

Abstract
Cocaine addiction is a chronic illness characterized by maladaptive drug-induced neuroplastic changes that confer lasting vulnerability to relapse. Over several weeks we observed the effects of the M1 receptor-selective agonist VU0364572 in adult male rats that self-administer cocaine in a cocaine vs. food choice procedure. The drug showed unusual long-lasting effects, as rats gradually stopped self-administering cocaine, reallocating behavior towards the food reinforcer. The effect lasted as long as tested and at least 4 weeks. To begin to elucidate how VU0364572 modulates cocaine self-administration, we then examined its long-term effects using dual-probe in vivo dopamine and glutamate microdialysis in nucleus accumbens and medial prefrontal cortex, and ex vivo striatal dopamine reuptake. Microdialysis revealed marked decreases in cocaine-induced dopamine and glutamate outflow 4 weeks after VU0364572 treatment, without significant changes in dopamine uptake function. These lasting and marked effects of M1 receptor stimulation reinforce our interest in this target as potential treatment of cocaine addiction. M1 receptors are known to modulate medium spiny neuron responses to corticostriatal glutamatergic signaling acutely, and we hypothesize that VU0364572 may oppose the addiction-related effects of cocaine by causing lasting changes in this system.
Subject terms: Reward, Addiction
Introduction
Drug addiction is a chronic relapsing illness characterized by complex changes in the biochemistry of the brain, many of which persist after weeks or months of abstinence and confer lasting susceptibility to relapse [1, 2]. Signaling in the prefrontal cortex (PFC), and from the PFC to the nucleus accumbens (NAc), and downstream dopamine signaling in NAc medium spiny neurons (MSNs), is considered a main pathway of the addicting effects of abused drugs, including cocaine [3]. Glutamatergic corticostriatal projections from PFC to NAc are important in drug-maintained behaviors, and it has been hypothesized that chronic drug use can alter this system leading to loss of control over drug use [3–6]. The complex long-term adaptations induced by cocaine are not fully understood, but clinical and preclinical studies indicate that basal PFC activity and glutamate levels are reduced, whereas responses to cocaine and cocaine cues are exaggerated [7, 8]. Less scrutinized in addictions is the muscarinic cholinergic system. Yet activity of cholinergic interneurons, which provide the major acetylcholine input in striatum/NAc, is related to stimuli that predict reward/reward omission, and indeed is more responsive to changes in reinforcement contingency than the mesolimbic dopaminergic neurons, whose reward prediction error encoding is well described [9–11]. Muscarinic acetylcholine M1 receptors are the most abundant muscarinic receptor in neocortex and striatum and are expressed, mainly postsynaptically, on most if not all neocortical pyramidal neurons and striatal MSNs including MSNs in NAc [12–17]. Activity at M1 receptors modulates corticostriatal glutamatergic signaling, integration of inputs by MSNs, and neuroplasticity, and appears to preferentially facilitate inhibition of behavior [18–21]. M1 receptors are thus well placed to modulate some of the processes affected by cocaine exposure.
We have previously shown that acute pharmacological stimulation of M1 receptors can fully suppress cocaine self-administration behavior in mice, while food-maintained behavior was unaffected [22]. The M1/M4 receptor-preferring agonist xanomeline reduced cocaine taking in rats given access to choose between cocaine or a palatable food reinforcer [23]. Unlike effects of dopamine receptor ligands in the same assay [24, 25], the effects of M1/M4 stimulation grew larger during subchronic dosing, and subsisted briefly after ended treatment [23]. In mice, M1/M4 stimulation facilitated extinction of cocaine seeking and prevented reinstatement of cocaine seeking when administered during extinction or between cocaine exposure and extinction [26]. Those findings suggested an unusual and possibly lasting modulation of the addiction-related effects of cocaine by M1/M4 stimulation, but the mechanisms are largely unknown. Here, we sought to delineate and better understand the contributions of M1 receptors to these effects. We assessed how the highly selective, brain-penetrant M1 receptor agonist VU0364572 [27, 28] affected cocaine self-administration behavior over time in rats, using the cocaine vs. food choice assay. To begin to understand how VU0364572 modulates cocaine self-administration, we then examined its long-term effects using in vivo dopamine and glutamate microdialysis in NAc and medial PFC (mPFC), and ex vivo striatal dopamine reuptake.
Materials and methods
Animals
Behavioral studies were conducted at McLean Hospital in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the McLean Hospital Institutional Animal Care and Use Committee, using experimentally naive male Sprague Dawley rats acquired at age 7–8 weeks (Charles River, Wilmington, MA). Rats were group-housed up to 4 per cage until catheter implantation, then housed individually. Water was available ad libitum, food was provided in amounts adjusted to maintain body weight at about 400–450 g (≈17 g/day Rat Diet 5001; PMI Feeds, St. Louis, MO). For enrichment, species-appropriate treats were provided once or twice weekly. Microdialysis and dopamine uptake study procedures were conducted at the Laboratory of Neuropsychiatry in an AAALAC-accredited facility, in accordance with the EU directive 2010/63/EU and were approved by the Animal Experiments Inspectorate under the Danish Ministry of Food, Agriculture, and Fisheries. Experimentally naive male Sprague Dawley rats aged 6 weeks were acquired from Taconic Denmark; they were pair-housed with ad libitum water and rodent chow (Altromin 1310, Brogaarden, Denmark). All rats were acclimated to the temperature- and humidity-controlled facilities for a week before experiments began. Lights were on from 07:00 to 19:00, testing occurred during the light phase. Rats were randomly assigned to experimental groups, and groups were evenly balanced between test stations and days/times. Whenever possible, the experimenter was blind to treatment group.
Behavioral studies: cocaine self-administration
Rats were trained and tested in a cocaine vs. food choice procedure we have previously employed [23, 24]. Rats were trained to self-administer intravenous cocaine (1.0 mg/kg/infusion) by pressing one lever five times, or receive a liquid food treat by pressing another lever five times, see Supplementary material for full details. At completed training, daily sessions consisted of five 20-min components during which cocaine dose increased for each component: 0, 0.056, 0.18, 0.56, 1.0 mg/kg/infusion, and food reward was held constant. Baseline behavior was recorded as the mean of three consecutive sessions, then, the effects of VU364572 were tested. First, acute effects of 0.032–1.8 mg/kg were assessed, initially within-subjects with at least three sessions of baseline between doses. It became apparent that higher doses of VU364572 had lasting effects, and we then tested saline, 0.1 and 1 mg/kg VU0364572 in individual subjects, with up to 4 weeks observation periods after single dosing, during which cocaine and food reinforcer were available in daily sessions as during baseline conditions, 5 days/week (Fig. 1a). The first rat treated with 1 mg/kg VU0364572 was only observed for 2 weeks. Three rats lost catheter patency during testing. Thus, group size varies over time (n = 6–9). Only data collected with demonstrated patent catheters were included.
Fig. 1. Experimental timelines.
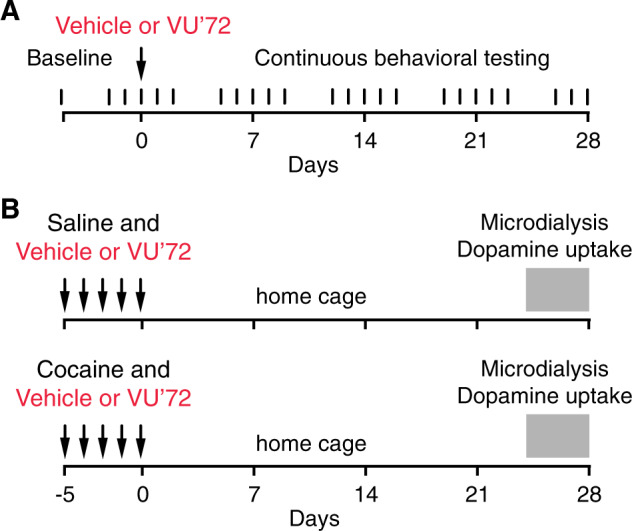
In the self-administration behavior experiment (a) rats were trained to baseline criteria over several weeks, then tested continuously 5 days per week for 4 weeks after administration of vehicle or either 0.1 or 1 mg/kg of the M1 agonist VU0364572 (VU’72). For microdialysis and dopamine uptake studies (b), rats received saline (“cocaine-naive” groups) or cocaine (“cocaine-experienced” groups), with vehicle or VU0364572.
Microdialysis
Rats were injected once daily for five consecutive days with 1 mg/kg VU0364572 or vehicle, and, 30 min later, with 15 mg/kg cocaine or saline intraperitoneally, and left for 1 h in a non home-cage enclosure. Treatment groups were: VU0364572 + saline, VU0364572 + cocaine, water + saline, water + cocaine (n = 6). All rats were then left in the home cage to allow the effect of VU0364572 to develop. Six age-matched controls, habituated to handling but otherwise naive, were administered saline on the microdialysis day. For practical feasibility, rats were treated and tested in two cohorts on consecutive weeks, and microdialysis was performed over 5 days for each cohort. Microdialysis was thus performed 24–28 days after the last injection (Fig. 1b).
Surgery, microdialysis sampling, and neurotransmitter analysis were performed as previously described [29] with minor modifications. Two microdialysis probes (CMA/12 2 mm, CMA/Microdialysis AB, Sweden) were implanted under sevoflurane anesthesia for simultaneous sampling of dopamine and glutamate in the mPFC (probe tip at AP, +2.5 mm; ML, +0.5 mm; DV, −5.4 mm relative to Bregma) and NAc (AP, +1.8 mm; ML, −1.4 mm; DV, −7.4 mm) in the same subjects. Anesthesia was maintained throughout testing. Probe placements were verified after ended experiments and are reported in Supplementary Fig. S1. The first four 20-min samples after starting perfusion were discarded, then, three samples were used to determine baseline level, and five further samples were collected after intraperitoneal administration of 15 mg/kg cocaine. Relative concentrations of dopamine in the dialysate were determined as previously by HPLC with electrochemical detection [29]. Relative glutamate concentrations were detected with fluorescence HPLC, see Supplementary material for full details.
Dopamine uptake
A separate cohort of rats (n = 4 per group, then repeated), was used for dopamine uptake, using the same treatment regimen as for microdialysis studies. Rats were sacrificed 24–27 days after the last injection (4 rats per day, counterbalanced by treatment group), and left ventral striatal tissue was dissected on ice from coronal sections using a brain matrix and 3 mm punch. Synaptosomal dopamine uptake was assessed as previously described [30, 31]. Background-subtracted fmol/min/µg protein counts were fitted by Michaelis–Menten kinetics using the median of triplicate determinations for each rat.
Drugs
Cocaine hydrochloride was provided by the National Institute on Drug Abuse (behavioral studies) or purchased from the Copenhagen University Hospital pharmacy (Copenhagen, Denmark; Copenhagen studies). VU0364572 was synthesized as previously described at Vanderbilt University [28], and was generously provided by Drs. P. Jeffrey Conn and Craig Lindsley (Vanderbilt University). Cocaine was dissolved in 0.9% saline. VU0364572 was dissolved in sterile water, fresh daily, and administered intraperitoneally 30 min before sessions.
Data analysis
Data were analyzed by analysis of variance with between- and within-subjects variables; for microdialysis, in addition, area under the curve (AUC) for 0–100 min after cocaine administration was calculated and analyzed. See supplemental materials for detailed descriptions and power analyses. Level of significance was set at α = 0.05. Data are presented as group means ± standard error of the mean (s.e.m.) unless noted otherwise (individual data).
Results
Behavior: cocaine self-administration
An initial study on acute effects indicated that VU0364572 produced a moderate reallocation of behavior away from cocaine self-administration towards food taking across a range of doses, with lower doses increasing food intake, whereas larger doses produced mixed effects that suggested some suppression of food taking early in the session (Supplementary Fig. S2). Acutely, VU0364572 only shifted behavior for the two lower cocaine doses, and did not result in overall reduction in cocaine intake over the session (Supplementary Table S1). We noticed that cocaine choice remained decreased for a day or two after administration of the low-intermediate doses of VU0364572. More strikingly, many rats that received 1.0 mg/kg VU0364572 subsequently failed to maintain cocaine-taking behavior at their baseline levels. We then explored this phenomenon systematically by recording cocaine vs. food choice behavior for up to 4 weeks after acute administration of saline or 0.1 or 1 mg/kg VU0364572.
Figure 2 shows choice behavior between cocaine injections and liquid food reinforcer at baseline and 4 weeks after acute administration of saline (Fig. 2a–c) or 1 mg/kg VU0364572 (Fig. 2d–f). Dose-effect functions for intervening time points are shown in Supplementary Fig. S3. Data are shown once weekly for efficiency, but the effect was continuous and the reported data are representative of the full test period. Rats receiving vehicle showed stable allocation of choice between cocaine and food over the observation period. Full statistical outcomes are reported in Supplementary Table S2A–D, only key comparisons are reported here for ease of reading. The effect of cocaine dose was always highly significant (p < 0.0001). Comparing percent cocaine choice at baseline, on the treatment day, and 7, 14, 21, and 28 days after VU0364572 administration, two-way ANOVA confirmed a significant effect of time after treatment [F(6,226) = 8.22, p = 0.0001]. Allocation of choice to cocaine-taking was significantly decreased in the VU0364572-treated animals on the treatment day and one, two, three, and 4 weeks later, relative to baseline (Fig. 2d). The change in percent choice reflected both a gradual decrease in cocaine injections taken ([F(6,232) = 5.38, p = 0.0007]; Fig. 2e) and an increase in food reinforcers taken ([F(6,232) = 6.87, p < 0.0001]; Fig. 2f). Decreases in cocaine-taking reached significance starting 1 week after treatment, the increase in food taking, from 2 weeks after treatment.
Fig. 2. Cocaine self-administration behavior was markedly reduced 4 weeks after acute VU0364572 (1 mg/kg) administration.
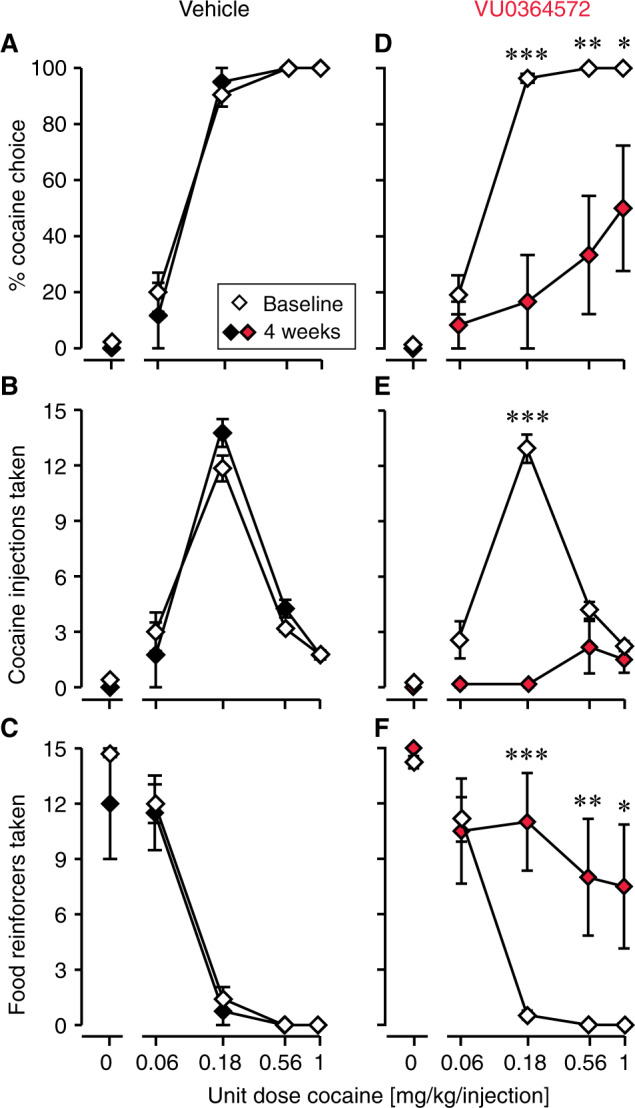
In vehicle-treated rats (n = 4), cocaine vs. food choice allocation (a), cocaine self-administration (b), and food taking (c) remained constant relative to baseline. In VU0364572-treated rats (n = 6–9), cocaine vs. food choice allocation (d) and cocaine self-administration (e) were significantly decreased, and behavior was reallocated to food taking (f) at 4 weeks post-treatment relative to baseline. *p < 0.05, **p < 0.01, ***p < 0.001 vs. baseline.
Also measured as session-wide total allocation of behavior to cocaine vs. food taking and as total intake, after administration of 1 mg/kg VU0364572 rats showed a gradual decrease in cocaine choice and cocaine intake as a function of time, while food intake increased (p < 0.02, see Supplementary Table S2D); see Fig. 3a–c for post-hoc significance levels. After administration of vehicle, behavior remained stable over 4 weeks (Fig. 3a–c). Figure 3d–i shows changes in behavior from baseline to day 28 (4 weeks) in individual rats: five of six VU0364572-treated rats showed marked decreases in cocaine choice and intake—and half completely stopped taking cocaine—whereas saline-treated rats maintained or increased cocaine choice and intake.
Fig. 3. Behavior was gradually reallocated from cocaine taking towards the food reinforcer.
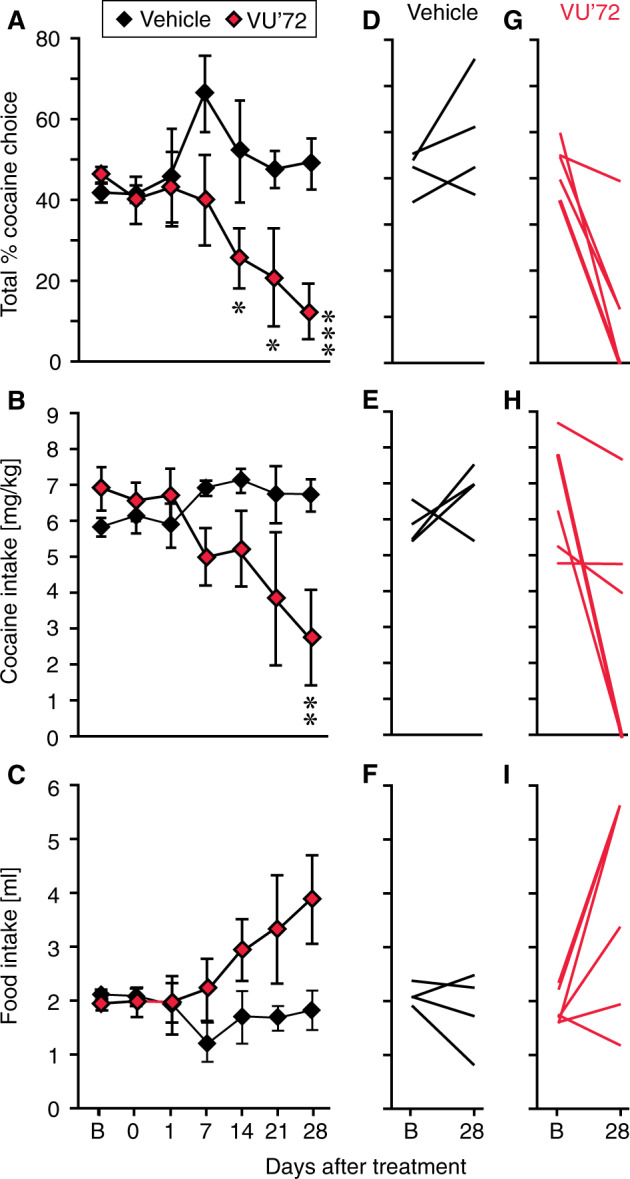
Session-wide cocaine vs. food choice allocation (a) and cocaine intake (b) were not significantly decreased on the VU0364572 treatment day, but started decreasing 1 or 2 weeks after treatment, while food intake (c) increased. In vehicle-treated rats, behavior remained stable. Changes from baseline (“B”) to day 28 post-treatment in individual subjects are shown for cocaine choice (d, g), cocaine intake (e, h), and food intake (f, i) in the vehicle group (d–f) and VU0364572 groups (g–i). *p < 0.05, **p < 0.01, ***p < 0.001 vs. baseline. Group sizes: saline (Vehicle), n = 4; VU0364572 (VU’72), n = 9 baseline to 7 days, n = 8 at 14 days, then n = 6.
A lower dose of 0.1 mg/kg VU0364572 produced a smaller, more transient decrease in cocaine-taking behavior, peaking 1–2 days after treatment and returning to baseline levels within 1 or 2 weeks (Supplementary Fig. S4).
Microdialysis
On the basis of evidence that M1 receptor stimulation modulates the integration of corticostriatal and dopaminergic signaling in NAc, we then asked whether VU0364572 decreased cocaine reinforcement by blunting NAc dopamine release and/or by altering corticostriatal glutamatergic signaling. Rats were treated with 1 mg/kg VU0364572 or vehicle in combination with saline or cocaine, and extracellular levels of dopamine and glutamate were measured 4 weeks later in vivo by dual-probe microdialysis in mPFC and NAc, after acute administration of 15 mg/kg cocaine. VU0364572 was never administered on the test day. Baseline extracellular dopamine and glutamate levels were comparable between groups (Supplementary Fig. S5).
Extracellular dopamine levels after cocaine administration were related to cocaine history, but importantly also to VU0364572 history both in the mPFC [F(1,20) = 6.64, p = 0.02] and in the NAc [F(1,20) = 43.2, p < 0.0001] (all statistical details in Supplementary Table S2E–G). In the mPFC, acute cocaine administration increased dopamine levels above baseline in the cocaine-experienced (sensitized, Fig. 4b) vehicle-treated animals but not in cocaine-naive animals (Fig. 4a; Supplementary Table S2F). The dopamine increase in the cocaine-experienced animals was completely blocked by VU0364572 pre-treatment (Fig. 4b). Figure 4c shows overall %increase in dopamine over baseline, which was related to VU0364572 history and cocaine history (p < 0.02, see Supplementary Table S2G). Vehicle-treated cocaine-experienced rats had higher cocaine-stimulated mPFC dopamine levels relative to vehicle-treated naive rats (p = 0.0006) and relative to VU0364572-treated cocaine-experienced rats (p = 0.001).
Fig. 4. VU0364572 treatment four weeks before testing prevented cocaine-induced increases in extracellular mPFC dopamine and NAc glutamate in cocaine-experienced rats.

Rats treated with VU0364572 (VU’72) 4 weeks earlier showed blunted dopamine responses to cocaine in PFC (a–c) and NAc (d–f). Acute cocaine administration (15 mg/kg, arrow) caused increased extracellular dopamine levels in NAc in both cocaine-naive and cocaine-experienced (d, e) rats, measured by in vivo dual-probe microdialysis. The VU0364572 treatment also blunted glutamate (Glu) responses to cocaine in NAc (j–l), but had no effect on glutamate levels in mPFC (g–i). Acute cocaine caused increased glutamate levels in NAc in cocaine-naive and cocaine-experienced (j) rats. Overall increases in dopamine (c, f) and glutamate (i, l) levels are depicted as area under the curve of 0–100 min after cocaine injection; points represent individual rats. n = 6, *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle (Veh).
In the NAc, cocaine administration increased dopamine levels above baseline in both cocaine-naive and cocaine-experienced animals treated with vehicle (Fig. 4d, e; Supplementary Table S2E). VU0364572 treatment significantly blunted the cocaine-stimulated increase in extracellular NAc dopamine in both cocaine-naive [F(1,10) = 62.9, p < 0.0001] and cocaine-experienced [F(1,10) = 4.56, p = 0.03] rats, but with different efficacy. In the cocaine-naive animals, prior VU0364572 treatment completely blocked the dopamine increase, whereas the cocaine-experienced animals still showed a dopamine increase. Figure 4f shows overall %increase in dopamine over baseline, which was related to VU0364572 treatment and cocaine history (p < 0.006; Supplementary Table S2G). Cocaine-stimulated NAc dopamine levels were decreased in VU0364572-treated rats relative to vehicle-treated rats in both cocaine-naive (p < 0.0001) and cocaine-experienced (p = 0.049) animals.
Extracellular glutamate levels in the mPFC were significantly related to cocaine history [F(1,20) = 23.5, p = 0.0001], although the effect of acute cocaine administration did not reach statistical significance relative to baseline levels in either group (Fig. 4g, h). There was no significant effect of VU0364572 history or interaction. There was a statistical outlier (over two standard deviations above the mean) in the naive/VU0364572 group, depicted with a gray point in Fig. 4i, but analyzing the data without this outlier did not reveal a significant effect of VU0364572 history. Figure 4i shows overall %increase in glutamate over baseline, which was again related to only cocaine (p = 0.0002; Supplementary Table S2G). Cocaine-experienced rats had significantly lower cocaine-stimulated mPFC glutamate levels than cocaine-naive rats whether they were treated with saline (p = 0.007) or VU0364572 (p = 0.01).
Acute cocaine administration increased NAc glutamate levels in vehicle- and VU0364572-treated cocaine-naive rats and in vehicle-treated cocaine-experienced rats, but the increase was completely abolished in the VU0364572-treated cocaine-experienced rats (Fig. 4j; Supplementary Table S2E). Extracellular glutamate levels in the NAc were lower in rats previously treated with VU0364572 [F(1,20) = 7.39, p = 0.01], whereas effects of cocaine history and interactions were not significant (Fig. 4j; Supplementary Table S2F). Data were therefore analyzed post-hoc with cocaine-naive and cocaine-experienced animals pooled (see Fig. 4k for significant time points). Figure 4l shows overall %increase in glutamate over baseline, which was related to only VU0364572 treatment (p = 0.01; Supplementary Table S2G). VU0364572-treated rats had lower cocaine-stimulated NAc glutamate levels than naive and experienced rats combined (p = 0.02).
Dopamine uptake
To determine whether the large effects of VU0364572 treatment on extracellular dopamine levels were due to altered dopamine transporter function, we assessed dopamine uptake in striatal tissue from rats that received the same treatments as in the microdialysis study. Dopamine uptake was related to dopamine concentration [F(5,60) = 59.2, p < 0.0001] with no significant effect of cocaine or VU0364572 history (Fig. 5; Supplementary Table S2H). Vmax and KM did not differ significantly between groups. There was a non-significant apparent decrease in dopamine uptake in the cocaine-experienced vehicle-treated rats relative to cocaine-naive rats, consistent with previous investigations. The VU0364572-treated cocaine-experienced rats showed dopamine uptake indistinguishable from naive rats. Because of this possible effect of VU0364572 treatment in the cocaine-experienced group, the experiment was subsequently repeated in cocaine-experienced animals only (n = 4, data not shown). This showed the same trend, and overall, Vmax was 39% higher in VU0364572-treated cocaine-experienced rats than in vehicle controls (p = 0.08), with no effect of VU0364572 treatment in cocaine-naive rats (Vmax 101.8% of controls, p > 0.9).
Fig. 5. VU0364572 treatment had no effect on dopamine uptake in cocaine-naïve rats, and produced only a non-significant trend of decreased uptake in cocaine-experienced rats.

Saturation curve for dopamine uptake in synaptosomes from accumbens of (a) cocaine-naive and (b) cocaine-experienced rats previously treated with vehicle or VU0364572 (n = 4). Best-fit values ± s.e.m. of the maximal dopamine uptake capacity Vmax in fmol/min/µg and apparent affinity KM in µM are indicated for each group.
Discussion
We discovered large effects on cocaine self-administration behavior and cocaine-induced dopamine and glutamate outflow weeks after treatment with the M1 agonist VU0364572. Although we previously observed effects delayed by a few days [23, 26], similar long-lasting delayed or progressive effects of M1 agonists have not been reported previously. Although much remains to be elucidated about the mechanism underlying these unusual delayed effects, our findings are consistent with M1-dependent modulation in the addiction pathway that comprises the glutamatergic corticostriatal projections.
Cocaine self-administration behavior
Baseline cocaine self-administration dose-response functions was consistent with previous reports, both choice and single-reinforcer [23, 24, 32–34]. Acutely, VU0364572 produced small decreases in cocaine self-administration consistent with functional antagonism of the reinforcing effects of cocaine: VU0364572 decreased self-administration of low to moderate doses of cocaine but not high doses, with no effect on overall intake levels. This is similar to the effects of the M1/M4-preferring agonist xanomeline in the same assay [23] and consistent with attenuation of cocaine discrimination by M1 and M4 stimulation [22, 35]. In the course of the experiment, it became apparent that VU0364572 could suppress cocaine self-administration in a much more pronounced manner, days to weeks after dosing. At a low dose, VU0364572 produced moderate decreases in cocaine-taking for 1–2 days after treatment, reminiscent of xanomeline [23]. These effects were magnified and prolonged with a higher VU0364572 dose, which over 4 weeks caused a remarkable decrease in cocaine-taking behavior—half the rats stopped taking cocaine entirely. We observed no adverse effects and rats continued to press the lever reinforced with food, indicating a lack of motor impairment or stimulus control disruption. Importantly, the delayed effects differed qualitatively from acute effects by showing a reduction in cocaine intake with no evidence of increased intake at the higher cocaine doses. Rather, the delayed effect is similar to the effect of removing the cocaine syringe in this assay [34], consistent with VU0364572 gradually reducing cocaine’s reinforcing effects. Acute effects of VU0364572 on cocaine self-administration in a single-reinforcer assay also support this interpretation (Supplementary Fig. S6). Increased reinforcing effect of the food is unlikely to account for the effects, since we never observed increased food-reinforced operant behavior after M1 receptor stimulation [22, 26, 35], and others reported increased food-reinforced behavior after blocking M1 receptors [36].
Microdialysis, dopamine uptake, and suggested mechanism
Vehicle-treated control rats yielded findings consistent with previous reports: cocaine significantly increased mPFC dopamine in cocaine-experienced rats only, increased NAc dopamine and glutamate in both cocaine-naive and -experienced rats, and failed to consistently increase mPFC glutamate [37–41]. VU0364572 treatment 4 weeks before testing completely prevented cocaine-induced increases in extracellular mPFC dopamine and NAc glutamate in cocaine-experienced rats. Both effects are consistent with suppressed cocaine self-administration: several studies indicate that PFC dopamine, but not glutamate, has an important role in cocaine seeking and taking in cocaine-experienced rats, whereas in NAc, glutamate, but not dopamine signaling, appears to mediate these behaviors [37, 42–47]. The VTA is the main source of dopamine in both mPFC and NAc [48], but VU0364572 did not affect dopamine response equally in both regions, perhaps making modulation in VTA dopamine neurons less likely. Further, M1 receptors are not expressed on VTA dopamine neurons [16, 49]. Taken together, we propose that M1-mediated modulation of corticostriatal glutamatergic pathways best explains our findings. Acutely, M1 receptor stimulation was shown to postsynaptically increase NMDA but not AMPA receptor-mediated responses in MSNs, and to modulate PFC and corticostriatal neuroplasticity [28, 50–54]. A balancing of relative AMPA/NMDA transmission, which is shifted following cocaine exposure [2, 46, 55], could be a way in which M1 stimulation opposed cocaine’s effect, although it is unclear how those acute M1 receptor-dependent actions and our delayed effects are related.
Cocaine-naive, VU0364572-treated rats showed no cocaine-induced NAc dopamine outflow. The fact that VU0364572 also blunted the cocaine-induced dopamine response in cocaine-naive rats indicates that at least some effects reflect modulation of the circuitry independent of cocaine-induced plasticity. These results extend our finding that M1/M4 receptor stimulation facilitated extinction of cocaine seeking also when not paired with cocaine exposure or extinction learning [26], although in those studies all animals were cocaine-experienced. It would be interesting to test whether M1 stimulation reduces subsequent acquisition of cocaine self-administration in drug-naive animals.
The large reductions in cocaine-induced dopamine outflow in VU0364572-treated rats cannot be attributed to upregulation of cell-surface DAT, as VU0364572 treatment had no effect on dopamine uptake in cocaine-naive rats, and only a non-significant trend in cocaine-experienced rats. Our results in the vehicle-treated rats are consistent with preclinical [56] and clinical [57] studies showing modest decreases in DAT availability weeks after cocaine self-administration or passive exposure, an effect that is variable in humans and likely depends on the duration of abstinence [58–61]. Although inconclusive, VU0364572 treatment may have prevented/attenuated this cocaine-induced change in DAT function; testing at intervals when cocaine-induced changes in DAT availability are more robust is needed to answer this question.
Limitations and future directions
Due to probe size, our microdialysis data cannot distinguish between effects in the prelimbic vs. infralimbic mPFC, and between the NAc core, NAc shell, or medioventral caudate putamen, although probe tips were located in the NAc core. Because prelimbic mPFC to NAc core projections and infralimbic mPFC to NAc shell projections are thought to modulate cocaine seeking differentially [2, 3], it would be interesting to extend our studies with more spatially focused methodologies. There are differences between effects of self-administered and experimenter-administered cocaine, the rats self-administering cocaine did not have a 4-week forced abstinence period, and VU0364572 dosing was a single dose vs. brief repeated daily administration over five days. This precludes a direct comparison between the microdialysis and uptake experiments and the behavioral experiments, limiting what conclusions can be drawn about mechanism. Conversely, the demonstration that M1 receptor stimulation opposed the effects of cocaine regardless of prolonged abstinence, extinction training, or mode and route of cocaine administration strengthens our confidence in the robustness of the effects and the potential clinical utility of the approach. Our findings may not extend to females, juveniles, awake animals, other species, and so forth.
We observed no adverse effects of VU0364572 at the doses tested. Excessive M1 stimulation may cause adverse effects including nausea, cognitive impairment, or pro-convulsant effects, which could be worrisome clinically especially when combined with cocaine use [62–67]. However, adverse effects may not be a great concern given that we obtained pronounced effects at relatively low doses of VU0364572, which was tolerated in rats at 10, 32, and even 56 mg/kg [28, 68, 69]. Although ongoing studies do not suggest VU0364572 has abuse potential (lack of conditioned place preference), this should be tested thoroughly. M1 receptor stimulation has shown promise improving cognitive performance in rodents and, in that context, it was suggested that M1 positive allosteric modulators (PAMs) may retain efficacy while lacking cholinergic toxicity [65, 70]. Whether pure M1-PAMs can replicate the current effects of the bitopic (allosteric and orthosteric) PAM/agonist VU0364572 [27] is worth exploring. It is possible that VU0364572 has a particularly useful pharmacological profile, as allosteric M1 agonists differ in activation of specific signaling pathways [66, 68, 71]. Nevertheless, we have observed acute “anti-cocaine” effects using several structurally distinct M1 agonists/PAMs, suggesting a common M1-dependent action [23, 26, 35].
Finally, corticostriatal glutamatergic transmission is also modulated (generally inhibited) by stimulation of presynaptic muscarinic M4 receptors in NAc [72, 73], and we have observed additive or synergistic effects on cocaine-induced behaviors by stimulating both M1 and M4 receptors, relative to either alone [22, 26, 35]. Thus, a combination of highly selective M1 and M4 receptor agonists or PAMs may show additional benefits in modulating addiction-related effects of cocaine.
Conclusions
These delayed effects of the muscarinic M1 receptor agonist may seem unusual in the context of typical preclinical pharmacology findings. However, they are in line with the clinically observed therapeutic effects of some types of medications in psychiatry, such as antidepressant and antipsychotic drugs. Viewed in that perspective, we suggest that any medication capable of ameliorating neurological maladaptations incurred in the development of a drug addiction would perhaps be unlikely to show its full effect acutely. Conversely, “classic” short-acting drugs require excellent adherence to treatment over time to be effective at minimizing relapse. The present findings not only reinforce our interest in M1 receptor agonists/PAMs as potential pharmacotherapies in cocaine addiction (and perhaps other addictions), but also suggest that it may be time to revisit the typical acute-treatment approach to evaluating new targets, focusing instead on longer windows of evaluation.
Funding and disclosure
The authors declare no competing interests.
Supplementary information
Acknowledgements
We thank professors P. Jeffery Conn and Craig W. Lindsley (Vanderbilt University) for the gift of VU0364572 for these studies. We thank Christopher Adam, Benoît Niclou, Kevin Stoll (behavior), and Saiy Kiasari, Dr. Gitta Wörtwein, Anne-Marie Paulsen (microdialysis) for technical assistance. This research was supported by National Institutes of Health National Institute on Drug Abuse (NIH-NIDA) grant DA027825 (MT), Independent Research Fund Denmark grant 8020-00110B (MT), the Mental health services in the Capital Region of Denmark (Anders Fink-Jensen) and Research Foundation Mental Health Services in the Capital Region of Denmark (MT), and the Ivan Nielsen Foundation (MT). Sponsors had no role in study design, data interpretation, or the decision to publish.
Author contributions
MT conceptualized the studies. MT, PW, and KLJ designed the experiments, analyzed the data, and interpreted the data. PW conducted the microdialysis studies, KLJ conducted the dopamine uptake experiments. MT directed the self-administration experiments. MT wrote the manuscript with assistance from KLJ and PW.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Pia Weikop, Kathrine L. Jensen
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-020-0684-1).
References
- 1.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3:760–73. doi: 10.1016/S2215-0366(16)00104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf ME. Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci. 2016;17:351–65. doi: 10.1038/nrn.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–72. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 4.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–9. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 5.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 6.Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith AC, et al. The nucleus accumbens: mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharm Rev. 2016;68:816–71. doi: 10.1124/pr.116.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchta WC, Riegel AC. Chronic cocaine disrupts mesocortical learning mechanisms. Brain Res. 2015;1628(Pt A):88–103. doi: 10.1016/j.brainres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–69. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994;14:3969–84. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atallah HE, McCool AD, Howe MW, Graybiel AM. Neurons in the ventral striatum exhibit cell-type-specific representations of outcome during learning. Neuron. 2014;82:1145–56. doi: 10.1016/j.neuron.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamanaka K, Hori Y, Minamimoto T, Yamada H, Matsumoto N, Enomoto K, et al. Roles of centromedian parafascicular nuclei of thalamus and cholinergic interneurons in the dorsal striatum in associative learning of environmental events. J Neural Transm. 2018;125:501–13. doi: 10.1007/s00702-017-1713-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12:3591–600. doi: 10.1523/JNEUROSCI.12-09-03591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14(5 Pt 2):3351–63. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA. 1996;93:13541–6. doi: 10.1073/pnas.93.24.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–26. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci USA. 1990;87:7050–4. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Z, Flores-Hernandez J, Surmeier DJ. Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience. 2001;103:1017–24. doi: 10.1016/s0306-4522(01)00039-2. [DOI] [PubMed] [Google Scholar]
- 18.Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg JA, Ding JB, Surmeier DJ. Muscarinic modulation of striatal function and circuitry. Handb Exp Pharmacol. 2012;208:223–41. doi: 10.1007/978-3-642-23274-9_10. [DOI] [PubMed] [Google Scholar]
- 20.Oldenburg IA, Ding JB. Cholinergic modulation of synaptic integration and dendritic excitability in the striatum. Curr Opin Neurobiol. 2011;21:425–32. doi: 10.1016/j.conb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, et al. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–66. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- 22.Thomsen M, Conn PJ, Lindsley C, Wess J, Boon JY, Fulton BS, et al. Attenuation of cocaine’s reinforcing and discriminative stimulus effects via muscarinic M1 acetylcholine receptor stimulation. J Pharm Exp Ther. 2010;332:959–69. doi: 10.1124/jpet.109.162057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomsen M, Fulton BS, Caine SB. Acute and chronic effects of the M1/M4-preferring muscarinic agonist xanomeline on cocaine vs. food choice in rats. Psychopharmacology. 2014;231:469–79. doi: 10.1007/s00213-013-3256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomsen M, Barrett AC, Butler P, Negus SS, Caine SB. Effects of acute and chronic treatments with dopamine D2 and D3 receptor ligands on cocaine versus food choice in rats. J Pharm Exp Ther. 2017;362:161–76. doi: 10.1124/jpet.117.241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomsen M, Fink-Jensen A, Woldbye DP, Wortwein G, Sager TN, Holm R, et al. Effects of acute and chronic aripiprazole treatment on choice between cocaine self-administration and food under a concurrent schedule of reinforcement in rats. Psychopharmacology. 2008;201:43–53. doi: 10.1007/s00213-008-1245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoll K, Hart R, Lindsley CW, Thomsen M. Effects of muscarinic M1 and M4 acetylcholine receptor stimulation on extinction and reinstatement of cocaine seeking in male mice, independent of extinction learning. Psychopharmacology. 2018;235:815–27. doi: 10.1007/s00213-017-4797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Digby GJ, Utley TJ, Lamsal A, Sevel C, Sheffler DJ, Lebois EP, et al. Chemical modification of the M(1) agonist VU0364572 reveals molecular switches in pharmacology and a bitopic binding mode. ACS Chem Neurosci. 2012;3:1025–36. doi: 10.1021/cn300103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lebois EP, Digby GJ, Sheffler DJ, Melancon BJ, Tarr JC, Cho HP, et al. Development of a highly selective, orally bioavailable and CNS penetrant M1 agonist derived from the MLPCN probe ML071. Bioorg Med Chem Lett. 2011;21:6451–5. doi: 10.1016/j.bmcl.2011.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brindisi M, Butini S, Franceschini S, Brogi S, Trotta F, Ros S, et al. Targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors for developing effective antipsychotics: synthesis, biological characterization, and behavioral studies. J Med Chem. 2014;57:9578–97. doi: 10.1021/jm501119j. [DOI] [PubMed] [Google Scholar]
- 30.Jensen KL, Runegaard AH, Weikop P, Gether U, Rickhag M. Assessment of dopaminergic homeostasis in mice by use of high-performance liquid chromatography analysis and synaptosomal dopamine uptake. J Vis Exp. 2017. [DOI] [PMC free article] [PubMed]
- 31.Runegaard AH, Jensen KL, Fitzpatrick CM, Dencker D, Weikop P, Gether U, et al. Preserved dopaminergic homeostasis and dopamine-related behaviour in hemizygous TH-Cre mice. Eur J Neurosci. 2017;45:121–28. doi: 10.1111/ejn.13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett AC, Miller JR, Dohrmann JM, Caine SB. Effects of dopamine indirect agonists and selective D1-like and D2-like agonists and antagonists on cocaine self-administration and food maintained responding in rats. Neuropharmacology. 2004;47(Suppl 1):256–73. doi: 10.1016/j.neuropharm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Caine SB, Bowen CA, Yu G, Zuzga D, Negus SS, Mello NK. Effect of gonadectomy and gonadal hormone replacement on cocaine self-administration in female and male rats. Neuropsychopharmacology. 2004;29:929–42. doi: 10.1038/sj.npp.1300387. [DOI] [PubMed] [Google Scholar]
- 34.Thomsen M, Barrett AC, Negus SS, Caine SB. Cocaine versus food choice procedure in rats: environmental manipulations and effects of amphetamine. J Exp Anal Behav. 2013;99:211–33. doi: 10.1002/jeab.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomsen M, Lindsley CW, Conn PJ, Wessell JE, Fulton BS, Wess J, et al. Contribution of both M1 and M4 receptors to muscarinic agonist-mediated attenuation of the cocaine discriminative stimulus in mice. Psychopharmacology. 2012;220:673–85. doi: 10.1007/s00213-011-2516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hailwood JM, Heath CJ, Phillips BU, Robbins TW, Saksida LM, Bussey TJ. Blockade of muscarinic acetylcholine receptors facilitates motivated behaviour and rescues a model of antipsychotic-induced amotivation. Neuropsychopharmacology. 2019;44:1068–75. doi: 10.1038/s41386-018-0281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hotsenpiller G, Wolf ME. Extracellular glutamate levels in prefrontal cortex during the expression of associative responses to cocaine related stimuli. Neuropharmacology. 2002;43:1218–29. doi: 10.1016/s0028-3908(02)00308-8. [DOI] [PubMed] [Google Scholar]
- 38.Moghaddam B, Bunney BS. Differential effect of cocaine on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens: comparison to amphetamine. Synapse. 1989;4:156–61. doi: 10.1002/syn.890040209. [DOI] [PubMed] [Google Scholar]
- 39.Williams JM, Steketee JD. Cocaine increases medial prefrontal cortical glutamate overflow in cocaine-sensitized rats: a time course study. Eur J Neurosci. 2004;20:1639–46. doi: 10.1111/j.1460-9568.2004.03618.x. [DOI] [PubMed] [Google Scholar]
- 40.Williams JM, Steketee JD. Time-dependent effects of repeated cocaine administration on dopamine transmission in the medial prefrontal cortex. Neuropharmacology. 2005;48:51–61. doi: 10.1016/j.neuropharm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Wu WR, Li N, Sorg BA. Prolonged effects of repeated cocaine on medial prefrontal cortex dopamine response to cocaine and a stressful predatory odor challenge in rats. Brain Res. 2003;991:232–9. doi: 10.1016/j.brainres.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–50. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 44.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–60. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–7. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, et al. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci. 2002;22:2916–25. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suto N, Elmer GI, Wang B, You ZB, Wise RA. Bidirectional modulation of cocaine expectancy by phasic glutamate fluctuations in the nucleus accumbens. J Neurosci. 2013;33:9050–5. doi: 10.1523/JNEUROSCI.0503-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–53. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- 49.Vilaro MT, Palacios JM, Mengod G. Localization of m5 muscarinic receptor mRNA in rat brain examined by in situ hybridization histochemistry. Neurosci Lett. 1990;114:154–9. doi: 10.1016/0304-3940(90)90064-g. [DOI] [PubMed] [Google Scholar]
- 50.Calabresi P, Centonze D, Gubellini P, Bernardi G. Activation of M1-like muscarinic receptors is required for the induction of corticostriatal LTP. Neuropharmacology. 1999;38:323–6. doi: 10.1016/s0028-3908(98)00199-3. [DOI] [PubMed] [Google Scholar]
- 51.Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Endogenous ACh enhances striatal NMDA-responses via M1-like muscarinic receptors and PKC activation. Eur J Neurosci. 1998;10:2887–95. doi: 10.1111/j.1460-9568.1998.00294.x. [DOI] [PubMed] [Google Scholar]
- 52.Ghoshal A, Moran SP, Dickerson JW, Joffe ME, Grueter BA, Xiang Z, et al. Role of mGlu5Receptors and inhibitory neurotransmission in M1 dependent muscarinic LTD in the prefrontal cortex: implications in schizophrenia. ACS Chem. Neurosci. 2017;8:2254–65. doi: 10.1021/acschemneuro.7b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, et al. Potentiation of M1 muscarinic receptor reverses plasticity deficits and negative and cognitive symptoms in a schizophrenia mouse model. Neuropsychopharmacology. 2016;41:598–610. doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–52. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–21. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson JM, Nobrega JN, Carroll ME, Niznik HB, Shannak K, Lac ST, et al. Heterogeneous subregional binding patterns of 3H-WIN 35,428 and 3H-GBR 12,935 are differentially regulated by chronic cocaine self-administration. J Neurosci. 1994;14(5 Pt 2):2966–79. doi: 10.1523/JNEUROSCI.14-05-02966.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Proebstl L, Kamp F, Manz K, Krause D, Adorjan K, Pogarell O, et al. Effects of stimulant drug use on the dopaminergic system: a systematic review and meta-analysis of in vivo neuroimaging studies. Eur Psychiatry. 2019;59:15–24. doi: 10.1016/j.eurpsy.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Ashok AH, Mizuno Y, Volkow ND, Howes OD. Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74:511–19. doi: 10.1001/jamapsychiatry.2017.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–69. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- 60.Little KY, McLaughlin DP, Zhang L, McFinton PR, Dalack GW, Cook EH, Jr., et al. Brain dopamine transporter messenger RNA and binding sites in cocaine users: a postmortem study. Arch Gen Psychiatry. 1998;55:793–9. doi: 10.1001/archpsyc.55.9.793. [DOI] [PubMed] [Google Scholar]
- 61.Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemannn R, Gatley SJ, et al. Cocaine uptake is decreased in the brain of detoxified cocaine abusers. Neuropsychopharmacology. 1996;14:159–68. doi: 10.1016/0893-133X(95)00073-M. [DOI] [PubMed] [Google Scholar]
- 62.Alt A, Pendri A, Bertekap RL, Jr, Li G, Benitex Y, Nophsker M, et al. Evidence for classical cholinergic toxicity associated with selective activation of M1 muscarinic receptors. J Pharm Exp Ther. 2016;356:293–304. doi: 10.1124/jpet.115.226910. [DOI] [PubMed] [Google Scholar]
- 63.Davoren JE, Garnsey M, Pettersen B, Brodney MA, Edgerton JR, Fortin JP, et al. Design and synthesis of gamma- and delta-Lactam M1 positive allosteric modulators (PAMs): convulsion and cholinergic toxicity of an M1-selective PAM with weak agonist activity. J Med Chem. 2017;60:6649–63. doi: 10.1021/acs.jmedchem.7b00597. [DOI] [PubMed] [Google Scholar]
- 64.Davoren JE, Lee CW, Garnsey M, Brodney MA, Cordes J, Dlugolenski K, et al. Discovery of the potent and selective M1 PAM-agonist N-[(3R,4S)-3-hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)ben zyl]pyridine-2-carboxamide (PF-06767832): evaluation of efficacy and cholinergic side effects. J Med Chem. 2016;59:6313–28. doi: 10.1021/acs.jmedchem.6b00544. [DOI] [PubMed] [Google Scholar]
- 65.Moran SP, Dickerson JW, Cho HP, Xiang Z, Maksymetz J, Remke DH, et al. M1-positive allosteric modulators lacking agonist activity provide the optimal profile for enhancing cognition. Neuropsychopharmacology. 2018;43:1763–71. doi: 10.1038/s41386-018-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rook JM, Abe M, Cho HP, Nance KD, Luscombe VB, Adams JJ, et al. Diverse effects on M1 signaling and adverse effect liability within a series of M1 Ago-PAMs. ACS Chem Neurosci. 2017;8:866–83. doi: 10.1021/acschemneuro.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vijayraghavan S, Major AJ, Everling S. Muscarinic M1 receptor overstimulation disrupts working memory activity for rules in primate prefrontal cortex. Neuron. 2018;98:1256–68. doi: 10.1016/j.neuron.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 68.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32:8532–44. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lebois EP, Schroeder JP, Esparza TJ, Bridges TM, Lindsley CW, Conn PJ, et al. Disease-modifying effects of M1 muscarinic acetylcholine receptor activation in an Alzheimer’s disease mouse model. ACS Chem Neurosci. 2017;8:1177–87. doi: 10.1021/acschemneuro.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rook JM, Bertron JL, Cho HP, Garcia-Barrantes PM, Moran SP, Maksymetz JT, et al. A novel M1 PAM VU0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem Neurosci. 2018;9:2274–85. doi: 10.1021/acschemneuro.8b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas RL, Langmead CJ, Wood MD, Challiss RA. Contrasting effects of allosteric and orthosteric agonists on m1 muscarinic acetylcholine receptor internalization and down-regulation. J Pharm Exp Ther. 2009;331:1086–95. doi: 10.1124/jpet.109.160242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pancani T, Bolarinwa C, Smith Y, Lindsley CW, Conn PJ, Xiang Z. M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses. ACS Chem Neurosci. 2014;5:318–24. doi: 10.1021/cn500003z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.