli M, Trimarco B and Iaccarino G. G protein-coupled receptor kinase 2 in patients with acute myocardial infarction. Am J Cardiol. 2011;107:1125–30. [DOI] [PubMed] [Google Scholar]
- 35.Izzo R, Cipolletta E, Ciccarelli M, Campanile A, Santulli G, Palumbo G, Vasta A, Formisano S, Trimarco B and Iaccarino G. Enhanced GRK2 expression and desensitization of betaAR vasodilatation in hypertensive patients. Clin Transl Sci. 2008;1:215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B and Iaccarino G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension. 2010;56:696–704. [DOI] [PubMed] [Google Scholar]
- 37.Santulli G, Trimarco B and Iaccarino G. G-protein-coupled receptor kinase 2 and hypertension: molecular insights and pathophysiological mechanisms. High Blood Press Cardiovasc Prev. 2013;20:5–12. [DOI] [PubMed] [Google Scholar]
- 38.Sorriento D, Ciccarelli M, Santulli G, Illario M, Trimarco B and Iaccarino G. Trafficking GRK2: Cellular and Metabolic consequences of GRK2 subcellular localization. Transl Med UniSa. 2014;10:3–7. [PMC free article] [PubMed] [Google Scholar]
- 39.Fusco A, Santulli G, Sorriento D, Cipolletta E, Garbi C, Dorn GW 2nd, Trimarco B, Feliciello A and Iaccarino G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell Signal. 2012;24:468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feierler J, Wirth M, Welte B, Schussler S, Jochum M and Faussner A. Helix 8 plays a crucial role in bradykinin B(2) receptor trafficking and signaling. J Biol Chem. 2011;286:43282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, Caballero T, Farkas H, Grumach A, Kaplan AP, Riedl MA, Triggiani M, Zanichelli A, Zuraw B and EAACI Hutpo. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602–16. [DOI] [PubMed] [Google Scholar]
- 42.Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J and Drouet C. Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy. 2011;66:76–84. [DOI] [PubMed] [Google Scholar]
- 43.Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J Jr., Lefkowitz RJ, Caron MG and Giros B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A. 1996;93:12974–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA and Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Q, Yang J, Santulli G, Reiken SR, Wronska A, Kim MM, Osborne BW, Lacampagne A, Yin Y and Marks AR. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc Natl Acad Sci U S A. 2016;113:8532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, Maione AS, Condorelli G, Puca A, Trimarco B, Illario M and Iaccarino G. CaMK4 Gene Deletion Induces Hypertension. J Am Heart Assoc. 2012;1:e001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carotenuto A, Cipolletta E, Gomez-Monterrey I, Sala M, Vernieri E, Limatola A, Bertamino A, Musella S, Sorriento D, Grieco P, Trimarco B, Novellino E, Iaccarino G and Campiglia P. Design, synthesis and efficacy of novel G protein-coupled receptor kinase 2 inhibitors. Eur J Med Chem. 2013;69:384–92. [DOI] [PubMed] [Google Scholar]
- 48.Cipolletta E, Gambardella J, Fiordelisi A, Del Giudice C, Di Vaia E, Ciccarelli M, Sala M, Campiglia P, Coscioni E, Trimarco B, Sorriento D and Iaccarino G. Antidiabetic and Cardioprotective Effects of Pharmacological Inhibition of GRK2 in db/db Mice. International journal of molecular sciences. 2019;20:1492 -DOI: 10.3390/ijms20061492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomez-Monterrey I, Sala M, Rusciano MR, Monaco S, Maione AS, Iaccarino G, Tortorella P, D’Ursi AM, Scrima M, Carotenuto A, De Rosa G, Bertamino A, Vernieri E, Grieco P, Novellino E, Illario M and Campiglia P. Characterization of a selective CaMKII peptide inhibitor. Eur J Med Chem. 2013;62:425–34. [DOI] [PubMed] [Google Scholar]
- 50.Maione AS, Cipolletta E, Sorriento D, Borriello F, Soprano M, Rusciano MR, D’Esposito V, Markabaoui AK, De Palma GD, Martino G, Maresca L, Nobile G, Campiglia P, Formisano P, Ciccarelli M, Marone G, Trimarco B, Iaccarino G and Illario M. Cellular subtype expression and activation of CaMKII regulate the fate of atherosclerotic plaque. Atherosclerosis. 2017;256:53–61. [DOI] [PubMed] [Google Scholar]
- 51.Santulli G, Xie W, Reiken SR and Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112:11389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amgalan D, Garner TP, Pekson R, Jia XF, Yanamandala M, Paulino V, Liang FG, Corbalan JJ, Lee J, Chen Y, Karagiannis GS, Sanchez LR, Liang H, Narayanagari SR, Mitchell K, Lopez A, Margulets V, Scarlata M, Santulli G, Asnani A, Peterson RT, Hazan RB, Condeelis JS, Oktay MH, Steidl U, Kirshenbaum LA, Gavathiotis E and Kitsis RN. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat Cancer. 2020;1:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lowry JL, Brovkovych V, Zhang Y and Skidgel RA. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J Biol Chem. 2013;288:4174–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, Washburn MP, Collazo-Lorduy A, Castillo-Martin M, Cordon-Cardo C, Sebti SM, Pinton P and Pagano M. PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature. 2017;546:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salcedo A, Mayor F Jr., and Penela P. Mdm2 is involved in the ubiquitination and degradation of G-protein-coupled receptor kinase 2. EMBO J. 2006;25:4752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murthy S, Koval OM, Ramiro Diaz JM, Kumar S, Nuno D, Scott JA, Allamargot C, Zhu LJ, Broadhurst K, Santhana V, Kutschke WJ, Irani K, Lamping KG and Grumbach IM. Endothelial CaMKII as a regulator of eNOS activity and NO-mediated vasoreactivity. PLoS One. 2017;12:e0186311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caccia S, Suffritti C and Cicardi M. Pathophysiology of Hereditary Angioedema. Pediatr Allergy Immunol Pulmonol. 2014;27:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bafunno V, Bova M, Loffredo S, Divella C, Petraroli A, Marone G, Montinaro V, Margaglione M and Triggiani M. Mutational spectrum of the c1 inhibitor gene in a cohort of Italian patients with hereditary angioedema: description of nine novel mutations. Ann Hum Genet. 2014;78:73–82. [DOI] [PubMed] [Google Scholar]
- 59.Evron T, Daigle TL and Caron MG. GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci. 2012;33:154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cipolletta E, Campanile A, Santulli G, Sanzari E, Leosco D, Campiglia P, Trimarco B and Iaccarino G. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc Res. 2009;84:407–15. [DOI] [PubMed] [Google Scholar]
- 61.Flick K and Kaiser P. Protein degradation and the stress response. Semin Cell Dev Biol. 2012;23:515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franco A, Sorriento D, Gambardella J, Pacelli R, Prevete N, Procaccini C, Matarese G, Trimarco B, Iaccarino G and Ciccarelli M. GRK2 moderates the acute mitochondrial damage to ionizing radiation exposure by promoting mitochondrial fission/fusion. Cell Death Discov. 2018;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gambardella J, Khondkar W, Morelli M, Wang B, Santulli G and Trimarco V. Arginine and Endothelial Function. Biomedicines. 2020;8:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manolis AJ, Marketou ME, Gavras I and Gavras H. Cardioprotective properties of bradykinin: role of the B(2) receptor. Hypertens Res. 2010;33:772–7. [DOI] [PubMed] [Google Scholar]
- 65.Terzuoli E, Meini S, Cucchi P, Catalani C, Cialdai C, Maggi CA, Giachetti A, Ziche M and Donnini S. Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation. PLoS One. 2014;9:e84358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ciccarelli M, Sorriento D, Franco A, Fusco A, Del Giudice C, Annunziata R, Cipolletta E, Monti MG, Dorn GW 2nd, Trimarco B and Iaccarino G. Endothelial G protein-coupled receptor kinase 2 regulates vascular homeostasis through the control of free radical oxygen species. Arterioscler Thromb Vasc Biol. 2013;33:2415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rivas V, Carmona R, Munoz-Chapuli R, Mendiola M, Nogues L, Reglero C, Miguel-Martin M, Garcia-Escudero R, Dorn GW 2nd, Hardisson D, Mayor F Jr. and Penela P. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest. 2013;123:4714–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aslan A, van Meurs M, Moser J, Popa ER, Jongman RM, Zwiers PJ, Molema G and Zijlstra JG. Organ-Specific Differences in Endothelial Permeability-Regulating Molecular Responses in Mouse and Human Sepsis. Shock. 2017;48:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sorriento D, Fusco A, Ciccarelli M, Rungi A, Anastasio A, Carillo A, Dorn GW 2nd, Trimarco B and Iaccarino G. Mitochondrial G protein coupled receptor kinase 2 regulates proinflammatory responses in macrophages. FEBS Lett. 2013;587:3487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ciccarelli M, Sorriento D, Fiordelisi A, Gambardella J, Franco A, Del Giudice C, Sala M, Monti MG, Bertamino A, Campiglia P, Oliveti M, Poggio P, Trinchese G, Cavaliere G, Cipolletta E, Mollica MP, Bonaduce D, Trimarco B and Iaccarino G. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020;7:1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sorriento D, Santulli G, Franco A, Cipolletta E, Napolitano L, Gambardella J, Gomez-Monterrey I, Campiglia P, Trimarco B, Iaccarino G and Ciccarelli M. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J Cardiovasc Transl Res. 2015;8:493–502. [DOI] [PubMed] [Google Scholar]
- 72.Rhaleb NE, Yang XP and Carretero OA. The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1:971–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yogi A, Callera GE, Tostes R and Touyz RM. Bradykinin regulates calpain and proinflammatory signaling through TRPM7-sensitive pathways in vascular smooth muscle cells. Am J Physiol Regul Integr Comp Physiol. 2009;296:R201–7. [DOI] [PubMed] [Google Scholar]
- 74.Duchene J and Ahluwalia A. The kinin B(1) receptor and inflammation: new therapeutic target for cardiovascular disease. Curr Opin Pharmacol. 2009;9:125–31. [DOI] [PubMed] [Google Scholar]
- 75.Brazy PC, Trellis DR and Klotman PE. Bradykinin stimulation of oxidative metabolism in renal cortical tubules from rabbit. Possible role of arachidonic acid. J Clin Invest. 1985;76:1812–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Chen JC and Yang CM. NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun Signal. 2012;10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petho G and Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92:1699–775. [DOI] [PubMed] [Google Scholar]
- 78.Kobayashi N, Honda T, Yoshida K, Nakano S, Ohno T, Tsubokou Y and Matsuoka H. Critical role of bradykinin-eNOS and oxidative stress-LOX-1 pathway in cardiovascular remodeling under chronic angiotensin-converting enzyme inhibition. Atherosclerosis. 2006;187:92–100. [DOI] [PubMed] [Google Scholar]
- 79.Othman R, Vaucher E and Couture R. Bradykinin Type 1 Receptor - Inducible Nitric Oxide Synthase: A New Axis Implicated in Diabetic Retinopathy. Front Pharmacol. 2019;10:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oeseburg H, Iusuf D, van der Harst P, van Gilst WH, Henning RH and Roks AJ. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension. 2009;53:417–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.