

Abstract
Traditional medicines contain natural products (NPs) as main ingredient which always give new direction and paths to develop new advanced medicines. In the COVID-19 pandemic, NPs can be used or can help to find new compound against it. The SARS coronavirus-2 main protease (SARS CoV-2 Mpro) enzyme, arbitrate viral replication and transcription, is target here. The study show that, from the electronic features and binding affinity of all the NPs with the enzyme, the compounds with higher hydrophobicity and lower flexibility can be more favorable inhibitor. More than fifty NPs were screened for the target and one terpenoid (T3) from marine sponge Cacospongia mycofijiensis shows excellent SARS CoV-2 Mpro inhibitory activity in comparison with known peptide based inhibitors. The molecular dynamics simulation studies of the terpenoids with the protein indicates that the complex is stable and hydrogen bonds are involved during the complexation. Considering binding affinity, bioavailability, pharmacokinetics and toxicity of the compounds, it is proposed that the NP T3 can act as a potential drug candidate against COVID-19 virus.
Keywords: Natural products, Sars cov-2 mpro, Docking, Molecular dynamics, ADME, Toxicity
Abbreviations: sars, severe acute respiratory syndrome; Dili, hepatotoxicity carcino carcinogenicity; Mutagen, mutagenicity; Cyto, cytotoxicity; Ahr, aryl hydrocarbon receptor; Ar, androgen receptor; Ar-lbd, androgen receptor ligand binding domain; Er, estrogen receptor alpha; Er-lbd, estrogen receptor ligand binding domain; PPAR-Gamma, Peroxisome Proliferator Activated Receptor Gamma; nrf2/ARE, Nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element; HSE, Heat shock factor response element; MMP, Mitochondrial Membrane Potential; ATAD5, ATPase family AAA domain-containing protein 5
Graphical abstract

1. Introduction
Rapidly changing world is stopped now due to COVID-19 epidemic in more than 212 countries of all the seven continents. At this moment, scientific community of various countries are trying to develop vaccine for the virus [1,2]. However, preparation of vaccine for RNA viruses is very difficult work which can be understand from case of HIV, Influenza, Hepatitis C etc. [3,4]. The world is suffering from the crisis of clinically effective anti-COVID-19 vaccines or medicines for prevention and treatment. Previously known medicines (Lopinavir/Ritonavir and Remdesivir) for virus or flu have been used as a different combinations but the success rate is very low with toxicity [5,6]. A safe and efficient drug to fight against COVID-19 is on demand.
Natural products (NPs) are the main ingredients in the traditional medicines. They are undoubtedly a major source of key compounds required for the development of new therapeutic agents. Almost top one third of the drugs in the market is NP derivatives [7]. However, their use has decreased in the last twenty five years due to the practical hurdles to investigate them in the molecular targets specific assays [7]. Now a days, these technical barriers for NP screening decreases with evolution of new advanced strategies [7]. Medicinal value of a NP is directly related to the mechanism of action with its target in the cell. The search of the target of a NP is one of the biggest challenges. In drug discovery it is most rational and important step. In the treatment of retrovirus (e.g. HIV), there are a number of drug targets to inhibit like viral envelope protein, RNA, reverse transcriptase, integrase and main protease enzyme (Mpro or 3CLpro) [8].
In silico identification of target for a NP is an efficient low-cost, time-economic and useful virtual screening tool [9]. There are two most widely accepted in silico techniques viz. quantitative structure activity relationship (QSAR) and molecular docking [9]. The last one can be easily utilized to recognize the target biomacromolecules (DNA, RNA and proteins) for synthetic compounds and NPs [10], [11], [12], [13]. The method also helps to understand the mechanism of action of NPs with its target proteins and provide a strategy for the development of new NP derived drugs [9]. The ligand-protein docking have been used to develop several NP based drugs [9].
Coronovirus is mainly a RNA viruse and genetically belongs to Betacoronavirus [14]. The genomic RNA of CoVs contains such 2 out of 6 open reading frames (ORFs) that translate into two polyproteins which produce four structural proteins and sixteen non-structural proteins. The main protease (Mpro) and papain-like proteases (PLPs) are the structural polyproteins of CoVs. All other constitutional and accessory CoV proteins comes from the non-structural proteins [15,16]. In the life-cycle of the virus, CoV Mpro plays an important role in viral replication. At this time, the protein of COVID-19 is mostly characterized [17]. Therefore, this can provide a lead to design decoy ligands or neutralizing antibodies for conquering of this viral epidemic.
Recently, some synthetic compounds having activities against COVID-19 were also developed and proposed by our group [18]. In this work, we screened 50 NPs, some of them already passed through high-throughput clinical assays for viruses, mainly HIV, against COVID-19 targeting SARS coronavirus-2 main protease (SARS CoV-2 Mpro) enzyme. We have also developed a strategy for choice of NPs on the basis of their structural similarity with SARS CoV-2 Mpro inhibitors, flexibility, hydrophilic and hydrophobic nature of NPs.
2. Computational section
2.1. Methods
The study focused on the SARC—CoV-2 Mpro, i.e. PDB ID: 5r7y, 5r7z, 5r80, 5r81, 5r82, 5r83, 5r84, 6lu7 and 6y7m for in silico studies to find some SARC—CoV-2 Mpro inhibitors from a library of natural products. The three dimensional structure of SARC—CoV-2 Mpro enzymes were collected from the RCSB website [19].
2.2. Density functional theory
The energy minimized structures of all the NPs were obtained with the help of the density functional theory (DFT). The optimization of ground state geometries of the NPs with B3LYP functional at 6–311 G level of theory in gaseous state. The job have been performed with the Gaussian 09 W Revision D.01 program [20] on the Windows platform.
2.3. Molecular docking
Energy minimized structure of all the investigated NPs which was obtained from DFT optimization were used for docking simulations with the SARC—CoV-2 Mpro (PDB ID: 6lu7) protein structure. The docking studies was performed using AutoDock 4.2.0 applications through Autodock tools at the Windows platform [21].The MGL Tools was utilized in the preparation of the structure of the NPs and the proteins in appropriate formats which were required for the calculations. In the case of SARC—CoV-2 Mpro enzyme with the NPs, the partial atomic charges (Gasteiger charges) have been allocate after putting hydrogens to all the atoms of the protein as well as the NPs, separately. Here, the NPs structures allowed as flexible moiety and the SARC—CoV-2 Mpro enzyme structure kept as rigid during the docking studies. The ten conformers of NPs inside the active site of the SARC—CoV-2 Mpro enzyme having minimum potential energy were obtained through subsequent 20 000 precise docking step with 1000 exhaustiveness parameter inside the 60×60×60 Å3 grid box using a Lamarckian genetic algorithm.
2.4. Protein structure modeling
The protein crystal structure data of all the SARC—CoV-2 Mpro enzymes were downloaded from Protein Data Bank (PDB ID: 5r7y, 5r7z, 5r80, 5r81, 5r82, 5r83, 5r84, 6lu7 and 6y7m). The protein structures utilized in all the further studies were prepared by Discovery studio 2017 R2 client. The pictures of the protein used here was made with MolSoft-ICM browser, Meastro 11.1, Samson core and Discovery studio 2017 R2 client. The SARC—CoV-2 Mpro protein-NPs docked complex with lowest potential energy structures also analyzed by the aforesaid software.
2.5. Molecular electrostatic potential (MEP) analysis
The energy minimized structures of the NPs gained from DFT were further utilized for MEP calculations. The same functional used for DFT has also been employed to generate the electrostatic potential map throughout the atomic framework of the NPs molecules. Here, the basis set was 6–31 g with the 0.03 isovalues. All these calculations were performed on the Windows version of Gaussian 09 W software with D1 revision [22].
2.6. ADME study
The SwissADME web server was utilized for all the ADME calculations [23] of the NPs showed top most binding affinity toward SARC—CoV-2 Mpro. The server have a strong data base to predict physicochemical properties like lipophilicity, water solubility, drug likeness, pharmacokinetics and medicinal properties with high accuracy.
2.7. Toxicity
The probability of Cardiac toxicity for all the NPs having high binding score in docking studies were calculated by Pred-hERG which is the only web-accessible computational server for this toxicity [24]. All the other type of toxicity of these NPs and FDA approved anti-viral drugs have been predicted using PROTOX-II [25]. In this case, we have considered the acute toxicity, organ toxicity, toxicological endpoint, nuclear receptor signaling pathways and stress response pathway.
2.8. Molecular dynamics simulation
Understanding the stability of protein upon ligand binding is significantly improved by molecular dynamics simulation studies. Molecular dynamics simulation of the SARS CoV2 Main Protease and the ligand T3 (from marine sponges) was performed with Groningen Machine for Chemical Simulation (GROMACS) version 2020.2. Topology parameters for protein and ligand were generated With GROMOS96 54a7 force field and Ligand topology was obtained from the PRODRG2 server. The protein -ligand system was embedded in a cubic box of approximate size with periodic boundary conditions using a simple point charge water solvation model (38,012 water molecules) [26]. The overall system was neutralized by adding 4 Na+ ions in solution and the SHAKE algorithm was used to constrain all bond lengths involving hydrogen atoms. Particle Mesh Ewald method with a cutoff of 12 Å was applied to treat the long range electrostatic interactions. The processed system was suitably minimized followed by the NPT and NVT ensemble equilibration steps at a uniform temperature and pressure of 300 K and 1 bar, respectively maintained for each system with Parrinello−Rahman barostat. The trajectories were saved at every 2 fs time step and the production MD simulation of the protein-ligand complex was performed for 95 nanoseconds [27].
3. Results and discussion
3.1. NPs selections
Chemically, the CoV Mpro is a cysteine proteases enzyme which can hydrolyze proteins with the help of its Cys-amino acid residues present in the active site [16]. Generally, this type of proteases enzymes are very much prone to suicide inhibitors which bind covalently to the cysteine residue [28]. Such inhibitors also found in the natural sources, containing functional groups like aldehyde, active ketones, α,β-unsaturated carbonyls, epoxide etc. [28]. In the active site of the SARC—CoV-2 Mpro, it have His41 and Cys145 amino acid residues (Fig. S1). The amino acid residue His-behave as common acid base and Cys-is very well known for its neucleophilic character. The Cys-is responsible for Michael addition reactions to the α,β-unsaturated ketones and neucleophilic attack to the ketones in biological reactions [28]. The crystal structure of first isolated SARC—CoV-2 Mpro revealed that the peptide based N3 ligand was attached inside its active site through a covalent bond by Cys145 (Fig. S2a). It was also important to note that the ligand with ketone groups can possess nucleophilic attack by C145 residue (compound 9, Fig. S2b).
At this point of time, we have several crystal structures of SARC—CoV-2 Mpro with different type of small ligands including N3 (some of them are shown in Fig. S3) [17,19,29,30]. All these ligands are able to stabilize the protein through strong binding interactions and crystallize with it. This means that these ligands can show inhibitory effect to the target proteins. We examine all the crystal structures of SARC—CoV-2 Mpro critically to find the degree of hydrophilicity and hydrophobicity required for strong binding. In this study, we found that the hydrophobic interactions of ligands with proteins is main though ligand contain sufficient hydrogen bonding acceptor and donor groups (Fig. S4). Therefore, a good SARC—CoV-2 Mpro inhibitor should contain either conjugated ketone (type-I) or active carbonyls (aldehydes or ketones; type-II) with sufficient hydrophobic parts for non-covalent interactions (Fig. 1 ).
Fig. 1.

The required structural features for a good covalent as well as non-covalent SARC—CoV-2 Mpro inhibitor.
Considering all this structural characteristics, we have chosen more than fifty NPs and some of which have been qualified high-throughput assays for different protein targets of different virus. In this study, NPs from the source of medicinal plant, marine origines and microbes have been considered. Chemically they are alkaloids (Fig. S5), terpenoid (Fig. S6), phenolic compounds (Fig. S7) and peptides (Fig. S8). Here, pyrrole, pyridine, indole, carbazole, quinoline, isoquinoline alkaloids with alcohol, phenolic, aldehyde, conjugated ketones, amide, ester functional groups was considered. For terpenes, macrocyclic α,β-unsaturated ketones, amides and esters and bridged compounds have been used. Most of them do not contain any aromatic groups but hydrophobic due to the presence of alkene groups. However, polyphenols are polar though they contain multiple aromatic rings. In all these sets of NPs, rigid to highly flexible compounds coexist. Although, the investigating peptide NPs are highly flexible and polar.
3.2. Docking studies
The previously known peptide based SARC—CoV-2 Mpro covalent inhibitor [17,29,30] N3 and Pep0 are docked into the protein and found that −4.47 and −5.34 are the docking score, respectively (Table 1 ). Interestingly, very few selected NPs are below of these values. The superposition of their docked structures with the corresponding X-ray crystallographic structure show that the docking is very efficient since there is well fitting of the experimental and theoretical structures (Fig. S9). Therefore, these docking scores can be considered as the control values and higher negative values from it will indicate the better inhibitor of SARC—CoV-2 Mpro enzyme.
Table 1.
Docking score of different types of NPs and the SARC—CoV-2 Mpro co-crystallized peptides with the enzyme.
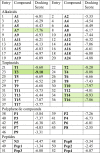 |
The molecular docking generally utilized to identify the target DNA, RNA and proteins for compounds from synthetic and natural sources. However, docking of all the selected anti-virals with the SARC—CoV-2 Mpro protein used to find the inhibitor molecule of coronavirus. The summarized docking results in Table 1 shows that, in comparison with the alkaloids, polyphenols and peptides, terpinoids have greater binding ability with SARC—CoV-2 Mpro. The terpinoids T3 have showed the highest docking score (i.e. ΔGo = −9.10) and the compound with the 15% of this value is also considered as good inhibitor of the compound. The only alkaloid compound A7 and terpinoids T1, T2, T3, T4, T10, T14 and T16 (Fig. 2 ) are able to qualify the conditions. The compound T14 also have similar binding ability like T3. It is interesting to note that polyphenols and peptides are low scoring.
Fig. 2.

The stereochemical features of inhibitor NPs show high binding affinity with SARC—CoV-2 Mpro.
The alkaloid A7 was isolated from root bark of decarine plant Zanthoxylum ailanthoides having potent anti-HIV, (EC50 <0.1 mg/mL) [31]. The terpinoids T1 to T4 are geometrical isomers of each other and obtained from the marine source Cacospongia mycofijiensis [32]. The oleanane triterpene T10 is anti-tumor agent (EC50: 17.9–20.6 μM on HepG2) found in apple peel [33]. The triterpene T14, isolated from a fungal source (mushroom) Ganoderma colossum, showed inhibition activity against HIV-1 protease enzyme (EC50 = 15.3 μM) [33]. The diterpenoids T16, micafungin, found in fungal cell walls, is available in market as the intravenous antifungal drug [34]. The source and activities of all other NPs are given in the Table S1.
The successful compounds contain at least one α,β-unsaturated carbonyl functionality except A7 (Fig. 2). The geometry of NPs T1-T4 are influence enough to the binding of them. They have three different types of α,β-unsaturated carbonyls, i.e., ketone, ester and amide with E/Z geometry. Inside the active site of the enzyme the α,β-unsaturated group was found to be covalently bound to the Cys145 residue. The DFT based energy calculations show that Z geometry of conjugated ketone (T2) destabilize the molecule (ΔG⁰ (T1-T2) = 152.28 kJ/mol). However, this destabilization is lower for the Z geometry of conjugated ester part (ΔG⁰ (T1-T4) = 126.024 kJ/mol) whereas the Z geometry of conjugated amide part stabilize the system (ΔG⁰ (T4-T3) = 23.89 kJ/mol) and destabilize overall (ΔG⁰ (T1-T3) = 102.39 kJ/mol). Therefore, the docking and DFT studies show that the unstable molecule with Z configuration favor the probability of Michael addition reaction. The rigid E,Z combination of double bonds also found in T14 like T3 and favor their binding.
To understand the behavior of these four classes of NPs in terms of the docking results (Table 1), we need to take their hydrophilicity, hydrophobicity and the flexibility in account. The first two parameters of NPs were interpreted with the help of the electrostatic potential at the molecular surface [35] and the last one by the number of rotatable bonds (Fig. S10). The alkaloids A7, having highest ΔGo value in their class, is planar aromatic system with only one rotatable bond. However, the compound A4 shows lowest value, have large hydrophobic surface and highest number of rotatable bond. Similar observation also found in the case of terpenoids with highest and lowest ΔGo values (T3 and T12, respectively). Although all the chosen phenolic NPs have lower number of rotatable bonds than the aforesaid classes but their comparatively higher hydrophilic parts are responsible for their overall binding affinity. Interestingly, peptides are high hydrophilic as well as most flexible and show lest binding affinity. Therefore, this combined conformation ability, molecular electrostatic potential and docking study indicates that higher hydrophobicity and less flexible NPs molecule required for the anti-SARC—CoV-2 Mpro, ultimately anti-COVID-19.
The CoV Mpro, a Cys-His-dyad containing three-domain protease enzyme, cleave polyprotein to produce mature proteins. The non-canonical dyad is situated at a groove connecting the domain I and II, is the active site which is structurally most conserved region among all the CoV Mpro with various common characteristics [36]. The active site is consists of four sites (i.e. S1, S1′, S2 and S4; Fig. S11) and the S1′ site, having the Cys-residue can get attached to the inhibitor NPs containing aldehyde, active ketone and α,β-unsaturated carbonyl groups by the covalent bond formation. This covalent attachment is essential to maintain irreversible anti-viral activities. The CoV Mpro is functionally and structurally non-homologous of the human protease enzyme and this is the reason why it is the ideal target for antiviral [37].
In the investigating NPs, all the high score compounds (T3, T14, T1, T2, T4, T10 and T16) are the α,β-unsaturated lactones which may anchor through covalent bond with C145. The docking results elucidate the SARC—CoV-2 Mpro inhibition mechanism of them. The docking pose clearly showed that all these compounds bind at the substrate binding pocket of the enzyme in elongated conformation (Fig. S12). The non-covalent docking reveals that the β-carbon of the α,β-unsaturated ester of T3 and the S atom of the C145 are just 3.5 Å apart from each other and corresponding ester carbonyl form 2.7 Å hydrogen bond with amide hydrogen of G143 (Fig. 3 a). It is 1.9 time higher distance of standard C-S bond. Therefore, the Michael addition reactions can be facilitated by the activation of ester carbonyl. The only amide H of T3 is hydrogen-bonded with peptide N of N142 (3.2 Å). The molecule have seven double bonds and long carbon chains which is also covered by the amino acid H41, M49, L141, N142, G143, E166, R188, Q189 and T190 through C—H… π and hydrophobic interactions. The compound T14, second highest and very close binding score to the T3, bind mainly through extensive hydrophobic interactions with H41, M49, L141, N142, M165, H172 and Q189. In this case, the Michael addition at the δ position of extended conjugated ester by the SH group of C145 can also be facile as they are just 3.0 Å apart from each other (Fig. 3b).
Fig. 3.

Docking pose and binding interactions of NPs (a) T3 and (b) T14 inside the SARC CoV-2 Mpro binding site.
3.3. Bioavailability studies
In order to elucidate drug-likeness of strong binders, we estimate their bioavailability (absorption, distribution, metabolism and excretion) which is an important criteria to approve any synthetic or NPs for clinical trial [23]. Lipophilicity and water solubility is one of the most important for absorption, distribution and excretion of the NPs. The ilogPo/w and consensus logPo/w (it is an average logPo/w values given in Table S2) values of all the NPs is in between +3 to +6 except T10 (Fig. 4 ). Such lipophilic NPs display good gastro intestinal absorption as well as distribution in tissues and thereby fulfill very essential criteria for drug molecules. All the NPs other than T10, have negative values from −5 to −7 of ESOL log S and Ali log S (Fig. 4). Therefore, they are moderate to poorly water soluble. This advises that the molecules can be body administrable through the oral or parenteral route.
Fig. 4.

Predicted lipophilicity (log P values) and water solubility (log S values) of NPs by different calculation models.
3.4. Pharmacokinetics
The pharmacokinetics study indicates that all the selected NPs from docking have high gastro intestinal absorption except T10. Only A7 and T16 can cross blood-brain barrier (Table S3). Hence, the lead NPs T3 and T14 have potential as a peripheral medicines. The liver and intestine cytochrome P450 enzymes (CYP1A2, CYP3A4, CYP2C19, CYP2D6, and CYP2C9) interact with drugs and are responsible for their metabolism. In this way the balance of drug concentration in blood is maintained. Inhibition of these enzymes can increase the drug concentration in the blood and some time it may causes adverse effect. Both the NPs T3 and T14 is predicted as capable to inhibit CYP1A2, CYP2D6 and CYP3A4 enzyme (Table S3). Therefore, the toxicity of NPs may a major concern and their toxicity have been discussed later.
3.5. Toxicity studies
The primary job in drug discovery is to find a least toxic or nontoxic compound as the limit of wide adequate usefulness of drugs is their toxicity in the body which have complicated relation with the patients. The toxicity of drugs very much connected with the stage of infection, the chemical nature of used drugs, its doses and their interaction with some important proteins. As for example, the marketing of terfenadine, sertindole and cisapride drugs have been stopped due to their inhibition ability of the human ether-á-go-go related gene (hERG) K + channels which is the cause of heart arrhythmia and ultimately death [24]. Therefore, the toxicity prediction of these NPs in different toxicity parameters is now an essential part of this study.
In this study, we have considered hERG K + channel, cardiotoxicity, organ toxicity (dili), toxicity endpoints (carcino., immune., mutagen. and cyto.), nuclear receptor signaling pathways (AhR, AR, AR-LBD, aromatase, ER, ER-LBD and PPAR-Gamma) and stress response pathways (nrf2/ARE, HSE, MMP, phosphoprotein (tumor suppressor) p53 and ATAD5) [25]. The prediction indicate that the NPs T10, T14 and T16 display potent cardiac toxic by the inhibition of hERG K + channel and eliminated from the lead compound list of the anti-SARC—CoV-2 Mpro though NP T14 showed second highest binding toward the protein (Table S4). The NPs A7, T14 as well as T16 can exhibit several toxic effect like carcino., immune., mutagenicity and AR inhibition (Table S5). The toxicity prediction also show that the NPs T3, T1, T2 and T4 are nontoxic in all aspects except immunotoxicity which was also observed in FDA approved drugs like paritaprevir, rilpivirine, glecaprevir, dolutegravir and grazoprevir (Table S6). Therefore, considering all the studies, it is our conclusion that the compound T3 is the lead for the inhibition of SARC—CoV-2 Mpro enzyme.
2.6. Molecular dynamics (MD)
MD simulations of 95 nanoseconds for the SARC—CoV-2 Mpro in complex with T3 revealed significant results about the stability of their binding [22]. The Root mean square deviations of the backbone atoms of SARC—CoV-2 Mpro and the ligand T3 are shown in Fig. 5 a. The protein backbone atoms exhibited an average RMSD of 0.46 nm and a higher fluctuation after 20 ns. The protein with ligand T3, as a complex was stable internally with fluctuatios less than 0.35 nm [38]. The RMSF fluctuations of Mpro in complex with T3. The average RMSF is 0.3 nm and represents the stability of the protein even after the ligand binding (Fig. 5b). Hydrogen bond analysis as seen in Fig. 5c, reveals there are at least two hydrogen bonds on an average and the minimum number of hydrogen bonds were found to be 0 and the maximum of 5 within 10 ns [39].
Fig. 5.

(a) Root mean square deviation (RMSD) of backbone atoms of SARS-CoV2 Mpro (Mpro) protein (red) and with T3 ligand (blue), (b) Root Mean Square fluctuations of Mpro in complex with T3, (c) Number of hydrogen bonds between Mpro and the natural compound T3.
From the results of MD simulations it is inferred that the binding of T3 with SARC—CoV-2 Mpro is stable throughout the simulation. The number of hydrogen bonds are favourable indicating better interactions of the ligand with protein. It is understood that the compound T3 with negligible fluctuations around the active site, exhibited good conformational changes and maintained close affinity with the protein target and hence it is capable of inhibiting SARC—CoV-2 Mpro with significant Hydrogen bond interactions.
4. Conclusion
In summary, we investigate the inhibitory activity of fifty NPs from different type of sources with structural and stereochemical diversity. The NPs have been chosen by finding the structural, stereochemical and electronic similarities with the inhibitors co-crystalized in SARS CoV-2 Mpro. The screening of all the NPs to the target protein shows that the terpenoid (T3) from marine sponge Cacospongia mycofijiensis can bind SARS CoV-2 Mpro excellently. Its inhibitory activity is almost twice in comparison with the known peptide based inhibitors. Observing the electronic features and the affinity towards SARS CoV-2 Mpro of all the NPs, we found that the higher hydrophobicity and lower flexibility favors the inhibition. The binding affinity, bioavailability, pharmacokinetics and toxicity of the NPs indicates T3 can act as a potential drug candidate against COVID-19 virus.
Author statement
N.S. and U.C.H. designed, evaluated and follow-up the calculations. N.S. short and optimized the structures of the NPs for the studies. A.S. performed the MD and wrote the part. A.A. and M.A. explain the toxicity analysis of the compounds under investigations. N.S. and U.C.H. wrote the manuscript with assistance and feedback from all other. All authors of the manuscript checked the theoretical results and approved of the manuscript as the final version. All authors agree the corresponding authors as the representative person for handling this manuscript.
Credit author statement
N.S. and U.C.H. designed, evaluated and follow-up the calculations. N.S. short and optimized the structures of the NPs for the studies. A.S. performed the MD and wrote the part. A.A. and M.A. explain the toxicity analysis of the compounds under investigations. N.S. and U.C.H. wrote the manuscript with assistance and feedback from all other.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The Department of Chemistry, Jadavpur University is gratefully acknowledged for all the research facilities. University Grant Commission (UGC)-CAS-II and DST-PURSE-II Program of Jadavpur University, Kolkata and King Saud University, Deanship of Scientific Research, Research Chair, Riyadh, KSA, are gratefully acknowledged for financially support. NS is thankful to RUSA 2.0 for his fellowship. The authors acknowledge BIOCHEMCOM (https://biochemcom.org/) for providing infrastructure support for the MD simulations.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2020.129433.
Appendix. Supplementary materials
References
- 1.Folegatti P.M., Ewer K.J., Aley P.K., Angus B., Becker S., Belij-Rammerstorfer S., Bellamy D., Bibi S., Bittaye M., Clutterbuck E.A., Dold C., Faust S.N., Finn A., Flaxman A.L., Hallis B., Heath P., Jenkin D., Lazarus R., Makinson R., Minassian A.M., Pollock K.M., Ramasamy M., Robinson H., Snape M., Tarrant R., Voysey M., Green C., Douglas A.D., Hill A.V.S., Lambe T., Gilbert S.C., Pollard A.J. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020 doi: 10.1016/S0140-6736(20)31604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Race for a COVID-19 vaccine. EBioMedicine. 2020;55 doi: 10.1016/j.ebiom.2020.102817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudometov A.P., Chikaev A.N., Rudometova N.B., Antonets D.V., Lomzov A.A., Kaplina O.N., Ilyichev A.A., Karpenko L.I. Artificial Anti-HIV-1 Immunogen Comprising Epitopes of Broadly Neutralizing Antibodies 2F5, 10E8, and a Peptide Mimic of VRC01 Discontinuous Epitope. Vaccines (Basel) 2019;7:83. doi: 10.3390/vaccines7030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duncan J.D., Urbanowicz R.A., Tarr A.W., Ball J.K. Hepatitis C Virus Vaccine: challenges and Prospects. Vaccines (Basel) 2020;8:90. doi: 10.3390/vaccines8010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., Xu J., Liu Z., Zhang Y., Li H., Shang L., Wang K., Li K., Zhou X., Dong X., Qu Z., Lu S., Hu X., Ruan S., Luo S., Wu J., Peng L., Cheng F., Pan L., Zou J., Jia C., Wang J., Liu X., Wang S., Wu X., Ge Q., He J., Zhan H., Qiu F., Guo L., Huang C., Jaki T., Hayden F.G., Horby P.W., Zhang D., Wang C. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harvey A.L., Edrada-Ebel R., Quinn R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 8.J.D.R, Piefer A.J. Emerging Drug Targets for Antiretroviral Therapy. Drugs. 2005;65:1747–1766. doi: 10.2165/00003495-200565130-00002. [DOI] [PubMed] [Google Scholar]
- 9.Chen X., Ung C.Y., Chen Y. Can an in silico drug-target search method be used to probe potential mechanisms of medicinal plant ingredients? Nat. Prod. Rep. 2003;20:432. doi: 10.1039/b303745b. [DOI] [PubMed] [Google Scholar]
- 10.Gorgulla C., Boeszoermenyi A., Wang Z.-.F., Fischer P.D., Coote P.W., Padmanabha Das K.M., Malets Y.S., Radchenko D.S., Moroz Y.S., Scott D.A., Fackeldey K., Hoffmann M., Iavniuk I., Wagner G., Arthanari H. An open-source drug discovery platform enables ultra-large virtual screens. Nature. 2020;580:663–668. doi: 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baxter C.A., Murray C.W., Clark D.E., Westhead D.R., Eldridge M.D. Flexible docking using tabu search and an empirical estimate of binding affinity. Proteins Struct. Funct. Genet. 1998;33:367–382. doi: 10.1002/(SICI)1097-0134(19981115)33:3<367::AID-PROT6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 12.Lorber D.M., Shoichet B.K. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J., Kollman P.A., Kuntz I.D. Flexible ligand docking: a multistep strategy approach. Proteins Struct. Funct. Genet. 1999;36:1–19. doi: 10.1002/(SICI)1097-0134(19990701)36. 1<1::AID−PROT1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramajayam R., Tan K.-.P., Liang P.-.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011;39:1371–1375. doi: 10.1042/BST0391371. [DOI] [PubMed] [Google Scholar]
- 16.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles’ heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 18.Sepay N., Sepay N., Al Hoque A., Mondal R., Halder U.C., Muddassir M. In silico fight against novel coronavirus by finding chromone derivatives as inhibitor of coronavirus main proteases enzyme. Struct. Chem. 2020;31:1831–1840. doi: 10.1007/s11224-020-01537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.https://www.rcsb.org/.
- 20.D.J. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L., No Title, (2009).
- 21.Sousa S.F., Fernandes P.A., Ramos M.J. Protein-ligand docking: current status and future challenges. Proteins Struct. Funct. Bioinforma. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 22.Bhardwaj V.K., Singh R., Sharma J., Rajendran V., Purohit R., Kumar S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1766572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braga R.C., Alves V.M., Silva M.F.B., Muratov E., Fourches D., Lião L.M., Tropsha A., Andrade C.H. Pred-hERG: a Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform. 2015;34:698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.No Title, (n.d.). http://tox.charite.de/protox_II/index.php?site=compound_input.
- 26.Mishra P., Günther S. New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 2018;8:9435. doi: 10.1038/s41598-018-27867-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gelpi J., Hospital A., Goñi R., Orozco M. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinforma. Chem. 2015;8:37–47. doi: 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaysser L. Built to bind: biosynthetic strategies for the formation of small-molecule protease inhibitors. Nat. Prod. Rep. 2019;36:1654–1686. doi: 10.1039/C8NP00095F. [DOI] [PubMed] [Google Scholar]
- 29.R. Zhang, L., Lin, D., Hilgenfeld, Crystal structure of the complex resulting from the reaction between the SARS-CoV main protease and tert-butyl (1-((S)-3-cyclohexyl-1-(((S)-4-(cyclopropylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyr, 2020. 10.2210/pdb6y7m/pdb. [DOI]
- 30.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;80-:eabb3405. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh I.P., Bodiwala H.S. Recent advances in anti-HIV natural products. Nat. Prod. Rep. 2010;27:1781. doi: 10.1039/c0np00025f. [DOI] [PubMed] [Google Scholar]
- 32.Carroll A.R., Copp B.R., Davis R.A., Keyzers R.A., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2020;37:175–223. doi: 10.1039/C9NP00069K. [DOI] [PubMed] [Google Scholar]
- 33.Kuo R.-.Y., Qian K., Morris-Natschke S.L., Lee K.-.H. Plant-derived triterpenoids and analogues as antitumor and anti-HIV agents. Nat. Prod. Rep. 2009;26:1321. doi: 10.1039/b810774m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanson J.R. Diterpenoids of terrestrial origin. Nat. Prod. Rep. 2016;33:1227–1238. doi: 10.1039/C6NP00059B. [DOI] [PubMed] [Google Scholar]
- 35.Sjoberg P., Politzer P. Use of the electrostatic potential at the molecular surface to interpret and predict nucleophilic processes. J. Phys. Chem. 1990;94:3959–3961. doi: 10.1021/j100373a017. [DOI] [Google Scholar]
- 36.Anand K. Coronavirus Main Proteinase (3CLpro) Structure: basis for Design of Anti-SARS Drugs. Science (80-.) 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 37.Kim Y., Liu H., Galasiti Kankanamalage A.C., Weerasekara S., Hua D.H., Groutas W.C., Chang K.-.O., Pedersen N.C. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLOS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan S.A., Zia K., Ashraf S., Uddin R., Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- 39.Paul S., Paul S. Molecular dynamics simulation study on the inhibitory effects of choline- O -sulfate on hIAPP protofibrilation. J. Comput. Chem. 2019;40:1957–1968. doi: 10.1002/jcc.25851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.